
Abstract
In chronic lymphocytic leukemia (CLL), the role of complex karyotype (CK) for prognostic stratification remains a topic of debate, and the impact of specific cytogenetic abnormalities is still unclear. This study aims to investigate the clinical and biological features of CLL with t(14;19)(q32;q13) (tCLL) involving the BCL3 gene. Patients with tCLL were younger and more commonly presented unmutated IGHV gene, subset #8 stereotypy, trisomy of chromosome 12, and complex karyotype than other patients without t(14;19) (oCLL). The presence of t(14;19) was associated with a shorter time to treatment and overall survival compared to oCLL. Gene expression analysis revealed a unique transcriptome profile in tCLL, characterized by the upregulation of BCL3 and the activation of B-cell receptor, PI3K-Akt. Conversely, apoptosis-related pathways were suppressed in tCLL. While the BTK gene was upregulated, the BCL2L11 gene, coding for the pro-apoptotic protein BIM, was downregulated. Notably, patients with tCLL were characterized by a trend (p = 0.058) for a longer time to the next treatment with BTK inhibitors (BTKi) compared to those treated with a venetoclax-based (Ven-based) regimen. We underscore the adverse outcomes of tCLL, its distinct molecular features and gene expression patterns. Therefore, our data suggest that identifying tCLL could help tailor therapeutic approaches.

Similar content being viewed by others

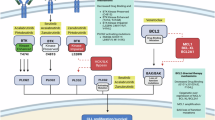

Background
Stimulated chromosome banding analysis (CBA) has recently been included in the biological criteria for stratification of patients with chronic lymphocytic leukemia (CLL) in some guidelines [1, 2] together with immunoglobulin heavy variable (IGHV) gene mutational analysis and TP53 abnormalities, i.e., 17p13 deletion assessed by interphase fluorescence in situ hybridization (FISH) and TP53 mutation. CBA can identify chromosome abnormalities that would have been missed by FISH [3,4,5,6]. A complex karyotype (CK), defined as the presence of at least 3 chromosomal abnormalities, can be identified in almost 20% of patients at the time of CLL diagnosis and up to 40% among relapsed/refractory cases [3, 7,8,9]. CK has been associated with TP53 abnormalities, chemo-refractoriness, early relapse, and a higher risk of Richter transformation [3,4,5,6,7]. At variance with other hematological disorders, a prognostic value has been associated only with the presence of at least 5 chromosomal abnormalities in the absence of TP53 aberrations (deletion of 17p and/or TP53 mutations). These subgroups of patients, generally defined as high-CK, had the worst outcome, even if managed with targeted therapies [4, 7, 8, 10,11,12]. Despite the reported data above, the role of CK remains a subject of debate.
Previous studies have reported that t(14;19)(q32;q13), which causes the relocalization of the B-cell leukemia/lymphoma 3 (BCL3) gene near regulatory elements of the immunoglobulin heavy chain (IGH), leads to BCL3 overexpression [13,14,15]. It is a very rare cytogenetic abnormality found in <1% of CLL and other B-cell malignancies. Some studies have indicated that CLLs with t(14;19) (tCLL) have an adverse prognosis [13,14,15,16,17,18,19,20]; however, these findings were not confirmed by others [21].
The function of BCL3 is not entirely known [22, 23]. BCL3 is an atypical member of the regulatory IκB family, which, unlike classical members, is a nuclear transcription cofactor. BCL3 can bind to NF-κB p50 and p52 homodimers, repressing or activating a subset of NF-κB-regulated genes. It can also inhibit the ubiquitination and subsequent proteasomal degradation of p50 homodimers, thereby stabilizing their DNA binding. However, it can also recruit a corepressor, inhibiting the transcription [22,23,24,25].
In this study, we aimed to define the clinical and biological features of tCLL, utilizing a large cohort of 101 patients from the European Research Initiative on CLL (ERIC) network, including 66 patients who were also treated with targeted agents. Furthermore, by sequencing the transcriptome of a sizable patient cohort, we aimed to characterize the expression profile of tCLL and gain a deeper understanding of the biological basis of this CLL subtype.
Materials and methods
Study design
The inclusion criteria for this study were a diagnosis of CLL according to the 2018 International Workshop on Chronic Lymphocytic Leukemia (iwCLL) criteria, age >18 years, CBA performed within one year from diagnosis, presence of t(14;19)(q32;q13) and Matutes score of 3 or above. Patients with a Matutes score of 3 were defined as atypical CLL. The data included in the comparative analysis were gender, age, Binet stage, need for treatment, cytogenetics detected by fluorescence in situ hybridization (FISH), IGHV gene mutational status, stereotyped B cell receptor (BCR), and TP53 abnormalities, including deletions of chromosome 17p and/or TP53 mutations [26]. Patients were treated according to the iwCLL guidelines. The primary endpoints were the time to first treatment (TTFT) and overall survival (OS) in patients with the t(14;19) translocation. The secondary endpoints included the time to the next treatment (TTNT) and a comparison of clinical-biological variables between tCLL and 540 karyotyped CLL patients without t(14;19) (oCLL) from the University of Padova. Patients were treated with fludarabine-cyclophosphamide-rituximab (FCR) / bendamustine-rituximab (BR), continuous BTK inhibitors (BTKi), and a time-limited venetoclax-based therapy (Ven-based). This study was approved by the local research ethics committee (4430/AO/18), and informed consent was obtained from all patients. Statistical analyses were done as reported in the Supplementary Materials.
Biological markers
IGHV gene mutational status, interphase FISH, and TP53 mutations were assessed in each of the collaborating centers following current ERIC recommendations as described in the Supplementary Materials.
RNA-seq sequencing
For RNA sequencing (RNA-seq), RNA was extracted from 106 leukemic B cells (RIN > 7) from 25 CLL patients with t(14;19), plus 22 CLL patients with normal karyotype/FISH or trisomy 12 ( + 12) and from mature B cells from 5 age-matched healthy individuals and sequenced by Illumina Novaseq (150 bp per end, 120 × 106 reads/sample). Four additional mature B-cell samples from healthy donor RNA-seq datasets were obtained from a previous study [27]. The expression of the BCL3 protein was assessed by Western blot analysis, as described in the Supplementary Methods. The expression of key selected genes or protein levels was validated by RT-qPCR or flow cytometry, as described in the supplementary methods. Primer sequences are listed in Table S1.
Bioinformatics analysis
RNA-seq data was analyzed with CirCompara2 v0.1.2.1 [28,29,30,31,32] using the GRCh38 human genome reference and ENSEMBL gene annotation v108. The differential expression was assessed using DESeq2 v1.40.1 [33], including removing batch effects using the sva algorithm v3.48.0 [34] and considering significant differences with Benjamini-Hochberg adjusted p values (p-adj) ≤ 0.01.
Gene set and pathway enrichment analysis were performed through the clusterProfiler v4.8.1 Bioconductor package. All analyses were performed with custom scripts in an R v4.3.0 environment. Results of gene set enrichment analysis [35,36,37,38] were considered significant for p-adj ≤ 0.05. Topology-based analyses were performed using the SPIA [39] package. The statistical significance of BCL3 target gene enrichment among overexpressed genes in tCLL was assessed using a hypergeometric test performed with R’s phyper function, utilizing parameters derived from the TFLink database and our gene expression data [40].
Results
Patient characteristics
We enrolled 101 CLL patients with t(14;19) (tCLL) from 23 centers (Table 1; Supplementary Fig. S1): 59% were male, and the median age was 57 ± 12 years. Of note, at CLL diagnosis, 30% of the patients were younger than 50 years, 59% were at Binet stage A, 28% at stage B, and 13% at stage C. IGHV mutational status was available for 85 patients, of which 91% carried unmutated IGHV genes (31% presented IGHV4-39). Patients were diagnosed between 1994 and 2022. The Sanger sequencing of IGHV analysis was available for 38 patients, and the stereotyped BCR subset #8 was found in 12 (31.6%) cases. FISH data were available from 97 patients: 5% harbored 13q-, 8% 11q-, 57% +12, 9% 17p, and 20% none of these abnormalities detected. TP53 abnormalities were detected in 14% of the patients (9 deletions and 5 mutations). Regarding karyotype, 52% displayed a CK3 (11 with and 36 without TP53 abnormalities, with 6 missing data), and 20% a CK5 (8 with and 10 without TP53 abnormalities, with 2 missing data). None had multiple trisomies. It is worth noting that the features of patients with tCLL were skewed compared to those with oCLL (Supplementary Table S2). Patients with tCLL were younger and more commonly showed unmutated IGHV genes, as well as +12 and BCR subset #8 (Supplementary Tables S2, S3).
The t(14;19) translocation has a negative prognostic impact
After a median follow-up of 6.92 years, 92% of the patients needed at least one treatment (median 2, range 1–11), 13% developed Richter transformation, and 43% died (23% infections, 12% CLL progression, 19% Richter transformation, 12% other causes, 26% unknown).
The time to first treatment (TTFT) in tCLL patients was shorter than in oCLL patients, with a median TTFT of 1.92 years and 6.94 years, respectively (p < 0.0001). After 7 years of follow-up, 97% and 50% of patients with and without t(14;19) started a second line of treatment, respectively (HR 3.55, 95% CI [2.46–5.12], Fig. 1A). The median overall survival (OS) was 12.6 years in tCLL, whereas it was not reached in oCLL (p < 0.0001). The 7-year OS was 66% and 97% for tCLL and oCLL, respectively (HR 2.53, 95% CI [1.59–4.03], Fig. 1B). The same differences were confirmed also when focusing only on patients at Binet A stage (60 tCLL and 407 oCLL) and with unmutated IGHV gene (75 tCLL and 309 oCLL), with the presence of t(14;19) being associated with a worse outcome (TTFT, HR 4.27, 95% CI [2.44–7.46], p < 0.0001; OS 2.21, HR 95% CI [1.22–4.01], p = 0.0008; Fig. 1C, D; TTFT, HR 2.01, 95% CI [1.48–3.00], p < 0.0001; OS 1.70, HR 95% CI [1.06–2.71], p = 0.0136; Fig. 1E, F).

The Kaplan-Meier curves of (A) time to first treatment and B overall survival of patients with t(14;19) (n = 101, tCLL) and other karyotyped CLL without t(14;19) (n = 450, oCLL), in patients at Binet A stage at diagnosis (C, D) and harboring unmutated IGHV genes (U-IGHV, E, F).
In univariate analysis, none of the variables assessed (age >65 years, male gender, U-IGHV, BcR subset 8, +12, TP53 abn, CK, CK5) predicted a shorter TTFT or OS in patients with t(14;19) (Supplementary Table 9).
To better understand the negative prognostic value associated with t(14;19), we compared the TTFT and OS with those of oCLL patients who had well-known negative prognostic markers such as TP53 abnormalities, CK, and CK5 [5]. Patients with t(14;19) and CK or +12 had a worse outcome compared to patients with CK or +12 without t(14;19); instead, their outcomes are superimposable to those of patients with TP53 abnormalities or CK5, despite being infrequent in patients with tCLL (Supplementary Figure S2).
The predictive value of t(14;19)
We also assessed whether t(14;19) influenced the response to treatment and compared the TTNT after different therapies of tCLL and oCLL. Among the 93 tCLLs requiring treatment, 33 (36%) received chemoimmunotherapy with FCR or BR as first-line therapy, 15 (16%) a BTKi (11 ibrutinib, 2 acalabrutinib, 2 zanubrutinib), 6 Ven-based therapy, and 39 other therapies, such as single-agent chemotherapy or monoclonal antibody. After frontline therapy with FCR/BR, the median TTNT was 3.98 and 5.48 for tCLL and oCLL (p = 0.0003, HR 2.47, 95% CI [1.38–4.44]; Fig. 2A). After 3 years, 46% of tCLLs started a second line of therapy. Instead, only 28% of oCLLs required further treatment (p < 0.001, Chi-square and p = 0.0003, Log-rank test). In addition, as shown in Fig. 2B, after 3 years from the start of Ven-based therapy, 33% of tCLLs needed further treatment as opposed to no need for treatment of oCLLs (p = 0.0235, HR 57, 95% CI [2.3–134]). However, the TTNT was not statistically different for patients managed with BTKi as the first-line therapy (p = 0.8340, HR 2.47, 95% CI [1.37–4.44]). At 3 years, none of the patients with tCLL and 88% of those with oCLL treated with BTKi needed further treatment (Fig. 2C). One patient received CAR T-cells and underwent six allogeneic stem cell transplants (Supplementary Results).

The Kaplan-Meier curves of time to the next treatment of patients treated with t(14;19) (tCLL) and other CLL without t(14;19) (oCLL) who received (A) fludarabine-cyclophosphamide-rituximab (FCR) / bendamustine-rituximab (BR) (n = 32 and n = 107, respectively), B venetoclax-based therapy (n = 6 and n = 39) and C BTK inhibitor (n = 14 and n = 180) as frontline therapy.
The distinct gene expression profile of CLL harboring t(14;19) points to the activation of proliferative pathways and suppression of cell death
To investigate the molecular characteristics of tCLL, we compared the gene expression profiles of 25 tCLL patients with those of 22 oCLL cases and B cells from 9 healthy donors by sequencing their transcriptomes at high depth with Illumina technology (RNA-seq). The clinical and biological features of the 25 sequenced tCLL patients and their outcomes were similar to those of the entire cohort of 101 patients (Supplementary Table S3; Supplementary Fig. S3A).
Unsupervised gene expression analysis (Fig. 3A) highlighted distinct profiles between CLL and normal B cells. Additionally, it revealed distinct signatures between tCLL and oCLL samples, with consistent expression patterns among patients within the same group.

A Dimensionality reduction by principal component analysis (PCA) performed on gene expression estimates. t(14;19) CLL patients (red crosses), control CLL (yellow triangles), and healthy donor B cell (blue circles) samples are displayed according to the first two principal components (PC1, horizontal axis; PC2, vertical axis) explaining the expression variability of the data (31.6% and 15.3%, respectively). B Volcano plot of the gene expression differences computed between t(14;19) CLL (tCLL) and other CLL without t(14;19) (oCLL). Dots and labels are colored according to the gene expression rank in tCLL (100% for the most expressed, 0% for the least expressed). C Western blotting analysis of Bcl-3 protein in tCLL and oCLL. The histogram in the bottom panel shows the densitometric analysis of Bcl-3/Actin, normalized to normal B cells, expressed as arbitrary units (A.I.). Expression levels, quantified by RNA-seq and RT-qPCR (DDCt method; GAPDH used as reference gene; Mean ± SD shown; A.U., Arbitrary Units) of BTK (D, E) and BCL2L11 (F, G) in patients with tCLL and oCLL. H Significant gene sets resulted from gene set enrichment analysis on the MSigDb hallmarks. Upward and downward arrows represent positive and negative normalized enrichment scores (NES), respectively, colored according to the magnitude of the NES (red indicates high positive, and blue indicates low negative). The arrow size indicates the statistical significance of the enrichment. I Heatmap of differentially expressed genes (rows) between t(14;19) CLL (tCLL), other CLL without t(14;19) (oCLL) samples, and B cells (columns). Pink and green cells represent gene-scaled expression, higher and lower than the gene mean of B cells, respectively. Columns are clustered according to the relative expression; rows are grouped according to differential expression significance, considering the three comparisons oCLL vs. B cells, tCLL vs. B cells, and tCLL vs. oCLL (dots in the panel mid sections indicate the significant comparisons). For each gene set, the enriched pathways are shown on the left side of the panel.
By comparing tCLL with oCLL (Supplementary Results), we confirmed the upregulation of the BCL3 mRNA (LFC = 0.9, p-adjusted <0.001; Fig. 3B; Table 2; Supplementary Fig. S3B), as expected from the t(14;19)(q32;q13) translocation [15], and observed an enrichment of its targets among the overexpressed genes (p < 0.001; Supplementary Results). This finding was supported by the observed overexpression of the BCL3 protein, as confirmed through Western blot analysis (Fig. 3C; Supplementary Results).
Noteworthy was the significantly higher expression of the CD79B and BTK genes in tCLL (Fig. 3B; Table 2; Supplementary Fig. S3B, Supplementary Fig. S6A), which are key elements of the BCR pathway. Among the differentially expressed immune checkpoint genes, the expression of PD-L1 and TIGIT was decreased, while LAG3 was highly expressed in tCLL (Fig. 3B, Supplementary Fig. S4, Supplementary Fig. S6B). Genes with high abundance in oCLL that were dramatically downregulated in tCLL included COL18A1, IGF2BP3, and LRRC32 (Fig. 3B). We also observed downregulation of the BCL2L11 gene (Table 2; Fig. 3B, D–G; Supplementary Figs. S3, S5A), which codes for the pro-apoptotic protein Bim.
Gene set enrichment and pathway analysis revealed that the oxidative phosphorylation (OXPHOS), E2F targets (Fig. 3H), and immunoglobulin complex (Supplementary Figs. S4, S6) were enriched in genes overexpressed in tCLL. Moreover, we observed the downregulation of genes in key pathways related to the suppression of immune processes, chemotaxis, and cytokine signaling (Supplementary Results, Supplementary Table S5, and Supplementary Fig. S7).
Topology-based pathway enrichment analysis [39] (TBA), which considers the interactions and functional outcomes of genes within pathways, indicated the activation of key cancer hallmark signaling pathways, including the PI3K-Akt, B-cell receptor, MAPK, and T-cell receptor signaling pathways (Supplementary Results, Supplementary Table S7, Supplementary Fig. S9).
Gene expression signature and pathway alteration patterns unique to tCLL
Comparison with normal B cells further clarified the distinctive dysregulation of gene expression and pathway activity in tCLL (Fig. 3I; Supplementary Results).
Interestingly, many genes deregulated in oCLL were not affected in tCLL compared to normal B cells (Supplementary Fig. S3B); these genes were primarily involved in cell cycle regulation, including BCL3, CD79B, TNPO2, SMC4, CENPE, AURKB, and UBASH3B (Fig. 3I; Supplementary Table S8). Genes differentially expressed uniquely in tCLL, which can be considered tCLL-related aberrancies, included lower expression of apoptosis regulators and important members of the IL-2/STAT5 signaling pathway, as well as overexpression of pro-proliferative genes (Fig. 3I; Supplementary Results). Accordingly, the ‘inflammatory response’ and multiple signaling pathways (TNFα, IL-2/STAT5, and IL-6/JAK/STAT3) were more downregulated in tCLL than oCLL, suggesting reduced immune capacity and altered cytokine responses. The interferon-gamma response was uniquely downregulated in tCLL, as well as the ‘UV response DN’ (Supplementary Figure S10), which suggests differences in the DNA damage response. Notably, mitotic spindle and G2M checkpoint pathways were downregulated in oCLL but not in tCLL, indicating more active proliferation in tCLL. Finally, the ‘chemokine signaling’ and ‘proteoglycans in cancer’ pathways were significantly activated only in tCLL (Supplementary Fig. S11).
Patients with tCLL showed a longer TTNT with BTKi than with venetoclax-based therapy
The distinct expression profile and cell state witnessed by the particular landscape of pathway activation in tCLL patients may influence the duration of response to treatment. Notably, reduced BCL2L11 expression can affect the duration of response after Ven-based therapy(62). We hypothesized that higher levels of BTK might impact the response to BTKi. Therefore, we analyzed TTNT after different treatments.
The median TTNT was 3.98 and 1.89 years for tCLL patients treated with FCR/BR and other chemotherapies, but not reached for those patients treated with BTKi or Ven-based frontline therapy (p < 0.0001; Fig. 4A). After 3 years from the start of treatment, 45%, 33%, 0%, and 62% of patients treated with FCR/BR, Ven-based therapy, BTKi, and other therapies, respectively, needed a second line of treatment (Fig. 4A). In particular, the TTNT was longer in patients treated with BTKi than FCR/BR (p = 0.0062, HR 5.19, 95% CI [2.4–7.9]), and there was a trend between BTKi vs. Ven-based therapy (p = 0.058, HR 1.7, 95% CI [1.01–12.99]).

The Kaplan-Meier curve of time to next therapy after different treatments, such as FCR/BR, venetoclax-based therapy (Ven-based), BTKi-based, or other chemo/chemo-immunotherapy (CIT), both as first-line (A) or further-line (B) therapy.
Regarding relapsed/refractory patients, 36 (44%) received chemo/chemoimmunotherapy (i.e., chlorambucil ± rituximab, BR, rituximab, etc.), 33 (41%) a BTKi (27 ibrutinib, 5 acalabrutinib, 1 zanubrutinib), and 12 (15%) a Ven-based therapy. The median TTNT was 2.19, 2.55, and 3.7 years for patients treated with CIT, Ven-based therapy, and BTKi, respectively (p = 0.0467; Fig. 4B). After 3 years, the rates of patients who started further treatment were 55%, 88% and 42%, respectively (Fig. 4B). The TTNT was significantly longer in patients treated with BTKi than those treated with CIT (p = 0.0253, HR 1.89, 95% CI [1.03–3.46]). However, it also tended to be more prolonged in patients managed with a Ven-based therapy, although this did not reach statistical significance (p = 0.1051, HR 2.15, 95% CI [0.96–5.64]).
In summary, tCLL patients showed durable responses if treated with continuous BTKi but experienced early relapse if treated with chemoimmunotherapy or with Ven-based time-limited therapy.
Discussion
In this study, we characterized the clinical and transcriptomic features in a large cohort of patients with CLL harboring the rare t(14;19) (tCLL), thanks to an extended international collaboration. We report evidence suggesting that identifying t(14;19), although a rare entity, by CBA in the era of continuous and fixed-duration therapy for CLL may help in tailoring treatment. Although karyotyping is now recommended by some CLL guidelines [1, 2] to identify patients with more than 3 or 5 chromosomal abnormalities (i.e., those harboring a CK) for treatment selection, its role remains controversial. However, it should not be limited to the number of chromosomal abnormalities, as some specific chromosomal rearrangements or lesions may have a particular prognostic or predictive impact. It is well known that patients harboring trisomies (i.e., +12, +19, +other numerical or structural aberrations) have a favorable outcome despite having a complex karyotype [7]. In our cohort, we demonstrated that patients with tCLL have an overall unfavorable prognosis independent of a complex karyotype, whether it is CK3 or CK5.
Most published studies on patients with tCLL reported an atypical immunophenotype and morphology with an increased frequency of trisomy 12, U-IGHV genes, and overexpression of the BCL3 gene [13,14,15,16,17,18,19,20,21]. We confirmed all these features, including the overexpression of the BCL3 protein. We also described a high rate of BCR stereotype #8 in tCLL, associating a chromosomal translocation with a specific stereotyped BCR. The transcriptomic analysis found that the TP63 gene, a member of the p53 transcription factor family, was overexpressed in our cohort of patients with tCLL. In line with our data, a previous study indicated that the TP63 gene was hypomethylated and overexpressed in stereotyped subset #8 [14, 41].
RNA sequencing enabled us to identify a distinct gene expression profile of tCLL compared to oCLL. In particular, we found a higher expression of the CD79B gene and protein, which justifies the atypical immunophenotype of the cells. Additionally, IGHG4 was overexpressed, leading to the expression of IgG on the surface of CLL cells. IgG–switched CLL is a rare variant of CLL that displays an overall distinct immunogenetic signature [42]. Notably, IgG-switched CLL is enriched in stereotype BCR #8 with an unmutated status of the IGHV gene and trisomy of chromosome 12 [42]. In our patient cohort, almost one-third of tCLLs were IgG-switched.
Among the most differently activated pathways, we found that the immune checkpoints TIGIT and PD-L1 were downregulated, while LAG3 was, conversely, overexpressed in tCLL. The dysregulated expression of immune checkpoints [41,42,43,44] and the low efficacy of the PD1 inhibitors have already been reported in CLL [45, 46]. Shapiro et al [47]. demonstrated a higher surface LAG3 expression associated with U-IGHV and a shorter time to first treatment. Interestingly, LAG3-blocking antibodies enhance in vitro T-cell activation [47]. The TIGIT pathway has also been studied in CLL [48], and its expression was inversely correlated with time to first treatment and the B cell receptor (BCR) signaling capacity, as determined by BTK activity and cell proliferation [48]. Along this line, a low baseline level of PD-L1 and TIGIT by tumor cells has been reported in patients with large B-cell lymphoma who have shown a better response to CAR T cells [44]. While these findings suggest potential clinical relevance, the exact roles of TIGIT and LAG3 in tCLL pathogenesis and treatment response require further investigation. The use of LAG3-blocking antibodies [49] and CAR T cells represents intriguing directions for future research but needs thorough clinical evaluation to determine their efficacy in tCLL.
Oxidative phosphorylation (OXPHOS) and E2F target pathways [50] were activated, whereas apoptosis-related pathways were suppressed specifically in tCLL. Furthermore, cancer hallmark signaling pathways, including the PI3K-Akt, B-cell receptor, MAPK, and T-cell receptor signaling pathways, were activated. While it is already known that CLL cells primarily use OXPHOS for generating energy [51], the OXPHOS enrichment in tCLL suggests higher oxidative stress related to an abundant generation of reactive oxygen species (ROS) by increased mitochondrial production [52], which can be linked to a more proliferative and aggressive phenotype in tCLL.
The gene expression comparison of the two CLL groups to normal B cells allowed us to highlight distinctive molecular characteristics for tCLL. Intriguingly, our data suggest possible mechanisms that could contribute to regulating the BCL3 abundance in tCLL, such as the GSK3β-mediated protein degradation pathway [53, 54] and miR-181a targeting BCL3 [55,56,57,58,59]. The GSK3β-mediated protein degradation is supported by the GSK3B expression level, which was very low in tCLL. Regarding miR-181a-mediated regulation, the high BCL3 abundance in tCLL matched with a lower miR-181a expression compared to B cells. Interestingly, tCLL displayed a specific upregulation of PAK1, previously shown to promote proliferation and sustain ibrutinib resistance in CLL.
Our results suggest that the translocation-induced overexpression of the BCL3 gene in tCLL triggers a complex and widespread dysregulation of gene expression. We identified molecular alterations specific to tCLL, which involved several cancer hallmarks signaling pathways (TNF-α, IL-2/STAT5, IL-6/JAK/STAT3, and PI3K-AKT) and metabolic processes that may affect apoptosis, cell proliferation, survival, antibody production, response to the microenvironment, homing and migration, and potentially drug resistance. Overall, this might help explain the worse outcome of patients with tCLL compared to those with oCLL. Notably, we demonstrated that circular RNAs are commonly dysregulated in tCLL, exhibiting a specific signature whose biological significance warrants further investigation [60].
A limitation of this study is its retrospective observational design, which encompasses over 20 years. During this timeframe, chromosome banding analysis was only recommended, not mandatory, and was therefore performed in only a few selected centers. For this reason, the prevalence of patients with tCLL might be inaccurate. To minimize selection bias and inaccurate data reporting, we asked physicians to report all consecutive patients with CLL harboring t(14;19) tested within 1 year of diagnosis. In addition, despite only a few patients receiving frontline Ven-based therapy, a statistical difference in the TTNT was found. Additionally, there may be biases influencing the samples collected for the transcriptomic analysis. However, as reported here, the TTFT and OS rates of the patients selected for RNA-seq did not significantly differ from those of the overall cohort, supporting the representativeness of our selection.
Furthermore, when comparing the TTNT between patients with tCLL or oCLL, we only included individuals who received similar treatments to minimize bias from differing therapy regimens. It should also be acknowledged that t(14;19) has been associated with a broad spectrum of B lymphoid neoplasms. Carbo-Meix A. et al. [14] recently reported that BCL3 breakpoints occur in two clusters at 5ʹ (n = 9) and 3ʹ (n = 4) regions of BCL3. While both breakpoints were mediated by aberrant class switch recombination of the IGH locus, only the 5’ breakpoints juxtaposed BCL3 upstream to an IGH enhancer, leading to the overexpression of the BCL3 gene. They found that the majority of upstream BCL3-rearranged tumors had unmutated IGHV, trisomy 12, an atypical CLL morphology, immunophenotype, DNA methylome, and expression profile that differ from CLL without t(14;19). For these reasons, we included only patients with a Matutes score of 3 or higher, mainly due to the expression of CD5, CD23, and higher expression of CD79b and sIg. Our study, which included only CLL with t(14;19), yielded similar results to those found in upstream BCL3-rearranged tumors.
By both RNA-seq and RT-PCR, we observed that tCLL was characterized by higher levels of BTK and the downregulation of the BCL2L11 gene, which encodes the pro-apoptotic protein BIM. Accordingly, the time to second-line therapy with a time-limited venetoclax-based therapy was shorter in patients with tCLL compared to those with oCLL, likely due to low mitochondrial priming and lower sensitivity to BCL2 inhibition, thereby impeding sustained remission. Conversely, continuous BTKi provides sustained remissions. It is noteworthy that in the exploratory post-hoc analyses of the CLL14 trial, which investigated venetoclax-obinutuzumab in elderly patients with CLL, achieving undetectable measurable residual disease (i.e., uMRD6) status was associated with higher pro-apoptotic BCL2L11 expression [61]. Furthermore, as in our study, the PI3K-Akt signaling pathway has been linked to BCL3 overexpression [62]. While targeting PI3K might be an alternative option for this subset of patients with CLL, the clinical use of PI3K inhibitors is limited by a wide range of adverse events [63].
Conclusions
In conclusion, we herein reported that patients with tCLL are younger than those with oCLL, enriched for +12, CK, U-IGHV, and stereotyped subset #8, and have a distinct gene expression profile featured by higher expression of BTK and lower apoptotic priming due to low BCL2L11 levels, driven by the overexpression of the BCL3 gene. In our retrospective cohort, continuous BTKi seems to be more effective than a time-limited strategy. Although a few compounds have recently been identified and tested in vitro [64,65,66], no clinically staged BCL3 inhibitor is currently available. Further biological and molecular studies are warranted to better understand the aggressive CLL that harbors the t(14;19) translocation .
Data availability
PRO-Seq data were deposited into the ArrayExpress repository under accession number E-MTAB-15004.
References
Eichhorst B, Ghia P, Niemann CU, Kater AP, Gregor M, Hallek M, et al. ESMO Clinical Practice Guideline interim update on new targeted therapies in the first line and at relapse of chronic lymphocytic leukaemia. Ann Oncol. 2024;35:762–8.
Wierda WG, Brown J, Abramson JS, Awan F, Bilgrami SF, Bociek G, et al. Chronic lymphocytic leukemia/small lymphocytic lymphoma, version 2.2024, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2024;22:175–204.
Rigolin GM, Saccenti E, Guardalben E, Cavallari M, Formigaro L, Zagatti B, et al. In chronic lymphocytic leukaemia with complex karyotype, major structural abnormalities identify a subset of patients with inferior outcome and distinct biological characteristics. Br J Haematol. 2018;181:229–33.
Serafin A, Cellini A, Cavarretta CA, Ruocco V, Angotzi F, Zatta I, et al. Exploring the prognostic role of complex karyotype in chronic lymphocytic leukaemia patients treated with venetoclax-based regimens. Br J Haematol. 2024;205:189–93.
Visentin A, Bonaldi L, Rigolin GM, Mauro FR, Martines A, Frezzato F, et al. The complex karyotype landscape in chronic lymphocytic leukemia allows the refinement of the risk of Richter syndrome transformation. Haematologica. 2022;107:868–76.
Visentin A, Bonaldi L, Rigolin GM, Mauro FR, Martines A, Frezzato F, et al. The combination of complex karyotype subtypes and IGHV mutational status identifies new prognostic and predictive groups in chronic lymphocytic leukaemia. Br J Cancer. 2019;121:150–6.
Baliakas P, Jeromin S, Iskas M, Puiggros A, Plevova K, Nguyen-Khac F, et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood. 2019;133:1205–16.
Baliakas P, Espinet B, Mellink C, Jarosova M, Athanasiadou A, Ghia P, et al. Cytogenetics in chronic lymphocytic leukemia: ERIC perspectives and recommendations. Hemasphere. 2022;6:e707.
Ramos-Campoy S, Puiggros A, Beà S, Bougeon S, Larráyoz MJ, Costa D, et al. Chromosome banding analysis and genomic microarrays are both useful but not equivalent methods for genomic complexity risk stratification in chronic lymphocytic leukemia patients. Haematologica. 2022;107:593–603.
Kittai AS, Miller C, Goldstein D, Huang Y, Abruzzo LV, Beckwith K, et al. The impact of increasing karyotypic complexity and evolution on survival in patients with CLL treated with ibrutinib. Blood. 2021;138:2372–82.
Thompson PA, O’Brien SM, Wierda WG, Ferrajoli A, Stingo F, Smith SC, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer. 2015;121:3612–21.
Fürstenau M, Thus YJ, Robrecht S, Mellink CHM, van der Kevie-Kersemaekers A-M, Dubois J, et al. High karyotypic complexity is an independent prognostic factor in patients with CLL treated with venetoclax combinations. Blood. 2023;142:446–59.
Michaux L, Mecucci C, Stul M, Wlodarska I, Hernandez JM, Meeus P, et al. BCL3 rearrangement and t(14;19)(q32;q13) in lymphoproliferative disorders. Genes Chromosom Cancer. 1996;15:38–47.
Carbo-Meix A, Guijarro F, Wang L, Grau M, Royo R, Frigola G, et al. BCL3-rearrangements in B-cell lymphoid neoplasms occur in two breakpoint clusters associated with different diseases. Haematol. 2023. https://doi.org/10.3324/haematol.2023.283209.
Chapiro E, Radford-Weiss I, Bastard C, Luquet I, Lefebvre C, Callet-Bauchu E, et al. The most frequent t(14;19)(q32;q13)-positive B-cell malignancy corresponds to an aggressive subgroup of atypical chronic lymphocytic leukemia. Leukemia. 2008;22:2123–7.
Nguyen-Khac F, Chapiro E, Lesty C, Grelier A, Luquet I, Radford-Weiss I, et al. Specific chromosomal IG translocations have different prognoses in chronic lymphocytic leukemia. Am J Blood Res. 2011;1:13–21.
Martín-Subero JI, Ibbotson R, Klapper W, Michaux L, Callet-Bauchu E, Berger F, et al. A comprehensive genetic and histopathologic analysis identifies two subgroups of B-cell malignancies carrying a t(14;19)(q32;q13) or variant BCL3-translocation. Leukemia. 2007;21:1532–44.
Kelly RJ, Wright D, Patil K, Chapple M, Jalihal SS, Barrans SL, et al. t(14;19)(q32;q13) incidence and significance in B-cell lymphoproliferative disorders. Br J Haematol. 2008;141:561–3.
Busschots AM, Mecucci C, Stul M, Vandenberghe E, Michaux J-L, Noel H, et al. Translocation (14;19)(q32;q13.1) in a young patient who developed a large cell lymphoma after an initial diagnosis of CLL. Leuk Lymphoma. 1991;5:281–6.
Fang H, Reichard KK, Rabe KG, Hanson CA, Call TG, Ding W, et al. IGH translocations in chronic lymphocytic leukemia: clinicopathologic features and clinical outcomes. Am J Hematol. 2019;94:338–45.
Rossi D, Deambrogi C, Monti S, Cresta S, De Paoli L, Fangazio M, et al. correspondence: BCL3 translocation in CLL with typical phenotype: assessment of frequency, association with cytogenetic subgroups, and prognostic significance. Br J Haematol. 2010;150:702–4.
Palmer S, Chen YH. Bcl-3, a multifaceted modulator of NF-κB-mediated gene transcription. Immunol Res. 2008;42:210–8.
Liu H, Zeng L, Yang Y, Guo C, Wang H. Bcl-3: a double-edged sword in immune cells and inflammation. Front Immunol. 2022;13:847699.
Ong ST, Hackbarth ML, Degenstein LC, Baunoch DA, Anastasi J, McKeithan TW. Lymphadenopathy, splenomegaly, and altered immunoglobulin production in BCL3 transgenic mice. Oncogene. 1998;16:2333–43.
Zhang X, Paun A, Claudio E, Wang H, Siebenlist U. The tumor promoter and NF-κB modulator Bcl-3 regulates splenic B cell development. J Immunol. 2013;191:5984–92.
Chatzikonstantinou T, Scarfò L, Karakatsoulis G, Minga E, Chamou D, Iacoboni G, et al. Other malignancies in the history of CLL: an international multicenter study conducted by ERIC, the European Research Initiative on CLL, in HARMONY. EClinicalMedicine. 2023;65:102307.
Gaffo E, Boldrin E, Dal Molin A, Bresolin S, Bonizzato A, Trentin L, et al. Circular RNA differential expression in blood cell populations and exploration of circRNA deregulation in pediatric acute lymphoblastic leukemia. Sci Rep. 2019;9:14670.
Gaffo E, Buratin A, Dal Molin A. Bortoluzzi S. Sensitive, reliable and robust circRNA detection from RNA-seq with CirComPara2. Brief Bioinforma. 2022;23:bbab418.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15.
Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019;20:278.
Love MI, Soneson C, Hickey PF, Johnson LK, Pierce NT, Shepherd L, et al. Tximeta: Reference sequence checksums for provenance identification in RNA-seq. PLoS Comput Biol. 2020;16:e1007664.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–3.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2nd ed. 2016. Springer International Publishing: Imprint: Springer: Cham, 2016. https://doi.org/10.1007/978-3-319-24277-4.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–40.
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25.
Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim J-S, et al. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82.
Liska O, Bohár B, Hidas A, Korcsmáros T, Papp B, Fazekas D, et al. TFLink: an integrated gateway to access transcription factor–target gene interactions for multiple species. Database. 2022;2022:baac083.
Papakonstantinou N, Ntoufa S, Tsagiopoulou M, Moysiadis T, Bhoi S, Malousi A, et al. Integrated epigenomic and transcriptomic analysis reveals TP63 as a novel player in clinically aggressive chronic lymphocytic leukemia. Int J Cancer. 2019;144:2695–706.
Vardi A, Agathangelidis A, Sutton L-A, Chatzouli M, Scarfò L, Mansouri L, et al. IgG-switched CLL has a distinct immunogenetic signature from the common MD variant: ontogenetic implications. Clin Cancer Res. 2014;20:323–30.
Ebrahimi N, Abdulwahid A-HRR, Mansouri A, Karimi N, Bostani RJ, Beiranvand S, et al. Targeting the NF-κB pathway as a potential regulator of immune checkpoints in cancer immunotherapy. Cell Mol Life Sci. 2024;81:106.
Vitale C, Griggio V, Perutelli F, Coscia M. CAR-modified cellular therapies in chronic lymphocytic leukemia: is the uphill road getting less steep?. HemaSphere. 2023;7:e988.
Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129:3419–27.
Visentin A, Frazzetto S, Trentin L, Chiarenza A. Innovative combinations, cellular therapies and bispecific antibodies for chronic lymphocytic leukemia: a narrative review. Cancers. 2024;16:1290.
Shapiro M, Herishanu Y, Katz B-Z, Dezorella N, Sun C, Kay S, et al. Lymphocyte activation gene 3: a novel therapeutic target in chronic lymphocytic leukemia. Haematologica. 2017;102:874–82.
Arruga F, Rubin M, Papazoglou D, Iannello A, Ioannou N, Moia R, et al. The immunomodulatory molecule TIGIT is expressed by chronic lymphocytic leukemia cells and contributes to anergy. Haematol. 2023;108:2101–15.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377:2545–54.
Müller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, et al. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15:267–85.
Jitschin R, Hofmann AD, Bruns H, Gießl A, Bricks J, Berger J, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014;123:2663–72.
Pagano MA, Frezzato F, Visentin A, Trentin L, Brunati AM. Protein phosphorylation and redox status: an as yet elusive dyad in chronic lymphocytic leukemia. Cancers. 2022;14:4881.
Woodgett JR. Judging a protein by more than its name: GSK-3. Sci STKE. 2001;2001:re12.
Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, et al. GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell. 2004;16:35–45.
He S, Zeng S, Zhou Z-W, He Z-X, Zhou S-F. Hsa-microRNA-181a is a regulator of a number of cancer genes and a biomarker for endometrial carcinoma in patients: a bioinformatic and clinical study and the therapeutic implication. Drug Des Devel Ther. 2015;9:1103–75.
Miranda KC, Huynh T, Tay Y, Ang Y-S, Tam W-L, Thomson AM, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–17.
Marisetty A, Wei J, Kong L-Y, Ott M, Fang D, Sabbagh A, et al. MiR-181 family modulates osteopontin in glioblastoma multiforme. Cancers (Basel). 2020;12:3813.
Ouyang Y-B, Lu Y, Yue S, Giffard RG. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012;12:213–9.
Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-β upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013;123:150–63.
Roncaglia E, Gaffo E, Calabretto G, Fürstenau M, Rogers KA, Baliakas P, et al. Circular RNA signature of aggressive CLL with t(14;19)(q32;q13). An ERIC study. J Hematol Oncol. 2025;18:74.
Al-Sawaf O, Zhang C, Jin HY, Robrecht S, Choi Y, Balasubramanian S, et al. Transcriptomic profiles and 5-year results from the randomized CLL14 study of venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab in chronic lymphocytic leukemia. Nat Commun. 2023;14:2147.
Seaton G, Smith H, Brancale A, Westwell AD, Clarkson R. Multifaceted roles for BCL3 in cancer: a proto-oncogene comes of age. Mol Cancer. 2024;23:7.
Visentin A, Frezzato F, Severin F, Imbergamo S, Pravato S, Romano Gargarella L, et al. Lights and shade of next-generation Pi3k inhibitors in chronic lymphocytic leukemia. OTT. 2020;ume 13:9679–88.
Saamarthy K, Ahlqvist K, Daams R, Balagunaseelan N, Rinaldo-Matthis A, Kazi JU, et al. Discovery of a small molecule that inhibits Bcl-3-mediated cyclin D1 expression in melanoma cells. BMC Cancer. 2024;24:103.
Soukupová J, Bordoni C, Turnham DJ, Yang WW, Seaton G, Gruca A, et al. The discovery of a novel antimetastatic Bcl3 inhibitor. Mol Cancer Ther. 2021;20:775–86.
Daams R, Tran TTP, Jemaà M, Sime W, Mickeviciute R, Ek S, et al. Enhancing cell death in B-cell malignancies through targeted inhibition of Bcl-3. Cell Death Dis. 2024;15:690.
Acknowledgements
We would like to thank BMR Genomics for the next-generation sequence service.
Funding
This work was supported by funding from the Italian Association for Cancer Research (AIRC) IG-25024 to LT and under 5 per Mille 2018 - ID. 21198 program to PG, Progetti di Rilevanza Nazionale PRIN PNRR (P2022PSMX4) to AV, the ONLUS ‘Ricerca per Credere nella Vita’ (RCV) odv, Padua, Italy, Supporting Talent in ReSearch@University of Padua - Starting Grant Be_CL3VER to AV. Fondazione AIRC per la Ricerca sul Cancro (IG 2017 #20052 and IG #2023 #28966 to SB), EU funding within the MUR PNRR “National Center for Gene Therapy and Drugs based on RNA Technology” (Project no. CN00000041 CN3 Spoke #6 “RNA chemistry”) and “National Center for HPC, Big Data and Quantum Computing” (Project no. CN00000013 CN1 Spoke #8 “In Silico Medicine and Omics Data”) and PRIN MIUR 2022 #20222EC7LA to SB. Towards Precision Medicine: Advanced cellular analytics in biomedical research–PureCell, funded by the Hellenic Foundation for Research and Innovation under the 1st call for research projects EL.ID.EK to KS. AZV NU21-08-00237 and DRO FNBr65269705 provided by the Ministry of Health, Czech Republic to KP. The funders had no role in the study design, data collection and analysis, publication decision, or manuscript preparation. Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement. Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
AVi conceived the study, provided data and samples, analyzed data, wrote the article, and acquired funding. EG conceived the study, analyzed transcriptomic data, and wrote the article. MF, KAR, PB, CC, CM, CH, KP, DO, ZD, FNK, GMR, AA, FBM, AVa, MJT, PA, BE, AP, AM, LB, FRM, LS, TC, ET, KAK, AK, FB, MD, PP, OK, VR, SO, AC, FA, CAD, AR, RF, SY, BE, JW, AC provided data and samples, gave intellectual input, revised the manuscript. ER analyzed transcriptomic data, gave intellectual input, and revised the manuscript. FF and GC performed Western blotting analysis, RT-PCR, and flow cytometry to validate data. KS, PG, LT, and SB conceived the study, provided intellectual input, contributed data, revised the manuscript, and secured funding.
Corresponding authors
Ethics declarations
Competing interests
AV, LT PG, LS, and FRM attended scientific boards organized by Johnson & Johnson, AstraZeneca, BeiGene, and AbbVie. PG received honoraria from AbbVie, AstraZeneca, BeiGene, BMS, Galapagos, Johnson & Johnson, Lilly/Loxo, MSD, and research funding from AbbVie, AstraZeneca, BeiGene, BMS, Johnson & Johnson, Lilly/Loxo, MSD. LyS received honoraria from AbbVie, AstraZeneca, BeiGene, Johnson & Johnson, Lilly, and MSD. G.M.R. received honoraria for participation in the speakers’ bureau and advisory board from AbbVie, AstraZeneca, and Janssen. A.C. received honoraria for participation in the speakers’ bureau and advisory board from AbbVie, AstraZeneca, BeiGene, and Janssen Lilly. MD Advisory boards and honoraria: AbbVie, AstraZeneca, Eli Lilly, Johnson and Johnson, Swixx Biopharma. The other authors declared no potential conflict of interest with this study.
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki, approved by the Ethics Committee of Padova University Hospital (protocol #4430/AO/18), and informed consent was obtained.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Visentin, A., Gaffo, E., Fürstenau, M. et al. Clinical and transcriptomic characterization of patients with chronic lymphocytic leukemia harboring t(14;19): an ERIC study. Leukemia 39, 2957–2967 (2025). https://doi.org/10.1038/s41375-025-02755-8
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-025-02755-8
