
Abstract
Pulmonary neuroendocrine neoplasms are classified by WHO as either typical or atypical carcinoids, large cell (LCNEC) or small cell (SCLC) neuroendocrine carcinoma based on mitotic count, morphology, and necrosis assessment. LCNEC with low mitotic count and sharing morphologic features with carcinoids are in a gray zone for classification and their rare prevalence and the paucity of studies precludes proper validation of the current grading system. In this study, we aim to investigate their clinicopathological and transcriptomic profiles. Lung resection specimens obtained from 18 patients diagnosed with carcinoids or LCNEC were selected. Four of them were characterized as borderline tumors based on a mitotic rate ranging between 10 and 30 mitoses per 2 mm2. Comprehensive morphological and immunohistochemical (IHC) evaluation was performed and tumor-based transcriptomic profiles were analyzed through unsupervised clustering. Clustering analysis revealed two distinct molecular groups characterized by low (C1) and high (C2) proliferation. C1 was comprised of seven carcinoids and three borderline tumors, while C2 was comprised of seven LCNEC and one borderline tumor. Furthermore, patients in cluster C1 had a better recurrence-free survival compared with patients in cluster C2 (20% vs 75%). Histological features, IHC profile, and molecular analysis showed that three out of four borderline tumors showed features consistent with carcinoids. Therefore, our findings convey that the current diagnostic guidelines are suboptimal for classification of pulmonary neuroendocrine tumors with increased proliferative index and carcinoid-like morphology. These results support the emerging concept that neuroendocrine tumors with carcinoid-like features and mitotic count of <20 mitoses per 2 mm2 should be regarded as pulmonary carcinoids instead of LCNEC.
Similar content being viewed by others

Introduction
The current WHO classification stratifies the pulmonary neuroendocrine tumors into pulmonary carcinoids, large cell neuroendocrine carcinoma (LCNEC) and small cell neuroendocrine carcinoma (SCLC) [1]. The diagnosis is based on overall neuroendocrine morphology (for instance, architectural patterns, nucleo-cytoplasmic ratio, “salt and pepper” chromatin, and nuclear molding) presence of necrosis, mitotic count, and immunohistochemistry profile (IHC) [1]. Lung carcinoids are further classified as typical tumors (<2 mitoses per 2 mm2 and absence of necrosis) and atypical tumors (2–10 mitoses per 2 mm2 with or without necrosis or <2 mitoses per 2 mm2 with necrosis) [2,3,4]. From a molecular point of view, despite the difference in the survival outcomes and metastatic potential of typical and atypical carcinoids, these two groups show striking similarities in gene expression [5]. A recent study carried out on DNA mutational profiles showed that similar gene alterations were shared between carcinoids and carcinomas, however with different prevalent rates among their subtypes [6]. Furthermore, alteration in MEN1 gene is found to be enriched in carcinoids, while TP53 and RB1 genes are found to be altered particularly in carcinomas [7,8,9,10]. Based on the current classification, LCNEC are tumors with mitotic count exceeding 10 mitoses per 2 mm2, extensive necrosis, and classical morphologic features such as an organoid growth pattern, solid nest of tumor cells with comedo necrosis, peripheral palisading, and rosette formation [4, 11].
One of the major challenges with the aforementioned WHO diagnostic criteria is their application to tumors that have classical carcinoid morphology but a proliferation index exceeding 10 mitoses per 2 mm2, which is still scarcely described in the literature [12, 13]. A few studies looking at the genomic profiling of such tumors revealed similarities to carcinoids rather than LCNEC [14,15,16,17]. A more recent work carried out by Alcala et al. showed that the separation between pulmonary carcinoids and LCNEC might be more subtle for a subset of patients, and identified a subgroup of atypical carcinoids, named supra-carcinoids, that has carcinoid-like morphological pattern but with molecular characteristics similar to LCNEC [18]. Altogether, these studies provide evidences that such borderline tumors need further classification and refinement including morphological criteria and biomarkers.
More importantly, the current diagnostic criteria are suboptimal for classification of pulmonary neuroendocrine tumors with carcinoid-like morphology and increased proliferation. These guidelines do not specifically address the classification of these tumors, which may potentially result in adverse consequences for patient management and prognosis. Efforts have been made to revise the WHO grading system to better characterize extra-pulmonary differentiated neuroendocrine tumors, using a three-tier grading system (G1–G3) and incorporating ki-67 as a classification criteria [18,19,20,21,22]. However, there are only a few studies to support similar recommendations in the grading criteria for lung neuroendocrine tumors with increased proliferation. Recognizing this gap in the literature and based on the aforementioned premise, we carried out this study to echo similar recommendations to lung neuroendocrine tumors as well. In order to do this, we aimed to investigate immunohistological and transcriptomic features of tumors with mitotic counts borderline between carcinoids and LCNEC. Particularly, we examined the morphology and immunohistochemical pattern of Ki-67, RB1, and p53 protein expression of all the samples. In addition, we also analyzed transcriptomic profiles to stratify neuroendocrine tumors based on molecular data.
Materials and methods
Cohort collection and selection
In this retrospective study, all the samples were retrieved from a single pulmonary pathology reference center with the approval of the institutional ethics board (#21045). Surgically resected samples of lung neuroendocrine tumors from 135 patients diagnosed between 2000 and 2016 were reviewed for this study. These specimens were part of the IUCPQ site of the Quebec Respiratory Health Network Tissue Bank (https://rsr-qc.ca/biobanque/). This cohort included 91 cases of carcinoids and 44 cases of pure LCNEC. The SCLC were excluded since the differential diagnosis of carcinoids with increased proliferative index is mainly with LCNEC, as stated in the manuscript. All these cases were reviewed to confirm the diagnosis. The availability of frozen tissue along with the quality and the amount of extracted RNA were assessed, upon which the cases of TC, AC, and LCNEC were randomly selected. However, for carcinoids, we wanted to select cases that cover the spectrum of 0–10 mitoses per 2 mm2 to have a representative group of carcinoids and to be in line with the clinically relevant groups of tumors. For LCNEC, representative cases were also randomly selected. All the original H&E slides were reviewed by thoracic pathologists to confirm the diagnostic characterization, including reevaluation of the mitotic count and Ki-67 index. Available data included primary diagnosis, pathology variables, clinical variables such as sex, tumor stage, smoking status, status of recurrence, and number of days of follow-up.
Histopathological evaluation and immunohistochemistry (IHC)
The histological analysis of tumors was carried out based on the 2015 WHO criteria [11]. Histological features such as the distinct morphological patterns, extent of necrosis, and cytological nuclear features were recorded. Mitotic count was calculated by two thoracic pathologists on H&E in most mitotically active areas evaluated on several blocks and expressed as number of mitoses per 2 mm2. The mean value of mitotic index for a single most proliferative area was calculated by averaging the pathologist values. Proliferation index was assessed using ki-67 IHC.
The WHO diagnostic criteria were used to classify the tumors as carcinoids or carcinomas. The mitotic count of <10 mitoses per 2 mm2 was strictly enforced to characterize the cases as carcinoids. In order to cover the complete spectrum of mitotic rate, 18 samples were chosen for this study. Tumors with mitotic counts lying on low end of the spectrum for LCNEC were defined as borderline, as some of the tumors in this mitotic range have shown genetic alterations more characteristic for carcinoids than LCNEC [18]. Based on a recent study, we used the mitotic range of 10–30 mitoses per 2 mm2 to select the tumors for this borderline group [23, 24]. All the cases with mitotic count >30 mitoses per 2 mm2 were characterized as LCNECs.
Neuroendocrine differentiation was confirmed by immunohistochemical markers including synaptophysin, chromogranin, and CD56. For immunohistochemical evaluation of RB1 (Retinoblastoma Gene Protein), p53, and Ki-67, a 4.0-μm-thick section was cut from a representative tumor block for each patient and deparaffinized. Heat-induced epitope retrieval was performed in a DAKO PT-Link (Dako-Agilent technologies) using high pH buffer for both Ki-67 and p53 and low pH buffer for RB1. IHC protocols were all carried out on the Autostainer Link 48 (Dako- Agilent Technologies, Santa Clara, CA). Ki-67 (clone MIB1) and p53 (clone DO-07) from Dako (Dako-Agilent Technologies) were used in the manufacturer’s prediluted format while RB1 (clone 13A10, Novocastra-Leica Biosystems, Nussloch, Germany) was used at a 1:80 dilution. Incubation time with all primary antibodies was 20 min and the remaining of the IHC procedure was a standard Autostainer protocol using Dako EnVision FLEX+ kit and ancillary’s reagents. After IHC, slides were counterstained using Harris hematoxylin. The evaluation of the Ki-67 index was performed according to the recommendations for GI well-differentiated NETs [25]. Ki-67 calculation was done manually by counting the percentage of cells with positive nuclear labeling. Briefly, 500 tumor cells were evaluated in the hot-spot region. The highest Ki-67 value was considered for the hot-spot, while the average value of Ki-67 was calculated as the mean across the slides.
RNA extraction and library preparation
After surgical removal, lung samples were immediately placed in 5-mL cryovials and snap frozen in liquid nitrogen. The cryovials were then transported in dry ice to the IUCPQ Biobank where they were stored at −80 °C until RNA processing. The time from surgical removal to storage was <30 min. RNA was extracted from 30 mg of frozen lung specimens using RNeasy Plus Universal Mini kit (Qiagen ON, Canada) following manufacturer’s instruction. RNA concentration and purity were verified by UV 260/280 nm ratio on the NanoVue spectrophotometer (GE Healthcare). RNA quality was checked using a TapeStation 2200 (Agilent Technologies, Santa Clara, CA, USA). RNA integrity number (RIN) for all samples was >7.0. The Illumina TruSeq stranded Total RNA library prep kit with Ribo-Zero Gold (Illumina Inc., San Diego, CA, USA) was used to prepare RNA sequencing libraries, according to manufacturer’s instruction. Briefly, 10 μg of total RNA was used. Ribosomal RNA (rRNA) including both cytoplasmic and mitochondrial rRNA was removed using biotinylated, target-specific oligos combined with Ribo-Zero rRNA removal beads. Following purification with Agencourt RNAClean XP beads (Beckman Coulter, Mississauga, Ontario, Canada), the RNA was fragmented using divalent cations under elevated temperature. The fragmented RNA was used as a template for cDNA synthesis by reverse transcriptase with random primers. Strand specificity was achieved by replacing dTTP with dUTP. This cDNA was further converted into double stranded DNA that was end-repaired to incorporate the specific index adapter for multiplexing. Following a purification with Agencourt AMPure XP beads (Beckman Coutler, Mississauga, Ontario, Canada), an amplification for 15 cycles was performed using a polymerase unable to incorporate dUTP. Therefore, the second strand is quenched during amplification. The quality of final amplified libraries was examined with a DNA screen tape D1000 on a TapeStation 2200 and the quantification was done on the Qubit 3.0 fluorometer (ThermoFisher Scientific, Canada). Subsequently, RNA-seq libraries were sequenced on an HiSeq 2500 V3 system at the Next-Generation Sequencing Platform, Genomics Center, CHU de Québec-Université Laval Research Center, Québec City, Canada for paired-end 100 bp sequencing.
Processing of RNA-seq and data analysis
RNA-seq data were processed using the Kallisto pipeline [26], similar to other studies in the literature [27, 28]. Using this method, the gene expression was computed from RNA-seq data to quantify expression using a pseudo-alignment method. The default analysis settings and parameters were used for the algorithm, and the Ensembl GRCh38 reference transcriptome was used for the workflow. Gene transcripts mapped data were normalized to transcript per million (TPM). The quantification was performed on the full transcriptome and gene-level TPM values were calculated by summing transcript-level TPM values. The expression values were calculated using the log2(TPM + 1), which was used for all the downstream analyses.
We used unsupervised hierarchical clustering with correlation measure as distance and complete linkage to cluster the gene expression for all patients. We repeated the clustering 18 times, each time leaving one sample out of the clustering analysis. This was carried out in order to make sure that defining two groups of samples as the two main branches of the dendrogram was reasonable. Defining these two groups would be considered unreasonable if the removal of a single sample could have a drastic impact on the clustering. To identify differentially expressed genes between the groups, we used Wilcoxon test, and corrected the p values using false discovery rate (FDR). A significant threshold of the corrected p < 0.05 was used for the analysis. The Wilcoxon signed-rank test was used to compare the continuous variables across two groups. All analyses were performed using the R software (R version 3.5.1).
Results
Demographic and clinical characteristics
The clinical characteristics of patients in the cohort are presented in Table 1. Eleven cases were diagnosed as LCNECs and seven as carcinoids on initial pathology evaluation based on 2015 WHO classification. Out of these 18 samples, four cases presenting a usually low mitotic index for a diagnosis of LCNEC and an increased mitotic count for a diagnosis of carcinoid were reclassified as borderline tumors [23, 24]. The original diagnosis of the four borderline samples were LCNEC. It should be noted that the prevalence of borderline tumors based on our results is 3.6%. However, it is worth noting that all samples used in this study were resection cases, although it is likely that the vast majority of patients diagnosed with pulmonary carcinoids will undergo lung resection in the course of their treatment.
Histopathological and immunohistochemical evaluation
In the group of pulmonary carcinoids, the average value of mitotic rate was 2.71 mitoses per 2 mm2 (range 1–5 mitoses per 2 mm2) and average value of Ki-67 proliferative index was 8.57% (range: 1–23%). Borderline cases had an average mitotic rate of 17.5 mitoses per 2 mm2 (range: 13–26 mitoses per 2 mm2) and average value of Ki-67 proliferative index was 28.5% (range: 15–58%). The average value of Ki-67 for the borderline cases S8, S9, S10, and S11 were 10.2, 6.25, 5, 37.5 respectively. The average value of mitotic rate of LCNECs was 73.7 mitoses per 2 mm2 (range: 42–128 mitoses per 2 mm2) and a proliferative index of 89.8% (range: 75–100%). The RB1 and p53 protein expression was preserved in both carcinoid and borderline tumors, while RB1 expression was altered in three out of seven LCNEC and p53 in five out of seven LCNEC cases. The spectrum of mitotic count is summarized in Table 2.
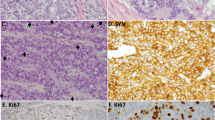
Histopathological analysis of carcinoids with increased proliferation rate revealed classical features of carcinoid tumors (Table 3 and Fig. 1). The Ki-67 labeling commonly often presented heterogeneity throughout the slide. All cases retained RB1 by IHC and had a wild-type P53 staining pattern. In general, cases with higher mitotic counts resulted in higher Ki-67 proliferative index. Case S11 exhibited high Ki-67 proliferation index comparable with that of LCNECs. Morphologically, borderline tumors displayed trabecular, solid, lobular, and nested architectural pattern and rosettes formation. Out of four tumors, three of them had focal necrosis, while we found no necrosis in Case S10. Cytologically, tumors displayed nuclei with salt and pepper chromatin with moderate to abundant cytoplasm. Case S11 had higher nucleo-cytoplasmic ratio and irregular nuclear borders, while others had relatively uniform or monotonous round nuclei, characteristic for pulmonary carcinoids (Fig. 1). The architecture of this case was also characterized by a lobular pattern with peripheral palisading and increased nucleo-cytoplasmic ratio compatible with LCNEC, however mitotic count remained on the low end of spectrum for a LCNEC (26 mitoses per 2 mm2) and evolution was compatible with carcinoid tumors with no relapse. The remaining of the borderline cases did not have the features of high-grade tumors, which were originally diagnosed as LCNEC based on the mitotic count only. Therefore, three tumors with increased proliferation had pulmonary carcinoid-like morphology, while one case displayed features of carcinoma. We hypothesize that these three carcinoid-like tumors should be regarded as pulmonary atypical carcinoids with increased proliferation rate. RB1 expression was preserved in all borderline tumors and p53 showed normal (wild-type) expression pattern.

H&E sections and Ki-67 marker expression.
Molecular evaluation of tumors
To test our hypothesis that borderline cases are more related to atypical carcinoids, we analyzed their molecular characteristics using gene expression profiling. Hierarchical clustering using the Ward algorithm with Pearson correlation distance identified two distinct clusters (Fig. 2a): low proliferative cluster (C1), which included seven carcinoids and three borderline tumors (S8, S9, and S10), and high proliferative cluster (C2), which included seven LCNEC and one borderline tumor (S11). C1 cluster was characterized by a lower mitotic count and Ki-67, while C2 cluster showed higher mitotic count and Ki-67. The leave-one-out analysis showed that individual samples do not switch from one cluster to the other, implying that the two clusters are reasonable and well-defined. The average values of mitotic counts for the C1 and C2 clusters were 6.1 mitoses per 2 mm2 (range: 1 and 17 mitoses per 2 mm2) and 67.7 mitoses per 2 mm2 (range: 26 and 128 mitoses per 2 mm2), respectively. In summary, our transcriptomic analysis revealed that a subset of borderline neuroendocrine carcinomas have genomic characteristics similar to pulmonary carcinoids. Moreover, we found that patients in low proliferation cluster (20%) had a better recurrence-free survival (see, Table 1) compared with patients in high proliferation cluster (75%).

a Molecular characterization of tumors using transcriptomic data: Hierarchical clustering of samples based on gene expression data. Three borderline cases (S8, S9, S10) highlighted in red color font were clustered with low proliferative group and one borderline case (S11) highlighted in red color font was clustered with high proliferative group; b Differences in mitotic rates between the two clustering groups; c Differences in Ki-67 values between the two clustering groups.
To identify the differences between the two groups obtained from the hierarchical clustering, we performed Wilcoxon test on the mitotic rates and Ki-67 values between the two clusters. We observed a statistically significant difference between the low and high proliferative clusters with a p value = 4e–04 (Fig. 2b) for mitotic rates. In addition, we also found a significant difference between the low and high proliferative groups with a p value = 4.6e–05 for Ki-67 values (Fig. 2c).
Next, we analyzed differentially expressed genes between the two groups, C1 and C2. For an FDR of 5%, we found 747 genes to be differentially expressed between the groups. Based on the p value, the top five genes that are differentially expressed are SYT13, CAPG, CAMK2B, OSBPL3, and IGF2BP2. The gene expression of differentially expressed genes is presented as a heatmap (Fig. 3), in which the rows are scaled to have mean zero and variance one. The row dendrogram was computed and reordered based on row means. In this way, samples with similar profiles are together and shared gene expression profiles become more apparent.

Heatmap of differentially expressed genes between the low and high proliferative groups (for an FDR < 5%).
Discussion
Lung neuroendocrine tumors represent a heterogeneous group of malignancies with distinct morphological patterns for which accurate diagnosis and classification has been a long-standing debate in clinic. The WHO classification criteria of 1999 and 2004 delineated pulmonary neuroendocrine tumors based on mitotic counts and absence/presence of necrosis. The thresholds for mitotic counts for pulmonary neuroendocrine tumors were largely established in epidemiologic studies using survival and tumor recurrence data [2, 29]. The cohorts included in those earlier studies, however, contained very limited number of cases borderline between carcinoid and LCNEC due to the rarity of these tumors [2, 29].
The International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus recently proposed a common framework for the classification of neuroendocrine tumors. According to these recommandations, neuroendocrine tumors were divided into well-differentiated neuroendocrine neoplasms (WD-neuroendocrine neoplasms) and neuroendocrine carcinomas. WD-neuroendocrine neoplasms at the pulmonary site (or pulmonary carcinoids) were classified as grade 1 (G1) and grade 2 (G2) tumors. The upper limit of mitotic counts was set at 10 mitoses per 2 mm2. However, the agency released a statement regarding the lack of evidence for the classification of borderline tumors, which exceeded the established mitotic cutoff. It was stated that the evidence of higher grade pulmonary WD-neuroendocrine neoplasms is currently limited to a few publications [17, 18], which precluded the introduction of grade 3 (G3) group with higher mitotic counts in the proposed classification. This contrasts with digestive WD-neuroendocrine neoplasms where three grades were introduced along with the incorporation of Ki-67 immunomarker in the grading system. The lack of studies on borderline neuroendocrine tumors is related to their rarity, which precludes most meaningful epidemiologic studies based on the clinical outcome data even though attempts were made along these lines [30]. IARC consensus concluded that existence and characterization of G3 WD-pulmonary neuroendocrine neoplasms, which warrants further research [31].
One of the challenging areas is the classification of atypical carcinoids that have increased proliferation with >10 mitoses per 2 mm2. One of the earliest study was done by Megyesi et al., where the authors described a carcinoid-type lung tumor with increased proliferation [12]. Klöppel et al. in their study on pancreatic neuroendocrine tumors have shown that few cases indeed exceeded the grade 2 proliferative criteria [32]. These samples were later recognized as grade 3 tumors by the WHO guidelines. In a study carried out by Rekhtman et al., the authors have reported that there are a few cases in lung neuroendocrine tumors that have carcinoid-like molecular features with elevated proliferation [14]. A study by Quinn et al. described 12 more cases and illustrated that their clinicopathological features were more similar to carcinoids [13]. In a more recent study by Rekhtman et al., the authors observed mitotic index exceeding 10 mitoses per 2 mm2 among 35% of patients with matched primary and metastatic samples [16]. In this regard, there has been a lack of studies that investigated the lung pulmonary borderline tumors defined by mitotic rates higher than 10 mitoses per 2 mm2, but lying on the lower end of spectrum for LCNEC. To obtain more insight into these rare tumors, we conducted a comprehensive clinical, pathological and transcriptomic characterization. It has been recently shown that WHO criteria are unable to robustly characterize the tumors that lie in the vast gray area between atypical carcinoids and LCNEC, for instance in thymus [24]. Hence, in this study, we defined borderline cases as those tumors that have mitotic count between 10 and 30 mitoses per 2 mm2 [23, 24]. We characterized four tumors in our cohort as borderline cases (i.e., S8, S9, S10, and S11). Firstly, we evaluated the histological features of these four borderline samples. Based on our findings, three of them exhibited morphology with solid, nested, trabecular architectural pattern, or displayed rosettes, had low-grade nuclei with salt and pepper chromatin, low nuclei-cytoplasmic ratio, and only focal necrosis, which are characteristics of atypical carcinoids. While, one tumor displayed features consistent with high-grade carcinoma. We also evaluated the tumors using p53 and RB1 immunohistochemical markers. In our cohort, four LCNEC’s expressed RB1 protein loss while all carcinoids showed preserved RB1 staining and patchy pattern of p53 expression corresponding to normal staining. All borderline tumors had preserved RB1 and p53 expression. These results concord with the existing literature showing RB1 and TP53 genetic alteration in a subset of LCNEC only and suggest that two markers alone are insufficient to rule out LCNEC [14, 30, 33]. However, a larger molecular panel would be a useful diagnostic tool in the evaluation of borderline tumors. Along these lines, efforts are currently underway to propose a clinically relevant molecular signature [34].
Secondly, we used transcriptomic data to test our hypothesis that a subset of borderline tumors belongs to atypical carcinoid subtype. Our analysis identified two distinct molecular groups with different clinical phenotypes. One cluster was characterized by a lower mitotic count and Ki-67 index, and another cluster was a representative of higher mitotic count and Ki-67 index. In total, three borderline cases were clustered with low proliferative index group and had mitotic counts of 10–20 mitoses per 2 mm2, while one borderline case was clustered with high proliferative group. Our findings are in line with other studies from the literature [17, 35, 36]. Similar findings were also recently reported by Vivero and Scholl, where they showed that genomic analysis carried out on a subset of carcinoid-like tumors with increased proliferation represent atypical carcinoids [35]. More recently, Alcala et al. showed that the separation between pulmonary carcinoids and LCNEC might be more subtle for a subset of patients, and they identified a subgroup of atypical carcinoids, named supra-carcinoids, that has carcinoid-like morphological pattern but with molecular characteristics similar to LCNEC [22]. Taken together, the above histological and molecular findings support the conclusion that a subset of borderline tumors with elevated proliferation resemble atypical carcinoids, and not LCNEC.
Our findings support to the emerging concept that that neuroendocrine tumors with carcinoid-like features and mitotic count of <20 mitoses per 2 mm2 should be regarded as pulmonary carcinoids instead of LCNEC. These observations were also made in pancreatic neuroendocrine tumors that exhibited morphologic and molecular characteristics similar to grade 1–2 neuroendocrine tumors. This has resulted in the recommendation to classify such tumors as grade 3 neuroendocrine tumors rather than carcinomas [15, 37]. Our study suggests that similar recommendations may be applicable in lung neuroendocrine tumors and should be incorporated to the WHO diagnostic grading system.
Although our results are of great interest from the clinical standpoint, there are some limitations associated with the current study, which includes, (i) Interobserver variability: there is a considerable interobserver variation in the histopathologic characterization of neuroendocrine lung tumors, in particular, samples with low to medium mitotic count [38]. Additional immunohistochemical markers are necessary to improve classification and prediction of prognosis. (ii) Sample size: due to the rarity of these borderline cases, the sample size of our cohort is small. However, the study design still allowed conclusive statements. Analysis on the larger cohort will potentially pave the way to build robust biomarkers using the molecular data for classification of lung neuroendocrine tumors. Overall, clustering analysis of the present study supports the evidence that low-grade neuroendocrine tumors are biologically distinct from neuroendocrine carcinomas. Our results illustrate that the spectrum of mitotic counts can be larger than the one presented in the WHO classification criteria. Although the upper value for the borderline tumors cannot be deduced from our study, however, our work will guide further research, to establish a robust mitotic range that will allow better classification of lung neuroendocrine tumors. This study also provides a basis to integrate molecular profiling into the histological classification, to achieve a more accurate stratification of lung neuroendocrine tumors. These findings have potential implications in the clinical management of lung neuroendocrine tumors. In addition, our findings also lay the foundation to build clinically relevant biomarkers, which can be augmented with conventional pathological variables for primary diagnosis.
References
Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10:1243–60.
Travis WD, Rush W, Flieder DB, Falk R, Fleming MV, Gal AA, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22:934–44.
Travis WD. Pathology and diagnosis of neuroendocrine tumors: lung neuroendocrine. Thorac Surg Clin. 2014;24:257–66.
Hendifar AE, Marchevsky AM, Tuli R. Neuroendocrine tumors of the lung: current challenges and advances in the diagnosis and management of well-differentiated disease. J Thorac Oncol. 2017;12:425–36.
Fernandez-Cuesta L, Peifer M, Lu X, Sun R, Ozretić L, Seidal D, et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat Commun. 2014;5:3518.
Simbolo M, Mafficini A, Sikora KO, Fassan M, Barbi S, Corbo V, et al. Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J Pathol. 2017;241:488–500.
Derks JL, Leblay N, Lantuejoul S, Dingemans AC, Speel EM, Fernandez-Cuesta L. New Insights into the molecular characteristics of pulmonary carcinoids and large cell neuroendocrine carcinomas, and the impact on their clinical management. J Thorac Oncol. 2018;13:752–66.
George J, Walter V, Peifer M, Alexandrov LB, Seidel D, Leenders F, et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat Commun. 2018;9:1048.
Umemura S, Mimaki S, Makinoshima H, Tada S, Ishii G, Ohmatsu H, et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J Thorac Oncol. 2014;9:1324–31.
Vollbrecht C, Werner R, Walter RFH, Christoph DC, Heukamp LC, Peifer M, et al. Mutational analysis of pulmonary tumours with neuroendocrine features using targeted massive parallel sequencing: a comparison of a neglected tumour group. Br J Cancer. 2015;113:1704–11.
Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. WHO classification of tumors of the lung, pleura, thymus and heart. 4th ed. Lyon: International Agency for Research on Cancer (IARC); 2015.
Megyesi M, Berta M, Khoor A. Endobronchial large cell neuroendocrine carcinoma. Pathol Oncol Res. 2003;9:198–200.
Quinn AM, Chaturvedi A, Nonaka D. High-grade neuroendocrine carcinoma of the lung with carcinoid morphology: a study of 12 cases. Am J Surg Pathol. 2017;41:263–70.
Rekhtman N, Pietanza MC, Hellmann MD, Naidoo J, Arora A, Won H, et al. Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin Cancer Res. 2016;22:3618–29.
Jones MH, Virtanen C, Honjoh D, Miyoshi T, Satoh Y, Okumura S, et al. Two prognostically significant subtypes of high-grade lung neuroendocrine tumours independent of small-cell and large-cell neuroendocrine carcinomas identified by gene expression profiles. Lancet. 2004;363:775–81.
Rekhtman N, Desmeules P, Litvak AM, Pietanza MC, Santos-Zabala ML, Ni A, et al. Stage IV lung carcinoids: spectrum and evolution of proliferation rate, focusing on variants with elevated proliferation indices. Mod Pathol. 2019;32:1106–22.
Simbolo M, Barbi S, Fassan M, Mafficini A, Ali G, Vicentini C, et al. Gene expression profiling of lung atypical carcinoids and large cell neuroendocrine carcinomas identifies three transcriptomic subtypes with specific genomic alterations. J Thorac Oncol. 2019;14:1651–61.
Alcala N, Leblay N, Gabriel AAG, Mangiante L, Hervas D, Giffon T, et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat Commun. 2019;10:3407.
Raj N, Valentino E, Capanu M, Tang LH, Basturk O, Untch BR, et al. Treatment response and outcomes of grade 3 pancreatic neuroendocrine neoplasms based on morphology: well differentiated versus poorly differentiated. Pancreas. 2017;46:296–301.
Heetfeld M, Chougnet CN, Olsen IH, Rinke A, Borbath I, Crespo G, et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2015;22:657–64.
Hijioka S, Hosoda W, Matsuo K, Ueno M, Furukawa M, Yoshitomi H, et al. Rb loss and mutation are predictors of the response to platinum-based chemotherapy in pancreatic neuroendocrine neoplasm with grade 3: a Japanese Multicenter Pancreatic NEN-G3 study. Clin Cancer Res. 2017;23:4625–32.
Vélayoudom-Céphise FL, Duvillard P, Foucan L, Hadoux J, Chougnet CN, Leboulleux S, et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr Relat Cancer. 2013;20:649–57.
Rindi G, Klersy C, Inzani F, Fellegara G, Ampollini L, Ardizzoni A, et al. Grading the neuroendocrine tumors of the lung: an evidence-based proposal. Endocr-Relat Cancer. 2014;21:1–16.
Dinter H, Bohnenberger H, Beck J, Bornemann-Kolatzki K, Schütz E, Küffer S, et al. Molecular classification of neuroendocrine tumors of the thymus. J Thorac Oncol. 2019;14:1472–83.
Rindi G, Arnold R, Bosman FT, Carlo C, Klimstra DS, Klöppel G, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: WHO Classification Tumours of the Digestive System, 4th ed, Bosman TF, Carneiro F, Hruban RH, Theise ND (Eds), International Agency for Reseach on Cancer (IARC), Lyon 2010. p. 13.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat Biotechnol. 2016;34:525–7.
Gendoo DMA, Zon M, Sandhu V, Manem VSK, Ratanasirigulchai N, Chen GM, et al. MetaGxData: Clinically Annotated Breast, Ovarian and Pancreatic Cancer Datasets and Their Use in Generating a Multi-Cancer Gene Signature. Sci Rep. 2019;9:8770.
Paquet ER, Lesurf R, Tofigh A, Dumeaux V, Hallett MT. Detecting Gene Signature Activation in Breast Cancer in an Absolute, Single-Patient Manner. Breast Cancer Res. 2017;19:32.
Beasley MB, Thunnissen FB, Brambilla E, Hasleton P, Steele R, Hammar SP, et al. Pulmonary Atypical Carcinoid: Predictors of Survival in 106 Cases. Hum Pathol. 2000;31:1255–65.
Kasajima A, Konukiewitz B, Oka N, Suzuki H, Sakurada A, Okada Y, et al. Clinicopathological Profiling of Lung Carcinoids with a Ki67 Index > 20. Neuroendocrinology. 2019;108:109–20.
Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A Common Classification Framework for Neuroendocrine Neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) Expert Consensus Proposal. Mod Pathol 2018;31:1770–86.
Klöppel GCA, Couvelard A, Hruban RH, Klimstra DS, Komminoth P, Osamura RY, et al. Neoplasms of the neuroendocrine pancreas, introduction. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J (Eds). WHO Classification of Tumours of Endocrine Organs. Lyon: IARC; 2017.
Beasley MB, Lantuejoul S, Abbondanzo S, Chu WS, Hasleton PS, Travis WD, et al. The P16/cyclin D1/Rb Pathway in Neuroendocrine Tumors of the Lung. Hum Pathol. 2003;34:136–42.
Laddha SV, da Silva EM, Robzyk K, Untch BR, Ke H, Rekhtman N, et al. Integrative Genomic Characterization Identifies Molecular Subtypes of Lung Carcinoids. Cancer Res. 2019;79:4339–47.
Vivero M, Scholl LM. “Borderline” neuroendocrine carcinomas of the lung are clinically and genomically distinct from large cell neuroendocrine carcinoma. Mod Pathol. 2016;29(suppl 2):485A.
Marchiò C, Gatti G, Massa F, Bertero L, Filosso P, Pelosi G, et al. Distinctive Pathological and Clinical Features of Lung Carcinoids with High Proliferation Index. Virchows Arch. 2017;471:713–20.
Tang LH, Untch BR, Reidy DL, O’Reilly E, Dhall D, Jih L, et al. Well-Differentiated Neuroendocrine Tumors with a Morphologically Apparent High-Grade Component: A Pathway Distinct from Poorly Differentiated Neuroendocrine Carcinomas. Clin Cancer Res. 2016;22:1011–17.
Swarts DR, van Suylen RJ, den Bakker MA, van Oosterhout MF, Thunnissen FB, Volante M, et al. Interobserver Variability for the WHO Classification of Pulmonary Carcinoids. Am J Surg Pathol. 2014;38:1429–36.
Acknowledgements
We would like to thank our colleagues, Christian Couture, Sylvain Trahan and Sylvain Pagé. Philippe Joubert is a member of the “Cancer, Environment and Genetic” axis of the Quebec Respiratory Health Network. Yohan Bossé holds a Canada Research Chair in Genomics of Heart and Lung Diseases. This project was supported by the Fonds J.D. Bégin. The authors would also like to thank the team at the Institut universitaire de cardiologie et de pneumologie de Québec (IUCPQ) site of the Respiratory Health Network (RHN) Tissue Bank of the Fonds de recherche Québec–Santé (FRQS) (www.tissuebank.ca) for their valuable assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sazonova, O., Manem, V., Orain, M. et al. Transcriptomic data helps refining classification of pulmonary carcinoid tumors with increased mitotic counts. Mod Pathol 33, 1712–1721 (2020). https://doi.org/10.1038/s41379-020-0538-8
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41379-020-0538-8
This article is cited by
-
Diagnostic relevance of p53 and Rb status in neuroendocrine tumors G3 from different organs: an immunohistochemical study of 465 high-grade neuroendocrine neoplasms
Virchows Archiv (2025)
-
Digital Immunophenotyping of Lung Atypical Carcinoids and Large Cell Neuroendocrine Carcinomas Identifies Three Subtypes With Specific Tumor-Immune Microenvironment Features
Endocrine Pathology (2025)
-
Altered splicing machinery in lung carcinoids unveils NOVA1, PRPF8 and SRSF10 as novel candidates to understand tumor biology and expand biomarker discovery
Journal of Translational Medicine (2023)
-
Clinic and genetic similarity assessments of atypical carcinoid, neuroendocrine neoplasm with atypical carcinoid morphology and elevated mitotic count and large cell neuroendocrine carcinoma
BMC Cancer (2022)
-
Prognostic significance of laterality in lung neuroendocrine tumors
Endocrine (2022)
