
Abstract
Amyloid accumulation in Alzheimer’s disease (AD) is associated with synaptic damage and altered connectivity in brain networks. While measures of amyloid accumulation and biochemical changes in mouse models have utility for translational studies of certain therapeutics, preclinical analysis of altered brain connectivity using clinically relevant fMRI measures has not been well developed for agents intended to improve neural networks. Here, we conduct a longitudinal study in a double knock-in mouse model for AD (AppNL-G-F/hMapt), monitoring brain connectivity by means of resting-state fMRI. While the 4-month-old AD mice are indistinguishable from wild-type controls (WT), decreased connectivity in the default-mode network is significant for the AD mice relative to WT mice by 6 months of age and is pronounced by 9 months of age. In a second cohort of 20-month-old mice with persistent functional connectivity deficits for AD relative to WT, we assess the impact of two-months of oral treatment with a silent allosteric modulator of mGluR5 (BMS-984923/ALX001) known to rescue synaptic density. Functional connectivity deficits in the aged AD mice are reversed by the mGluR5-directed treatment. The longitudinal application of fMRI has enabled us to define the preclinical time trajectory of AD-related changes in functional connectivity, and to demonstrate a translatable metric for monitoring disease emergence, progression, and response to synapse-rescuing treatment.
Similar content being viewed by others

Introduction
It can take decades for the hallmarks of Alzheimer’s disease (AD) to manifest clinically. Optimistically, this is evidence of a long therapeutic window during which treatment could interrupt or reverse disease processes, provided we have access to ways of identifying patients, staging disease, and effective therapeutics. Functional magnetic resonance imaging (fMRI) is a strong candidate for helping to address these needs, given that it is non-invasive and can assay the entire brain. BOLD (blood-oxygen-level dependent) fMRI yields measures of functional connectivity (FC, or inter-regional BOLD signal synchrony). In AD, robust changes in FC are observed, with brain-network specificity, years prior to the clinical manifestation of neurodegenerative signatures of disease [1,2,3,4,5,6,7,8,9,10]. Changes in FC progress in concert with AD-related declines in executive function and cognition, lending confidence to the notion that aberrant FC is a harbinger of neurodegeneration. Further, changes in FC follow a similar spatial trajectory as subsequent brain atrophy, Aβ-pathology, neurofibrillary tangle deposition [11, 12], and synapse losses [13,14,15,16]. In sum, BOLD-fMRI FC may be sensitive to the varied stages of AD emergence and progression. However, despite tremendous effort, and investment, human fMRI research has yet to produce actionable neuroimaging markers of AD [4]. In part, this is due to the long time-scale and complexity of AD pathology, the difficulty of relating fMRI markers to underlying mechanisms, and substantial heterogeneity within the population.
To help address these gaps, the application of BOLD-fMRI in animal models of AD is fundamental for improving our understanding of the imaging correlates of disease and treatment response [17,18,19]. Studies in animals provide a well-controlled environment for measuring disease phenotypes and testing therapeutics on a tractable timescale, as well as the freedom to obtain invasive measurements. The short lifespan, low-cost, and genetic malleability of mice, together with recent improvements in rodent BOLD-fMRI data quality and processing techniques [20,21,22,23,24] (including inter-species translation) [25, 26], mean the field is well-positioned to make new inroads into AD research. Advances in how we model AD in mice, where it has been hard to faithfully recapitulate human disease [27], lends further cause for enthusiasm. Here, we use the double-knock-in (DKI) AppNL-G-F/hMapt (amyloid-precursor-protein/human microtubule-associated protein tau) mouse model [28, 29], which critically avoids overexpression artifacts. With this model, we seek to establish BOLD-fMRI FC correlates of AD emergence, progression, and treatment response. We collect longitudinal resting-state BOLD-fMRI data spanning a wide-range of AD-relevant timepoints, from 4 to 22 months of age (M) or the rough equivalent of 20–70 years of age in humans [30]. In late-stage disease (20–22 M), we treat a subgroup of mice with a metabotropic glutamate receptor 5 (mGluR5) silent allosteric modulator (SAM, BMS-984923/ALX001) [31]. Vitally, treatment is given during very late-stage disease which is in stark contrast to existing anti-amyloid drugs which have only shown efficacy during early (asymptomatic) illness [32]. Further, this compound is known to modify AD course by rescuing synapses and improving memory [33], and has recently entered clinical trials (ClinicalTrials.gov; NCT05804383). However, the treatment-elicited effects on BOLD-fMRI FC are currently unknown. We seek to gain this knowledge given the potential usefulness of BOLD-fMRI FC for monitoring disease and treatment response.
We find that FC in AD mice follows a distinct age-related trajectory relative to wild-type (WT) littermates. In AD mice, changes emerge later and show more widespread decreases in FC with time, compared to WT mice. The age and AD-related brain networks we identify using data-driven approaches show significant enrichment for (i.e., overlapped representation that is above chance with) established circuits including the default mode and lateral cortical networks (DMN and LCN, respectively) [34, 35]. Further, oral treatment with BMS-984923 normalizes or reverses FC deficits in AD mice. In sum, this work identifies age- and AD-related phenotypes in BOLD-fMRI data across the mouse lifespan in a relevant model of disease [28, 29]. We also find timely evidence that BOLD-fMRI FC may prove useful for assessing the effectiveness of a novel synapse-rescuing treatment.
Materials and methods
All procedures were approved by the Yale Institutional Animal Care and Use Committee (IACUC) and followed the National Institute of Health Guide for the Care and Use of Laboratory Animals. Both biological sexes were included. Mice were group housed on a 12-hour light/dark cycle with ad libitum food and water. Cages were individually ventilated as well as temperature and humidity controlled.
Model of Alzheimer’s disease
Among available AD models there are amyloid-precursor-protein (APP) knock-in (KI) strains [28, 29]. Here, we used AppNL-G-F KI mice where aβ-containing exons from human APP with Swedish, Arctic, and Iberian mutations were included [28]. In addition, mice were crossed with a strain in which the human wild-type microtubule-associated protein tau (MAPT) locus, with its full sequence and splice sites, replaced the murine locus to generate AppNL-G-F/hMapt homozygous double-knock-in animals [28]. This strain has been back-crossed with C57Bl6J for >10 generations, with strain matching confirmed by genetic markers. Male and female AD and WT littermate controls were included.
Dataset overview
A total of N = 74 animals were included from across three experiments (Fig. 1, Supplementary Table 1). In the first experiment, N = 27 animals underwent transverse sinus injections on postnatal day zero (P0) which induced whole-brain fluorescence (adeno-associated virus, AAV, purchased from https://www.addgene.org/; AAV.Syn.GCaMP6s.WPRE.SV40 - #100843-AAV9) [36]. These mice then underwent head-post surgery at 3 M and multimodal imaging at 4, 6, and 9 M where wide-field calcium (WF-Ca2+) and fMRI data were collected simultaneously [37,38,39]. The WF-Ca2+ imaging data are the subject matter of a separate publication. In the second experiment, N = 16 animals underwent head-post surgery at 5 or 8 M and unimodal imaging (fMRI-only) at 6 or 9 M, respectively. These data were collected to address attrition in the first experiment. In the final experiment, N = 30 animals underwent head-post surgery at 19 M and unimodal imaging (fMRI-only) at 20 and 22 M. Between these imaging sessions, AD mice were split between a treatment (N = 11) and vehicle (N = 9) subgroup.

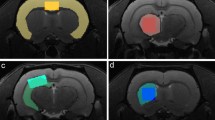
a A 172 ROIs-atlas is used to generate the connectomes. Color-coded subdivision in the three networks of the triple network model: default-mode network (DMN) in green, salience network (SN) in pink, lateral-cortical network (LCN) in orange; regions in common across all three networks (light blue), regions shared by DMN and LCN (light purple) are also shown. b Example of average connectomes shown (2 regions removed from the connectomes images—but kept for all analyses—solely for visualization purposes, i.e. smaller than 10 voxels). c AD-like pathology trajectory illustrated on the timeline in warm colors [28, 29, 31]: β-amyloid accumulation (yellow), synapse loss (orange) and phosphorylated-tau accumulation (red). Decreasing performance on memory tests (e.g., Y- maze test) [28] in blue. Imaging timepoints (MRI, orange) indicated by black dotted arrows. A month before the first imaging timepoint (in each group, Supplementary Table 1), all mice undergo a minimally invasive surgical procedure [37] where a head-plate (HP) is affixed to the skull to reduce susceptibility artifacts and motion (yellow). The treatment (Tx) period is indicated on the timeline (purple). Average Z-scored connectomes/group/timepoint are shown above the timeline. Data from WT mice are outlined in grey (lower left). Data from AD (and ADvehicle/ADveh) groups are outlined in black (upper right). Data from the ADTx group (at 22 M) are outlined in purple. The number of mice (‘N’) and runs (‘n’) indicated above each connectome. ROIs: regions of interest. Animal breakdown: 4 M: NWT = 5/5 M/F; NAD = 7/8 M/F; 6 M: NWT = 5/5 M/F; NAD = 5/7 M/F; 9 M: NWT = 6/4 M/F; NAD = 6/5 M/F; 20 M: NWT = 5/3 M/F; NAD = 12/7 M/F; 22 M: NWT = 5/0 M/F; NADveh = 6/3 M/F; NADTx = 5/4 M/F.
Head-implant surgery
Surgical details are provided in the Supplementary Material.
Data acquisition
All mice underwent imaging under anesthesia (free-breathing low-dose isoflurane, 0.5–0.75%, in 50/50 medical air/O2). The MRI data were acquired on an 11.7 T preclinical magnet (Bruker, Billerica, MA), using ParaVision version 6.0.1 software. We used an in-house built saddle coil which dovetailed with the implanted head-plates [37]. Body temperature was monitored (Neoptix fiber), maintained with a circulating water bath at 36.6–37 °C, and recorded (Spike2, Cambridge Electronic Design Limited, Master-8 A.M.P.I.). Breathing rate was measured using a foam respiration pad (SAM-32, Starr), but was not used to trigger imaging.
Structural MRI data acquisition
At every imaging session, three structural images were acquired. Two of which were needed for multi-modal data registration [37]. For unimodal data registration (and the present study), only one was needed [39]: a whole-brain isotropic 3D-image using a multi spin multi echo (MSME) sequence, with 0.2 × 0.2 × 0.2 mm3 resolution, repetition time and echo time (TR/TE) of 5500/20 ms, 78 slices, and 2 averages (11 mins and 44 s). For the remaining two structural images see Supplementary Material.
Functional MRI data acquisition
Data were acquired using a gradient-echo, echo-planar-imaging (GE-EPI) sequence with a TR/TE of 1.8 s/11 ms. Data were collected across 25 slices without gaps at 0.31 × 0.31 × 0.31 mm3; resulting in near whole-brain coverage. Each run had 334 repetitions, corresponding to 10 min of data per run. We acquired 3 runs per session for a total of 30 mins of data per mouse per imaging session. For examples of raw images and other data quality metrics see Supplementary Fig. 1. An example ‘method’ file (native Bruker format) containing all parameters is included in our supplementary documentation for both EPI and MSME sequences (BrukerMethods_EPI, BrukerMethods_MSME, respectively).
Data pre-processing and analysis
Functional MRI pre-processing and registration to common space
Data registration and pre-processing were performed using RABIES (Rodent automated BOLD improvement of EPI sequences) v0.4.8 (https://rabies.readthedocs.io/en/stable/) [40]. All functional and isotropic MSME structural scans (from all imaging sessions and experiments, Supplementary Table 1) were input and pre-processed together. Slice time correction was applied on native space timeseries, and head motion parameters were estimated.
The isotropic MSME structural images (one per mouse per session) were corrected for inhomogeneities (N3 nonparametric nonuniform intensity normalization) [41, 42], non-linearly registered and then averaged to create a within-sample template. The within-sample template was non-linearly registered to an out-of-sample template we created from N = 162 datasets (collected on the same scanner from N = 64 mice from a separate longitudinal study which used the same isotropic MSME sequence), see Supplementary Material [39]. The out-of-sample template was registered to the Allen Atlas reference space Common Coordinate Framework Reference Atlas (CCfv3) [39, 43, 44]. For more details on pre-processing steps, registrations, and denoising see Supplementary Material.
Exclusion criteria
Of all N = 74, N = 6 animals and n = 2 runs were excluded. N = 1 for enlarged ventricles, N = 1 for a brain tumor, N = 2 for anaesthesia malfunction (i.e., tubes were improperly connected, N = 1 at 4 M and N = 1 at 6 M), and N = 2 due to implant detachment (caused by a glass coating that was not present in other surgeries). An additional n = 2 runs were excluded (n = 1 at 6 M and n = 1 at 9 M) due to image registration failures (see, Supplementary Material). Data quality was inspected using the RABIES --data diagnosis toolkit [40]. No data were excluded due to a low signal to noise ratio (SNR) or motion (framewise displacement, FD > 0.075 mm in over 1/3rd of frames per run).
Connectomes
The BOLD functional scans of individual animals were transformed into a common template space, where the template was derived based on separate cohort of animals. In this template space, we defined a 172-regions of interest (ROIs) atlas from Allen CCvf3. The ROIs were selected based on the resolution of our fMRI data (a minimum of 6 voxels per ROI) (Fig. 1a). To compute connectomes, BOLD signals within each ROI were spatially averaged, and the inter-regional Pearson’s correlation computed (Fig. 1b). Results were computed for each run using data in the out-of-sample template space. Connectomes were converted to z-scores using the fisher r to z transform. For a description of the data-derived networks and the triple network model see Supplementary Material. The 172-ROIs atlas and MRI template space are shared as part of BioImage Suite (https://bioimagesuiteweb.github).
Treatment
Drug synthesis, incorporation into diet, feeding, and validation were based on previous work. See summary in Supplementary Material.
Analyses and statistics
Three major contrasts were examined in this work: (i) aging (in WT and AD mice) whereby older animals (6, 9, 20 and 22 M) were compared to the youngest animals (4 M) to investigate changes in FC during aging; (ii) pathology whereby changes in FC during AD progression were investigated at each timepoint, by comparing AD to age-matched WT controls; and (iii) treatment, whereby the effects of treatment in AD mice (ADtreated) were compared to both age-matched WT mice (WTvehicle), and age-matched AD mice (ADvehicle) that were given the vehicle (regular chow).
For each comparison (i–iii), group-averaged connectomes (Fig. 1c) were subtracted (e.g., average WT connectome at 6 M – average WT connectome at 4 M) resulting in t-value maps. Significant changes in connectomes and the associated t-statistic maps were shown in each related figure, for each comparison (i–iii) as red/blue matrices of t-values (e.g. WT connectome at 6 M vs WT connectome at 4 M, in Fig. 2 top left matrix with bottom half showing unthresholded t-values, top half showing t-values thresholded with p <0.05 corrected for multiple comparison, see below).

Two-sample t-tests are applied to uncover age-related changes in FC within WT and AD mice. In both groups, the earliest timepoint (4 M) is used as a baseline for all subsequent imaging sessions. Four stages of ‘aging’ are captured: early (6 M vs. 4 M), mid (9 M vs. 4 M), late (20 M vs. 4 M) and very late (22 M vs. 4 M). For connectomes, t-test results are displayed without (bottom-half), and with (top-half) statistical threshold (corrected, p <0.05), for WT (top row) and AD (bottom row). All other displays, vertical histograms of node degree (WT, top row; AD, bottom row), circle plots and node degree maps (WT, left panels in each pair; AD, right panels in each pair), are shown after the statistical thresholds. For the brain region labels (rainbow) color-scale, see Fig. 1a. Positive t-values (red) indicate greater FC strength in older vs. younger mice. Negative values (blue) indicate the opposite. Node degree maps are displayed on anatomical reference images (greyscale). Clear and distinct aging effects are observed in both groups. The WT group shows an overall increase in FC strength in older vs. younger mice which emerges at 9 M, persists at 20 M, and shows some evidence of waning at 22 M. Conversely, the AD group shows an overall decrease in FC strength in older vs. younger mice which emerges at 20 M and persists until 22 M. As a data reduction strategy, we summarize these data at the indicated timepoints for each group (Fig. 3, Supplementary Fig. 3). MB midbrain, VIS visual, MO motor, AUD auditory, SS somatosensory, ACA anterior cingulate, RSP retrosplenial, TH thalamus, HY hypothalamus, CBX cerebellum, PERI perirhinal, ENT entorhinal, PIR piriform, CP caudoputamen, ACB nucleus accumbens, AI agranular insula. Triple network color-coding: default-mode network (DMN) in green, salience network (SN) in pink, lateral-cortical network (LCN) in orange; regions in common across all three networks (light blue), regions shared by DMN and LCN (light purple) are also shown.
Statistical significance tests were computed in MATLAB (R2021bV5 and R2023b). Differences between individual edges were tested using a two-sample t-test (MATLAB, ttest2), and multiple comparison correction was applied using the Benjamini-Hochberg method, for all tests. For assessing treatment effects, a one-way ANOVA (MATLAB, anova1) was used to test if there were any pair of groups (WTvehicle vs. ADvehicle, WTvehicle vs ADtreated, and ADvehicle vs ADtreated) that were significantly different from the others. A follow-up t-test was applied to identify which two groups were significantly different.
The statistically significant edges in each t-value map formed two data-derived maps: one composed of hyper-connected edges (i.e., a positive map or network), and the other hypo-connected edges (i.e., a negative map or network). The significance of each map was confirmed with permutation testing (×1000 iterations) where the test statistics of the null distribution were the size of the network of a given comparison, and where the group labels of the data were randomly shuffled. The two-sample t-test statistical threshold was set to p <0.05, two-tailed. The false discovery rate for the Benjamini-Hochberg test was set to 0.05. The test shows that the true group labels led to significantly more different edges than random labelling. For each of these contrasts (i–iii), the two timepoints showing the strongest effects, i.e. the highest number of edges surviving (e.g. in WT aging, 9 M–4 M and 20 M–4 M) were combined to establish data-derived networks, (both by sum and by intersection, more in ‘Results’), per each contrast.
The overlap of each pair of timepoint inputs to each data-derived network were assessed with a Jaccard similarity test and compared to a null distribution (×1000 iterations) to confirm the meaningfulness of the significant overlap found.
Subsequent analyses summarized these data by investigating the over/under enrichment between data-derived networks and a priori networks within the triple-network model. Descriptive statistics were given as mean differences and 25/75th percentiles unless stated otherwise, and graphically represented as box plots (MATLAB, boxplot).
To assess the robustness of our findings to sample size, we randomly selected subsets (N/2) of our full sample (×100 iterations) and repeated our analyses, computing data-derived connectomes for (i) aging, WT and AD, (ii) pathology, and (iii) treatment. As with the full sample, the resultant data-derived networks were characterized in terms of their over/under enrichment for a priori networks in the triple network model. Overlap between the N/2 subsample data-derived networks were compared to the full sample using Dice overlap (MATLAB, dice) and compared to a null distribution.
Results
Averaged connectomes from each group at each timepoint are shown in Fig. 1. Stereotypical patterns including high FC between bilateral brain regions were observed across all groups, at all timepoints (Fig. 1c). Relatively stable temporal signal-to-noise ratio (tSNR) was found across both groups, and at all timepoints (Supplementary Fig. 1). For each contrast (aging, pathology, and treatment), differences between connectomes were displayed with and without thresholding for significance (p <0.05) at the edge-level and correcting for multiple comparisons (‘Material and Methods’). In all results, the test statistic of the null distribution was the number of edges (region-to-region measures of FC) remaining after correcting for multiple comparisons. Identified brain circuits that showed significant effects were summarized as connectomes, histograms, circle plots, and node-degree maps. In each display, ROIs and the triple network model [34, 35] (DMN, LCN, and salience network, SN) are color-coded as shown in Fig. 1a, b.
Age-related changes in functional brain organization diverged between WT and AD mice
AD progression occurs atop a background of age-related changes in behavior and brain function [45, 46]. Here, we assessed how aging affects FC within each genotype (WT or AD) to investigate our hypothesis that age-related changes in FC would diverge between groups beginning in mid-life. We examined age-related changes by quantifying differences between the earliest timepoint (4 M) and all other subsequent timepoints (contrast (i), see ‘Material and Methods’). Four stages of aging were defined: (1) early: 4 vs. 6 M, (2) mid: 4 vs. 9 M, (3) late: 4 vs. 20 M, and (4) very late: 4 vs. 22 M.
Few early-life age-related changes in FC were identified in either group (Fig. 2, permutation test p <0.05). Beginning at mid-life, WT mice showed a widespread increase in FC (Fig. 2, permutation test p <0.001), that was not observed in AD mice (although, a trend in this direction was noticeable in unthresholded and uncorrected data, Fig. 2, permutation test p <0.05). This increase, in the WT group, persisted until the study endpoint. At 20 and 22 M, some decreases in FC were also observed in ventral areas alongside more widespread increases (permutation test p <0.001). AD mice showed a more modest FC increase with age, which emerged later, in conjunction with a pronounced decrease in FC within subcortical regions (Fig. 2). Overall, as hypothesized, WT and AD groups followed distinct patterns of age-related changes in brain functional organization that unfolded along different timescales. As a control, to account for any differences between WT and AD mice at 4 M, we repeated our analyses using all mice (WT and AD) combined in a single group, at 4 M, as a reference (Supplementary Fig. 2). No differences in outcome patterns were observed.
Regions in WT mice that showed pronounced age-related increases in FC beginning at 9 M included somatosensory and motor regions (SS/MO), visual cortices (VIS), midbrain (MB), hypothalamus (HY) and the retrosplenial areas (RSP). AD mice showed more modest increases in the same areas but starting at 20 M. Moreover, AD mice showed a more pronounced decrease in FC in the ventral parts of the brain encompassing the hippocampal formation (HF), entorhinal cortex (ENT), ectorhinal and perirhinal regions (ECT, PERI), ventral striatal areas including the caudoputamen (CP), and piriform and insular regions (PIR, INS).
Aging trajectories in WT and AD mice effected overlapping and distinct networks
In addition to examining which regions were most affected by age, we assessed whether the implicated data-derived networks overlapped with established canonical networks (DMN, LCN, and SN) as well as the edges that connect them (e.g., DMN to LCN). Our hypothesis was that our data-driven networks would contain a high representation of edges within a priori networks that have been previously found to change with age (specifically, the DMN) [47, 48].
Above (Fig. 2), we established that WT and AD mice showed distinct patterns of age-related changes in FC. Here, we defined a data-derived ‘WT-aging’ and an ‘AD-aging’ (Fig. 3a; Supplementary Fig. 3a) network to facilitate a comparison with a priori networks. To do so, we selected both the sum of all edges surviving correction (Fig. 3a, left), and the intersect of the common edges surviving correction (Fig. 3a, right), at the two (most affected, i.e. number of edges involved) timepoints (as indicated in Fig. 2). This established two versions of our data-derived networks; one that was comprehensive (the sum, Fig. 3a left matrices) and one that was stringent (the intersect, Fig. 3a right matrices). To create both versions, we collapsed across the two timepoints that showed the most pronounced effects in each group: mid & late for WT and late & very late for AD (as indicated in Fig. 2). Note that the edges identified in both groups at the chosen stages were highly overlapping (Supplementary Fig. 3, observed Jaccard similarity significantly different from a null distribution, p <0.001, for both groups).

a We consider both the sum (all edges across two stages, left matrices) and intersect (common edges across two stages, right matrices) as a comprehensive and stringent definition of our data-driven aging networks for WT (top matrices) and AD (bottom matrices). Edges at chosen stages (Fig. 2) are highly overlapping (difference from null distribution, p <0.001) for both groups. Aging networks are displayed as connectomes (172 × 172 ROIs, see Fig. 1). Greater FC in older relative to younger mice is shown in red (upper right half matrices in connectomes). Greater FC in younger relative to older mice is shown in blue (lower left half matrices in connectomes). Data are also shown as vertical histograms of node degree (on the right of each matrix); node degree maps displayed on anatomical reference images (greyscale), and circle plots are shown in the bottom half of (a), for WT and AD (left and right respectively). All results have a statistical threshold (corrected, p <0.05) applied. b, c Fisher’s exact test (MATLAB) is used to assess (over/under) enrichment for edges within data-derived networks and the triple-network model: DMN, LCN, and SN, as well as their inter-network pairs (corrected, permutation test ×1000 iterations). These results are depicted using histogram diagrams for WT (b) and AD (c). Positive ( + ve) edges are shown in red (for older > younger) and negative (−ve, younger > older) are shown in blue, top and bottom of (b, c). Real data results are indicated with a grey vertical line and a black triangle. Null distribution is color-coded according to the network investigated (see Fig. 1a). A left-shift from the null distribution indicates under enrichment, a right-shift from the null distribution indicates over enrichment in the networks analyzed. Histograms indicate results from the sum. Significance is displayed as * for p <0.05, ** for p <0.01 and *** for p <0.001. Equivalent findings are uncovered for triple network representation when we use the sum or intersect definition. ENT entorhinal, HF hippocampal formation, RSP retrosplenial, TH thalamus, HY hypothalamus, SS somatosensory, CP caudoputamen, ACB nucleus accumbens, MO motor, VIS visual, ACA anterior cingulate, MB midbrain, PERI perirhinal, AUD auditory, TEa temporal association area, AI agranular insula, OLF olfactory, CBX cerebellar. Triple network color-coding: default-mode network (DMN) in green, salience network (SN) in pink, lateral-cortical network (LCN) in orange; regions in common across all three networks (light blue), regions shared by DMN and LCN (light purple) are also shown.
We computed enrichment (or depletion, under enrichment) for edges within the triple-network model and our aging networks (Fig. 3b, c) using Fisher’s exact test as well as permutation testing (×1000 iterations). Results were corrected for multiple comparisons. We found that the WT-aging positive ( + ve) network (older>younger, Fig. 3b, top) was enriched for edges in the DMN and LCN as well as edges that connect all three networks. The WT-aging negative (−ve) network (older < younger; Fig. 3b, bottom) showed under-enrichment (depletion) for edges within the DMN and between DMN and LCN. Further, the AD-aging ( + ve) network (older > younger; Fig. 3c, top) showed enrichment in the DMN, in the LCN, and DMN to LCN and DMN to SN, and under-enrichment in the SN. While the AD-aging (−ve) network (older < younger; Fig. 3c, bottom) showed under-enrichment in the LCN, DMN to LCN and a trend towards under-enrichment in the DMN. In sum, WT and AD mice showed some common trends for which networks in the triple-network model were implicated in age-related changes in connectivity strength, but also some clear differences.
Decreased FC strength in AD relative to WT mice emerged at 9 M
After establishing aging trajectories for both genotypes (WT and AD), resulting in the formation of two aging networks, WT-aging, and AD-aging, we then computed differences between WT and AD connectomes at each timepoint (contrast (ii), see ‘Material and Methods’). This analysis highlighted the effects of pathology-related changes in FC using age-matched groups (Fig. 4a). As above (for aging) we established a pathology-related data-derived network; hereon the AD-like network (Supplementary Fig. 3b). Our hypothesis was that differences in FC between WT and AD mice would emerge already in mid-life and that, in-line with previous work, e.g., [17, 49, 50], regions that have been previously found to be affected by AD would emerge (including the hippocampal formation and the default-mode regions, e.g., [51, 52]).

a Similar to Fig. 2 we compare WT to AD groups at each timepoint. Positive t-values (red) indicate greater FC strength in AD vs. WT mice. Negative values (blue) indicate the opposite. Vertical histograms summarizing the number of positive/negative edges are displayed next to each matrix. b AD-like network for sum (left matrix) and intersect (right matrix), collapsed across 9 and 20 M timepoints (as indicated in a). AD-like network is also displayed as vertical histograms (right of each matrix), circle plot (middle) and node degree maps (bottom). Right: Fisher’s exact test (MATLAB) is used to assess (over/under) enrichment for edges within data-derived networks and the triple-network model: DMN, LCN, and SN, as well as their inter-network pairs (corrected, permutation test ×1000 iterations). Negative (−ve) edges are shown in blue (for AD < WT). No positive edges survive. Real data results are indicated with a grey vertical line and a black triangle. Null distribution is color-coded according to the network investigated (see Fig. 1a). A left-shift from the null distribution indicates under enrichment, a right-shift from the null distribution indicates over enrichment in the networks analyzed. Histograms indicate results from the sum. Significance is displayed as * for p <0.05, ** for p <0.01 and *** for p <0.001. ENT entorhinal, VIS visual, HF hippocampal formation, RSP retrosplenial, AUD auditory, TH thalamus, Tea temporal association area, SS somatosensory, ACB nucleus accumbens, MO motor, MB midbrain. Triple network color-coding: default-mode network (DMN) in green, salience network (SN) in pink, lateral-cortical network (LCN) in orange; regions in common across all three networks (light blue), regions shared by DMN and LCN (light purple) are also shown.
Edges where FC in AD was greater than WT (AD > WT) were plotted as positive t-values (red). Edges where FC in AD < WT were plotted as negative t-values (blue). Permutation testing was applied (as above). Some very sparse but significant differences between WT and AD mice were observed at early timepoints (4 and 6 M, p <0.05) with strong effects emerging by 9 M (permutation test, p <0.001). Overwhelmingly, AD mice showed lower FC compared to WT (Fig. 4). Notably, affected areas were bilaterally symmetric and appeared throughout the brain but were concentrated in the cortex. Identified regions include the HF, and regions of the DMN as expected. In addition, decreased FC in AD mice was also found in SS, MO, VIS, and RSP areas, as well as the temporal association areas (TeA), thalamus (TH), nucleus accumbens (ACB), auditory (AUD) and MB areas.
The networks most strongly affected in AD relative to WT were the DMN and LCN
As above, we investigated the network composition [35, 53] of the brain circuits most affected in AD relative to WT mice. The most pronounced differences were observed at 9 and 20 M (permutation test, p <0.001, Fig. 4a) and the edges identified at these timepoints were highly overlapping (observed jaccard similarity vs. null distribution, p <0.001). As above, we considered the sum (comprehensive) and intersect (stringent) in our definition of a data-derived ‘AD-like network’ (Fig. 4b, left/middle) to assess its over/under enrichment for edges within the triple network model. Our hypothesis was that we would uncover a high-degree of overlap between our data-derived AD-network and the DMN [7, 52, 54].
As expected, we found that the AD-like (−ve) network (AD < WT) was enriched for edges in the DMN (Fig. 4b, right). Additionally, we found enrichment for edges in the LCN as well as edges that connect the DMN to the LCN, and LCN to SN (Fisher’s exact test; Fig. 4b, right). A trend was noted for under enrichment for edges between the DMN and the SN. There were no significant AD-like ( + ve) network (AD > WT) findings, as such AD-like ( + ve) is not investigated further.
Comparison of mouse aging and AD networks with human literature meta-analysis (Neurosynth)
In an exploratory analysis, we used Neurosynth (http://neurosynth.org) to compare our mouse-model data-derived aging and AD-like networks to human neuroimaging studies using the queries ‘Age’ and ‘Alzheimer’ (Supplementary Fig. 4). Neurosynth accepted one-word queries and provided ‘activation maps’ proportional to ‘hits’ in the literature without directionality (increases/decreases) [55]. As such, our aim was to visualize Age/Alzheimer maps across species for a purely qualitative comparison. Our hypothesis was that regions which have previously been implicated in both species – e.g., HF, dorsolateral areas, visual areas, SS, MO, TH, ACA [17, 46, 49, 56] – would emerge.
Supplementary Fig. 4 shows the association test maps (z-scored, corrected for false discovery rate, FDR = 0.01) [55] from human fMRI data next to the node degree maps we obtained from mouse fMRI data. Encouragingly, in each comparison, homologous regions emerged. Specifically, somatomotor, SS, and dorsolateral-prefrontal areas were implicated in (healthy/WT) aging across species (Supplementary Fig. 4, top), as were VIS, insula and RSP cortices (i.e., including RSP and precuneus in humans) [25]. In AD, subcortical areas, including the HF and TH, appeared in both species (Supplementary Fig. 4, bottom).
Treatment with BMS-984923 reverses FC deficits in AD mice at 22 M
Next, we assessed the effects of 2-months of treatment with BMS-984923 on FC in a subgroup of AD mice (ADtreated, or ADTx; contrast (iii), see ‘Material and Methods’). As this drug has previously been shown to rescue synapses and memory deficits (in AD animals) [31, 33], we hypothesized some degree of reversal (or pseudo-normalization) of AD-related FC deficits. We characterized treatment effects using three comparisons: (1) 22 M WTvehicle (or WTveh) vs. 22 M ADTx (Fig. 5a), (2) 22 M ADTx vs. 20 M ADveh (Fig. 5b, left), and (3) 22 M ADveh vs. 22 M ADTx (Fig. 5b, right). Across all comparisons we observed evidence of treatment effects. In comparison (1), there was a lack of FC differences between 22 M WTveh and 22 M ADTx (Fig. 5a right, p > 0.5 for the permutation test), even though there was a persistent difference between 22 M WTveh and 22 M ADveh (Fig. 5a, left, reproduced from Fig. 4, p <0.001 for the permutation test). Comparisons (2) and (3) highlighted edges whose strengths changed with treatment. We selected the brain circuits identified in comparisons (2) and (3) to generate, as before, a comprehensive and a stringent data-derived ‘treatment network’ (Fig. 5c, left/middle; summary of all data-derived networks in Supplementary Fig. 3c).

a Differences in FC strength between WTveh and ADveh (left, replicated from Fig. 4a) as well as WTveh and ADTx (right) at 22 M. Positive t-values (red) indicate increased FC strength in ADveh vs. WTveh mice (left) or increased FC strength in ADTx vs. WTveh mice (right). Negative t-values (blue) indicated decreased FC strength in ADveh vs. WTveh mice (left) or decreased FC strength in ADTx vs. WTveh mice (right). Treatment effects are also displayed as vertical histograms of number of positive/negative edges (right side of each matrix), node degree maps and circle plots (below each matrix). BMS-984923 treatment appears to globally reduce FC strength differences between groups at 22 M (two-sampled t-test, MATLAB, ttest2). b Treatment effects in ADTx (22 M) relative to age-matched ADveh mice (right) or ADveh mice at 20 M (left). Positive t-values (red) indicate increased FC in ADTx vs. ADveh mice. Negative t-values (blue) indicate decreased FC strength in ADTx vs. ADveh mice. c. As in Figs. 2, 4a except here, we collapse across treatment effects (as indicated in b). Histograms indicate results from the sum. Significance is displayed as * for p <0.05, ** for p <0.01 and *** for p <0.001. Tx treatment, Veh vehicle, VIS visual cortex, AUD auditory cortex, HF hippocampal formation, HY hypothalamus, AMY amygdala, CP caudoputamen, RSP retrosplenial, ORB orbital, STR striatum, MB midbrain, TT taenia tecta, OLF olfactory, ACB nucleus accumbens, ENT entorhinal, TH thalamus. Triple network color-coding: default-mode network (DMN) in green, salience network (SN) in pink, lateral-cortical network (LCN) in orange; regions in common across all three networks (light blue), regions shared by DMN and LCN (light purple) are also shown.
Regions within this network included the VIS, AUD, MB, HY, HF, TH and CP areas. The ( + ve) network (treated > vehicle) was under-enriched for edges in the LCN, and edges between the LCN and DMN (Fig. 5c, right). There were no significant (−ve) network (treated<vehicle) findings. Of note, the difference in FC at 22 M between WTveh and ADveh showed a smaller effect size relative to the 20 M timepoint. We found that this was likely due to a smaller sample size at the 22 M timepoint. We tested this by randomly selecting N/2 (number of subjects) and therefore half number of scans at 20 M, to match the number available at 22 M, and we found a similarly weakened effect size (data not shown).
Regions that showed the highest degree of association with treatment included the VIS, MB, AUD, HY (Supplementary Fig. 5). Notably, these regions also showed high-degree representation in the AD-aging and AD-like networks (AD-aging and AD-like in Supplementary Fig. 5) suggesting treatment specific targeting of regions involved in AD (as opposed to indirect or off-target effects of drug treatment).
Mapping the shared functional landscape of data-derived networks
Eight data-derived networks were identified: AD-aging (−ve/+ve), WT-aging (−ve/+ve), AD-like (−ve/+ve) and treatment (−ve/+ve; −ve treatment was no longer analyzed in the rest of the work because it comprised only one edge), all summarized in Supplementary Fig. 3. To assess whether shared patterns were uncovered across our analyses, we quantified the amount of cross-network (under) enrichment (as above), i.e., whether there was a significant number of shared edges, (or lack of shared edges), using Fisher’s exact test (and permutation testing, ×1000 iteration), between all pairs of data-derived networks (Fig. 6). Our hypothesis was that the aging and AD-like networks would share a common pattern as would the AD-like and treatment networks. We also anticipated that the DMN might be well-represented within both.

a Greyscale lower-left: the fraction of shared edges between data-derived networks computed as the fraction of the smaller network contained within the larger network (e.g., 85% of the 643 edges within the AD-like (−ve) network are within the 2313 edges within the WT-aging (+ve) network). Color upper-right: Using Fisher’s exact test (corrected, permutation test ×1000), we determine if a significant number of shared edges (hot-colors), or a significant (p < 0.008) lack of shared edges (cool-colors), are found between data-derived network pairs. Number of shared edges is shown as white in each cell. b Overlap (red arrows) and lack of overlap (blue lines with caps) between data-derived networks. Arrow size proportional to significance. c Edges shared between AD-aging(+ve), WT-aging(+ve), and AD-like(−ve) pairs–green: 1–3 shown as vertical ROI plots (upper half) and histograms (lower half) with null distribution (colored histogram) and real data (gray line and black triangle) . d Bilateral regions that are identified consistently (in 1–3) with the highest degree. e Overlap between two groupings of edges shared across data-derived networks: Age, AD (left of dotted line, green: 1–3), and treatment (right of dotted line, purple: 4–6). No significant representation of edges shared across data-derived networks including the treatment network with the triple-network system. f A layered histogram of degree for the 1–3 grouping with a bird’s eye view of a degree color-map (red), in-lay. For significance see Figs. 3, 4, 5.
By definition, there was under-enrichment between AD-aging ( + ve) vs. (−ve), and WT-aging ( + ve) vs. (−ve) (Fig. 6a, b). Similarly, we also found under-enrichment between AD-aging ( + ve) vs. WT-aging (−ve), AD-aging (−ve) vs. WT-aging ( + ve), and WT-aging (−ve) vs. AD-like (−ve). In-line with our hypothesis, two patterns of enrichment emerged: the first between aging (AD and WT, +ve) and AD-like (-ve), (Fig. 6e, left) and the second between treatment ( + ve) and AD-aging (-ve), WT aging ( + ve), and AD-like (−ve) (Fig. 6e, right). The second pattern was weaker, without as many edges, and showed no representation in the triple-network model (i.e., shared edges did not belong to DMN, LCN, SN or their inter-network pairings). Conversely, the first pattern showed a higher proportion of shared edges across network pairs (Fig. 6a,e, left). Edges shared between AD-aging ( + ve) and AD-like (−ve) included 30% of the smaller, AD-like (−ve), network (proportion of shared edges, Fig. 6a, computed as the fraction of the smaller network). Further, this 30% was fully contained within the other two inter-network pairings (Fig. 6e, left). The remaining pairings: WT-aging ( + ve) vs. AD-aging ( + ve) as well as WT-aging ( + ve) vs. AD-like (−ve), showed an even higher proportion of shared edges (67 and 85%, respectively, Fig. 6a), but a more modest inter-network pairing overlap (35%, Fig. 6e, left). Edges within the three pairings in the second pattern showed a high representation of DMN and LCN, as well as edges that link DMN to LCN, and LCN to SN. Two of the three, AD-aging ( + ve) vs. WT-aging ( + ve) and AD-aging ( + ve) vs. AD-like (−ve), showed under-enrichment for the SN (Fig. 6c). Brain regions with high-degree that appeared within these inter-network pairings (Fig. 6d, f) included primary SS, MO, VIS and RSP areas.
Mean FC within data-derived networks across the mouse lifespan
To assess how networks established at the group-level highlighted differences in FC strength at the individual-level, the product of each data-derived network and each mouse’s connectome was computed at each timepoint. Each mouse’s connectome was masked using each of the data-derived networks (binary), so that edges not included in the networks were set to zero. The mean FC of nonzero elements were plotted for the six major networks identified: WT/AD aging (+ ve and −ve), AD-like (−ve) and treatment (+ ve) (Supplementary Fig. 6). The WT-aging ( + ve) network associated with healthy brain maturation highlighted an increase in mean FC in WT relative to AD mice at 9 M and 20 M (Supplementary Fig. 6a, left/middle). The key metric of disease was the AD-like (−ve) network (Supplementary Fig. 6b, right). Mean FC in this network was decreased in AD relative to WT mice at 9 M and 20 M (p <0.01), but not at earlier timepoints. At 22 M, the ADveh mice showed decreased mean FC in the AD-like (−ve) network relative to WTveh and relative to ADTx mice, but ADTx were indistinguishable from WTveh. The treatment ( + ve) network by definition showed increased mean FC in ADTx mice relative to ADveh at 22 M (Supplementary Fig. 6a, right). There was a significant treatment effect on AD pathology at p <0.001 for the two conditions (pathology and treatment; ANOVA, [F(2,20) = 10.48, p = 0.0008]). Importantly, this drug-responsive network exhibited statistically significant decreases for AD mice relative to WT mice preceding treatment at 9 M and 20 M. Overall, connectivity in these data-driven networks across groups was well-aligned with the specific comparisons above.
Replication analysis
Our data-derived networks: aging (WT and AD), AD-like, and treatment, showed a range of effect sizes. To investigate how sample-size effected our findings, we randomly sub-sampled half of our dataset (N/2) and re-derived the data-driven networks (×100 iterations). Supplementary Figs. 7 and 8 show the resultant networks (N/2) (summed across iterations as indicated) for uncorrected and corrected findings, respectively, next to our original results (full-N). The spatial overlap between N/2 and full-N were compared (using dice overlap). Similarly, the N/2 results were compared to a null distribution. Overall, a high degree of overlap was uncovered across conditions. As expected, the most robust findings included aging (WT and AD, +ve and −ve) and pathology (−ve). Treatment ( + ve) results were also recapitulated, but to a lesser extent (as expected, given the lower initial N). As above, N/2 data-derived networks were characterized by triple network representation (Supplementary Figs. 9 and 10). Summary break-down of data-derived networks from N/2 and full dataset are shown in Supplementary Figs. 11, 12, uncorrected and corrected, respectively. Overall, similar patterns were uncovered compared to the full-N findings.
Discussion
Non-invasive imaging technologies, including fMRI, applied in animal models of human disease allow aspects of pathology to be characterized in a controlled environment, on a tractable timescale, and for the assessment of clinically accessible bioimaging markers of disease emergence, progression, and response to treatment [57]. Here, we investigated the BOLD-fMRI correlates of AD, throughout the mouse lifespan, in an optimized model of disease [31, 58]. In addition, we investigated the neuroimaging correlates of a novel synapse rescuing treatment (BMS-984923) administered late in life, once AD-like pathology is well established. We uncovered networks which characterized aging (with and without disease), disease-associated changes in FC strength, as well as treatment-response. Analyses were conducted using data-driven approaches (i.e., no a priori assumptions were made about the roles established brain circuits, e.g., DMN, or regions play in these processes). We quantified the presence (or absence) of classic large-scale networks following the discovery phase and examined correspondences across species.
We explored changes in FC strength with age in WT and AD mice at three life-stages: young-adulthood (6 M or 20–30 years-of-age in humans), middle-age (9 M or 38–47 years), and older-adulthood (20–22 M or 56–69 years) [30]. We found FC strength in WT mice followed an inverted-U-shaped profile (Fig. 2, Supplementary Fig. 6a, left). This pattern was roughly in-line with a small pool of existing work using fMRI in mice [49, 59, 60] including a study by Morrissey et al. using a mouse model very similar to the one used in this work [49] (Supplementary Table 2). Across studies, relative to baseline (2–4 M), there was good agreement that circuit-specific increases in FC strength emerge and peak within 5–9 M [49, 59, 60]. Between 10 and 18 M (which we do not examine), fMRI [59, 60] shows a return to baseline, in one case a steady increase [49] (notably, Morrissey et al., was focused on hippocampal regions only), and optical imaging methods [61] showed a dip below baseline (at 17–18 M). At 20 M, we observed a persistent increase in FC strength, followed by a reduction at 22 M. Differences in these collective observations in later-life could be due to a mismatch between timepoints, modalities (optical vs. fMRI), anaesthesia (Egimendia et al. [60] and Vasilkovska et al. [59] used medetomidine and isoflurane, Morrissey et al. [49] used isoflurane, whereas optical data in the Albertson et al. [61] study was acquired from awake animals), or the specific brain circuits being examined. It is also possible that the inverted-U-shaped profile is incomplete, and the brain follows an even more complex double-humped profile. Despite the small number of existing studies, increased FC strength in mid-life seems to be a relatively reproducible observation. Encouragingly, this inverted-U-shaped profile in fMRI FC strength is also reported in cognitively healthy human subjects. As in mice, middle-aged people show greater FC strength, relative to the young and elderly [47, 48].
Further, in mice, we found good agreement across studies in terms of which brain regions show age-related changes in FC strength (Supplementary Table 3). Universally, the DMN—which includes RSP and VIS areas among others—shows age-related changes [47]. Motor (MO), anterior cingulate (ACA) as well as auditory and somatosensory (SS) areas also show good-moderate cross-study agreement (given the methodological differences between studies). Notably, these implicated areas include regions that are not typically associated with cognition (e.g., SS and MO areas). We also note strong alignment across species. In humans, the DMN [47, 48], as well as ‘cognitive’ and ‘motor’ circuits [62,63,64] all show age-related changes in FC strength (that generally follow the inverted-U-shaped profile). In the present study, we also observed the LCN (including SS, Temporal association, and MO areas), midbrain, hypothalamus, and thalamus, to be involved in WT-aging. Given inter-study difference in how networks are defined, and analyses conducted (data-driven, vs. a priori network or ROI driven), it is unclear whether these findings are unique. Finally, our Neurosynth findings in the human brain associated with age (Supplementary Fig. 4) show considerable overlap (including the TH and TeA) with the regions and networks identified in mice (Supplementary Table 3).
This emerging cross-study (and species) convergence is promising. Yet, there are several competing theories on the neurobiological drivers of healthy aging which have proven difficult to reconcile (see reviews) [65, 66] especially when using fMRI in isolation. Although it is beyond the scope of the present work to interrogate the mechanisms which give rise to these fMRI-observable age-related changes, we are well-positioned to interrogate their origins as part of our future work using multimodal approaches [37, 38, 67]. More broadly, interpreting BOLD-based FC changes in isolation must be done with caution due to the nature of the BOLD signal itself. BOLD fMRI is an indirect measure of brain activity which relies on changes in blood flow, volume, and oxygenation [68, 69]. Vascular effects, in particular, which are known to be altered by AD [70, 71], alter the BOLD signal independently of changes in neural activity. We cannot rule out the possibility that these effects are evident within our findings. In addition, the present work uses anesthesia which is known to affect both neural and vascular systems [24], complicating the relationship between neural activity and BOLD, as well as how each are shaped by disease [72]. On the other hand, the use of anesthesia is still the gold-standard in rodent-fMRI, to minimize motion and stress; thus, our results are more easily related to the current literature [73]. However, recent efforts to move the field towards performing experiments in awake mice are underway (see our own work in awake mice [74], and our recent systematic review) [72]. As discussed in detail within our review, there is still much we can learn from experiments in anesthetized animals, and from moving methodically towards performing experiments on awake animals. Future work will interrogate both the underlying neural changes which support BOLD-fMRI FC and the role of anesthesia.
Further, it is noteworthy that this study does not include a group of WTtreated mice. This decision was made based on our previous work which showed no changes in synaptic density or single cell transcriptomics in a WTtreated cohort [31]. Similarly, although we included equal distributions of both biological sexes, we were not adequately powered for sex-based comparisons [57]. This will be addressed as part of our future work. Likewise, although we use a preprocessing pipeline which has undergone a significant amount of development and testing (considering that it is specific to rodent fMRI data [40, 73]), it is relatively new and we do not interrogate the effects of different preprocessing approaches (e.g., top-up [75]) on our findings. Finally, the present work is limited to measures of static FC. Future work will consider dynamic measure such as quasi-periodic patterns or co-activation patterns [76].
Decreases in FC strength (AD vs. control) is an established hallmark of neuropathology in mouse models [17, 49, 50, 59, 77, 78]. Likewise, fMRI measures from humans show a tight association between cognitive decline and lower FC strength [79, 80]. Yet, these AD-related changes occur against a non-trivial background of aging that can be overlooked when a single timepoint is considered. Is lower FC strength (in AD vs. control) a result of a decrease in the AD group? Or (as our data suggest, Figs. 2, 4, 6, Supplementary Fig. 6), a failure to increase with maturation? This is likely an important distinction that will require further investigation.
In addition to examining healthy aging, we examined age-related changes in FC strength in AD mice alongside differences between WT and AD groups at each timepoint. Changes in FC strength with age in AD mice (Fig. 2) emerged later in life (relative to WT aging) and showed more widespread decreases (Supplementary Fig. 3a). Yet, there were also preserved common elements between WT and AD aging which was reflected by the high proportion of common circuitry (Fig. 6, Supplementary Fig. 7). From our inter-group (AD vs. WT) comparison (at each timepoint), we observed a clear decrease in FC strength by 9 M in AD. The directionality of these FC changes (decrease in AD-like models compared to WT) is in-line with previous fMRI work in other mouse models of AD [17,18,19, 77]. Notably, the FC deficits reported here appeared later in life (9 M) compared to the literature (as early as ~3 M) [17], which may be a result of the milder nature of the pathology in this mouse model. Critically, the DKI AppNL-G-F/hMapt model [28, 29] investigated here has not previously been examined using fMRI. This is important, as this new model represents a step-forward in our ability to emulate AD-like pathology via its milder genetic profile [81].
The circuits and regions we found to be involved in aging (Figs. 2,3 Supplementary Fig. 3), WT vs. AD (Fig. 4), and their common elements (Fig. 6) are summarized in Supplementary Table 4. Only two regions (accumbens and hypothalamus) showed age-related changes uniquely in WT mice, whereas more widespread age-related changes were unique to AD mice (insula, ectorhinal, perirhinal, caudoputamen and piriform). Regions that did not show age-related changes but were identified when comparing WT vs. AD included temporal and posterior-parietal association areas (TeA, PPA) and regions within the hippocampal formation (HF, including ENT). Most circuits and regions, however, were shared across at least 2/3 comparisons (as expected based on our analyses, Fig. 6). Roughly, in order of frequency across comparisons (columns in Supplementary Table 4), we observed involvement of the DMN-to-LCN and LCN (including SS and MO), the DMN (particularly visual areas, as well as retrosplenial (RSP), MO, and to a lesser extent ACA), TH, LCN-to-SN and MB. These findings, particularly the involvement of the DMN and LCN, were in-line with what has been observed in AD patients [51, 54], individuals with mild cognitive impairment (MCI) [2, 82], the amyloid-positive but cognitively normal elderly [7], and healthy people of an advanced age [83]. Further, these patterns align (in time and space) with established neurobiological correlates of AD including Aβ-plaques [84] and synapse losses [85] across species. Specifically, the DMN shows increased FC in healthy adults where future preferential deposition of Aβ-plaques occur in (older) AD patients [86] in parallel with decreases in FC [51]. Further, regions in the DMN (including the RSP or PREC) are among the earliest to show both Aβ-pathology [54] and changes in FC in individuals with MCI [82]. Mouse models show Aβ-pathology and synapse density changes in the DMN (RSP and VIS) and LCN (SS and MO), and HF [28]. Mice also show alterations in FC that arise slightly before (at 6–9 M) substantial increases in Aβ-pathology (which plateau—in the cortex—within the 8–10 M window) [28]. Together, these findings hint at a relationship between changes in FC strength that precede Aβ-pathology and synapse loss. Altogether, changes which position fMRI as a potentially useful tool for both AD identification and management.
In this regard, AD-associated cognitive deficits are attributable, at least in part, to synaptic failure triggered by Aβ and tau accumulation [9, 87]. Targeting a critical link in this cascade (see ‘Supplementary Discussion’), our group has shown that treatment with BMS-984923 elicits behavioral recovery, normalizes neuronal transcriptomics, prevents synaptic loss (if administered in early stages) and rescues synapse losses (when administered at late stages of the pathology, like in our study) in two mouse models of AD (inclusive of the DKI AppNL-G-F/hMapt) [31, 33]. Here, we conducted the first fMRI investigation of BMS-984923 treatment effects. To further characterize the therapeutic potential of this drug, we administered to older mice (9–19 vs. 20–22 M), after AD-pathology was well established. In BMS-984923 ADTx vs. WTveh mice (at 22 M), we observed pseudo-normalized FC (Fig. 5a, Supplementary Fig. 6a). In ADTx vs. ADveh (at 20 or 22 M) we observed significant increases in FC (Fig. 5b). Although a simple triple-network model representation was not emergent for these treatment-elicited changes in FC, regions highly involved in the SN (TH, HY and CP) were detected. Additionally, there was significant overlap with areas implicated in our AD-aging (−ve), WT-aging ( + ve), and AD-like (−ve) results (Fig. 6e, right).
Using FC in the identified AD-like (−ve) network as a disease measure (Fig. 6), the deficits observed in ADvehicle relative to WT mice were reversed in ADtreated mice at 22 M after 2 months of intervention. Tracking connectivity in this network, or its human ortholog, provides a potential tool to monitor treatment benefit during translational development of this or other AD therapeutics with putative synapse rescuing benefit.
In conclusion, we present a comprehensive investigation of the fMRI-FC correlates of AD in an optimized mouse model of the disease across the lifespan (4–22 M). A detailed consideration of aging in both healthy-WT mice and their AD-counterparts was included alongside more classic groupwise comparisons. Overall, our findings align well with the existing literature from other mouse models and the human fMRI literature. We observed progressive AD-related dysconnectivity that emerged in known AD-vulnerable areas (sites of Aβ-accumulation, Tauopathy, and synaptic loss). Administration of a synapse-targeting treatment (BMS-984923) reversed (or pseudo-normalized) the observed FC deficits in late-stage disease indicating the potential utility of this therapeutic as a synapse rescuing treatment and the role neuroimaging with fMRI could play in therapeutic development and patient management.
Data availability
Data will be made available upon reasonable request to the corresponding authors.
References
Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D’Angelo G, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. J Neurosci. 2010;30:17035–40.
Wang J, Zuo X, Dai Z, Xia M, Zhao Z, Zhao X, et al. Disrupted functional brain connectome in individuals at risk for Alzheimer’s disease. Biol Psychiatry. 2013;73:472–81.
Kim J, Jeong M, Stiles WR, Choi HS. Neuroimaging modalities in Alzheimer’s disease: diagnosis and clinical features. Intern jour mol sci. 2022;23:6079.
Marquez F, Yassa MA. Neuroimaging biomarkers for Alzheimer’s disease. Mol Neurodegener. 2019;14:21.
Wang X, Huang W, Su L, Xing Y, Jessen F, Sun Y, et al. Neuroimaging advances regarding subjective cognitive decline in preclinical Alzheimer’s disease. Mol Neurodegener. 2020;15:55.
Chetelat G. Multimodal neuroimaging in Alzheimer’s disease: early diagnosis, physiopathological mechanisms, and impact of lifestyle. J Alzheimers Dis. 2018;64:S199–S211.
Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67:584–7.
Morris JC, Storandt M, McKeel DW Jr., Rubin EH, Price JL, Grant EA, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996;46:707–19.
Brier MR, Thomas JB, Ances BM. Network dysfunction in Alzheimer’s disease: refining the disconnection hypothesis. Brain Connect. 2014;4:299–311.
Aggleton JP, Pralus A, Nelson AJ, Hornberger M. Thalamic pathology and memory loss in early Alzheimer’s disease: moving the focus from the medial temporal lobe to Papez circuit. Brain. 2016;139:1877–90.
Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38.
Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–84.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80.
DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64.
Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–44.
Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–50.
Mandino F, Yeow LY, Bi RZ, Sejin L, Bae HG, Baek SH, et al. The lateral entorhinal cortex is a hub for local and global dysfunction in early Alzheimer’s disease states. J Cereb Blood Flow Metab. 2022;42:1616–31.
Grandjean J, Derungs R, Kulic L, Welt T, Henkelman M, Nitsch RM, et al. Complex interplay between brain function and structure during cerebral amyloidosis in APP transgenic mouse strains revealed by multi-parametric MRI comparison. Neuroimage. 2016;134:1–11.
Shah D, Jonckers E, Praet J, Vanhoutte G, Delgado YPR, Bigot C, et al. Resting state FMRI reveals diminished functional connectivity in a mouse model of amyloidosis. PLoS ONE. 2013;8:e84241.
Mandino F, Cerri DH, Garin CM, Straathof M, van Tilborg GAF, Chakravarty MM, et al. Animal functional magnetic resonance imaging: trends and path toward standardization. Front Neuroinform. 2019;13:78.
Markicevic M, Savvateev I, Grimm C, Zerbi V. Emerging imaging methods to study whole-brain function in rodent models. Transl Psychiatry. 2021;11:457.
Gorges M, Roselli F, Muller HP, Ludolph AC, Rasche V, Kassubek J. Functional connectivity mapping in the animal model: principles and applications of resting-state fMRI. Front Neurol. 2017;8:200.
Zerbi V, Grandjean J, Rudin M, Wenderoth N. Mapping the mouse brain with rs-fMRI: an optimized pipeline for functional network identification. Neuroimage. 2015;123:11–21.
Grandjean J, Schroeter A, Batata I, Rudin M. Optimization of anesthesia protocol for resting-state fMRI in mice based on differential effects of anesthetics on functional connectivity patterns. Neuroimage. 2014;102:838–47.
Xu N, LaGrow TJ, Anumba N, Lee A, Zhang X, Yousefi B, et al. Functional connectivity of the brain across rodents and humans. Front Neurosci. 2022;16:816331.
Gozzi A, Zerbi V. Modeling brain dysconnectivity in rodents. Biol Psychiatry. 2023;93:419–29.
Sharma H, Chang KA, Hulme J, An SSA. Mammalian Models in Alzheimer's Research: An Update. Cells. 2023;12:2459.
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, et al. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17:661–3.
Saito T, Mihira N, Matsuba Y, Sasaguri H, Hashimoto S, Narasimhan S, et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J Biol Chem. 2019;294:12754–65.
Dutta S, Sengupta P. Men and mice: relating their ages. Life Sci. 2016;152:244–8.
Spurrier J, Nicholson L, Fang XT, Stoner AJ, Toyonaga T, Holden D, et al. Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci Transl Med. 2022;14:eabi8593.
Huang LK, Kuan YC, Lin HW, Hu CJ. Clinical trials of new drugs for Alzheimer disease: a 2020–2023 update. J Biomed Sci. 2023;30:83.
Haas LT, Salazar SV, Smith LM, Zhao HR, Cox TO, Herber CS, et al. Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing alzheimer’s mouse phenotypes. Cell Rep. 2017;20:76–88.
Menon V. Large-scale brain networks and psychopathology: a unifying triple network model. Trends Cogn Sci. 2011;15:483–506.
Mandino F, Vrooman RM, Foo HE, Yeow LY, Bolton TAW, Salvan P, et al. A triple-network organization for the mouse brain. Mol Psychiatr. 2022;27:865–72.
Hamodi AS, Martinez Sabino A, Fitzgerald ND, Moschou D, Crair MC. Transverse sinus injections drive robust whole-brain expression of transgenes. Elife. 2020;9:e53639.
Lake EMR, Ge X, Shen X, Herman P, Hyder F, Cardin JA, et al. Simultaneous cortex-wide fluorescence Ca(2+) imaging and whole-brain fMRI. Nat Methods. 2020;17:1262–71.
Vafaii H, Mandino F, Desrosiers-Gregoire G, O’Connor D, Markicevic M, Shen X, et al. Multimodal measures of spontaneous brain activity reveal both common and divergent patterns of cortical functional organization. Nat Commun. 2024;15:229.
Mandino F, Horien C, Shen X, Desrosiers-Gregoire G, Luo W, Markicevic M, et al. Multimodal identification of the mouse brain using simultaneous Ca (2+) imaging and fMRI. bioRxiv. 2024;2024.2005. 2024.594620.
Desrosiers-Grégoire G, Devenyi GA, Grandjean J, Chakravarty MM. A standardized image processing and data quality platform for rodent fMRI. Nat Comm 2024;15:6708.
Manjon JV, Coupe P, Marti-Bonmati L, Collins DL, Robles M. Adaptive non-local means denoising of MR images with spatially varying noise levels. J Magn Reson Imaging. 2010;31:192–203.
Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging. 1998;17:87–97.
Atlas AMB Allen institute for brain science.
Wang Q, Ding SL, Li Y, Royall J, Feng D, Lesnar P, et al. The Allen mouse brain common coordinate framework: a 3D reference atlas. Cell. 2020;181:936–53.e920.
Bondi MW, Houston WS, Salmon DP, Corey-Bloom J, Katzman R, Thal LJ, et al. Neuropsychological deficits associated with Alzheimer’s disease in the very-old: discrepancies in raw vs. standardized scores. J Int Neuropsychol Soc. 2003;9:783–95.
Betzel RF, Byrge L, He Y, Goni J, Zuo XN, Sporns O. Changes in structural and functional connectivity among resting-state networks across the human lifespan. Neuroimage. 2014;102:345–57.
Mak LE, Minuzzi L, MacQueen G, Hall G, Kennedy SH, Milev R. The default mode network in healthy individuals: a systematic review and meta-analysis. Brain Connect. 2017;7:25–33.
Staffaroni AM, Brown JA, Casaletto KB, Elahi FM, Deng J, Neuhaus J, et al. The longitudinal trajectory of default mode network connectivity in healthy older adults varies as a function of age and is associated with changes in episodic memory and processing speed. J Neurosci. 2018;38:2809–17.
Morrissey ZD, Gao J, Zhan L, Li W, Fortel I, Saido T, et al. Hippocampal functional connectivity across age in an App knock-in mouse model of Alzheimer’s disease. Front Aging Neurosci. 2022;14:1085989.
Zerbi V, Wiesmann M, Emmerzaal TL, Jansen D, Van Beek M, Mutsaers MP, et al. Resting-state functional connectivity changes in aging apoE4 and apoE-KO mice. J Neurosci. 2014;34:13963–75.
Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci USA. 2004;101:4637–42.
Damoiseaux JS, Prater KE, Miller BL, Greicius MD. Functional connectivity tracks clinical deterioration in Alzheimer’s disease. Neurobiol Aging. 2012;33:828 e819–830.
Whitesell JD, Liska A, Coletta L, Hirokawa KE, Bohn P, Williford A, et al. Regional, layer, and cell-type-specific connectivity of the mouse default mode network. Neuron. 2021;109:545–59.e548.
Sheline YI, Raichle ME. Resting state functional connectivity in preclinical Alzheimer’s disease. Biol Psychiatry. 2013;74:340–7.
Yarkoni T, Poldrack RA, Nichols TE, Van Essen DC, Wager TD. Large-scale automated synthesis of human functional neuroimaging data. Nat Methods. 2011;8:665–70.
Cantero JL, Atienza M, Gomez-Herrero G, Cruz-Vadell A, Gil-Neciga E, Rodriguez-Romero R, et al. Functional integrity of thalamocortical circuits differentiates normal aging from mild cognitive impairment. Hum Brain Mapp. 2009;30:3944–57.
Ozalay O, Mediavilla T, Giacobbo BL, Pedersen R, Marcellino D, Oradd G, et al. Longitudinal monitoring of the mouse brain reveals heterogenous network trajectories during aging. Commun Biol. 2024;7:210.
Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T, et al. Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer’s disease. J Neurosci. 2015;35:13275–86.
Vasilkovska T, Adhikari MH, Van Audekerke J, Salajeghe S, Pustina D, Cachope R, et al. Resting-state fMRI reveals longitudinal alterations in brain network connectivity in the zQ175DN mouse model of Huntington’s disease. Neurobiol Dis. 2023;181:106095.
Egimendia A, Minassian A, Diedenhofen M, Wiedermann D, Ramos-Cabrer P, Hoehn M. Aging reduces the functional brain networks strength-a resting state fMRI study of healthy mouse brain. Front Aging Neurosci. 2019;11:277.
Albertson AJ, Landsness EC, Tang MJ, Yan P, Miao H, Rosenthal ZP, et al. Normal aging in mice is associated with a global reduction in cortical spectral power and network-specific declines in functional connectivity. Neuroimage. 2022;257:119287.
Bo J, Lee CM, Kwak Y, Peltier SJ, Bernard JA, Buschkuehl M, et al. Lifespan differences in cortico-striatal resting state connectivity. Brain Connect. 2014;4:166–80.
Jolles DD, van Buchem MA, Crone EA, Rombouts SA. A comprehensive study of whole-brain functional connectivity in children and young adults. Cereb Cortex. 2011;21:385–91.
Li Z, Petersen IT, Wang L, Radua J, Yang G-c, Liu X. The Lifespan Trajectories of Brain Activities Related to Cognitive Control. bioRxiv. 2024;2023.2008.2020.554018.
McDonough IM, Nolin SA, Visscher KM. 25 years of neurocognitive aging theories: what have we learned? Front Aging Neurosci. 2022;14:1002096.
Sala-Llonch R, Bartres-Faz D, Junque C. Reorganization of brain networks in aging: a review of functional connectivity studies. Front Psychol. 2015;6:663.
O’Connor D, Mandino F, Shen X, Horien C, Ge X, Herman P, et al. Functional network properties derived from wide-field calcium imaging differ with wakefulness and across cell type. Neuroimage. 2022;264:119735.
Logothetis NK, Wandell BA. Interpreting the BOLD signal. Annu Rev Physiol. 2004;66:735–69.
Logothetis NK, Pfeuffer J. On the nature of the BOLD fMRI contrast mechanism. Magn Reson Imaging. 2004;22:1517–31.
Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 2016;1862:887–900.
Thal DR, Attems J, Ewers M. Spreading of amyloid, tau, and microvascular pathology in Alzheimer’s disease: findings from neuropathological and neuroimaging studies. J Alzheimers Dis. 2014;42:S421–29.
Mandino F, Vujic S, Grandjean J, Lake EMR Where do we stand on fMRI in awake mice? Cereb Cortex. 2024;34:bhad478.
Grandjean J, Canella C, Anckaerts C, Ayrancı G, Bougacha S, Bienert T, et al. Common functional networks in the mouse brain revealed by multi-centre resting-state fMRI analysis. Neuroimage. 2020;205:116278.
Mandino F, Shen X, O’Connor D, Mukherjee B, DeLuca K, et al. Longitudinal simultaneous cortex-wide Ca2+ imaging and whole-brain functional magnetic resonance imaging in awake mice. J Cereb Blood Flow Metab (Vol 42, No 1_ SUPPL, pp 75–75) 2455 Teller Rd, Thousand Oaks, CA 91320 USA: Sage Publications Inc 2022.
Andersson JL, Skare S, Ashburner J. How to correct susceptibility distortions in spin-echo echo-planar images: application to diffusion tensor imaging. Neuroimage. 2003;20:870–88.
Adhikari MH, Belloy ME, Van der Linden A, Keliris GA, Verhoye M. Resting-state co-activation patterns as promising candidates for prediction of Alzheimer’s disease in aged mice. Front Neural Circuits. 2020;14:612529.
Shah D, Praet J, Latif Hernandez A, Hofling C, Anckaerts C, Bard F, et al. Early pathologic amyloid induces hypersynchrony of BOLD resting-state networks in transgenic mice and provides an early therapeutic window before amyloid plaque deposition. Alzheimers Dement. 2016;12:964–76.
Green C, Sydow A, Vogel S, Anglada-Huguet M, Wiedermann D, Mandelkow E, et al. Functional networks are impaired by elevated tau-protein but reversible in a regulatable Alzheimer’s disease mouse model. Mol Neurodegener. 2019;14:13.
Damoiseaux JS, Beckmann CF, Arigita EJ, Barkhof F, Scheltens P, Stam CJ, et al. Reduced resting-state brain activity in the “default network” in normal aging. Cereb Cortex. 2008;18:1856–64.
Onoda K, Ishihara M, Yamaguchi S. Decreased functional connectivity by aging is associated with cognitive decline. J Cogn Neurosci. 2012;24:2186–98.
Sasaguri H, Hashimoto S, Watamura N, Sato K, Takamura R, Nagata K, et al. Recent advances in the modeling of Alzheimer’s disease. Front Neurosci. 2022;16:807473.
Vann SD, Aggleton JP, Maguire EA. What does the retrosplenial cortex do? Nat Rev Neurosci. 2009;10:792–802.
Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, et al. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–35.
Hampel H, Hu Y, Hardy J, Blennow K, Chen C, Perry G, et al. The amyloid-beta pathway in Alzheimer’s disease: a plain language summary. Neurodegener Dis Manag. 2023;13:141–9.
Zhang J, Wang J, Xu X, You Z, Huang Q, Huang Y, et al. In vivo synaptic density loss correlates with impaired functional and related structural connectivity in Alzheimer’s disease. J Cereb Blood Flow Metab. 2023;43:977–88.
Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–17.
Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91.
Acknowledgements
Dr. Carolyn Fredericks for a close read of the manuscript and helpful feedback. Drs. Joel Greenwood, Omer Mano, and Paul Shamble from the Neurotechnology Core of the Yale Kavli Institute for Neuroscience for their technological expertise. Drs. Michael Crair and Ali Hamodi for help with optimizing and implementing transverse sinus injection experiments. Dr. Yonghyun Ha for his technical expertise and coil mending abilities. Dr. Corey Horien for critical discussion and coding advice. Kristin DeLuca for mouse husbandry. Ludwig Family Foundation for research support.
Funding
Yale Alzheimer’s Disease Research Center P30AG066508 (SMS & EMRL). Wu Tsai Institute at Yale (SMS & EMRL). National Institutes of Health R21AG075778-01 (EMRL), National Institutes of Health RF1NS130069 (EMRL & SMS). National Institutes of Health R01AG034924 (SMS). National Institutes of Health RF1AG070926 (SMS). Ludwig Family Foundation (SMS).
Author information
Authors and Affiliations
Contributions
Conceptualization: FM, SMS, EMRL. Methodology: FM, SMS, EMRL, GDG, DO, XS, XP, AQ, JO. Investigation: FM, BM, AO. Visualization: FM, EMRL. Funding acquisition: SMS, EMRL. Project administration: SMS, EMRL. Supervision: SMS, EMRL, MC supervised GDG. Writing—original draft: FM, EMRL. Writing—review & editing: all co-authors.
Corresponding authors
Ethics declarations
Competing interests
SMS is a Founder, Consultant, Equity Holder and Director of Allyx Therapeutics seeking to develop mGluR5-directed therapies for Alzheimer’s and Parkinson’s Disease. XP is a consultant for the Brain Electrophysiology Laboratory Company. XP also consults and has an ownership stake in Veridat.
Ethics approval
All procedures were approved by the Yale Institutional Animal Care and Use Committee (IACUC; IACUC #2023-20284; 2023-07281) and followed the National Institute of Health Guide for the Care and Use of Laboratory Animals. Human analyses were carried out via data from the publicly available NeuroSynth platform.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mandino, F., Shen, X., Desrosiers-Grégoire, G. et al. Aging-dependent loss of functional connectivity in a mouse model of Alzheimer’s disease and reversal by mGluR5 modulator. Mol Psychiatry 30, 1730–1745 (2025). https://doi.org/10.1038/s41380-024-02779-z
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41380-024-02779-z
This article is cited by
-
Multimodal identification of the mouse brain using simultaneous Ca2+ imaging and fMRI
Communications Biology (2025)
