
Abstract
Aberrant type 2 responses underlie the pathologies in allergic diseases like asthma, yet, our understanding of the mechanisms that drive them remains limited. Recent evidence suggests that dysregulated innate immune factors can perpetuate asthma pathogenesis. In susceptible individuals, allergen exposure triggers the activation of complement, a major arm of innate immunity, leading to the aberrant generation of the C3a anaphylatoxin. C3 and C3a have been shown to be important for the development of Th2 responses, yet remarkably, the mechanisms by which C3a regulates type 2 immunity are relatively unknown. We demonstrate a central role for C3a in driving type 2 innate lymphoid cells (ILC2)-mediated inflammation in response to allergen and IL-33. Our data suggests that ILC2 recruitment is C3a-dependent. Further, we show that ILC2s directly respond to C3a, promoting type 2 responses by specifically: (1) inducing IL-13 and granulocyte-macrophage colony-stimulating factor, whereas inhibiting IL-10 production from ILC2; and (2) enhancing their antigen-presenting capability during ILC-T-cell cross-talk. In summary, we identify a novel mechanism by which C3a can mediate aberrant type 2 responses to aeroallergen exposure, which involves a yet unrecognized cross-talk between two major innate immune components—complement and group 2 innate lymphoid cells.
Similar content being viewed by others


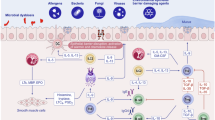
Introduction
Allergic airway diseases like asthma are increasing in prevalence worldwide, which poses a serious public health challenge.1 These diseases are considered to be primarily driven by aberrant type 2 immune responses, but much remains unknown about the underlying causes promoting these processes.
Recent research has outlined the importance of innate immunity in regulating the development of allergic diseases. Complement activation and generation of anaphylatoxins (C3a, C5a) at mucosal surfaces appears to be a common pathway for the induction and regulation of Th2-mediated inflammatory responses to a variety of triggers (virus, allergen, fungus, etc). In particular, the C3 pathway is required for the development of Th2 responses in allergic inflammation.2,3 Consistent with its importance for Th2 immunity, levels of airway C3/C3a are highly induced in patients with allergic airway diseases4,5,6,2,7 including asthma, with a positive correlation between C3/C3a levels and asthma severity.8 These observations are supported by the discovery of single-nucleotide polymorphisms in the C3 and C3AR1 loci that increase the susceptibility to develop asthma.9,10,11 Despite the importance of the C3 pathway to allergic disease, the mechanism(s) by which C3a promotes type 2 immunity are unclear.
C3a signaling targets many immune cells and has a wide range of effects from increasing cytokine production,12,13,14 to promoting cell migration,15,16 differentiation,13,17,18 and proliferation.19 C3a-mediated mast cell degranulation, one of the earliest documented effects of C3a,20 may contribute to the pro-asthmatic effects of C3a, though this remains controversial.21,22,23 C3a also regulates dendritic cell (DC) function,24,25 like antigen uptake and co-stimulatory molecule expression.24 However, DC-expressed C3aR does not appear to play a role in house dust mite (HDM)-driven allergy, as the transfer of HDM-pulsed C3ar1−/− or C3ar1+/+ bone marrow-derived DCs (BMDCs) drives similar Th2 responses.25 T cells also respond to C3a. Whereas C3a responsiveness is central to maintain their Th1 polarization13,26 thereby preventing Treg differentiation,17,18,27 intrinsic T-cell C3a signaling seems to have a less-potent effect on Th2 differentiation.13,26 These data suggest that other C3a-responsive cells may also be involved in the development of type 2 responses.
Type 2 innate lymphoid cells (ILC2) play a crucial role in the initiation and exacerbation of type 2 responses. They are enriched in the blood and tissues of allergic individuals,28,29,30 and airway exposure to allergen readily induces the recruitment of IL-13-producing ILC2 into the lungs.31,32,33,34,35 In addition to their cytokine−secreting function, recent reports have identified ILC2 as antigen-presenting cells.36,37,38 ILC2-driven antigen presentation has been shown to be necessary for the development of anti-helminth Th2 responses37 and significantly enhances allergic airway inflammation in response to ovalbumin.39 However, the pathways that regulate these ILC2 functions remain unclear. Based on this, we investigate the hypothesis that C3a signaling regulates ILC2 function, thereby initiating aberrant type 2 immune responses in the lung.
Results
C3a signaling is required for the development of allergen-induced type 2 responses in the lungs
To determine whether C3a modulates allergen-driven immune responses, we first assessed whether allergen exposure itself could modulate C3a production in the airways. To this end, we exposed BALB/c mice to HDM and assessed the levels of C3a in the BAL fluid. Although C3a was present at baseline in PBS-exposed mice, its levels were highly increased upon allergen exposure at the airway surface (Fig. 1a). In the airways, many cell types can generate C3 including epithelial cells, fibroblasts, macrophages, and dendritic cells, but as the primary point of contact with the environment the epithelium is poised to respond to allergen via production of C3a. Accordingly, primary human bronchial epithelial cells treated with HDM for 24 h secreted significant amounts of C3a (Fig. 1b). We then determined whether C3a signaling was required for type-2 immune responses in response to allergen inhalation. Wildtype (wt) and C3ar1−/− mice were treated with PBS or HDM intratracheally (i.t.) (on days 0 and 14) and the allergic phenotype was assessed 72 h after the second HDM inhalation. In response to nebulized methacholine, C3ar1−/− mice had significantly lower airway hyperresponsivness (AHR) than wt mice (Fig. 1c), along with decreases in allergen-induced lung eosinophils (Fig. 1d) and HDM-induced IgE (Fig. 1e). However, lack of C3a signaling had no effect on HDM-induced mucus accumulation (Figure S1a, b) or HDM-induced Muc5ac or Muc5b mRNA (Figure S1c, d). Nonetheless, in line with our AHR and eosinophil data, we observed a significant reduction in HDM-induced IL-5 and IL-13 protein (Fig. 1f, g) and mRNA (Figure S1e, f) levels from the lungs of C3ar1−/− mice. The impaired HDM-induced IgE production in C3ar1−/− mice is consistent with a decrease in HDM-induced Il4 message, which is known to promote IgE class switching (Figure S1g). However, C3a signaling in the context of allergen exposure seemed to specifically modulate Th2 responses, as levels of IFNγ protein and Ifng message were not altered between wt and C3ar1−/− mice (Fig. 1h and S1h).

C3a signaling is required to develop allergen-driven type 2 immunity. a Wildtype C57BL/6 mice were given PBS or HDM intraperitoneally (i.p.) (days 0 and 7), followed by intratracheal (i.t.) PBS or HDM (100 µg) on days 14 and 21, on day 24 BAL C3a was determined by ELISA. b Normal human bronchial epithelial (NHBE) cells were exposed to media or HDM for 24 h and C3a was measured in the supernatant. Wildtype BALBc/J and C3ar1−/− mice were exposed to PBS or HDM (100 µg) i.t. on days 0 and 14. On day 17, c airway hyperresponsiveness, d levels of lung eosinophils, e serum IgE were determined. Lung single cell suspensions were restimulated with 30 µg/ml HDM for 3 days and supernatant levels of f IL-5, g IL-13, and h IFNγ were determined by ELISA. Lung cells (from non-lavaged mice) were analyzed for i CD4+ T cells j IL-13+CD4+ T cells, k CD8+ T cells, l Lin− (CD11b, CD11c, Gr1, B220, CD19, TCRb, TCRgd, CD49b, CD4, CD8, FcER1) ICOS+IL-33R+ ILC2, and m IL-13+ICOS+IL-33R+ ILC2 were enumerated by flow cytometry. Data represents means + SEM. Data are representative from two to three independent experiments *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Consistent with these data, we observed that C3a signaling was necessary for the recruitment of HDM-induced Th2 cells and ILC2 in the lungs. Specifically, we show that the numbers of HDM-induced CD4+ T cells (Fig. 1i) and IL-13+CD4+ T cells (Fig. 1j), but not CD8+ T cells (Fig. 1k) were significantly abrogated in the absence of C3a signaling.
We further wanted to determine whether lung ICOS+IL-33R+ ILC2 (see gating scheme, Figure S2a) were affected by lack of C3a signaling. We observed that both the amount and percentage of HDM-induced ILC2 (Fig. 1l and S2b) and IL-13+ ILC2 (Fig. 1m and S2c) were profoundly reduced in C3ar1−/− mice as compared with control animals. Interestingly, in the context of cigarette smoke and viral infection, lung ILC2 have been shown to exhibit some functional plasticity that allows them to switch into IFNγ+ ILCs.40 For this reason, we examined if decrements in HDM-elicited IL-13+ILC2 in C3ar1−/− mice could be owing to a shift away from IL-13 and toward IFNγ-producing ILC1-like cells. However, we did not find that decreased IL-13+ ILC2 levels in C3ar1−/− mice were accompanied by a concomitant shift favoring IFNγ production (Figure S3a, b).
Collectively, these data not only demonstrate the integral role of C3a signaling in the development of dust mite-driven Th2 immunity, but also expands its role as a driver of type 2 innate responses.
C3a is required for lung IL-33-driven ILC2 responses
ILC2s rapidly accumulate at mucosal surfaces in response to a variety of triggers (allergens, virus), which depends on epithelial-derived innate mediators like IL-33, a central driver of type 2 responses in the lungs. Thus, we investigated whether C3a signaling was necessary for IL-33-driven responses. To this end, we exposed wt and C3ar1−/− mice to PBS or 0.5 µg rIL-33 intranasally (i.n.) on days 0, 2, and 4, and analyzed the response on day 6. rIL-33-treated mice developed significant AHR to cholinergic stimulation (Fig. 2a), and consistent with our observations with allergen, C3ar1−/− mice were significantly protected against IL-33-driven AHR as compared with wt mice (Fig. 2a). In addition, the IL-33-mediated total BAL cell, as well as eosinophil and neutrophil influx to the airways were reduced in mice lacking C3a signaling (Fig. 2b–d).

C3a signaling is necessary for IL-33-dependent ILC2 responses in the lungs. Wildtype BALBc/J and C3ar1−/− mice were given PBS and rIL-33 on days 0, 2, and 4. On day 6, a airway hyperresponsiveness was determined, following which BAL was collected for enumeration of b total BAL cells, c eosinophils, and d neutrophils. Lung cells (from non-lavaged mice) were analyzed by flow cytometry for Lin− (CD11b, CD11c, Gr1, B220, CD19, TCRb, TCRgd, CD49b, CD4, CD8, FcER1) ICOS+IL-33R+ ILC2 e, f frequency and g numbers. h Frequency and i numbers of IL-13+ICOS+IL-33R+ ILC2 were determined. Levels of lung j Il5 and k Il13 mRNA. Data represent means + SEM. Data are representative of two independent experiments with 4–6 mice/group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Because IL-33 is critical for the activation of ILC2 effector functions, we investigated whether C3a signaling played a significant role in driving IL-33-elicited ILC2 responses. As described above, mice were exposed to PBS or IL-33, and lung ILC2 were identified by flow cytometry (see gating scheme, Fig. 2e). We observed that although C3ar1-sufficient and -deficient mice had a similar frequency of IL-33R+ICOS+ ILC2 (Fig. 2f), their numbers were profoundly reduced in the absence of C3a signaling (Fig. 2g). Consistently, we found that the frequency (Fig. 2h) and numbers (Fig. 2i) of IL-33-induced IL-13+ ILC2 showed no significant increase in C3ar1−/− mice, as compared with controls. This was accompanied by reduced levels of IL-33-induced Il5 (Fig. 2j) and Il13 (Fig. 2k) mRNA in the lungs of C3ar1−/− mice.
As lung ILC2 are known to proliferate locally,41,42 we tested whether the decreased numbers of ILC2 in C3ar1−/− mice were due to diminished proliferative potential. We observed that C3ar1−/− ILC2 are functionally similar to wt ILC2 in their capacity to proliferate. We show an equivalent increase in the frequency of EdU+ILC2 (Figure S4a, b) in response to IL-33. Based on this, and consistent with the chemotactic function of C3a,15,16,43,44 these data suggest that instead, ILC2 may require C3a for their trafficking to the lungs. Taken together, our results indicate that C3a signaling is required for optimal IL-33-dependent ILC2 responses in the lungs.
ILC2 can respond to C3a
Our data show that C3a signaling is necessary for the recruitment of ILC2 in vivo, suggesting that ILC2 can directly respond to C3a. Indeed, freshly flow-sorted lung ILC2 from naive mice displayed constitutive expression of C3ar1 mRNA in (Fig. 3a). Consistent with our findings, analysis of microarray data from the ImmGen Consortium revealed that, within various lymphoid lineages, resting small intestine lamina propria ILC2 expressed some of the highest levels of both anaphylatoxin receptors C3ar1 and C5ar1 (Figure S5a). ILC2 had ~1.4-fold greater levels of C3ar1 and C5ar1 than CD4 + T cells (Figure S5b,c). We found that C3ar1 expression was enhanced by IL-33 in culture, suggesting that IL-33 may increase ILC2 responsiveness to C3a (Fig. 3b). Based on this, we tested whether ILC2 could directly respond to C3a. We observed that, on its own, C3a can induce modest IL-13 levels (Fig. 3c) and enhances IL-33-mediated IL-13 production from cultured ILC2 (Fig. 3d). Moreover, we found that cultured ILC2 can consitituvely generate C3a and that it is dose-dependently increased by IL-33 (Fig. 3e).

ILC2 respond to C3a. C3ar1 mRNA expression in a fresh flow-sorted lung Lin− (CD11b, CD11c, Gr1, B220, CD19, TCRb, TCRgd, CD49b, CD4, CD8, FcER1) ICOS+IL-33R+ ILC2 from naive BALB/c and C3ar1−/− mice, and b wt ILC2 cultured in IL-2 (10 ng/ml) alone or in addition to IL-33 (0.5 ng/ml) for 3 days. IL-13 in the supernatant of sorted lung ILC2 cultured in c IL-2 (10 ng/ml) or IL-2 with C3a (1 µg/ml) or d media, IL-2 + IL-33 or IL-2 + IL-33 + C3a after 3 days. e C3a in the supernatant of lung ICOS+IL-33R+ ILC2 cultured in IL-2 (10 ng/ml) containing 0, 0.5, or 1.0 ng/ml IL-33 for 3 days. Lung ICOS+IL-33R+ ILC2 cultured in IL-2 (10 ng/ml) or IL-2 in combination with IL-33 (0.5 ng/ml) for 3 days, and levels of f IL-13 and g GM-CSF in the supernatant was measured, and cells were harvested for h Il10 mRNA determination. Rag1−/− mice exposed to PBS, C3a (1 µg), IL-33 (0.5 µg) or C3a + IL-33 i.n. on days 0, 2, and 4, and lungs were analyzed on day 6 for i numbers and j frequency of IL-13+ ILCs (Lin-CD45+IL-33R+IL-13+), as well as k IL-13 median fluorescence intensity (MFI) in ILC2. Data represent means + SEM. Data are representative of two to three independent experiments with four replicate wells or 3–5 mice/group. *p < 0.05, **p < 0.01, ***p < 0.001
Based on that, we cultured wt and C3ar1−/− ILC2 to determine whether autocrine C3a responsiveness could alter cytokine secretion. We found that C3ar1−/− ILC2 secreted significantly less IL-13 (Fig. 3f) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Fig. 3g) in response to IL-33 than wt ILC2, along with significantly reduced IL-33-induced Il5 and Il13 mRNAs (Figure S6a,b). This effect was specific as C3a signaling was inhibitory to IL-33-induced Il10 mRNA (Fig. 3h), and amphiregulin (Figure S6c). This observation is consistent with previously published reports by us and others showing an inhibitory effect of C3a on IL-10 production.14,45
We further wanted to determine whether C3a could directly modulate IL-33-elicited ILC2 recruitment and activation in vivo independently of CD4+ T cells. Rag1−/− mice, received PBS, IL-33 (0.5 µg), or IL-33 + C3a (1 µg) i.n. every other day (days 0, 2, and 6). Twenty-four hours afterwards, the numbers of lung IL-13 + ILC2s were enumerated. Mice exposed to IL-33 + C3a had significantly higher numbers (Fig. 3i) and frequency (Fig. 3j) of IL-13+ILC2 than those exposed to IL-33 alone, suggesting a synergistic effect between C3a and IL-33 on ILC2 recruitment in vivo. Moreover, and consistent with our in vitro data, IL-13 production per cell, as represented by median fluorescence intensity (MFI), was also significantly increased by C3a (Fig. 3k). These data support the concept that C3a can enhance IL-33-induced ILC2 number and function.
C3a signaling to ILC2 is necessary for ILC-T-cell cross-talk
The biology of ILCs has expanded beyond its role as a cytokine-producing effector cell. Recent reports have shown that ILC2s can act as antigen-presenting cells and cross-talk with CD4+ T cells.36,37,39 Based on this, we wanted to establish whether C3a signaling could impact this aspect of ILC2 effector function. To this end, we flow-sorted lung IL-33R+ ILC2 and co-cultured them with CFSE-labeled lymph node CD4+ DO11.10 OVATg T cells (Fig. 4a). In C3ar1-sufficient ILC-T-cell co-cultures, addition of the major histocompatibility complex II (MHCII)-restricted OVA 323–339 peptide significantly increased proliferation and numbers of T cells (Fig. 4b, c), recapitulating the observation that OVA-loaded ILCs can drive CD4+ T cells proliferation and activation.37,39 However, T cells cultured with C3ar1-deficient ILC2 displayed impaired proliferation and accumulation (Fig. 4b, c). Consistent with these findings, OVA-induced levels of IL-2 seen in wt ILC-T-cell co-cultures were abrogated in C3ar1−/− ILC + T-cell conditions (Fig. 4d).

C3a regulates ILC-T-cell cross-talk. 1 × 104 Flow-sorted lung Lin- (CD11b, CD11c, Gr1, B220, CD19, TCRb, TCRgd, CD49b, CD4, CD8, FcER1) ICOS+IL-33R+ ILC2 from wildtype and C3ar1−/− mice were co-cultured with 1 × 104 CFSE-labeled OVA-transgenic (D011.10Tg) lymph node CD4+ T cells in the presence of 1 µg/ml OVA 323–339 peptide for 5 days. a Representative flow plot showing pre- and post-sort purity of sorted lung ILC2. b CFSE-labeled DO11.10Tg T cells proliferation. c Total T-cell numbers (CD3+CD4+), d IL-2 production, e frequency IL-13+CD4+ cells, and supernatant levels of f IL-13 g GM-CSF, and h IFNγ production. Data are means + SEM, representative of two independent experiments. All flow cytometry gating is based on FMO controls and, non-CFSE-labeled T cells. *p < 0.05, ***p < 0.001
We then hypothesized that lack of C3a signaling within the ILC compartment would also impact Th2 cell polarization. We show that although C3ar1-sufficient ILC2 promoted IL-13 + T cells in the presence of OVA peptide, however, this was decreased when T cells were incubated with C3ar1-deficient ILC2 (Fig. 4e). Further, co-cultures containing C3ar1−/− ILC2 showed decreased OVA-driven IL-13 (Fig. 4f) and GM-CSF (Fig. 4g) as compared with wt ILC2-T-cell co-cultures, whereas basal IFNγ levels were not modulated in these cultures (Fig. 4h). These data are supported by the previous observation that autocrine C3a signaling in DCs is necessary for T-cell cytokine production,24 and this is in part thought to be owing to the role of C3a in enhancing MHCII and co-stimulatory molecule expression.16,46,47 However, the frequency and expression of MHCII (Figure S7a–c), or the co-stimulatory molecules CD86 (Figure S7d–e) and CD80 (Figure S7f–h) remained relatively unchanged or only marginally lower in C3ar1−/− ILC2, suggesting that other aspects of ILC biology that impact ILC-T-cell cross-talk are regulated by C3a signaling. In contrast, when we used DCs, in co-culture experiments C3a signaling had no effect on Th2 polarization. Addition of rC3a to DC-T-cell co-cultures did not yield differences in OVA-induced IL-5 or IL-13 production (Figure S8a, b). This is consistent with reports demonstrating that C3a signaling to either DCs or T cells does not appear to modulate Th2 differentiation.13,25,26 However, addition of exogenous C3a increased IL-17A levels (Figure S8c), in line with previous studies showing that C3a signaling is required for DC-derived Th17-driving cytokines like IL-23 and IL-1β.12,14,25 In addition, we found that co-culturing CD4+ DO11.10 OVATg T cells with either wt or C3ar1−/− lung CD11b+ DCs resulted in equivalent T-cell proliferation (Figure S8d–f). This is in contrast with previous work, where C3ar1-deficient DCs triggered less OVA-driven T-cell proliferation than C3ar1-sufficient BMDCs.24 This discrepancy could be explained by several factors including: (1) our use of flow-sorted lung CD11b+ DCs as opposed to BMDCs; (2) OVA 323–339 peptide versus full-length OVA protein24; (3) as well as the use DO11.10 OVATg mice in contrast to OT-II OVATg mice,24 which have different MHC backgrounds.
Taken together, these data suggest that, ILC2 need C3a signaling to optimally function as antigen-presenting cells in order to support Th2 differentiation and proliferation. This identifies a previously unrecognized mechanism by which C3a further fuels Th2 immunity.
C3a signaling to ILC2 is necessary for ILC-T-cell-driven airway responses
We further wanted to confirm whether the role of C3a signaling in ILC2-T-cell cross-talk could have a wider implication, and directly modulate lung function, the cardinal feature of the asthmatic response. To this end, we pulsed C3ar1-sufficient or C3ar1-deficient lung ILC2 with 1 µg/ml OVA 323–339 peptide for 1 h at 37°C. These ILC2s were then transferred with lymph node CD3+CD4+ DO11.10 OVATg T cells to alymphoid Rag2−/−Il2rg−/− mice. Mice were exposed to OVA 323–339 peptide (40 µg, i.t.) and lung function was determined 3 days later. We found that mice receiving wt ILC2 and T cells developed significant AHR to methacholine as compared with mice receiving only T cells but not significantly different than T cells + OVA (Fig. 5a). However, and in accordance with our in vitro data, mice receiving C3ar1−/− ILC2 along with T cells developed significantly less AHR (Fig. 5a). Concomitant with this, we observed a decreased number of CD4+ T cell (Figure S9a) in the lungs following OVA challenge in mice receiving C3ar1−/− ILC2 as compared with mice receiving wt ILC2. This also correlated with a decreased frequency of IL-13+ DO11.10Tg CD4+ T cells (Fig. 5b, c), number of IL-13+CD4+ T cells (Fig. 5d), IL-13 MFI (Fig. 5e), Il13 (Fig. 5c), and Il2 (Fig. 5d) mRNA in the lungs of mice that received C3ar1−/− ILC2 as compared with mice receiving wt ILC2. These results were not owing to a decrease in survival of transferred CD4+ T cells or ILC2 (Figure S9b, c, d). suggesting that CD4+ T cells proliferation and activation in the lungs after airway challenge requires intact C3a signaling to ILC2.

C3a is necessary for ILC-T-cell-driven airway responses to OVA. In total, 2 × 105 flow-sorted DO11.10 OVATg T cells (CD3+CD4+) were transferred with 1 × 104 flow-sorted lung IL-33R+ ILC2 from wildtype or C3ar1−/− mice i.v. to Rag2−/−Il2rg−/− recipient mice. After 24 h, mice were challenged i.t. with 40 µg OVA 323–339 peptide. Three days after OVA challenge, a airway hyperresponsiveness to nebulized methacholine was determined. Lungs were isolated for analysis of b, c frequency and d numbers of IL-13+CD4+ T cells, e IL-13 MFI in T cells, and levels of f Il13 and g Il2 mRNA. Data are means + SEM, and pooled from two independent experiments with 6–12 mice per group, *p < 0.05
C3a signaling is necessary for optimal ILC2-mediated antigen presentation
In their function as antigen-presenting cells, ILC2 can take up and process antigen.37 As C3a signaling plays a role in antigen uptake in DCs,24 we investigated whether antigen uptake and/or processing could be altered in C3ar1−/− ILC2 as compared to wt ILC2. We establish, consistent with others,37 that lung ILC2 can endocytose and degrade simple protein antigens like ovalbumin-DQ (OVA-DQ) (Fig. 6a). OVA-DQ is a quenched protein that only fluoresces when cleaved by intracellular proteases. We found that the frequency of OVA-DQ+ ILC2 (Fig. 6b) and per-cell quantity of processed OVA (Fig. 6c) was similar between wt and C3ar1−/− ILC2.

C3a drives optimal antigen presentation in ILC2. 0.5–1.0 × 104 flow-sorted lung Lin− (CD11b, CD11c, Gr1, B220, CD19, TCRb, TCRgd, CD49b, CD4, CD8, FcER1) ICOS+IL-33R+ ILC2 from wildtype or C3ar1−/− mice were cultured in 96-well round-bottom dishes. a Detection of fluorescence resulting from OVA-DQ degradation by ILC2, b frequency of OVA-DQ+ ILC2, and c OVA-DQ median fluorescence intensity (MFI). d Detection of fluorescence resulting from pHrodo-HDM phagosome-mediated degradation e frequency of pHrodo-HDM+ ILC2, and f pHrodo-HDM median fluorescence intensity (MFI). g Detection of processed and presented Ea peptide on ILC2, and frequency of h EaGFP+ and i YAe+ ILC2. Data are mean + SEM and representative of two independent experiments.*p < 0.05
We next tested whether C3a could influence the processing of a more complex source of antigen like HDM extract. We found that ILC2 can endocytose and degrade HDM labeled with pHrodo (Fig. 6d), which is a pH-sensitive dye that exploits the acidification of endocytic vesicles and only fluoresces when internalized. Much like with OVA-DQ, the percentage of pHrodo-HDM+ ILC2 (Fig. 6e) and quantity of phagosome-associated pHrodo-HDM (Fig. 6f) was comparable in both wt and C3ar1−/− ILC2. Although these data clearly demonstrate that C3a does not influence uptake and processing of antigen, it does not rule out the possibility that loading and presentation of processed antigen may be regulated by C3a. To analyze this, we cultured ILC2 in the presence of the E-alpha GFP fusion protein (EαGFP)48 and used the YAe antibody to detect Eα-derived peptide bound to MHCII (Fig. 6g). Our data show that although uptake of the EαGFP protein was similar (Fig. 6h), MHCII-mediated presentation of the Eα peptide was significantly lower in C3ar1−/− ILC2 as compared with wt ILC2 (Fig. 6i). In sum, our data indicate that in ILC2 C3a signaling facilitates MHC peptide loading, which is necessary to drive optimal ILC-T-cell cross-talk and MHCII-dependent T-cell activation.
Discussion
The anaphylatoxin C3a has been repeatedly shown to be essential for the development of several manifestations of asthma including AHR, eosinophilia, and the production of type 2 cytokines. In mouse and humans, allergen exposure readily induces C3a production,2,7 and although C3a is central to the development of allergic responses,2,3 the mechanisms by which it promotes type 2 immunity are unclear. Here we demonstrate that C3a plays a yet unregognized role in ILC2 biology: (1) C3a controls the numbers of lung ILC2 likely via C3aR-mediated chemotaxis, and (2) intrinsic C3a ILC2 signaling facilitates ILC antigen-presentation through increased MHCII-peptide loading and enhanced ILC-T-cell cross-talk.
We show here that allergen-induced airway responses and inflammation are dependent on C3a signaling, and this is consistent with many reports from us and others that show the importance of C3a in driving allergen-induced asthma,2,3,14 yet the mechanism driving this effect has remained unclear. A major tipping point for the development of allergic airway diseases is the aberrant production of IL-33 in response to allergen, and here we establish that IL-33-induced AHR and airway inflammation also require C3a, signifying that events downstream of IL-33 are dependent on C3a signaling to operate. Given the central role of IL-33 in driving ILC2 responses, we investigated whether C3a could regulate their biology.
We find that ILC2 express the C3a receptor and directly respond to C3a. Although expression of the C3aR is classically associated with myeloid cells, our data support other reports showing a critical role for C3a responsiveness in cells of lymphoid origin.13,49,50 In addition to promoting Th2 cell responses, C3a signaling has a profound impact on the numbers of both HDM- and IL-33-elicited ILC2 in the lungs. Accumulation of ILC2 in mucosal tissues is thought to occur via several mechanism including local proliferation,41,42 survival,51 as well as recruitment from the bone marrow52 or blood.53 Although C3a has been shown to enhance the proliferation of human T cells in vitro,19 we find that, in vivo, C3a signaling does not impact ILC2 proliferation in response to IL-33. This suggests that C3a-dependent ILC2 accumulation is driven by mechanisms other than enhanced proliferation. Although it is unclear how C3a signaling promotes ILC2 accumulation in response to allergen or IL-33, we speculate that it may be required for recruitment of ILC2 into the lungs. As C3a is a well-established chemoattractant,16,43,44 and is substantially upregulated in the airways by allergen, it is therefore likely that C3ar1−/− ILC2 maybe have impaired trafficking to the lungs.
In addition to driving ILC2 recruitment, C3a can directly modulate the effector functions of ILC2. C3a enhances IL-13 and GM-CSF from ILC2 in vivo and in vitro, whereas decreasing anti-inflammatory cytokines like IL-10 and amphiregulin, suggesting that C3a promotes the allergic response by increasing type 2 mediators, whereas simultaneously downregulating protective signals.
Although ILC2 have been shown to independently drive allergic inflammation and promote worm expulsion by virtue of the cytokines they secrete, recent reports have demonstrated that ILC2-mediated MHCII-dependent antigen presentation enhances adaptive immune responses and is necessary for the development of Th2 responses in vivo.37,39 We demonstrate that ILC2 can drive T cells to proliferate and produce cytokines, consistent with previous reports demonstrating a role for ILC2 in maintaining T-cell fitness.37,39 In contrast, ILC2 that cannot respond to C3a are defective in their ability to optimally drive T-cell proliferation and cytokine production. These findings clearly demonstrate that C3a not only enhances IL-33-induced cytokines from ILC2, but is necessary for proper ILC-T-cell cross-talk. Interestingly, C3a signaling in DC-T-cell co-cultures did not seem to play a role in driving enhanced T-cell proliferation or Th2 cytokine production, but enhanced IL-17A production. This is in line with previous reports that show that C3a signaling to DCs promotes Th17,12,14,25 but not Th2 responses.25 Although the connection between C3a signaling and the development of Th2 responses in the lungs is well recognized,2,3,54 we demonstrate that this may be through enhanced ILC-T-cell interaction.
Consistent with our ILC-T-cell co-culture data, we show that C3a signaling to ILC2 is also necessary to drive in vivo allergic responses. Co-transfer of OVATg CD4+ T cells with wildtype ILC2 to alymphoid mice is sufficient to drive AHR in response to OVA, and this is accompanied by an increase frequency of IL-13+ T cells and lung Il13 and Il2 mRNA. In contrast, these parameters were significantly decreased when OVATg CD4+ T cells were co-transferred with C3ar1−/− ILC2. Our data demonstrate the importance of C3a in ILC2-driven Th2 responses, but it appears that C3a signaling is also important for Th1 and Th17 immunity as well. Several reports also show a role for C3a signaling to DC in mediating Th1 and Th17 responses.12,14,24,25
C3a responsiveness in DCs is thought to mediate Th1 and Th17 immunity in part through superior co-stimulatory function, via increased expression of CD80 and CD86.55 Although we found, consistent with others37 that, in addition to MHCII, lung ILC2 express CD80 and CD86, their expression was not significantly altered by C3a signaling. Also, we found that C3a does not affect ILC2 antigen uptake and processing, unlike what is seen in BMDCs.24 However, we make the novel observation that C3a unresponsiveness in ILC2 is associated with impaired MHCII-peptide loading, necessary for optimal MHCII-dependent antigen presentation and T-cell activation.
We have uncovered a new pathophysiologically important pathway whereby allergen-driven C3a targets ILC2s and is required for the full magnitude of type 2 responses in the lungs. We establish that C3a signaling to ILC2 is essential for their recruitment and function as antigen-presenting cells. Our findings suggest that the elevated levels of C3a in asthmatics airways2,7,8 may contribute to asthma pathogenesis by driving enhanced ILC2 numbers and function, thus revealing a new mechanism by which C3a drives type 2 immunity.
Materials and methods
Mice
C57BL/6 J, BALB/cJ, C3ar1−/−, Rag1−/−, and Rag2−/−Il2rg−/− were obtained from Jackson and bred in our facility. Mice were housed in a specific pathogen free animal facility in micro-isolator cages. Mice were provided autoclaved food (Lab diet 5010) and water ad libitum. All procedures were approved by the Animal Care and Use Committee of Johns Hopkins University. Experiments were performed with mice that have been bred in the same facility for over a year.
HDM, rIL-33, and rC3a in vivo administrations
Seven to 10-week-old mice were given PBS (40 µl) or HDM (100 µg/40 µl) intratracheally (i.t.) days 0 and 14. After 72 h, the last challenge the allergic phenotype was assessed. Alternatively, mice received either PBS or 0.5 µg rIL-33 (eBioscience) intranasally (i.n.) on days 0, 2, and 4, and mice were analyzed on day 6. Some mice received 1 µg recombinant mouse C3a (RnD Systems) alone or in combination with 0.5 µg rIL-33 i.n. on days 0, 2, and 4.
In vitro ILC2 cultures
Lung ILC2 were sorted as lineage− (CD3, CD4, CD8, CD11b, Gr1, CD11c, TCRb, TCRgd, FceR1, CD49b, CD19) CD45+ICOS+IL-33R+ from naive male wildtype of C3ar1−/− mice. A total of 1 × 103 ILC2 were seeded at the bottom of round-bottom 96-well dishes and cultured in Roswell Park Memorial Institute (RPMI) containing 10% fetal bovine serum (FBS), l-glutamine, penicillin/streptomycin, and 55 µM 2-mercaptoethanol, 10 ng/ml IL-2 (RnD Systems) combined with IL-33 (RnD Systems), with or without 1–2 µg/ml C3a (RnD Systems). Cultures were maintained for 3–5 days, and supernatants were harvested for enzyme-linked immunosorbent assay (ELISA).
In vitro ILC-T-cell co-culture
Flow-sorted lung ILC2, defined as lineage− (CD3, CD4, CD8, CD11b, Gr1, CD11c, TCRb, TCRgd, FceR1, CD49b, CD19) CD45+ICOS+IL-33R+, were isolated from naive male BALB/c and C3ar1−/− mice. CD3+CD4+ OVATg T cells were sorted from lymph nodes of DO11.10Tg mice and labeled with 4 µM CFSE for 10 min at RT (Thermo). A total of 0.5 × 104 ILC2 were co-cultured with 0.5 × 104 DO11.10Tg CFSE-labeled T cells in RPMI containing 10% FBS, l-glutamine, penicillin/streptomycin and 55 µM 2-mercaptoethanol and incubated with 1 µg/ml MCHII-restricted OVA 323–339 peptide (Anaspec) for 5 days. Supernatants were collected for ELISA and cells were analyzed by flow cytometry.
In vitro DC-T-cell co-culture
CD4+ and CD11c+ cells were magnetically (Miltenyi) purified from lymph nodes and spleen of DO11.10Tg mice. In total, 3.0 × 104 CD11c+ cells were co-cultured with 1.5 × 105 CD4+ DO11.10Tg CD4+ T cells in RPMI containing 10 % FBS, l-glutamine, penicillin/streptomycin, and 55 µM 2-mercaptoethanol, and incubated with 1 µg/ml MCHII-restricted OVA 323–339 peptide (Anaspec) for 5 days with or without recombinant mouse C3a (0.2 or 1 µg/ml). Supernatants were collected for ELISA.
In vivo ILC2-T-cell transfer
Lung ILC2 (Lin-CD45+IL-33R+) from naive male BALB/c and C3ar1−/− mice, and CD3+CD4+ T from lymph nodes of DO11.10Tg male mice were flow-sorted. ILC2 were pre-loaded with 1 µg/ml OVA 323–339 peptide for 1 h at 37°C/5% CO2, after 1 h, ILCs were washed in PBS. A total of 2 × 105 DO11.10Tg CD3+CD4+ T cells were transferred alone or in combination with 1 × 104 OVA-loaded lung ILC2 to Rag2−/−Il2rg−/− mice i.v. 24 h later, mice were challenged i.t. with PBS or 40 µg OVA 323–339 peptide. Three days after, AHR was determined, and lungs collected for flow cytometry and RNA.
Airway measurements
In brief, mice were anesthetized by i.p. administration of ketamine/xylazine and tracheotomized before insertion of an 18-gauge cannula into the trachea. Mice were paralyzed with suxamethonium chloride (3 mg/kg), intubated, and respirated at a rate of 120 breaths per minute with a constant tidal volume (0.2 ml). After a stable baseline was achieved, mice were exposed to 30 mg/ml nebulized methacholine (Sigma). After 10 seconds, dynamic airway pressure (cm H20 × s) was recorded for 3 min. Following airway reactivity measurements, serum, BAL fluid, and lungs were collected, and processed as previously described.14,56
ELISA
Mouse IL-13, IL-10, GM-CSF, IL-2, IL-5, IL-17A, IFNγ, and amphiregulin were detected using DuoSets (RnD Systems). Human C3a was detected using a DuoSet kit (RnD Systems). Mouse C3a was detected using antibodies from BD Biosciences, in brief, C3a was captured using rat anti-mouse C3a clone 187–1162 (2 µg/ml) and detected with biotinylated rat anti-mouse C3a clone 187–419 (1 µg/ml). Serum IgE was measured by ELISA using the BD OptEIA kit (BD Biosciences).
Real-time PCR
Total RNA was extracted from lung tissue using TRIzol RNA isolation reagent (Invitrogen). The reverse transcription reaction was performed using a high capacity cDNA synthesis kit (Applied Biosystems). Quantitative PCR analyses of mouse genes were performed by using TaqMan real-time PCR assays (Applied Biosystems), where primers spanned exons to avoid co-amplification of genomic DNA. Rps13 and Actb were used for normalization.
Flow cytometry
Mouse lung cells were obtained by digestion of lung tissue with 0.05 mg/ml Liberase TL (Roche) and 0.5 mg/ml DNaseI (Sigma) for 45 min at 37 °C in 5% CO2. Digested tissue was filtered through a 70-µm nylon mesh (BD Biosciences) and centrifuged. Pellet was resuspended in red blood cell lysis buffer (Ammonium-Chloride-Potassium lysis buffer). Recovered cells were counted (trypan blue exclusion), plated at 4–5 × 106 cells/ml. For intracellular staining, cells were stimulated with phorbol 12-myristate 13-acetate (PMA, 50 ng/ml) and ionomycin (1 µg/ml) for 16 h, then Brefeldin A and monensin (eBioscience) were added for the last 3–4 h. All cells were filtered using a 40-µm nylon mesh (BD Biosciences), washed with PBS and labeled with live/dead dye (Zombie Aqua, Biolegend) for 10 min at RT, and blocked with anti-CD16/32 (BioLegend) for an additional 20 min at RT. To identify ILC2 cells were stained with PerCP-Cy5.5-conjugated lineage antibodies: CD3ε (145-2C11, BD Biosciences), αβTCR (H57-597, BD Biosciences), CD11b (M1/70, BD Biosciences), CD11c (N418, eBioscience), Gr1 (RB6-8C5, eBioscience), CD19 (6D5, BioLegend), γδTCR (GL3, BioLegend), FceR1 (MAR-1, BioLegend), and TER-119 (TER-119, BioLegend), CD4 (RM4-5, BioLegend), CD8a (53-6.7, BioLegend), CD49b (DX5, BioLegend), APC-Fire750 anti-CD45 (30-F11, BioLegend), FITC-anti-IL-33R (T1/ST2, MD Bioproducts), and Brilliant Violet 786 ICOS (15F9, BioLegend). Alternatively, when ILC2 were co-identified along with CD4+ and CD8+ T cells, cells were stained with PerCP-Cy5.5-conjugated lineage antibodies (CD11b, CD11c, Gr1, γδTCR, FceR1, CD49b, CD19, B220) in addition to PE-Cy7 anti-CD45 (30-F11, BioLegend), FITC-anti-IL-33R (T1/ST2, MD Bioproducts), Brilliant Violet 421 anti-CD3, Brilliant Violet 605 anti-CD4, PE-Dazzle 594 CD8a (53-6.7, BioLegend), and Brilliant Violet 786 ICOS. For intracellular cytokine staining, cells were first surface stained, then fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 10 min at RT and permeabilized in 0.1% saponin (Sigma) for 20 min at RT prior to staining with eFluor 660-conjugated anti-IL-13 (eBio13A, eBioscience) and Alexa Fluor 700-conjugated anti-IFNγ (XMG1.2, BioLegend). Data were acquired on an LSRII flow cytometer (BD Biosciences), and gated to exclude debris and to select single cells (FSC-W/FSC-A + SSC-W/SSC-A). Data were analyzed using FACSDiVa (BD Biosciences) and FlowJo (Treestar).
EαGFP fusion protein
A plasmid encoding the EαGFP fusion protein, a kind gift of Dr. Marc K. Jenkins, was used to generate the EαGFP protein. To assess whether wt and C3ar1−/− ILC2s can present processed antigen differently, we cultured sorted lung ILC2s (ICOS+IL-33R+) in the presence of 100 μg/ml EαGFP protein for 16 h at 37 °C, 5% CO2. Cells were incubated with a biotin-conjugated antibody (1:100) specific to the Eα52-68 bound to MHC (I-Ab) (clone eBio YAe-eBiosciences) for 20 mins at RT, washed, then followed by Brilliant Violet 421-conjugated streptavidin (BioLegend). As negative control, cells were incubated with EαGFP, where the YAe antibody was omitted, but Brilliant Violet 421-conjugated streptavidin was added.
In vitro OVA-DQ uptake
In vitro OVA-DQ endocytosis and degradation assay was performed by incubating 20 µg/ml DQ-conjugated OVA (Thermo) with sorted lung isolated ILC2 (ICOS+IL-33R+) for 16 h at 37 °C, 5% CO2.
In vitro pHrodo-HDM uptake
HDM extract (Greer) was labeled with pHrodo Red succinimidyl ester (Thermo), followed by dyalisis against PBS overnight at 4 °C using a 3.5 K MWCO SnakeSkin tubing (Thermo). In vitro pHrodo-HDM endocytosis and degradation assay was performed by incubating 100 µg/ml pHrodo-HDM with sorted lung isolated ILC2 (ICOS+IL-33R+) for 16 h at 37 °C, 5% CO2.
NHBE culture
Normal human bronchial epithelial cells (Lonza) were cultured in Bronchial Epithelial Cell Growth Medium (BEGM) media (Lonza). Cells were placed in collagen-coated flat-bottom 96 dishes. At confluency, cells were washed and media was replaced with BEGM minus bovine pituitary extract and starved overnight. Cells were stimulated with HDM (100 µg/ml) for 4 h and supernatants were harvested for determination of C3a.
References
Pawankar, R., Canonica, G. W., Holgate, S. T. & Lockey, R. F. Allergic diseases and asthma: a major global health concern. Curr. Opin. Allergy Clin. Immunol. 12, 39–41 (2012).
Humbles, A. A. et al. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature 406, 998–1001 (2000).
Drouin, S. M., Corry, D. B., Hollman, T. J., Kildsgaard, J. & Wetsel, R. A. Absence of the complement anaphylatoxin C3a receptor suppresses Th2 effector functions in a murine model of pulmonary allergy. J. Immunol. 169, 5926–5933 (2002).
Kapp, A., Wokalek, H. & Schopf, E. Involvement of complement in psoriasis and atopic dermatitis--measurement of C3a and C5a, C3, C4 and C1 inactivator. Arch. Dermatol. Res. 277, 359–361 (1985).
Jun, S. W. et al. Overexpression of the anaphylatoxin receptors, complement anaphylatoxin 3a receptor and complement anaphylatoxin 5a receptor, in the nasal mucosa of patients with mild and severe persistent allergic rhinitis. J. Allergy Clin. Immunol. 122, 119–125 (2008).
Kapp, A. & Schopf, E. Involvement of complement in atopic dermatitis. Acta Derm. Venereol. Suppl. (Stockh.). 114, 152–154 (1985).
Krug, N., Tschernig, T., Erpenbeck, V. J., Hohlfeld, J. M. & Kohl, J. Complement factors C3a and C5a are increased in bronchoalveolar lavage fluid after segmental allergen provocation in subjects with asthma. Am. J. Respir. Crit. Care. Med. 164, 1841–1843 (2001).
Nakano, Y. et al. Elevated complement C3a in plasma from patients with severe acute asthma. J. Allergy Clin. Immunol. 112, 525–530 (2003).
Barnes, K. C. et al. Variants in the gene encoding C3 are associated with asthma and related phenotypes among African Caribbean families. Genes Immun. 7, 27–35 (2006).
Hasegawa, K. et al. Variations in the C3, C3a receptor, and C5 genes affect susceptibility to bronchial asthma. Hum. Genet. 115, 295–301 (2004).
Inoue, H. et al. Association study of the C3 gene with adult and childhood asthma. J. Hum. Genet. 53, 728–738 (2008).
Asgari, E. et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 122, 3473–3481 (2013).
Liszewski, M. K. et al. Intracellular complement activation sustains T-cell homeostasis and mediates effector differentiation. Immunity 39, 1143–1157 (2013).
Lajoie, S. et al. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat. Immunol. 11, 928–935 (2010).
Carmona-Fontaine, C. et al. Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev. Cell 21, 1026–1037 (2011).
Gutzmer, R. et al. Human monocyte-derived dendritic cells are chemoattracted to C3a after up-regulation of the C3a receptor with interferons. Immunology 111, 435–443 (2004).
Kwan, W. H., van der Touw, W., Paz-Artal, E., Li, M. O. & Heeger, P. S. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J. Exp. Med. 210, 257–268 (2013).
Strainic, M. G., Shevach, E. M., An, F., Lin, F. & Medof, M. E. Absence of signaling into CD4( + ) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3( + ) regulatory T cells. Nat. Immunol. 14, 162–171 (2013).
Cravedi, P. et al. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am. J. Transplant. 13, 2530–2539 (2013).
Johnson, A. R., Hugli, T. E. & Muller-Eberhard, H. J. Release of histamine from rat mast cells by the complement peptides C3a and C5a. Immunology 28, 1067–1080 (1975).
Zaidi, A. K., Amrani, Y., Panettieri, R. A. & Ali, H. Response to C3a, mast cells, and asthma. FASEB J. 20, 199 (2006).
Bradding, P. C3a, mast cells, and asthma. FASEB J. 19, 1585 (2005).
Thangam, E. B. et al. Airway smooth muscle cells enhance C3a-induced mast cell degranulation following cell-cell contact. FASEB J. 19, 798–800 (2005).
Li, K. et al. Cyclic AMP plays a critical role in C3a-receptor-mediated regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood 112, 5084–5094 (2008).
Engelke, C. et al. Distinct roles of the anaphylatoxins C3a and C5a in dendritic cell-mediated allergic asthma. J. Immunol. 193, 5387–5401 (2014).
Ghannam, A., Fauquert, J. L., Thomas, C., Kemper, C. & Drouet, C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol. Immunol. 58, 98–107 (2014).
van der Touw, W. et al. Cutting edge: Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J. Immunol. 190, 5921–5925 (2013).
Mjosberg, J. M. et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 12, 1055–1062 (2011).
Bartemes, K. R., Kephart, G. M., Fox, S. J. & Kita, H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J. Allergy Clin. Immunol. 134, 671–678 e674 (2014).
Walford, H. H. et al. Increased ILC2s in the eosinophilic nasal polyp endotype are associated with corticosteroid responsiveness. Clin. Immunol. 155, 126–135 (2014).
Gold, M. J. et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J. Allergy Clin. Immunol. 133, 1142–1148 (2014).
Bartemes, K. R. et al. IL-33-responsive lineage- CD25 + CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J. Immunol. 188, 1503–1513 (2012).
Kamijo, S. et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J. Immunol. 190, 4489–4499 (2013).
Halim, T. Y. et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014).
Van Dyken, S. J. et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity 40, 414–424 (2014).
Hepworth, M. R. et al. Innate lymphoid cells regulate CD4 + T-cell responses to intestinal commensal bacteria. Nature 498, 113–117 (2013).
Oliphant, C. J. et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4( + ) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41, 283–295 (2014).
von Burg, N. et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl. Acad. Sci. USA 111, 12835–12840 (2014).
Drake, L. Y., Iijima, K. & Kita, H. Group 2 innate lymphoid cells and CD4 + T cells cooperate to mediate type 2 immune response in mice. Allergy 69, 1300–1307 (2014).
Silver, J. S. et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 17, 626–635 (2016).
Doherty, T. A. et al. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J. Allergy Clin. Immunol. 132, 205–213 (2013).
Gasteiger, G., Fan, X., Dikiy, S., Lee, S. Y. & Rudensky, A. Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985 (2015).
Fernandez, H. N., Henson, P. M., Otani, A. & Hugli, T. E. Chemotactic response to human C3a and C5a anaphylatoxins. I. Evaluation of C3a and C5a leukotaxis in vitro and under stimulated in vivo conditions. J. Immunol. 120, 109–115 (1978).
Ward, P. A. & Zvaifler, N. J. Complement-derived leukotactic factors in inflammatory synovial fluids of humans. J. Clin. Invest. 50, 606–616 (1971).
Wang, Y. et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. 6, 1022–1035 (2016).
Peng, Q. et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood 111, 2452–2461 (2008).
Sheen, J. H. et al. TLR-induced murine dendritic cell (DC) activation requires DC-intrinsic complement. J. Immunol. 199, 278–291 (2017).
Pape, K. A., Catron, D. M., Itano, A. A. & Jenkins, M. K. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 26, 491–502 (2007).
Fischer, W. H. & Hugli, T. E. Regulation of B cell functions by C3a and C3a(desArg): suppression of TNF-alpha, IL-6, and the polyclonal immune response. J. Immunol. 159, 4279–4286 (1997).
Werfel, T. et al. Activated human T lymphocytes express a functional C3a receptor. J. Immunol. 165, 6599–6605 (2000).
Robinette, M. L. et al. IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat. Commun. 8, 14601 (2017).
Stier, M. T. et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J. Exp. Med. 215, 263–281 (2018).
Karta, M. R. et al. beta2 integrins rather than beta1 integrins mediate Alternaria-induced group 2 innate lymphoid cell trafficking to the lung. J. Allergy Clin. Immunol. 141, 329–338 e312 (2018).
Hawlisch, H., Wills-Karp, M., Karp, C. L. & Kohl, J. The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol. Immunol. 41, 123–131 (2004).
Strainic, M. G. et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4 + T cells. Immunity 28, 425–435 (2008).
Lajoie, S. et al. IL-21 receptor signalling partially mediates Th2-mediated allergic airway responses. Clin. Exp. Allergy 44, 976–985 (2014).
Acknowledgements
This work was supported by R56AI118791 (S.L.), R01AI127644 (S.L.), Parker B. Francis fellowship (S.L.), AI083315 (MWK), NIEHS 5T32ES007141 (N.G). We are grateful to the flow sorting expertize of Dr. Hao Zhang and the cell sorting service from the Johns Hopkins Bloomberg School of Public Health.
Author information
Authors and Affiliations
Contributions
N.G., U.S., M.W.K. and S.L. designed the study. N.G., and U.S. performed the experiments and analyzed the data with help from H-M.Y., I.P.L., N.Y. and A.S. E.G. performed histological analsysis. N.G. and S.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Gour, N., Smole, U., Yong, HM. et al. C3a is required for ILC2 function in allergic airway inflammation. Mucosal Immunol 11, 1653–1662 (2018). https://doi.org/10.1038/s41385-018-0064-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41385-018-0064-x
This article is cited by
-
A computational approach for the identification of key genes and biological pathways of chronic lung diseases: a systems biology approach
BMC Medical Genomics (2023)
-
The Role of Innate Immune Cells in Allergen Immunotherapy
Current Treatment Options in Allergy (2023)
-
New insights into the immune functions of complement
Nature Reviews Immunology (2019)
