
Abstract
Epithelial-mesenchymal transition (EMT) and breast cancer stem cells (BCSCs) are pivotal in breast cancer mechanism research. It was demonstrated that Sine oculis homeobox homolog 1 (SIX1) orchestrates breast cancer EMT and BCSCs, concurrently activating the Signal transducer and activator of transcription 3 (STAT3) signaling pathway. Yet, the mechanism by which SIX1 modulates STAT3 and its potential to regulate EMT and BCSCs through STAT3 signaling remain unexplored. Here, cellular, animal, organoid models, and integrated single-cell transcriptomic and ST-seq of human breast cancer specimens were conducted. The results revealed that SIX1 can enhance Zinc finger E-box binding homeobox 1 (ZEB1) expression and translation, which in turn binds to the Interleukin-6 (IL6) promoter (1138bp–1148bp) to stimulate its transcription, translation, and secretion. Subsequently, IL6 can activate the cell’s own STAT3 signaling pathway, promote the phosphorylation of STAT3, promote the downstream signal c-Myc and Cyclin D1 transduction, and promote the expression of stem cell-related transcription factors such as ALDH1A1, OCT4, and SOX2, thereby promoting EMT and stemness. In addition, this study found a new cell interaction model, in which the above-mentioned secreted IL6 can promote the activation of STAT3 signaling pathway, EMT and stemness transformation in the surrounding cells with low expression of SIX1 in a paracrine manner. Our data favored that SIX1/ZEB1/IL6 axis activated the STAT3 signaling pathway of the breast cells themselves and surrounding cells with low SIX1 expression, thus promoting EMT and stemness transformation, activating the malignant progression of the whole breast cancer.

Similar content being viewed by others


Background
Breast cancer stands out as the most frequently diagnosed cancer and the second leading cause of cancer-related mortality in women [1,2,3,4]. A substantial body of research underscores a significant connection between epithelial-mesenchymal transition (EMT) and the presence of cancer stem cells (CSCs) within breast cancer. The plasticity of cancer cells can induce EMT, alongside aberrations in epigenetic modifications, the expression of stemness-associated genes, and corresponding signaling pathways, all of which are crucial for maintaining CSC characteristics. Conversely, CSCs contribute to sustaining this cellular plasticity, EMT, and epigenetic adaptability [5].
Ford et al. were the first to elucidate that Sine oculis homeobox homolog 1 (SIX1) promotes EMT in breast cancer cells, which subsequently activates GLI in neighboring non-EMT cells, thereby enhancing cancer progression [6]. Our previous studies have demonstrated that breast cancer cells overexpressing SIX1 through EMT can stimulate the NRP2-GLI signaling pathway in adjacent non-EMT cells via VEGF-C secretion, thereby promoting cancer proliferation, invasion, and metastasis [7]. In thyroid cancer, SIX1 has been shown to activate the phosphorylation of Signal transducer and activator of transcription 3 (STAT3), facilitating the malignant progression of thyroid cancer [8]. Additionally, our preliminary work indicates that SIX1 can activate the STAT3 signaling pathway in breast cancer [9]. However, the exact mechanism by which SIX1 activates STAT3 remains to be fully elucidated.
In the context of breast cancer, STAT3 stands out within the STAT protein family as a subject of extensive inquiry, with a wealth of literature detailing ligands capable of activating it [10]. Interleukin-6 (IL6) emerges as a critical cytokine in this activation process, being the most archetypal and influential player. Elevated serum levels of IL6 enhance STAT3 activity, while STAT3 can bind directly to the IL6 promoter, promoting its transcription and establishing a positive feedback loop that drives tumorigenesis and metastasis [11]. However, the role of IL6 in the activation of the STAT3 pathway by SIX1 remains unclear. Furthermore, whether SIX1 can regulate IL6 expression needs to be explored.
The production and secretion of IL6 are regulated by a cohort of transcription factors. Studies have shown that the overexpression of Zinc finger E-box binding homeobox 1 (ZEB1) in breast cancer cells significantly increases the transcriptional activity of IL6 and IL8 [12]. Similarly, in human osteosarcoma cells, ZEB1 has been implicated in enhancing IL6 expression, thereby boosting the invasive potential and chemoresistance of these cancer cells [13]. Additionally, in cholangiocarcinoma, ZEB1 has been identified as a key player in promoting the paracrine secretion of IL6 by both cancer cells and myofibroblasts [14].
Previous research has indicated that in colon cancer, SIX1 elevates ZEB1 protein levels primarily through post-transcriptional modifications rather than altering the transcriptional activity of ZEB1 [15]. However, Cieply et al. demonstrated that SIX1 can accumulate within the promoter region of the endogenous ZEB1, thereby facilitating EMT in breast cancer [16]. Moreover, SIX1 can enhance ZEB1 expression indirectly by promoting the transcription of lncATB and inhibiting the activity of miR-200c [17]. Nevertheless, experimental evidence linking SIX1 regulation of IL6 through ZEB1 in breast cancer is currently limited.
In this study, we employed an integrative approach, combining cellular and animal models, organoid cultures, and advanced omics techniques such as single-cell transcriptomic sequencing (scRNA-seq) and spatial transcriptomic sequencing (ST-seq). Our findings revealed that SIX1 upregulates the transcription and translation of ZEB1, which subsequently binds to the promoter of IL6, thereby enhancing its expression. This cascade activation triggers the STAT3 signaling pathway, culminating in EMT and stemness transformation in breast cancer cells. Furthermore, we observed that EMT cells with elevated SIX1 expression can export IL6 through paracrine signaling, inducing malignant properties, including stemness transformation, in neighboring cells with low SIX1 expression. Notably, we have identified the therapeutic potential of IL6 blockade in SIX1-high breast cancer patients, providing a novel treatment target for addressing the heterogeneous nature of this disease.
Methods
Cell culture
MCF7 and 66cl4 derivative cell lines were previously described [9]. The 66cl4 is a murine-derived triple-negative breast cancer cell (TNBC) line, generously provided by Prof. Kongming Wu from Tongji Medical College, Huazhong University of Science and Technology. MCF7 is a human luminal A subtype breast cancer cell line. The 293T cell line was obtained from the National Collection of Authenticated Cell Cultures. Short tandem repeat authentication of the cells used in this study confirmed no misidentification or contamination with other cells. All cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in 5% CO2. The cells were also free from Mycoplasma contamination. Comprehensive details on all treatments, including shRNA, siRNA, overexpression lentiviruses, and drugs, were provided in the Supplemental Methods. The sequences of siRNA and shRNA were listed in Supplementary Data S1.
Preparation and intervention of conditioned medium (CM)
At a cell density of 20–30%, the existing medium in the culture bottle was replaced with fresh complete medium supplemented with 10% FBS, and the cells were incubated for an additional 48 h. The medium was then transferred to a 10 ml syringe and filtered through a 0.45 μm filter to obtain CM, which was used in subsequent experiments. If neutralizing antibodies were required, they were added to the control group. CM intervention was initiated when the cell density reached 40–50%. The culture medium was replaced with CM, and the cells were incubated for another 48 h. Following incubation, western blotting and other biological function tests were conducted.
RT-qPCR and Western blot
RT-qPCR and Western blot were performed as previously described [9]. Total RNA was prepared using the RNAeasy™ Animal RNA Isolation Kit with Spin Column (R0026, Beyotime). cDNA was reverse transcribed from 1 μg total RNA using the HiScript® III All-in-one RT SuperMix Perfect for qPCR (R333-01, Vazyme). RT-qPCR was performed with the Taq Pro Universal SYBR qPCR Master Mix (Q712-02, Vazyme). And gene expressions were normalized to β-actin. The quantitative analysis was calculated by using 2−ΔΔCt method. Primer sequences were listed in Supplementary Data S2. Total proteins were extracted with RIPA buffer (IN-B001, Invent). Protein concentration was measured by BCA protein assay kit (P0010, Beyotime). Then proteins were separated on 10% SDS-PAGE gels. Then we electrophoresed equal amounts of proteins onto polyvinylidene difluoride membranes. Subsequently, the membranes were blocked using TBST with 5% milk and probed with primary antibodies overnight at 4 °C. The membranes were then incubated with secondary antibodies for 2 h at room temperature. Finally, scanned the protein bands. Antibodies were listed in Supplementary Data S3.
Enzyme-linked immunosorbent assay (ELISA)
At a cell density of 20–30%, the existing medium in the culture bottle was replaced with fresh complete medium supplemented with 10% FBS, followed by a 48-h incubation period. Subsequently, the medium was collected and filtered through a 0.45 μm membrane using a 10 ml syringe. IL6 levels in the medium were measured using ELISA kits (human: RK00004, Abclonal; mouse: RK00008, Abclonal) according to the manufacturer’s instructions. Incubated the ELISA plate with samples and enzyme-conjugated detection antibodies. Then washed the plate to remove unbound components, and measuring enzymatic substrate conversion via optical density detection.
Migration assay
After intervention, cells were seeded into six-well plates. Upon reaching over 90% confluence, the cells were gently washed twice with PBS, and scratch wounds were generated using a sterile white pipette tip. The cells were gently washed twice with PBS, after which they were imaged under a light microscope. The cells were then cultured in either serum-free medium or CM for 24 h, after which they were imaged under a light microscope again.
Invasion assay
Following treatments, 1 × 10^5 cells in serum-free medium or CM were plated in the upper chambers of a transwell apparatus with Matrigel (356234, Corning). DMEM containing 10% FBS was used in the bottom chambers as an attractant. After 24 h of incubation, the cells that crossed the inserts were fixed with 4% paraformaldehyde and stained with 0.1% crystal-violet. Three random fields of stained cells were photographed by microscope.
Immunofluorescence
Cells were inoculated into a Petri dish with a cover slip glass at a density of 1 × 10^5 cells/ml and cultured in an incubator for 48 h. The culture medium was removed, and the cells were washed twice with PBS. The cells were fixed in 4% paraformaldehyde for 15 min. After washing twice with PBS, the cells were permeabilized with 0.1% Triton X-100 and then blocked with 10% goat serum. The cells were incubated with primary antibody, followed by fluorescent secondary antibody, TSA, and DAPI, and then washed three times with PBST. The cells were mounted and observed under fluorescence microscopy. Antibodies were listed in Supplementary Data S3.
Mammosphere formation assay and Extreme Limiting Dilution Analysis (ELDA)
These assays were performed as previously described [9]. Single-cell suspensions were prepared using 0.05% trypsin-EDTA and cultured in an ultralow attachment culture dish (3471, Corning) at 2 × 10^3 cells/ml. The serum-free medium contained DMEM supplemented with B27 (1×, 17504044, Gibco), EGF (20 ng/mL, 315-09/AF-100-15, PeproTech), bFGF (20 ng/mL, AF-450-33/100-18B, PeproTech), and heparin (2 μg/mL, HY-17567, MCE). Mammosphere cultures were maintained for 7 days (MCF7) or 5 days (66cl4). Spheres >50 μm in diameter were counted under a microscope. Mammosphere-forming efficiency was calculated as: (mammospheres per well/initial cell count) ×100%. As for ELDA, the cells were seeded into 96-well ultralow attachment culture plates (7007, Corning) at a density of 50, 100, 200, or 400 cells/well, with 8 replicates for each density. Then the mammosphere number was counted after 5 days (66cl4) or 7 days (MCF7). The mammosphere formation efficiency was calculated via the ELDA (http://bioinf.wehi.edu.au/software/elda).
Flow cytometry analysis, alcohol dehydrogenase (ALDH), and cell proliferation assay
A total of 1 × 10^6 cells in each group were harvested and then were incubated with antibodies in the dark at 4 °C for 30 min. Antibodies were listed in Supplementary Data S3. Cells were washed and re-suspended in 500 µl of PBS and analyzed using a flow cytometer.
As for ALDH detection, cells were first placed on ice then ALDH was detected by an ALDH test kit (BC0755, Solarbio) as indicated by the manufacturer. All ALDH activities were evaluated using a microplate reader at 340 nm by measuring the production of NAD + . Higher optical density values indicate stronger activity.
For proliferation detection, Cell Counting Kit 8 (CCK-8, BS350B, Biosharp) was used. 1 × 10^3 cells of various 66cl4 and 2 × 10^3 cells of various MCF7 were plated in 96-well plates. Day 0 time point was measured 6 h post plating. Following 24-, 48-, 72-, or 96-h of incubation, day 1–4 time points were analyzed. 10 μl of CCK-8 was added to each well and incubated at room temperature for 60 min and luminescence was measured by using a microplate reader at 450 nm.
Prediction of transcription factor binding sites in JASPAR database
We utilized the JASPAR database (https://jaspar.elixir.no/) to identify DNA promoter binding sequences for ZEB1, specifying “Homo sapiens” as the species. The promoter sequence of IL6 was accessed via the NCBI website (https://www.ncbi.nlm.nih.gov/). This sequence was uploaded into the JASPAR database, and using the Scan tool, we searched for all potential ZEB1 binding sites on the IL6 promoter. Sequences with binding site scores exceeding 10 were selected for further analysis.
Dual-luciferase reporter assay (Dual-LUC)
The luciferase assay kit (E1960, Promega) was used for Dual-LUC. The IL6 promoter region was amplified via PCR and cloned into the pGL4.10-basic luciferase reporter vector (Firefly luciferase) to generate the IL6 promoter-driven reporter construct. At 70–80% confluency, 293T cells were co-transfected with the IL6-pGL4.10 reporter plasmid, the pcDNA3.1(+)-ZEB1 overexpression plasmid (or empty pcDNA3.1(+) vector as control), and the pRL-TK Renilla luciferase internal control plasmid (E1960, Promega) using Lipofectamine 3000, following the manufacturer’s protocol. Twenty-four hours post-transfection, cells were washed with PBS and lysed using Passive Lysis Buffer. Firefly and Renilla luciferase activities were sequentially measured on a luminometer. Briefly, 20 µL of lysate was mixed with 100 µL Luciferase Assay Reagent II to quantify Firefly luciferase activity, followed by the addition of 100 µL Stop & Glo Reagent to quench Firefly activity and activate Renilla luciferase. Relative luciferase activity was calculated as the ratio of Firefly to Renilla luminescence values to normalize transfection efficiency.
Chromatin immunoprecipitation (ChIP)
SimpleChIP® Enzymatic Chromatin IP Kit (9002, Cell Signaling Technology) was used for this assay. According to the manufacturer’s instructions, MCF7 transfected with OE SIX1 were incubated with formaldehyde at a final concentration of 1% for 10 min to stabilize protein-DNA interactions. After terminating the cross-linking, the cells were washed twice with ice-cold PBS. The cells were collected using a cell scraper into a 15-ml centrifuge tube and collected by centrifugation. Protease inhibitor complex was added and the cells were sonicated to achieve DNA fragments ranging from 200 to 1000 bp. For immunoprecipitation, chromatin lysates were incubated with ZEB1-specific antibody (ab155249, Abcam) or control IgG and incubated overnight at 4 °C, followed by the addition of ChIP-grade Protein G magnetic beads to capture antibody-chromatin complexes. Beads were washed sequentially with low-salt, high-salt, and LiCl buffers to remove nonspecific binding. DNA-protein crosslinks were reversed by incubation with 5 M NaCl at 65 °C for 4 h, and DNA was purified. Enriched DNA fragments were analyzed by RT-qPCR with primers targeting specific genomic regions. Data were expressed as fold enrichment relative to IgG controls.
In vivo experiments
Prior to experiments, female BALB/c nude mice (6–8 weeks old) were provided with ad libitum access to food and water. All animal studies were reviewed and ethically approved by the Laboratory Animal Welfare & Ethics Committee of Renmin Hospital at Wuhan University. All animal experiments adhered to the guidelines in the Guide for the Care and Use of Laboratory Animals developed by the Institute of Laboratory Animal Research. Specific details about individual animal experiments can be found in the Supplementary Methods.
Hematoxylin and eosin (HE) staining
HE staining was performed as previously reported [18]. Briefly, paraffin-embedded tissue sections were deparaffinized through xylene I (15 min), xylene II (15 min), and xylene III (15 min), followed by rehydration in graded ethanol concentrations (100, 95, 85, and 70%, 5 min each). Nuclear staining was achieved using Harris hematoxylin (5–10 min), where oxidized hematoxylin binds to DNA via electrostatic interactions. Differentiation with 1% acid ethanol removed excess stain, while bluing in alkaline solution (Scott’s tap water substitute) restored nuclear contrast. Cytoplasmic counterstaining employed 0.5–1% eosin Y (1–3 min) to highlight cellular and extracellular matrix components. Finally, sections were dehydrated through graded alcohols, cleared in xylene, and mounted with neutral resin for microscopic evaluation.
Cultivation and proliferation of organoids
Breast cancer specimens were obtained from Renmin Hospital of Wuhan University. The tumor samples were meticulously cleaned of surface mucus and necrotic tissue before washing with PBS. The tissues were then immersed in active iodine for 1 min, followed by three rinses with penicillin/streptomycin solution. The tissue was minced into 0.5-mm cubes and digested for 15 min at 37°C using a 50:50 mixture of TrypLE™ Express (12605028, Gibco) and DMEM. The digestion was neutralized with an equal volume of Ca2 + -containing Hanks’ Balanced Salt Solution. The cell suspension was allowed to settle for 3–5 min, after which the supernatant was carefully aspirated, and the pellet was centrifuged. The resulting pellet was washed twice with advanced DMEM/F12 medium, counted, and combined with growth factor-reduced Matrigel. In a pre-warmed 24-well plate, 8000–10,000 cells were seeded per well using 30 µl of Matrigel. After a 30-min incubation at 37 °C to allow Matrigel to solidify, 600 µl of breast cancer organoid medium was added, and the plate was placed in a 37 °C incubator with 5% CO2.
As the breast cancer organoids entered the logarithmic growth phase, they were dissociated mechanically and enzymatically by incubating them with TrypLE (12604013, Gibco) for 10–14 min, followed by resuspension in Matrigel at a concentration of 3 × 10^5 cells/ml on ice. For proliferation analysis, the cell suspensions were inoculated into pre-warmed 96-well plates at a density of 3 × 10^3 cells per well and maintained in complete culture medium (100 µl per well). After the designated treatment, cell viability was determined using 50 µl of CellTiter-Glo 3D (G9681, Promega) mixed with 50 µl of culture medium, according to the manufacturer’s protocol. This mixture replaced the culture medium, and viability quantification was conducted using an Infinite 200 Pro plate reader (Tecan Life Sciences).
Immunohistochemistry (IHC)
Breast cancer specimens were fixed with 4% paraformaldehyde at 4 °C for 24 h, followed by paraffin embedding and sectioning into 5 µm thick slices. IHC staining protocols were performed according to the manufacturer’s recommendations (PK10009, Proteintech). Antibodies were listed in Supplementary Data S3.
ScRNA-seq and ST-seq
Breast cancer samples were collected from two patients. Each patient’s sample was divided into two parts, one for single-cell transcriptome sequencing and the other for spatial transcriptome sequencing. The research involving human participants received ethical approval from the Clinical Research Ethics Committee of Renmin Hospital, Wuhan University (WDRY-2022-K002). All patients provided written informed consent for their participation in this study. The detailed analysis steps were described in the Supplementary Methods.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9.4 software (GraphPad Software, CA, USA). The student’s t test was employed for two-group comparisons, while one-way ANOVA non-parametric was used for comparisons involving three or more groups. Proliferation data and tumor growth curves were subjected to two-way ANOVA analysis. Kaplan–Meier curves for metastasis-free-survival were subjected to the log-rank test. All in vitro experiments were conducted three times. Statistical significance was indicated by p values < 0.05. Detailed p values can be found in the figures.
Results
SIX1 upregulates the transcription, translation, and secretion of IL6
Our previous work demonstrated that SIX1 expression could activate the STAT3 signaling. However, how SIX1 regulate STAT3 signaling is unclear [9]. Given that IL6 is the most prevalent cytokine that activates STAT3 signaling, we initially examined alterations in IL6 expression following SIX1 modulation. We systematically evaluated SIX1 expression profiles across multiple breast cancer cell lines. Through RT-qPCR, 66cl4 cells were identified as exhibiting constitutively elevated SIX1 expression, whereas MCF7 cells displayed minimal endogenous SIX1 expression (Supplementary Fig. S1A). The 66cl4 is a murine-derived TNBC line characterized by high invasiveness and metastatic potential, whereas MCF7 represents a human luminal A subtype breast cancer cell model with comparatively indolent biological behavior. Thus, we selected 66cl4 to construct SIX1-silenced cell lines, MCF7 to construct SIX1 overexpressing cell lines. The experimental groups comprised: (1) 66cl4-SCR (scramble control transfected with non-targeting shRNA), (2) 66cl4-shSix1#1 and 66cl4-shSix1#2 (stable knockdown clones targeting distinct SIX1 loci through lentiviral transduction of validated shRNAs), (3) MCF7-OE Ctrl (empty vector-transfected control), and (4) MCF7-OE SIX1 (stably overexpressing human SIX1 via pcDNA3.1(+) vector). To explore the role of SIX1 in regulating IL6 expression, we quantified IL6 mRNA levels in breast cancer cells using RT-qPCR. Knockdown of Six1 markedly reduced IL6 mRNA compared to control cells (Fig. 1A). Conversely, overexpression of SIX1 increased IL6 mRNA levels (Fig. 1A). To further investigate whether SIX1 influences breast cancer malignancy via IL6, we established stable cell lines using lentiviral overexpression of IL6 and lentiviral IL6 knockdown (Fig. 1B, Supplementary Fig. S1B). The shNT served as the non-targeting control for shIL6. RT-qPCR results indicated that the mRNA level of SIX1 did not significantly change with the increase or decrease of IL6 mRNA (Fig. 1C, Supplementary Fig. S1C). Western blot analysis revealed no significant change in SIX1 protein levels following overexpression or knockdown of IL6 (Fig. 1D–G). Additionally, knockdown of Six1 decreased IL6 levels, while SIX1 overexpression increased IL6 (Fig. 1D–G). These findings demonstrated that SIX1 regulates IL6 expression, and IL6 does not affect SIX1 expression. ELISA analysis of IL6 content in cell supernatants showed that IL6 secretion decreased upon Six1 knockdown and increased with SIX1 overexpression (Fig. 1H, I and Supplementary Fig. S1D). This also confirmed the successful construction of stable cell lines with IL6 overexpression and knockdown.

A RT-qPCR analysis of IL6 mRNA levels in 66cl4 transfected with SCR or shSix1 and MCF7 transfected with OE Ctrl or OE SIX1, n = 3. B RT-qPCR analysis of IL6 mRNA levels in indicated groups, n = 3. C RT-qPCR analysis of SIX1 mRNA levels in indicated groups, n = 3. D Western blot analysis of SIX1 and IL6 protein levels in Six1-knockdown 66cl4, followed by IL6 overexpression. E Quantitation of Western blot in (D), n = 4. F Western blot analysis of SIX1 and IL6 protein levels in SIX1 overexpressed MCF7, followed by IL6 knockdown. G Quantitation of Western blot in (F), n = 4. H ELISA analysis of IL6 content in the cellular supernatant of Six1-knockdown 66cl4, followed by IL6 overexpression, n = 3. I ELISA analysis of IL6 content in the cellular supernatant of SIX1 overexpressed MCF7, followed by IL6 knockdown, n = 3. J Proliferation curves of Six1-knockdown 66cl4, followed by IL6 overexpression, n = 4. K Proliferation curves of SIX1 overexpressed MCF7, followed by IL6 knockdown, n = 4. L Representative migration assay (0–24 h) on Six1-knockdown 66cl4, followed by IL6 overexpression, scale bar is 100 μm. M Quantitation of cell migration in (L), n = 3. N Representative invasion assay on Six1-knockdown 66cl4, followed by IL6 overexpression, scale bar is 50 μm. O Quantitation of cell invasion in (N), n = 3. P Representative IF of E-cadherin (red) and N-cadherin (green) in SIX1 overexpressed MCF7, followed by IL6 knockdown. DAPI (blue), scale bar is 20 μm. Q Representative IF of Vimentin (pink) in SIX1 overexpressed MCF7, followed by IL6 knockdown. DAPI (blue), scale bar is 20 μm. R Quantitation of relative fluorescence intensity in (P) and (Q), n = 4. Data were presented as means ± SD; ANOVA (B, C, E, G, H, I, M, O, R, and the left panel of A) or two-sided Student’s t-test (the right panel of A) were used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns P > 0.05. SCR scramble, OE Ctrl overexpressing control, shNT shRNA of negative control, sh#1 #2 #3 three independent shRNA constructs targeting genes.
SIX1 promotes proliferation, migration, and invasion of breast cancer cells through IL6
To assess the impact of SIX1 on breast cancer cell proliferation, migration, and invasion via IL6, we performed CCK-8, wound healing, and transwell assays. Figure 1J and Supplementary Fig. S1E shows that Six1 knockdown inhibited cell proliferation, an effect partially reversed by IL6 overexpression. Conversely, SIX1 overexpression enhanced proliferation, which was moderately reversed by IL6 knockdown (Fig. 1K, Supplementary Fig. S1F).
Wound healing assays indicated that Six1 knockdown reduced migration by approximately 50%, with IL6 overexpression counteracting this effect and increasing migration rates (Fig. 1L, M). Transwell assays demonstrated that Six1 knockdown decreased invasiveness, and IL6 overexpression reversed this, increasing the number of invasive cells (Fig. 1N, O).
Given MCF7 cells’ limited migration and invasion capabilities, we stained E-cadherin, N-cadherin, and Vimentin by immunofluorescence. SIX1 overexpression decreased E-cadherin fluorescence intensity and increased that of N-cadherin and Vimentin. Knockdown of IL6 reversed these changes, leading to a slight increase in E-cadherin and a slight decrease in N-cadherin and Vimentin fluorescence intensity (Fig. 1P–R).
SIX1 promotes stemness of breast cancer cells through IL6
Next, we investigated the role of IL6 in the regulation of stemness transformation mediated by SIX1. Supplementary Fig. S2A, B shows that silencing Six1 reduced stemness related protein levels of Itgb1, Aldh1a1, Oct4, and Sox2. However, IL6 overexpression partially reversed this effect. A similar pattern was observed in MCF7 cells, where SIX1 overexpression significantly increased stemness-related protein levels of ITGB1, ALDH1A1, OCT4, and SOX2, and this upregulation was partially reversed by subsequent IL6 downregulation (Fig. 2A, B). ALDH is an essential enzyme involved in aldehyde detoxification and retinoic acid synthesis, and its activity is linked to drug resistance—a characteristic of CSCs. ALDH activity assays revealed that IL6 overexpression partially reversed the decrease in ALDH activity induced by SIX1 silencing (Fig. 2C). In MCF7 cells, IL6 knockdown partially reversed the increase in ALDH activity induced by SIX1 overexpression (Fig. 2D).

A Western blot analysis of stemness-related proteins in SIX1 overexpressed MCF7, followed by IL6 knockdown. B Quantitation of Western blot in (A), n = 4. C ALDH activity in Six1-knockdown 66cl4, followed by IL6 overexpression, n = 3. D ALDH activity in SIX1 overexpressed MCF7, followed by IL6 knockdown, n = 3. E Representative images of mammospheres in SIX1 overexpressed MCF7, followed by IL6 knockdown, scale bar is 100 μm. F Quantitation of mammosphere-formation efficiencies in (E), n = 3. G Quantitation of relative mammosphere size in (E), n = 4. H ELDA assays showed the mammosphere-formation abilities in SIX1 overexpressed MCF7, followed by IL6 knockdown. I Representative flow cytometry plots of CD24highCD49fhigh subset in Six1-knockdown 66cl4, followed by IL6 overexpression. J Quantitation of CD24highCD49fhigh subset in (I), n = 3. K Representative images of tumor formation ability in Six1-knockdown 66cl4, followed by IL6 overexpression at three different concentrations, scale bar is 1 cm. L, M Quantitation of tumor formation rate in (K). N Tumor volume of Six1-knockdown 66cl4, followed by IL6 overexpression at the end of the limiting dilution experiment. O Tumor weight of Six1-knockdown 66cl4, followed by IL6 overexpression at the end of the limiting dilution experiment. P Representative images of surgery. Q Kaplan– Meier curve for metastasis-free-survival in indicated groups, n = 10. R Representative luminescent images of organs in indicated groups taken at the same time point (4 weeks after surgery). S HE staining of lung tissue of mice in indicated groups, scale bar is 100 μm. Data were presented as means ± SD; ANOVA or log-rank test were used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns P > 0.05. SCR scramble, OE Ctrl overexpressing control, shNT shRNA of negative control, sh#1 #2 #3 three independent shRNA constructs targeting genes.
We also performed mammosphere formation experiments and ELDA assays, and demonstrated that SIX1 overexpression significantly enhanced mammosphere size and formation efficiency, effects that were abrogated by additional IL6 knockdown (Fig. 2E-H). SIX1 silencing reduced mammosphere size and formation efficiency, which was partially reversed by IL6 overexpression (Supplementary Fig. S2C-F). Recent studies suggest that CD24highCD49fhigh murine breast cancer cells and CD24lowCD44high human breast cancer cells exhibit stem cell properties, also known as BCSCs or breast cancer stem-like cells [19, 20]. What’s more, flow cytometry analysis showed that SIX1 silencing decreased the proportion of CD24highCD49fhigh cell subsets, an effect completely reversed by IL6 overexpression (Fig. 2I, J). SIX1 overexpression in MCF7 cells increased the proportion of CD24lowCD44high subsets, and this increase was partially attenuated by further IL6 knockdown (Supplementary Fig. S2G, H).
SIX1 promotes tumorigenesis and metastasis via IL6 in vivo
To investigate whether SIX1 regulates the malignant phenotype of breast cancer cells through IL6 in vivo, we established stable cell lines and developed animal models. To further confirm the role of Six1/IL6 in enhancing the stemness of breast cancer cells, we injected different amounts of 66cl4 cells with lentiviruses to assess their tumorigenic ability. Cells with a higher capacity for tumor formation exhibit stronger stemness even at lower cell numbers. Through orthotopic inoculation with decreasing cell doses (5 × 10^5 to 2 × 10^4 cells/mouse, 5-fold serial dilutions), we observed a dosage-dependent impairment of tumorigenesis. Limiting dilution analysis revealed significantly reduced tumor-initiating capacity after silencing Six1. Strikingly, at the critical threshold of 2 × 10^4 cells, Six1-deficient implants almost failed to establish detectable tumors (1/8 and 0/8 mice) within 14 days post-inoculation compared to 37.5% (3/8) tumor formation in controls. IL6 overexpression rescued tumorigenic potential, achieving 37.5% (3/8) engraftment success at the 2 × 10^4 dose (Fig. 2K–M). However, we found that IL6 overexpression did not significantly rescue the reduced tumor volume and tumor weight caused by Six1 knockdown (Fig. 2N, O). To further investigate whether SIX1-mediated IL6 signaling orchestrates tumor growth and metastasis in vivo, we performed complementary orthotopic transplantation assays by injecting 1 × 10^6 cells/mouse into the mammary fat pad. Experimental timelines were extended. It was demonstrated a significant reduction in the growth rate of breast cancer cells in mice following Six1 knockdown, with this effect becoming more pronounced over time (Supplementary Fig. S3A–C). Conversely, IL6 overexpression did not significantly accelerate cancer cell growth (Supplementary Fig. 3SA–C). Upon removal of the primary tumor and subsequent monitoring of metastases (Fig. 2P), we observed a marked decrease in the rate of metastasis after Six1 knockdown, with a delayed onset of metastatic events (Fig. 2Q–S). IL6 overexpression partially reversed this, elevating the metastatic rate and advancing the timing of metastasis (Fig. 2Q–S).
SIX1 promotes transcriptional translation and secretion of IL6 through ZEB1
Our findings suggested SIX1 promotes IL6 transcription. As a canonical transcription factor, SIX1 typically binds target gene promoters. However, transcription factor binding sites prediction analysis by using JASPAR revealed no SIX1-binding motif in the IL6 promoter, implying indirect regulation through intermediate factors. Literature review suggests ZEB1 as a potential mediator of SIX1-mediated IL6 regulation. Using JASPAR, we identified potential ZEB1 binding sites on the human IL6 promoter, as corroborated in Fig. 3A–C. From NCBI, we obtained the IL6 promoter sequence and used JASPAR’s online scanning function to identify 42 possible binding sites (Supplementary Data S4). Eight loci with scores above 10 were selected and clustered into three regions (Fig. 3D, E). The Dual-LUC assay using 293T cells showed that ZEB1 bound to the IL6-WT promoter. However, this binding was abolished upon mutation of regions 1–3, indicating that ZEB1 interacts with the IL6 promoter through these regions (Fig. 3F). To validate the exact binding loci, we performed ChIP assays using two primer sets: primer 1 targeting region 1 and primer 2 spanning regions 2–3. This experiment using MCF7 transfected with OE SIX1 confirmed the amplification of primers targeting these regions, with primer 1 showing superior performance (Fig. 3G), inferring that region 1 (1138bp–1148bp, “ATGCAGGTGCC”) is the primary site. To explore the SIX1-ZEB1-IL6 relationship, we found that IL6 mRNA levels did not significantly affect ZEB1 mRNA levels, yet SIX1 mRNA levels correlated with ZEB1 in the same direction (Supplementary Fig. S4A). Stable cell lines and siRNA knockdown experiments were established to assess the impact of ZEB1 and SIX1 expression. The siNT served as the non-targeting control for siZEB1. It was demonstrated that SIX1 controls ZEB1 expression levels, as SIX1 silencing caused a decrease and SIX1 enforced expression caused an increase in ZEB1 levels (Fig. 3H). SIX1-induced IL6 transcription requires ZEB1, as ZEB1 overexpression can revert IL6 reduction induced by SIX1 silencing, and ZEB1 silencing inhibited SIX1-induced IL6 transcription (Fig. 3I). Finally, SIX1 levels were not affected by ZEB1 modulation (Fig. 3J-N), indicating that SIX1 is upstream ZEB1 and IL6. And we found that Zeb1 ectopic expression in Six1-silenced cells reverted IL6 secretion, silencing ZEB1 in SIX1-overexpressed cells rescued IL6 secretion (Fig. 3O).

A–C Bioinformatic analysis of the binding site of ZEB1 and promoter. D DNA sequences, position, and corresponding positive and negative chains of IL6 promoters to which ZEB1 may bind obtained from JASPAR (Score > 10). E ZEB1 and IL6 promoter binding site and mutant construction. F Dual-LUC verified the activation of IL6 promoter by ZEB1. CONTROL (transfected control plasmid), ZEB1 (transfected ZEB1 overexpression plasmid), WT (wild-type IL6 promoter), and MUT (IL6 promoter with deletion mutation), n = 3. G ChIP analysis of the direct interaction between ZEB1 and the IL6 promoter, n = 3. H RT-qPCR analysis of ZEB1 mRNA levels in indicated groups, n = 3. I RT-qPCR analysis of IL6 mRNA levels in indicated groups, n = 3. J RT-qPCR analysis of SIX1 mRNA levels in indicated groups, n = 3. K Western blot analysis of SIX1, ZEB1, IL6 protein levels in Six1-knockdown 66cl4, followed by Zeb1 overexpression. L Quantitation of Western blot in (K), n = 4. M Western blot analysis of SIX1, ZEB1, IL6 protein levels in SIX1 overexpressed MCF7, followed by ZEB1 silencing. N Quantitation of Western blot in (M), n = 4. O ELISA analysis of IL6 content in the cellular supernatant in indicated groups, n = 3. P Representative migration assay (0–24 h) on Six1-knockdown 66cl4, followed by Zeb1 overexpression, scale bar is 100 μm. Q Quantitation of cell migration in (P), n = 3. R Representative invasion assay on Six1-knockdown 66cl4, followed by Zeb1 overexpression, scale bar is 50 μm. S Quantitation of cell invasion in (R), n = 3. T Proliferation curves of Six1-knockdown 66cl4, followed by Zeb1 overexpression, n = 4. Data were presented as means ± SD; ANOVA (F, H–J, L, N, O, Q, and S) or two-sided Student’s t test (G) were used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns P > 0.05. SCR scramble, OE Ctrl overexpressing control, sh#1 #2 two independent shRNA constructs targeting genes, siNT siRNA of negative control, si#1 #2 #3 three independent siRNA constructs targeting genes.
SIX1 promotes proliferation, migration, invasion, and stemness of breast cancer cells through ZEB1/IL6
To investigate whether SIX1 regulates breast cancer cell proliferation, migration, and invasion via the ZEB1/IL6 axis, we performed CCK-8 assays, wound healing assays, and transwell invasion assays. We found that SIX1 can upregulate breast cancer cells’ migration (Fig. 3P, Q), invasion (Fig. 3R, S), proliferation (Fig. 3T, Supplementary Fig. S4B), and EMT-related markers (Supplementary Fig. S4C–E) through ZEB1/IL6. The expression of stemness-related proteins (Fig. 4A, B and Supplementary Fig. S5A, B), ALDH activity (Fig. 4C, D), stem cell subsets, mammosphere size, and formation efficiency (Fig. 4E–J and Supplementary Fig. S5C–H) were also upregulated by SIX1/ZEB1/IL6.

A Western blot analysis of stemness-related proteins in SIX1 overexpressed MCF7, followed by ZEB1 silencing. B Quantitation of Western blot in (A), n = 4. C ALDH activity in Six1-knockdown 66cl4, followed by Zeb1 overexpression, n = 3. D ALDH activity in SIX1 overexpressed MCF7, followed by ZEB1 silencing, n = 3. E Representative images of mammospheres in SIX1 overexpressed MCF7, followed by ZEB1 silencing, scale bar is 100 μm. F Quantitation of mammosphere-formation efficiencies in (E), n = 3. G Quantitation of relative mammosphere size in (E), n = 4. H ELDA assays showed the mammosphere-formation abilities in SIX1 overexpressed MCF7, followed by ZEB1 silencing. I Representative flow cytometry plots of CD24highCD49fhigh subset in Six1-knockdown 66cl4, followed by Zeb1 overexpression. J Quantitation of CD24highCD49fhigh subset in (I), n = 3. K Representative images of tumor formation ability in Six1-knockdown 66cl4, followed by Zeb1 overexpression at three different concentrations, scale bar is 1 cm. L, M Quantitation of tumor formation rate in (K). N Tumor volume of Six1-knockdown 66cl4, followed by Zeb1 overexpression at the end of the limiting dilution experiment. O Representative luminescent images of nude mice in indicated groups taken at the same time point (3 weeks after injection of cells). P Quantitation of luminescent signal of primary tumor in indicated groups, n = 10. Q Kaplan–Meier curve for metastasis-free-survival in indicated groups, n = 10. R Representative luminescent images of organs in indicated groups taken at the same time point (4 weeks after surgery). S HE staining of lung tissue of mice in indicated groups, scale bar is 100 μm. Data were presented as means ± SD; ANOVA or log-rank test were used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns P > 0.05. SCR scramble, OE Ctrl overexpressing control, sh#1 #2 two independent shRNA constructs targeting genes, siNT siRNA of negative control, si#1 #2 #3 three independent siRNA constructs targeting genes.
SIX1 promotes tumorigenesis, proliferation, and metastasis via ZEB1/IL6 in vivo
To investigate whether SIX1 regulates the malignant phenotype of breast cancer cells through the ZEB1/IL6 axis in vivo, we established stable cell lines and developed animal models. Limiting dilution analysis revealed Zeb1 overexpression rescued tumorigenic potential caused by Six1 knockdown, achieving 37.5% (3/8) and 25.0% (2/8) engraftment success at the 2 × 10^4 dose (Fig. 4K–M). It was showed that Zeb1 overexpression significantly rescue the reduced tumor volume and tumor weight caused by Six1 knockdown (Fig. 4N and Supplementary Fig. S6A). Furthermore, ZEB1 overexpression mitigated the in vivo proliferation slowdown induced by Six1 knockdown (Fig. 4O, P and Supplementary Fig. S6B). Then we detected the Ki67 and N-cad in primary tumors. We found that Six1 knockdown reduced Ki67 and N-cad in primary tumors, an effect partially reversed by Zeb1 overexpression (Supplementary Fig. S6C, D). Upon removal of the primary tumor and subsequent monitoring of metastases, the result showed ZEB1 overexpression augmented the metastatic rate, as well as the timing of metastasis, countering the inhibitory effect of Six1 knockdown on metastasis (Fig. 4Q, R). HE staining of lung tissue sections revealed reduced lung metastasis after Six1 knockdown, whereas ZEB1 overexpression increased metastasis (Fig. 4S). SIX1 promotes the malignant progression of breast cancer cells in vivo by enhancing tumorigenesis, proliferation and metastasis through ZEB1/IL6, while ZEB1 upregulates the transcription, translation, and secretion of IL6. Collectively, these findings suggest that SIX1 promotes the expression of IL6 through ZEB1, thereby facilitating the malignant progression of breast cancer in vivo.
SIX1 can activate the STAT3 signaling pathway and downstream signal transduction via ZEB1/IL6
Immunofluorescence analysis revealed robust nuclear p-Stat3 accumulation in the control group, with significantly higher fluorescence intensity in the nucleus than in the cytoplasm. p-Stat3 nucleation was notably attenuated upon Six1 knockdown, and nuclear p-Stat3 fluorescence intensity was markedly enhanced with IL6 overexpression (Fig. 5A, B). Furthermore, Zeb1 overexpression, following Six1 knockdown, significantly elevated nuclear p-Stat3 fluorescence (Fig. 5C, D). The fluorescence intensity of Stat3 was not influenced after Six1 knockdown, IL6 overexpression, and Zeb1 expression (Fig. 5A–D). Western blot analysis revealed that Six1 knockdown decreased p-Stat3 expression and the expression of c-Myc and Cyclin D1, downstream molecules of the STAT3 signaling pathway, while IL6 or Zeb1 overexpression increased their expression (Fig. 5E–L). Moreover, SIX1 overexpression upregulated p-STAT3 and c-MYC and CYCLIN D1 expression, whereas IL6 or ZEB1 knockdown reduced their expression (Fig. 5M–T). Collectively, these data suggest that SIX1 controls IL6 transcription and translation via ZEB1, thereby modulating STAT3 signaling pathway activation and promoting downstream signaling transduction.

A Representative IF of p-Stat3 (green) and Stat3 (red) in Six1-knockdown 66cl4, followed by IL6 overexpression. DAPI (blue), scale bar is 10 μm. B Quantitation of nuclear/cytoplasmic p-Stat3 fluorescence intensity ratio in (A), n = 4. C Representative IF of p-Stat3 (green) and Stat3 (red) in Six1-knockdown 66cl4, followed by Zeb1 overexpression. DAPI (blue), scale bar is 10 μm. D Quantitation of nuclear/cytoplasmic p-Stat3 fluorescence intensity ratio in (C), n = 4. E Western blot analysis of STAT3 pathway related protein levels in Six1-knockdown 66cl4, followed by IL6 overexpression. F–H Quantitation of Western blot in (E), n = 3. I Western blot analysis of STAT3 pathway related protein levels in Six1-knockdown 66cl4, followed by Zeb1 overexpression. J–L Quantitation of Western blot in (I), n = 3. M Western blot analysis of STAT3 pathway related protein levels in SIX1 overexpressed MCF7, followed by IL6 knockdown. N–P Quantitation of Western blot in (M), n = 3. Q Western blot analysis of STAT3 pathway related protein levels in SIX1 overexpressed MCF7, followed by ZEB1 silencing. R–T Quantitation of Western blot in (Q), n = 3. Data were presented as means ± SD; ANOVA test were used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. SCR scramble, OE Ctrl overexpressing control, shNT shRNA of negative control, sh#1 #2 #3 three independent shRNA constructs targeting genes, siNT siRNA of negative control, si#1 #2 #3 three independent siRNA constructs targeting genes.
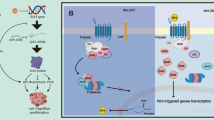
EMT cells secrete IL6 by SIX1/ZEB1/IL6 signaling axis to activate STAT3 signaling pathway and downstream signal transduction of non-EMT cells
Breast cancer exhibits marked heterogeneity, with variable SIX1 expression and malignancy among cancer cells. The dynamic tumor microenvironment (TME) further drives functional plasticity through intercellular interactions. This raises the question: do high-SIX1 and low-SIX1 cells interact dynamically to influence tumorigenesis? Our previous study established that breast cancer cells overexpressing SIX1, such as MCF7-OE SIX1, can be classified as EMT cells due to their enhanced EMT-related phenotypes, and their culture medium can be designated as EMT CM. Conversely, MCF7-OE Ctrl cells, exhibiting weaker EMT-related phenotypes, were classified as non-EMT cells, and the medium they produced was designated as non-EMT CM [7]. It has been demonstrated that EMT cells with elevated SIX1 expression stimulate GLI pathway activation through VEGF-C/NRP2 axis, promoting oncogenic phenotypes. IL6’s potential analogical role remains to be elucidated.
To explore whether EMT cells with high SIX1 expression can activate the STAT3 signaling pathway in non-EMT cells via the SIX1/ZEB1/IL6 axis and promote downstream signaling in breast cancer, we utilized the cell lines stabilized by lentivirus mentioned above. Proliferation, migration, and invasion capabilities of the stable transmissible cell lines in each group, along with other EMT-related phenotypes, were validated in the previous results. As for 66cl4, compared with the shSix1#1 + OE Ctrl group, the SCR + OE Ctrl and shSix1#1 + OE IL6 groups showed stronger EMT-related phenotypes such as proliferation, migration, and invasion. Therefore, we defined the SCR + OE Ctrl and shSix1#1 + OE IL6 groups as the EMT cell group and the shSix1#1 + OE Ctrl group as the non-EMT cell group. Consequently, the supernatant from the SCR + OE Ctrl and shSix1#1-OE IL6 groups was collected and used as EMT CM, while the shSix1#1 + OE Ctrl group supernatant served as non-EMT CM. Based on the result that knockdown of Six1 impeded proliferation, migration, and invasion, we defined cells from the shSix1#1 group as non-EMT group and exposed to CMs. Similar procedures were employed to prepare MCF7-OE Ctrl and MCF7-OE SIX1 group supernatants, with IgG added to the OE Ctrl group to generate non-EMT CM and the OE SIX1 group supernatant split into two, one for EMT CM with IgG and another for non-EMT CM with IL6 neutralizing Ab. Subsequently, non-EMT cells from the MCF7-OE Ctrl group were intervened with CM. IL6 content in each medium was presented in prior results (Fig. 6A).

A Representative image of the co-culture experiment. B Western blot analysis of STAT3 pathway related protein levels in 66cl4 transfected with shSix1#1 grown for 48 h in indicated CM. C Quantitation of Western blot in (B), n = 3. D Western blot analysis of STAT3 pathway related protein levels in 66cl4 transfected with shSix1#2 grown for 48 h in indicated CM. E Quantitation of Western blot in (D), n = 3. F Western blot analysis of STAT3 pathway related protein levels in MCF7 transfected with OE Ctrl grown for 48 h in indicated CM. G Quantitation of Western blot in (F), n = 3. H Representative IF of p-StaT3 (green) and Stat3 (red) in 66cl4 transfected with shSix1#2 grown for 48 h in indicated CM. DAPI (blue), scale bar is 10 μm. I Quantitation of nuclear/cytoplasmic p-Stat3 fluorescence intensity ratio in (H), n = 4. J Representative IF of p-STAT3 (green) and STAT3 (red) in MCF7 transfected with OE Ctrl grown for 48 h in indicated CM. DAPI (blue), scale bar is 10 μm. K Quantitation of nuclear/cytoplasmic p-STAT3 fluorescence intensity ratio in (J), n = 4. L Representative invasion assay performed on 66cl4 transfected with shSix1#1 grown for 48 h in indicated CM, scale bar is 50 μm. M Quantitation of cell invasion in (L), n = 3. N Representative invasion assay performed on 66cl4 transfected with shSix1#2 grown for 48 h in indicated CM, scale bar is 50 μm. O Quantitation of cell invasion in (N), n = 3. P Representative IF of E-cadherin (red) and N-cadherin (green) of MCF7 transfected with OE Ctrl grown for 48 h in indicated CM with 1 μg/ml IL6 antibody or an equal amount of IgG. DAPI (blue), scale bar is 20 μm. Q, R Quantitation of relative fluorescence intensity in (P), n = 4. S Western blot analysis of Vimentin in 66cl4 transfected with shSix1#1 grown for 48 h in indicated CM. Data were presented as means ± SD; ANOVA were used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. SCR scramble, OE Ctrl overexpressing control, sh#1 #2 two independent shRNA constructs targeting genes, CM conditioned medium.
Western blot results revealed that c-Myc and Cyclin D1 protein levels were reduced in the shSix1#1 + OE Ctrl CM (non-EMT CM) group compared to the SCR + OE Ctrl CM (EMT CM) group. Intervention with shSix1#1 + OE IL6 CM (EMT CM), rich in IL6, partially restored c-Myc and Cyclin D1 levels and significantly activated Stat3 phosphorylation (Fig. 6B, C). Similar trends were observed in the 66cl4-shSix1#2 group (Fig. 6D, E). As for MCF7, OE SIX1+IgG CM (EMT CM) intervention significantly upregulated C-MYC and CYCLIN D1, and p-STAT3 expression trended upwards. However, the addition of IL6 neutralizing Ab markedly downregulated C-MYC and CYCLIN D1 contents and dampened p-STAT3 expression (Fig. 6F, G). Immunofluorescence results indicated nuclear p-Stat3 accumulation was reduced in the shSix1#2 + OE Ctrl CM (non-EMT CM) group compared to the SCR + OE Ctrl CM (EMT CM) group. Intervention with shSix1#2 + OE IL6 CM (EMT CM), rich in IL6, partially restored it (Fig. 6H, I). Similar trends were observed in the 66cl4-shSix1#1 group (Supplementary Fig. S7A, B). As for MCF7, OE SIX1+IgG CM (EMT CM) intervention significantly activated nuclear p-STAT3 accumulation compared to the OE Ctrl+IgG CM (non-EMT CM). However, the addition of IL6 neutralizing Ab to MCF7-OE SIX1 CM markedly abolished this effect (Fig. 6J, K).
EMT cells secrete IL6 by SIX1/ZEB1/IL6 signaling axis to promote the proliferation, metastasis, and stemness of non-EMT cells
Figure 6L, M demonstrated that the number of invading cells decreased following intervention with shSix1#1 + OE Ctrl CM (non-EMT CM) compared to SCR + OE Ctrl CM (EMT CM). This reduction was partially reversed by shSix1#1 + OE IL6 CM (EMT CM). A corresponding pattern was observed in the shSix1#2 group (Fig. 6N, O). CCK-8 results revealed that non-EMT cells exhibited reduced proliferation rates following non-EMT CM intervention, contrasting with EMT CM intervention. However, increased IL6 content in the CM reversed this effect, enhancing non-EMT cell proliferation (Supplementary Fig. S7C). OE SIX1+IgG CM (EMT CM) intervention decreased E-cadherin fluorescence intensity, while N-cadherin and Vimentin fluorescence intensity and the protein level of Vimentin increased, compared to OE Ctrl+IgG CM (non-EMT CM) (Fig. 6P–S and Supplementary Fig. S7D, E). The addition of IL6 neutralizing antibody partially reversed this effect (Fig. 6P–S and Supplementary Fig. S7D, E) in MCF7.
Western blot analysis (Fig. 7A, B) revealed that the protein levels of Itgb1, Aldh1a1, and Epcam were lower in the shSix1#2 + OE Ctrl CM (non-EMT CM) group compared to the SCR + OE Ctrl CM (EMT CM) group. Intervention with shSix1#2 + OE IL6 CM (EMT CM) partially upregulated these proteins. In the OE SIX1+IgG CM (EMT CM) group, ITGB1, ALDH1A1, and EPCAM protein levels were significantly elevated. However, the addition of IL6 neutralizing antibody downregulated ITGB1, ALDH1A1, and EPCAM (Fig. 7C–E). Mammosphere size and formation efficiency decreased in the shSix1#2 + OE Ctrl CM (non-EMT CM) group relative to the SCR + OE Ctrl CM (EMT CM) group, whereas shSix1#2 + OE IL6 CM (EMT CM) enhanced these parameters (Fig. 7F–I). A similar trend was observed in the shSix1#1 group (Supplementary Fig. S8A–D). The OE SIX1+IgG CM (EMT CM) group exhibited increased mammosphere size and formation efficiency compared to the OE Ctrl+IgG CM (non-EMT CM) group, and the IL6 neutralizing antibody reduced these parameters (Supplementary Fig. S8E–H). Additionally, we found that IL6 secreted by EMT cells promoted the proportion of stem cell subsets, such as CD24lowCD44high and CD24highCD49fhigh, in non-EMT cells (Fig. 7J, K and Supplementary Fig. S8I–L).

A Western blot analysis of stemness related protein levels in 66cl4 transfected with shSix1#2 grown for 48 h in indicated CM. B Quantitation of Western blot in (A), n = 3. C Western blot analysis of stemness related protein levels in MCF7 transfected with OE Ctrl grown for 48 h in indicated CM with 1 μg/ml IL6 antibody or an equal amount of IgG. D, E Quantitation of Western blot in (C). F Representative images of mammospheres of 66cl4 transfected with shSix1#2 grown for 48 h in indicated CM, scale bar is 100 μm. G Quantitation of mammosphere-formation efficiencies in (F), n = 3. H Quantitation of relative mammosphere size in (F), n = 4. I ELDA assays showed the mammosphere-formation abilities in 66cl4 transfected with shSix1#2 grown for 48 h in indicated CM. J Representative flow cytometry plots of CD24lowCD44high subset in MCF7 transfected with OE Ctrl grown for 48 h in indicated CM with 1 μg/ml IL6 antibody or an equal amount of IgG. K Quantitation of CD24lowCD44high subset in (J), n = 3. Correlation plots showing the mRNA relative expression levels of SIX1 and ZEB1 (L) and IL6 (M) in human breast cancer samples, quantified by RT-qPCR, n = 48. N Correlation plot showing the mRNA relative expression levels of ZEB1 and IL6 in human breast cancer samples, quantified by RT-qPCR, n = 48. Correlation plots showing the mRNA relative expression levels of SIX1 and OCT4 (O), SOX2 (P), CD44 (Q), and CD24 (R) in human breast cancer samples, quantified by RT-qPCR, n = 48. Representative IHC images of breast cancer tissues from two organoids, showing the expression of SIX1 (S), ZEB1 (T), IL6 (U), and IL6R (V), scale bar is 100 μm. W Viability curves of organoids treated with neutralizing antibodies of different concentrations of IL6. The blue curve represents organoids with lower SIX1 expression, while the red curve represents organoids with higher SIX1 expression, n = 3. X Representative images of organoids treated with different concentrations of IL6-neutralizing antibodies at days 0, 2, 4, and 6, scale bar is 100 μm. Data were presented as means ± SD; ANOVA were used for statistical analysis. Correlation analysis was performed using Pearson’s correlation coefficient. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. SCR scramble, OE Ctrl overexpressing control, sh#1 #2 two independent shRNA constructs targeting genes, CM conditioned medium.
RNA relationship and treatment efficiency of IL6 blocking in organoids
The relative mRNA content of SIX1, ZEB1, and IL6 in breast cancer tissues from 48 patients was assessed, with regression curves generated to examine correlations. The basic information of these two patients was listed in Supplementary Table S1. The results showed a significant correlation coefficient of 0.78 between SIX1 and ZEB1 (Fig. 7L), and a high correlation of 0.97 with IL6 (Fig. 7M). Additionally, a correlation coefficient of 0.78 was observed between the relative mRNA content of ZEB1 and IL6 (Fig. 7N), indicating a strong positive correlation among SIX1, ZEB1, and IL6. The data also suggested a positive correlation between the mRNA relative contents of SIX1, ZEB1, IL6, and those of ALDH1A1, NANOG, OCT4, SOX2, and CD44 (Fig. 7O–Q and Supplementary Fig. S9A–L). Finally, we found SIX1, ZEB1, IL6 were negative correlated with CD24 (Fig. 7R and Supplementary Fig. S9M, N).
Subsequently, two breast cancer organoid models were developed from patient samples, displaying different SIX1 expression levels. The basic information of these two patients was listed in Supplementary Table S2. Tissues and organoids from patients with higher SIX1 expression were designated SIX1-high, while those with lower expression were termed SIX1-low (Fig. 7S). Immunohistochemical analysis revealed that the expression levels of ZEB1, IL6, and IL6R were higher in SIX1-high breast cancer tissues compared to SIX1-low tissues (Fig. 7T–V). When IL6 neutralizing antibodies of varying concentrations were applied to the organoids, a gradient of inhibition and reduced growth rate was observed, with the response varying between the two organoid types. SIX1-high organoids exhibited less inhibition, whereas SIX1-low organoids were more significantly inhibited. Half inhibition was achieved with an IL6 neutralizing antibody concentration of 16 μg/ml (Fig. 7W, X).
Further validation by scRNA-seq and ST-seq
Specimens from two untreated breast cancer patients, BC1 and BC2, were collected and divided into pairs for scRNA-seq and ST-seq analysis. The basic information of these two patients was listed in Supplementary Table S3. Dimensionality reduction and cell annotation were performed on the UMAP results (Fig. 8A and Supplementary Fig. S10A). Cell cluster marker genes are displayed in Fig. 8B and Supplementary Fig. S10B. Breast cancer cells were annotated based on marker genes and verified using inferCNV values (Fig. 8C–E and Supplementary Fig. S10C–E). Cells with SIX1 expression above 0 were classified as SIX1+, and those with expression ≤0 as SIX1-. ScRNA-seq data revealed that SIX1+ cells exhibited higher expression of IL6 downstream genes, including STAT3, MYC, and CCND1, compared to SIX1- cells (Fig. 8F–K and Supplementary Fig. S10F–I). EPCAM expression was also elevated in SIX1+ cells (Supplementary Fig. S10J). The expression of SIX1 colocalized spatially with these genes (Fig. 8L and Supplementary Fig. S10K–N). Differential gene analysis identified 64 upregulated genes in SIX1+ cells (Supplementary Data S5). GO and KEGG enrichment analyses were conducted on these genes, with results presented in Supplementary Fig. S11 and detailed in Supplementary Data S6–9. Upregulated genes in SIX1+, such as ARSB, STAT5A, and FZD3, were found to regulate EMT and stem cell-related biological processes (Fig. 8M). The JAK-STAT pathway was involved in the enrichment (Fig. 8N). Spatial colocalization of these genes with SIX1 is shown in Supplementary Fig. S12, further supporting the regulatory role of SIX1 on EMT and CSC via scRNA-seq and ST-seq.

A UMAP representation of eight distinct cell subtypes in BC1 from scRNA-seq data. B Marker genes of eight cell subtypes. C Hierarchical heatmap of CNV scores of eight cell subtypes estimated by InferCNV. UMAP representation of CNV scores (D) and CNV status (E) in BC1. UMAP representation of SIX1 (F), STAT3 (G), and MYC (H) expression in BC1. Violin plot showing the expression of SIX1 (I), STAT3 (J), and MYC (K) in SIX1- and SIX1+ cancer cells in BC1. L ST-seq data showing expression of SIX1 and STAT3 in BC1. M Selective display of GO: BP enrichment results of SIX1+ and SIX1- cancer cells’ differential genes related to EMT and CSCs regulation in BC1. N KEGG enrichment results of SIX1+ and SIX1- cancer cells’ differential genes in BC1. O ST-seq data showing localization of indicated subgroups in BC1. P Violin plot showing the expression of IL6R of indicated subgroups in BC1 from ST-seq data. Q Ripley’s function showing the distribution of indicated subgroups in BC1 from ST-seq data. R Co-occurrence score among indicated subgroups in BC1 at increasing distance threshold across the tissue from ST-seq data. S Identification of breast cancer cell compartments using NMF is depicted in a dot plot, illustrating the estimated NMF weights of different cell types (rows) across NMF components (columns) in BC1 from ST-seq data.
To confirm that SIX1+ cancer cells promote EMT and stemness transformation in SIX1- cancer cells within the spatial transcriptome, cell subpopulations were defined based on SIX1, CD24, and CD44 expression levels (Fig. 8O). IL6R, IL6, CDH2, and VIM expressions were higher in SIX1+CD24-CD44+ and SIX1-CD24-CD44+ subgroups compared to SIX1-CD24+CD44- (Fig. 8P and Supplementary Fig. S13). Distribution patterns varied, with SIX1-CD24-CD44+ tending to cluster, while SIX1+CD24-CD44+ and SIX1-CD24+CD44- were more scattered (Fig. 8Q). Spatially, SIX1-CD24-CD44+ was closer to SIX1+CD24-CD44+ (Fig. 8R). Spatial transcriptome data were deconvolved with single-cell transcriptome data to infer cell type abundance at each spot, and cancer cells were analyzed individually (Supplementary Fig. S14A-D). The spatial distribution abundance of SIX1+CD24-CD44+, SIX1-CD24-CD44+, and SIX1-CD24+CD44- subgroups is presented in Supplementary Fig. S14E, F. NMF decomposition revealed a higher co-localization of SIX1-CD24-CD44+ with SIX1+CD24-CD44+ (Fig. 8S and Supplementary Fig. S14G). Epithelial cells, endothelial cells, and T cells were found to co-occur with these three subgroups (Fig. 8S and Supplementary Fig. S15).
Discussion
For the first time, we demonstrated that SIX1 enhanced the transcription and translation of IL6, a factor known to contribute to tumorigenesis by activating signaling pathways such as JAK-STAT3. In breast cancer, IL6 promotes invasion through the downregulation of E-cadherin and influences cell proliferation by sustaining STAT3 activation, which affects proteins including BCL-2, BCL-XL, SURVIVIN, CYCLIN D1, c-MYC, and MCL-1. IL6 also modulates angiogenesis (HIF-1α, VEGF), EMT (Vimentin, Twist, MMP7/9), and other processes that facilitate breast cancer progression [10, 21]. Our findings corroborate the regulatory role of IL6 in breast cancer malignancy, showing that the inhibitory effects of SIX1 knockdown on proliferation, migration, and invasion can be partially reversed by IL6 overexpression. While the effects of IL6 knockdown may vary, overall, the enhanced malignancy associated with SIX1 overexpression is ameliorated. Experimentally, both SIX1 and IL6 activate the STAT3 signaling pathway and transmit downstream signals such as c-MYC and CYCLIN D1 in TNBC and Luminal A breast cancer cell models, aligning closely with previous literature reports.
IL6 has been reported to bind to IL6R, activate STAT3 signaling, and drive the generation and maintenance of BCSC subpopulations [22, 23]. Furthermore, IL6 can reprogram differentiated tumor cells into CSC-like cells across various breast cancer molecular subtypes [24]. Treatment with exogenous IL6 in MCF7 cells upregulates CD44 expression, enhances cell self-renewal, and promotes stemness and EMT traits. Similarly, IL6 treatment increases stemness in the human TNBC line MDA-MB-231 [25]. Our findings corroborate these reports, demonstrating that IL6 silencing in MCF7 cells overexpressing SIX1 reduced the proportion of CD24lowCD44high cells, while IL6 overexpression in 66cl4 rescued the loss of stemness caused by Six1 suppression. In lung cancer, IL6 promotes the stemness of lung CSC-like cells and enhances ALDH1A1 and NANOG expression. However, the role of IL6 in regulating ALDH1A1 in breast cancer remains unexplored. Our study reveals that IL6 upregulates ALDH1A1 in breast cancer cells, yet the underlying mechanism remains unclear [26]. It is known that ITGB1 and STAT3 engage in feedback loops, mutually enhancing each other’s expression [27]. This finding highlights the relevance of these interactions for our study. We observed that modulating IL6 expression correlated with changes in ITGB1 expression levels in the same direction. Although the regulation of ITGB1 by IL6 in breast cancer is underexplored, it is hypothesized that IL6 activates STAT3 signaling to promote ITGB1 expression. Such alterations in ITGB1 expression may, in turn, further stimulate STAT3 signaling activation.
Tumor-derived IL6 and IL6 secreted by tumor-associated macrophages (TAMs) both play critical roles in breast cancer stemness. IL6 from TAMs activates the STAT3 signaling pathway, fostering the production of breast cancer stem cells (BCSCs) and upregulating stem cell-associated transcription factors, including SOX2, OCT4, and NANOG, in BCSCs. Concurrently, this process enhances the migratory and angiogenic capabilities of breast cancer cells [28]. Suppression of TAM-derived IL6 led to a decrease in the CD24lowCD44high cell subpopulation, ALDH-positive cells, and the expression of stem cell-related factors such as NANOG, SOX2, OCT4, and CD44 in BCSCs, corroborating our findings. Furthermore, adipocytes and fibroblasts within the TME also contribute to IL6 secretion, which activates STAT3 signaling in breast cancer cells and exacerbates their malignancy [21].
Our investigation into the regulatory mechanism of SIX1 on IL6 revealed that alterations in IL6 expression do not modulate SIX1 expression in a feedback manner. Collectively, IL6 appears to be a downstream effector of SIX1. Previous research from our lab has demonstrated that SIX1 can modulate the activation of the STAT3 signaling pathway, thereby influencing stem cell transformation [9]. In the current study, we observed that the manipulation of IL6 in the context of SIX1 modulation reversed this effect, with the activation of the STAT3 pathway and stemness conversion showing a tight correlation. This suggested that SIX1 can activate the STAT3 pathway by enhancing the transcription and translation of IL6, and subsequent promotion of breast cancer cell stemness. Additionally, the predictions of transcription factor binding sites indicated the absence of a SIX1 binding site in the IL6 promoter region.
ZEB1, a key transcription factor in embryonic development and tumor invasion, belongs to the zinc finger E-box binding protein family. It directly inhibits or enhances the transcription of target genes by binding to their promoter regions [29]. Numerous studies have linked the overexpression of ZEB1 to the malignant progression of breast cancer, viewing it as a central player in EMT [29]. Our data corroborated that Zeb1 overexpression reversed the reduction in proliferation, migration, and invasion prompted by Six1 knockdown. Conversely, ZEB1 knockdown elevated E-cadherin expression, possibly due to attenuated transcriptional inhibition by ZEB1. Furthermore, we observed that ZEB1 knockdown incremented Vimentin expression, aligning with the literature reporting ZEB1’s ability to bind Vimentin’s promoter and augment its transcription [30].
In patients with multiple sclerosis, ZEB1 inhibits miR-101-3p, which in turn, can impede the phosphorylation of STAT3 and the expression of JAK2. Consequently, ZEB1 indirectly activates the STAT3 signaling pathway, thereby enhancing the expression of IL17 and IFN-γ [31]. Furthermore, ZEB1 stimulates the transcription and translation of IL6, a pivotal cytokine that activates the STAT3 signaling pathway. Hence, ZEB1 could potentially activate the STAT3 signaling pathway through IL6, a mechanism that has not been previously reported in breast cancer. However, the regulation of STAT3 by ZEB1 is not one-directional. Activation of STAT3 can also induce the expression of ZEB1, which, in a feedback loop, can further upregulate ITGB1, promote the transduction of the Integrin α3β1 signaling pathway via JNK, and inhibit the expression of the gemcitabine transporter ENT1, thereby reducing drug uptake by tumor cells and contributing to drug resistance [32]. RT-qPCR analysis at the mRNA level revealed a trend toward increased Zeb1 expression following overexpression of IL6, suggesting the potential for a positive feedback loop involving ZEB1, IL6, and STAT3 in breast cancer, which warrants further investigation. In lung cancer, ZEB1 can enhance the transduction of the Integrin α3β1 signaling pathway by inhibiting the activity of PP2A, which may lead to increased growth and metastasis of lung cancer cells [33].
The role of SIX1 in enhancing the transcription and translation of ZEB1 through binding to its promoter has been previously established [16]. Nevertheless, the precise regulatory mechanism by which ZEB1 modulates IL6 expression remains elusive. Our study reveals that ZEB1 interacts with the promoter of IL6, thereby facilitating its transcription and translation, and has identified a specific binding site. Region 1 (1138bp–1148bp) appears to be the primary site for this interaction.
In vivo demonstrated that knockdown of SIX1 reduced the metastatic rate and delayed the onset of metastasis in cancer cells. Conversely, overexpression of IL6 partially reversed these effects, consistent with the pro-metastatic role of IL6 as described in the literature. Contrary to this, our animal study findings indicated that IL6 expression had no significant impact on the in vivo growth rate of breast cancer cells, contrasting with previous reports. In vitro results from our study showed that IL6 overexpression accelerated cancer cell growth. Two hypotheses may explain this discrepancy: Firstly, the high efficiency of IL6 overexpression may result in excessive IL6 secretion during in vivo growth, creating an acute inflammatory microenvironment that could counteract the proliferation of 66cl4 cells following IL6 overexpression. Secondly, the animal models used, specifically nude mice which lack a thymus and thereby T lymphocyte maturation, but retain neutrophils and antigen-presenting cells like dendritic cells, may account for the IL6’s role in promoting dendritic cell differentiation into macrophages and neutrophil recruitment, potentially antagonizing the promotional effect of IL6 on tumor growth, thus masking its in vivo growth-promoting effects [34, 35]. In the context of breast cancer, ZEB1’s facilitation of tumor cell proliferation and metastasis in vivo [36, 37] which is consistent with our animal experiment results. We confirmed that SIX1 can modulate IL6 expression through ZEB1 and subsequent regulation of the STAT3 signaling pathway and its downstream signaling. Consequently, we infer that SIX1 promotes the in vivo proliferation and metastasis of breast cancer cells by regulating the activation and downstream signaling of the STAT3 pathway through ZEB1 and IL6.
The complex interplay within the TME drives the emergence of breast cancer heterogeneity, which, in turn, mirrors the dynamic nature of the TME. Signaling interactions occur among adipocytes, fibroblasts, immune cells, and tumor cells themselves, characterized by both cooperation and competition [38]. Here, we unveil a novel cooperative interaction paradigm involving tumor cells. EMT cells, expressing high levels of SIX1, secrete IL6 that targets non-EMT cells with lower SIX1 expression via the SIX1/ZEB1/IL6 signaling axis. Predominantly, the IL6R on the surface of these non-EMT cells is likely to bind IL6. Subsequent activation of the STAT3 signaling cascade within these cells enhances their proliferative and invasive capabilities. Prior research has demonstrated that high SIX1-expressing EMT cells can activate the GLI signaling pathway by secreting VEGF-C, which binds to the NRP2 receptor on non-EMT cells, thereby stimulating breast cancer cell proliferation, migration, and invasion [7]. Additionally, the IL6/STAT3 axis can upregulate VEGF expression [39]. Furthermore, IL6 can upregulate VEGF expression in the circulatory system of breast cancer patients, promoting the rapid formation of blood vessels [35]. Targeting the IL6/STAT3 pathway may enhance the efficacy of anti-VEGF therapies [40]. Given the strong correlation between IL6 and VEGF, it is plausible that EMT cells facilitate their own VEGF-C secretion via the SIX1/ZEB1/IL6 axis.
By administering neutralizing antibodies against IL6 at varying concentrations to intercept IL6-mediated stemness signaling between cells, we intriguingly observed that IL6 neutralization effectively curbed the proliferation of tumor organoids. The inhibitory effect was found to intensify with increasing antibody concentrations. Notably, breast cancer organoids with higher SIX1 expression exhibited a lower inhibition rate and greater resistance compared to those with reduced SIX1 expression, further corroborating the notion that breast cancers with elevated SIX1 expression harbor enhanced stemness properties. Moreover, immunohistochemical assays revealed a higher IL6 expression in breast cancer tissues with increased SIX1 expression. Based on these findings, we speculate that organoids with high SIX1 expression may secrete IL6 as a compensatory mechanism to partially counteract the neutralizing antibody’s blocking effect, thereby maintaining stemness signaling. The expression level of IL6R is relatively higher in SIX1-high organoids, which implies that the surface of SIX1-high organoids possesses a greater number of receptors capable of receiving IL6 signals. These may account for the less effective growth inhibition observed in the presence of IL6-neutralizing antibodies in organoids with high SIX1 expression.
Interference with the STAT3 signaling pathway holds significant clinical promise, with the therapeutic potential of the JAK1/2 inhibitor rusolitinib being investigated in clinical trials. A Phase I/II trial evaluating rusolitinib in combination with trastuzumab for the treatment of trastuzumab-resistant HER2+ metastatic breast cancer yielded disappointing results, as the addition of rusolitinib did not enhance patient outcomes [41]. Similarly, a Phase II trial that employed rusolitinib in conjunction with paclitaxel for triple-negative inflammatory breast cancer patients reduced p-STAT3 expression but did not demonstrate a therapeutic benefit [42]. In contrast, targeting IL6 or IL6R has shown promise in clinical trials for other pathologies, such as reducing the adverse effects of immune checkpoint inhibitors and bolstering the antitumor immune response in melanoma patients [43]. However, targeted therapy involving IL6 or IL6R in breast cancer remains largely in the preclinical stage, with limited clinical studies. The findings of this study suggest that precision therapy targeting IL6 could have significant clinical relevance, offering an alternative treatment option for patients with high SIX1 expression or more aggressive breast cancer progression.
Utilizing the conjunction of scRNA-seq and ST-seq, we substantiated the role of SIX1 in governing the downstream IL6 via the JAK-STAT pathway, thereby facilitating EMT and stemness conversion. Notably, genes including CDH1 and VIM, which are emblematic of EMT, were found to be more highly expressed in the SIX1-CD24-CD44+ subgroup compared to the SIX1-CD24+CD44- subgroup. The CD24-CD44+ cohort is a well-investigated and broadly acknowledged subset of breast cancer associated with increased stemness. Deconvolution analyses confirmed that, spatially, the SIX1-CD24-CD44+ subgroup is nearer to the SIX1+CD24-CD44+ subgroup, exhibits a higher frequency of co-occurrence, and shows elevated expression of IL6 and IL6R. This finding offers an alternative perspective supporting the notion that breast cancer cells with high SIX1 expression can augment EMT and stemness in cells with low SIX1 expression, likely through the paracrine action of IL6 binding to IL6R.
Conclusions
Our study reveals that the SIX1/ZEB1/IL6 axis not only activates the STAT3 pathway to promote malignant phenotypes such as EMT and stemness transformation within breast cancer cells but also exerts effects on neighboring cells with low SIX1 expression through paracrine IL6, activating their STAT3 pathway and enhancing malignant traits. This creates a cascade that amplifies stemness across the TME.
Data availability
The datasets supporting the conclusions of this article are included within the article and its additional files. Additional data related to this paper may be requested from the corresponding author.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33.
Yi M, Li T, Niu M, Luo S, Chu Q, Wu K. Epidemiological trends of women’s cancers from 1990 to 2019 at the global, regional, and national levels: a population-based study. Biomark Res. 2021;9:55.
Miller KD, Nogueira L, Devasia T, Mariotto AB, Yabroff KR, Jemal A, et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J Clin. 2022;72:409–36.
Fatma H, Siddique HR. Cancer cell plasticity, stem cell factors, and therapy resistance: how are they linked? Cancer Metastasis Rev. 2023;43:423–40.
Neelakantan D, Zhou H, Oliphant MUJ, Zhang X, Simon LM, Henke DM, et al. EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells. Nat Commun. 2017;8:15773.
Kong D, Zhou H, Neelakantan D, Hughes CJ, Hsu JY, Srinivasan RR, et al. VEGF-C mediates tumor growth and metastasis through promoting EMT-epithelial breast cancer cell crosstalk. Oncogene. 2021;40:964–79.
Kong D, Li A, Liu Y, Cui Q, Wang K, Zhang D, et al. SIX1 activates STAT3 signaling to promote the proliferation of thyroid carcinoma via EYA1. Front Oncol. 2019;9:1450.
Guo L, Li F, Liu H, Kong D, Chen C, Sun S. SIX1 amplification modulates stemness and tumorigenesis in breast cancer. J Transl Med. 2023;21:866.
Wong GL, Manore SG, Doheny DL, Lo HW. STAT family of transcription factors in breast cancer: Pathogenesis and therapeutic opportunities and challenges. Semin Cancer Biol. 2022;86:84–106.
Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15:848–62.
Katsura A, Tamura Y, Hokari S, Harada M, Morikawa M, Sakurai T, et al. ZEB1-regulated inflammatory phenotype in breast cancer cells. Mol Oncol. 2017;11:1241–62.
Han X, Liu F, Zhang C, Ren Z, Li L, Wang G. SIAH1/ZEB1/IL-6 axis is involved in doxorubicin (Dox) resistance of osteosarcoma cells. Biol Chem. 2019;400:545–53.
Lobe C, Vallette M, Arbelaiz A, Gonzalez-Sanchez E, Izquierdo L, Pellat A, et al. Zinc finger E-Box binding homeobox 1 promotes cholangiocarcinoma progression through tumor dedifferentiation and tumor-stroma paracrine signaling. Hepatology. 2021;74:3194–212.
Ono H, Imoto I, Kozaki K, Tsuda H, Matsui T, Kurasawa Y, et al. SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene. 2012;31:4923–34.
Cieply B, Farris J, Denvir J, Ford HL, Frisch SM. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013;73:6299–309.
Zhu L, Jiang S, Yu S, Liu X, Pu S, Xie P, et al. Increased SIX-1 expression promotes breast cancer metastasis by regulating lncATB-miR-200s-ZEB1 axis. J Cell Mol Med. 2020;24:5290–303.
Liu J, Huang B, Rao Y, Guo L, Cai C, Gao D, et al. Intraductal photothermal ablation: a noninvasive approach for early breast cancer treatment and prevention. Theranostics. 2024;14:3997–4013.
Vassilopoulos A, Chisholm C, Lahusen T, Zheng H, Deng CX. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. 2014;33:5477–82.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8.
Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer. 2016;138:2570–8.
Ma Y, Zhu Y, Shang L, Qiu Y, Shen N, Wang J, et al. LncRNA XIST regulates breast cancer stem cells by activating proinflammatory IL-6/STAT3 signaling. Oncogene. 2023;42:1419–37.
Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer. 2019;18:42.
Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011;108:1397–402.
Ortiz-Montero P, Londoño-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. 2017;15:17.
Xie C, Liang C, Wang R, Yi K, Zhou X, Li X, et al. Resveratrol suppresses lung cancer by targeting cancer stem-like cells and regulating tumor microenvironment. J Nutr Biochem. 2023;112:109211.
Dittmer A, Dittmer J. A CAF-fueled TIMP-1/CD63/ITGB1/STAT3 feedback loop promotes migration and growth of breast cancer cells. Cancers. 2022;14:4983.
Radharani NNV, Yadav AS, Nimma R, Kumar TVS, Bulbule A, Chanukuppa V, et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via Stat-3 pathway. Cancer Cell Int. 2022;22:122.
Wu HT, Zhong HT, Li GW, Shen JX, Ye QQ, Zhang ML, et al. Oncogenic functions of the EMT-related transcription factor ZEB1 in breast cancer. J Transl Med. 2020;18:51.
Qin Y, Yu J, Zhang M, Qin F, Lan X. ZEB1 promotes tumorigenesis and metastasis in hepatocellular carcinoma by regulating the expression of vimentin. Mol Med Rep. 2019;19:2297–306.
Qian Y, Arellano G, Ifergan I, Lin J, Snowden C, Kim T, et al. ZEB1 promotes pathogenic Th1 and Th17 cell differentiation in multiple sclerosis. Cell Rep. 2021;36:109602.
Liu M, Zhang Y, Yang J, Cui X, Zhou Z, Zhan H, et al. ZIP4 Increases expression of transcription factor Zeb1 to promote integrin α3β1 signaling and inhibit expression of the gemcitabine transporter Ent1 in pancreatic cancer cells. Gastroenterology. 2020;158:679–692.e671.
Tan X, Banerjee P, Liu X, Yu J, Lee S, Ahn YH, et al. Transcriptional control of a collagen deposition and adhesion process that promotes lung adenocarcinoma growth and metastasis. JCI Insight. 2022;7:e153948.
Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173:3844–54.
Habanjar O, Bingula R, Decombat C, Diab-Assaf M, Caldefie-Chezet F, Delort L. Crosstalk of inflammatory cytokines within the breast tumor microenvironment. Int J Mol Sci. 2023;24:4002.
Jiang H, Wei H, Wang H, Wang Z, Li J, Ou Y, et al. Zeb1-induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis. 2022;13:206.
Mohammadi Ghahhari N, Sznurkowska MK, Hulo N, Bernasconi L, Aceto N, Picard D. Cooperative interaction between ERα and the EMT-inducer ZEB1 reprograms breast cancer cells for bone metastasis. Nat Commun. 2022;13:2104.
Guo L, Kong D, Liu J, Zhan L, Luo L, Zheng W, et al. Breast cancer heterogeneity and its implication in personalized precision therapy. Exp Hematol Oncol. 2023;12:3.
Xiao L, Wang Z, Lu N, He Y, Qiao L, Sheng X, et al. GPER mediates the IL6/JAK2/STAT3 pathway involved in VEGF expression in swine ovary GCs. J Mol Endocrinol. 2021;68:23–33.
Eichten A, Su J, Adler AP, Zhang L, Ioffe E, Parveen AA, et al. Resistance to anti-VEGF therapy mediated by autocrine IL6/STAT3 signaling and overcome by IL6 blockade. Cancer Res. 2016;76:2327–39.
Kearney M, Franks L, Lee S, Tiersten A, Makower DF, Cigler T, et al. Phase I/II trial of ruxolitinib in combination with trastuzumab in metastatic HER2 positive breast cancer. Breast Cancer Res Treat. 2021;189:177–85.
Lynce F, Stevens LE, Li Z, Brock JE, Gulvady A, Huang Y, et al. TBCRC 039: a phase II study of preoperative ruxolitinib with or without paclitaxel for triple-negative inflammatory breast cancer. Breast Cancer Res. 2024;26:20.
Hailemichael Y, Johnson DH, Abdel-Wahab N, Foo WC, Bentebibel SE, Daher M, et al. Interleukin-6 blockade abrogates immunotherapy toxicity and promotes tumor immunity. Cancer Cell. 2022;40:509–23.e506.
Funding
This work was funded by the National Natural Science Foundation of China (No. 82103671). We are grateful to all the editors and reviewers for their hard work, and all the authors for their efforts.
Author information
Authors and Affiliations
Contributions
Liantao Guo: Conceptualization, Methodology, Project administration, Data curation, Formal analysis, Writing—original draft, Writing—review & editing. Yan Rao: Methodology, Project administration Data curation and Formal analysis. Yawen Song: Methodology, Data curation and Formal analysis. Jiawei Hu: Methodology and Project administration. Zixuan Luo: Methodology and Project administration. Shengrong Sun: Funding acquisition. Chuang Chen: Funding acquisition, Conceptualization, Methodology, Project administration. Deguang Kong: Funding acquisition, Conceptualization, Methodology, Project administration, Writing—review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The animal experiment of the study was approved by the Laboratory Animal Welfare & Ethics Committee of Renmin Hospital at Wuhan University (Issue No. 20200702). The maximal tumor size/burden was not exceeded. The study was approved by the ethics committee of Renmin Hospital, Wuhan University (approved number: WDRY2022-K002). Written informed consent was obtained from all participants or their families in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, L., Rao, Y., Song, Y. et al. SIX1 transmits signals for breast cancer progression via the ZEB1/IL6/STAT3 signaling axis. Oncogene 44, 3493–3511 (2025). https://doi.org/10.1038/s41388-025-03499-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41388-025-03499-6
