
Abstract
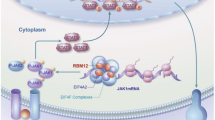
Programmed death ligand 1 (PD-L1) is a protein expressed in hepatocellular carcinoma (HCC) that drives immune evasion by binding to programmed death receptor 1 (PD-1) on activated T cells. Understanding PD-L1 regulation is essential to understand the immunosuppressive microenvironment for antitumor immunity. We screened ribonucleic acid (RNA)-binding motif proteins (RBMs). RBM30 can enhance PD-L1 expression in HCC cells. In this study, we found that high RBM30 expression in tumor tissues can drive HCC tumor immune evasion and accelerate disease progression via increased PD-L1 transcription. We conducted multiple molecular and high-throughput assays to elucidate the intrinsic molecular mechanisms by which RBM30 upregulates PD-L1 expression in HCC. RBM30 binds to DNA near the transcriptional start site of STAT1 and recruits DOT1L to promote H3K79me3 enrichment, enhancing its accessibility to upregulate STAT1 transcription, consequently activating the PD-L1 transcription. This enhances PD-L1 expression to facilitate immune evasion. These findings reveal the vital role of RBM30 in HCC immune evasion.

This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others


Data availability
Raw and processed sequencing data have been deposited at SRA database (PRJNA1089826) and are publicly available as of the date of publication. The rest of the data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. All data generated or analyzed during this study are included in this published article and its supplementary information files, The high-throughput sequencing data generated in this study are publicly available at SRA datebase (PRJNA1089826).
References
Endo Y, Sasaki K, Moazzam Z, Woldesenbet S, Yang J, Araujo Lima H, et al. The impact of a liver transplant program on the outcomes of hepatocellular carcinoma. Ann Surg. 2023;278:230–8.
Brown Z, Tsilimigras D, Ruff S, Mohseni A, Kamel I, Cloyd J, et al. Management of hepatocellular carcinoma: a review. JAMA Surg. 2023;158:410–20.
He M, Huang Y, Du Z, Lai Z, Ouyang H, Shen J. et al. Lenvatinib, Toripalimab plus FOLFOX Chemotherapy in Hepatocellular Carcinoma Patients with Extrahepatic Metastasis: A Biomolecular Exploratory, Phase II Trial (LTSC). Clin Cancer Res. 2023;29:5104–5115. https://doi.org/10.1158/1078-0432.CCR-23-0060.
Zanuso V, Rimassa L, Braconi C. The rapidly evolving landscape of HCC: Selecting the optimal systemic therapy. Hepatology. 2025;81:1365–1386. https://doi.org/10.1097/HEP.0000000000000572.
Yu S. Immunotherapy for hepatocellular carcinoma: recent advances and future targets. Pharmacol Ther. 2023;244:108387.
El Zarif T, Nassar A, Adib E, Fitzgerald B, Huang J, Mouhieddine T, et al. Safety and activity of immune checkpoint inhibitors in people living with HIV and cancer: a real-world report from the cancer therapy using checkpoint inhibitors in people living with HIV-international (CATCH-IT) consortium. J Clin Oncol. 2023;41:3712–23.
Gao X, Xu N, Li Z, Shen L, Ji K, Zheng Z, et al. Safety and antitumour activity of cadonilimab, an anti-PD-1/CTLA-4 bispecific antibody, for patients with advanced solid tumours (COMPASSION-03): a multicentre, open-label, phase 1b/2 trial. Lancet Oncol. 2023;24:1134–46.
Sharma P, Goswami S, Raychaudhuri D, Siddiqui B, Singh P, Nagarajan A, et al. Immune checkpoint therapy-current perspectives and future directions. Cell. 2023;186:1652–69.
Sánchez-Magraner L, Gumuzio J, Miles J, Quimi N, Martínez Del Prado P, Abad-Villar M, et al. Functional engagement of the PD-1/PD-L1 complex but not PD-L1 expression is highly predictive of patient response to immunotherapy in non-small-cell lung cancer. J Clin Oncol. 2023;41:2561–70.
Wang T, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature. 2022;611:358–64.
Huang Q, Wu X, Wang Z, Chen X, Wang L, Lu Y, et al. The primordial differentiation of tumor-specific memory CD8 T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell. 2022;185:4049–66.e4025.
Callahan M, Chapman P. PD-1 or PD-L1 blockade adds little to combination of BRAF and MEK inhibition in the treatment of BRAF V600-mutated melanoma. J Clin Oncol. 2022;40:1393–5.
Liu Y, Wang L, Song Q, Ali M, Crowe W, Kucera G, et al. Intrapleural nano-immunotherapy promotes innate and adaptive immune responses to enhance anti-PD-L1 therapy for malignant pleural effusion. Nat Nanotechnol. 2022;17:206–16.
Zhao J, Yap D, Chan Y, Tan B, Teo C, Syn N, et al. Low programmed death-ligand 1-expressing subgroup outcomes of first-line immune checkpoint inhibitors in gastric or esophageal adenocarcinoma. J Clin Oncol. 2022;40:392–402.
Kang Y, Chen L, Ryu M, Oh D, Oh S, Chung H, et al. Nivolumab plus chemotherapy versus placebo plus chemotherapy in patients with HER2-negative, untreated, unresectable advanced or recurrent gastric or gastro-oesophageal junction cancer (ATTRACTION-4): a randomised, multicentre, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022;23:234–47.
Qin S, Chen Z, Fang W, Ren Z, Xu R, Ryoo B, et al. Pembrolizumab versus placebo as second-line therapy in patients from asia with advanced hepatocellular carcinoma: a randomized, double-blind, phase III trial. J Clin Oncol. 2023;41:1434–43.
Cha J, Chan L, Li C, Hsu J, Hung M. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359–70.
Cerezo M, Guemiri R, Druillennec S, Girault I, Malka-Mahieu H, Shen S, et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat Med. 2018;24:1877–86.
Ladányi A, Tímár J. Immunologic and immunogenomic aspects of tumor progression. Semin Cancer Biol. 2020;60:249–61.
Philips R, Wang Y, Cheon H, Kanno Y, Gadina M, Sartorelli V, et al. The JAK-STAT pathway at 30: much learned, much more to do. Cell. 2022;185:3857–76.
Xie W, Medeiros L, Li S, Tang G, Fan G, Xu J. PD-1/PD-L1 Pathway: A Therapeutic Target in CD30+ Large CellLymphomas. Biomedicines. 2022;10:1587. https://doi.org/10.3390/biomedicines10071587.
Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield V, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–78.
Copeland R. Molecular pathways: protein methyltransferases in cancer. Clin Cancer Res. 2013;19:6344–50.
Chen S, Zhao Y, Liu S, Zhang J, Assaraf Y, Cui W, et al. Epigenetic enzyme mutations as mediators of anti-cancer drug resistance. Drug Resist Updates 2022;61:100821.
Deshpande A, Bradner J, Armstrong S. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33:563–70.
Saygin C, Carraway H. Emerging therapies for acute myeloid leukemia. J Hematol Oncol. 2017;10:93.
Liu Y, Wu Q, Sun T, Huang J, Han G, Han H. DNAAF5 promotes hepatocellular carcinoma malignant progression by recruiting USP39 to improve PFKL protein stability. Front Oncol. 2022;12. https://doi.org/10.3389/fonc.2022.1032579.
Han H, Lin T, Wang Z, Song J, Fang Z, Zhang J, et al. RNA-binding motif 4 promotes angiogenesis in HCC by selectively activating VEGF-A expression. Pharmacol Res. 2023;187:106593.
Han H, Lin T, Fang Z, Zhou G. RBM23 drives hepatocellular carcinoma by activating NF-κB signaling pathway. BioMed Res Int. 2021;2021:6697476.
Han H, Yuan Y, Li C, Liu L, Yu H, Han G, et al. RNA-binding motif protein 28 enhances angiogenesis by improving STAT3 translation in hepatocellular carcinoma. Cancer Lett. 2024;604. https://doi.org/10.1016/j.canlet.2024.217191.
Zhao W, Lin T, Fang Z, Zhou H, Du Y, Han H. NAT10 promotes the malignant progression of hepatocellular carcinoma through upregulating RelA/p65 acetylation. J Biol Regul Homeost Agents. 2023;37:2935–46.
Pinter M, Jain R, Duda D. The current landscape of immune checkpoint blockade in hepatocellular carcinoma: a review. JAMA Oncol. 2021;7:113–23.
Han H, Shi Q, Zhang Y, Ding M, He X, Liu C. et al. RBM12 drives PD-L1-mediated immune evasion in hepatocellular carcinoma by increasing JAK1 mRNA translation. Oncogene. 2024;43:3062–3077. https://doi.org/10.1038/s41388-024-03140-y.
Willemsen L, Prange K, Neele A, van Roomen C, Gijbels M, Griffith G, et al. DOT1L regulates lipid biosynthesis and inflammatory responses in macrophages and promotes atherosclerotic plaque stability. Cell Rep. 2022;41:111703.
Gilan O, Talarmain L, Bell C, Neville D, Knezevic K, Ferguson D, et al. CRISPR-ChIP reveals selective regulation of H3K79me2 by menin in MLL leukemia. Nat Struct Mol Biol. 2023;30:1592–606.
Sangro B, Chan S, Meyer T, Reig M, El-Khoueiry A, Galle P. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J Hepatol. 2020;72:320–41.
Johnson P, Zhou Q, Dao D, Lo Y. Circulating biomarkers in the diagnosis and management of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2022;19:670–81.
Kanwal F, Singal A. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology. 2019;157:54–64.
Pinter M, Scheiner B, Peck-Radosavljevic M. Immunotherapy for advanced hepatocellular carcinoma: a focus on special subgroups. Gut. 2021;70:204–14.
Rimassa L, Finn R, Sangro B. Combination immunotherapy for hepatocellular carcinoma. J Hepatol. 2023;79:506–15.
Shen Y, Hsu C, Jeng Y, Ho M, Ho C, Yeh C, et al. Reliability of a single-region sample to evaluate tumor immune microenvironment in hepatocellular carcinoma. J Hepatol. 2020;72:489–97.
Kang H, Oh J, Chun S, Kim D, Ryu Y, Hwang H, et al. Immunogenomic landscape of hepatocellular carcinoma with immune cell stroma and EBV-positive tumor-infiltrating lymphocytes. J Hepatol. 2019;71:91–103.
Chen Y, Hao X, Sun R, Wei H, Tian Z. Natural killer cell-derived interferon-gamma promotes hepatocellular carcinoma through the epithelial cell adhesion molecule-epithelial-to-mesenchymal transition axis in hepatitis B Virus transgenic mice. Hepatology. 2019;69:1735–50.
Li Y, Zhang C, Martincuks A, Herrmann A, Yu H. STAT proteins in cancer: orchestration of metabolism. Nat Rev Cancer. 2023;23:115–34.
Han H, Zhu W, Lin T, Liu C, Zhai H. N4BP3 promotes angiogenesis in hepatocellular carcinoma by binding with KAT2B. Cancer Sci. 2022;113:3390–404.
Ivashkiv L. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18:545–58.
Subramani A, Hite M, Garcia S, Maxwell J, Kondee H, Millican G, et al. Regulation of macrophage IFNγ-stimulated gene expression by the transcriptional coregulator CITED1. J Cell Sci. 2023;136.
Chang W, Luo Q, Wu X, Nan Y, Zhao P, Zhang L, et al. OTUB2 exerts tumor-suppressive roles via STAT1-mediated CALML3 activation and increased phosphatidylserine synthesis. Cell Rep. 2022;41:111561.
Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, et al. Methyltransferase SETD2-mediated methylation of STAT1 Is critical for interferon antiviral activity. Cell. 2017;170:492–506.e414.
Xiao Z, Wang S, Tian Y, Lv W, Sheng H, Zhan M, et al. METTL3-mediated m6A methylation orchestrates mRNA stability and dsRNA contents to equilibrate γδ T1 and γδ T17 cells. Cell Rep. 2023;42:112684.
Xu H, Jiang Y, Xu X, Su X, Liu Y, Ma Y. et al.Inducible degradation of lncRNA Sros1 promotes IFN-γ-mediated activation of innate immune responses by stabilizing Stat1 mRNA.Nat Immunol. 2019;20:1621–1630. https://doi.org/10.1038/s41590-019-0542-7.
Jiang Y, Zheng Y, Zhang YW, Kong S, Dong J, Wang F, et al. Reciprocal inhibition between TP63 and STAT1 regulates anti-tumor immune response through interferon-γ signaling in squamous cancer. Nat Commun. 2024;15.
Zietse M, van Leeuwen RWF, Barjesteh van Waalwijk van Doorn-Khosrovani S, de Boer JE, Dupree R, Mathijssen RHJ, et al. Interchangeability of immune checkpoint inhibitors: an urgent need for action. Lancet Oncol. 2024;25:e611–e616. https://doi.org/10.1016/S1470-2045(24)00212-2.
Zhao S, Allis C, Wang G. The language of chromatin modification in human cancers. Nat Rev Cancer. 2021;21:413–30.
Yi Y, Ge S. Targeting the histone H3 lysine 79 methyltransferase DOT1L in MLL-rearranged leukemias. J Hematol Oncol. 2022;15:35.
Campbell R, Tummino P. Cancer epigenetics drug discovery and development: the challenge of hitting the mark. J Clin Invest. 2014;124:64–9.
Alexandrova E, Salvati A, Pecoraro G, Lamberti J, Melone V, Sellitto A, et al. Histone methyltransferase DOT1L as a promising epigenetic target for treatment of solid tumors. Front Genet. 2022;13:864612.
Wood K, Tellier M, Murphy S. DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules. 2018;8. https://doi.org/10.3390/biom8010011.
Gu X, Hua Y, Yu J, Yang L, Ge S, Jia R, et al. Epigenetic drug library screening reveals targeting DOT1L abrogates NAD synthesis by reprogramming H3K79 methylation in uveal melanoma. J Pharm Anal. 2023;13:24–38.
Acknowledgements
We sincerely thank the central Laboratory of Taizhou People’s Hospital Affiliated to Nanjing Medical University for the help of instruments and equipment. We thank Home for Researchers editorial team (www.home-for-researchers.com) for language editing service and drawing the pattern diagram.
Funding
This project was supported by National Natural Science Foundation of China (82303115), The Youth Fund of Taizhou People’s Hospital Affiliated to Nanjing Medical University (TZKY20240109), Scientific research start-up fund of Taizhou People’s Hospital (QDJJ202106), Taizhou Society Development Project, Jiangsu, China (TS202308), General Project of Jiangsu Provincial Health Commission (H2023030), China Postdoctoral Science Foundation (2024M751162), Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (HB2023060).
Author information
Authors and Affiliations
Contributions
Zhicheng Gong and Hexu Han performed study concept and design; Hexu Han and Chen Gong completed the experimental part of the project; Yue Zhang and Cuixia Liu performed development of methodology and writing, review and revision of the paper; Junxing Huang provided acquisition, analysis and interpretation of data, and statistical analysis; Yifan Wang and Dakun Zhao provided technical and material support. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The experimental protocol was approved by the Institutional Ethics Review Board of The Affiliated Taizhou People’s Hospital of Nanjing Medical University (2022-008-01). Animal ethical certificates were approved by the Experimental Center of Jiangsu Hanjiang Biotechnology Co., Ltd (HJSW-23050302), and the animals were raised according to animal welfare laws.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Han, H., Gong, C., Zhang, Y. et al. RBM30 recruits DOT1L to activate STAT1 transcription and drive immune evasion in hepatocellular carcinoma. Oncogene 44, 3955–3973 (2025). https://doi.org/10.1038/s41388-025-03550-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41388-025-03550-6
