
Abstract
As essential micronutrients, metal ions such as iron, manganese, copper, and zinc, are required for a wide range of physiological processes in the brain. However, an imbalance in metal ions, whether excessive or insufficient, is detrimental and can contribute to neuronal death through oxidative stress, ferroptosis, cuproptosis, cell senescence, or neuroinflammation. These processes have been found to be involved in the pathological mechanisms of neurodegenerative diseases. In this review, the research history and milestone events of studying metal ions, including iron, manganese, copper, and zinc in neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD), will be introduced. Then, the upstream regulators, downstream effector, and crosstalk of mental ions under both physiologic and pathologic conditions will be summarized. Finally, the therapeutic effects of metal ion chelators, such as clioquinol, quercetin, curcumin, coumarin, and their derivatives for the treatment of neurodegenerative diseases will be discussed. Additionally, the promising results and limitations observed in clinical trials of these metal ion chelators will also be addressed. This review will not only provide a comprehensive understanding of the role of metal ions in disease development but also offer perspectives on their modulation for the prevention or treatment of neurodegenerative diseases.
Similar content being viewed by others

Introduction
Neurodegenerative diseases are characterized by neuronal death and loss-of function, which typically result in a gradual decline in cognitive, motor, and sensory functions. Metal ions, such as iron, manganese, copper, zinc, etc., play crucial roles in various physiological processes in the central nervous system (CNS), including energy metabolism, protein synthesis, DNA replication, membrane protein construction, myelin and neurotransmitter synthesis, and so on. The homeostasis of metal ions in the brain is regulated by multiple proteins and molecular mechanisms, which work together to control the process of absorption, storage, and release, thereby maintaining the appropriate concentration and distribution among different brain regions, cells, and organelles. However, once the homeostasis of these metal ions is disrupted, either depletion or accumulation, they can affect the activity of enzymes involved in neurodegenerative diseases since they serve as important cofactors for enzymes. Additionally, an imbalance in metal ions can contribute to the development of neurodegenerative diseases through a variety of mechanisms, including promoting the production and aggregation of pathological proteins, inducing oxidative stress, ferroptosis, cuproptosis, cell senescence, or neuroinflammation. Since iron deposits in the brains of patients with PD and AD were first observed in 1924 and 1953, respectively, the relationship between iron dyshomeostasis and neurodegenerative diseases has attracted more and more attention. Abnormal iron deposition in special brain regions has been proven to be positively correlated with progress development and disease severity in neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD). Additionally, other metal ions, such as manganese, copper, and zinc, are also found to participate in the development of neurodegenerative diseases by increasing the risk of neurodegenerative diseases or promoting aggregation of pathological proteins. Notably, the identification of metal ions dependent cell death forms, such as ferroptosis and cuproptosis, have provided new pathological mechanisms in neurodegenerative disease. Although there are many challenges in the development of new drugs for neurodegenerative diseases, such as complex pathogenesis, irreversibility of the disease course, difficulty in penetrating the blood–brain barrier (BBB) and clinical trials, therapeutic strategies targeted metal ions have achieved promising results and offers valuable insights into the prevention and treatment of neurodegenerative diseases.
This review will introduce the research history and milestone events of the study on mental ions (including iron, manganese, copper, and zinc) in neurodegenerative diseases (including PD, AD, ALS, and HD). It will also discuss upstream regulators, downstream effector, and crosstalk of mental ions homeostasis in physiology and neurodegenerative diseases. Given their ability to selectively capture metal ions and dissociate them from target sites implicated in disease progression, chelators offer the potential to minimize side effects associated with broad-spectrum treatments. In this review, we provide a comprehensive summary of the therapeutic effects of various chelating compounds, including clioquinol (CQ), quercetin, curcumin, coumarin, and their derivatives, in the pathology of neurodegenerative diseases. Additionally, we discuss the promising results and limitations observed in clinical trials involving deferiprone (DFP), Cu2+-diacetylbis (4-methylthiosemicarbazone)--CuII(atsm), and PBT. This review provides a comprehensive overview of the critical roles redox-active metal ions play in the emergence and progression of neurodegenerative diseases. It emphasizes the need for further research into their mechanisms and the development of effective interventions targeting metal homeostasis as a promising approach for the prevention and treatment of neurodegenerative diseases. Through understanding and modulation of these processes, future strategies could open new avenues for therapeutic intervention.
Research history of metal ions in neurodegenerative diseases
As early as 1924, iron deposition was first observed in the globus pallidus (GP) of PD patients through Perls’ and Turnbull staining (Fig. 1).1 In 1987, Dexter et al. reported significant iron deposition in the substantia nigra (SN) of postmortem PD patient brains,2 and subsequent research confirmed increased total iron content in the SN of postmortem PD patients using inductively coupled plasma spectroscopy (ICP-MS).3,4,5 Besides observing higher total iron levels, there was also an increase in ferric iron in the SN of PD patients.6,7,8 In 1993, in vivo magnetic resonance imaging (MRI) revealed a higher iron content in the SN of patients with PD.9 In 2000, the application of detecting redox-active iron in situ demonstrated that iron aggregated in the neocortical Lewy bodies of PD patients.10 As the main pathological feature of PD, Lewy bodies are composed of a large amount of misfolded α-synuclein, and the toxic couple between iron deposition and α-synuclein aggregation accelerates the progression of PD.11 Nigral iron deposition and hyperechogenicity were found in the 6-OHDA-induced PD rat model in 1999.12 Subsequently, nigral hyperechogenicity showed higher iron levels prior to the diagnosis of PD in 2002.13 In 2003, genetic or pharmacological methods demonstrated that iron chelator presented the neuroprotection in 1-methyl-4-phenyl-1, 2, 3, 6-tetrapyridine (MPTP)-induced PD model.14 Furthermore, a clinical trial conducted until 2014 revealed that oral administration of DFP exhibited neuroprotection on early-stage PD patients through chelation of labile iron.15 In 2007, a new finding revealed that the iron levels were increased in individual nigral dopaminergic neurons in postmortem PD patients using sensitive and specific wavelength dispersive electron probe x-ray microanalysis coupled with cathodoluminescence spectroscopy.16 Idiopathic rapid eye movement sleep behavior disorder (iRBD) is considered a prodromal stage of α-synucleinopathies, such as PD. Within 5 years, 41% of iRBD cases will convent to neurodegenerative diseases, and this rate increases to 73.4% within 10 years.17 In 2019, elevated iron was observed in the bilateral substantia nigra of iRBD patients compared to healthy controls by quantitative susceptibility mapping (QSM), and the level of iron in the substantia nigra PD is even higher than that in iRBD, indicating that abnormal nigral iron deposition may be an important factor for accelerating the conversion from prodromal to clinical stage of neurodegenerative diseases.18 Furthermore, T2*-weighted magnetic resonance imaging confirmed a positive correlation between nigral iron deposition and the progression of disease, as well as motor and cognitive dysfunction in PD patients.19 Although nigral iron deposition has been proven to be positively correlated with the progression of PD, decreased ferritin has been found in several brain regions in PD patients.3 In 2021, the ratio of iron to ferritin was first reported to be increased in the cerebrospinal fluid (CSF) of PD patients, which may serve as a potential progression marker for disease progression.20 Ferroptosis, a newly named form of programmed cell death in 2012 characterized by iron accumulation and lipid peroxidation, was first observed in an MPTP-induced PD mouse model in 2016.21 Acyl-CoA synthetase long-chain family member 4 (ACSL4) can esterify polyunsaturated fatty acids (PUFAs) and trigger ferroptosis.22 Increased levels of ACSL4 have been observed in the SN of both MPTP-induced PD mouse model and PD patients, while genetic or pharmacologic inhibition of ACSL4 can specifically prevent the elevation of lipid ROS and ameliorate Parkinsonism phenotypes,22 suggesting that interventions in the ferroptosis pathway may become a treatment strategy for PD. Recently, we reported that the toxic interaction between α-synuclein and iron induces cell senescence in a PD mouse model, preceding the loss of nigral dopaminergic neurons.23

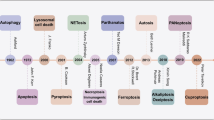
Timeline and milestone events of study on iron and other metal ions in neurodegenerative diseases. The timeline begins at 1924 and expands to 2024. Milestone events of studying metal ions, including iron, manganese, copper, and zinc in neurodegenerative diseases, such as PD, AD, ALS, and HD, are listed in the figure. This figure was created with BioRender.com/d84k316
Iron deposition was first observed in the brain of AD patients with the method of Prussian blue reaction in 1953,24 and then subsequently identified in the cerebral cortex of AD patients by the Turnbull blue method in 1960.25 A single-blind clinical trial in 1991 demonstrated that desferrioxamine (DFO) treatment could significantly reduce the decline of daily living skills, indicating a positive slowing effect on dementia associated with AD by the iron chelator.26 After that, studies on regional brain trace elements received increasing attention. In 1994, in vivo evaluation of brain iron by MRI revealed a higher iron content in the caudate and GP of AD patients.27 Additionally, increased ferritin iron accompanied by decreased tissue integrity was identified in the hippocampus of in vivo AD patients by MRI.28 In 1997, the associations between redox-active iron and senile plaques and neurofibrillary tangles in the hippocampal tissue of AD patients were reported, and redox-active iron not only participated in the in situ oxidation, but also catalyzed H2O2-dependent oxidation, implicating iron accumulation as a source of free radicals in AD.29 And then redox-iron, which participates in lipid peroxidation and oxidative stress, was reported to mediate the toxicity of Aβ.30 According to the neuroimaging using QSM in 2017, brain iron deposition was found to be consistent with the rate of cognitive decline and fibrillar amyloid accumulation, suggesting that brain iron may interact with Aβ to accelerate the clinical development of AD.31 Additionally, a study conducted in 2020 reported an association between brain iron and accelerated cognitive decline in AD patients.32 Meanwhile, a positive association between iron deposition and insoluble tau aggregates in the inferior temporal gyrus of AD patients was identified by MR-based QSM and tau-PET in 2020.33 Recently, DNAzyme-based fluorescent turn-on sensors have been developed, which are selective for either ferrous iron or ferric iron and enable the monitoring of different redox states of iron in living cells.34 Using these sensors, increased levels of both ferric and ferrous iron were observed in ferroptotic cells, while a decreased ratio of ferric iron to ferrous iron was observed. Additionally, an elevated ratio of ferric iron to ferrous iron was observed in the cortex regions with Aβ plaque deposition of AD mice.34 With a label-free and nanoscale chemical imaging using synchrotron X-ray spectromicroscopy, higher iron level, which presented as chemically reduced and low-oxidation-state phases, was observed in the amyloid plaques of human AD brain tissue,35 providing an approach for analyzing the chemical composition of amyloid plaques.
In early 1967, it was first observed that exposure to excessive manganese increased the risk of PD among miners.36 In 2003, a study reported a combined effect of iron and manganese intake from diet on increasing the risk of PD.37 In 1977, a case report was published on a patient with elevated level of manganese, dementia and an extrapyramidal syndrome.38 Both neurotic plaques and neurofibrillary tangles were observed in the brain of this patient. Another case in 1986 reported chronic manganese poisoning causing neuropsychiatric symptoms and neurodegeneration in the basal ganglia, particularly in the pallidum.39 In non-human primates exposed to manganese, although the nigrostriatal dopamine system remained intact, subtle motor function deficits were observed, which were associated with decreased dopamine release.40 In 2017, exposure to welding fumes containing manganese was reported to induce dose-dependent progression of Parkinsonism.41 Recently, it has been proposed that manganese diffuses along white matter tracts. Following manganese exposure, manganese deposition was observed in the cerebellum and frontal cortex as well as the hippocampus using whole-brain MRI relaxometry methods.42 After imaging the in situ secondary structure of the amyloid plaques using synchrotron fourier transform infrared micro-spectroscopy (FTIRM) and detecting the metal ions accumulation using synchrotron X-ray fluorescence (SXRF) microprobe in the same brain tissue of AD patients, co-localization of accumulated copper and zinc with Aβ deposits was first observed in 2006.43 It wasn’t until 2022 that a copper-dependent form of cell death called cuproptosis was first described, which is mitochondrial respiration- dependent and distinct from known cell death mechanisms.44,45 This mechanism has also been found to be involved in neurodegenerative diseases.
In 1991, after analyzing frozen postmortem brain tissue using inductively coupled plasma spectroscopy, researchers observed increased total iron levels in the striatum (putamen and/or caudate nucleus) and elevated copper levels in the putamen and SN of HD patients.3 Additionally, increased zinc levels were observed in the SN, caudate nucleus, and lateral putamen of PD patients. In 2016, QSM results revealed significant iron accumulation in the basal ganglia (including pallidum, putamen and caudate) of both premanifest and symptomatic HD patients.46 Furthermore, iron accumulation in both putamen and caudate was significantly associated with the disease severity. Notably, significant iron deposition was also observed in the left precentral gyrus and the thalamus, and iron deposition in the thalamus is associated with disease severity in ALS patients in 2022.47 Copper/zinc superoxide dismutase, also known as SOD1, exhibits altered reactivity in catalyzing oxidative reactions, which has been suggested to initiate the neuropathologic changes in ALS in 1996.48 Additionally, the enzyme activity of mutant SOD1 was found to be reduced by approximately 50% in patients in 1997.49 In a recent study conducted in 2021, analysis of metal levels in erythrocytes using ICP-MS revealed an association between decreased ALS risk and zinc, while cadmium and lead were associated with an increased risk of developing ALS.50
Brain metal ions homeostasis in physiological state
Iron
As an essential trace element, iron functions as a cofactor for many physiological processes, including oxygen transport, DNA synthesis, mitochondrial respiration, and phospholipid synthesis in the brain. One of the main functions of iron in living organisms is to participate in oxygen transport. In red blood cells, iron is a component of hemoglobin, which binds to oxygen and forms oxygenated hemoglobin that transports oxygen from the lungs to the rest of the body. This process is essential for cellular energy metabolism. Iron is an essential component of various crucial enzymes in the enzyme complex of the mitochondrial respiratory chain, particularly the iron-sulfur clusters and cytochromes present in Complex I and Complex II. Iron-sulfur clusters are nonheme cofactors composed of iron and sulfur atoms that play a vital role in electron transport within the electron transport chain. Inadequate levels of iron directly impact the synthesis and stability of these iron-sulfur clusters, thereby hindering the activity of the respiratory streptase complex. Iron is also a component of certain enzymes that are directly involved in key steps in the DNA replication and repair process. For example, iron-sulfur clusters are active centers for enzymes involved in electron transport chains that provide the energy necessary for DNA synthesis. In addition, iron is involved in redox reactions within cells, which are essential for maintaining the reducing environment within cells and preventing oxidative stress damage, which can damage DNA and affect its normal synthesis. In the process of DNA synthesis, iron’s role is not limited to energy supply, but also involves direct participation in the formation of nucleic acid chains. Iron-dependent enzymes such as DNA polymerase play a catalytic role in the synthesis of new DNA strands, helping to link nucleotides together into long strands. These functions of iron are essential for cell growth, division, and the transmission of genetic information. Iron deficiency or excess can affect DNA synthesis and normal cell function. Iron deficiency may lead to a decrease in the rate of DNA synthesis, affecting cell proliferation and differentiation, while iron excess may lead to DNA damage through the production of free radicals. Therefore, maintaining a balance of iron is essential for normal brain function and neurodegenerative diseases.
Iron influx into the brain
BBB is one of the major barriers preventing peripheral iron from entering the brain.51 During the process of crossing the BBB, endothelial cells, which are the core anatomical structure of BBB, have been suggested to function as gatekeepers. Iron is initially imported by the microvascular endothelial cells at the luminal membrane. In this process, transferrin (Tf) bound ferric iron binds with transferrin receptor 1(TfR1) and enters the cell through endocytosis, while non-transferrin bound iron (known as NTBI) or ferrous iron enters the cell through divalent metal transporter 1 (DMT1).51,52 Furthermore, both H-ferritin and Tf could also serve as iron source cross the BBB.53 The iPSC-derived brain endothelial cells have been found to uptake H-ferritin through T-cell immunoglobulin and mucin receptor 1, and then secrete H-ferritin into the brain, which process could be affected by DMT1.53 In addition to H-ferritin, brain endothelial cells also secrete Tf.53 Inhibition of DMT1 by XEN602 could alter the transport of both Tf and iron across the endothelial cells.54 With aging, the level of serum ferritin has shown an age-related tendency to rise, and it is higher in males than in females.55 Recently, serum ferritin has been suggested to be associated with cognitive performance in aging.56 In subjects aged 65 years or older, the level of serum ferritin was found to be positively associated with executive function and language function in males, with evidence of increased cognitive scores of total digits span (TDS), phonemic verbal fluency (PVF), and semantic verbal fluency (SVF).56 However, there were no significant associations found between serum ferritin and cognitive scores in subjects aged 50-64 years or in female subjects.56 Furthermore, the association of serum ferritin with cognition was found to be regulated by the gut microbiota through microbial-derived metabolites.57 Although in a bi-chamber cell culture model of BBB, Tf-mediated transport of radiolabeled iron (59Fe) was found to correlate positively with the concentration of plasma hemoglobin but not serum ferritin level,58 however, considering that H-ferritin could cross the BBB,53 increased serum ferritin may contribute to an increased iron level in some brain regions with aging. In a Belgrade rat model with brain iron deficiency, altered distribution of Tf receptors in the microvasculature was observed in luminal, intracellular, and abluminal membranes which depended on brain iron status.59 Ferroportin 1 (FPN1) functions as a gateway for iron release into the brain interstitial spaces at the abluminal membrane of brain microvascular endothelial cells. In mice with deletion of FPN1 from the brain vascular endothelial cells, although the level of iron in the serum was lower, elevated iron levels were observed in the brain vascular endothelial cells, along with evidence of l-ferritin accumulation, implicating the important role of FPN1 in exporting iron from vascular endothelial cells in the brain.60 In an in vitro BBB model, iron release from endothelial cells has been found to be stimulated by the iron chelator deferoxamine (DFO) and apo-transferrin (apo-Tf, iron-poor Tf).54 The level of FPN in the brain microvascular endothelial cells has been found to be controlled by hepcidin (Hp), which was secreted by astrocytes.61 In cultured microvascular endothelial cells, hepcidin peptide significantly reduced Tf-Fe and NTBI uptake and iron release accompanied by downregulation of TfR1, DMT1, and FPN1, whereas knockdown of hepcidin generated opposite results.62 In addition, it has been confirmed that human brain microvasculature endothelial cells can express Hp protein and soluble ceruloplasmin (Cp) transcript, and FPN-mediated iron efflux from human brain microvasculature endothelial cells requires endogenous Hp or extracellular Cp, which act as exocytoplasmic ferroxidase.63 Recently, it was discovered that holo-Tf (iron-bound) directly interacts with FPN and induces its internalization, while apo-Tf (iron-free) directly interacts with hephaestin in iPSC-derived endothelial cells or HEK293 cells.64 Moreover, pathophysiological levels of Hp only disrupt the interaction between holo-Tf and FPN by causing the internalization of FPN, indicating a potential mechanism for apo-Tf and holo-Tf in regulating iron release from endothelial cells.
CSF is another major barrier that prevents peripheral iron from entering the brain.51 The blood-CSF barrier is located within the choroid plexus and established by the tight junctions between choroidal epithelial cells, which serve as an important interface to separate the blood from the CSF. Since there is no structural impediment between CSF and interstitial fluid, their components can freely exchange and reach equilibrium. The choroid plexus plays a crucial role in regulating iron homeostasis due to its large surface area and high velocity of blood flow.65 Additionally, choroid plexus cells have the ability to synthesize various types of iron transport proteins such as Tf, TfR1, DMT1, FPN1, Cp, and hephaestin.66,67,68 There are two distinct isoforms of Tf, namely the brain-specific Tf and the serum-derived Tf, which demonstrate variations in their glycan configurations.69 FPN1 is widely distributed in the cytoplasm but less polarized, whereas TfR is predominantly concentrated in the vicinity of nuclei in the form of clusters and bilateral distributed in choroidal cells.70 With the cooperation of DMT1, FPN1/Cp, and FPN1/hephaestin, iron can cross the blood-CSF barrier mediated by choroidal epithelial cells and bind to apo-Tf after entering the CSF.71 Besides, due to the presence of fenestrated capillaries on the choroid plexus, Tf-Fe readily combines with TfR1 and enters the choroidal epithelium via endocytosis. Subsequently, it is dissociated in the acidic endosome and transformed into a divalent state through the activity of Steap3, ultimately being transported into CSF via DMT1 and FPN1. However, an alternative perspective on the blood-CSF barrier is that it is also responsible for the clearance and detoxification of iron from the brain. DMT1 is predominantly localized at the apical plasma membrane, exhibiting a polarized pattern that determines the tendency of free iron to flow from CSF to the blood. Experiments have also demonstrated that the total iron content flowing from the CSF to the blood is 128% higher than its influx.72 Iron, either bound to Tf or in its free state, is presented in the CSF. Subsequently, the apical microvilli on the choroidal epithelia respectively absorb them, utilizing DMT1 for free iron and TfR1 for Tf-bound Fe, transporting it into the bloodstream. Therefore, it has been established that iron crosses the BBB for entry and exits from the brain through the blood-CSF barrier.73
After peripheral iron enters the brain, axonal iron transport may contribute to the distribution of iron among different brain regions. To date, two pathways of axonal iron transport has been reported: one from the ventral hippocampus (vHip) to the medial prefrontal cortex (mPFC) to the SN, and another from the thalamus (Tha) to the AMG to the mPFC.74 Notably, the axonal iron transport pathway of vHip-mPFC modulated anxiety-related behaviors in the brain, while the Tha-AMG-mPFC pathway did not. All these pieces of evidence support the hypothesis that dysregulated axonal iron transport can lead to abnormal distribution of iron among different brain regions, thereby causing disease-related symptoms.75
Iron metabolism in glial cells
Physiological iron levels are not uniform among different types of cells in the brain. In the neocortex, the iron concentration, which was analyzed using a nuclear microprobe and scanning proton-induced X-ray emission spectrometry, is fivefold higher in the oligodendrocytes, threefold higher in the microglia, and twofold higher in the astrocyte than that in the neurons, indicating that glial cells are the most iron-rich cells in the brain.76 Therefore, glial cells are considered to be responsible for maintaining iron homeostasis and providing buffering and protection in the brain.76,77,78
As the most abundant glial cells in the CNS, astrocytes perform various crucial functions, including structural support, neurotransmitter transport, injury repair, and inflammation response modulation.79 Astrocytes express almost all proteins related to iron metabolism; however, they appear to have a lower metabolic demand for iron but can efficiently store it in ferritin.80 Most importantly, once iron crosses the BBB, astrocytes are able to absorb it and then mediate its distribution directly to neurons. Therefore, astrocytes are ideally positioned for iron absorption, distribution, and metabolism.81 Hepcidin is an iron-regulatory hormone with widespread distribution in the brain, and its expression in astrocytes has been indicated to regulate iron transport across the BBB and play an essential role in controlling the overall brain iron level. Overexpression of hepcidin in the astrocytes was found to decrease brain iron load, possibly by regulating the FPN1 on the brain microvascular endothelial cells.61,82 In the primary cultured astrocytes, hepcidin was also found to regulate iron-related proteins, including TfR1, DMT1, and FPN1, and control iron import. Importantly, hepcidin directly inhibited the expression of TfR1 through a cyclic AMP-protein kinase Α dependent manner.83 All these evidences implicate a potential neuroprotective role of astrocyte hepcidin in maintaining iron homeostasis.84 Astrocytes can release ferritin. In our recent studies, the process of ferritin release by astrocytes was found to be enhanced by iron overload to buffer extracellular iron.85 Notably, the ferritin released by astrocytes can enter and protect dopaminergic neurons by inhibiting the increase of the labile iron pool (LIP) and reducing reactive oxygen species (ROS).85 Furthermore, we have reported that ferritin is secreted through transient receptor potential channel 1(TRPML1, mucolipin subfamily)-mediated exocytosis in primary cultured astrocytes.86 This secretion can be enhanced by iron treatment and inhibited by autophagy inhibitors 3-MA or chloroquine.
Microglia are the major immune cells in the CNS, constantly moving and removing pathogens and damaged cells, thus protecting neurons from damage. However, when microglia are activated, they can release pro-inflammatory factors that promote neuroinflammation and trigger neuronal damage. Microglia also function as the most efficient iron-absorbing glia cell, which plays an essential role in iron homeostasis.87,88 In a tri-culture system consisting of astrocytes, microglia, and neurons from both primary cultured cells and human-induced pluripotent stem cells, microglia were observed to be highly responsive to iron and more susceptible to ferroptosis compared to astrocyte and neurons, which may be due to the different regulation of iron metabolism and the ability to handle iron.89,90 The cells reveals more resistance to ferroptosis in tri-culture system compared to that in the monoculture.89 Through genome-wide CRISPR screening, SEC24B was identified as a regulator of ferroptosis in microglia in addition to ACSL4, and knockout cells lacking SEC24B showed high resistance to ferroptosis.90 Although ferritinophagy, a selective degradation of ferritin through autophagy, has been implicated in promoting ferroptosis, and SEC24B has been indicated to regulate ferroptosis by altering the labile iron pool rather than affecting ferritinophagy, it is also involved in autophagosome formation by binding SEC24A and SEC23B in response to starvation.90 Specifically, only activated microglia are capable of synthesizing lactoferrin (Lf).91 Although Lf was observed in both human nigral dopaminergic neurons and microglia, only the activated microglia contained the messenger of Lf.91 The release of Lf by activated microglia could be enhanced under the treatment with tumor necrosis factor alpha (TNF-α), 1-methyl-4-phenylpyridinium (MPP+) or iron overload.91,92 Lf could bind with the lactoferrin receptor (LfR) and enter into the nigral dopaminergic neurons through an endocytosis mechanism. In the cellular, there are two forms of Lf: iron-free Lf (apo-Lf) and iron-saturated Lf (holo-Lf). In addition to chelating cellular iron by apo-Lf, both apo-Lf and holo-Lf exhibit neuroprotective benefits by improving Cu/Zn-superoxide dismutase activity, enhancing the mitochondrial transmembrane potential, and increasing the level of Bcl-2.92
Oligodendrocytes, which play a critical role in myelination and iron-dependent metabolic enzyme activities, have been found to harbor the highest concentration of iron within the CNS. Cell type-specific iron detection using micro particle-induced X-ray emission (µPIXE) coupled with nickel-enhanced immunocytochemical methods has confirmed that both oligodendrocytes and astrocytes hold the highest level of iron in the SN of non-neurodegenerative control individuals.93 Ferritin heavy chain is considered as the major source of iron in oligodendrocytes, which can bind to its receptor, Tim-1(in human) or Tim-2 (in mice), on the membrane of oligodendrocytes and then be imported into cytosol through clathrin-dependent endocytosis.94,95,96 In mice, oligodendrocytes have been identified as expressing a high level of ferritin heavy chain, which can also secrete ferritin heavy chain through extracellular vesicles. Once the secretion or expression of ferritin heavy chain were disrupted in oligodendrocytes, it would cause oxidative damage and neuronal loss, suggesting an antioxidant effect of oligodendrocytes.97 DMT1 has been identified as necessary for iron import and development in oligodendrocyte progenitor cells.98 Although TfR1 has been found on the membrane of cultured oligodendrocytes, both TfR1 and DMT1 were absent in oligodendrocytes in the adult brain of mice and rats, indicating that the Tf-TfR1 system and DMT1 may only participate in iron import during the immature age of oligodendrocytes.80,99,100 The iron export of oligodendrocytes is also facilitated by FPN1, with the assistance of the ferroxidase hephaestin.67,101 Although the level and requirement of iron are higher in oligodendrocytes, they are also susceptible to oxidative stress.102,103 It has been found that mobilization of iron from ferritin through copper chelation in oligodendrocytes induces demyelination and leads to loss of oligodendrocytes through ferroptosis.103
Cellular iron metabolism in neurons
Neurons are most vulnerable to iron dysregulation in the CNS. As we summarized before,104,105 metabolism of iron in neurons includes the uptake, storage, and export. Briefly, the Tf-TfR system mediates the uptake of ferric iron and DMT1 mediates the uptake of ferrous iron are two major pathways for neuronal iron influx. Ferritin serves as the primary cellular iron storage protein, which can store excess cytoplasmic iron or release it for the synthesis of iron-containing structures. FPN1 is currently known as the only responsible protein for exporting ferrous iron.106 With the assistance of ferroxidases, such as Cp, hephaestin, and APP, ferrous iron exported by FPN1 is oxidized to ferric iron and recycled by the Tf-TfR system. As we mentioned previously,104 iron regulatory proteins (IRPs) can post-transcriptionally regulate the mRNAs of iron-related proteins (including TfR1, DMT1, ferritin, and FPN1) that contain IREs in the 3′-or 5′-UTRs, and then maintain the cellular iron homeostasis. Lysosomes are critical organelles for intracellular iron storage and have gained increasing recognition.105,107 As we summarized previously, lysosomal iron mainly comes from two sources: the degradation of iron-containing substrates through the autophagy-lysosome pathway and endocytosis.105 In the acidic and reducing environment of the lysosome, ferric iron is reduced to ferrous iron and then released into cytosolic through DMT1, TRPML1, natural resistance-associated macrophage protein 1 (Nramp1), or two-pore channels (TPCNs).105 DMT1, which is localized in the early or late endosome, is mainly responsible for the release of lysosomal iron from Tf-TfR1 recycling. When the lysosomal iron comes from the Tf-Fe2 complex or iron-containing cargos, they will be released by TRPML1, which is localized on the late endosome and lysosome membranes, or Nramp1. The selective degradation of ferritin through autophagy-lysosome pathway is known as ferritinophagy, which serves as the exclusive identified mechanism for releasing iron bound to ferritin. Transcription factor EB (TFEB) acts as a master regulator of both lysosomal biogenesis and autophagy. Our recent study has reported that overexpression of TFEB can upregulate TfR1 synthesis through the FBXL5-IRP2 pathway and increase the localization of TfR1 in lysosomes, which facilitates lysosomal iron import and transient lysosomal iron storage.108 TRPML1 also serves as a lysosomal calcium release channel, which process is involved in the clearance of α-synuclein. In cultured cells expressing TRPML1/2, iron overload triggers an increase in cytosolic ferrous iron and cytotoxicity, and overexpression of TFEB increases the number of iron-positive endolysosomes and promotes lysosomal exocytosis, a process that depends on TRPML1/2 mediated calcium release and can rescue apoptosis induced by iron overload.109,110 Mitochondria are another important organelle involved in iron metabolism. Under normal conditions, voltage-dependent anion channel (VDAC), Tf-TfR2, and DMT1, all located on the outer mitochondrial membrane, are responsible for the iron cross outer mitochondrial membrane. The transport of iron across the inner mitochondrial membrane is mediated by mitoferrin 1/2 (Mfrn1/2). In the mitochondrial matrix, iron can be utilized for the biogenesis of Fe/S cluster and heme or stored in mitochondrial ferritin (FtMt). The transport of both Fe/s cluster and iron from mitochondria into cytosol is mediated by the ATP-binding cassette subfamily B member 7 (ABCB7/8).111
Other metal ions
Manganese
As a crucial micronutrient, manganese functions as an essential cofactor for various proteins, especially for manganese metalloenzymes, such as arginase, pyruvate carboxylase, glutamate synthetase and manganese superoxide dismutase (MnSOD, also called SOD2), and plays a significant role in maintaining the normal physiological function of CNS.112 Glutamine synthetase is considered the most prominent Mn-rich protein, predominantly expressed in astrocytes and catalyzing the conversion of glutamate to glutamine.113 And pyruvate carboxylase is necessary for interacting with manganese to produce oxaloacetate, which then undergoes the TCA cycle.114 In the reactive catalytic center, SOD2 can prevent cells from oxidative stress by mitigating the generation of ROS within mitochondria. Additionally, manganese has the capacity to activate ATM protein kinase and tumor suppressor p53, which regulate cell cycles and reduce DNA damage.115 To achieve a delicate balance between its indispensability and neurotoxic implications, the uptake of manganese in neurons and glial cells is rigorously regulated, including both receptor-mediated endocytosis and non-transferrin-mediated uptake. Trivalent manganese can bind with Tf, however, the transfer velocity is relatively slow. Once manganese is internalized through TfR, it is converted into its divalent form and subsequently transported to the cytosol via DMT1. After absorption, manganese primarily accumulates in the basal ganglia region, particularly in the striatum and SN. The majority of manganese binds to manganese metalloproteins, particularly glutamine synthetase within astrocytes. manganese was also found adjacent to the nucleus of dopaminergic neurons in SN.116 SLC30A10 is a cell surface protein involved in the efflux of manganese, and deficiency in manganese efflux transporter SLC30A10 causes ~20–60-fold higher level of manganese level in the brain, indicating a protection role of SLC30A10 against neurotoxicity.117,118 Patients with a mutation in SLC30A10 exhibit significantly elevated levels of manganese in their blood and all of them present with Dystonia or Parkinsonism. However, an effective treatment option is oral iron supplementation, which may enhance the competition between iron and manganese for transporters SLC30A10.119 The iron exporter FPN is also involved in the efflux of manganese, which could reduce manganese cytotoxicity and accumulation.120 However, this process can be inhibited by low extracellular pH and high K(+) in the medium.121
Copper
The brain has the second highest amount of copper (Cu), which is essential for respiratory functions and defense itself by generating radicals, as well as generating neuroendocrine peptides and hormones. Additionally, due to the variable oxidation form of Cu+ and Cu2+, copper also serves as a cofactor for several enzymes, such as copper/zinc superoxide dismutase (CnZnSOD, also called SOD1), Cp, and so on. SOD1 serves as the first protector against damage from reactive oxygen species (ROS) and superoxide anion (O2•−) radicals in both the cytosol and mitochondrial intermembrane space.122 Cp belongs to the multi-copper oxidase family of enzymes and primarily functions as ferroxidase in vivo, which can convert toxic ferrous iron into nontoxic ferric form while playing an important role in iron homeostasis.123 Both BBB and CSF also function as barrier systems for the transport of copper and restrict the permeability of copper into the brain strictly, which has been summarized recently.124 The cellular uptake, efflux, and distribution of copper are regulated by membrane-integrated copper transporters. Copper transporter 1 (CTR1) is the primary mechanism for copper uptake, which occurs independently of ATP hydrolysis and can be stimulated by acidic extracellular pH and elevated concentrations of K+ ions.125,126 CTR1 heterozygous and homozygous deletion mice exhibit defects in the activities of copper-dependent enzymes, as well as growth and development.127 In mice with knockdown of CTR2, significant copper accumulation has been observed, which may depend on the cleavage function of CTR2 to CTR1.128 This is because a truncated form of CTR1 protein can facilitate the export of copper from the endosome. DMT1 also participates in the transportation of bivalent copper, and there is a competitive relationship between the uptake of copper and iron by DMT1. In cells with knockdown of DMT1, increased uptake of Cu1+ and reduced uptake of Cu2+ were observed.129 Meanwhile, Cu1+ inhibits the transport of Fe2+, while Fe2+ can inhibit the uptake of Cu1+. The ATPase proteins ATP7A and ATP7B play an important role in copper export mediated by copper chaperone antioxidant protein 1 (Atox1).130 The absence of ATP7B could cause hepatic copper accumulation and subsequently increase the concentration of copper in the brain.131,132,133
Under normal conditions, cells contain extremely low levels of free copper due to the sequestration by GSH, storage in metallothioneins complex, and transportation by copper chaperones such as Atox1 and copper chaperone for superoxide dismutase (CCS). Firstly, the most important intracellular copper chelator is GSH, which forms complexes and serves as a reservoir of copper. Cu (I)–GSH can deliver copper to metallothioneins, SOD1, and Atox1 while preventing damage to host cells caused by free copper-induced ROS. However, the Cu (I)–GSH complex is redox-active and also has the ability to generate Fe2+ that is redox-active and facilitate a superoxide-driven Fenton reaction.134 Moreover, GSH could affect the metabolism of copper. Although overexpression of GSH upregulated both the level of CTR1 and the rate of copper transport, it also decreased the bioavailable copper pool.135 In addition, GSH could also regulate the activity of both ATP7A and ATP7B.136 Metallothioneins also function as a detoxification and neuroprotective substance by binding with copper or zinc, which plays a crucial role in the safe storage of copper.137 As a copper chaperone, Atox1 functions as a carrier for copper and facilitates its transfer to the ATPase-mediated secretory pathway.130 Atox1 also regulates the activity of ATP7B through modulating domain dynamics.138 Recently, evidence has shown that Atox1 plays a pro-inflammatory role in a mouse model of intestinal inflammation, with increased levels of pro-inflammatory cytokines and regulation of the ROS-NLRP3 pathway.139 CCS, another copper chaperone serves as a transporter and activator of copper for superoxide dismutase, also functions as a target of the X-linked inhibitor of apoptosis (XIAP), which could be ubiquitinated by XIAP and enhances its chaperone activity to SOD1.140The acquisition and distribution of CCS-dependent copper were found to largely occur at membrane interfaces.141
Zinc
Zinc (Zn), the second most abundant essential transition metal, plays a fundamental role in the immune system, acts as a cofactors for enzymes, functions as intracellular second messengers, and has a neuroprotective role in the brain. The metabolism and signaling pathway of zinc have been summarized by Chen et al. recently,142 mainly focusing on peripheral and cancer. The concentration of zinc in the brain is also strictly regulated by three substances: metallothioneins, zinc transporters (ZnTs), and Zrt- and Irt-like proteins (ZIPs). In addition to copper, metallothioneins can also bind and release zinc. However, abnormal release of zinc from metallothioneins-3 could cause neuronal death in the thalamus and CA1 region of hippocampus.143 The ZnT family comprises ten distinct members (ZnT1-10), characterized by six transmembrane domains and intracellular termini of NH2 and COOH. These proteins play a distinct role in removing intracellular zinc by transporting it into various intracellular compartments and effluxing it from the cytosol. In the monkey spinal cord, ZnT1-10 can be observed in both motor and sensory neurons, but not in glial cells, indicating its potential role in motor and sensory functions.144 ZnT1 is responsible for transporting zinc out of the neurons,145,146 and also functions as a neuronal Zn2+/Ca2+ exchanger, playing an important role in neuronal signaling.147 ZnT3 is an indispensable component of the activity-dependent release of zinc from neurons, residing on synaptic vesicles and serving as the sole pathway for transporting zinc into these vesicles. Mice lacking ZnT3 exhibit increased vulnerability to epileptic episodes and cognitive loss, indicating the potential role of ZnT3 in regulating changes in zinc levels.148,149,150 ZIP family consists of four subfamilies, namely ZIP I, ZIP II, gufA, and the LIV-1 subfamily. These subfamilies are responsible for importing zinc from the extracellular into the cytoplasm. FPN1 is also involved in zinc transport, and zinc can induce binding between transcription factor-1 (MTF-1) and the FPN1 promoter, thereby affecting its expression.151
Crosstalk and downstream effectors of iron and other metal ions
The most common downstream effector of metal ions accumulation is oxidative stress (Fig. 2). Both ferric iron and copper can promote the process of Fenton reaction, resulting in generation of damaging ROS and initiating oxidative stress, which leads to damage cell membrane, proteins, as well as nuclei acids.152 Due to the similar structure between iron and manganese, there is a competitive relationship between iron and manganese in DMT1. After treating with 500 µM manganese in Caco2 cells, a conspicuously reduction in the absorption of iron by DMT1 was observed.153 Therefore, iron significantly impacts manganese homeostasis. Expanding into clinical application, oral iron therapy could attenuate symptoms of excessive manganese through competition for transporters.119 On the other hand, exposure to manganese translational inhibited the 5′-untranslated regions of H-ferritin and APP in a dose- and time-dependent manner, thereby resulting in iron accumulation and neurotoxic oxidative stress.154 The exposure to a high level of manganese also induces oxidative stress by oxidizing dopamine or interfering with normal mitochondrial respiration.155 Copper can also engage in redox reactions, which is a double-faced sword. The advantage lies in its participation in biochemical reactions through its redox activity. On the contrary, excessive copper can trigger the production of free radicals through the Fenton and Haber–Weiss reactions, leading to oxidative stress and neuronal death.156 Although there is no direct connection between zinc and ROS, a complex composed of zinc, ROS, and protein thiols has been linked zinc to the redox-signaling pathway.157 Additionally, zinc has been found to reduce the overproduction of ROS and improve oxidative stress.158

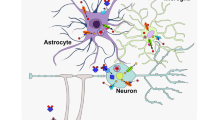
Downstream effectors of iron and other metal ions. a The import of iron mainly depends on TfR1-mediated endocytosis or DMT1, while FPN1 is the sole known exporter of iron. The import of copper mainly depends on CTR1, which can be upregulated by Cu(I)-GSH. DMT1 also participates in the import of copper. The export of copper is mediated by ATP7A/7B with the assistance of Atox1. As a copper-binding protein, Cp also functions as a ferroxidase to convert toxic ferrous iron into nontoxic ferric iron. Excessive copper leads to the aggregation of lipoylated proteins and the loss of iron-sulfur cluster proteins, resulting in cuproptosis. The import of manganese relies on the DAT, ZIP8, Tf/TfR system, or DMT1, while the export of manganese depends on SLC30A70 and FPN1. Manganese can activate ATM/ p53, which regulates cell cycles and reduces DNA damage. In the mitochondria, Mn2+ can bind to intermediate products of the TCA cycle and promote the generation of ROS, while Mn3+ can help SOD2 to mitigate the generation of ROS and prevent cells from undergoing apoptosis. ZIPs are the main channels for transporting zinc into the cytoplasm from extracellular or ER, while ZnTs are responsible for transporting zinc out of the cytoplasm or into synaptic vesicles, lysosomes, and Golgi. Both ferrous iron and copper can promote the Fenton reaction, leading to the generation of ROS and initiating oxidative stress. However, zinc can compete with copper or iron, thereby preventing the generation of ROS. Both excessive Fe2+ and Mn2+ iron can lead to the accumulation of lipid peroxidation and trigger ferroptosis, which can be inhibited by copper chelators. b High levels of metal ions, including iron, manganese, copper, and zinc have been found to be involved in cell senescence. This process can be rescued by the iron chelator DFO. c Excessive metal ions such as iron, manganese, copper, and zinc can also activate microglia and astrocytes to release pro-inflammatory cytokines, thereby triggering neuroinflammation. This figure was created with BioRender.com/j25f189
As an iron-dependent cell death form, ferroptosis is first described in 2012 and characterized by iron accumulation, depletion of GSH, and lipid peroxidation.159 Recently, the signaling pathways and involvement of iron in ferroptosis has been detailed summarized by Carsten Berndt et al.160 The selective degradation of ferritin through autophagy-lysosome pathway is known as ferritinophagy, which is mediated by the selective cargo receptor NCOA4.161 As ferritinophagy could release iron from ferritin, it has been reported that autophagy promotes the process of ferroptosis, which is also known as an autophagy-dependent cell death form.162,163 However, autophagy has two sides, and self-protective autophagy has been demonstrated to be beneficial for neurodegenerative diseases. Iron overload can disrupt the fusion of autophagosomes and lysosomes by decreasing Rab7, which causes autophagosome accumulation and blocks autophagic flux in microglia.164 In the L6 skeletal muscle cells, iron overload promotes the production of ROS, inhibits the self-protective autophagy, ultimately leading to apoptotic cell death.165 Our recent study has reported that TFEB-mediated autophagy maintains cellular labile iron at a low level and prevents ferroptosis in a TfR1-dependent manner.108 In addition, cell density can modulate iron regulatory protein 1 (IRP1) to affect the levels of FPN and TfR1, resulting in changes in cellular iron levels that are critical for susceptibility to ferroptosis.166 Zinc is also involved in ferroptosis, and zinc addition can promote ferroptosis even when iron chelation is present.167 With genome-wide RNAi screening, SLC39A7 (ZIP7), which is responsible for transporting zinc from the endoplasmic reticulum (ER) to the cytosol, has been identified as a genetic determinant of ferroptosis.167 In the vascular endothelial cells, zinc oxide nanoparticles induce ferroptosis through NCOA4-mediated ferritinophagy.168 Recently, mitophagy was also found to be involved in zinc-induced ferroptosis in porcine testis cells.169 Manganese was found to induce ferroptosis by inhibiting the mitochondrial dihydroorotate dehydrogenase in tumor cells.170 Both in vivo and in vitro results support that exposure to manganese causes increases in lipid peroxidation, ferrous iron, as well as ROS, which triggers ferroptosis and neurotoxicity.171 Additionally, manganese-induced ferroptosis in dopaminergic neurons is also mediated through the pathway of HIF-1α/p53/SLC7A11.171
Described for the first time in 2022, cuproptosis is a novel form of regulated cell death that is distinct from known cell death mechanisms and relies on copper and mitochondrial respiration.44,45 During the process of cuproptosis, copper directly binds to lipoylated components of the tricarboxylic acid (TCA) cycle, resulting in aggregation of lipoylated proteins and loss of iron-sulfur cluster proteins.44 Copper exposure was also found to cause cognitive impairment in mice, which was related to the modulation of cuproptosis, damaged synaptic plasticity, and inhibition of the CREB/BDNF pathway.172 Copper has been found to bind with GPX4 at the cysteines C107 and C148 and promote the degradation of GPX4 through Tax1 binding protein 1 mediated autophagy, thereby driving ferroptosis.173 Additionally, copper chelators tetrathiomolybdate and tetraethylenepentamine could block ferroptosis induced by erastin but not other types of cell death.173 Furthermore, there is a profound interplay between iron and copper that can influence their transport under abnormal concentrations. In an iron-deficient rat model, a significant increase (+55%) in the level of copper in the CSF and brain parenchyma is observed. Under the ventriculo-cisternal perfusion, the clearance of copper from the CSF is remarkably augmented in iron-deficient rats. This may be attributed to an upregulation of DMT1 expression due to the deficiency of iron rather than CTR1.174
Cell senescence, a process characterized by gradual declines in cell proliferation, differentiation, or physiological function, was first proposed by Hayflick and Moorhead in 1961.175 The main characteristics of cell senescence include (1) increased activity of senescence-related β-galactosidase (SA-β-gal), lipofuscin accumulation caused by lysosomal dysfunction, and mitochondrial dysfunction; (2) activation of senescence-related signal pathways in p53-p21-pRB or p16-pRB; (3) the appearance of senescence-related secretory phenotype; (4) macromolecular damages in DNA, protein or lipid, and so on. Although cell senescence is a mechanism of mitotic cell cycle arrest, it also occurs in post-mitotic cells, such as terminally differentiated neurons.176 As a hallmark of aging, cell senescence occurs in many types of cells in the CNS, including neurons, astrocytes, microglia, oligodendrocytes, and neural stem cells.177,178 Recently, iron, whether in its free form or released by lysed erythrocytes, has been found to induce ROS-mediated cell senescence.179 Furthermore, iron accumulation in the senescent cells could drive the development of senescence-related secretory phenotype. Additionally, iron overload induces cell senescence in both the brain vasculature and brain tissue itself, which phenomenon is associated with the downregulation of Robo4 in the cerebral endothelial cells derived from aged female mice.180 Iron overload resulting from NCOA4-mediated ferritinophagy could cause mitochondrial dysfunction and trigger mitochondrial DNA release, leading to cell senescence through the cGAS-STING pathway.181 Meanwhile, the iron chelator DFO could significantly rescue retinal pigment epithelial senescence induced by ferric ammonium citrate or D-galactose in mice.181 Notably, iron accumulation in senescent cells has been found to be coupled with impaired ferritinophagy and inhibition of ferroptosis.182 Elevated copper was also observed in senescent MEF cells, which may be caused by an increase in CTR1 and a decrease in ATP7A accompanied by enhanced antioxidant defense.183
In addition, neuroinflammation also acts as a downstream effector of metal ions, mainly mediated by the activation of microglia or astrocytes in the CNS, which promotes the release of pro-inflammatory cytokines. As microglia are the most efficient iron-absorbing glial cells in the CNS, it is well known that iron can activate microglia and promote the secretion of pro-inflammatory cytokines, resulting in neuroinflammation. Other metal ions, such as manganese, copper, and zinc, also exert a regulatory effect on neuroinflammation. Exposure to copper increases the microglial secretion of pro-inflammatory cytokines, including IL-1β, TNF-α, and IL-6, thereby elevating neuroinflammation both in vitro and in vivo.184 Copper-induced neuroinflammation is mediated through the ROS/NF-κB pathway and autophagy impairment.185 Although zinc, in a state of homeostasis, inhibits microglia-mediated neuroinflammation, both zinc depletion and zinc accumulation can promote neuroinflammation, which has been detailed and summarized.186 Manganese exposure induces neuroinflammation not only in microglia,187 but also in astrocytes through impairing mitochondrial dynamics.188 Manganese dose-dependently increases the levels of pro-inflammatory cytokines and chemokines, such as IL-6, TNF, CCL2, and CCL5, in microglia, and the pro-inflammatory cytokines released by microglia could dramatically enhance the mRNA levels of TNF, IL-1β, IL-6 in astrocytes, while inhibiting the NF-κB pathway in the microglia could block microglial-induced astrocyte activation.187 This indicates that manganese induces inflammatory responses in microglia, which amplifies the inflammatory activation of astrocytes through the NF-κB pathway. In addition, manganese could activate the cGAS-STING pathway in microglia, increase the expression of proinflammatory mediators, and induce neuroinflammation, which could be reduced by sesamol.189 Intranasal exposure to a high dose of manganese induces neuroinflammation, which is accompanied by disruptions in dopamine metabolism in both the striatum and hippocampus of rat.190 Autophagy is also involved in the manganese-induced neuroinflammation, including SIRT1/FOXO3-mediated autophagy signaling,191 glycogen synthase kinase-3β (GSK-3β) signaling,192 and NLRP2-CASP1 signaling.193 Manganese could increase the level and activity of LRRK2, a kinase that has recently been found to be involved in manganese-induced neuroinflammation in microglia.194 The activation of RAB10 by manganese-LRRK2, which is exacerbated by the LRRK2 mutation G2019S, dysregulates the microglial autophagy-lysosome pathway and NLRP3 inflammasome.194
The role and mechanism of iron and other metal ion dysregulation in neurodegenerative diseases
Brain regional redistribution of iron and other metal ions
Methods of detecting metal ions in the brain
MRI is a non-invasive and sensitive method that has been widely used to detect the distribution and very low concentration of metal ions in the brain, especially in neurodegenerative diseases.9,28 T2*-weighted MRI is a specific sequence in MRI techniques, and QSM is a sensitive MRI technique. Both T2*-weighted MRI and QSM are widely used to evaluate the levels of metal ions in the brain of individuals with neurodegenerative diseases.18,19 Furthermore, T2*-weighted magnetic resonance imaging confirmed a positive correlation between nigral iron deposition and the progression of the disease, as well as motor and cognitive dysfunction in PD patients.19 Transcranial sonography (TCS) is also employed as a non-invasive method for detecting iron deposition in the midbrain. It has been suggested that TCS and MRI parameters should be considered complementary in the detection of iron deposition in PD.195,196 Magnetic Sensitivity weighted imaging (SWI) is a special MRI technique that is highly sensitive to magnetic differences in tissues. By combining the high spatial resolution and phase information of the gradient echo sequence, SWI can detect subtle changes in magnetic susceptibility more effectively, enabling better visualization of vascular structures, bleeding, and iron deposition.197 X-ray fluorescence microscopy (XFM) is a technique used to detect and image the distribution of elements in a sample, specifically metal ions. Due to its ability to provide high spatial resolution information about the types and concentrations of elements in a sample, XFM has been employed for detecting the distribution of metal ions in the brain, which is crucial for understanding their role in neurodegenerative diseases.198 Furthermore, the combination of synchrotron X-ray fluorescence (SXRF) microprobe and synchrotron Fourier transform infrared micro-spectroscopy (FTIRM) allows for the assessment of metal ions co-localization with aggregated proteins.43
Mass spectrometry (MS) is a technique that can be used for the quantitative analysis of metal ions and the detection of their concentration in biological samples, such as CSF and brain tissue. ICP-MS is a technique developed based on MS, which has high sensitivity and a wide dynamic range. It is capable of detecting elements from ultra-trace to major levels. ICP-MS operates by introducing the sample into an inductively coupled plasma, ionizing the elements in the sample into ions, which are then separated and detected by MS.3,4,5 Laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) combines laser ablation technology with ICP-MS. In this technique, a laser beam is used to remove tiny materials from the surface of a solid sample, and the resulting aerosols are directly transmitted to the ICP source for ionization and analysis. LA-ICP-MS enables in situ microzone analysis of solid samples, allowing for element and isotope imaging as well as quantitative analysis at the micron or even nanoscale.199 Atomic absorption spectroscopy (AAS) is another technique used to detect the concentration of metal ions. It is based on the principle that atoms absorb specific wavelengths of light and can be utilized for quantifying the metal ions present in a brain sample.200,201,202 Inductively coupled plasma atomic emission spectrometry (ICP-AES) has been used to detect the levels of iron in the blood and serum,.203
Histopathological methods, such as Perls’ and Turnbull staining,1 allow for the observation of metal ion deposition in postmortem brain tissue through microscopic examination of tissue sections. Recently, fluorescent turn-on sensors based on DNAzymes have been developed that are selective for either ferrous or ferric iron and enable the monitoring of different redox states of iron in living cells.34
Brain regional metal ions redistribution in PD
ICP-MS results have shown that although there were a significant increase in iron level and a decrease in ferritin level in the CSF samples from PD patients (Table 1), the ratio of iron/ferritin was significantly increased, indicating that iron-ferritin ratio in the CSF may serve as a potential progression marker for PD.20 In a postmortem study, significantly increased permeability of BBB has been confirmed in the striatum of patients through several methods, including erythrocyte extravasation, perivascular hemosiderin, and leakage of various serum proteins outside UEA-staining vessel walls.204 In the SN of 6-hydroxydopamine (6-OHDA) induced PD rat model, increased permeability of BBB was observed by both gadolinium-enhanced MRI and immunohistochemistry after injection of 6-OHDA into the medial forebrain bundle for 2 days.205 However, this increased permeability was restored after 1 week of injection. At the same time, decreased immunoreactivity of tyrosine hydroxylase was observed in the SN of 6-OHDA rats at both 2 days and 4 weeks, while iron deposition was observed at 1 and 4 weeks,205 suggesting that alteration of BBB might contribute to nigral iron deposition in the PD rat model. In the high-iron and PD mice models, knockout of the transcription factor NF-E2-related factor 2 (Nrf2) prevented iron deposition in the SN and striatum.206 The mechanism is likely achieved by decreasing the level of FPN1 on microvascular endothelial cells, which hinders the process of iron entry into the brain.
The iron distribution in the brain regions is heterogeneous, and selective iron accumulation occurs in several brain regions with aging, such as the SN, caudate putamen, and GP. However, the degree of iron accumulation is particularly severe in the corresponding brain regions of patients with PD. With the development of imaging technology, an increased number of brain regions have been identified to contain abnormal iron levels in PD patients or PD animal models. Furthermore, abnormal accumulation of iron in specific brain regions may not only contribute to motor dysfunction, but also be associated with the non-motor symptoms in PD. In addition to a significant increase of nigral iron levels in all Hoehn and Yahr (H&Y) stages of PD patients without significant difference within stages, compared to healthy, age-matched controls, there was also an observed increase in iron level QSM in the red nucleus in stage II and combined stages III and IV, whereas no significant change of iron levels in caudate putamen, and GP between all stages of PD and controls.207 However, another report employing susceptibility-weighted imaging (SWI) has revealed iron depositions in the putamen and GP in idiopathic PD patients, suggesting an association with mitochondrial impairment.197 Accumulation of misfolded α-synuclein is one of the most important factors in the pathological development of PD. Intranasal administration of human α-synuclein preformed fibrils (PFFs) was found to cause iron deposition in the SN and GP in a time-dependent manner from 1 to 17 months in the male Macaca fascicularis.208 Susceptibility MRI data has reported dynamics changes of nigral iron in PD, which are lower before dopaminergic medication and then increase throughout the disease, eventually plateauing at the late stages.209 In drug-naive PD patients, lower iron levels were identified in the SN, caudate nucleus, and GP compared to controls, but not in the red nucleus or putamen, however, higher nigral iron were found in drug-treated PD patients compared to controls or drug-naive PD patients.209 Notably, PD medications may result in differential association with nigral iron deposition. Higher nigral iron was found to be associated with levodopa usage, while lower nigral iron was correlated with selegiline usage.209
Anxiety is a common neuropsychiatric manifestation of PD, and the prevalence of anxiety disorders in PD is higher than that in other chronic neurodegenerative diseases.210,211 Anxiety and fear are considered to share the fear circuit, which is composed of the amygdala (AMG), medial prefrontal cortex (mPFC), anterior cingulate cortex, hippocampus, insula, and striatum.212 QSM data has revealed that, compared to the healthy controls, increased brain iron accumulation was observed in the fear circuit (including mPFC and anterior cingulate cortex), supplementary motor area, precuneus, angular gyrus, and middle occipital gyrus of PD patients with anxiety, however, increased brain iron accumulation was observed in the parahippocampal gyrus and superior temporal gyrus of PD patients without anxiety.213 Specifically, compared to PD patients without anxiety, significant iron deposition was observed in the hippocampus of PD patients with anxiety. Pain is another common non-motor symptom of PD, however, it is still unclear whether iron is involved in PD-related pain. A recent clinical report has found increased iron accumulation in the putamen, caudate, and nucleus accumbens (NAC) of migraineurs compared to the controls, and the degree of iron deposition in NAC could be employed to distinguish the patients with chronic migraine from episodic migraine,214 indicating that abnormal iron accumulation in related nuclei may account for the pain of PD. In addition, QSM data has shown increased iron content in the prefrontal cortex and putamen (p < 0.05 corrected for multiple comparisons) of individuals with PD compared to controls.215 Additionally, within the PD group, higher levels of iron were associated with (1) lower cognitive performance in the hippocampus and thalamus; (2) poorer visual function and higher dementia risk scores in parietal, frontal, and medial occipital cortices; (3) worse motor performance in the putamen. Furthermore, compared to the healthy controls, increased magnetic susceptibility was observed in the frontal, posterior parietal, and insular cortices of PD patients when analyzed by QSM, whereas slightly decreased susceptibility values were observed in the occipital cortex of PD patients.216 In addition to iron accumulation in the aforementioned brain regions, our group has previously reported decreased levels of iron in the temporal cortex of postmortem PD patients brains compared to the age-matched healthy controls, which were accompanied by decreased levels of iron-related proteins, including DMT1 (+IRE), TfR1, FPN1, and IRP1.217 Conversely, no significant changes in iron levels were observed in the temporal cortex of AD patients. These findings suggest that abnormal distribution of iron may exist in different brain regions in PD and contribute to PD-related symptoms. In addition to the imaging methods mentioned above, transcranial sonography (TCS) is also employed as a non-invasive method for detecting iron deposition in the midbrain. It has been suggested that TCS and MRI parameters should be considered complementary in the detection of iron deposition in PD.195,196
Brain regional metal ions redistribution in AD
The redistribution of iron also exists in the brain of AD patients. Several brain regions associated with the pathogenesis of AD (Table 1), such as the cerebral cortex, hippocampus, and basal ganglia, exhibit higher levels of iron in AD patients.28,218,219,220 Within the CA1 region of the hippocampus, iron deposition was observed in the stratum molecular-radial and stratum oriens of AD patients, as indicated by LA-ICP-MS results.199 However, a reduced iron concentration was observed in the GP of AD patients compared to that in age-matched control participants, which differed from the results in PD patients.221 Additionally, higher levels of iron were also observed in the neocortical regions, deep gray matter, and putamen of AD patients compared to healthy controls.222,223 Due to the complexity of the cerebral cortex, there was a diverse iron content in AD patients. In comparison to the controls, there were significantly higher correlations between iron concentrations and Aβ plaques as well as tau pathology in the frontal cortex and temporal cortex of AD group,224,225,226 while iron levels remained unchanged in the cingulate cortex, parietal cortex, and entorhinal cortex of AD patients.226,227 Importantly, iron deposition in the temporal lobe was closely associated with cognitive decline in AD patients.222 It was a common phenomenon that there were significant spatial differences in the distribution of cortical iron in AD patients, which may indicate a redistribution of iron within the cerebral cortex.224,225,228 QSM results has shown that cortical iron accumulation is associated with both cognitive decline and cerebral atrophy in AD.229 Both ICP-MS and atomic absorption spectrometry (AAS) results showed no significant changes in the level of iron in the CSF of AD patients.200,201,202 However, the iron level in the blood of AD patients varied depending on the methods used. Inductively coupled plasma atomic emission spectrometry (ICP-AES) results showed that serum iron levels were higher in control subjects compared to patients with AD,203 which was consistent with the ICP-MS results.230,231,232 AAS results indicated no significant change in serum iron content between the control group and AD patients.202,233,234 However, higher serum iron levels were also reported in AD patients.235,236
Brain regional metal ions redistribution in ALS
As a fatal neurodegenerative disease, ALS is characterized by the loss of motor neurons and muscular atrophy.237 Significant iron deposition was observed in the left precentral gyrus and the thalamus, with iron deposition in the thalamus being associated with disease severity in ALS patients (Table 1).47 QSM results revealed increased iron levels in the motor cortex, SN, GP, and red nucleus, while decreased iron levels were observed in the white matter of corticospinal tract in ALS patients.238 Elevated levels of serum ferritin was also found in ALS patients.239 Meta-analysis revealed that ALS patients had lower total iron-binding capacity, and elevated serum ferritin levels were associated with reduced survival as indicated by pooled hazard ratios.240 Increased levels of iron and copper were also observed in the blood ALS patients, and disease severity was positively correlated with levels of copper, calcium, cadmium, and lead.241 In the cortical region, which is first affected in the ALS, although significantly increased iron was observed in ALS patients by ultra-high field (7 T) MRI, calcium was selectively accumulated at the low myelin borders, indicating the role of calcium in monitoring demyelination in ALS patients.242 Increased levels of iron, manganese, copper and zinc were detected in the CSF of ALS patients with a disease duration less than 19 months using ICP-MS.243 Furthermore, higher levels of copper, iron, manganese, zinc were observed in the CSF of ALS with spinal onset compared to those with bulbar onset.243 Ion chromatography-inductively coupled plasma mass spectrometry has also revealed a potential positive correlation between increased copper in the CSF and genetic ALS.244 However, another set of ICP-MS results showed lower levels of copper in the CSF of ALS patients.245 Additionally, elevated levels of copper, iron and zinc were observed in the muscle tissue of SOD1G93A mice.246 Although an increased expression level of Cp was observed in the CSF of ALS patients, the ferroxidase activity of Cp was comparable between ALS patients and controls, indicating impaired function of Cp in ALS.247 With high-resolution ICP-MS, significantly higher levels of metals, including manganese, copper, zinc et al., were observed in the CSF of ALS patients, and the levels of these metal ions were also found to be higher in the CSF of ALS patients than in their blood,248 which may be attributed to the disruption of BBB permeability.249
The spinal cords of ALS patients were also observed to have increased levels of manganese through radiochemical neutron activation analysis.250 Metabolomic analysis of the CSF revealed that copper and manganese were the most significant redox metals for ALS patients.251 Increased levels of zinc and decreased levels of magnesium were observed in the brain of ALS transgenic SOD1G93A mice, particularly in the motor cortex, the prelimbic, and infralimbic areas of the frontal cortex, and nucleus of the vertical limb of the diagonal band.252 After analyzing the metal levels in the erythrocyte using ICP-MS, it was found that zinc was associated with a decreased risk of ALS, while cadmium and lead were associated with an increased risk of ALS.50 Increased levels of zinc were also observed in the white matter of several mutant SOD1 mice.253 Dysregulation and exposure to metal ions, including manganese and zinc, among others, in early life have been shown to contribute to ALS.252 A recent ecological study conducted in the province of Ferrara, northern Italy found a strong and direct correlation between ALS density and copper concentrations in air pollutants, which correlation was higher in the urban sector, particularly among women in the overall population and urban population, indicating a potential toxic effect of copper on ALS.254
Brain regional metal ions redistribution in HD
As an autosomal-dominant neurological condition, HD is characterized by a gradual deterioration of psychiatric, cognitive, and motor functions. The pathogenesis of HD is caused by an abnormal expansion of the polyglutamine repeat sequence at the N-terminus of the mutant Huntingtin (HTT) protein, which ultimately leads to atrophy in key brain regions, particularly the striatum and cerebral cortex.255 After analyzing frozen postmortem brain tissue using inductively coupled plasma spectroscopy, increased levels of total iron were observed in the striatum (putamen and/or caudate nucleus), as well as elevated copper levels in the putamen and SN of HD patients in 1991 (Table 1).3 QSM results showed that iron accumulation in the basal ganglia, including the pallidum, putamen, and caudate of both premanifest and symptomatic HD patients, and iron accumulation in both the putamen and caudate was significantly associated with the severity of HD.46 Elevated levels of copper, manganese and zinc were observed in the CSF prior to alterations in canonical biomarkers of HD, and elevated iron was also observed in the CSF of manifest HD patients.256 Additionaly, increased levels of iron and zinc were detected in the blood of individuals with HD.257 QSM revealed increased iron concentration in the striatum and GP of patients who were closer to onset or had early HD, which were directly correlated with their HD CAG-age product score and brain atrophy.258 However, decreased iron levels in the GP were reported in the brain tissues from nine HD cases using ICP-MS.259 Meanwhile, decreased copper levels were observed in the cerebellum, while decreased manganese levels were found in the SN. Additionally, increased zinc concentrations were detected in the putamen, globus pallidus, and middle frontal gyrus by ICP-MS. Furthermore, elevated levels of Cp, a copper transport protein, were identified in the hippocampus, parietal cortex, and SN of HD cases.260
Cellular iron dysregulation
Cellular iron dysregulation in PD
Enrichment analyses for genes associated with cortical iron deposition in PD patients and expression-weighted cell-type enrichment analysis have shown that the top 20% of up-weighted genes (324 genes) related to brain iron were significantly enriched in astrocytes, glutamatergic and GABAergic neurons, as well as oligodendrocyte precursor cells, whereas the top 20% of down-weighted genes (214 genes) showed significantly increased expression in GABAergic and glutamatergic neurons.216 Although PD is also associated with an increase in NTBI concentration in the brain,261 in the cultured cells exposed to ferric ammonium citrate, astrocytes, microglia, and neurons are all able to safely store NTBI without significant changes in viability, suggesting that dysregulation of iron transport or storage may mainly contribute to iron-induced toxicity in PD (Fig. 3).87

Iron dysregulation among different cells occurs in the state of PD. The increased iron level and decreased ferritin level have been found in the CSF under the state of PD. The increased permeability of BBB facilitates the import of iron through TfR1 by microvascular endothelial cells, which is then released into the brain through FPN1. Once iron crosses the BBB, astrocytes absorb it and mediate its transfer to neurons. Astrocytes have shown a neuroprotective role in PD through the secretion of hepcidin and ferritin, which not only decreases brain iron load by regulating FPN1 on microvascular endothelial cells but also controls iron import by regulating iron-related proteins. With exposure to α-synuclein, iron, or neurotoxins, astrocytes also exhibit an iron-releasing phenotype that is detrimental to neighboring neurons. Microglia are the most susceptible neuronal cells to ferroptosis. Upon stimulation, activated microglia release proinflammatory factors, iron, as well as α-synuclein, which interact and exacerbate dopaminergic neurotoxicity. On the other hand, activated microglia can synthesize lactoferrin and protect vulnerable dopaminergic neurons. Oligodendrocytes harbor the highest concentration of iron in the CNS, which can secrete a ferritin heavy chain and protect dopaminergic neurons from oxidative stress. Abnormal import and export of iron-mediated by iron-related proteins cause iron deposition in nigral dopaminergic neurons, resulting in the loss of dopaminergic neurons in PD. This figure was created with BioRender.com/f23j195
Neuron
Selective loss of nigral dopaminergic neurons is one of the most common pathological hallmarks in PD, and increasing evidences have proven that iron deposition induced by dysregulated iron-related proteins contributes to the aforementioned process by inducing iron deposition. DMT1 is the most well-investigated iron transport involved in neuronal iron dysregulation in PD. In the substantia nigra pars compacta (SNpc) of postmortem tissue from PD patients, both increased DMT1 labeling and iron accumulation were observed in the remaining neuromelanin-positive dopaminergic neurons compared with age-matched control.262 As DMT1 labeling also appeared in activated microglia, the increased expression of DMT1 (+IRE) and iron deposition in the SNpc of postmortem tissue from PD patients may be the results of elevated levels of DMT1 in the remaining dopaminergic neurons and activated microglia.262 Furthermore, increased DMT1 in the SN has also been observed in animal models of PD, including MPTP-induced mice model,262 6-OHDA induced rat model,263 rotenone-induced mice model,264 PD mice with overexpression of human α-synuclein-A53T in the SNpc, PD transgenic mice with overexpression of human α-synuclein-A53T,265 and related PD cell models. Nedd4 family–interacting protein 1 (Ndfip1) is an adapter for the Nedd4 family of E3 ligases, which can bind to DMT1 in response to iron exposure and facilitate the degradation of DMT1 through the ubiquitin-proteasome system.266 In our previous study, decreased Ndfip1 was observed in 6-OHDA-induced PD rat and cell models, and overexpression of Ndfip1 downregulated the level of DMT1(+IRE), reduced iron accumulation, and attenuated neurotoxicity induced by 6-OHDA.267 The iron import ability of DMT1 could be enhanced by its S-nitrosylation, which may contribute to the loss of dopaminergic neurons, and S-nitrosylated DMT1 has also been observed in the SN of postmortem tissue from individuals with PD, as well as in PD mice induced by lipopolysaccharide.268 Recently, it has been found that increased DMT1 is induced by HMGB1-mediated inflammation, which contributes to dopaminergic neurodegeneration in the early stage of PD.269 Notably, mutations in DMT1, which impaired the iron transport, have shown neuroprotection for nigral dopaminergic neurons in both MPTP- and 6-OHDA-induced PD mice models.262 Also, the inhibition of S-nitrosylation of DMT1 by either the NO synthase inhibitor l-NAME or the DMT1-selective blocker ebselen was able to prevent lipopolysaccharide-induced loss of dopaminergic neuron.268 All these evidences suggest that regulating DMT1 through genic or pharmacologic methods may be an effective strategy for PD prevention or treatment. The Tf-TfR1 system is the major pathway for importing ferric iron from the extracellular environment. In cultured neurons, treatment with Tf was found to correct iron accumulation, and in the MPTP-induced PD mice model, Tf treatment ameliorated iron accumulation and improved iron deficits.270 Our recent report showed that α-synuclein-A53T and iron function as a toxic couple, inducing cell senescence in both mice and cell models of PD, which precedes the loss of nigral dopaminergic neuron.23 Additionally, reducing the iron load through DFO or knockdown of TfR1 significantly improves the phenotypes of cell senescence induced by α-synuclein-A53T (Figs. 4, 5).

Iron dysregulation in the lysosome and mitochondria of PD. The Tf-TfR system mediates the uptake of Fe3+ and DMT1 mediates the uptake of Fe2+, which are the two major pathways for the neuronal iron influx. FPN1 is responsible for exporting Fe2+ with the help of APP, Cp, or hephaestin (Hep). Endosome contains abundant Fe3+ through Tf-TfR1 uptake, while autophagosome contains ferritin bound to NCOA4, which are the main source of iron in lysosomes. In the acidic and reducing environment of the lysosome, Fe3+ is reduced to Fe2+ and released into the cytosolic through DMT1, TRPML1, Nramp1, or TPCNs. The overexpression of TFEB can upregulate the synthesis of TfR1 through the FBXL5-IRP2 pathway and increase the localization of TfR1 in lysosomes, thereby facilitating the import and temporary storage of iron in lysosomes. VDAC, Tf-TfR2, and DMT1, all of which are located on the outer mitochondrial membrane, are responsible for the iron transport across the outer mitochondrial membrane. The transport of iron across the inner mitochondrial membrane is mediated by Mfrn1/2. In the mitochondrial matrix, iron is stored in FtMt. Rotenone induces an increased level of Tf, while MPTP or 6-OHDA induces increased levels of VDAC in mitochondria. Additionally, excessive iron induces the generation of ROS and lipid peroxidation through the Fenton reaction, further leading to ferroptosis. However, overexpression of TFEB or FtMt has the ability to suppress α-synuclein aggregation and decrease the cellular labile iron pool. All these processes contribute to the nigral dopaminergic neuronal death. This figure was created with BioRender.com/u48z218

Crosstalk between dysregulation of metal ions and pathological proteins in neurodegenerative diseases. Excessive iron affects α-synuclein aggregation, while misfolded α-synuclein accelerates iron deposition. There are two copper-binding sites in the N-terminus of α-synuclein that increase its aggregation. Silencing CTR1 can decrease the phosphorylation and aggregation of α-synuclein. PARK9 deficiency induces increased zinc levels and α-synuclein aggregation. Manganese can indirectly stimulate the expression of α-synuclein through activating the ERK1/2 MAPK pathway. On the other hand, α-synuclein aggregation could enhance manganese-induced neurotoxicity through the NF-κB pathway. The binding of IRP to iron inhibits the IRE, which increases the expression of APP. APP can be cleaved by β- and γ-secretase in early endosomes to form Aβ in the amyloidogenic pathway. On one hand, excessive metal ions induce increased levels of Aβ monomers; on the other hand, metal ions can bind to Aβ and promote its aggregation. Iron, copper, and zinc enhance tau phosphorylation by activating CDK5, GSK-3β, or MAPKs and inactivating PP-2A activity to accelerate the formation of NFTs. Manganese exposure promotes the development of ALS. Additionally, partial deficiency of SOD2 significantly exacerbates motor deficits. Iron dysregulation induced by SOD1G93A is mediated by impairment of the Akt/FOXO3a signaling pathway. Meanwhile, increased levels of copper, zinc, and metallothioneins-I/II/III are observed in SOD1G93A mice. Copper has the ability to bind with the N-terminus of HTT proteins, thereby promoting their aggregation. Mutant HTT can lead to a deficiency in neuronal manganese, which affects arginase activity. Additionally, mutant HTT inhibits the binding of Sp1 to the promoter of the ZnT3 gene, resulting in decreased levels of synaptic vesicular zinc in the hippocampus, cortex, and striatum. This figure was created with BioRender.com/q53d457
As the sole known iron exporter, decreased FPN1 also contributes to the accumulation of iron in nigral dopaminergic neurons in PD.106 Recently, significant decreases in both mRNA and protein expression of FPN1 have been observed in the brains of PD patients compared with age-matched controls.271 In primary cultured ventral mesencephalic neurons and MES23.5 dopaminergic cells with 6-OHDA treatment, decreased FPN1 but not hephaestin contributed to the iron accumulation.272 Both hepcidin and Cp function as ferroxidase, which converts the highly toxic ferrous iron into nontoxic ferric form and collaborates with FPN1 to facilitate iron export.273 In the N27 dopaminergic neuronal cells, the knockdown of hepcidin significantly upregulates FPN1 and decreases intracellular iron, which also attenuates 6-OHDA-induced apoptosis.274 In the primary cultured cortical neurons, increased hepcidin by adenovirus resulted in inhibited iron uptake and release, as well as decreased levels of iron transport proteins including TfR1, DMT1 (+IRE), DMT1 (−IRE), and FPN1.62 Conversely, knockdown of hepcidin yielded opposite results. In the 6-OHDA-induced PD rat model, acupuncture was found to reduce iron accumulation in the SN by balancing the ratio of DMT1/FPN1, thereby protecting nigral dopaminergic neurons.275 In PC12 cells, aspirin has been reported to increase the expression of FPN1 by inhibiting hepcidin through the IL-6/JAK/STAT3 pathway, which promotes FPN1-mediated iron release and reduces neuronal iron levels.276 All these evidences suggest that modulating FPN1 is promising manner to reduce iron level in nigral dopaminergic neurons. The level of Cp in the serum is suggested to be a potential biomarker for PD, but it appears to vary across different reports. A significantly lower level of Cp in the serum was observed in PD patients using I-FP-CIT-SPECT in 2017277; however, no significant change of Cp in the serum was reported in PD patients in 2018.278 Interestingly, a study conducted in 2020 found higher serum Cp levels specifically among females with PD, which were associated with increased impulsivity.279 Therefore, it may be necessary to consider using more sensitive detection methods and increasing the number of samples from PD patients to confirm changes in Cp levels. In the Cp knockout mice, a loss of approximately 30% nigral dopaminergic neurons was observed at the age of 5–6 months, accompanied by nigral iron deposition, and peripheral administration of Cp could attenuate both the nigral iron deposition and neurodegeneration in MPTP-induced PD mice model.280,281 In a clinical trial, PD patients with higher levels of Cp-ferroxidase activity in their serum and CSF were found to be more sensitive to iron chelator deferiprone treatment, which showed greater reduction in UPDRS scores and nigral iron levels after 6 to 12 months of therapy,282 suggesting that targeting Cp may have the potential to improve iron accumulation in nigral dopaminergic neurons.
Microglia
Microglia, as the most efficient iron-absorbing glial cells, play an essential role in maintaining iron homeostasis.87,88 Additionally, microglia are susceptible to ferroptosis, which is dependent on the vesicle trafficking gene SEC24B; however, their transcriptional state changes significantly after iron overload.90,283 Compared to astrocytes and neurons, human-induced pluripotent stem cell-derived microglia have been recognized as highly responsive to iron and susceptible to ferroptosis. Additionally, iron overload resulted in a PD-like shift in the transcriptional state of microglia.90 Microglia activation has been proposed to be an early event in PD. Idiopathic rapid eye movement sleep behavior disorder (iRBD) is considered a prodromal stage of α-synucleinopathies, such as PD, with a conversion rate to α-synucleinopathies reaching 41% within five years.284 Positron emission computed tomography has shown increased microglia activation in the SN of patients with iRBD, which is accompanied by reduced dopaminergic function in the putamen.285 In the SN of rotenone-induced PD mice model, microglial-mediated neuroinflammation was found to be triggered by gut microbiota-induced elevation of CXCL1.264 In the rat neuron-microglia-astrocyte, iron induces selective toxicity in dopaminergic neurons, and microglia activation exacerbates the neurotoxicity of dopaminergic neurons.286 Proinflammatory factors released by activated microglia, such as interleukin (IL)-6, IL-1β, and TNF-α, have been found to upregulate the expression of iron import-related proteins, including DMT1, IRP1, and TfR1, while downregulating the expression of the iron export protein FPN1, which leads to neuronal iron accumulation.286,287,288,289 Conversely, iron overload further enhances microglial activation and proinflammatory factor release, creating a vicious circle. LRRk2 is indicated to be downstream of cellular proinflammatory signals, and it has been found to be activated in the microglia of postmortem PD tissue. In iPSC-derived microglia from PD patients carrying LRRK2-G2019S, Tf recycling was found to be dysregulated under proinflammatory conditions, and this dysregulation was observed in the lysosomes located near the nucleus.290 Additionally, both iron and ferritin accumulation were found in the inflammatory microglia of LRRK2-G2019S knock-in mice.290 Microglia can interact directly with α-synuclein, thereby promoting the occurrence and development of PD. Microglia can engulf α-synuclein released by neurons and degrade it through autophagy. When microglia are unable to degrade intracellular α-synuclein, they can release α-synuclein through exosomes, promoting the transmission of α-synuclein.291,292,293 Furthermore, aggregated α-synuclein can inhibit microglial phagocytosis and autophagy, while impaired microglial autophagy can promote inflammation, aggravating the accumulation of α-synuclein and loss of nigral dopaminergic neurons.292,294,295 In male Macaca fascicularis, nigral iron deposition induced by nasal mucosal delivery of human PFFs was specifically localized in microglia rather than dopaminergic neurons and other types of glial cells.208 However, increased levels of Tf, TfR1, TfR2, and FPN1 were observed in the nigral dopaminergic neurons. In the 6-OHDA induced PD rat model, microglia activation, L-ferritin and iron deposits were found to be co-localized in the SN.205 Specifically, only activated microglia can synthesize Lf, which is able to bind with iron and protect vulnerable dopaminergic neurons, and an increase in iron overload could enhance the release of Lf by activated microglia.92,296,297
Astrocytes
Astrocytes have attracted increasing attention in PD.298 In both rotenone- and 6-OHDA-induced PD mice, overexpression of hepcidin using a virus-based strategy has been found to suppress major pathologies of parkinsonism and motor deficits,299 suggesting that further studies could explore the protective role of astrocyte-derived hepcidin in PD. In a co-cultured system of primary astrocytes and MES23.5 cells, it was observed that ferritin released by astrocytes entered MES23.5 cells and protected them against MPP+-induced ferroptosis and neurotoxicity.85 Estrogen is considered to play a neuroprotective role in women with a lower risk of PD, which displays a different regulatory role in iron metabolism between astrocytes and neurons.300 Estrogen increases the levels of FPN1 and DMT1 via HIF-1α in astrocytes, while decreasing DMT1 and increasing FPN1 in the neurons, suggesting that estrogen promotes the iron transport in astrocyte, which may transfer iron to other tissues such as blood vessels, thereby reducing neuronal iron accumulation.300 The homeostatic iron regulator (HFE) gene variant (H63D in humans, H67D in mice) acts as a disease-modifier in PD, while also playing an important role in regulating cellular iron uptake. With the treatment of paraquat to induce PD-like phenotype, H67D HFE mice showed an increased level of ferritin light chain and exhibited resistance to paraquat-induced nigral neuronal toxicity.301 Furthermore, increased levels of ferritin light chain and activation of Nrf2 were observed in the H67D-HEF astrocytes following paraquat treatment, which provided protection against paraquat-induced neurotoxicity for both astrocytes and neighboring neurons.302 On the other hand, astrocytes also have a negative effect on neuronal iron accumulation in PD. When exposed to both α-synuclein and iron, the ratio of hepcidin to ferritin decreased in primary culture astrocytes, indicating an iron-releasing phenotype that may be detrimental to neighboring neurons.303 Both DMT1 and TfR1 are responsible for the iron uptake in astrocytes.304 In primary cultured astrocytes, increased iron transporters, such as DMT1( + IRE) and FPN1, IRP1 and ferritin light chain were observed after 6-OHDA treatment, indicating that 6-OHDA promotes the trafficking of iron in astrocytes, which may contribute to the accumulation of iron in neurons.263,305 In primary cultured astrocytes from IRP2 knockout mice, MPP+ increased the levels of ferritin and DMT1(-IRE) while decreasing the level of TfR1, which resulted in an elevated iron level in the astrocytes and may reduce their ability to buffer iron from neuron, thereby contributing to nigral iron deposition and neuronal apoptosis.306 The accumulation of iron gradually increases in astrocytes with age, which may lead to the development of neurodegenerative diseases.81,307 Heme oxygenase-1 (HO-1) is a 32 kDa enzyme that controls the cellular heme catabolism, leading to the production of carbon monoxide, iron, and biliverdin, which process is involved in gliopathy and contributes to neuronal vulnerable to oxidative injury in PD.308 In the GFAP.HMOX1 transgenic mice aged between 8.5 and 19 months, overexpression of heme oxygenase-1 (HO-1) in astrocytes induced parkinsonism-like phenotypes, such as nigrostriatal hypodopaminergia, altered gait, locomotor incoordination, reduced olfaction, increased α-synuclein by downregulating miR-153 and/or miR-223, iron deposition in the basal ganglia, mitochondrial damage, and oxidative stress.309,310,311
Oligodendrocytes
Iron dyshomeostasis in the oligodendrocytes is also involved in PD; however, the understanding of this process is limited. In the SN of PD patients, although there is no significant change in iron levels in ferritin-positive oligodendrocytes, the level of ferritin in these cells was reduced by almost half compared to that of non-neurodegenerative control individuals.93 In the striatum of MPTP-induced PD mice model, ginsenoside Rg1 has been reported to regulate iron balance by increasing the level of ferritin heavy chain and decreasing the level of ferritin light chain, which plays a positive role in protecting oligodendrocytes against lipid peroxidation stress.312 Meanwhile, ginsenoside Rg1 also revealed a neuroprotective effect on nigral dopaminergic neurons in the MPTP-induced PD mice model.312 Considering the highest concentration of iron in oligodendrocytes within the CNS, it is worth exploring the contribution of iron dysregulation in oligodendrocytes to PD in further studies.
Subcellular
Lysosome damage or dysfunction has been implicated in the pathology of PD, mainly focusing on an autophagy-lysosome pathway. However, as a major cellular iron storage organelle, there is a large amount of ferrous iron in the lysosome (Fig. 4). Nevertheless, an overload of ferrous iron could trigger oxidative stress in the cytosolic and cytotoxicity. Genetic variations in LRRK2 have been implicated in their association with PD, which also participates in lysosomal functions. LRRK2 is capable of phosphorylating a specific group of Rab GTPases, such as Rab8a and Rab29, which are responsible for the spatiotemporal regulation and correct transportation of vesicles.313,314 In iPSC-derived inflammatory microglia with mutant LRRK2-G2019S, Rab8a was sequestered, leading to lysosome damage and mistrafficking of Tf into lysosome closer to the nucleus, resulting in iron accumulation.290 Ferritinophagy functions as the exclusively identified mechanism for the liberation of iron bound to ferritin. Aggregated α-synuclein can bind and inhibit the v-SNAREs, which blocks fusion between lysosomes and autophagosomes, inhibiting the release of iron from ferritin in PD.315 Visual symptoms related to retina degeneration are prominent in PD, and iron dyshomeostasis has been indicated to be involved in the degeneration of the retina.316,317 Both in vivo and in vitro evidences has shown that α-synuclein suppresses the process of ferritinophagy and causes accumulation of iron-rich ferritin in the retinal pigment epithelium, which may be responsible for retinal iron dyshomeostasis and PD-associated retinal degeneration.317 As the autophagy-lysosome pathway is the primary pathway for clearing abnormal aggregated α-synuclein, and has been implicated as a promising strategy for PD prevention or treatment,318 we have attempted to identify a selective autophagy, which can not only degrade α-synuclein but also inhibit ferroptosis in our recent study.108 TFEB-mediated autophagy not only promotes the clearance of aggregated α-synuclein but also maintains cellular labile iron at a low level and prevents ferroptosis.108 Iron treatment was also found to promote the aggregation and transmission of α-synuclein through TFEB-mediated autophagosome-lysosome fusion.319 TRPML1 is a lysosomal release channel for both calcium and iron. It has been reported that activation of TRPML1 could facilitate the maturation of autophagosomes containing α-synuclein, increase the lysosomal exocytosis, and protect dopaminergic neurons from α-synuclein toxicity.320,321 Therefore, the activator of TRPML1, such as Artemisia argyi Levl. et Vant., has been suggested to be neuroprotective in PD.322 In the postmortem striatum of sporadic PD patients, the level of microglial Nramp1 was found to be significantly increased, accompanied by iron deposition and α-synuclein aggregation.323 Under conditions of iron overload, functional Nramp1 is involved in the degradation of α-synuclein aggregates with lysosomal cathepsin D in microglia.323
As ROS is mainly produced in mitochondria, which induces oxidative stress and damages dopaminergic neurons, mitochondria dysfunction is considered a causative factor of PD.324 Mitochondria are also an important organelle involved in iron metabolism, and dysregulation of mitochondrial iron has attracted increased attention in PD (Fig. 4).111 Disruption of mitochondrial iron in PD is mainly caused by the dysregulation of proteins related to mitochondrial iron. Although the mRNA levels of three forms of VDAC (VDAC1/2/3) and TH were significantly decreased, the mRNA ratios between the three forms of VDAC to TH were increased in the SN of PD patients.21 Meanwhile, increased VDAC was observed in MPTP- or 6-OHDA-induced PD models.325,326 Although VDAC-mediated mitochondrial iron transport has not been directly detected in PD, increased VDAC levels may lead to mitochondrial iron overload, thereby inducing mitochondrial dysfunction in PD. In the rotenone-induced PD cell and rat models, increased Tf levels were found in the nigral dopaminergic neurons, particularly accumulating in the mitochondria.327 In the SN of PD patients, increased levels of Tf and oxidized Tf were observed, which proteins were found to co-localize with TfR2 on the mitochondrial of remaining nigral dopaminergic neurons, suggesting that the Tf-TfR2 mediated mitochondrial iron transport system is disrupted in PD, which may be involved in the development of this disease.327 Recently, DMT1 has been identified as maintaining the mitochondrial membrane potential. Knockdown of DMT1 was found to enhance the activity of mitochondrial complex while decreasing the activity of complex III, thereby reducing erastin-induced ferroptosis.328 In the C. elegans model of AD, the knockdown of mitochondrial iron importer mitoferrin was found to decrease the concentration of mitochondrial iron and levels of ROS.329 In Drosophila, overexpression of the mitochondrial iron importer mitoferrin, or knockdown of the iron binder Fer2HCH, changes iron homeostasis in the mitochondrial, which rescues phenotypes associated with PINK1 loss-of-function, including mitochondrial morphology defects, reduced mitochondrial aconitase activity, and flight deficits.330 These findings indicate that targeting mitoferrin or Fer2HCH and modulating mitochondrial iron may be a potential strategy to improve PD. As an important iron storage protein in the mitochondrial, FtMt is expressed in nigral dopaminergic neurons, which has been proven to protect mitochondria from oxidative stress. Overexpression of FtMt was found to suppress α-synuclein expression, sequester iron in the mitochondrial, decrease the cellular labile iron pool, and combat oxidative stress induced by H2O2 or 6-OHDA.331,332 Furthermore, overexpression of FtMt was found to suppress the increase in labile iron pool and inhibit mitochondrial damage, thereby attenuating the Parkinsonian phenotype induced by MPTP in mice.333
Cellular iron dysregulation in AD
AD is characterized by two pathognomonic protein aggregates, including extracellular Aβ plaques and intracellular tau neurofibrillary tangles. Abnormal iron deposition is involved in the pathological progress of AD, leading to excessive iron accumulation in cells and plaques, such as neurons, glia, as well as Aβ plaques and neurofibrillary tangles.29,334,335 It has been reported that redox-active iron is co-localized with senile plaques and neurofibrillary tangles in the hippocampal tissue of AD, and redox-active iron not only participates in on-site oxidation but also catalyzes H2O2-dependent oxidation.29 Chelated removal of metal can abolish the H2O2-dependent oxidation catalyzed by senile plaques and neurofibrillary tangles, while incubating of iron or copper after the above process can re-induce the catalytic redox reactivity by the lesions.336 Recently, with a label-free and nanoscale chemical imaging by synchrotron X-ray spectromicroscopy, higher levels of iron was observed in the amyloid plaques of human AD brain tissue, and this iron appeared to be chemically reduced and in a low-oxidation state.35 Cp plays a crucial role in converting ferrous iron to ferric iron. In the AD brains, although the level of Cp was significantly increased in the neuropil, the immunoreactivity of Cp in the neurons was similar to that in the age-matched controls, implying the important role of Cp in the accumulation of redox-active iron in neurons of AD.337
APP, a major source of Aβ in AD, can be cleaved by α- and γ-secretase at the plasma membrane to form soluble APP α (sAPPα) through non-amyloidogenic pathway.338 After endocytosis, APP can be cleaved by β- and γ-secretase at the early endosomes to form Aβ through an amyloidogenic pathway. It has been reported that IRP can bind to the IRE-mRNA of APP, which not only regulates iron homeostasis,166,339 but also downregulates the levels of APP, Aβ, and protein aggregation.340 However, in the presence of iron accumulation, the levels of APP, Aβ, and protein aggregation can be upregulated. These pieces of evidence establish a link between IRP-mediated iron homeostasis and AD. APP has been reported to function as an analogous iron-exporting chaperone for neurons and other types of cells, playing an essential role in stabilizing FPN on the cell surface and supporting the export of iron from neurons.341,342 In the APP knockout mice, decreased FPN was observed in the brain, and age-dependent iron elevation was observed in both the brain and liver, which correlated with increased ferritin levels and decreased TfR1 expression.343 Both genetic and pharmacological methods have demonstrated that the endocytotic amyloidogenic processing of APP reduces iron export by disrupting FPN stabilization on the cell surface, leading to cellular iron retention.344 In contrast, the preferential non-amyloidogenic processing of APP on the cell surface enhances FPN stabilization and decreases neuronal iron levels. In the rat primary cortical neurons, iron overload significantly increased the non-amyloidogenic carboxy-terminal fragment α (CTFα) derived from α-secretase cleavage of APP, while decreasing the amyloidogenic products sAPPβ and Aβ. This may be attributed to direct inhibition of β-secretase activity by iron overload.345 Additionally, iron overload caused significant alterations in the distribution of neuronal secreted sAPPα, with evidence indicating an increase in cellular levels of sAPPα.345 Ferric iron can interact with Aβ and promote the aggregation of both Aβ40 and Aβ42.346,347 During the process of fibril formation, Aβ42 can act as a ferritin-specific metallochaperone-like molecule and reduce ferric iron from the ferrihydrite core of ferritin, resulting in a concentration of ferrous iron that is two times higher than that produced by ferritin itself.348 X-ray spectromicroscopy and electron microscopy revealed that co-aggregation of Aβ and ferritin leads to the conversion of ferritin into reactive low-oxidation states, which may contribute to increased oxidative stress in AD.349 Furthermore, Aβ42 can induce the accumulation of ferric iron within the amyloid aggregates and lead to the reduction of ferric iron into ferrous iron, which process can be enhanced by aluminum.350 Increased labile ferrous iron produced during the aforementioned process may induce ferroptosis or enhance oxidative stress, thereby contributing to the development of AD. Tau protein has been suggested as the downstream effector of Aβ toxicity in AD,351 which could also promote the transport of APP to the neuronal surface and facilitate FPN-mediated iron export. In the tau-knockout mice injected with Aβ oligomers into the hippocampus, tau ablation was found to reduce the iron accumulation induced by Aβ in the hippocampus.352 Iron also has the ability to bind to tau protein, which triggers both tau phosphorylation and aggregation.353,354 A positive association between iron deposition and insoluble tau aggregates in the inferior temporal gyrus of AD patients has been confirmed through MR-based QSM and tau-PET.33 In the APP/presenilin 1 (APP/PS1) double transgenic mice treated with deionized water containing ferric iron, tau phosphorylation induced by iron overload can be abolished by DFO through cyclin-dependent kinase 5 (CDK5) and GSK-3β pathway.355 Meanwhile, high dietary iron could also induce the expression of Aβ and phospho-τ in the hippocampus of both wild-type and APP/PS1 transgenic mice.356 The upregulation of HO-1 and its co-localization with tau in the brain is another phenomenon in AD, which can lead to cognitive decline.357 In transgenic mice and N2a cells with overexpressing HO-1, long-term overexpression of HO-1 significantly increases tau aggregation, tau phosphorylation, as well as iron accumulation in the brain, and the induction of tau phosphorylation by HO-1 is mediated by iron accumulation.358 Insulin resistance is an important hallmark in the brain of AD. In addition to causing aberrant phosphorylation of tau, iron overload also disrupts insulin signaling, as evidenced by decreased levels of tyrosine phosphorylation in insulin receptor β (IRβ), insulin signal substrate 1 (IRS-1), and phosphoinositide 3-kinase p85α (PI3K p85α) in primary cultured neurons, as well as in the brains of iron-overload mice accompanied by impaired learning and memory.359
As the inherent immune cells in the CNS, microglia also play an essential role in iron dyshomeostasis in AD, and activated microglia are characterized by both iron accumulation and infiltration of Aβ plaques in AD.360 Immunohistochemical analysis revealed that the activation and proliferation of microglia were distributed in Aβ plaques, which were also co-localized with iron, in both AD patients and animal models.361 According to the report, excessive iron was found deposited in the activated microglia and plaques, as well as in the mid-cortical layers along myelinated fibers of AD patients using Perl’s histochemical procedure.225 Microglia were identified as the main cells containing ferritin, which was associated with senile plaques and blood vessels.334 Meanwhile, MRI results showed a higher presence of iron-positive microglia in the hippocampus of AD patients compared to healthy control subjects.362 Aβ can upregulate the level of DMT1 and enhance the uptake of NTBI by microglia.363 Iron accumulation in microglia induces a rapid transformation from an M2 to a harmful M1 phenotype, exacerbating the process of Aβ-induced microglial IL-1β secretion.364,365 In the cingulate cortex of AD patients, Aβ plaques can upregulate hepcidin by increasing microglial secretion of IL-6, which subsequently downregulates FPN and leads to iron deposition in the brain of AD.366 Both apo-Tf and holo-Tf regulate the release of iron from endothelial cells at the BBB.64 In iPSC-derived astrocytes and endothelial cells, Aβ has been reported to promote iron uptake by astrocytes and increase the level of apo-Tf in the media, which further stimulates iron transport from endothelial cells.367 In APP/PS1 mice, the overexpression of Hp in astrocytes significantly decreased iron levels and reduced the formation of Aβ plaques in the cortex and hippocampus, which alleviated oxidative stress and neuroinflammation and enhanced cognitive decline.368 Neuroimaging researches have demonstrated that micro- and macrostructural abnormalities in white matter are closely related to the progression of AD.369,370,371 The changes in myelin and oligodendrocytes mainly feature in the abnormal white matter of AD.372,373 As the glial cell with the highest iron content, it is worth investigating the role of iron in the aforementioned oligodendrocyte-mediated changes in AD.
Cellular iron dysregulation in ALS
In SH-SY5Y cells overexpressing SOD1G93A, the mRNA levels of TfR1 and DMT1, as well as the expression levels of mitoferrin 1 and 2, frataxin, and iron-sulfur cluster scaffold protein were all increased, suggesting that dysregulation of iron-related proteins may contribute to the iron imbalance induced by SOD1G93A in ALS.374 Abnormal iron is deposited in the motor neurons and glia, accompanied by increased levels of DMT1, FPN, and Cp in the cervical cord,375 as well as increased ferritin in glial cells, suggesting the contribution of iron the disease progression of SOD1G93A mice.375 The iron dysregulation induced by SOD1G93A is mediated through impairment of Akt/FOXO3a signaling pathway,376 and may also be related to damage of iron-sulfur cluster.377 Iron accumulation induced by SOD1G93A increases the activity of TNF-α converting enzyme (TACE), which promotes the secretion of TNF-α and induces oxidative stress.378 Tf was identified as being localized in Bunina bodies and some of the basophilic inclusions in ALS patients.379 The disruption of the blood–spinal cord barrier leads to the accumulation of iron derived from blood and neurotoxic hemoglobin in the spinal cord, which contributes to early motor-neuron degeneration in SOD1G93A mice.380 GPX4 depletion and ferroptosis were observed in the spinal cords and brains of transgenic mice models of ALS, including mutant superoxide dismutase 1 (SOD1G93A), TDP-43Q331K, and C9orf72500 mice models, which observations were associated with impairment of Nrf2 signaling pathway and decreased H-ferritin.381 Overexpression of GPX4 in SOD1G93A mice can improve locomotor dysfunction and delay disease onset, indicating the involvement of ferroptosis in motor neuronal degeneration in ALS. Additionally, activating Nrf2 could also improve neurodegeneration in the SOD1G93A mice model of ALS.382 Decreased Speedy/RINGO cell cycle regulator family member A (SPY1), which was caused by MDM2-mediated ubiquitination degradation, was also involved in ferroptosis in the SOD1G93A mice, and overexpression of SPY1 could inhibit ferroptosis by regulating GCH1/BH4 axis and TfR1, thereby delaying the occurrence and prolonging the survival SOD1G93A mice.383
Cellular iron dysregulation in HD
In order to investigate the iron changes in the white matter of HD, myelin breakdown was observed with no significant change in iron level in the PreHD stage, and decreased iron was observed in the isthmus in early HD stage using MRI.384 Increased iron was found in the putamen, GP and external capsule of premanifest HD individuals from the HD Young Adult Study, indicating that iron accumulation in subcortical structures and the surrounding white matter is an early feature of HD.385 Although decreased ferritin levels in the serum of HD patients was reported in 1991,386 increased iron was also observed in the striatum and cortex of N171-82Q HD transgenic mice, accompanied by elevated levels of IRP1, Tf, ferritin and TfR.387 Furthermore, it was reported that increased nuclear levels of signal transducer and activator of transcription 5 (STAT5) in the brains of N171-82Q mice and 160Q HEK293 cells enhanced the expression of IRP1 and initiated iron deposition.388 QSM results showed that iron accumulation in the basal ganglia, including the pallidum, putamen and caudate of both premanifest and symptomatic HD patients, and iron accumulation in both putamen and caudate was significantly associated with the severity of disease.388 Iron levels were reported to be inversely correlated with the volume of putamen, globus pallidus and the anterior cingulate; and directly correlated with the volume of cortical structures.389 Quantitative susceptibility MRI results revealed higher iron levels in the striatum and GP in individuals with closer-to-onset and early HD, which were directly correlated with the HD CAG-age product score and brain atrophy.258 After a longitudinal 1-year follow-up test, a higher rate of iron deposition was found in the caudate and GP, indicating that brain iron might server as a maker for monitoring the progression of HD.258 In neonatal R6/2 HD mice, elevated iron uptake caused oxidative stress, energetic dysfunction, and potentiated the disease phenotype.390 Additionally, in female YAC128 HD mice, neonatal-iron supplement led to increased striatal degeneration at 1 year old.391 Iron accumulation was observed in the perinuclear cytoplasm of striatal neurons in R6/2 HD mice along with decreased IRP1/2 and TfR1 levels and increased FPN expression.392 Furthermore, increased ferritin levels were primarily observed in microglia of R6/2 HD mice, suggesting it as an early event of HD.393 In the N171-82Q mice model of HD, iron was found to activate microglia and directly stimulate the activity of indoleamine-2, 3-dioxygenase, which catalyzes the initial step of kynurenine pathway.394 Additionally, accumulated iron was also observed in the astrocytes of the striatum in the postmortem tissue from HD patients.395 In the brain of both 12-week R6/2 and 12-month YAC128 HD mouse models, accumulated mitochondrial iron, increased iron uptake protein mitoferrin 2, and decreased iron-sulfur cluster synthesis protein frataxin were observed, which changes were accompanied by increased lipid peroxidation and mitochondrial dysfunction.396 It has been reported that APP is involved in the iron accumulation in HD, with evidence showing that knockdown of APP significantly increases cerebral and striatal iron levels in the YAC128 HD mouse model.397 HTT has been reported as an iron-regulated protein,398 and the elimination of HTT expression alters brain iron homeostasis in adult mice,399 suggesting that HTT may play a crucial role in the dysregulation of iron in HD.
Dysregulation of other metal ions
Dysregulation of manganese
PD
Exposure to excess manganese could increase the risk of PD, which was first observed in 1967 among miners who exhibited symptoms of an increasing incidence of dystonia and non-resting tremor.36 Additionally, a study in 2003 reported a combined effect of dietary intake of iron and manganese in increasing the risk of PD.37 The increased manganese in the whole blood was observed in PD patients without depression compared to both PD patients with depression and the controls.400 Recently, it was found that intraperitoneal injection of manganese can lead to Parkinson-like symptoms in C57BL/6J mice, accompanied by disorders in lipid metabolism, oxidative stress, and damage to nigral dopaminergic neurons.401 In the MitoPark PD mice model, exposure to manganese was found to impair mitochondrial function and worsen progressive motor deficits.402 In 6-OHDA-induced rat models, a significantly increased level of manganese was observed in the SN, GP, putamen, and amygdala over time following 6-OHDA treatment,403 which may contribute to neurodegeneration in the basal ganglia of PD. Additionally, manganese could enhance the effects of 6-OHDA on histamine in the brain.404 However, in an MPTP-treated PD monkey model, the level of manganese in the brain was not affected by MPTP.405 In the DJ-1 knockout PD model mice with MnCl2 saline treatment, manganese mainly deposits in subcortical regions, such as ventricles, hippocampus, medial preoptic nucleus (MPO), lateral septal nucleus (LS), and ventromedial hypothalamic nucleus (VMH) as observed by manganese-enhanced MRI and LA-ICP-MS.406 Meanwhile, manganese treatment disrupted the homeostasis of iron, zinc, copper, and calcium in the DJ-1 knockout PD model mice, resulting in more severe symptoms of PD. In developing rats, exposure to manganese also induced motor deficits and striatal oxidative stress.407 Chronic exposure to MnCl2 also establishes a PD zebrafish model involving oxidative stress, neuroinflammation, and apoptosis pathway.408 Although no significant changes were observed in markers of dopamine terminal integrity or dopamine receptors in the striatum, manganese exposure caused a decrease in in vivo dopamine release, which may account for manganese-induced motor deficits.40 Increased dopamine transporter (DAT) levels were observed in the living non-human primate brain after acute manganese administration using positron emission tomography (PET), which may indicate a compensatory response to its inhibitory action on DAT.409 Additionally, manganese can dysregulate the activity of dopamine neurons410 and indirectly stimulate the expression of α-synuclein in PC12 cells through activating ERK1/2 MAPK; on the other hand, expression of α-synuclein could enhance neurotoxicity induced by manganese through NF-κB pathway.411,412,413
AD
Although some evidence indicates that abnormal manganese levels can play a role in the development of AD, this connection is relatively limited. Despite its capability to bind to the N-terminal part of the Aβ (1–40) peptide, however, manganese exhibits weak binding affinity within the millimolar to micromolar range.414 After the subcutaneous injection of MnCl2, manganese bound to plaques and enhanced the MRI signal in the 5xFAD mice, indicating a potential role of manganese in imaging amyloid plaques in AD.415 A meta-analysis revealed a significant reduction in serum manganese levels in AD patients, which may be associated with cognitive impairment.416 The level of plasma Aβ peptides was found to increase with elevated manganese, and high manganese was correlated with Aβ-related cognitive impairment.417 The concentrations of manganese were found to be higher in the plasma and lower in the CSF in AD patients compared to controls.418 An autopsy of AD patients showed decreased mRNA and protein levels of SOD2 in the hippocampus, which were restored by PKCε activation.419 In Cynomologous macaques after intravenous injection of manganese (3.3–5.0 mg/kg/w) for 10 months, increased manganese was found in frontal cortex, accompanied by increased iron, copper and zinc.420 Meanwhile, increased Aβ plaques were observed in the frontal cortex, along with degenerative cortical neurons, which may contribute to the cognitive deficits induced by manganese exposure.420 Drinking water containing manganese chloride (200 mg/L) for 5 weeks resulted in increased production of Aβ1-40 and Tau in the rat brain, companied by hippocampal degeneration, necrosis, as well as inflammation in peripheral blood and CNS.421 Transplantation of gut microbiota from normal rats could not only reduces the levels of Aβ and tau, but also attenuates the neuroinflammation by inhibiting cerebral NLRP3 inflammasomes induced by manganese exposure.421 Acute manganese exposure increased the levels of manganese in the cortex, hippocampus, and live in both wild-type control mice and APP/PS1 mice, and impaired glutamatergic function by increasing the level of cortical GLAST protein.422 In addition to inducing neuroinflammation in microglia in the hippocampal region, manganese exposure also led to impairment of learning and memory ability, which implicates its neurotoxicity in AD.193
ALS
A case report has indicated that exposure to manganese after traditional medicine procedures in Kenya promotes the development of ALS.423 SOD2, also known as MnSOD, is a homotetrameric enzyme that protects mitochondria against oxidative stress. Immunohistochemical results showed a higher ratio of SOD2-positive neurons to total neurons in the oculomotor nucleus and Onuf’s nucleus compared to normal controls, while a lower ratio was observed in the hypoglossal nucleus of sporadic ALS patients, indicating that sufficient expression of SOD2 could protect neurons from toxic superoxide radicals in the sporadic ALS.424 Increased immunoreactivities of both SOD1 and SOD2 were observed in the brain stems of ALS patients, particularly in the terminal phase, in motor neurons and glia.425 In a study with ten sporadic ALS patients, increased SOD2 was found in three patients, which also exhibited lower levels of superoxide and decreased Bcl-2.426 In the ALS transgenic SOD1G93A mice, partial deficiency of SOD2 significantly reduces survival and exacerbates motor deficits.427 In a cell model of ALS, neuronal cell death caused by human SOD1G37R could be attenuated by overexpression of SOD2.428 Elevated levels of nitrated SOD2 were also observed in the CSF of ALS patients, which may serve as a biomarker for peroxynitrite-mediated oxidative stress.429
HD
In comparison to wild-type mice, the HD model mice of YAC128Q exhibited a decreased level of manganese in the striatum, which is the brain region most affected by HD and is highly vulnerable, after exposure to manganese.430 XK is responsible for the import of manganese, which is trafficked together with Rab11. It has been reported that impaired XK recycling by Rab11 onto cell surfaces contributes to the decreased vulnerability of the striatum to manganese-induced damage in HD.431 The decreased accumulation of manganese was found in both HD human neuroprogenitors and HD mouse striatal cells, which was also associated with an altered manganese-dependent ATM-p53 pathway.432 The resistance to manganese sensitivity in HD is different between lineages and developmental stages. It was observed that the HD genotype increased the sensitivity of early post-mitotic midbrain neurons to manganese, but it had no significant influence to post-mitotic cortical neurons and HD human-induced pluripotent stem cells-derived neuroprogenitor cells.433 After exposure to manganese, premanifest YAC128 mice exhibited suppressed transcriptional and protein changes, while manifest YAC128 mice showed a diminished response to metabolic changes.434 In wild-type mice exposed to subtle manganese, behavioral changes were induced and neuron density in the striatum was reduced; however, YAC128 mice were able to protect against these aforementioned changes.435 Manganese exposure also induces hyperactivity and dopaminergic dysfunction, with the effects being dependent on sex, age, and YAC128 genotype.436 Manganese also functions as an insulin/IGF receptor to phosphorylate Akt and participate in glucose uptake, and decreased manganese levels may contribute to impaired IGF signaling and glucose uptake in HD.437 Acute manganese treatment could restore the reduced autophagic cargo loading in HD cells, indicating that manganese deficiency may contribute to the impaired autophagy flux in HD.438 At the early stage of HD, mutant HTT could cause a deficiency of neuronal manganese, which affects arginase activity and contributes to the pathophysiology of urea cycle dysfunction in the striatum of HD.439
Dysregulation of copper
PD
Occupational exposure to copper also increases the risk of PD. It has been found that combining chronic copper exposure with aging induces PD features, including altered motor function, dopaminergic neuronal loss, increased α-synuclein accumulation, and aggregation, as well as alterations in proteasome and autophagy.440 Recently, dietary intake of copper has also been reported to be associated with the risk of PD.441 In PD patients, an increase in concentration of copper in CSF has been observed, and this copper concentration has been found to be correlated with severity and progression of PD.442,443 Although a significantly increased level of copper was observed in the GP, putamen, and amygdala of rats over time after 6-OHDA treatment,403 a significant decrease in copper was observed in the SN and locus coeruleus of PD patients using synchrotron-based x-ray fluorescence microscopy (SRXFM), as well as in their blood concentration of copper and Cp.443,444,445 Widespread copper decreases in copper levels were observed in the primary motor cortex (MCX), cingulate gyrus (CG), primary visual cortex (PVC), hippocampus, SN, medulla oblongata (MED), and middle temporal gyrus (MTG) of individuals with PD dementia, as determined by ICP-MS.446 All these pieces of evidences indicate a potential relationship between decreased copper levels and PD.447,448 The bridge linking copper deficiency with PD may be the aberrant iron, as copper predominately binds with Cp, triggering its ferroxidase activity to convert toxic ferrous iron into nontoxic ferric form and maintain iron homeostasis; however, insufficient copper can lead to iron deposition and neuronal death.449 Copper can directly bind to α-synuclein and promote the aggregation of α-synuclein.450,451,452 Additionally, both copper and iron have been suggested to accelerate the prion-like propagation of α-synuclein fibrils in PD because α-synuclein fibrils formed in the presence of copper or iron were more cytotoxic.453 The folded conformation is stabilized by the combination of copper, which alleviates the electrostatic repulsion among the negative charges.454 However, acetylation at the N-terminus or pathological H50Q mutation of α-synuclein can disturb its binding with copper and inhibit copper-induced aggregation.455 After aggregation, copper also facilitates the release of mature fibrils of α-synuclein into the extracellular environment.453 The combination of copper leads to an increased neurotoxicity caused by α-synuclein.456 While the overexpression of α-synuclein for a short time had no toxic effect on dopaminergic neurons, cell death occurs after exposure to copper due to increased CTR1 and depleted GSH, and the oxidative stress induced by copper is mediated through modulation of autophagy or the ubiquitin-proteasome system.457 In transgenic human α-synuclein (A53T) mice and SH-SY5Y cells overexpressing of human α-synuclein (A53T), low-dose copper exposure was found to enhance the accumulation of α-synuclein, which was associated with mitochondrial impairments, including excessive ROS production and reduced mitochondrial ATP production.458 In addition, defects in copper transportation may be involved in PD, as evidenced by a reduction of CTR1 in neurons of low-copper concentration encephalic regions, which is crucial for copper uptake.445,458,459 Furthermore, the aggregation of α-synuclein caused by copper stimulation can be inhibited by silencing CTR1, the primary pathway for copper uptake.460 This finding is consistent with the results showing a reduction in α-synuclein aggregates after knockdown of CTR1 in yeast and mammalian cell models expressing human α-synuclein.461 Furthermore, deficiency of CTR1 could significantly reduce the phosphorylation of α-synuclein at S129 and alleviate nigrostriatal degeneration in an AAV-based mouse model of PD.461
AD
Replication studies and meta-analysis have shown that serum copper levels are higher in AD patients that in healthy controls.462 In the APPNL-G-F knock-in mice, both age and AD development were found to affect copper level in the blood plasma; however, only AD was found to have an impact on the blood plasma copper isotope ratio, and a significant increase in copper was also observed in the brain stem of both young and aged AD mice compared with healthy controls.463 Elevated labile copper was also observed in the postmortem cortical tissue from AD patients, which was associated with oxidative pathology of AD.464 APP, which can produce neurotoxic Aβ through sequential protease cleavage and contribute to the pathology of AD, has a high affinity with copper at its N-terminal and facilitates its reduction from Cu2+ to Cu1+, which process could promote the production of hydroxyl radicals and induce AD-related toxicity.465 On the other hand, this binding between APP and copper also affects copper levels, as evidenced by studies on transgenic mouse brains. Overexpression of AAP containing Aβ significantly reduces the levels of copper and iron, while overexpression of APP in Tg2567 transgenic mice only reduces the level of copper.466 Increased copper was observed in the cerebral cortex and liver of APP and APLP2 knockout mice.467 Additionally, copper can enhance the localization of APP on the cell surface by increasing exocytosis and reducing endocytosis.468 Transthyretin can directly bind to Aβ and modulate its accumulation, which was lower in the CSF of AD patients, and it has been reported that copper regulates the binding between Aβ and transthyretin.469,470 Copper can enhance the proportion of beta-sheet and alpha-helix compositions within the Aβ proteins, thereby facilitating the formation and accumulation of Aβ in the brain plaques.471 Additionally, the Cu-Aβ complex not only activates microglia and reduces the release of TNF-α and nitric oxide through NF-κB mediation,472 but also downregulates the expression of LRP1, impeding the clearance of neurotoxic Aβ and triggering the buildup of brain deposits.184 Recently, different impacts of copper on the assembly of Aβ40 and Aβ42 isoforms have been reported.473 Copper increases primary nucleation and promotes the fibril formation of Aβ40, resulting in enhanced generation of neurotoxic oligomers; on the other hand, copper causes the disassembly of Aβ42 fibrils and induces protofibrils and oligomers.473,474 Oxidative stress can influence the process of Aβ assembling into β-sheet rich fibrils in AD by causing oxidation of Aβ. In the presence of copper, oxidized Aβ was found to assemble into weakly-structured and untangled Cu-Aβ fibrils.475 Copper can also bind with tau peptide and modulate the aggregation of tau.476,477 In addition, excess copper was found to enhance the phosphorylation of tau proteins in human neuroblastoma cells, and lowering the level of copper by oral zinc could significantly decrease tau phosphorylation in the transgenic mice model expressing wild-type human tau protein478 and attenuate spatial memory impairment in female PS19 mice.479 Although there is ample evidence of copper’s involvement in Aβ accumulation and tau phosphorylation, compelling evidence also indicates that increasing copper levels in the brain has the potential to mitigate amyloid pathology and offer positive effects for AD. For example, increasing the bioavailability of copper with compound Cu(II)(gtsm), which can remain at high level in the brain after uptake, could inhibit Aβ oligomers and tau phosphorylation while restoring cognitive function in APP/PS1 transgenic AD model mice.480,481
ALS
Increased copper in the ALS cells co-localized with total lipids, leading to an increase in oxidized lipids and potentially directly inducing oxidative stress.482,483 The increased influx of copper, upregulation of copper chaperones, and decreased efflux of copper resulted in the accumulation of copper in spinal motor neurons, which was associated with age-dependent increases in copper in the spinal cord.484 As a copper-regulating protein, metallothioneins can bind copper ions and decrease oxidative toxicity. SOD1, also known as copper/zinc SOD, can protect mitochondria from oxidative damage.485 Mutations in SOD1, including D90A, A4V, G93A, and G37R, have been associated with familial ALS.486 In the SOD1G93A mice, increased levels of metallothioneins-I and metallothioneins-II were observed within astrocytes in both white and gray matter as mice age, and increased levels of metallothioneins-III were observed in neurons, while elevated metallothioneins-III levels were observed in glial cells at the later stages of disease.487 In the SOD1 Tg mice, increased levels of copper and lipid peroxides were found in the spinal cord at the age of 8 and 16 weeks, while zinc levels were decreased.488 However, motor paralysis and increased metallothionein-III were only observed at 16 weeks of age, indicating that metallothioneins-I and metallothioneins-II function at the early stage of ALS, whereas metallothioneins-III function during the late stage. The reduction of metallothioneins promotes disease expression,489 while the overexpression of metallothionein-I can significantly improve copper dyshomeostasis and extend the lifespan of SOD1G93A mice.490 Mutation in the copper transporter ATP7AM1311V, which was found in ALS patients, also caused intracellular copper accumulation and impaired motor neurons.491 Increased hydroxyl radical-generating activity was found in the G93A mutant SOD1, which facilitates copper release and induces oxidative stress through the Fenton reaction.48,492 Furthermore, mutant SOD1 loses its specificity in metal ion binding, which may play a role in ALS.493 Mutant SOD1 aggregates have the ability to bind and aggregate with anti-apoptotic protein Bcl-2 in spinal cord mitochondria,494 and misfolded mutant SOD1 has been found to deposit onto the cytoplasmic face of the outer mitochondrial membrane.495 Mutant SOD1 could activate caspase-1 and caspase-3, which were observed in the ALS transgenic mice and cell model, triggering apoptosis,496,497 suggesting that inhibition of caspase is protective in the ALS.498 In the primary cultured astrocytes overexpressing wild-type SOD1 or mutant SOD1G93A, both wild-type and G93A mutant SOD1 can be secreted by astrocyte through exosome in varying amount.499 Moreover, the secreted SOD1G93A through exosome is transferred from astrocyte to spinal neurons, resulting in selective motor neuron death. Increasing the total and mitochondrial NAD (+) content in astrocytes expressing mutant SOD1 can protect co-cultured motor neurons from death.500 Motor neurons can secrete active endogenous SOD1; however, this secretion is impaired by mutant SOD1, which causes cytoplasmic inclusions and neuronal toxicity.501 The interaction between mutant SOD1 and dynein-dynactin complex has been found to contribute to inclusion formation.502,503 Additionally, oxidative modification of Tryptophan 32, occurring in both wild-type and mutant SOD1, can promote the aggregation and toxicity of SOD1G93A.504 As the disease progression and endpoint were significantly delayed following chronic intraspinal injection of exogenous wild-type hSOD1 in the SOD1G93A rat, it was suggested that extracellular SOD1 has neuroprotective effects in ALS.501 Mutant TAR DNA binding protein-43 (TDP-43) is also associated with the development of frontotemporal lobar degeneration and ALS. Increased levels of copper, zinc, and manganese in the spinal cords, but not in the brain, may explain the decline in locomotion observed in TDP-43 (A315T) mice.505
HD
Copper has the ability to bind with the N-terminus of HTT proteins and further stimulate their aggregation.506 In a Drosophila model of HD with HTT exon 1-polyQ, knockdown of Ctr1B and DmATP7 genes, which are involved in copper metabolism, can affect the progression of HD, and the level of HTT aggregation significantly decreases after reducing copper.507 Meanwhile, the copper-enhanced toxicity of HTT dissipated after substituting Met8 and His82, which are potential copper-binding residues in HTT.507 In a Drosophila model of HD, copper dose-dependently increased the aggregation of mutant HTT, promoted the accumulation of Thioflavin S positive β-amyloid structures in HTT aggregates, and altered the autophagy-lysosome pathway.508 Furthermore, the progression of HD may be exacerbated by copper’s inhibitory effect on mitochondrial dehydrogenases, particularly succinate dehydrogenase (SDH) and lactate dehydrogenase (LDH), which are sensitive to copper and become inactivated. LDH plays a pivotal role in the astrocyte-neuron lactate shuttle mechanism, utilizing lactate and providing energy for neurons.509,510 The blockage of lactate metabolism in the brain can lead to an insufficient energy supply, which contributes to HD.506,510 Although elevated copper levels have been found in HD patients and models, in a QUIN-induced HD rat model, supplement of copper in drinking water (90 ppm Cu, 28 days) reduced lipid peroxidation and ROS, possibly due to increased SOD1 activity.511
Dysregulation of zinc
PD
The mainstream view suggests a reduction in zinc concentration among PD patients, and a comprehensive meta-analysis has demonstrated a significant downward trend in zinc levels in plasma, serum, and CSF.512,513,514,515 Serum zinc deficiency has been proposed as a risk factor for the development of PD dementia.516 Zinc deficiency also exacerbated movement disorders and dopaminergic neurodegeneration in MPTP-induced PD mice.517 In a Drosophila model of PD, providing sufficient zinc significantly enhances both lifespan and motor functions.518 As we know, oxidative stress plays a significant role in the development of PD, including lipid peroxidation and nucleic acid oxidation. It’s worth noting that zinc possesses antioxidative properties. Therefore, the decrease in zinc in PD patients may contribute to excessive antioxidative reactions. In a rotenone-induced PD rat model, adequate zinc supplementation alleviated neuronal damage by inhibiting lipid peroxidation, suggesting a protective role of zinc against oxidative stress.519 However, zinc also plays a toxic role in promoting the degeneration of dopaminergic neurons within the SN.520 In the 6-OHDA-induced PD rat model, a significantly increased level of zinc was observed in the GP, putamen, and amygdala over time after 6-OHDA treatment.403 Additionally, increased zinc levels were observed in the SNpc of paraquat-treated rats.521 Mutations within the ATP13A2 gene (PARK9) are the underlying cause of Kufor-Rakeb syndrome (KRS), which leads to juvenile-onset Parkinsonism. PARK9, encoded by the ATP13A2 gene, is abundantly expressed in nigral dopaminergic neurons, and mutations within ATP13A2 can cause hereditary Parkinsonism with dementia.522 Deficiency in ATP13A2 results in dysregulation of lysosomes and aggregation of α-synuclein,523 possibly due to zinc dyshomeostasis.524,525
AD
Due to its ability to combine with amyloid plaques and accelerate the aggregation of Aβ peptides and tau protein, zinc is also implicated in the progress of AD. In clinical detection of AD patients, elevated levels of zinc have been observed in the cortex, hippocampus, and amygdala, which are all highly vulnerable brain regions in AD.526,527 In AD animal models such as APP/PS1 mice and macaques, zinc was found to accumulate in the plaques.528,529 Certain zinc-bearing transcription factors, including NF-κB and the tumor suppressor protein p53, are involved in the synthesis of APP. Zinc can interact with Aβ nonspecifically, and the Aβ–zinc complex exhibits high resilience against proteolysis, thereby enhancing the persistence of Aβ aggregations.530,531 In addition to copper, oxidized Aβ was also found to assemble into long untangled zinc-Aβ fibrils in the presence of zinc.475 Furthermore, Aβ aggregation can stimulate the release of zinc from metallothionein, potentially disrupting mitochondrial function and stimulating the generation of ROS, which in turn promotes zinc release and triggers apoptosis.532,533 In relation to the tau protein, accumulating evidence indicates that elevated zinc may potentially contribute to the excessive phosphorylation of the tau protein by activating extracellular signal-regulated kinase such as GSK-3β and MAPKs, while inactivating the main serine/threonine phosphatase in brain tissues, protein-phosphatase 2A (PP-2A), thereby disrupting the homeostasis of tau protein.534,535,536,537 Excessive phosphorylation is more likely to accumulate in neurons, leading to the formation of non-degradable neurofibrillary tangles that resist proteolytic degradation. This accumulation subsequently disrupts axonal transport and microtubule synthesis, resulting in neurotoxicity.538,539 However, some literature reports a significant decrease in zinc level in AD patients and suggests that zinc deficiency is a potential risk factor of AD.540,541 The depletion of intracellular zinc destabilizes microtubules, triggering a chain reaction that includes the release of tau protein, hyperphosphorylation, and the formation of neurofibrillary tangles. Overall, a plethora of experiments support the improvement of cognitive performance after zinc supplementation.542 Since 1992, zinc has been permitted as a potential therapeutic approach for AD, administered both orally and parenterally. On the one hand, zinc supplementation restores the serum zinc level; however, it simultaneously decreases the level of serum-free copper by impeding its absorption in the intestine. This leads to an upregulation of metallothionein for binding copper. Nevertheless, the supplementation of zinc has side effects that exacerbate tauopathy in both behavioral and biochemical impairments, suggesting that zinc should be avoided unless absolutely necessary.543 Variations in intracellular zinc levels potentially arise from modifications of ZIP or ZnT proteins, which are respectively responsible for the influx of zinc from the extracellular environment and the redistribution of zinc to either the extracellular milieu or intracellular components. In neurons, the ZnT3 transporter is responsible for pumping free zinc into synaptic vesicles. Knocking down ZnT3 in mice leads to cognitive difficulties occurring earlier than expected.149 An interesting discovery revealed a decline in ZnT3 levels in the brains of mice and humans with aging; however, an even more significant reduction was observed in AD patients.544 Despite the downregulation of ZnT3, compensatory upregulations of ZnT4, ZnT6, and ZnT1 were observed in the hippocampus of AD.545,546
ALS
A high level of labile zinc was observed in the neurons and astrocytes in the spinal cords of SOD1G93A mice, which may be due to increased HNE levels.547 The native SOD1 protein is expressed as a homodimer in the cytosol, capable of binding with either four copper or four zinc ions.548 However, mutations in SOD1 impair dimerization and promotes its aggregation in ALS.549 Zinc can stabilize the native structure of the SOD1 monomer and promote its homo-dimerization. Copper chaperone (Ccs) binds to SOD1 and facilitates its binding with zinc, thereby stabilizing the conformation of SOD1; however, this process is disrupted by mutant SOD1.550 The absence of zinc but not copper, significantly impacted the membrane attachment of SOD1 and facilitated its aggregation in an in vitro ALS model.551 However, initiation and seed growth in the fibrillation SOD1 were controlled by the disulfide bond rather than zinc or dimerization.552 The loss of zinc in the SOD1 could induce motor neuronal death, which plays a causal role in ALS.553 Under zinc-deficient conditions, SOD1 functions as a molecular switch to initiate homeostatic ER stress.554 Aberrant zinc binding to SOD1 also triggers amorphous aggregation.555 Decreased levels of zinc transporters ZnT3 and ZnT6 were observed in the spinal cords of sporadic ALS patients.556 Zinc can bind to RRM2 peptide of TDP-43.557 Additionally, zinc can bind to the RNA recognition motif of TDP-43, which induces the formation of amyloid-like aggregates.558 Zinc was found to specifically induce the depletion and aggregation of endogenous TDP-43, which was not observed in copper, iron or H2O2.559 Although the induction of apoptosis in motor neuronal death by human mutant SOD1G93A depends on copper and the absence of zinc, chronic oral administration of zinc sulfate decreased the survival of SOD1G93A mice, indicating that zinc amplifies the SOD1G93A-mediated toxicity.560 Zinc pre-treatment also enhanced the NMDAR-mediated excitotoxicity in cultured cortical neurons derived from SOD1G93A mice.561 Additionally, zinc migration and subunit swapping in SOD1 may contribute to its neurotoxicity in ALS.562 Cytoplasmic accumulation and aggregation of SFPQ are hallmarks of ALS. Zinc was found to bind with SFPQ, a ubiquitous nuclear RNA-binding protein, and induce infinite polymerization of SFPQ in primary cortical neurons.563 Aggregation of FUS protein is another hallmark of ALS.564 Zinc could enhance liquid-liquid phase separation of FUS protein and promote its aggregation.565
HD
In R6/1 HD mice model, a significant reduced level of zinc was observed in the hippocampus and cortex, where typically have a high concentration of zinc.566 This deficiency exacerbated the deficit in hippocampal long-term potentiation (LTP) and diminished AMPA receptors. In the N171-82Q HD transgenic mice, decreased levels of synaptic vesicular zinc and declined transcriptional activity of ZnT3 were observed in the hippocampus, cortex, and striatum.567 These changes were caused by mutant HTT inhibiting Sp1 from binding to the promoter of the ZnT3 gene. In a C. elegans-based HD model, chronic exposure to copper, zinc, or their mixture can cause neurodegeneration by increasing the aggregation of polyQ protein in the muscles and neurons.568 Neuronal zinc finger protein (ZFP) transcriptional repressors can significantly reduce mutant HTT and rescue HD-associated behavioral and molecular phenotypes.569
Crosstalk between metal ions dysregulation and pathological proteins
Lewy bodies are composed of a large amount of misfolded α-synuclein, which co-localizes with redox-active iron (Fig. 5). The toxic interaction between iron deposition and α-synuclein aggregation has been demonstrated to accelerate the progression of PD.10,11 We have also reported that the toxic combination of α-synuclein and iron induces cell senescence in a PD mice model, which occurs prior to the loss of nigral dopaminergic neurons.23 Reports from our lab and other labs have demonstrated that iron can induce α-synuclein aggregation. In addition to APP, IRP can also bind to the IRE-mRNA of α-synuclein and downregulate its protein level and aggregation.340 However, in the presence of iron overload, the protein levels of α-synuclein and its aggregation can be upregulated by IRP, establishing a link between IRP-mediated iron homeostasis and PD. Conversely, α-synuclein also affects iron in the process of PD. Nasal mucosal delivery of human PFFs caused time-dependent iron deposition in the SN and GP in male Macaca fascicularis.208 Aggregated α-synuclein could inhibit the iron release from ferritin by inhibiting v-SNAREs in PD.315 Manganese can indirectly stimulate the expression of α-synuclein, which contributes to the aggregation of α-synuclein in glial cells and neurons, and α-synuclein can also enhance the manganese-induced neurotoxicity.411,412,413 As a copper-binding protein, copper can directly bind to α-synuclein and promote its aggregation.450,451,452 Additionally, both copper and iron have been suggested to accelerate the prion-like propagation of α-synuclein fibrils in PD, because α-synuclein fibrils formed in the presence of copper or iron exhibit were more cytotoxic.453 Overexpressed α-synuclein was also found to exacerbate copper toxicity through the autophagy-lysosome pathway or ubiquitin-proteasome system.457 Furthermore, zinc deficiency is also implicated in the aggregation of α-synuclein.523
Dysregulation of cerebral transition metal ions, such as iron, copper, and zinc, has been implicated as a precursor event for Aβ aggregation in AD (Fig. 5). Ferric iron can interact with Aβ and promote the aggregation of both Aβ40 and Aβ42.346,347 Additionally, Aβ42 can induce the accumulation of ferric iron within amyloid aggregates and lead to the reduction of ferric iron into ferrous iron.350 Redox-active iron was co-localized with both senile plaques and neurofibrillary tangles in the hippocampal tissue of AD.29With the LA-ICP-MS, significantly higher concentrations of iron, copper and zinc have been observed in the hippocampus, cortex and retina of 9-month-old wild-type mice compared to the APP/PS1 mice.570 Analysis of the isotope ratios of iron, copper, and zinc in transgenic tau mice and 5×FAD mice showed a significant pathology-specific distribution of both iron and zinc in the mouse models of AD.571 Tau can traffic APP to facilitate iron efflux. Lowered levels of brain tau caused by lithium has been found to elevate nigral and cortical iron levels through attenuating iron efflux, which may be associated with the adverse effect of lithium such as PD-like hand tremor and limit its application for the treatments of neurodegenerative or neuropsychiatric disorders.572 Overexpression of the carboxyl-terminal fragment of APP, which contains Aβ, could significantly reduce the levels of both iron and copper in the transgenic mouse brain; however, overexpression of APP only reduced the level of copper in Tg2576 transgenic mice.466 Concomitant increased levels of manganese were observed in the brains of both transgenic mice.466 Although excessive copper and zinc may induce neocortical Aβ production in AD, both copper and zinc at physiological concentrations could promote the degradation of soluble Aβ.573 However, the mechanisms by which they reduce the secreted level of Aβ differ between copper and zinc.574 Zinc induces dimerization of APP-C99 and prevents its cleavage by γ-secretase, while copper directly targets the subunits presenilin and nicastrin in the γ-secretase complex, reducing the production of Aβ.574 In addition, oral zinc could lower the level of copper, significantly reducing tau phosphorylation in a transgenic mouse model expressing wild-type human tau protein478 and alleviating spatial memory impairment in female PS19 mice.479 By using synchrotron Fourier transform infrared micro-spectroscopy (FTIRM) to image the in situ secondary structure of amyloid plaques and synchrotron X-ray fluorescence (SXRF) microprobe to detect the accumulation of metal ions in the same brain tissue of AD patients, accumulations of both copper and zinc were first observed in Aβ deposits.43 Recently, after comparing the images of Aβ detection by synchrotron X-ray phase-contrast tomography (XPCT) among four transgenic AD animal models, including APPPS1 mice, ArcAβ mice, J20 mice (zinc and iron accumulation), and TgF344 rat (copper accumulation), which had similar β-sheet content but varying metal levels, hyper-density of Aβ plaques was observed in both J20 mice and TgF344 rats.575 In contrast, hypo-density with a hyperdense core was observed in APPPS1 and ArcAβ mice. These findings suggest that metal accumulation may be the primary factor enabling XPCT to map the distribution of Aβ plaques throughout the entire brain without labeling.
Cell membranes also play a crucial role in the metal ion-mediated amyloid aggregation in the brain environment.576 The effect of metal ions, such as copper and zinc, on Aβ toxicity, complicates the interaction between Aβ peptide and cell membrane.577 Studies from the lab of Pappalardo have investigated the Aβ amyloidosis and fibrillogenic properties of Tau/Aβ in the presence of copper and zinc.578,579 Using large unilamellar vesicles (LUVs) to mimic the lipid composition of neuronal membranes, the conformational changes of Tau26-33, but not Tau9-16, were found to be enhanced by copper and zinc in the presence of membrane surfaces. Notably, the aggregation rate of Tau26-33 was decreased by LUVs/copper and LUVs/zinc; however, the aggregation rate of Tau9-16 was increased by LUVs/copper and LUVs/zinc. Additionally, both copper and zinc were found to enhance the interaction between Aβ40 and membrane surfaces and cause the formation of amyloid aggregates. All these pieces of evidence indicate the interplay between metal ions and Tau/ Aβ on the cell membrane, which may play an important role in AD by affecting amyloid formation. Amyloid fibril aggregates, such as senile plaques, also provide large surface areas for metal ions. When combined with metal ions, these bio-interfaces not only induce misfolding of peptides but also modulate both the primary and secondary nucleation of peptide monomers from the microscopic step.580 The surface-chelated copper has been reported to dynamically interact with Aβ chains, restricting their two-dimensional diffusivity on the surface and slowing down their fibrillation.581 In contrast, surfaces without copper facilitate the two-dimensional diffusivity of Aβ chains, promoting improved interpeptide interaction and accelerating Aβ fibrillation. This highlights the critical role of surface-chelated copper in both Aβ fibrillation and AD progression.
Aggregation of mutant SOD1 is a common hallmark of ALS. In transgenic SOD1G93A mice, the overexpression of Aβ promotes the onset of motor impairment and aggregation of SOD1G93A, accompanied by elevated levels of copper and zinc in the brain and spinal cord.582 Upregulated APP in muscle fibers coincides with symptom onset in both sporadic ALS patients and SOD1G93A mice, while genetic ablation of APP significantly improves multiple disease parameters in the SOD1G93A mice.583 Although high levels of SOD1 are also found in the nigrostriatal dopaminergic neurons, SOD1 mutations have a greater cytotoxic effect on motor and dopaminergic neurons.584 The absence of zinc, but not copper, significantly affects the membrane attachment of SOD1 and promotes its aggregation in an in vitro ALS model.551 Aberrant binding of zinc to SOD1 also triggers amorphous aggregation.555
HTT has been reported as an iron-regulated protein,398 and the elimination of HTT expression alters brain iron homeostasis in adult mice,399 indicating that HTT might play an important role in the dysregulation of iron in HD. In the early stages of HD, mutant HTT could cause a deficiency of neuronal manganese, which affects arginase activity and contributes to the pathophysiology of urea cycle in HD striatum.439 Copper has the ability to bind with the N-terminus of HTT proteins and further stimulate their aggregation.506 In a Drosophila model of HD, copper increases the aggregation of mutant HTT in a dose-dependent manner.508 In N171-82Q HD transgenic mice, mutant HTT was found to inhibit the binding of Sp1 to the promoter of ZnT3 gene, resulting in decreased levels of synaptic vesicular zinc in the hippocampus, cortex and striatum.567
Treatment targeting mental ions in neurodegenerative diseases
Metal chelators, capable of sequestering metal ions and dissociating them from their target sites through the formation of multiple coordinate bonds, have emerged as promising therapeutic targets for modulating redox processes and mitigating abnormal protein aggregation. In this part, we summarize the treatment targeting mental ions for neurodegenerative diseases.
PD
Metal ions chelation
DFO and CQ derivative
DFO, an outstanding molecule widely used to treat diseases caused by excess iron, has been approved by the Food and Drug Administration for many years.585 DFO has been shown to be strongly neuroprotective in several PD animal models, improving motor defects and enhancing the survival of dopaminergic neurons following treatment with MPTP,586 rAAV-α-synuclein,587 6-OHDA,588,589,590 and rotenone591 (Table 2). In addition to chelating iron, increasing the expression levels of GPX4 and ferritin heavy chain, DFO has been shown to directly reduce the aggregation of α-synuclein.592 Furthermore, it can activate insulin signaling and glucose metabolism,593 accumulates HIF-1α to induce autophagy,594 exerting its roles through multiple targets. Intranasal delivery, a non-invasive method of bypassing the BBB to deliver therapeutics along the olfactory and trigeminal nerve pathways to the brain, improves the application and efficiency of DFO.593 Furthermore, a polymeric nanoparticle system capable of intracerebrally delivering DFO could also overcome the disadvantages of its short half-life in vivo and poor penetration through the BBB. You et al. report a nanoparticle system loaded RVG29 peptide through specific receptor-mediated endocytosis, resulting in significant decreases in iron content and oxidative stress levels in the SN of PD mice without any adverse effects.595 This DFO-based nanoformulation proposes another potential strategy for delivering DFO into the brain. CQ has been extensively utilized as both an antifungal and antiprotozoal agent, exhibiting emerging efficacy in the treatment of neurodegenerative disorders. One of its mechanisms involves its ability to interact with metals.596 It has been reported that oral administration of CQ provides protection against MPTP-induced damage in mice by inhibiting iron overload.14,597 Furthermore, CQ significantly improves motor and non-motor deficits in a stabilized monkey model of PD induced by MPTP, primarily targeting iron content and ROS levels in the SN.598 The Akt/mTOR pathway plays a crucial role in this process. Additionally, CQ attenuates zinc-mediated cytotoxicity in mice injected with LB-extracts derived from PD patients containing toxic α-synuclein aggregates, resulting in reduced lysosomal alterations and dopaminergic neurodegeneration.599 A novel quinazolinone compound, named PBT434, is being developed for the treatment of PD. In vitro studies have demonstrated that PBT434 can inhibit iron-mediated redox activity and α-synuclein aggregation.600 In vivo, PBT434 rescued motor performance and reduced nigral α-synuclein accumulation in 6-OHDA and MPTP-treated or transgenic mice (hA53T α-synuclein).600 Importantly, PBT434 did not deplete tissue iron stores; instead, it increased levels of FPN and DJ-1. Furthermore, PBT434 modulates the uptake of iron by human brain microvascular endothelial cells through chelation of interstitial iron and inhibition of iron re-uptake by endothelial cells of the BBB.601 Importantly, not only does PBT434 inhibit hyposmia and accumulation of iron and copper in the bulbar region in young tau-/-mice, but it also attenuates motor impairment and neurodegeneration in the SN in aged tau-/-mice, suggesting its potential as a therapeutic agent for both prodromal and clinical stages of PD.602
Natural compounds and derivatives
Numerous natural compounds and their corresponding synthetic derivatives, which possess multifunctional neuroprotective properties, also exhibit an affinity towards metal ions. DL-3-n-butylphthalide, isolated from Apium graveolens seeds, has been approved for the treatment of acute ischemic stroke. A recent study demonstrated its ability to alleviate rotenone-induced motor impairment and dopaminergic neuron loss in rats. Notably, there was an improvement in both iron deposition within SN and serum iron levels, which is consistent with the observed alterations in the expression of the iron metabolism-related proteins, suggesting that DL-3-n-butylphthalide may have potential as a ferroptosis inhibitor.603 Paeoniflorin, a water-soluble monoterpene glycoside extracted from Paeonia lactiflora Pall, can prevent ferroptosis by activating the Akt/Nrf2/Gpx4 pathway in MPP+-induced primary dopaminergic neurons.604 Quercetin (QCT), a natural flavonoid with diverse pharmacological activities, exhibits potential anti-ferroptotic effects in erastin/RSL3 and MPP+-induced cells as well as MPTP-induced PD mouse model, primarily through the activation of Nrf2.605 Furthermore, QCT can attenuate copper-induced apoptotic cell death and ER stress in SH-SY5Y cells through autophagic modulation.606 Moreover, QCT effectively inhibited manganese-induced apoptosis and inflammatory response in SK-N-MC cells and SD rats, potentially involving the iNOS/NF-κB and HO-1/Nrf2 pathways.607 Epigallocatechin-3-gallate (EGCG) possesses a number of pharmacological activities and demonstrates potential neuroprotective properties in PD models. By acting as a metal chelator, EGCG attenuates the Fe3+-induced conformational transition of α-synuclein and protects AS-PC12 cells against Fe3+-induced death.608 It also exhibits a neurorescue effect in both a mouse model and a Drosophila model of PD.609,610 Inhibition of ferroptosis was identified as the underlying mechanism of EGCG-mediated protection.610 Furthermore, EGCG can bind to metal ions [Cu2+ and Zn2+]-Aβ monomers and dimers, resulting in compact peptide conformations and the formation of ternary EGCG-metal-Aβ complexes.611 Emodin, a natural anthraquinone derivative, also inhibits zinc-induced neurotoxicity in SH-SY5Y cells.612 Thonningianin A, a polyphenolic compound found in natural plant foods, attenuated ferroptosis induced by 6-OHDA in zebrafish and dopaminergic neurons. It exerts its effects by targeting the Kelch domain of Keap1 and promoting its degradation in an Atg7-dependent manner, leading to the translocation of Nrf2 into nucleus, suggesting it as a Keap1-Nrf2 PPI inhibitor to inhibit ferroptosis.613 (-)-Clausenamide Clau, an alkaloid isolated from the plant Clausena lansium (Lour.), exhibits both in vivo and in vitro neuroprotective activities as a scavenger of lipid peroxide products. Data analysis reveals that (-)-Clausenamide directly targets Ser663 on ALOX5, which is the PKCα-phosphorylation site.614 The results propose a new idea that targeting ALOX5 and preventing ferroptosis in dopaminergic neurons may be a strategy for PD therapy. Some extracts have also shown protective roles against metal-induced toxicity. Oral administration of E. amoenum extract increased catecholamine content and improved depression-like behavior in the Mn2+-treated hippocampus of rats.615 Anthocyanin-rich açaí (Euterpe oleracea Mart.) extract attenuates manganese-induced oxidative stress in primary astrocyte cultures of rats.616 The polyphenolic extract of Euphorbia supina could enhance antioxidant activity by regulating ER stress and mediating apoptosis to attenuate manganese-induced neurotoxicity.617 The similar effects were also observed in the extract of Melissa officinalis aqueous and Peumus boldus (Boldo) Aqueous e in manganese-injured mice or D. melanogaster.618,619
Multifunctional iron chelators
In recent years, a series of multifunctional iron chelators has been reported. One such example is the multimodal iron chelating drug M30, which has been shown to facilitate numerous neuroprotective-adaptive mechanisms and pro-survival signaling pathways. It has also been reported to increase striatal dopamine levels and enhance dopaminergic and transferrin receptor cell populations in the SNpc.620,621 Recently, Bar-Am et al. synthesized a novel multipotent compound called VAR10303, which possesses brain-permeable iron-chelation properties. This compound demonstrated significant improvement in PD symptoms in rat models induced by 6-OHDA as well as mouse models treated with MPTP.622 Gutbier et al. designed a series of small molecules capable of transporting iron to the brain via the neutral amino acid transporter, LAT1 (SLC7A5), which was found to effectively block neurite loss and cell death induced by MPP+ and 6-OHDA in LUHMES cells.623 In an MPTP-induced PD mouse model, GW501516, known as a PPARδ agonist, showed potential for alleviating motor impairment and reducing NLRP3 inflammasomes-mediated neuroinflammation.624 Furthermore, it was discovered that it regulates the expression of FPN1 and ferritin, while modulating cellular iron homeostasis without affecting TfR1 in the context of 6-OHDA.625 Additionally, α-Lipoic acid has been reported to decrease the occurrence of neuropathy, and it is possible that the PI3K/Akt/Nrf2 pathway could serve as a potential target.626 In the context of severe manganese-induced Parkinsonism in humans, Para-aminosalicylic acid (PAS), an FDA-approved anti-tuberculosis drug, has demonstrated neuroprotective effects. PAS effectively reduced manganese levels in the striatum, hippocampus, and frontal cortex of Sprague-Dawley rats, along with iron and copper levels.627,628
Clinical trials targeting metal ions
In certain clinical practices for the treatment of PD, chelators have demonstrated favorable efficacy. DFP, as a membrane-permeant bidentate chelator, has shown neuroprotective effects in both experimental models and clinical trials of PD (Table 3). DFP effectively reduces labile iron levels and attenuates oxidation products of lipids and DNA in cells and animals subjected to oxidative stress, leading to improvements in motor functions while mitigating dopamine depletion.15 A case report revealed that DFP treatment (30 mg/kg/day for 32 months) resulted in decreased iron accumulation in the bilateral dentate nuclei and SN of a PD patient with dysarthria and orofacial dystonia, as well as iron accumulation across various brain regions including the dentate nuclei, SN, and red nuclei.629 Despite significant improvement in iron overload, the patient’s best T2* values had not fully recovered. A double-blind placebo clinical trial involving 40 patients was conducted to evaluate the efficacy and safety of DFP (30 mg/kg/day).15 The results indicated that DFP significantly reduced iron content in the SN, with slight improvement observed in motor signs after 1 year of treatment. Although three cases of neutropenia occurred, symptoms rapidly disappeared upon discontinuation of oral therapy (ClinicalTrials.gov reference: NCT00943748). Another study also demonstrated that DFP therapy effectively suppressed iron accumulation in specific brain regions in PD patients for 6 months (either 20 or 30 mg/kg), with the exception of two patients who developed neutropenia (ClinicalTrials.gov reference: NCT01539837).630 Iron chelation may serve as a potential alternative treatment option for individuals suffering from neurodegeneration associated with brain iron accumulation. However, it is important to consider the significant decrease in plasma ferritin levels following long-term treatment, as this could potentially lead to body iron depletion and subsequent iron deficiency. Therefore, when evaluating the positive effects of chelators, one must carefully assess the risk of agranulocytosis and determine an optimal dosage.629,630 Additionally, Grolez et al. demonstrated that patients with lower Cp activity displayed a more favorable response to iron chelation therapy (ClinicalTrials.gov reference: NCT00943748).282 However, it is crucial to conduct large-scale trials in order to establish the relationship between Cp activity and PD. A FAIRPARK-II clinical trial of DFP investigated the use of DFP in early PD patients and revealed that no clinical benefit from this therapy; instead, it resulted in a significant decrease in MDS-UPDRS scores at 9 months and worse scores on measures of Parkinsonism compared to those receiving placebo over a 36-week period (ClinicalTrials.gov reference: NCT02655315).631 These findings suggest that caution should be exercised when considering iron-chelating agents as potential therapeutic interventions for PD.
CuII(atsm) exhibited neuroprotective effects in four distinct animal models of PD by enhancing dopamine metabolism and restoring cognitive and motor function. In vitro, it demonstrated the inhibition of peroxynitrite-mediated formation of α-synuclein oligomers.632 Encouraging results from separate Phase 1 studies in patients with ALS and PD indicate that CuII(atsm) inhibits Fe2+-induced lipid peroxidation, suggesting its potential as a disease-modifying mechanism against ferroptosis.633 With comparable potency to liproxstatin-1 in vitro, CuII(atsm) possesses favorable properties such as oral bioavailability and brain penetration, which makes it an attractive investigational product for clinical trials targeting ferroptosis-related diseases.634 Additionally, N-acetylcysteine (NAC) and GSH have shown improved clinical symptoms in PD patients during clinical trials (ClinicalTrials.gov reference: NCT02424708; NCT02445651).635,636 Omega fatty acids and CoQ10 have demonstrated some efficacy in treating PD patients637,638; however, the therapeutic response depends on baseline levels of ubiquinol and the dosage of CoQ10 (ClinicalTrials.gov reference: NCT00740714). Furthermore, vitamin E co-supplemented with omega-3 fatty acids has also been found to have favorable effects on UPDRS and markers of insulin metabolism in individuals with PD.639 Limited treatment options are available for manganese-induced neurotoxicity; chelation therapy is preferred. Calcium disodium ethylenediaminetetraacetate (CaNa2EDTA), which replaces toxic metals with calcium within the EDTA core, thereby inhibiting dopaminergic auto-oxidation, has been reported to improve seven workers affected by manganese-induced Parkinsonism.640,641 However, controversial results were observed due to poor BBB permeability leading to low bioavailability and efficacy.628
AD
Metal ions chelation
DFO and CQ derivative
The dysregulation of metal homeostasis contributes to the pathogenesis of AD. Therefore, metal chelators that can reduce extracellular metal bioavailability or compete with endogenous ligands for metal ions may be a beneficial choice in the treatment of AD642 (Table 2). Intranasal DFO has been reported to reduce cognitive decline and inhibit amyloid beta deposits in P301L tau transgenic mice643 and APP/PS1 mice.355,644 Intranasal DFO can act on multiple processes to slow disease progression, with the most significant improvements observed in reduced Aβ aggregation and hyperphosphorylation of tau.355 DFO is not only a metal-chelator that suppress ferroptosis, including iron-associated oxidative stress,645 but also a potent inhibitor of GSK-3β, which may connect Aβ accumulation and subsequent tau hyperphosphorylation in AD.643,644 DFO also targets HIF-1α to activate glucose transporters, including glucose transporter 1 (GLUT1), and plays a role in the activation of insulin signaling pathway.646,647,648 The N-acylhydrazones group is a precursor for many compounds with chelating properties. An example of such a compound is 8-hydroxyquinoline (8-HQ), which is a natural lipophilic molecule that exhibits strong anti-AD effects by chelating metal ions.649 Based on the structural framework of 8-HQ, a series of nitroxoline-based analogs were designed, including 8- H2QH, 8-H2QT, and 8-H2QS. These compounds exhibited inhibitory effects on self-induced aggregation of the amyloid beta peptide (Aβ1–42) and selectively formed complexes with Cu2+.597,650,651 CQ is a nonspecific copper-zinc chelator derived from 8-HQ, which not only improves cognitive behavior and decreases Aβ deposits in an APP transgenic mouse model of AD,652 but also prevents age-dependent brain atrophy, iron accumulation, and concomitant cognitive deficits in tau-knockout mice.653 Another mechanism of CQ against AD involves the resumption of Cu2+-suppressed fibril growth of Aβ (1–40). Interestingly, when combined with Zn2+ ions, it synergistically retards fibril growth.654 Furthermore, the combination therapy involving CQ and dihydropyrimidine-thiones demonstrates enhanced amelioration of Aβ toxicity by reducing Aβ levels and restoring functional vesicle trafficking, highlighting the potential value of synergistic compounds for enhancing targeted activities.655 Novel polymeric derivatives based on the structure of 8-HQ were designed with N and O donor atoms similar to those present in CQ, and these derivatives exhibit a superior capacity for disaggregating Aβ plaques due to their strong affinity for copper ions.656 The structure of CQ was used as a basis for synthesizing a series of selenium-containing derivatives. Importantly, compound 8a exhibited predominant antioxidant activity and effectively inhibited Cu2+-induced Aβ1–42 aggregation, suggesting its potential as a multifunctional agent for treating AD.657 The compounds X1TMP and X1Benz, which possess the ability to diminish ROS generation by the copper2+-Aβ system, exhibit promising potential against Aβ fibril formation in AD.658 Additionally, a series of adamantane-based semicarbazones and hydrazones compounds were designed and demonstrated as novel therapeutic agents by inhibiting Cu2+-mediated Aβ aggregation and effectively chelating iron.659 Furthermore, certain synthetic hydrazone ligands presented efficient inhibition of Aβ fragments aggregation and efficiently chelated copper and zinc.660
Polyphenols compounds
Besides, polyphenols have been proven to be effective in preventing the death of dopaminergic neurons. Curcumin has been reported to reduce the aggregation of amyloid and oxidized proteins, as well as improve cognitive deficits. As a metal chelating agent, its alternative mechanism may involve reducing metal ions to prevent Aβ amyloid fibril formation.661 Curcumin displayed different affinity for copper, zinc, and iron. At least two curcumin molecules can bind to Cu2+ or Fe2+ ion, and exhibited positive or negative cooperativity, with little bind to Zn2+,.662 A curcumin analog named CRANAD-17, which utilizes curcumin as an anchoring moiety to H13 and H14 of Aβ, has the capability to inhibit copper-induced cross-linking of Aβ42, suggesting that the curcumin scaffold holds potential as a tool for AD research.663 (-)-epigallocatechin (EGC) and (-)-epicatechin-3-gallate (ECG), the main polyphenolic compounds derived from tea, have been found to alleviate the toxicity of Aβ oligomers and fibrils induced by Cu2+ or Zn2+.664 Mandel et al. proposed the use of a cocktail of multimodal brain-permeable iron-chelating drugs that possess neuroprotective-neurorescue and APP-processing regulatory activities as therapeutic agents, with a specific focus on EGCG and curcumin.665
Flavonoids compounds
Flavonoids, a class of polyphenolic compounds, have been shown to independently chelate metal ions and interact with Aβ, indicating their potential bifunctionality toward metal–Aβ species. Eriodictyol, a natural flavonoid compound, effectively ameliorated cognitive deficits in APP/PS1 mice and suppressed Tau phosphorylation in Aβ1–42 induced HT22 cells and APP/PS1 mice. Moreover, eriodictyol exerted an anti-ferroptosis effect by activating the Nrf2/HO-1 signaling pathway.666 The biflavonoid compound mentoflavone, which possesses good metal-chelating properties, can regulate the formation of neurotoxic soluble Aβ42 oligomers and reduce the Cu2+-ascorbate redox cycling and ROS formation.667 Luteolin, derived from Elsholtzia rugulosa, exhibits inhibitory effects on copper-induced neurotoxicity in SH-SY5Y cells overexpressing APP by reducing Aβ secretion, maintaining mitochondrial function, and suppressing the caspase-mediated apoptosis.668 Similar effects were observed with apigenin and vitegnoside.669,670 Myricetin has been identified as another natural regulator of metal-induced Aβ aggregation and neurotoxicity.671 Silibinin, a clinically used hepatoprotectant isolated from Silybum marinum, attenuates memory impairment induced by Aβ by reducing oxidative stress and inflammation in the mouse brain.672 It has been reported that silymarin, delivered through silymarin-encapsulated liposome nanoparticles, can protect against zinc-induced dopaminergic neurodegeneration, improve spatial memory, and reduce depression induced by copper.673,674 Additionally, silymarin protects the cerebral cortex against manganese-induced neurotoxicity in adult rats.675 Due to three potential metal-binding sites mediated by the catechol moieties, quercetin possesses natural chelating activity, which can combat copper-induced toxicity in SH-SY5Y cells by regulating apoptotic cell death and ER stress involving PI3K/Akt and ERK1/2 signaling.606,676 Liu et al. developed a nanomedicine by modifying quercetin with triphenylphosphonium (TQCN), which efficiently chelates iron to ameliorate various neurodegenerative manifestations associated with brain iron deposition, thus rescuing severe cognitive decline in AD mice and demonstrating the promising potential of intelligent nanotherapeutics against AD progression.677 Sterubin and fisetin, two flavonoids derived from natural pharmacopeia, have shown strong inhibition on oxytosis/ferroptosis. Recently, it has been reported that sterubin and fisetin have diverse homeostatic impacts on mitochondrial physiology. Although they can enhance bioenergetic efficiency by restoring mitochondrial homeostasis in terms of redox regulation, calcium uptake, biogenesis, fusion/fission dynamics, and modulation of respiration, they do not require mitochondria to exert neuroprotective effects.678 The intricate interplay and targets should be evaluated when considering these compounds as agents. The assessment of structure-activity relationships revealed specific binding sites for metal ions, including the catechol moiety in the B-ring, the 3-hydroxy and 4-oxo groups in the C-ring, and the 4-oxo and 5-hydroxyl groups between the C- and A-rings.679 Genistein-O-alkylbenzylamine derivatives emerged as Cu2+ chelators and significantly inhibited copper-induced Aβ aggregation.680 Another noteworthy example based on flavonoids is FLV2 5, which exhibited copper chelating properties along with enhanced AChE inhibitory activity and inhibition of self-induced Aβ aggregation.681
Alkaloids and terpene compounds
Several other natural alkaloids, lactones and terpenes with neuroprotective effects also demonstrate the ability to chelate metal ions. Berberine, a natural alkaloid, exhibited neuroprotective value in triple transgenic AD mice by inhibiting iron levels and ferroptosis while activating Nrf2 signaling in RSL3-induced ferroptosis.682 Recent reports have found that coumarin and its analogs and derivatives possess many properties targeting AD, such as chelation of metal ions, inhibition of cholinesterases (ChEs) or monoamine oxidase B, proposing coumarin as a potential scaffold for improving the structure of anti-AD therapeutics.683,684 Naringin has been reported to efficiently decrease nonheme iron, thereby reducing the formation of amyloid plaques in iron-overloaded mice.685 Tetrahydroxy stilbene glycoside (TSG) dose-dependently mitigates Aβ-induced brain damage by activating GSH/GPX4/ROS and Keap1/Nrf2/ARE signaling pathways. Additionally, TSG restores mitochondrial function to reverse Aβ-induced injury, suggesting its potential as a multifunctional candidate against AD.686 Qian et al. synthesized a derivative of TSG, called Mito-TSG, and demonstrated its superior effects in mitigating mitochondrial free radical damage and apoptosis in APP695V717I transgenic model mice.687 1, 6-O, O-diacetylbritannilactone (OABL), a 1,10-seco-eudesmane sesquiterpene lactone isolated from the herb Inula britannica L., attenuates the impairments in cognitive function and the overactivation of microglia and astrocytes in the brains of 5xFAD mice, which also inhibits oxytosis and ferroptosis in PC12 cells.688 Ginkgolide B, a terpene lactone derivative of Ginkgo biloba, also alleviates AD-induced cognitive defects by mitigating oxidative stress, neuroinflammation, and ferroptosis, and the inhibition of ferroptosis has been found to be essential for the beneficial effects of Ginkgolide B in AD using SAMP8 mice as models.689 Hinokitiol, a potent small molecule inhibitor of ferroptosis, dynamically transports iron from areas with high-iron concentration to those with low concentration in order to dissipate iron buildup. It has been reported to alleviate neurotoxicity and rescue the neurobehavioral impairment caused by paclitaxel or 6-OHDA in vitro and in vivo.690 Senegenin (Sen), the major component of Radix Polygala, exhibits diverse pharmacological activities in neurodegenerative disorders by enhancing cognitive function, exerting anti-aging effects, and acting as an antioxidant. It effectively reverses mitochondrial depolarization in Aβ25–35-induced PC12 cells, demonstrating potent neuroprotective properties against oxidative damage and lipid metabolite associated with ferroptosis.691 Considering the necessity for specific metal-ion binding affinity coupled with antioxidant functionalities, yclen (1) acts as a metal ion passivation and antioxidant agent to prevent and disrupt copper-induced aggregation of Aβ (1-40).692 Furthermore, the alkaloid extract from African Jointfir (Gnetum africanum) leaves counteracted manganese-induced elevation in acetylcholinesterase enzyme (AChE) activity, as well as levels of nitric oxide (NO) and ROS, in Drosophila melanogaster.693
Amino acids and polypeptides
The α-carbon of amino acids, such as methionine, tyrosine, histidine, and cysteine, can chelate metals to form metal complexes through their amine and carboxylate moieties. As a result, peptides composed of these amino acids also possess the ability to chelate metals. A notable example is the histidine-bearing dipeptide. Additionally, certain peptides have been identified as potent inhibitors of Aβ peptide aggregation, including tryptophantyrosine, glycine-arginine dipeptide, methioninetryptophan dipeptide, muramyl dipeptide, and hydroxyethylene-containing diphenyl alaninedipeptide.694 In a study by Suchita et al., bifunctional backbone-modified squaramide dipeptides were synthesized, which have the ability to inhibit Aβ aggregation and selectively chelate metal ions (Cu2+, Zn2+, and Fe3+).694 Additionally, these dipeptides exhibit antioxidant properties. Therefore, they hold potential as novel chemical entities for AD treatment. GHK (Gly-His-Lys), a bioactive matricryptin released from extracellular proteins via limited proteolysis, suppresses ROS production catalyzed by the copper-Aβ complex system.695 Its conjugate (BioGHK) exhibits similar effects while also displaying antiglycating activity toward carbonylated species Aβ. Asadbegi et al.’s research led to the discovery of a novel multifunctional ligand that can simultaneously inhibit Aβ42 and chelation zinc.696 The inhibitor domain binds to the C-terminal hydrophobic region of Aβ while another domain is responsible for zinc chelation, as confirmed by molecular docking and molecular dynamics evaluation.
Compounds based on lead compounds
The lead compounds were modified to enhance their metal chelating effect, leading to the development of numerous new compounds. Gunesch et al. synthesized a series of 7-O-Esters of taxifolin, named 7-O-cinnamoyltaxifolin and 7-O-feruloyltaxifolin, which exhibits significant synergistic neuroprotective effects in an HT22 cell model by effectively inhibiting ferroptosis.697 Additionally, administration of these compounds improved short-term memory in an AD mouse model induced by oligomerized Aβ25–35 peptides, potentially through modulation of Nrf2/GSH signaling as a therapeutic target. Three catechol-based ligands demonstrated selective chelation abilities towards redox-active metal ions, particularly Cu2+ and Fe3+, with a lower affinity for Zn2+.698 These ligands effectively inhibited the reduction of dioxygen of Cu-II-Aβ (1–16). To investigate the role of metal-Aβ interactions in AD pathogenesis, ligands were designed that incorporate a metal binding site into a framework known to interact with Aβ.699 These ligands have been shown to attenuate ROS generation and mitigate oxidative stress. Based on natural-origin compound scaffolds, several multitarget compounds have been designed and applied for the treatment of various neurodegenerative diseases. Among them, chromone-lipoic acid conjugate exhibits acceptable butyrylcholinesterase inhibition activity, as well as antioxidant and copper-chelation properties, making it a promising anti-AD agent.700 SNH6, a hydrazone of 6-methoxysalicylaldehyde nicotinoyl, exhibits significant inhibition of copper-mediated Aβ aggregation and effectively donated NAD+ to NAD-dependent metabolic processes.701 Additionally, it demonstrated efficacy in chelation iron, suggesting its potential as a multifunctional precursor for AD treatment. The multitarget-directed ligands (MTDLs) strategy was employed to synthesize a class of chrysin derivatives. These compounds demonstrated effective inhibition of self-, Cu2+-, and AChE-induced Aβ1–42 aggregation, with favorable BBB penetration and predicted drug-like properties in silico. Notably, compound 1 exhibited highly selective inhibition of butyrylcholinesterase (BuChE).702,703 Bolognesi et al. modified a lead compound bis(7)-tacrine and endowed it with triple function as a dual-binding site AChE inhibitor and chelating metals, which presented inhibited effects on the AChE-induced Aβ aggregation.704 Triethylene tetramine dihydrochloride (trientine), a selective copper chelator, was found to mitigate amyloidosis in the brain of APP/PS1 mice by inhibiting the RAGE/NF-κB/BACE1 pathway.705 Subsequently, Li et al. designed a novel compound named melatonin-trientine by covalently synthesizing melatonin with trientine. The compound effectively maximized the therapeutic efficacy by targeting multiple mechanisms associated with AD pathology by not only inhibiting metal ion dyshomeostasis but also decreasing Aβ deposition.706 Resveratrol, a multitarget compound, exerts an anti-apoptotic effect and reduces Aβ and SOD1 aggregation through its antioxidant activity and mitochondrial protection.707,708 By virtue of its stilbene structure, a series of imine resveratrol derivatives was synthesized with the ability to chelate copper and inhibit Aβ aggregation.709 Tenuazonic acid and its conjugates with donepezil were shown to be a potent chelator of Fe3+, Cu2+, and Zn2+ with the potential effects on AChE and Aβ aggregation inhibition, proving to be noncytotoxic against SH-SY5Y cells.710 Another group of mental chelators reported by Kilic et al. includes a series of thiourea and benzamide derivatives that exhibit ChEs inhibition.711 Results from a pilot placebo-controlled trial on AD patients revealed that although d-penicillamine, a copper-chelating agent, decreased oxidative stress, no difference was observed in the rate of cognitive decline.712 Further studies involving larger cohorts are required to elucidate the drug’s efficacy in relation to the clinical progression of AD. Synthesized hydroxylated chalcones with dual-functional inhibitors targeting Aβ aggregation and ferroptosis, along which chalcones 14a-c exhibited a more significant neuroprotection against Aβ1–42 induced toxicity.713 The compounds also demonstrated favorable ADMET properties and BBB penetration. Iminodiacetic acid-modified human serum albumin (I-HSA) effectively inhibits Zn2+ and Cu2+-associated Aβ42 aggregation, making it the first multifunctional macromolecule for reducing metal-induced Aβ42 aggregation.714 This finding provides valuable insights for the development of macromolecular drugs targeting AD. Furthermore, a series of DFP derivatives has been synthesized as dual-binding site AChE inhibitors, allowing simultaneous interaction with catalytic active site and peripheral anionic site of the enzyme, and these compounds chelate bio-metals co-localized in Aβ plaques, such as iron, in some cases with copper and zinc.715
Other compounds
Edaravone, a drug targeted mitochondria and recognized for its ability to scavenge hydroxyl radicals (∙OH) and inhibit lipid peroxidation, has emerged as a promising therapeutic agent against various neurological disorders.716,717 Recently, it has been demonstrated that it can reduce Aβ1–42-induced apoptosis of HT22 cells by inhibiting the TLR4/NF-κB /NLRP3 signaling pathway and ferroptosis.718 Additionally, the compound 1,1’-xylyl bis-1,4,8,11-tetraaza cyclotetradecane was reported as an effective drug for clearing Aβ by specifically reducing the transcription of APP and targeting copper concentration in the brain cortex region.719 In a search for neuroprotective agents, GIF-0726-r, an oxindole compound, presented neuroprotective roles against glutamate-induced oxytosis and erastin-induced ferroptosis in the hippocampal HT22 cell line. Additionally, GIF-0726-r was found to effectively inhibit the accumulation of ROS and Ca2+ influx. Several synthesized derivatives of GIF-0726-r, were identified as potent activators of the antioxidant response element.720 Many compounds with positive treated effects also exhibit anti-AD roles, such as M30, which can effectively reduce Aβ accumulation and tau phosphorylation, and alleviate memory deficits.721,722 The metal chelator H2dqpyca and H2bqch exhibited significant neuroprotection against both Aβ peptide- and H2O2-induced toxicities in SH-SY5Y cells, indicating a strong interaction with the catalytic active site of AChE.723 The administration of salidroside alleviated cognitive impairment and inhibited neuronal ferroptosis in SAMP8 mice, possibly through activating Nrf2/GPX4 axis and reducing the infiltration of CD8 + T cells.724 β-hydroxybutyrate (BHB) alleviated oxidative stress and ferroptosis induced by MPP+/MPTP in SN4741 cells and C57BL/6 mice by modulating zinc finger protein 36 (ZFP36)/ACSL4 axis, with ZFP36 being identified as a target of BHB.725 Virtual screening technology has been recognized and applied in the screening targeted compounds.726 Soriano-Castell et al. discovered a small set of anti-oxytotic/ferroptotic quinones from a comprehensive library of over 900 natural compounds, which protect against intracellular Aβ toxicity in nerve cells by modulating quinone oxidoreductase 1 (NQO1) and ferroptosis suppressor protein 1 (FSP1).727 An imidazolylacetophenone oxime ether (IOE) was discovered against neuroinflammation based on the screening (∼1400 compounds). Its derivatives, specifically compound 12i, exhibited prominent metal-chelating and inhibition of AChE properties.728 It inhibited H2O2-induced PC12 cells damage and ferroptosis, while also showing potential for chelating copper with BBB permeability. Two small molecules, VU0063088 and VU0026921, were selected from a high throughput screen of 40,167 compounds, which play protective roles against manganese toxicity in C. elegans by modulating manganese levels in the worms independently of SMF-2, a regulator of manganese transport.729 Another molecule, known as MESM, has been identified as a manganese-selective ionophore, which can effectively reduce excessive cellular manganese without altering levels of calcium, iron, cobalt, copper, zinc or molybdenum.730 This discovery holds promise for patients with elevated manganese levels.
Clinical trials targeting metal ions
A large number of metal chelator compounds have been developed to treat AD; however, only a few of them have progressed to clinical trials. The clinical trials involving metal chelated therapeutics are presented in Table 3.
Curcumin not only inhibits Aβ aggregation and the activities of β-secretase in copper-induced cells, but also decreases Aβ deposition and oligomer formation, reduces tau phosphorylation, and improves behavioral impairment in AD animal models.731 The potential mechanisms may be related to oxidative stress and mitochondrial apoptosis.732 Chelation with copper and iron suggests one possible mechanism of action in AD animal models.662 Currently, several clinical trials investigating the effects of curcumin on AD has been conducted (ClinicalTrials.gov reference: NCT04149639; NCT01716637; NCT01001637; NCT00164749; NCT00099710).731,733,734 However, no significant differences in changes in cognitive function were found between placebo and curcumin groups. Additional trials are necessary to evaluate the roles in AD, while considering its bioavailability and ability to penetrate the BBB. Based on the structure-activity relationship, many curcumin derivatives have been designed to potentially play a more effective role.709 A 2-year, single-blind study showed that 24 months of treatment with DFO (125 mg intramuscularly twice daily, 5 days per week) reduced the rate of decline in daily living skills of 48 patients with probable AD.26 Slight side effects, including weight (1/48) loss and appetite (4/48) were observed. An ongoing clinical trial (NCT03234686) on DFP in AD has yielded several outcomes, including performance on a neuropsychological test battery and measurement of brain iron levels using MRI. PBT2 (5, 7-dichloro-2-(dimethylamino)-methyl-8-hydroxyquinoline), a derivative of CQ, is in clinical trials for the treatment of AD and HD with well safety and tolerability (ClinicalTrials.gov reference: NCT00471211 and NCT01590888). It exerts a central effect on amyloid plaque metabolism by binding excesses of copper and zinc, possibly iron, to inhibit the formation of toxic oligomers. Patients treated with PBT2 at a dose of 250 mg showed significant improvement in two executive functions and a dose-dependent reduction in CSF Aβ concentration.630,735,736 Despite PBT2 appearing to improve cognitive function in animal models and in clinical trials, it did not have a significant effect on the burden of amyloid plaque in larger-scale clinical trials.737 Furthermore, CQ exhibited severe neurotoxic side effects, and PBT2 did not meet the predetermined endpoints; therefore, the clinical trial was terminated. The hydroxyl group and the nitrogen atom was postulated as the important mental binding.738 Subsequently, several other copper chelators derived from CQ have been developed based on the ion properties and structure-activity relationship. Among them, bis-8-hydroxyquinoline derivatives (PA1637, with undisclosed structure) showed superior therapeutic effects and significantly lower toxicity compared to CQ in an AD mouse model; further clinical trials are required to validate its effects.597,739
ALS
Iron accumulation has been observed in animal models and in both sporadic and familial forms of ALS patients. DFP was reported to extend the mean lifespan of Sod1G86R mice compared with placebo. In a pilot clinical trial lasting 12 months (ClinicalTrials.gov reference: NCT 00868166), DFP has been proven to be good safety without anemia. Additionally, DFP treatment (30 mg/kg/day) for the first 3 months significantly improved the ALS Functional Rating Scale and body mass index compared to the initial treatment-free period. After DFP treatment, iron levels in multiple sites and oxidative stress in CSF decreased, suggesting that moderate iron chelation may be a novel therapeutic approach for neuroprotection in ALS.740 It was reported that oral treatment with CuII(atsm) improved locomotor function and the survival of the SOD1G37R mice through improving the metal content of mutant SOD1 in vivo, rather than reducing levels of misfolded SOD1.633,741,742 Furthermore, administration of CuII(atsm) showed improvements on the lifespan and preservation of motor neurons in transgenic SOD1G93A mice, and the activation of astrocyte and microglial was also been attenuated.743,744 A clinical trial (ClinicalTrials.gov reference: NCT02870634), completed in 2020, demonstrated that disease severity and cognitive function relative to historical controls were improved after treatment with CuII(atsm) in both phase 1 and phase 2a cohorts. However, another clinical trial (ClinicalTrials.gov reference: NCT04082832) found that CuII(atsm) has no significant effect on motor neuronal pathology in patients with ALS.745 Therefore, further studies are needed to evaluate the effects of CuII(atsm). In another study, the SOD1-G73R mice treated with ZnII(atsm) presented similar improvements in locomotor and survival. Interestingly, this treatment did not affect the content of zinc levels, but increased the levels of copper in SOD1 and overall, suggesting that the neuroprotective effects of ZnII(atsm) may in fact be due to subsequent delivery of copper.746 These findings suggest that improving copper content represents a promising therapeutic strategy for treating ALS. In contrast, many copper-chelating drugs have been reported to be beneficial in mutant SOD1 mouse models. For example, ammonium tetrathiomolybdate (TTM), a copper chelator, prolonged the survival of both presymptomatic and symptomatic SOD1-G93A mice by removing copper from the copper thiolate cluster of SOD1, which led to the degeneration of mutant SOD1.747,748,749 Tardiff et al. found that different 8-HQ protect models of TDP-43 protein, α-synuclein, and polyglutamine proteotoxicity through distinct mechanisms.750 HQ-415 and CQ exhibited stronger chelation activities than HQ-161. Each model can rely on at least two metal chelation pathways to be involved.750 The lipophilic metal chelators DP-109 and DP-460, which chelate calcium, copper, and zinc, significantly improved motor performance, significantly extended survival, and dramatically reduced cell loss in the lumbar spinal cord of G93A-transgenic ALS mouse model.751 Brain-permeable iron chelators VK28 and M30 have shown to prevent motor neuron degeneration in SOD1G93A transgenic mice of ALS.752 Additionally, other chelators such as metformin and cyclodipeptides have been effectively applied to treat neurodegenerative diseases, including AD, despite being originally designed for other purposes.683,753
HD
Many metal chelator compounds also exhibit neuroprotective effects and resistance against HD. CQ, a metal-binding compound, decreased the expression of mutant protein with olyglutamine-expanded huntingtin exon 1 in PC12 cells and improved behavioral and pathologic phenotypes in transgenic R6/2 HD mice.754 PBT2 was also reported to improve motor ability, increase body and brain weight, reduce striatal atrophy of R6/2 mouse, and ameliorate toxicity in a C. elegans model of polyQ aggregation.755 In 2014, a randomized, double-blind, placebo-controlled trial was conducted for 26 weeks (ClinicalTrials.gov reference: NCT01590888). The early/mid-stage HD patients were administered PBT2 (250 or 100 mg) or placebo once daily, however, neither dose significantly improved cognition on the cognitive tests until the end of the trail. PBT2 was generally safe and well tolerated, a larger study was needed to evaluate potential benefits.756 Chen et al. found that intra-ventricular administration of DFO results in an improvement in the motor phenotype in R6/2 HD mice.392 Additionally, oral DFP treatment for 10 days on 9-week R6/2 HD mice not only removed mitochondrial iron to decrease lipid peroxidation but also improved motor endurance.396
Conclusion and perspective
Although the contribution of dysregulated metal ions, especially iron deposition, has been implicated in the pathological progression of neurodegenerative diseases and proposed as a promising target strategy for prevention or treatment, the exact distribution map of metal ions among different brain regions, cells, and organelles is still unclear. In patients with PD, AD, ALS, and HD, the iron levels in specific brain regions are significantly elevated, and these iron levels are closely associated to the severity of the disease.46,47,757,758 At the brain regional level, the identification of the axonal iron transport pathway from the vHip to the mPFC to the SN may support the hypothesis that dysregulated axonal iron transport results in abnormal iron distribution among different brain regions, particularly in the SN, thereby causing PD-related symptoms.74,75,759 It is worth investigating whether abnormal axonal iron transport contributes to iron deposition and subsequently results in motor or non-motor symptoms of neurodegenerative diseases. Further investigations focusing on this hypothesis may provide a new perspective and mechanism for understanding the pathogenesis at the regional level. As demands for iron and other metal ions vary among neurons, glial cells, and different organelles, further investigations focusing on the crosstalk of metal ion distribution among cells and organelles in the CNS may provide a comprehensive understanding of their role in the development of neurodegenerative diseases.
The early diagnosis of neurodegenerative diseases faces multiple challenges. Firstly, the pathological processes of these diseases often initiate many years before clinical symptoms manifest, making it difficult to diagnose them through traditional clinical assessments prior to symptom onset. Secondly, early symptoms are frequently nonspecific and can be mistaken for various other diseases, resulting in misdiagnosis or missed diagnoses. Furthermore, the identification of biomarkers for neurodegenerative diseases remains incomplete, thereby limiting the accuracy and reliability of early detection. Considering the crucial role of metal ions in the development of neurodegenerative diseases, the levels and distribution of these ions can serve as biomarkers, aiding in the early diagnosis of such conditions. Currently, researchers are exploring how these metal ions change in biological samples such as CSF and blood, and their relationship to the pathological features of neurodegenerative diseases. By developing more sensitive biochemical and molecular biology techniques, researchers can quantitatively analyze these metal ions and assess their potential as diagnostic markers. Additionally, higher sensitivity and specificity in MRI imaging, along with the development of specific probes, will also contribute to evaluating changes in metal ion levels in vivo, providing technical support for early disease diagnosis.
Due to complex causative factors, the precise pathophysiological mechanisms underlying neurodegenerative diseases have yet to be fully elucidated. This lack of understanding complicates drug design aimed at targeting specific molecules or pathways. Furthermore, limited specificity and sensitivity in biomarkers present challenges not only for early diagnosis and monitoring disease progression but also result in delays in timely treatment for these conditions. The slow progression of neurodegenerative diseases coupled with specific barriers within the brain makes it challenging to effectively distribute, metabolize, and evaluate new drugs within the body, consequently affecting the efficiency of drug screening and verification. The advantage of metal ion chelators in neurodegenerative diseases lies in their ability to regulate the bioavailability of metal ions, reduce oxidative stress and protein aggregation, thereby providing neuroprotective effects. These advantages make metal ions to be promising targets for the treatment of neurodegenerative diseases. Metal ions, such as iron, manganese, copper, and zinc, summarized in this review, play important roles in numerous biological processes in the brain. However, their accumulation can lead to ferroptosis, cuproptosis, oxidative stress, cell senescence, and neuroinflammation. Therefore, an imbalance of metal ion levels is detrimental. Imbalances in iron metabolism contribute to neuronal death through the ferroptosis pathway. Imbalances in iron metabolism contribute to neuronal death through the ferroptosis pathway, while imbalances in copper metabolism lead to neuronal death via the cuproptosis pathway. Therefore, interventions targeting the metabolism of iron or copper, particularly the ferroptosis and cuproptosis pathways, may become treatment strategies for neurodegenerative diseases.22,152,760,761 Genetic or pharmacological strategies targeting metal ion distribution at the levels of brain region, cell or organelle may be more effective in modulating motor or non-motor behaviors in neurodegenerative diseases. Chinese herbal medicine contains a large number of natural active compounds, which often exhibit unique pharmacological effects.762 Screening natural compounds from Chinese herbal medicine that chelate metal ions and structurally modifying them to improve their selectivity, reduce toxicity, enhance their ability to penetrate the BBB, and develop multimodal therapeutic strategies may be an effective approach for developing new drugs for neurodegenerative diseases.
References
Lhermitte, J., Kraus, W. M. & McAlpine, D. Original Papers: On the occurrence of abnormal deposits of iron in the brain in parkinsonism with special reference to its localisation. J. Neurol. Psychopathol. 5, 195–208 (1924).
Dexter, D. T. et al. Increased nigral iron content in postmortem parkinsonian brain. Lancet 2, 1219–1220 (1987).
Dexter, D. T. et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114, 1953–1975 (1991).
Dexter, D. T. et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem 52, 1830–1836 (1989).
Mann, V. M. et al. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol. 36, 876–881 (1994).
Sofic, E. et al. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Transm. 74, 199–205 (1988).
Sofic, E. et al. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J. Neurochem 56, 978–982 (1991).
Riederer, P. et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem 52, 515–520 (1989).
Antonini, A. et al. T2 relaxation time in patients with Parkinson’s disease. Neurology 43, 697–700 (1993).
Castellani, R. J., Siedlak, S. L., Perry, G. & Smith, M. A. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 100, 111–114 (2000).
Chen, B. et al. Interactions between iron and alpha-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 141, 253–260 (2019).
Berg, D. et al. Iron accumulation in the substantia nigra in rats visualized by ultrasound. Ultrasound Med. Biol. 25, 901–904 (1999).
Berg, D. et al. Echogenicity of the substantia nigra: association with increased iron content and marker for susceptibility to nigrostriatal injury. Arch. Neurol. 59, 999–1005 (2002).
Kaur, D. et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron 37, 899–909 (2003).
Devos, D. et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal 21, 195–210 (2014).
Oakley, A. E. et al. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 68, 1820–1825 (2007).
Iranzo, A. et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. 12, 443–453 (2013).
Sun, J. et al. Quantitative evaluation of iron content in idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 35, 478–485 (2020).
Tambasco, N. et al. T2*-weighted MRI values correlate with motor and cognitive dysfunction in Parkinson’s disease. Neurobiol. Aging 80, 91–98 (2019).
Maass, F. et al. Cerebrospinal fluid iron-ferritin ratio as a potential progression marker for Parkinson’s disease. Mov. Disord. 36, 2967–2969 (2021).
Do Van, B. et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 94, 169–178 (2016).
Tang, F. et al. Inhibition of ACSL4 alleviates Parkinsonism phenotypes by reduction of lipid reactive oxygen species. Neurotherapeutics 20, 1154–1166 (2023).
Shen, Q. Q. et al. Cell senescence induced by toxic interaction between alpha-synuclein and iron precedes nigral dopaminergic neuron loss in a mouse model of Parkinson’s disease. Acta Pharm. Sin. 45, 268–281 (2024).
Goodman, L. Alzheimer’s disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. J. Nerv. Ment. Dis. 118, 97–130 (1953).
Hallgren, B. & Sourander, P. The non-haemin iron in the cerebral cortex in Alzheimer’s disease. J. Neurochem. 5, 307–310 (1960).
Crapper McLachlan, D. R. et al. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308 (1991).
Bartzokis, G. et al. In vivo evaluation of brain iron in Alzheimer’s disease and normal subjects using MRI. Biol. Psychiatry 35, 480–487 (1994).
Raven, E. P. et al. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimers Dis. 37, 127–136 (2013).
Smith, M. A., Harris, P. L., Sayre, L. M. & Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl Acad. Sci. USA 94, 9866–9868 (1997).
Rottkamp, C. A. et al. Redox-active iron mediates amyloid-beta toxicity. Free Radic. Biol. Med. 30, 447–450 (2001).
Ayton, S. et al. Cerebral quantitative susceptibility mapping predicts amyloid-beta-related cognitive decline. Brain 140, 2112–2119 (2017).
Ayton, S. et al. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 25, 2932–2941 (2020).
Spotorno, N. et al. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 143, 1341–1349 (2020).
Wu, Y. et al. Simultaneous Fe(2+)/Fe(3+) imaging shows Fe(3+) over Fe(2+) enrichment in Alzheimer’s disease mouse brain. Sci. Adv. 9, eade7622 (2023).
Everett, J. et al. Label-free in situ chemical characterization of amyloid plaques in human brain tissues. ACS Chem. Neurosci. 15, 1469–1483 (2024).
Mena, I., Marin, O., Fuenzalida, S. & Cotzias, G. C. Chronic manganese poisoning. clinical picture and manganese turnover. Neurology 17, 128–136 (1967).
Powers, K. M. et al. Parkinson’s disease risks associated with dietary iron, manganese, and other nutrient intakes. Neurology 60, 1761–1766 (2003).
Banta, R. G. & Markesbery, W. R. Elevated manganese levels associated with dementia and extrapyramidal signs. Neurology 27, 213–216 (1977).
Yamada, M. et al. Chronic manganese poisoning: a neuropathological study with determination of manganese distribution in the brain. Acta Neuropathol. 70, 273–278 (1986).
Guilarte, T. R. et al. Nigrostriatal dopamine system dysfunction and subtle motor deficits in manganese-exposed non-human primates. Exp. Neurol. 202, 381–390 (2006).
Racette, B. A. et al. Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology 88, 344–351 (2017).
Monsivais, H. et al. Whole-brain mapping of increased manganese levels in welders and its association with exposure and motor function. Neuroimage 288, 120523 (2024).
Miller, L. M. et al. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155, 30–37 (2006).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254–1261 (2022).
Li, S. R., Bu, L. L. & Cai, L. Cuproptosis: lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal Transduct. Target Ther. 7, 158 (2022).
Dominguez, J. F. et al. Iron accumulation in the basal ganglia in Huntington’s disease: cross-sectional data from the IMAGE-HD study. J. Neurol. Neurosurg. Psychiatry 87, 545–549 (2016).
Li, Q. et al. Beyond the motor cortex: thalamic iron deposition accounts for disease severity in amyotrophic lateral sclerosis. Front. Neurol. 13, 791300 (2022).
Wiedau-Pazos, M. et al. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 271, 515–518 (1996).
Orrell, R. W. et al. Clinical and functional investigation of 10 missense mutations and a novel frameshift insertion mutation of the gene for copper-zinc superoxide dismutase in UK families with amyotrophic lateral sclerosis. Neurology 48, 746–751 (1997).
Peters, S. et al. Blood metal levels and amyotrophic lateral sclerosis risk: a prospective cohort. Ann. Neurol. 89, 125–133 (2021).
McCarthy, R. C. & Kosman, D. J. Iron transport across the blood-brain barrier: development, neurovascular regulation and cerebral amyloid angiopathy. Cell Mol. Life Sci. 72, 709–727 (2015).
Halcrow, P. W., Lynch, M. L., Geiger, J. D. & Ohm, J. E. Role of endolysosome function in iron metabolism and brain carcinogenesis. Semin. Cancer Biol. 76, 74–85 (2021).
Chiou, B. et al. Endothelial cells are critical regulators of iron transport in a model of the human blood-brain barrier. J. Cereb. Blood Flow. Metab. 39, 2117–2131 (2019).
Duck, K. A., Simpson, I. A. & Connor, J. R. Regulatory mechanisms for iron transport across the blood-brain barrier. Biochem. Biophys. Res. Commun. 494, 70–75 (2017).
Casale, G. et al. Serum ferritin and ageing. Age Ageing 10, 119–122 (1981).
Rosell-Diaz, M. et al. Lower serum ferritin levels are associated with worse cognitive performance in aging. J. Nutr. Health Aging 28, 100190 (2024).
Rosell-Diaz, M. et al. Gut microbiota links to serum ferritin and cognition. Gut. Microbes. 15, 2290318 (2023).
Connor, J. R. et al. Evidence for communication of peripheral iron status to cerebrospinal fluid: clinical implications for therapeutic strategy. Fluids Barriers CNS 17, 28 (2020).
Simpson, I. A. et al. A novel model for brain iron uptake: introducing the concept of regulation. J. Cereb. Blood Flow. Metab. 35, 48–57 (2015).
Baumann, B. H. et al. Ferroportin-mediated iron export from vascular endothelial cells in retina and brain. Exp. Eye Res. 187, 107728 (2019).
You, L. et al. Astrocyte-derived hepcidin controls iron traffic at the blood-brain-barrier via regulating ferroportin 1 of microvascular endothelial cells. Cell Death Dis. 13, 667 (2022).
Du, F. et al. Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron-overloaded rats. Mol. Neurobiol. 52, 101–114 (2015).
McCarthy, R. C. & Kosman, D. J. Ferroportin and exocytoplasmic ferroxidase activity are required for brain microvascular endothelial cell iron efflux. J. Biol. Chem. 288, 17932–17940 (2013).
Baringer, S. L. et al. Apo- and holo-transferrin differentially interact with hephaestin and ferroportin in a novel mechanism of cellular iron release regulation. J. Biomed. Sci. 30, 36 (2023).
Maktabi, M. A., Heistad, D. D. & Faraci, F. M. Effects of angiotensin II on blood flow to choroid plexus. Am. J. Physiol. 258, H414–H418 (1990).
Rouault, T. A., Zhang, D. L. & Jeong, S. Y. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab. Brain Dis. 24, 673–684 (2009).
Wu, L. J. et al. Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res. 1001, 108–117 (2004).
Dickson, P. W. et al. High prealbumin and transferrin mRNA levels in the choroid plexus of rat brain. Biochem. Biophys. Res. Commun. 127, 890–895 (1985).
Murakami, Y., Saito, K., Ito, H. & Hashimoto, Y. Transferrin isoforms in cerebrospinal fluid and their relation to neurological diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 95, 198–210 (2019).
Wang, X., Miller, D. S. & Zheng, W. Intracellular localization and subsequent redistribution of metal transporters in a rat choroid plexus model following exposure to manganese or iron. Toxicol. Appl. Pharm. 230, 167–174 (2008).
Leitner, D. F. & Connor, J. R. Functional roles of transferrin in the brain. Biochim. Biophys. Acta 1820, 393–402 (2012).
Wang, X., Li, G. J. & Zheng, W. Efflux of iron from the cerebrospinal fluid to the blood at the blood-CSF barrier: effect of manganese exposure. Exp. Biol. Med. 233, 1561–1571 (2008).
Zheng, W. & Monnot, A. D. Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharm. Ther. 133, 177–188 (2012).
Wang, Z. et al. Axonal iron transport in the brain modulates anxiety-related behaviors. Nat. Chem. Biol. 15, 1214–1222 (2019).
Chen, L., Li, C. & Xie, J. Axonal iron transport might contribute to iron deposition in Parkinson’s disease. Neurosci. Bull. 37, 275–277 (2021).
Reinert, A. et al. Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neurosci. 20, 25 (2019).
Dusek, P. et al. Cerebral iron deposition in neurodegeneration. Biomolecules. 12, 714 (2022).
Zecca, L. et al. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 5, 863–873 (2004).
Verkhratsky, A. & Nedergaard, M. Physiology of astroglia. Physiol. Rev. 98, 239–389 (2018).
Qian, Z. M. & Ke, Y. Brain iron transport. Biol. Rev. Camb. Philos. Soc. 94, 1672–1684 (2019).
Dringen, R. et al. The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem. Res. 32, 1884–1890 (2007).
Zhang, X. et al. Hepcidin overexpression in astrocytes alters brain iron metabolism and protects against amyloid-beta induced brain damage in mice. Cell Death Discov. 6, 113 (2020).
Du, F. et al. Hepcidin directly inhibits transferrin receptor 1 expression in astrocytes via a cyclic AMP-protein kinase A pathway. Glia 59, 936–945 (2011).
Qian, Z. M. & Ke, Y. Hepcidin and its therapeutic potential in neurodegenerative disorders. Med. Res. Rev. 40, 633–653 (2020).
Zhang, N. et al. Ferritin confers protection against iron-mediated neurotoxicity and ferroptosis through iron chelating mechanisms in MPP(+)-induced MES23.5 dopaminergic cells. Free Radic. Biol. Med. 193, 751–763 (2022).
Yu, X., Xiao, Z., Xie, J. & Xu, H. Ferritin is secreted from primary cultured astrocyte in response to iron treatment via TRPML1-mediated exocytosis. Cells. 12, 2519 (2023).
Bishop, G. M., Dang, T. N., Dringen, R. & Robinson, S. R. Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox. Res. 19, 443–451 (2011).
Gao, C., Jiang, J., Tan, Y. & Chen, S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct. Target Ther. 8, 359 (2023).
Jiao, L. et al. Iron metabolism mediates microglia susceptibility in ferroptosis. Front. Cell Neurosci. 16, 995084 (2022).
Ryan, S. K. et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 26, 12–26 (2023).
Fillebeen, C. et al. Lactoferrin is synthesized by activated microglia in the human substantia nigra and its synthesis by the human microglial CHME cell line is upregulated by tumor necrosis factor alpha or 1-methyl-4-phenylpyridinium treatment.Mol. Brain Res. 96, 103–113 (2001).
Wang, J. et al. The protective effect of lactoferrin on ventral mesencephalon neurons against MPP + is not connected with its iron binding ability. Sci. Rep. 5, 10729 (2015).
Friedrich, I. et al. Cell specific quantitative iron mapping on brain slices by immuno-microPIXE in healthy elderly and Parkinson’s disease. Acta Neuropathol. Commun. 9, 47 (2021).
Todorich, B., Zhang, X. & Connor, J. R. H-ferritin is the major source of iron for oligodendrocytes. Glia 59, 927–935 (2011).
Todorich, B. et al. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem. 107, 1495–1505 (2008).
Han, J. et al. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS ONE 6, e23800 (2011).
Mukherjee, C. et al. Oligodendrocytes provide antioxidant defense function for neurons by secreting ferritin heavy chain. Cell Metab. 32, 259–272.e210 (2020).
Cheli, V. T. et al. The divalent metal transporter 1 (DMT1) is required for iron uptake and normal development of oligodendrocyte progenitor cells. J. Neurosci. 38, 9142–9159 (2018).
Han, J., Day, J. R., Connor, J. R. & Beard, J. L. Gene expression of transferrin and transferrin receptor in brains of control vs. iron-deficient rats. Nutr. Neurosci. 6, 1–10 (2003).
Skjorringe, T., Burkhart, A., Johnsen, K. B. & Moos, T. Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 8, 19 (2015).
Zarruk, J. G. et al. Expression of iron homeostasis proteins in the spinal cord in experimental autoimmune encephalomyelitis and their implications for iron accumulation. Neurobiol. Dis. 81, 93–107 (2015).
Connor, J. R. & Menzies, S. L. Relationship of iron to oligodendrocytes and myelination. Glia 17, 83–93 (1996).
Jhelum, P. et al. Ferroptosis mediates cuprizone-induced loss of oligodendrocytes and demyelination. J. Neurosci. 40, 9327–9341 (2020).
Jiang, H., Wang, J., Rogers, J. & Xie, J. Brain iron metabolism dysfunction in Parkinson’s disease. Mol. Neurobiol. 54, 3078–3101 (2017).
Chen, L. L. et al. Iron dysregulation in Parkinson’s disease: focused on the autophagy-lysosome pathway. ACS Chem. Neurosci. 10, 863–871 (2019).
Qian, Z. M., Li, W. & Guo, Q. Ferroportin1 in the brain. Ageing Res. Rev. 88, 101961 (2023).
Zhang, Y. et al. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid. Redox Signal 13, 999–1009 (2010).
Chen, L. et al. TFEB regulates cellular labile iron and prevents ferroptosis in a TfR1-dependent manner. Free Radic. Biol. Med. 208, 445–457 (2023).
Rizzollo, F., More, S., Vangheluwe, P. & Agostinis, P. The lysosome as a master regulator of iron metabolism. Trends Biochem. Sci. 46, 960–975 (2021).
Fernandez, B. et al. Iron-induced cytotoxicity mediated by endolysosomal TRPML1 channels is reverted by TFEB. Cell Death Dis. 13, 1047 (2022).
Xiao, Z. et al. Mitochondrial iron dyshomeostasis and its potential as a therapeutic target for Parkinson’s disease. Exp. Neurol. 372, 114614–114623 (2024).
Balachandran, R. C. et al. Brain manganese and the balance between essential roles and neurotoxicity. J. Biol. Chem. 295, 6312–6329 (2020).
Wedler, F. C. & Denman, R. B. Glutamine synthetase: the major Mn(II) enzyme in mammalian brain. Curr. Top. Cell Regul. 24, 153–169 (1984).
Mildvan, A. S., Scrutton, M. C. & Utter, M. F. Pyruvate carboxylase. VII. A possible role for tightly bound manganese. J. Biol. Chem. 241, 3488–3498 (1966).
Horning, K. J. et al. Manganese is essential for neuronal health. Annu. Rev. Nutr. 35, 71–108 (2015).
Robison, G., Sullivan, B., Cannon, J. R. & Pushkar, Y. Identification of dopaminergic neurons of the substantia nigra pars compacta as a target of manganese accumulation. Metallomics 7, 748–755 (2015).
Hutchens, S. et al. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J. Biol. Chem. 292, 9760–9773 (2017).
Taylor, C. A. et al. SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity. J. Biol. Chem. 294, 1860–1876 (2019).
DeWitt, M. R., Chen, P. & Aschner, M. Manganese efflux in Parkinsonism: insights from newly characterized SLC30A10 mutations. Biochem. Biophys. Res. Commun. 432, 1–4 (2013).
Yin, Z. et al. Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J. Neurochem. 112, 1190–1198 (2010).
Madejczyk, M. S. & Ballatori, N. The iron transporter ferroportin can also function as a manganese exporter. Biochim. Biophys. Acta 1818, 651–657 (2012).
Wang, L. et al. Mitochondrial respiratory chain inhibitors involved in ROS production induced by acute high concentrations of iodide and the effects of SOD as a protective factor. Oxid. Med. Cell Longev. 2015, 217670 (2015).
Jeong, S. Y. & David, S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J. Neurosci. 26, 9810–9819 (2006).
Roy, S. & Lutsenko, S. Mechanism of Cu entry into the brain: many unanswered questions. Neural Regen. Res. 19, 2421–2429 (2024).
Lee, J., Pena, M. M., Nose, Y. & Thiele, D. J. Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 277, 4380–4387 (2002).
Nose, Y., Kim, B. E. & Thiele, D. J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 4, 235–244 (2006).
Lee, J., Prohaska, J. R. & Thiele, D. J. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc. Natl Acad. Sci. USA 98, 6842–6847 (2001).
Ohrvik, H. et al. Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain. Proc. Natl Acad. Sci. USA 110, E4279–E4288 (2013).
Arredondo, M., Munoz, P., Mura, C. V. & Nunez, M. T. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am. J. Physiol. Cell Physiol. 284, C1525–1530 (2003).
Hamza, I., Prohaska, J. & Gitlin, J. D. Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc. Natl Acad. Sci. USA 100, 1215–1220 (2003).
Lutsenko, S., Efremov, R. G., Tsivkovskii, R. & Walker, J. M. Human copper-transporting ATPase ATP7B (the Wilson’s disease protein): biochemical properties and regulation. J. Bioenerg. Biomembr. 34, 351–362 (2002).
Buiakova, O. I. et al. Null mutation of the murine ATP7B (Wilson disease) gene results in intracellular copper accumulation and late-onset hepatic nodular transformation. Hum. Mol. Genet. 8, 1665–1671 (1999).
Washington-Hughes, C. L. et al. Atp7b-dependent choroid plexus dysfunction causes transient copper deficit and metabolic changes in the developing mouse brain. PLoS Genet. 19, e1010558 (2023).
Aliaga, M. E. et al. Superoxide-dependent reduction of free Fe(3+) and release of Fe(2+) from ferritin by the physiologically-occurring Cu(I)-glutathione complex. Bioorg. Med. Chem. 19, 534–541 (2011).
Chen, H. H. et al. Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by up-regulation of copper transporter hCtr1. Mol. Pharm. 74, 697–704 (2008).
Singleton, W. C. J. et al. Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B. J. Biol. Chem. 285, 27111–27121 (2010).
Tapia, L. et al. Metallothionein is crucial for safe intracellular copper storage and cell survival at normal and supra-physiological exposure levels. Biochem. J. 378, 617–624 (2004).
Yu, C. H. et al. The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics. J. Biol. Chem. 292, 18169–18177 (2017).
Chen, M. et al. Atox1 regulates macrophage polarization in intestinal inflammation via ROS-NLRP3 inflammasome pathway. J. Transl. Med. 22, 497 (2024).
Brady, G. F. et al. Regulation of the copper chaperone CCS by XIAP-mediated ubiquitination. Mol. Cell Biol. 30, 1923–1936 (2010).
Pope, C. R., De Feo, C. J. & Unger, V. M. Cellular distribution of copper to superoxide dismutase involves scaffolding by membranes. Proc. Natl Acad. Sci. USA 110, 20491–20496 (2013).
Chen, B. et al. Cellular zinc metabolism and zinc signaling: from biological functions to diseases and therapeutic targets. Signal Transduct. Target Ther. 9, 6 (2024).
Lee, J. Y., Kim, J. H., Palmiter, R. D. & Koh, J. Y. Zinc released from metallothionein-iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp. Neurol. 184, 337–347 (2003).
Zong, N., Ma, S. X. & Wang, Z. Y. Localization of zinc transporters in the spinal cord of cynomolgus monkey. J. Chem. Neuroanat. 82, 56–59 (2017).
Palmiter, R. D. & Findley, S. D. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 14, 639–649 (1995).
Qin, Y., Thomas, D., Fontaine, C. P. & Colvin, R. A. Silencing of ZnT1 reduces Zn efflux in cultured cortical neurons. Neurosci. Lett. 450, 206–210 (2009).
Gottesman, N. et al. ZnT1 is a neuronal Zn(2+)/Ca(2+) exchanger. Cell Calcium 101, 102505 (2022).
Cole, T. B. et al. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 39, 153–169 (2000).
Adlard, P. A., Parncutt, J. M., Finkelstein, D. I. & Bush, A. I. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 30, 1631–1636 (2010).
Choi, S., Hong, D. K., Choi, B. Y. & Suh, S. W. Zinc in the brain: friend or foe? Int. J. Mol. Sci. 21, 8941 (2020).
Troadec, M. B. et al. Induction of FPN1 transcription by MTF-1 reveals a role for ferroportin in transition metal efflux. Blood 116, 4657–4664 (2010).
Chen, L., Min, J. & Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target Ther. 7, 378 (2022).
Bannon, D. I., Abounader, R., Lees, P. S. & Bressler, J. P. Effect of DMT1 knockdown on iron, cadmium, and lead uptake in Caco-2 cells. Am. J. Physiol. Cell Physiol. 284, C44–C50 (2003).
Venkataramani, V. et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 147, 831–848 (2018).
Chib, S. & Singh, S. Manganese and related neurotoxic pathways: A potential therapeutic target in neurodegenerative diseases. Neurotoxicol. Teratol. 94, 107124 (2022).
Uriu-Adams, J. Y. & Keen, C. L. Copper, oxidative stress, and human health. Mol. Asp. Med. 26, 268–298 (2005).
Hubner, C. & Haase, H. Interactions of zinc- and redox-signaling pathways. Redox Biol. 41, 101916 (2021).
Zhang, H., Sun, K., Gao, M. & Xu, S. Zinc inhibits lead-induced oxidative stress and apoptosis of ST cells through ROS/PTEN/PI3K/AKT axis. Biol. Trace Elem. Res. 202, 980–989 (2024).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Berndt, C. et al. Ferroptosis in health and disease. Redox Biol. 75, 103211 (2024).
Mancias, J. D. et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 (2014).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Gao, M. H. et al. Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016).
Fu, D. et al. Iron blocks autophagic flux and induces autophagosomes accumulation in microglia. Food Chem. Toxicol. 181, 114054 (2023).
Sung, H. K., Murugathasan, M., Abdul-Sater, A. A. & Sweeney, G. Autophagy deficiency exacerbates iron overload induced reactive oxygen species production and apoptotic cell death in skeletal muscle cells. Cell Death Dis. 14, 252 (2023).
Yan, H. F., Tuo, Q. Z. & Lei, P. Cell density impacts the susceptibility to ferroptosis by modulating IRP1-mediated iron homeostasis. J. Neurochem. 168, 1359–1373 (2024).
Chen, P. H. et al. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis. 12, 198 (2021).
Qin, X. et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy 17, 4266–4285 (2021).
Li, Q. et al. Mitophagy contributes to zinc-induced ferroptosis in porcine testis cells. Food Chem. Toxicol. 179, 113950 (2023).
Zhang, S. et al. Manganese induces tumor cell ferroptosis through type-I IFN dependent inhibition of mitochondrial dihydroorotate dehydrogenase. Free Radic. Biol. Med. 193, 202–212 (2022).
Wang, C. et al. Neurotoxicity of manganese via ferroptosis induced by redox imbalance and iron overload. Ecotoxicol. Environ. Saf. 278, 116404 (2024).
Zhang, Y. et al. Copper induces cognitive impairment in mice via modulation of cuproptosis and CREB signaling. Nutrients. 15, 972 (2023).
Xue, Q. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 19, 1982–1996 (2023).
Monnot, A. D., Behl, M., Ho, S. & Zheng, W. Regulation of brain copper homeostasis by the brain barrier systems: effects of Fe-overload and Fe-deficiency. Toxicol. Appl. Pharm. 256, 249–257 (2011).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Baker, D. J. & Petersen, R. C. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Invest. 128, 1208–1216 (2018).
Jurk, D. et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004 (2012).
Holloway, K., Neherin, K., Dam, K. U. & Zhang, H. Cellular senescence and neurodegeneration. Hum. Genet. 142, 1247–1262 (2023).
Maus, M. et al. Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat. Metab. 5, 2111–2130 (2023).
Noh, B. et al. Iron overload induces cerebral endothelial senescence in aged mice and in primary culture in a sex-dependent manner. Aging Cell 22, e13977 (2023).
Li, H. Y. et al. Iron derived from NCOA4-mediated ferritinophagy causes cellular senescence via the cGAS-STING pathway. Cell Death Discov. 9, 419 (2023).
Masaldan, S. et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 14, 100–115 (2018).
Masaldan, S. et al. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 16, 322–331 (2018).
Kitazawa, M., Hsu, H. W. & Medeiros, R. Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol. Sci. 152, 194–204 (2016).
Zhou, Q. et al. Copper induces microglia-mediated neuroinflammation through ROS/NF-kappaB pathway and mitophagy disorder. Food Chem. Toxicol. 168, 113369 (2022).
Liu, S. et al. Zinc homeostasis: an emerging therapeutic target for neuroinflammation related diseases. Biomolecules. 13, 416 (2023).
Kirkley, K. S. et al. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflammation 14, 99 (2017).
Sarkar, S. et al. Manganese exposure induces neuroinflammation by impairing mitochondrial dynamics in astrocytes. Neurotoxicology 64, 204–218 (2018).
Wu, J. et al. Sesamol alleviates manganese-induced neuroinflammation and cognitive impairment via regulating the microglial cGAS-STING/NF-kappaB pathway. Environ. Pollut. 319, 120988 (2023).
Ivleva, I., Pestereva, N., Zubov, A. & Karpenko, M. Intranasal exposure of manganese induces neuroin fl ammation and disrupts dopamine metabolism in the striatum and hippocampus. Neurosci. Lett. 738, 135344 (2020).
Yan, D. et al. SIRT1/FOXO3-mediated autophagy signaling involved in manganese-induced neuroinflammation in microglia. Ecotoxicol. Environ. Saf. 256, 114872 (2023).
Yang, Y. et al. Manganese activates autophagy and microglia M2 polarization against endoplasmic reticulum stress-induced neuroinflammation: Involvement of GSK-3beta signaling. Biomed. Pharmacother. 170, 116053 (2024).
Wang, D. et al. The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 13, 914–927 (2017).
Pajarillo, E. et al. The role of microglial LRRK2 kinase in manganese-induced inflammatory neurotoxicity via NLRP3 inflammasome and RAB10-mediated autophagy dysfunction. J. Biol. Chem. 299, 104879 (2023).
Prasuhn, J. et al. Neuroimaging correlates of substantia nigra hyperechogenicity in Parkinson’s disease. J. Parkinsons Dis. 12, 1191–1200 (2022).
Zhu, Y. et al. Iron accumulation and microglia activation contribute to substantia nigra hyperechogenicity in the 6-OHDA-induced rat model of Parkinson’s disease. Parkinsonism Relat. Disord. 36, 76–82 (2017).
Prasuhn, J. et al. Relationship between brain iron deposition and mitochondrial dysfunction in idiopathic Parkinson’s disease. Mol. Med. 28, 28 (2022).
Davies, K. M. et al. Comparative study of metal quantification in neurological tissue using laser ablation-inductively coupled plasma-mass spectrometry imaging and X-ray fluorescence microscopy. Anal. Chem. 87, 6639–6645 (2015).
Junceda, S. et al. Iron dysregulation in Alzheimer’s disease: LA-ICP-MS bioimaging of the distribution of iron and ferroportin in the CA1 region of the human hippocampus. Biomolecules. 14, 295 (2024).
Gerhardsson, L. et al. Concentrations of metals, beta-amyloid and tau-markers in cerebrospinal fluid in patients with Alzheimer’s disease. Dement Geriatr. Cogn. Disord. 28, 88–94 (2009).
Lavados, M. et al. Mild cognitive impairment and Alzheimer patients display different levels of redox-active CSF iron. J. Alzheimers Dis. 13, 225–232 (2008).
Molina, J. A. et al. Cerebrospinal fluid levels of transition metals in patients with Alzheimer’s disease. J. Neural Transm. 105, 479–488 (1998).
Alimonti, A. et al. Serum chemical elements and oxidative status in Alzheimer’s disease, Parkinson disease and multiple sclerosis. Neurotoxicology 28, 450–456 (2007).
Gray, M. T. & Woulfe, J. M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow. Metab. 35, 747–750 (2015).
Olmedo-Diaz, S. et al. An altered blood-brain barrier contributes to brain iron accumulation and neuroinflammation in the 6-OHDA rat model of Parkinson’s disease. Neuroscience 362, 141–151 (2017).
Han, K. et al. Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice. Free Radic. Biol. Med. 162, 592–602 (2021).
Li, K. R. et al. Quantitative evaluation of brain iron accumulation in different stages of Parkinson’s disease. J. Neuroimaging 32, 363–371 (2022).
Guo, J. J. et al. Intranasal administration of alpha-synuclein preformed fibrils triggers microglial iron deposition in the substantia nigra of Macaca fascicularis. Cell Death Dis. 12, 81 (2021).
Du, G. et al. Dynamics of nigral iron accumulation in Parkinson’s disease: from diagnosis to late stage. Mov. Disord. 37, 1654–1662 (2022).
Martinez-Martin, P. & Damian, J. Parkinson disease: depression and anxiety in Parkinson disease. Nat. Rev. Neurol. 6, 243–245 (2010).
Pontone, G. M. et al. Prevalence of anxiety disorders and anxiety subtypes in patients with Parkinson’s disease. Mov. Disord. 24, 1333–1338 (2009).
Penninx, B. W., Pine, D. S., Holmes, E. A. & Reif, A. Anxiety disorders. Lancet 397, 914–927 (2021).
Chen, K. et al. The impact of iron deposition on the fear circuit of the brain in patients with Parkinson’s disease and anxiety. Front. Aging Neurosci. 15, 1116516 (2023).
Xu, X. et al. Increased iron deposition in nucleus accumbens associated with disease progression and chronicity in migraine. BMC Med. 21, 136 (2023).
Thomas, G. E. C. et al. Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 91, 418–425 (2020).
Thomas, G. E. C. et al. Regional brain iron and gene expression provide insights into neurodegeneration in Parkinson’s disease. Brain 144, 1787–1798 (2021).
Yu, X. et al. Decreased iron levels in the temporal cortex in postmortem human brains with Parkinson disease. Neurology 80, 492–495 (2013).
Ding, B. et al. Correlation of iron in the hippocampus with MMSE in patients with Alzheimer’s disease. J. Magn. Reson. Imaging 29, 793–798 (2009).
Ramos, P. et al. Iron levels in the human brain: a post-mortem study of anatomical region differences and age-related changes. J. Trace Elem. Med. Biol. 28, 13–17 (2014).
Corrigan, F. M., Reynolds, G. P. & Ward, N. I. Hippocampal tin, aluminum and zinc in Alzheimer’s disease. Biometals 6, 149–154 (1993).
Griffiths, P. D. & Crossman, A. R. Distribution of iron in the basal ganglia and neocortex in postmortem tissue in Parkinson’s disease and Alzheimer’s disease. Dementia 4, 61–65 (1993).
Damulina, A. et al. Cross-sectional and Longitudinal Assessment of Brain Iron Level in Alzheimer Disease Using 3-T MRI. Radiology 296, 619–626 (2020).
Hare, D. J. et al. Laser ablation-inductively coupled plasma-mass spectrometry imaging of white and gray matter iron distribution in Alzheimer’s disease frontal cortex. Neuroimage 137, 124–131 (2016).
Bulk, M. et al. Postmortem T2*- weighted MRI imaging of cortical iron reflects severity of Alzheimer’s disease. J. Alzheimers Dis. 65, 1125–1137 (2018).
van Duijn, S. et al. Cortical iron reflects severity of Alzheimer’s disease. J. Alzheimers Dis. 60, 1533–1545 (2017).
House, M. J., St Pierre, T. G. & McLean, C. 1.4T study of proton magnetic relaxation rates, iron concentrations, and plaque burden in Alzheimer’s disease and control postmortem brain tissue. Magn. Reson. Med. 60, 41–52 (2008).
Srivastava, R. A. & Jain, J. C. Scavenger receptor class B type I expression and elemental analysis in cerebellum and parietal cortex regions of the Alzheimer’s disease brain. J. Neurol. Sci. 196, 45–52 (2002).
Bulk, M. et al. Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early- and late-onset Alzheimer’s disease. Neurobiol. Aging 62, 231–242 (2018).
Yang, A. et al. Associations of cortical iron accumulation with cognition and cerebral atrophy in Alzheimer’s disease. Quant. Imaging Med. Surg. 12, 4570–4586 (2022).
Baum, L. et al. Serum zinc is decreased in Alzheimer’s disease and serum arsenic correlates positively with cognitive ability. Biometals 23, 173–179 (2010).
Bocca, B. et al. Monitoring of chemical elements and oxidative damage in patients affected by Alzheimer’s disease. Ann. Ist. Super. Sanita 41, 197–203 (2005).
Bostrom, F. et al. CSF Mg and Ca as diagnostic markers for dementia with Lewy bodies. Neurobiol. Aging 30, 1265–1271 (2009).
Huang, C. W. et al. Potential blood biomarker for disease severity in the Taiwanese population with Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 28, 75–83 (2013).
Squitti, R. et al. Free copper distinguishes mild cognitive impairment subjects from healthy elderly individuals. J. Alzheimers Dis. 23, 239–248 (2011).
Ozcankaya, R. & Delibas, N. Malondialdehyde, superoxide dismutase, melatonin, iron, copper, and zinc blood concentrations in patients with Alzheimer disease: cross-sectional study. Croat. Med. J. 43, 28–32 (2002).
Vural, H. et al. Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer’s disease. J. Trace Elem. Med. Biol. 24, 169–173 (2010).
Feldman, E. L. et al. Amyotrophic lateral sclerosis. Lancet 400, 1363–1380 (2022).
Acosta-Cabronero, J. et al. Quantitative susceptibility MRI to detect brain iron in amyotrophic lateral sclerosis. Radiology 289, 195–203 (2018).
Goodall, E. F., Haque, M. S. & Morrison, K. E. Increased serum ferritin levels in amyotrophic lateral sclerosis (ALS) patients. J. Neurol. 255, 1652–1656 (2008).
Cheng, Y., Chen, Y. & Shang, H. Aberrations of biochemical indicators in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Transl. Neurodegener. 10, 3 (2021).
Qin, X. et al. Comparative assessment of blood Metal/metalloid levels, clinical heterogeneity, and disease severity in amyotrophic lateral sclerosis patients. Neurotoxicology 89, 12–19 (2022).
Northall, A. et al. Multimodal layer modelling reveals in vivo pathology in amyotrophic lateral sclerosis. Brain 147, 1087–1099 (2024).
Patti, F. et al. CSF neurotoxic metals/metalloids levels in amyotrophic lateral sclerosis patients: comparison between bulbar and spinal onset. Environ. Res. 188, 109820 (2020).
Violi, F. et al. The study of levels from redox-active elements in cerebrospinal fluid of amyotrophic lateral sclerosis patients carrying disease-related gene mutations shows potential copper dyshomeostasis. Metallomics 12, 668–681 (2020).
Chen, Q. Y. et al. Association of cerebral spinal fluid copper imbalance in amyotrophic lateral sclerosis. Front. Aging Neurosci. 14, 970711 (2022).
Enge, T. G. et al. Longitudinal assessment of metal concentrations and copper isotope ratios in the G93A SOD1 mouse model of amyotrophic lateral sclerosis. Metallomics 9, 161–174 (2017).
Conti, A. et al. Differential expression of ceruloplasmin isoforms in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Proteom. Clin. Appl. 2, 1628–1637 (2008).
Roos, P. M. et al. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res. 151, 159–170 (2013).
Stamenkovic, S. et al. In vivo EPR pharmacokinetic evaluation of the redox status and the blood brain barrier permeability in the SOD1(G93A) ALS rat model. Free Radic. Biol. Med. 108, 258–269 (2017).
Miyata, S., Nakamura, S., Nagata, H. & Kameyama, M. Increased manganese level in spinal cords of amyotrophic lateral sclerosis determined by radiochemical neutron activation analysis. J. Neurol. Sci. 61, 283–293 (1983).
Solovyev, N. et al. Interplay of metallome and metabolome in amyotrophic lateral sclerosis: a study on cerebrospinal fluid of patients carrying disease-related gene mutations. ACS Chem. Neurosci. 14, 3035–3046 (2023).
Figueroa-Romero, C. et al. Early life metal dysregulation in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 7, 872–882 (2020).
Lelie, H. L. et al. Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J. Biol. Chem. 286, 2795–2806 (2011).
Antonioni, A. et al. Amyotrophic lateral sclerosis and air pollutants in the province of Ferrara, Northern Italy: an ecological study. Int. J. Environ. Res. Public Health. 20, 5591 (2023).
Bates, G. P. et al. Huntington disease. Nat. Rev. Dis. Prim. 1, 15005 (2015).
Pfalzer, A. C. et al. Alterations in metal homeostasis occur prior to canonical markers in Huntington disease. Sci. Rep. 12, 10373 (2022).
Squadrone, S., Brizio, P., Abete, M. C. & Brusco, A. Trace elements profile in the blood of Huntington’ disease patients. J. Trace Elem. Med. Biol. 57, 18–20 (2020).
Chen, L. et al. Altered brain iron content and deposition rate in Huntington’s disease as indicated by quantitative susceptibility MRI. J. Neurosci. Res. 97, 467–479 (2019).
Scholefield, M., Patassini, S., Xu, J. & Cooper, G. J. S. Widespread selenium deficiency in the brain of cases with Huntington’s disease presents a new potential therapeutic target. EBioMedicine 97, 104824 (2023).
Loeffler, D. A. et al. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 738, 265–274 (1996).
Berg, D. et al. Brain iron pathways and their relevance to Parkinson’s disease. J. Neurochem. 79, 225–236 (2001).
Salazar, J. et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl Acad. Sci. USA 105, 18578–18583 (2008).
Jiang, H. et al. Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Res. 20, 345–356 (2010).
Ma, X. Z. et al. Gut microbiota-induced CXCL1 elevation triggers early neuroinflammation in the substantia nigra of Parkinsonian mice. Acta Pharm. Sin. 45, 52–65 (2024).
Bi, M. et al. alpha-Synuclein regulates iron homeostasis via preventing Parkin-mediated DMT1 ubiquitylation in Parkinson’s disease models. ACS Chem. Neurosci. 11, 1682–1691 (2020).
Howitt, J. et al. Divalent metal transporter 1 (DMT1) regulation by Ndfip1 prevents metal toxicity in human neurons. Proc. Natl Acad. Sci. USA 106, 15489–15494 (2009).
Jia, W. et al. Ndfip1 attenuated 6-OHDA-induced iron accumulation via regulating the degradation of DMT1. Neurobiol. Aging 36, 1183–1193 (2015).
Liu, C. et al. S-nitrosylation of divalent metal transporter 1 enhances iron uptake to mediate loss of dopaminergic neurons and motoric deficit. J. Neurosci. 38, 8364–8377 (2018).
Liang, T. et al. HMGB1 mediates inflammation-induced DMT1 increase and dopaminergic neurodegeneration in the early stage of Parkinsonism. Mol. Neurobiol. 61, 2006–2020 (2024).
Ayton, S. et al. Transferrin protects against Parkinsonian neurotoxicity and is deficient in Parkinson’s substantia nigra. Signal Transduct. Target Ther. 1, 16015 (2016).
Raha, A. A. et al. Interplay of ferritin accumulation and ferroportin loss in ageing brain: implication for protein aggregation in Down syndrome dementia, Alzheimer’s, and Parkinson’s diseases. Int. J. Mol. Sci. 23, 1060 (2022).
Song, N., Wang, J., Jiang, H. & Xie, J. Ferroportin 1 but not hephaestin contributes to iron accumulation in a cell model of Parkinson’s disease. Free Radic. Biol. Med. 48, 332–341 (2010).
Ganz, T. Cellular iron: ferroportin is the only way out. Cell Metab. 1, 155–157 (2005).
Xu, Q., Kanthasamy, A. G., Jin, H. & Reddy, M. B. Hepcidin plays a key role in 6-OHDA induced iron overload and apoptotic cell death in a cell culture model of Parkinson’s disease. Parkinsons Dis. 2016, 8684130 (2016).
Li, L., Lu, J., Sun, Y. & Jin, X. Acupuncture protects from 6-OHDA-induced neuronal damage by balancing the ratio of DMT1/Fpn1. Saudi J. Biol. Sci. 26, 1948–1955 (2019).
Huang, S. N. et al. Aspirin increases ferroportin 1 expression by inhibiting hepcidin via the JAK/STAT3 pathway in interleukin 6-treated PC-12 cells. Neurosci. Lett. 662, 1–5 (2018).
Song, Y. S. et al. Serum ceruloplasmin and striatal dopamine transporter density in Parkinson disease: comparison with 123I-FP-CIT SPECT. Clin. Nucl. Med. 42, 675–679 (2017).
Xu, W. et al. Correlations between abnormal iron metabolism and non-motor symptoms in Parkinson’s disease. J. Neural Transm. 125, 1027–1032 (2018).
Bakeberg, M. C. et al. Elevated serum ceruloplasmin levels are associated with higher impulsivity in people with Parkinson’s disease. Parkinsons Dis. 2020, 8296203 (2020).
Ayton, S. et al. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 73, 554–559 (2013).
Ayton, S. et al. Iron accumulation confers neurotoxicity to a vulnerable population of nigral neurons: implications for Parkinson’s disease. Mol. Neurodegener. 9, 27 (2014).
Grolez, G. et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol. 15, 74 (2015).
Kenkhuis, B., Bush, A. I. & Ayton, S. How iron can drive neurodegeneration. Trends Neurosci. 46, 333–335 (2023).
Postuma, R. B. et al. Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: a multicenter study. Ann. Neurol. 77, 830–839 (2015).
Stokholm, M. G. et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol. 16, 789–796 (2017).
Zhang, W. et al. Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 49, 1153–1165 (2014).
Wang, J. et al. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 1832, 618–625 (2013).
Ward, R. J., Dexter, D. T. & Crichton, R. R. Iron, neuroinflammation and neurodegeneration. Int. J. Mol. Sci. 23, 7267 (2022).
Urrutia, P. et al. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549 (2013).
Mamais, A. et al. Mutations in LRRK2 linked to Parkinson disease sequester Rab8a to damaged lysosomes and regulate transferrin-mediated iron uptake in microglia. PLoS Biol. 19, e3001480 (2021).
Song, N., Chen, L. & Xie, J. Alpha-synuclein handling by microglia: activating, combating, and worsening. Neurosci. Bull. 37, 751–753 (2021).
Choi, I. et al. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 11, 1386–1399 (2020).
Guo, M. et al. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 143, 1476–1497 (2020).
Tu, H. Y. et al. alpha-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell. 20, e13522–e13538 (2021).
Choi, I., Heaton, G. R., Lee, Y. K. & Yue, Z. Regulation of alpha-synuclein homeostasis and inflammasome activation by microglial autophagy. Sci. Adv. 8, eabn1298–eabn1306 (2022).
Rousseau, E., Michel, P. P. & Hirsch, E. C. The iron-binding protein lactoferrin protects vulnerable dopamine neurons from degeneration by preserving mitochondrial calcium homeostasis. Mol. Pharm. 84, 888–898 (2013).
Wang, B., Timilsena, Y. P., Blanch, E. & Adhikari, B. Lactoferrin: structure, function, denaturation and digestion. Crit. Rev. Food Sci. Nutr. 59, 580–596 (2019).
Wang, C. et al. Astrocyte dysfunction in Parkinson’s disease: from the perspectives of transmitted alpha-synuclein and genetic modulation. Transl. Neurodegener. 10, 39 (2021).
Liang, T. et al. Brain hepcidin suppresses major pathologies in experimental Parkinsonism. iScience 23, 101284 (2020).
Xu, M. et al. Differential regulation of estrogen in iron metabolism in astrocytes and neurons. J. Cell Physiol. 234, 4232–4242 (2019).
Nixon, A. M. et al. HFE genotype restricts the response to paraquat in a mouse model of neurotoxicity. J. Neurochem. 145, 299–311 (2018).
Song, I. Y. et al. The Nrf2-mediated defense mechanism associated with HFE genotype limits vulnerability to oxidative stress-induced toxicity. Toxicology 441, 152525 (2020).
Cui, J. et al. Hepcidin-to-ferritin ratio is decreased in astrocytes with extracellular alpha-synuclein and iron exposure. Front. Cell Neurosci. 14, 47 (2020).
Tulpule, K., Robinson, S. R., Bishop, G. M. & Dringen, R. Uptake of ferrous iron by cultured rat astrocytes. J. Neurosci. Res. 88, 563–571 (2010).
Zhang, H. Y. et al. 6-Hydroxydopamine promotes iron traffic in primary cultured astrocytes. Biometals 26, 705–714 (2013).
Ci, Y. Z. et al. Iron overload induced by IRP2 gene knockout aggravates symptoms of Parkinson’s disease. Neurochem. Int. 134, 104657 (2020).
Block, M. L., Zecca, L. & Hong, J. S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69 (2007).
Schipper, H. M., Song, W., Tavitian, A. & Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 172, 40–70 (2019).
Song, W. et al. Parkinsonian features in aging GFAP.HMOX1 transgenic mice overexpressing human HO-1 in the astroglial compartment. Neurobiol. Aging 58, 163–179 (2017).
Cressatti, M. et al. Glial HMOX1 expression promotes central and peripheral alpha-synuclein dysregulation and pathogenicity in parkinsonian mice. Glia 67, 1730–1744 (2019).
Tavitian, A. et al. Strategic timing of glial HMOX1 expression results in either schizophrenia-like or Parkinsonian behavior in mice. Antioxid. Redox Signal 32, 1259–1272 (2020).
Chen, Y. et al. Ginsenoside Rg1 plays a neuroprotective role in regulating the iron-regulated proteins and against lipid peroxidation in oligodendrocytes. Neurochem. Res. 47, 1721–1735 (2022).
Steger, M. et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 5, e12813 (2016).
Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 (2009).
Boag, M. K. et al. Ferritinophagy and alpha-synuclein: pharmacological targeting of autophagy to restore iron regulation in Parkinson’s disease. Int. J. Mol. Sci. 23, 2378 (2022).
Chen, H. et al. Dysfunction of the retinal pigment epithelium with age: increased iron decreases phagocytosis and lysosomal activity. Invest. Ophthalmol. Vis. Sci. 50, 1895–1902 (2009).
Baksi, S. & Singh, N. alpha-Synuclein impairs ferritinophagy in the retinal pigment epithelium: Implications for retinal iron dyshomeostasis in Parkinson’s disease. Sci. Rep. 7, 12843 (2017).
Hou, X., Watzlawik, J. O., Fiesel, F. C. & Springer, W. Autophagy in Parkinson’s disease. J. Mol. Biol. 432, 2651–2672 (2020).
Xiao, Y. et al. Iron promotes alpha-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J. Neurochem. 145, 34–50 (2018).
Tsunemi, T. et al. Increased lysosomal exocytosis induced by lysosomal Ca(2+) channel agonists protects human dopaminergic neurons from alpha-synuclein toxicity. J. Neurosci. 39, 5760–5772 (2019).
Pollmanns, M. R. et al. Activated endolysosomal cation channel TRPML1 facilitates maturation of alpha-synuclein-containing autophagosomes. Front. Cell Neurosci. 16, 861202 (2022).
Wu, L. K. et al. Artemisia Leaf Extract protects against neuron toxicity by TRPML1 activation and promoting autophagy/mitophagy clearance in both in vitro and in vivo models of MPP+/MPTP-induced Parkinson’s disease. Phytomedicine 104, 154250 (2022).
Wu, K. C. et al. The critical role of Nramp1 in degrading alpha-synuclein oligomers in microglia under iron overload condition. Neurobiol. Dis. 104, 61–72 (2017).
Henrich, M. T., Oertel, W. H., Surmeier, D. J. & Geibl, F. F. Mitochondrial dysfunction in Parkinson’s disease - a key disease hallmark with therapeutic potential. Mol. Neurodegener. 18, 83–102 (2023).
Burte, F., De Girolamo, L. A., Hargreaves, A. J. & Billett, E. E. Alterations in the mitochondrial proteome of neuroblastoma cells in response to complex 1 inhibition. J. Proteome Res. 10, 1974–1986 (2011).
Magalingam, K. B. et al. 6-Hydroxydopamine induces neurodegeneration in terminally differentiated SH-SY5Y neuroblastoma cells via enrichment of the nucleosomal degradation pathway: a global proteomics approach. J. Mol. Neurosci. 72, 1026–1046 (2022).
Mastroberardino, P. G. et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol. Dis. 34, 417–431 (2009).
Tan, Q. et al. DMT1 differentially regulates mitochondrial complex activities to reduce glutathione loss and mitigate ferroptosis. Free Radic. Biol. Med. 207, 32–44 (2023).
Huang, J. et al. Mitoferrin-1 is involved in the progression of Alzheimer’s disease through targeting mitochondrial iron metabolism in a Caenorhabditis elegans model of Alzheimer’s disease. Neuroscience 385, 90–101 (2018).
Wan, Z. et al. Elevating bioavailable iron levels in mitochondria suppresses the defective phenotypes caused by PINK1 loss-of-function in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 532, 285–291 (2020).
Guan, H. et al. Mitochondrial ferritin protects SH-SY5Y cells against H(2)O(2)-induced oxidative stress and modulates alpha-synuclein expression. Exp. Neurol. 291, 51–61 (2017).
Shi, Z. H. et al. Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine-induced dopaminergic cell damage: implication for neuroprotection in Parkinson’s disease. Antioxid. Redox Signal 13, 783–796 (2010).
You, L. H. et al. Mitochondrial ferritin suppresses MPTP-induced cell damage by regulating iron metabolism and attenuating oxidative stress. Brain Res. 1642, 33–42 (2016).
Connor, J. R., Menzies, S. L., St Martin, S. M. & Mufson, E. J. A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J. Neurosci. Res. 31, 75–83 (1992).
Rao, S. S. & Adlard, P. A. Untangling tau and iron: exploring the interaction between iron and tau in neurodegeneration. Front. Mol. Neurosci. 11, 276–288 (2018).
Sayre, L. M. et al. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J. Neurochem. 74, 270–279 (2000).
Castellani, R. J. et al. Is increased redox-active iron in Alzheimer disease a failure of the copper-binding protein ceruloplasmin? Free Radic. Biol. Med. 26, 1508–1512 (1999).
Al-Kuraishy, H. M. et al. Evaluation and targeting of amyloid precursor protein (APP)/amyloid beta (Abeta) axis in amyloidogenic and non-amyloidogenic pathways: a time outside the tunnel. Ageing Res. Rev. 92, 102119 (2023).
Khan, M. A. et al. Iron response elements (IREs)-mRNA of Alzheimer’s amyloid precursor protein binding to iron regulatory protein (IRP1): a combined molecular docking and spectroscopic approach. Sci. Rep. 13, 5073 (2023).
Zhou, Z. D. & Tan, E. K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 12, 75 (2017).
Duce, J. A. et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142, 857–867 (2010).
Wong, B. X. et al. beta-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 9, e114174 (2014).
Belaidi, A. A. et al. Marked age-related changes in brain iron homeostasis in amyloid protein precursor knockout mice. Neurotherapeutics 15, 1055–1062 (2018).
Tsatsanis, A. et al. Amyloidogenic processing of Alzheimer’s disease beta-amyloid precursor protein induces cellular iron retention. Mol. Psychiatry 25, 1958–1966 (2020).
Chen, Y. T. et al. Iron dysregulates APP processing accompanying with sAPPα cellular retention and β-secretase inhibition in rat cortical neurons. Acta Pharm. Sin. 39, 177–183 (2018).
House, E. et al. Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer’s disease. J. Alzheimers Dis. 6, 291–301 (2004).
Tahmasebinia, F. & Emadi, S. Effect of metal chelators on the aggregation of beta-amyloid peptides in the presence of copper and iron. Biometals 30, 285–293 (2017).
Balejcikova, L., Siposova, K., Kopcansky, P. & Safarik, I. Fe(II) formation after interaction of the amyloid beta-peptide with iron-storage protein ferritin. J. Biol. Phys. 44, 237–243 (2018).
Everett, J. et al. Iron stored in ferritin is chemically reduced in the presence of aggregating Abeta(1-42). Sci. Rep. 10, 10332 (2020).
Everett, J. et al. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1-42). J. R. Soc. Interface 11, 20140165 (2014).
Roberson, E. D. et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754 (2007).
Li, X. et al. Enduring elevations of hippocampal amyloid precursor protein and iron are features of beta-amyloid toxicity and are mediated by tau. Neurotherapeutics 12, 862–873 (2015).
Lovell, M. A. et al. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimers Dis. 6, 659–671 (2004).
Yamamoto, A. et al. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 82, 1137–1147 (2002).
Guo, C. et al. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 62, 165–172 (2013).
Chen, M. et al. High dietary iron disrupts iron homeostasis and induces amyloid-beta and phospho-tau expression in the hippocampus of adult wild-type and APP/PS1 transgenic mice. J. Nutr. 149, 2247–2254 (2019).
Wang, D. et al. Overexpression of heme oxygenase 1 causes cognitive decline and affects pathways for tauopathy in mice. J. Alzheimers Dis. 43, 519–534 (2015).
Hui, Y. et al. Long-term overexpression of heme oxygenase 1 promotes tau aggregation in mouse brain by inducing tau phosphorylation. J. Alzheimers Dis. 26, 299–313 (2011).
Wan, W. et al. Iron deposition leads to hyperphosphorylation of tau and disruption of insulin signaling. Front. Neurol. 10, 607 (2019).
Kenkhuis, B. et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 9, 27 (2021).
Hansen, D. V., Hanson, J. E. & Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472 (2018).
Zeineh, M. M. et al. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging 36, 2483–2500 (2015).
McCarthy, R. C. et al. Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 293, 7853–7863 (2018).
Kroner, A. et al. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 83, 1098–1116 (2014).
Nnah, I. C., Lee, C. H. & Wessling-Resnick, M. Iron potentiates microglial interleukin-1beta secretion induced by amyloid-beta. J. Neurochem. 154, 177–189 (2020).
Chaudhary, S. et al. Upregulation of local hepcidin contributes to iron accumulation in Alzheimer’s disease brains. J. Alzheimers Dis. 82, 1487–1497 (2021).
Baringer, S. L. et al. Amyloid-beta exposed astrocytes induce iron transport from endothelial cells at the blood-brain barrier by altering the ratio of apo- and holo-transferrin. J. Neurochem. 167, 248–261 (2023).
Xu, Y. et al. Astrocyte hepcidin ameliorates neuronal loss through attenuating brain iron deposition and oxidative stress in APP/PS1 mice. Free Radic. Biol. Med. 158, 84–95 (2020).
Brickman, A. M. et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch. Neurol. 69, 1621–1627 (2012).
Brickman, A. M. et al. Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer’s disease incidence. Neurobiol. Aging 36, 27–32 (2015).
Lee, S. et al. White matter hyperintensities are a core feature of Alzheimer’s disease: evidence from the dominantly inherited Alzheimer network. Ann. Neurol. 79, 929–939 (2016).
Cai, Z. & Xiao, M. Oligodendrocytes and Alzheimer’s disease. Int. J. Neurosci. 126, 97–104 (2016).
Nasrabady, S. E., Rizvi, B., Goldman, J. E. & Brickman, A. M. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6, 22–31 (2018).
Hadzhieva, M. et al. Dysregulation of iron protein expression in the G93A model of amyotrophic lateral sclerosis. Neuroscience 230, 94–101 (2013).
Jeong, S. Y. et al. Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 610–619 (2009).
Halon-Golabek, M. et al. hmSOD1 gene mutation-induced disturbance in iron metabolism is mediated by impairment of Akt signalling pathway. J. Cachexia Sarcopenia Muscle 9, 557–569 (2018).
Popović-Bijelić, A. et al. Iron-sulfur cluster damage by the superoxide radical in neural tissues of the SOD1(G93A) ALS rat model. Free Radic. Biol. Med. 96, 313–322 (2016).
Lee, J. K., Shin, J. H., Gwag, B. J. & Choi, E. J. Iron accumulation promotes TACE-mediated TNF-α secretion and neurodegeneration in a mouse model of ALS. Neurobiol. Dis. 80, 63–69 (2015).
Mizuno, Y. et al. Transferrin localizes in Bunina bodies in amyotrophic lateral sclerosis. Acta Neuropathol. 112, 597–603 (2006).
Winkler, E. A. et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl Acad. Sci. USA 111, E1035–E1042 (2014).
Wang, T. et al. Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ. 29, 1187–1198 (2022).
Yang, B. et al. NRF2 activation suppresses motor neuron ferroptosis induced by the SOD1. Neurobiol. Dis. 184, 106210 (2023).
Wang, D. et al. SPY1 inhibits neuronal ferroptosis in amyotrophic lateral sclerosis by reducing lipid peroxidation through regulation of GCH1 and TFR1. Cell Death Differ. 30, 369–382 (2023).
Di Paola, M. et al. MRI measures of corpus callosum iron and myelin in early Huntington’s disease. Hum. Brain Mapp. 35, 3143–3151 (2014).
Johnson, E. B. et al. Altered iron and myelin in premanifest Huntington’s disease more than 20 years before clinical onset: evidence from the cross-sectional HD Young Adult Study. EBioMedicine 65, 103266 (2021).
Bonilla, E. et al. Serum ferritin deficiency in Huntington’s disease patients. Neurosci. Lett. 129, 22–24 (1991).
Niu, L. et al. Mutant huntingtin induces iron overload via up-regulating IRP1 in Huntington’s disease. Cell Biosci. 8, 41 (2018).
Niu, L., Zhou, Y., Wang, J. & Zeng, W. Nuclear translocation of STAT5 initiates iron overload in huntington’s disease by up-regulating IRP1 expression. Metab. Brain Dis. 39, 559–567 (2024).
Sánchez-Castañeda, C. et al. The role of iron in gray matter degeneration in Huntington’s disease: a magnetic resonance imaging study. Hum. Brain Mapp. 36, 50–66 (2015).
Berggren, K. L. et al. Neonatal iron supplementation potentiates oxidative stress, energetic dysfunction and neurodegeneration in the R6/2 mouse model of Huntington’s disease. Redox Biol. 4, 363–374 (2015).
Berggren, K. L. et al. Neonatal iron supplementation induces striatal atrophy in female YAC128 Huntington’s disease mice. J. Huntingt. Dis. 5, 53–63 (2016).
Chen, J. et al. Iron accumulates in Huntington’s disease neurons: protection by deferoxamine. PLoS ONE 8, e77023 (2013).
Simmons, D. A. et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 55, 1074–1084 (2007).
Donley, D. W., Realing, M., Gigley, J. P. & Fox, J. H. Iron activates microglia and directly stimulates indoleamine-2,3-dioxygenase activity in the N171-82Q mouse model of Huntington’s disease. PLoS ONE 16, e0250606 (2021).
Bulk, M. et al. Pathological characterization of T2*-weighted MRI contrast in the striatum of Huntington’s disease patients. Neuroimage Clin. 28, 102498 (2020).
Agrawal, S., Fox, J., Thyagarajan, B. & Fox, J. H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 120, 317–329 (2018).
Berggren, K. et al. Amyloid precursor protein haploinsufficiency preferentially mediates brain iron accumulation in mice transgenic for the Huntington’s disease mutation. J. Huntingt. Dis. 6, 115–125 (2017).
Hilditch-Maguire, P. et al. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 9, 2789–2797 (2000).
Dietrich, P., Johnson, I. M., Alli, S. & Dragatsis, I. Elimination of Huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 13, e1006846 (2017).
Fukushima, T. et al. Heavy metals in blood and urine and its relation to depressive symptoms in Parkinson’s disease patients. Fukushima J. Med. Sci. 59, 76–80 (2013).
Lu, M. et al. Manganese overexposure induces Parkinson-like symptoms, altered lipid signature and oxidative stress in C57BL/6 J mouse. Ecotoxicol. Environ. Saf. 263, 115238 (2023).
Langley, M. R. et al. Manganese exposure exacerbates progressive motor deficits and neurodegeneration in the MitoPark mouse model of Parkinson’s disease: relevance to gene and environment interactions in metal neurotoxicity. Neurotoxicology 64, 240–255 (2018).
Tarohda, T. et al. Regional distributions of manganese, iron, copper, and zinc in the brains of 6-hydroxydopamine-induced parkinsonian rats. Anal. Bioanal. Chem. 383, 224–234 (2005).
Brus, R. et al. Effect of pre- and postnatal manganese exposure on brain histamine content in a rodent model of Parkinson’s disease. Neurotox. Res. 21, 143–148 (2012).
Li, S. J. et al. The role of iron in Parkinson’s disease monkeys assessed by susceptibility weighted imaging and inductively coupled plasma mass spectrometry. Life Sci. 240, 117091 (2020).
Fang, T. et al. LA-ICP-MS bioimaging demonstrated disturbance of metal ions in the brain of Parkinson’s disease model mouse undergoing manganese-enhanced MRI. Anal. Bioanal. Chem. 414, 5561–5571 (2022).
Cordova, F. M. et al. Manganese-exposed developing rats display motor deficits and striatal oxidative stress that are reversed by Trolox. Arch. Toxicol. 87, 1231–1244 (2013).
Nadig, A. P. R. et al. Manganese chloride (MnCl(2)) induced novel model of Parkinson’s disease in adult zebrafish; involvement of oxidative stress, neuroinflammation and apoptosis pathway. Biomed. Pharmacother. 155, 113697 (2022).
Chen, M. K. et al. Acute manganese administration alters dopamine transporter levels in the non-human primate striatum. Neurotoxicology 27, 229–236 (2006).
Lin, M. et al. Mechanism of manganese dysregulation of dopamine neuronal activity. J. Neurosci. 40, 5871–5891 (2020).
Cai, T. et al. Manganese induces the overexpression of alpha-synuclein in PC12 cells via ERK activation. Brain Res. 1359, 201–207 (2010).
Prabhakaran, K., Chapman, G. D. & Gunasekar, P. G. alpha-Synuclein overexpression enhances manganese-induced neurotoxicity through the NF-kappaB-mediated pathway. Toxicol. Mech. Methods 21, 435–443 (2011).
Li, Y. et al. alpha-Synuclein overexpression during manganese-induced apoptosis in SH-SY5Y neuroblastoma cells. Brain Res. Bull. 81, 428–433 (2010).
Wallin, C. et al. Characterization of Mn(II) ion binding to the amyloid-beta peptide in Alzheimer’s disease. J. Trace Elem. Med. Biol. 38, 183–193 (2016).
Kim, E. et al. In vivo multi-parametric manganese-enhanced MRI for detecting amyloid plaques in rodent models of Alzheimer’s disease. Sci. Rep. 11, 12419 (2021).
Du, K. et al. Association of serum manganese levels with Alzheimer’s disease and mild cognitive impairment: a systematic review and meta-analysis. Nutrients. 9, 231 (2017).
Tong, Y. et al. High manganese, a risk for Alzheimer’s disease: high manganese induces amyloid-beta related cognitive impairment. J. Alzheimers Dis. 42, 865–878 (2014).
Gerhardsson, L., Lundh, T., Minthon, L. & Londos, E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement Geriatr. Cogn. Disord. 25, 508–515 (2008).
Millien, G. et al. PKCε activation restores loss of PKCε, manganese superoxide dismutase, vascular endothelial growth factor, and microvessels in aged and Alzheimer’s disease hippocampus. Front. Aging Neurosci. 14, 836634 (2022).
Guilarte, T. R. et al. Increased APLP1 expression and neurodegeneration in the frontal cortex of manganese-exposed non-human primates. J. Neurochem. 105, 1948–1959 (2008).
Wang, H. et al. The gut microbiota attenuate neuroinflammation in manganese exposure by inhibiting cerebral NLRP3 inflammasome. Biomed. Pharmacother. 129, 110449 (2020).
Spitznagel, B. D. et al. Acute manganese exposure impairs glutamatergic function in a young mouse model of Alzheimer’s disease. Neurotoxicology 95, 1–11 (2023).
Roos, E. et al. Amyotrophic lateral sclerosis after exposure to manganese from traditional medicine procedures in Kenya. Biol. Trace Elem. Res. 199, 3618–3624 (2021).
Wakai, M., Mokuno, K., Hashizume, Y. & Kato, K. An immunohistochemical study of the neuronal expression of manganese superoxide dismutase in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 88, 151–158 (1994).
Liu, Y., Brooks, B. R., Taniguchi, N. & Hartmann, H. A. CuZnSOD and MnSOD immunoreactivity in brain stem motor neurons from amyotrophic lateral sclerosis patients. Acta Neuropathol. 95, 63–70 (1998).
McEachern, G. et al. Manganese superoxide dismutase levels are elevated in a proportion of amyotrophic lateral sclerosis patient cell lines. Biochem. Biophys. Res. Commun. 273, 359–363 (2000).
Andreassen, O. A. et al. Partial deficiency of manganese superoxide dismutase exacerbates a transgenic mouse model of amyotrophic lateral sclerosis. Ann. Neurol. 47, 447–455 (2000).
Flanagan, S. W., Anderson, R. D., Ross, M. A. & Oberley, L. W. Overexpression of manganese superoxide dismutase attenuates neuronal death in human cells expressing mutant (G37R) Cu/Zn-superoxide dismutase. J. Neurochem. 81, 170–177 (2002).
Aoyama, K. et al. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann. Neurol. 47, 524–527 (2000).
Williams, B. B. et al. Disease-toxicant screen reveals a neuroprotective interaction between Huntington’s disease and manganese exposure. J. Neurochem. 112, 227–237 (2010).
Chhetri, G. et al. Impaired XK recycling for importing manganese underlies striatal vulnerability in Huntington’s disease. J. Cell Biol. 221, e202112073 (2022).
Tidball, A. M. et al. A novel manganese-dependent ATM-p53 signaling pathway is selectively impaired in patient-based neuroprogenitor and murine striatal models of Huntington’s disease. Hum. Mol. Genet. 24, 1929–1944 (2015).
Joshi, P. et al. Huntington’s disease associated resistance to Mn neurotoxicity is neurodevelopmental stage and neuronal lineage dependent. Neurotoxicology 75, 148–157 (2019).
Pfalzer, A. C. et al. Huntington’s disease genotype suppresses global manganese-responsive processes in pre-manifest and manifest YAC128 mice. Metallomics 12, 1118–1130 (2020).
Wilcox, J. M. et al. YAC128 mouse model of Huntington disease is protected against subtle chronic manganese (Mn)-induced behavioral and neuropathological changes. Neurotoxicology 87, 94–105 (2021).
Wilcox, J. M. et al. Manganese-induced hyperactivity and dopaminergic dysfunction depend on age, sex and YAC128 genotype. Pharm. Biochem. Behav. 213, 173337 (2022).
Bryan, M. R. et al. Manganese acts upon insulin/IGF receptors to phosphorylate AKT and increase glucose uptake in Huntington’s disease cells. Mol. Neurobiol. 57, 1570–1593 (2020).
Bryan, M. R. et al. Acute manganese treatment restores defective autophagic cargo loading in Huntington’s disease cell lines. Hum. Mol. Genet. 28, 3825–3841 (2019).
Bichell, T. J. V. et al. Reduced bioavailable manganese causes striatal urea cycle pathology in Huntington’s disease mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1596–1604 (2017).
Gonzalez-Alcocer, A. et al. Combined chronic copper exposure and aging lead to neurotoxicity in vivo. Neurotoxicology 95, 181–192 (2023).
Zeng, Z. et al. Dietary copper intake and risk of Parkinson’s disease: a cross-sectional study. Biol. Trace Elem. Res. 202, 955–964 (2024).
Pall, H. S. et al. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 2, 238–241 (1987).
Scolari Grotto, F. & Glaser, V. Are high copper levels related to Alzheimer’s and Parkinson’s diseases? A systematic review and meta-analysis of articles published between 2011 and 2022. Biometals 37, 3–22 (2024).
Genoud, S., Senior, A. M., Hare, D. J. & Double, K. L. Meta-analysis of copper and iron in Parkinson’s disease brain and biofluids. Mov. Disord. 35, 662–671 (2020).
Davies, K. M. et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol. Aging 35, 858–866 (2014).
Scholefield, M. et al. Widespread decreases in cerebral copper are common to Parkinson’s disease dementia and Alzheimer’s disease dementia. Front. Aging Neurosci. 13, 641222 (2021).
Ilyechova, E. Y. et al. A low blood copper concentration is a co-morbidity burden factor in Parkinson’s disease development. Neurosci. Res. 135, 54–62 (2018).
Kim, M. J. et al. Association of metals with the risk and clinical characteristics of Parkinson’s disease. Parkinsonism Relat. Disord. 55, 117–121 (2018).
Patel, B. N. et al. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 22, 6578–6586 (2002).
Dudzik, C. G. et al. Coordination of copper to the membrane-bound form of alpha-synuclein. Biochemistry 52, 53–60 (2013).
Binolfi, A. et al. Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 128, 9893–9901 (2006).
Binolfi, A. et al. Site-specific interactions of Cu(II) with alpha and beta-synuclein: bridging the molecular gap between metal binding and aggregation. J. Am. Chem. Soc. 130, 11801–11812 (2008).
Li, Y. et al. Copper and iron ions accelerate the prion-like propagation of alpha-synuclein: A vicious cycle in Parkinson’s disease. Int. J. Biol. Macromol. 163, 562–573 (2020).
McDowall, J. S. & Brown, D. R. Alpha-synuclein: relating metals to structure, function and inhibition. Metallomics 8, 385–397 (2016).
Mason, R. J., Paskins, A. R., Dalton, C. F. & Smith, D. P. Copper binding and subsequent aggregation of alpha-synuclein are modulated by N-terminal acetylation and ablated by the H50Q missense mutation. Biochemistry 55, 4737–4741 (2016).
Choi, T. S. et al. Supramolecular modulation of structural polymorphism in pathogenic alpha-synuclein fibrils using copper(II) coordination. Angew. Chem. Int. Ed. Engl. 57, 3099–3103 (2018).
Anandhan, A. et al. Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol. Dis. 81, 76–92 (2015).
Chen, J. et al. Low-dose Cu exposure enhanced α-synuclein accumulation associates with mitochondrial impairments in mice model of Parkinson’s disease. Toxicol. Lett. 387, 14–27 (2023).
Larner, F. et al. High precision isotope measurements reveal poor control of copper metabolism in parkinsonism. Metallomics 5, 125–132 (2013).
Kuo, Y. M., Zhou, B., Cosco, D. & Gitschier, J. The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc. Natl Acad. Sci. USA 98, 6836–6841 (2001).
Gou, D. H. et al. Inhibition of copper transporter 1 prevents alpha-synuclein pathology and alleviates nigrostriatal degeneration in AAV-based mouse model of Parkinson’s disease. Redox Biol. 38, 101795 (2021).
Wang, Z. X. et al. Serum iron, zinc, and copper levels in patients with Alzheimer’s disease: a replication study and meta-analyses. J. Alzheimers Dis. 47, 565–581 (2015).
Hobin, K. et al. Alzheimer’s disease and age-related changes in the Cu isotopic composition of blood plasma and brain tissues of the APP(NL-G-F) murine model revealed by multi-collector ICP-mass spectrometry. Biology 12, 857 (2023).
James, S. A. et al. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic. Biol. Med. 52, 298–302 (2012).
Multhaup, G. et al. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I). Science 271, 1406–1409 (1996).
Maynard, C. J. et al. Overexpression of Alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277, 44670–44676 (2002).
White, A. R. et al. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 842, 439–444 (1999).
Acevedo, K. M. et al. Copper promotes the trafficking of the amyloid precursor protein. J. Biol. Chem. 286, 8252–8262 (2011).
Ciccone, L. et al. Copper mediated amyloid-beta binding to Transthyretin. Sci. Rep. 8, 13744 (2018).
Cao, Q. et al. The inhibition of cellular toxicity of amyloid-beta by dissociated transthyretin. J. Biol. Chem. 295, 14015–14024 (2020).
Dai, X. L., Sun, Y. X. & Jiang, Z. F. Cu(II) potentiation of Alzheimer Abeta1-40 cytotoxicity and transition on its secondary structure. Acta Biochim Biophys. Sin. 38, 765–772 (2006).
Yu, J. et al. Identification of the key molecules involved in chronic copper exposure-aggravated memory impairment in transgenic mice of Alzheimer’s disease using proteomic analysis. J. Alzheimers Dis. 44, 455–469 (2015).
Tian, Y., Shang, Q., Liang, R. & Viles, J. H. Copper(II) can kinetically trap Arctic and Italian amyloid-beta(40) as toxic oligomers, mimicking Cu(II) binding to wild-type amyloid-beta(42): implications for familial Alzheimer’s disease. JACS Au 4, 578–591 (2024).
Abelein, A. et al. Molecular structure of Cu(II)-bound amyloid-beta monomer implicated in inhibition of peptide self-assembly in Alzheimer’s disease. JACS Au 2, 2571–2584 (2022).
Cheignon, C., Collin, F., Sabater, L. & Hureau, C. Oxidative damages on the Alzheimer’s related-Aβ peptide alters its ability to assemble. Antioxidants 12, 472 (2023).
Ma, Q. et al. Copper binding properties of a tau peptide associated with Alzheimer’s disease studied by CD, NMR, and MALDI-TOF MS. Peptides 27, 841–849 (2006).
Zhou, L. X. et al. Copper (II) modulates in vitro aggregation of a tau peptide. Peptides 28, 2229–2234 (2007).
Voss, K. et al. Modulation of tau phosphorylation by environmental copper. Transl. Neurodegener. 3, 24 (2014).
Harris, C. J. et al. Copper modulation and memory impairment due to hippocampal tau pathology. J. Alzheimers Dis. 78, 49–60 (2020).
Crouch, P. J. et al. Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc. Natl Acad. Sci. USA 106, 381–386 (2009).
Andreozzi, E. M. et al. Studies of copper trafficking in a mouse model of Alzheimer’s disease by positron emission tomography: comparison of (64)Cu acetate and (64)CuGTSM. Metallomics 9, 1622–1633 (2017).
Barros, A., Dourado, M. E. T. Jr., Pedrosa, L. F. C. & Leite-Lais, L. Association of copper status with lipid profile and functional status in patients with amyotrophic lateral sclerosis. J. Nutr. Metab. 2018, 5678698 (2018).
Kreuzer, M. et al. Lipids status and copper in a single astrocyte of the rat model for amyotrophic lateral sclerosis: correlative synchrotron-based X-ray and infrared imaging. J. Biophotonics 13, e202000069 (2020).
Tokuda, E., Okawa, E. & Ono, S. Dysregulation of intracellular copper trafficking pathway in a mouse model of mutant copper/zinc superoxide dismutase-linked familial amyotrophic lateral sclerosis. J. Neurochem. 111, 181–191 (2009).
Sturtz, L. A. et al. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276, 38084–38089 (2001).
Xu, X. et al. Integrative analysis of metabolomics and proteomics unravels purine metabolism dysregulation in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 181, 106110 (2023).
Gong, Y. H. & Elliott, J. L. Metallothionein expression is altered in a transgenic murine model of familial amyotrophic lateral sclerosis. Exp. Neurol. 162, 27–36 (2000).
Tokuda, E. et al. Metallothionein proteins expression, copper and zinc concentrations, and lipid peroxidation level in a rodent model for amyotrophic lateral sclerosis. Toxicology 229, 33–41 (2007).
Nagano, S. et al. Reduction of metallothioneins promotes the disease expression of familial amyotrophic lateral sclerosis mice in a dose-dependent manner. Eur. J. Neurosci. 13, 1363–1370 (2001).
Tokuda, E., Okawa, E., Watanabe, S. & Ono, S. Overexpression of metallothionein-I, a copper-regulating protein, attenuates intracellular copper dyshomeostasis and extends lifespan in a mouse model of amyotrophic lateral sclerosis caused by mutant superoxide dismutase-1. Hum. Mol. Genet. 23, 1271–1285 (2014).
Bakkar, N. et al. The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol. Dis. 149, 105228 (2021).
Eum, W. S. & Kang, J. H. Release of copper ions from the familial amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutants. Mol. Cells 9, 110–114 (1999).
Goto, J. J. et al. Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 275, 1007–1014 (2000).
Pasinelli, P. et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43, 19–30 (2004).
Vande Velde, C., Miller, T. M., Cashman, N. R. & Cleveland, D. W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl Acad. Sci. USA 105, 4022–4027 (2008).
Pasinelli, P. et al. Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc. Natl Acad. Sci. USA 95, 15763–15768 (1998).
Pasinelli, P., Houseweart, M. K., Brown, R. H. & Cleveland, D. W. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA 97, 13901–13906 (2000).
Li, M. et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 288, 335–339 (2000).
Basso, M. et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 288, 15699–15711 (2013).
Harlan, B. A. et al. Enhancing NAD+ salvage pathway reverts the toxicity of primary astrocytes expressing amyotrophic lateral sclerosis-linked mutant superoxide dismutase 1 (SOD1). J. Biol. Chem. 291, 10836–10846 (2016).
Turner, B. J. et al. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 25, 108–117 (2005).
Strom, A. L. et al. Interaction of amyotrophic lateral sclerosis (ALS)-related mutant copper-zinc superoxide dismutase with the dynein-dynactin complex contributes to inclusion formation. J. Biol. Chem. 283, 22795–22805 (2008).
Zhang, F. et al. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 282, 16691–16699 (2007).
Taylor, D. M. et al. Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 282, 16329–16335 (2007).
Dang, T. N. et al. Increased metal content in the TDP-43(A315T) transgenic mouse model of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Front. Aging Neurosci. 6, 15 (2014).
Fox, J. H. et al. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS ONE 2, e334 (2007).
Xiao, G., Fan, Q., Wang, X. & Zhou, B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc. Natl Acad. Sci. USA 110, 14995–15000 (2013).
Lobato, A. G. et al. Copper enhances aggregational toxicity of mutant huntingtin in a Drosophila model of Huntington’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1870, 166928 (2024).
Kasischke, K. A. et al. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305, 99–103 (2004).
Sheline, C. T. & Choi, D. W. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann. Neurol. 55, 645–653 (2004).
Martínez-Lazcano, J. C. et al. Sub-chronic copper pretreatment reduces oxidative damage in an experimental Huntington’s disease model. Biol. Trace Elem. Res. 162, 211–218 (2014).
Hegde, M. L. et al. Serum trace element levels and the complexity of inter-element relations in patients with Parkinson’s disease. J. Trace Elem. Med. Biol. 18, 163–171 (2004).
Du, K., Liu, M. Y., Zhong, X. & Wei, M. J. Decreased circulating Zinc levels in Parkinson’s disease: a meta-analysis study. Sci. Rep. 7, 3902 (2017).
Zhao, H. W. et al. Assessing plasma levels of selenium, copper, iron and zinc in patients of Parkinson’s disease. PLoS ONE 8, e83060 (2013).
Sun, H. et al. Association between serum zinc levels and the risk of Parkinson’s disease: a meta-analysis. Biol. Trace Elem. Res. 179, 45–51 (2017).
Lee, J., Park, S. & Jang, W. Serum zinc deficiency could be associated with dementia conversion in Parkinson’s disease. Front. Aging Neurosci. 15, 1132907 (2023).
Zhang, X. et al. Zinc deficiency exacerbates behavioral impediments and dopaminergic neuron degeneration in a mouse model of Parkinson disease. J. Nutr. 153, 167–175 (2023).
Saini, N. & Schaffner, W. Zinc supplement greatly improves the condition of parkin mutant Drosophila. Biol. Chem. 391, 513–518 (2010).
Mbiydzenyuy, N. E., Ninsiima, H. I., Valladares, M. B. & Pieme, C. A. Zinc and linoleic acid pre-treatment attenuates biochemical and histological changes in the midbrain of rats with rotenone-induced Parkinsonism. BMC Neurosci. 19, 29 (2018).
Tamano, H. et al. AMPA-induced extracellular Zn(2+) influx into nigral dopaminergic neurons causes movement disorder in rats. Neurotoxicology 69, 23–28 (2018).
Tamano, H. et al. Blockade of rapid influx of extracellular Zn(2+) into nigral dopaminergic neurons overcomes paraquat-induced Parkinson’s disease in rats. Mol. Neurobiol. 56, 4539–4548 (2019).
Ramirez, A. et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191 (2006).
Usenovic, M. et al. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 32, 4240–4246 (2012).
Tsunemi, T. & Krainc, D. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 23, 2791–2801 (2014).
Park, J. S. et al. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 23, 2802–2815 (2014).
Danscher, G. et al. Increased amount of zinc in the hippocampus and amygdala of Alzheimer’s diseased brains: a proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. J. Neurosci. Methods 76, 53–59 (1997).
Religa, D. et al. Elevated cortical zinc in Alzheimer disease. Neurology 67, 69–75 (2006).
James, S. A. et al. Iron, copper, and zinc concentration in Aβ plaques in the APP/PS1 mouse model of Alzheimer’s disease correlates with metal levels in the surrounding neuropil. ACS Chem. Neurosci. 8, 629–637 (2017).
Ichinohe, N., Hayashi, M., Wakabayashi, K. & Rockland, K. S. Distribution and progression of amyloid-beta deposits in the amygdala of the aged macaque monkey, and parallels with zinc distribution. Neuroscience 159, 1374–1383 (2009).
Bush, A. I., Pettingell, W. H. Jr., Paradis, M. D. & Tanzi, R. E. Modulation of A beta adhesiveness and secretase site cleavage by zinc. J. Biol. Chem. 269, 12152–12158 (1994).
Cuesta, A., Zambrano, A., Royo, M. & Pascual, A. The tumour suppressor p53 regulates the expression of amyloid precursor protein (APP). Biochem. J. 418, 643–650 (2009).
Sensi, S. L., Rapposelli, I. G., Frazzini, V. & Mascetra, N. Altered oxidant-mediated intraneuronal zinc mobilization in a triple transgenic mouse model of Alzheimer’s disease. Exp. Gerontol. 43, 488–492 (2008).
Sensi, S. L. et al. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl Acad. Sci. USA 100, 6157–6162 (2003).
Brewer, G. J. et al. Subclinical zinc deficiency in Alzheimer’s disease and Parkinson’s disease. Am. J. Alzheimers Dis. Other Demen. 25, 572–575 (2010).
Kim, I. et al. Zinc stimulates tau S214 phosphorylation by the activation of Raf/mitogen-activated protein kinase-kinase/extracellular signal-regulated kinase pathway. Neuroreport 22, 839–844 (2011).
Liu, F., Grundke-Iqbal, I., Iqbal, K. & Gong, C. X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 22, 1942–1950 (2005).
Zhang, H. L., Wang, X. C. & Liu, R. Zinc in regulating protein kinases and phosphatases in neurodegenerative diseases. Biomolecules. 12, 785 (2022).
Lei, P. & Ayton, S. TRIMming the tangles. Sci. Bull. 68, 2507–2509 (2023).
Chen, L. L. et al. The metal ion hypothesis of Alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators. Bioorg. Chem. 131, 106301 (2023).
Stoltenberg, M. et al. Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 150, 357–369 (2007).
Panayi, A. E. et al. Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J. Neurol. Sci. 195, 1–10 (2002).
Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 5, 33 (2013).
Craven, K. M., Kochen, W. R., Hernandez, C. M. & Flinn, J. M. Zinc exacerbates tau pathology in a tau mouse model. J. Alzheimers Dis. 64, 617–630 (2018).
Beyer, N. et al. Zinc transporter mRNA levels in Alzheimer’s disease postmortem brain. J. Alzheimers Dis. 29, 863–873 (2012).
Lovell, M. A., Smith, J. L., Xiong, S. & Markesbery, W. R. Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage Alzheimer’s disease. Neurotox. Res. 7, 265–271 (2005).
Olesen, R. H. et al. Obesity and age-related alterations in the gene expression of zinc-transporter proteins in the human brain. Transl. Psychiatry 6, e838 (2016).
Kim, J. et al. Accumulation of labile zinc in neurons and astrocytes in the spinal cords of G93A SOD-1 transgenic mice. Neurobiol. Dis. 34, 221–229 (2009).
Tajiri, M. et al. Metal distribution in Cu/Zn-superoxide dismutase revealed by native mass spectrometry. Free Radic. Biol. Med. 183, 60–68 (2022).
Kim, J. et al. Dimerization, oligomerization, and aggregation of human amyotrophic lateral sclerosis copper/zinc superoxide dismutase 1 protein mutant forms in live cells. J. Biol. Chem. 289, 15094–15103 (2014).
Boyd, S. D. et al. Mutations in superoxide dismutase 1 (Sod1) linked to familial amyotrophic lateral sclerosis can disrupt high-affinity zinc-binding promoted by the copper chaperone for Sod1 (Ccs). Molecules. 25, 1086 (2020).
Sannigrahi, A. et al. The metal cofactor zinc and interacting membranes modulate SOD1 conformation-aggregation landscape in an in vitro ALS model. Elife. 10, e61453 (2021).
Chattopadhyay, M. et al. The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu,Zn superoxide dismutase (SOD1) fibrillation. J. Biol. Chem. 290, 30624–30636 (2015).
Franklin, R., Hare, M. & Beckman, J. S. Determining copper and zinc content in superoxide dismutase using electron capture dissociation under native spray conditions. Methods Mol. Biol. 2500, 201–210 (2022).
Homma, K. et al. SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol. Cell 52, 75–86 (2013).
Leal, S. S. et al. Aberrant zinc binding to immature conformers of metal-free copper-zinc superoxide dismutase triggers amorphous aggregation. Metallomics 7, 333–346 (2015).
Kaneko, M. et al. Zinc transporters ZnT3 and ZnT6 are downregulated in the spinal cords of patients with sporadic amyotrophic lateral sclerosis. J. Neurosci. Res. 93, 370–379 (2015).
Golovin, A. V. et al. Zinc binds to RRM2 peptide of TDP-43. Int. J. Mol. Sci. 21, 9080 (2020).
Garnier, C. et al. Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates. Sci. Rep. 7, 6812 (2017).
Caragounis, A. et al. Zinc induces depletion and aggregation of endogenous TDP-43. Free Radic. Biol. Med. 48, 1152–1161 (2010).
Groeneveld, G. J. et al. Zinc amplifies mSOD1-mediated toxicity in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 352, 175–178 (2003).
Nutini, M. et al. Zinc pre-treatment enhances NMDAR-mediated excitotoxicity in cultured cortical neurons from SOD1(G93A) mouse, a model of amyotrophic lateral sclerosis. Neuropharmacology 60, 1200–1208 (2011).
Dashnaw, C. M. et al. Metal migration and subunit swapping in ALS-linked SOD1: Zn(2+) transfer between mutant and wild-type occurs faster than the rate of heterodimerization. J. Biol. Chem. 298, 102610 (2022).
Huang, J. et al. Structural basis of the zinc-induced cytoplasmic aggregation of the RNA-binding protein SFPQ. Nucleic Acids Res. 48, 3356–3365 (2020).
Patel, A. et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 (2015).
Chen, Y., Pei, X., Chen, L. & Chen, L. A dynamic regulatory switch for phase separation of FUS protein: Zinc ions and zinc finger domain. Biochem. Biophys. Res. Commun. 710, 149862 (2024).
Ayton, S. et al. Brain zinc deficiency exacerbates cognitive decline in the R6/1 model of Huntington’s disease. Neurotherapeutics 17, 243–251 (2020).
Niu, L. et al. Disruption of zinc transporter ZnT3 transcriptional activity and synaptic vesicular zinc in the brain of Huntington’s disease transgenic mouse. Cell Biosci. 10, 106 (2020).
Cordeiro, L. M. et al. Toxicity of copper and zinc alone and in combination in Caenorhabditis elegans model of Huntington’s disease and protective effects of rutin. Neurotoxicology 97, 120–132 (2023).
Gangwani, M. R. et al. Neuronal and astrocytic contributions to Huntington’s disease dissected with zinc finger protein transcriptional repressors. Cell Rep. 42, 111953 (2023).
Hosseinpour Mashkani, S. M. et al. Distribution of copper, iron, and zinc in the retina, hippocampus, and cortex of the transgenic APP/PS1 mouse model of Alzheimer’s disease. Cells. 12, 1144 (2023).
Solovyev, N. et al. Cu, Fe, and Zn isotope ratios in murine Alzheimer’s disease models suggest specific signatures of amyloidogenesis and tauopathy. J. Biol. Chem. 296, 100292 (2021).
Lei, P. et al. Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol. Psychiatry 22, 396–406 (2017).
Strozyk, D. et al. Zinc and copper modulate Alzheimer Aβ levels in human cerebrospinal fluid. Neurobiol. Aging 30, 1069–1077 (2009).
Gerber, H. et al. Zinc and copper differentially modulate amyloid precursor protein processing by gamma-secretase and amyloid-beta peptide production. J. Biol. Chem. 292, 3751–3767 (2017).
Chourrout, M. et al. Virtual histology of Alzheimer’s disease: biometal entrapment within amyloid-beta plaques allows for detection via X-ray phase-contrast imaging. Acta Biomater. 170, 260–272 (2023).
Feng, J., She, Y., Li, C. & Shen, L. Metal ion mediated aggregation of Alzheimer’s disease peptides and proteins in solutions and at surfaces. Adv. Colloid Interface Sci. 320, 103009 (2023).
Lau, T. L. et al. Cholesterol and Clioquinol modulation of A beta(1-42) interaction with phospholipid bilayers and metals. Biochim. Biophys. Acta 1768, 3135–3144 (2007).
Sciacca, M. F. M., Di Natale, G., Milardi, D. & Pappalardo, G. Tau/Aβ chimera peptides: a thioflavin-T and MALDI-TOF study of Aβ amyloidosis in the presence of Cu(II) or Zn(II) ions and total lipid brain extract (TLBE) vesicles. Chem. Phys. Lipids 237, 105085 (2021).
Sciacca, M. F. M. et al. Tau/Abeta chimera peptides: evaluating the dual function of metal coordination and membrane interaction in one sequence. J. Inorg. Biochem. 205, 110996 (2020).
Cohen, S. I. et al. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl Acad. Sci. USA 110, 9758–9763 (2013).
Wen, L. & Shen, L. Effect of surface-chelated Cu(2+) on amyloid-beta peptide fibrillation. Langmuir 38, 174–181 (2022).
Li, Q. X. et al. Overexpression of Abeta is associated with acceleration of onset of motor impairment and superoxide dismutase 1 aggregation in an amyotrophic lateral sclerosis mouse model. Aging Cell 5, 153–165 (2006).
Bryson, J. B. et al. Amyloid precursor protein (APP) contributes to pathology in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 21, 3871–3882 (2012).
Przedborski, S. et al. Nigrostriatal dopaminergic function in familial amyotrophic lateral sclerosis patients with and without copper/zinc superoxide dismutase mutations. Neurology 47, 1546–1551 (1996).
Farr, A. C. & Xiong, M. P. Challenges and opportunities of deferoxamine delivery for treatment of Alzheimer’s disease, Parkinson’s disease, and intracerebral hemorrhage. Mol. Pharm. 18, 593–609 (2021).
Guo, C. et al. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 280, 13–23 (2016).
Febbraro, F. et al. Chronic intranasal deferoxamine ameliorates motor defects and pathology in the α-synuclein rAAV Parkinson’s model. Exp. Neurol. 247, 45–58 (2013).
Fine, J. M. et al. Intranasally-administered deferoxamine mitigates toxicity of 6-OHDA in a rat model of Parkinson׳s disease. Brain Res. 1574, 96–104 (2014).
Haleagrahara, N., Siew, C. J. & Ponnusamy, K. Effect of quercetin and desferrioxamine on 6-hydroxydopamine (6-OHDA) induced neurotoxicity in striatum of rats. J. Toxicol. Sci. 38, 25–33 (2013).
Lv, H. et al. Ameliorating effects of combined curcumin and desferrioxamine on 6-OHDA-induced rat mode of Parkinson’s disease. Cell Biochem. Biophys. 70, 1433–1438 (2014).
Mursaleen, L., Somavarapu, S. & Zariwala, M. G. Deferoxamine and curcumin loaded nanocarriers protect against rotenone-induced neurotoxicity. J. Parkinsons Dis. 10, 99–111 (2020).
Zeng, X. et al. Benefits of iron chelators in the treatment of Parkinson’s disease. Neurochem. Res. 46, 1239–1251 (2021).
Kosyakovsky, J., Fine, J. M., Frey, W. H., 2nd & Hanson, L. R. Mechanisms of intranasal deferoxamine in neurodegenerative and neurovascular disease. Pharmaceuticals 14, 95 (2021).
Wu, Y. et al. Neuroprotection of deferoxamine on rotenone-induced injury via accumulation of HIF-1 alpha and induction of autophagy in SH-SY5Y cells. Neurochem. Int. 57, 198–205 (2010).
You, L. et al. Targeted brain delivery of rabies virus glycoprotein 29-modified deferoxamine-loaded nanoparticles reverses functional deficits in Parkinsonian mice. ACS Nano 12, 4123–4139 (2018).
Perez, D. R., Sklar, L. A. & Chigaev, A. Clioquinol: to harm or heal. Pharmacol. Ther. 199, 155–163 (2019).
Deraeve, C. et al. Preparation and study of new poly-8-hydroxyquinoline chelators for an anti-Alzheimer strategy. Chemistry 14, 682–696 (2008).
Shi, L. et al. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging 12, 9515–9533 (2020).
Teil, M. et al. The zinc ionophore clioquinol reduces Parkinson’s disease patient-derived brain extracts-induced neurodegeneration. Mol. Neurobiol. 59, 6245–6259 (2022).
Finkelstein, D. I. et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 5, 53 (2017).
Bailey, D. K., Clark, W. & Kosman, D. J. The iron chelator, PBT434, modulates transcellular iron trafficking in brain microvascular endothelial cells. PLoS ONE 16, e0254794 (2021).
Beauchamp, L. C. et al. ATH434 rescues pre-motor hyposmia in a mouse model of Parkinsonism. Neurotherapeutics 19, 1966–1975 (2022).
Hu, C. B. et al. DL-3-n-butylphthalide alleviates motor disturbance by suppressing ferroptosis in a rat model of Parkinson’s disease. Neural Regen. Res. 18, 194–199 (2023).
Wang, L. et al. The neuroprotective effects of paeoniflorin against MPP(+)-induced damage to dopaminergic neurons via the Akt/Nrf2/GPX4 pathway. J. Chem. Neuroanat. 122, 102103 (2022).
Lin, Z. H. et al. Quercetin protects against MPP(+)/MPTP-induced dopaminergic neuron death in Parkinson’s disease by inhibiting ferroptosis. Oxid. Med. Cell. Longev. 2022, 7769355 (2022).
Chakraborty, J. et al. Quercetin attenuates copper-induced apoptotic cell death and endoplasmic reticulum stress in SH-SY5Y cells by autophagic modulation. Biol. Trace Elem. Res. 200, 5022–5041 (2022).
Bahar, E., Kim, J. Y. & Yoon, H. Quercetin attenuates manganese-induced neuroinflammation by alleviating oxidative stress through regulation of apoptosis, iNOS/NF-κB and HO-1/Nrf2 pathways. Int. J. Mol. Sci. 18, 1989 (2017).
Zhao, J. et al. Metal chelator EGCG attenuates Fe(III)-induced conformational transition of α-synuclein and protects AS-PC12 cells against Fe(III)-induced death. J. Neurochem. 143, 136–146 (2017).
Xu, Q., Langley, M., Kanthasamy, A. G. & Reddy, M. B. Epigallocatechin gallate has a neurorescue effect in a mouse model of Parkinson disease. J. Nutr. 147, 1926–1931 (2017).
Xia, Y. et al. Inhibition of ferroptosis underlies EGCG mediated protection against Parkinson’s disease in a Drosophila model. Free Radic. Biol. Med. 211, 63–76 (2024).
Hyung, S. J. et al. Insights into antiamyloidogenic properties of the green tea extract (-)-epigallocatechin-3-gallate toward metal-associated amyloid-β species. Proc. Natl Acad. Sci. USA 110, 3743–3748 (2013).
Liu, W. et al. Emodin inhibits zinc-induced neurotoxicity in neuroblastoma SH-SY5Y cells. Biosci. Rep. 39, BSR20182378 (2019).
Sun, Y. et al. Activation of Atg7-dependent autophagy by a novel inhibitor of the Keap1-Nrf2 protein-protein interaction from Penthorum chinense Pursh. attenuates 6-hydroxydopamine-induced ferroptosis in zebrafish and dopaminergic neurons. Food Funct. 13, 7885–7900 (2022).
Li, K. et al. ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharm. Res. 193, 106779 (2023).
Sadeghi, L., Tanwir, F. & Yousefi Babadi, V. Physiological and biochemical effects of Echium amoenum extract on Mn(2+)-imposed Parkinson like disorder in rats. Adv. Pharm. Bull. 8, 705–713 (2018).
da Silva Santos, V. et al. Anthocyanin-rich açaí (Euterpe oleracea Mart.) extract attenuates manganese-induced oxidative stress in rat primary astrocyte cultures. J. Toxicol. Environ. Health A. 77, 390–404 (2014).
Bahar, E. et al. Polyphenolic extract of Euphorbia supina attenuates manganese-induced neurotoxicity by enhancing antioxidant activity through regulation of ER stress and ER stress-mediated apoptosis. Int. J. Mol. Sci. 18, 300 (2017).
Martins, E. N. et al. Protective effect of Melissa officinalis aqueous extract against Mn-induced oxidative stress in chronically exposed mice. Brain Res. Bull. 87, 74–79 (2012).
Bianchini, M. C. et al. Peumus boldus (Boldo) aqueous extract present better protective effect than boldine against manganese-induced toxicity in D. melanogaster. Neurochem. Res. 41, 2699–2707 (2016).
Kupershmidt, L. et al. Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 189, 345–358 (2011).
Youdim, M. B. M30, a brain permeable multitarget neurorestorative drug in post nigrostriatal dopamine neuron lesion of parkinsonism animal models. Parkinsonism Relat. Disord. 18, S151–S154 (2012).
Bar-Am, O. et al. Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-A/monoamine oxidase-B inhibitor in animal models of Parkinson’s disease and aging. Neurobiol. Aging 36, 1529–1542 (2015).
Gutbier, S. et al. Design and evaluation of bi-functional iron chelators for protection of dopaminergic neurons from toxicants. Arch. Toxicol. 94, 3105–3123 (2020).
Chen, L. et al. PPARß/δ agonist alleviates NLRP3 inflammasome-mediated neuroinflammation in the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 356, 483–489 (2019).
Lee, W. J. et al. PPARδ activation mitigates 6-OHDA-induced neuronal damage by regulating intracellular iron levels. Antioxidants 11, 810 (2022).
Liu, L., Yang, S. & Wang, H. α-Lipoic acid alleviates ferroptosis in the MPP(+) -induced PC12 cells via activating the PI3K/Akt/Nrf2 pathway. Cell Biol. Int. 45, 422–431 (2021).
Zheng, W. et al. Chelation therapy of manganese intoxication with para-aminosalicylic acid (PAS) in Sprague-Dawley rats. Neurotoxicology 30, 240–248 (2009).
Martins, A. C. Jr. et al. Manganese-induced neurodegenerative diseases and possible therapeutic approaches. Expert Rev. Neurother. 20, 1109–1121 (2020).
Kwiatkowski, A. et al. Long-term improvement under deferiprone in a case of neurodegeneration with brain iron accumulation. Parkinsonism Relat. Disord. 18, 110–112 (2012).
Ward, R. J., Dexter, D. T. & Crichton, R. R. Neurodegenerative diseases and therapeutic strategies using iron chelators. J. Trace Elem. Med. Biol. 31, 267–273 (2015).
Devos, D. et al. Trial of deferiprone in Parkinson’s disease. N. Engl. J. Med. 387, 2045–2055 (2022).
Hung, L. W. et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 209, 837–854 (2012).
Southon, A. et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br. J. Pharm. 177, 656–667 (2020).
Hilton, J. B. W. et al. Evidence for disrupted copper availability in human spinal cord supports CuII(atsm) as a treatment option for sporadic cases of ALS. Sci. Rep. 14, 5929 (2024).
Monti, D. A. et al. N‐acetyl cysteine is associated with dopaminergic improvement in Parkinson’s disease. Clin. Pharmacol. Ther. 106, 884–890 (2019).
Mischley, L. K. et al. Phase IIb study of intranasal glutathione in Parkinson’s disease. J. Parkinsons Dis. 7, 289–299 (2017).
Seet, R. C. et al. Does high-dose coenzyme Q10 improve oxidative damage and clinical outcomes in Parkinson’s disease? Antioxid. Redox Signal 21, 211–217 (2014).
Parkinson Study Group, Q. E. I. et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 71, 543–552 (2014).
Taghizadeh, M. et al. The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: a randomized, double-blind, placebo-controlled trial. Neurochem. Int. 108, 183–189 (2017).
Nachtman, J. P., Delor, S. & Brennan, C. E. Manganese neurotoxicity: effects of varying oxygen tension and EDTA on dopamine auto-oxidation. Neurotoxicology 8, 249–253 (1987).
Herrero Hernandez, E. et al. Follow-up of patients affected by manganese-induced Parkinsonism after treatment with CaNa2EDTA. Neurotoxicology 27, 333–339 (2006).
Mazur, T., Malik, M. & Bienko, D. C. The impact of chelating compounds on Cu(2+), Fe(2+)/(3+), and Zn(2+) ions in Alzheimer’s disease treatment. J. Inorg. Biochem. 257, 112601 (2024).
Fine, J. M. et al. Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1alpha, and phosphorylates GSK3beta in P301L tau transgenic mice. Exp. Brain Res. 219, 381–390 (2012).
Fine, J. M. et al. Intranasal deferoxamine engages multiple pathways to decrease memory loss in the APP/PS1 model of amyloid accumulation. Neurosci. Lett. 584, 362–367 (2015).
Masaldan, S. et al. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 133, 221–233 (2019).
Guo, C. et al. Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1α pathway on the brain of APP/PS1 transgenic mice. Front. Aging Neurosci. 7, 104 (2015).
Semenza, G. L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 13, 167–171 (2001).
Siddiq, A. et al. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J. Neurosci. 29, 8828–8838 (2009).
Bica, L., Crouch, P. J., Cappai, R. & White, A. R. Metallo-complex activation of neuroprotective signalling pathways as a therapeutic treatment for Alzheimer’s disease. Mol. Biosyst. 5, 134–142 (2009).
Yang, X. et al. Novel 8-hydroxyquinoline derivatives targeting beta-amyloid aggregation, metal chelation and oxidative stress against Alzheimer’s disease. Bioorg. Med. Chem. 26, 3191–3201 (2018).
Knez, D. et al. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 23, 4442–4452 (2015).
Cherny, R. A. et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30, 665–676 (2001).
Lei, P. et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295 (2012).
Raman, B. et al. Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid {beta} peptide. J. Biol. Chem. 280, 16157–16162 (2005).
Tardiff, D. F. et al. Dihydropyrimidine-thiones and clioquinol synergize to target β-amyloid cellular pathologies through a metal-dependent mechanism. ACS Chem. Neurosci. 8, 2039–2055 (2017).
Baswar, D., Sharma, A. & Mishra, A. In silico screening of pyridoxine carbamates for anti-Alzheimer’s activities. Cent. Nerv. Syst. Agents Med. Chem. 21, 39–52 (2021).
Wang, Z. et al. Design, synthesis, and evaluation of multitarget-directed selenium-containing clioquinol derivatives for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 5, 952–962 (2014).
Carvalho, A. et al. New mescaline-related N-acylhydrazone and its unsubstituted benzoyl derivative: Promising metallophores for copper-associated deleterious effects relief in Alzheimer’s disease. J. Inorg. Biochem. 238, 112033 (2023).
Palanimuthu, D. et al. Novel chelators based on adamantane-derived semicarbazones and hydrazones that target multiple hallmarks of Alzheimer’s disease. Dalton Trans. 47, 7190–7205 (2018).
Wegermann, C. A. et al. Interaction studies of oxindole-derivatives with β-amyloid peptides inhibiting its aggregation induced by metal ions. J. Inorg. Biochem. 245, 112227 (2023).
Yang, H. et al. Curcumin scaffold as a multifunctional tool for Alzheimer’s disease research. Molecules 27, 3879 (2022).
Baum, L. & Ng, A. Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J. Alzheimers Dis. 6, 367–377 (2004).
Zhang, X. et al. Design and synthesis of curcumin analogues for in vivo fluorescence imaging and inhibiting copper-induced cross-linking of amyloid beta species in Alzheimer’s disease. J. Am. Chem. Soc. 135, 16397–16409 (2013).
Chen, T. et al. Inhibition of Aβ aggregates in Alzheimer’s disease by epigallocatechin and epicatechin-3-gallate from green tea. Bioorg. Chem. 105, 104382 (2020).
Mandel, S., Amit, T., Bar-Am, O. & Youdim, M. B. Iron dysregulation in Alzheimer’s disease: multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 82, 348–360 (2007).
Li, L. et al. Eriodictyol ameliorates cognitive dysfunction in APP/PS1 mice by inhibiting ferroptosis via vitamin D receptor-mediated Nrf2 activation. Mol. Med. 28, 11 (2022).
Sun, L., Sharma, A. K., Han, B. H. & Mirica, L. M. Amentoflavone: a bifunctional metal chelator that controls the formation of neurotoxic soluble Aβ(42) oligomers. ACS Chem. Neurosci. 11, 2741–2752 (2020).
Liu, R. et al. Luteolin isolated from the medicinal plant Elsholtzia rugulosa (Labiatae) prevents copper-mediated toxicity in β-amyloid precursor protein Swedish mutation overexpressing SH-SY5Y cells. Molecules 16, 2084–2096 (2011).
Zhao, L., Wang, J. L., Wang, Y. R. & Fa, X. Z. Apigenin attenuates copper-mediated β-amyloid neurotoxicity through antioxidation, mitochondrion protection and MAPK signal inactivation in an AD cell model. Brain Res. 1492, 33–45 (2013).
Wang, Q. et al. Vitegnoside mitigates neuronal injury, mitochondrial apoptosis, and inflammation in an Alzheimer’s disease cell model via the p38 MAPK/JNK pathway. J. Alzheimers Dis. 72, 199–214 (2019).
DeToma, A. S., Choi, J. S., Braymer, J. J. & Lim, M. H. Myricetin: a naturally occurring regulator of metal-induced amyloid-β aggregation and neurotoxicity. Chembiochem 12, 1198–1201 (2011).
Tota, S., Kamat, P. K., Shukla, R. & Nath, C. Improvement of brain energy metabolism and cholinergic functions contributes to the beneficial effects of silibinin against streptozotocin induced memory impairment. Behav. Brain Res. 221, 207–215 (2011).
Maryam, T. et al. Silymarin encapsulated liposomal formulation: an effective treatment modality against copper toxicity associated liver Dysfunction and neurobehavioral abnormalities in Wistar rats. Molecules. 28, 1514 (2023).
Singh, G., Mittra, N. & Singh, C. Tempol and silymarin rescue from zinc-induced degeneration of dopaminergic neurons through modulation of oxidative stress and inflammation. Mol. Cell Biochem. 478, 1705–1718 (2023).
Chtourou, Y. et al. Silymarin, a natural antioxidant, protects cerebral cortex against manganese-induced neurotoxicity in adult rats. BioMetals 23, 985–996 (2010).
Zubčić, K. et al. PI3K/Akt and ERK1/2 signalling are involved in quercetin-mediated neuroprotection against copper-induced injury. Oxid. Med. Cell Longev. 2020, 9834742 (2020).
Liu, Y. et al. In situ self-assembled phytopolyphenol-coordinated intelligent nanotherapeutics for multipronged management of ferroptosis-driven Alzheimer’s disease. ACS Nano 18, 7890–7906 (2024).
Goujon, M. et al. The neuroprotective flavonoids sterubin and fisetin maintain mitochondrial health under oxytotic/ferroptotic stress and improve bioenergetic efficiency in HT22 neuronal cells. Antioxidants 13, 460 (2024).
Giampietro, R., Spinelli, F., Contino, M. & Colabufo, N. A. The pivotal role of copper in neurodegeneration: a new strategy for the therapy of neurodegenerative disorders. Mol. Pharm. 15, 808–820 (2018).
Qiang, X. et al. Design, synthesis and evaluation of genistein-O-alkylbenzylamines as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 76, 314–331 (2014).
Li, S. Y. et al. Multifunctional tacrine-flavonoid hybrids with cholinergic, β-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 69, 632–646 (2013).
Li, X. et al. Berberine ameliorates iron levels and ferroptosis in the brain of 3 × Tg-AD mice. Phytomedicine 118, 154962 (2023).
Gucký, A. & Hamuľaková, S. Targeting biometals in Alzheimer’s disease with metal chelating agents including coumarin derivatives. CNS Drugs 38, 507–532 (2024).
Karcz, D. et al. Novel coumarin-thiadiazole hybrids and their Cu(II) and Zn(II) complexes as potential antimicrobial agents and acetylcholinesterase inhibitors. Int. J. Mol. Sci. 22, 9709 (2021).
Jahanshahi, M., Khalili, M. & Margedari, A. Naringin chelates excessive iron and prevents the formation of amyloid-beta plaques in the hippocampus of iron-overloaded mice. Front. Pharm. 12, 651156 (2021).
Gao, Y. et al. Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int. Immunopharmacol. 99, 108002 (2021).
Qian, J. et al. Effects of tetrahydroxy stilbene glycoside derivatives on free radical damage and apoptosis in APP695V717I transgenic mice. Redox Rep. 28, 2259246 (2023).
Tang, J. J. et al. Cognitive enhancement and neuroprotective effects of OABL, a sesquiterpene lactone in 5xFAD Alzheimer’s disease mice model. Redox Biol. 50, 102229 (2022).
Shao, L. et al. Ginkgolide B protects against cognitive impairment in senescence-accelerated P8 mice by mitigating oxidative stress, inflammation and ferroptosis. Biochem. Biophys. Res. Commun. 572, 7–14 (2021).
Xi, J. et al. Hinokitiol functions as a ferroptosis inhibitor to confer neuroprotection. Free Radic. Biol. Med. 190, 202–215 (2022).
Zhang, H. et al. Senegenin rescues PC12 cells with oxidative damage through ihibition of ferroptosis. Mol. Neurobiol. 59, 6983–6992 (2022).
Lincoln, K. M. et al. An N-heterocyclic amine chelate capable of antioxidant capacity and amyloid disaggregation. ACS Chem. Neurosci. 3, 919–927 (2012).
Oboh, G., Ogunsuyi, O. B., Awonyemi, O. I. & Atoki, V. A. Effect of alkaloid extract from African jointfir (Gnetum africanum) leaves on manganese-induced toxicity in Drosophila melanogaster. Oxid. Med. Cell. Longev. 2018, 8952646 (2018).
Dattatray Shinde, S. et al. Bifunctional backbone modified squaramide dipeptides as amyloid beta (Aβ) aggregation inhibitors. Bioorg. Med. Chem. 97, 117538 (2024).
Tosto, R., Vecchio, G. & Bellia, F. New biotinylated GHK and related copper(II) complex: antioxidant and antiglycant properties in vitro against neurodegenerative disorders. Molecules 28, 6724 (2023).
Asadbegi, M. & Shamloo, A. Identification of a novel multifunctional ligand for simultaneous inhibition of amyloid-beta (Aβ(42)) and chelation of zinc metal ion. ACS Chem. Neurosci. 10, 4619–4632 (2019).
Gunesch, S. et al. 7-O-Esters of taxifolin with pronounced and overadditive effects in neuroprotection, anti-neuroinflammation, and amelioration of short-term memory impairment in vivo. Redox Biol. 29, 101378 (2020).
Nguyen, M., Meunier, B. & Robert, A. Catechol‐based ligands as potential metal chelators inhibiting redox activity in Alzheimer’s disease. Eur. J. Inorg. Chem. 2017, 3198–3204 (2017).
Savelieff, M. G., DeToma, A. S., Derrick, J. S. & Lim, M. H. The ongoing search for small molecules to study metal-associated amyloid-β species in Alzheimer’s disease. Acc. Chem. Res. 47, 2475–2482 (2014).
Jalili-Baleh, L. et al. Chromone-lipoic acid conjugate: Neuroprotective agent having acceptable butyrylcholinesterase inhibition, antioxidant and copper-chelation activities. Daru 29, 23–38 (2021).
Wu, Z. et al. Novel multifunctional iron chelators of the aroyl nicotinoyl hydrazone class that markedly enhance cellular NAD(+) /NADH ratios. Br. J. Pharm. 177, 1967–1987 (2020).
Yang, A. et al. A multifunctional anti-AD approach: design, synthesis, X-ray crystal structure, biological evaluation and molecular docking of chrysin derivatives. Eur. J. Med. Chem. 233, 114216 (2022).
Yang, A. et al. Study on the novel usnic acid derivatives: design, synthesis, X-Ray crystal structure of Cu(II) complex and anti-AD activities. J. Mol. Struct. 1263, 133018 (2022).
Bolognesi, M. L. et al. Multi-target-directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer’s disease. J. Med. Chem. 50, 6446–6449 (2007).
Wang, C. Y. et al. Trientine reduces BACE1 activity and mitigates amyloidosis via the AGE/RAGE/NF-kappaB pathway in a transgenic mouse model of Alzheimer’s disease. Antioxid. Redox Signal 19, 2024–2039 (2013).
Li, L. B. et al. Novel melatonin-trientine conjugate as potential therapeutic agents for Alzheimer’s disease. Bioorg. Chem. 128, 106100 (2022).
Tellone, E. et al. Resveratrol: a focus on several neurodegenerative diseases. Oxid. Med. Cell. Longev. 2015, 392169 (2015).
Jang, J. H. & Surh, Y. J. Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic. Biol. Med. 34, 1100–1110 (2003).
Chen, S. Y. et al. Design, synthesis, and biological evaluation of curcumin analogues as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 19, 5596–5604 (2011).
Poliseno, V. et al. Derivatives of tenuazonic acid as potential new multi-target anti-Alzheimer’s disease agents. Biomolecules. 11, 111 (2021).
Kilic, B. et al. New thiourea and benzamide derivatives of 2-aminothiazole as multi-target agents against Alzheimer’s disease: design, synthesis, and biological evaluation. Bioorg. Chem. 131, 106322 (2023).
Squitti, R. et al. d-penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur. J. Clin. Invest. 32, 51–59 (2002).
Cong, L. et al. On the role of synthesized hydroxylated chalcones as dual functional amyloid-β aggregation and ferroptosis inhibitors for potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 166, 11–21 (2019).
Xie, B. et al. Iminodiacetic acid-modified human serum albumin: a multifunctional agent against metal-associated amyloid β-protein aggregation and cytotoxicity. ACS Chem. Neurosci. 8, 2214–2224 (2017).
Bortolami, M. et al. New deferiprone derivatives as multi-functional cholinesterase inhibitors: design, synthesis and in vitro evaluation. Eur. J. Med. Chem. 198, 112350 (2020).
Sucha, M. et al. Experimental treatment with edaravone in a mouse model of spinocerebellar ataxia 1. Int. J. Mol. Sci. 24, 10689–10717 (2023).
Cha, S. J. & Kim, K. Effects of the edaravone, a drug approved for the treatment of amyotrophic lateral sclerosis, on mitochondrial function and neuroprotection. Antioxidants. 11, 195 (2022).
Guo, S. et al. Edaravone attenuates Aβ 1-42-induced inflammatory damage and ferroptosis in HT22 cells. Neurochem. Res. 48, 570–578 (2023).
Moret, V. et al. 1,1’-Xylyl bis-1,4,8,11-tetraaza cyclotetradecane: a new potential copper chelator agent for neuroprotection in Alzheimer’s disease. Its comparative effects with clioquinol on rat brain copper distribution. Bioorg. Med. Chem. Lett. 16, 3298–3301 (2006).
Hirata, Y. et al. Novel oxindole derivatives prevent oxidative stress-induced cell death in mouse hippocampal HT22 cells. Neuropharmacology 135, 242–252 (2018).
Kupershmidt, L. et al. The novel multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer’s disease. Antioxid. Redox Signal 17, 860–877 (2012).
Li, K. & Reichmann, H. Role of iron in neurodegenerative diseases. J. Neural Transm. 123, 389–399 (2016).
Pavlidis, N. et al. Synthesis, characterization and pharmacological evaluation of quinoline derivatives and their complexes with copper(ΙΙ) in in vitro cell models of Alzheimer’s disease. J. Inorg. Biochem. 217, 111393 (2021).
Yang, S. et al. Salidroside alleviates cognitive impairment by inhibiting ferroptosis via activation of the Nrf2/GPX4 axis in SAMP8 mice. Phytomedicine 114, 154762 (2023).
Yu, X. et al. Ketone body β-hydroxybutyric acid ameliorates dopaminergic neuron injury through modulating zinc finger protein 36/Acyl-CoA synthetase long-chain family member four signaling axis-mediated ferroptosis. Neuroscience 509, 157–172 (2023).
Wang, Y., Lv, M. N. & Zhao, W. J. Research on ferroptosis as a therapeutic target for the treatment of neurodegenerative diseases. Ageing Res. Rev. 91, 102035 (2023).
Soriano-Castell, D., Liang, Z., Maher, P. & Currais, A. Profiling the chemical nature of anti-oxytotic/ferroptotic compounds with phenotypic screening. Free Radic. Biol. Med. 177, 313–325 (2021).
Ren, B. et al. Imidazolylacetophenone oxime-based multifunctional neuroprotective agents: discovery and structure-activity relationships. Eur. J. Med. Chem. 228, 114031 (2022).
Peres, T. V. et al. Small molecule modifiers of in vitro manganese transport alter toxicity in vivo. Biol. Trace Elem. Res. 188, 127–134 (2019).
Horning, K. J. et al. Identification of a selective manganese ionophore that enables nonlethal quantification of cellular manganese. J. Biol. Chem. 295, 3875–3890 (2020).
Hamaguchi, T., Ono, K. & Yamada, M. REVIEW: curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 16, 285–297 (2010).
Xiang, B. et al. Curcumin ameliorates copper-induced neurotoxicity through inhibiting oxidative stress and mitochondrial apoptosis in SH-SY5Y cells. Neurochem. Res. 46, 367–378 (2021).
Baum, L. et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 28, 110–113 (2008).
Ringman, J. M. et al. Oral curcumin for Alzheimer’s disease: tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 4, 43 (2012).
Lannfelt, L. et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786 (2008).
Faux, N. G. et al. PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J. Alzheimers Dis. 20, 509–516 (2010).
Summers, K. L. et al. Copper(II) binding to PBT2 differs from that of other 8-hydroxyquinoline chelators: implications for the treatment of neurodegenerative protein misfolding diseases. Inorg. Chem. 59, 17519–17534 (2020).
Arbiser, J. L. et al. Clioquinol-zinc chelate: a candidate causative agent of subacute myelo-optic neuropathy. Mol. Med. 4, 665–670 (1998).
Ceccom, J. et al. Copper chelator induced efficient episodic memory recovery in a non-transgenic Alzheimer’s mouse model. PLoS ONE 7, e43105 (2012).
Moreau, C. et al. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid. Redox Signal 29, 742–748 (2018).
Roberts, B. R. et al. Oral treatment with CuII(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 34, 8021–8031 (2014).
McAllum, E. J. et al. Therapeutic effects of CuII(atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 586–590 (2013).
Soon, C. P. W. et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 286, 44035–44044 (2011).
Hilton, J. B. et al. Cu(II)(atsm) improves the neurological phenotype and survival of SOD1(G93A) mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci. Rep. 7, 42292 (2017).
Yang, Y. et al. Treatment with the copper compound CuATSM has no significant effect on motor neuronal pathology in patients with ALS. Neuropathol. Appl. Neurobiol. 49, e12919 (2023).
McAllum, E. J. et al. ZnII(atsm) is protective in amyotrophic lateral sclerosis model mice via a copper delivery mechanism. Neurobiol. Dis. 81, 20–24 (2015).
Tokuda, E. et al. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp. Neurol. 213, 122–128 (2008).
Tokuda, E. et al. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol. Dis. 54, 308–319 (2013).
Gil-Bea, F. J. et al. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert Rev. Mol. Med. 19, e7 (2017).
Tardiff, D. F. et al. Different 8-hydroxyquinolines protect models of TDP-43 protein, alpha-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J. Biol. Chem. 287, 4107–4120 (2012).
Petri, S. et al. The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurochem. 102, 991–1000 (2007).
Wang, Q. et al. Prevention of motor neuron degeneration by novel iron chelators in SOD1(G93A) transgenic mice of amyotrophic lateral sclerosis. Neurodegener. Dis. 8, 310–321 (2011).
Singh, N. et al. Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities. Antioxid. Redox Signal 20, 1324–1363 (2014).
Nguyen, T., Hamby, A. & Massa, S. M. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc. Natl Acad. Sci. USA 102, 11840–11845 (2005).
Cherny, R. A. et al. PBT2 reduces toxicity in a C. elegans model of polyQ aggregation and extends lifespan, reduces striatal atrophy and improves motor performance in the R6/2 mouse model of Huntington’s disease. J. Huntingt. Dis. 1, 211–219 (2012).
Huntington Study Group Reach, H. D. I. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 14, 39–47 (2015).
Ayton, S. & Lei, P. Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed. Res. Int. 2014, 581256 (2014).
Lei, P., Ayton, S. & Bush, A. I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 296, 100105 (2021).
Lei, P., Ayton, S. & Bush, A. I. Axonal dispatch of iron in neuronal signaling. Nat. Chem. Biol. 15, 1135–1136 (2019).
Yan, H. F. et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct. Target Ther. 6, 49–64 (2021).
Huang, M., Zhang, Y. & Liu, X. The mechanism of cuproptosis in Parkinson’s disease. Ageing Res. Rev. 95, 102214 (2024).
Chen, L., Liu, Y. & Xie, J. The beneficial pharmacological effects of Uncaria rhynchophylla in neurodegenerative diseases: focus on alkaloids. Front. Pharm. 15, 1436481 (2024).
Molina-Holgado, F. et al. Neuroprotective actions of deferiprone in cultured cortical neurones and SHSY-5Y cells. J. Neurochem. 105, 2466–2476 (2008).
Prasanthi, J. R. et al. Deferiprone reduces amyloid-β and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J. Alzheimers Dis. 30, 167–182 (2012).
Zou, D. J. et al. Chromone-deferiprone hybrids as novel MAO-B inhibitors and iron chelators for the treatment of Alzheimer’s disease. Org. Biomol. Chem. 22, 6189–6197 (2024).
Fischer, W. et al. Old age-associated phenotypic screening for Alzheimer’s disease drug candidates identifies sterubin as a potent neuroprotective compound from Yerba santa. Redox Biol. 21, 101089 (2019).
Martin-Bastida, A. et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 7, 1398 (2017).
Monti, D. A. et al. N-acetyl cysteine may support dopamine neurons in Parkinson’s disease: preliminary clinical and cell line data. PLoS ONE 11, e0157602 (2016).
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (32471049 and 32170984) to Prof. Junxia Xie, Natural Science Foundation of Shandong Province- Outstanding Youth Foundation (ZR2020YQ23 and ZR2024MC153) and Qingdao Natural Science Foundation (24-4-4-zrjj-133-jch) to Dr. Leilei Chen, and National Natural Science Foundation of China (32200802) to Dr. Yingjuan Liu.
Author information
Authors and Affiliations
Contributions
L.C., Q.S., Y.L., Y.Z., and L.S. reviewed the literature, developed the concept, and wrote the manuscript. L.C., Q.S., Y.L., and X.M. drafted and polished the figures and tables. J.X., L.C., and N.S. revised the manuscript. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Shen, Q., Liu, Y. et al. Homeostasis and metabolism of iron and other metal ions in neurodegenerative diseases. Sig Transduct Target Ther 10, 31 (2025). https://doi.org/10.1038/s41392-024-02071-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-024-02071-0
This article is cited by
-
NLRP3 facilitates α-synuclein-induced dopaminergic neuronal senescence in a mouse model of Parkinson’s disease through SATB1/DNA damage/p21 signaling pathway
Acta Pharmacologica Sinica (2026)
-
Lipocalin 2: a double-edged sword in cellular ferroptosis
Cell Biology and Toxicology (2026)
-
Endogenous lithium deficiency: a potential early contributor to Alzheimer’s disease onset
Neuroscience and Behavioral Physiology (2026)
-
Combined metallomics and metabolomics reveal impact of metal homeostasis on biological pathways in C. elegans
Analytical and Bioanalytical Chemistry (2026)
-
Metallothioneins in Neurodegenerative Diseases: Metal Homeostasis, Autoimmunity, and Therapeutic Potential
Molecular Neurobiology (2026)
