
Abstract
As a highly complex organ with digestive, endocrine, and immune-regulatory functions, the liver is pivotal in maintaining physiological homeostasis through its roles in metabolism, detoxification, and immune response. Various factors including viruses, alcohol, metabolites, toxins, and other pathogenic agents can compromise liver function, leading to acute or chronic injury that may progress to end-stage liver diseases. While sharing common features, liver diseases exhibit distinct pathophysiological, clinical, and therapeutic profiles. Currently, liver diseases contribute to approximately 2 million deaths globally each year, imposing significant economic and social burdens worldwide. However, there is no cure for many kinds of liver diseases, partly due to a lack of thorough understanding of the development of these liver diseases. Therefore, this review provides a comprehensive examination of the epidemiology and characteristics of liver diseases, covering a spectrum from acute and chronic conditions to end-stage manifestations. We also highlight the multifaceted mechanisms underlying the initiation and progression of liver diseases, spanning molecular and cellular levels to organ networks. Additionally, this review offers updates on innovative diagnostic techniques, current treatments, and potential therapeutic targets presently under clinical evaluation. Recent advances in understanding the pathogenesis of liver diseases hold critical implications and translational value for the development of novel therapeutic strategies.
Similar content being viewed by others
Introduction
The liver, a multifaceted organ, is central to regulating physiological processes including metabolism, detoxification, protein synthesis, and immune response.1 These functions are primarily mediated by hepatocytes, the major parenchymal cells within the liver. Supporting these are liver non-parenchymal cells (NPCs)— liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), cholangiocytes, Kupffer cells (KCs), and other immune cell types that maintain liver homeostasis.2 Liver sinusoids are lined by LSECs with characteristic fenestrations, which facilitate substantial exchanges between sinusoids and hepatocytes. HSCs reside in the space of Disse, secreting cytokines and growth factors that nurture neighboring cells. Cholangiocytes line the intra- and extrahepatic ducts of the biliary tree and contribute to the modification of hepatocyte-derived bile. KCs, along with other immune cells, play a pivotal role in defending against pathogens from the portal circulation.2
Liver diseases represent a wide array of disorders characterized by hepatocyte injury, inflammatory cell infiltration, and HSC activation, which cumulatively impair liver function and disrupt its architecture.3 Annually, liver diseases are linked to approximately 2 million deaths and account for 4% of global mortality.4 Acute liver diseases often result from hepatotropic virus infections, though drug-induced liver injury (DILI) is also becoming increasingly prevalent worldwide. Chronic liver conditions, on the other hand, typically arise from factors like alcohol consumption, hepatitis B virus (HBV), and hepatitis C virus (HCV) infections, along with a rising incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) globally.5 Progression from such chronic conditions to end-stage liver diseases, including cirrhosis and liver cancer, contributes significantly to morbidity and mortality.4
Despite often presenting similar clinicopathological features—ranging from asymptomatic stages to nonspecific digestive symptoms—these liver diseases share biochemical and histological profiles that complicate their differentiation based on a single diagnostic parameter.6 Accurate diagnosis typically requires a combination of clinical presentation, specific biomarkers, and liver biopsy. Currently, clinical management of liver diseases largely focuses on hepatocyte protection, cause elimination, and symptom alleviation.7 The removal of causative agents such as ethanol and viruses does not always prevent progression to cirrhosis, suggesting that the underlying mechanisms driving disease onset and progression are incompletely understood.8 Thus, this review aims to provide an updated, comprehensive overview of the epidemiology and characteristics of liver diseases, highlight the complex pathogenetic mechanisms involved, and summarize the current clinical treatments and investigational drugs in clinical trials that hold potential for future therapeutic management.
Epidemiology of liver diseases
Global mortality
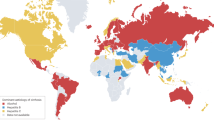
Liver disease stands as a leading cause of global mortality. The Global Burden of Disease 2019 study reported that 1.26 million individuals succumbed to cirrhosis and other chronic liver diseases in 2019, marking a 13% increase since 1990 (Fig. 1 and Table 1).9 Liver cancer, a terminal outcome of liver disease, accounted for approximately 830,000 deaths in 2020, representing 8.3% of global cancer-related deaths.10 Viral hepatitis, especially HBV and HCV, annually leads to around 1.3 million deaths.11 Moreover, approximately 3.3 million people are diagnosed with alcohol-associated liver disease (ALD) annually, accounting for 5.9% of global deaths.12 The rising fatalities from MASLD are also noteworthy, with an estimated 280,000 deaths in 2019.13 Notably, liver disease mortality rates show significant regional disparities; for example, Mongolia reports the highest liver cancer mortality rate at 71.0 per 100,000 individuals, compared to 6.6 in the United States (U.S.).10 This stark contrast arises primarily from the higher prevalence of HBV and HCV, limited healthcare resources, and elevated levels of alcohol consumption in Mongolia. Conversely, the U.S. benefits from effective hepatitis vaccination programs, comprehensive screening, and advanced treatment options, resulting in significantly lower mortality rates. While global trends indicate an increase in liver disease mortality, some high-income countries such as the U.S. have observed a decline since peaking in 2013, with an annual decrease of 3.2%.14 These variances underscore differences in disease burden, healthcare access, and public health strategies across different regions and countries.

Worldwide distribution of liver disease prevalence. Prevalence of (a) drug-induced liver injury (DILI), (b) hepatitis B virus infection (HBV), (c) hepatitis C virus infection (HCV), (d) metabolic dysfunction-associated steatotic liver disease (MASLD), (e) alcohol-associated liver disease (ALD), (f) primary sclerosing cholangitis (PSC), (g) liver cirrhosis, and (h) hepatocellular carcinoma (HCC) are displayed
Global morbidity
Liver disease incidence is on the rise worldwide, posing an increasing risk of morbidity. Despite intensified global public health interventions, liver diseases continue to represent a significant portion of the global disease burden, underscoring the complexity and multidimensionality of liver disease epidemiology. The shift towards lifestyle-associated liver diseases, such as MASLD and ALD, is particularly alarming. This trend is closely linked to changes in global dietary habits, sedentary behavior, and rising obesity rates.15,16 Recent meta-analyses have identified MASLD as the most common chronic liver disease, affecting 38.0% of the global adult population between 2016-2019.17 Additionally, the incidence of ALD is climbing, paralleling increases in global alcohol consumption.18 This trend is evident in regional data that show significant correlations between increasing alcohol consumption and ALD rates. In the U.S., the age-adjusted death rate from ALD increased by 34.4% from 2009 to 2016, rising from 9.6 to 12.9 per 100,000 population, corresponding with a 7% increase in per capita alcohol consumption.19 Similarly, the United Kingdom (U.K.) experienced a 43% increase in hospital admissions for ALD between 2002 and 2019, accompanied by a 10% increase in alcohol sales.20 In South Korea, the age-standardized prevalence of ALD nearly doubled from 3.8% in 1998 to 7.4% in 2016, corresponding with a 43% increase in per capita alcohol consumption.21 Moreover, China has also seen a significant surge, the prevalence of ALD among patients with chronic liver diseases increased from 4.3% in 2000 to 8.7% in 2015, concurrent with a 70% increase in recorded alcohol consumption between 2005 and 2016.22 While new infections of HBV and HCV are declining in many regions due to effective public health interventions, chronic infections continue to pose a global challenge. The reduction in new infections can be attributed primarily to several key health measures beyond vaccination. Enhanced screening protocols for blood and organ donors have significantly reduced the risk of transfusion-associated hepatitis.23 Furthermore, harm reduction programs targeting high-risk populations, such as needle and syringe exchange programs, have been instrumental in preventing transmission among intravenous drug users.24 Moreover, the introduction of direct-acting antivirals (DAAs) for HCV has revolutionized treatment, achieving cure rates over 95% across all HCV genotypes.25 Improved infection control practices in healthcare settings have also minimized iatrogenic transmissions, underscoring the effectiveness of a comprehensive approach to combating these viral infections. The World Health Organization (WHO) estimates that there are 1.5 million new HBV and HCV infections annually.26 Acute hepatitis forms, such as hepatitis A (HAV) and hepatitis E (HEV), remain prevalent in developing countries, with approximately 10 million and 20 million new infections each year, respectively.27,28 Socioeconomic factors exacerbate disease progression, as evidenced by the rising global incidence of cirrhosis from 20.7 to 23.4 per 100,000 people between 2000 and 2015.9 The global incidence of cirrhosis, the end-stage of various chronic liver conditions, increased from 20.7 per 100,000 people in 2000 to 23.4 per 100,000 in 2015.9 Moreover, the incidence of liver cancer continues to escalate, with around 20 million new cases reported globally in 2022.29 Although improved diagnostics have enhanced early detection, they may also contribute to apparent increases in incidence. Emerging risk factors, such as environmental pollution and hepatotoxic drug use, further complicate efforts to reduce the global burden of liver diseases.30,31
Acute liver disease
Acute viral hepatitis
HAV remains a significant global health concern despite strong vaccination recommendations, with an estimated 10 million new infections annually.27 In 2019, around 159 million acute HAV infections were reported worldwide.32 HAV prevalence varies considerably across different regions, with developing countries and low-income areas experiencing higher seroprevalence rates, particularly in sub-Saharan Africa and South Asia. In these regions, almost all children encounter HAV early in life, reducing susceptibility in adulthood.33 Conversely, in middle-income areas such as Latin America, the Middle East, North Africa, Eastern Europe, and parts of Asia, the prevalence exhibits transitional characteristics, posing a risk to unexposed adolescents and adults.34 High-income nations such as Western Europe, Australia, New Zealand, Canada, the U.S., Japan, and Singapore have very low HAV seroprevalence, with infections primarily confined to specific high-risk groups including travelers, men who have sex with men, drug users, the homeless, and the incarcerated.35,36
HEV leads to approximately 3.3 million symptomatic cases of acute hepatitis globally. The distribution of HEVs by genotype and geographic location is uneven and particularly prevalent in developing regions like South Asia, Africa, rural China, and Latin America. Here, genotypes 1 and 2 of HEV, which infect only humans and are transmitted through contaminated water sources, precipitate major outbreaks in resource-limited settings.37,38 In recent decades, there has been a significant increase in HEV cases in Europe, with seropositivity rates ranging from 20% to 30% in countries including France, Germany, and the Netherlands.39,40,41 While the overall mortality from HEV is low, specific high-risk groups, such as immunocompromised individuals, face greater health threats and higher case fatality rates.42 Targeted monitoring and protective measures for these vulnerable groups remain crucial.
Drug-induced liver injury
Determining the precise incidence of DILI is challenging due to diagnostic complexities and widespread underreporting. The estimates of DILI incidence fluctuate significantly, from 1 in 10,000 to 1 in 1,000,000 cases, influenced by variable factors such as diagnostic criteria, detection capabilities, population demographics, drug types, cultural factors, and reporting practices.42,43
In Europe, retrospective studies from the U.K. and Sweden suggest an annual incidence of DILI around 2.3-2.4 per 100,000 individuals.44,45 France reported a higher figure from a prospective study, at 13.9 cases per 100,000, equating to over 8,000 cases annually.46 In Iceland, prospective data revealed a rate of 19.1 cases per 100,000 people.47 In the U.S., a Delaware-based study noted an annual incidence of 2.7 cases per 100,000 adults, with nearly 43% linked to herbal and dietary supplements.48 A comparative analysis shows that DILI incidence is generally higher in Asian countries; a nationwide prospective study in South Korea documented an annual rate of 12 hospitalizations per 100,000 due to DILI,49 while a retrospective study in China reported 23.8 cases per 100,000.43 Notably, the proportion of DILI cases attributed to herbal and dietary supplements is on the rise globally, indicating an evolving trend in the epidemiology of DILI and pointing towards the need for enhanced awareness and regulation.
Chronic liver diseases
Chronic hepatitis B/D
Chronic hepatitis B (CHB) remains a significant public health challenge globally. As per the 2019 WHO data, the worldwide seroprevalence of hepatitis B surface antigen (HBsAg) is 3.8%, accounting for approximately 296 million people living with CHB.50 The GBD study from the same year estimated CHB prevalence at around 4.1%, translating to approximately 316 million cases globally.51 Over the three decades leading up to 2019, CHB prevalence saw a notable decrease of 31.3%.21 This notable decline is primarily due to the global implementation of universal HBV vaccination programs, which have significantly reduced new infections among newborns and children, the most vulnerable to chronic infection.52 Additionally, targeted public health initiatives, including enhanced maternal and perinatal healthcare services, have effectively prevented vertical transmission from mothers to infants. Increased public awareness through education campaigns, along with improved access to healthcare, has further contributed to the reduction in CHB prevalence.53 The highest CHB prevalence rates occur in the Western Pacific region (5.26%) and Africa (8.83%), with the disease burden in West and Central Africa accounting for 82.8% and 17.2%, respectively.54,55 The countries most afflicted by CHB are China, India, and Nigeria, harboring 74 million, 17 million, and 15 million cases respectively.56 Europe exhibits significant intra-regional variation in CHB prevalence, with less than 1% in Western and Northern Europe, contrasting sharply with 4-8% in Eastern Europe. Prevalence rates in the Americas are also diversified, with Mexico at 0.20% and Haiti at 13.55%.57
Among specific populations, approximately 2.7 million people living with human immunodeficiency virus (HIV) are co-infected with HBV, 71% of whom are in sub-Saharan Africa. Furthermore, around 0.5% (1.3 million) of injection drug users globally are HBV-positive.58 Hepatitis delta (HDV) co-infection has been significantly impacted by vaccination efforts59; although an estimated 15-20 million people worldwide are infected with HDV, the exact prevalence rates are elusive due to a dearth of comprehensive studies. The most affected countries include Benin, Gabon, Mauritania, Nauru, and Mongolia, with the latter exhibiting the highest rate at 36.9% of HBsAg-positive individuals co-infected with HDV. Despite a global decline in HDV due to vaccination initiatives, persistent high prevalence in regions like Moldova and parts of Africa and Asia underscore an ongoing health burden.60,61
Chronic hepatitis C
In 2020, approximately 57 million people were estimated to be living with chronic HCV infection globally, exhibiting a viremia prevalence rate of 0.7%. Over 70% of these cases were concentrated in low- and middle-income countries. The highest burdens of HCV are seen in China, India, Pakistan, Russia, and the U.S., with 30 countries collectively accounting for 80% of the global HCV burden.62
High HCV prevalence regions include Central Asia, East Asia, and the North Africa-Middle East, each with over 3.5% of the population affected. Moderate prevalence rates (1.5-3.5%) are noted in Southeast Asia, the Andes, Central and South Latin America, Oceania, the Caribbean, Europe, and sub-Saharan Africa. Contrastingly, some Western European nations like the U.K., Denmark, France, Germany, Sweden, and Switzerland report HCV seroprevalence rates of less than 1%. The Asia-Pacific region, tropical Latin America, and North America each have relatively low prevalence rates, under 1.5%.63 Despite a decreasing global trend in HCV prevalence, there is a critical need for focused interventions and increased attention in high-burden countries and regions.
Chronic metabolic liver diseases—MASLD and ALD
MASLD has rapidly become the most prevalent chronic liver disease worldwide. Its global prevalence escalated from 25.3% between 1990-2006 to 38.0% between 2016-2019, heavily influenced by the rising rates of obesity and type 2 diabetes mellitus (T2DM).64 MASLD exhibits substantial regional variations; in the Americas, Latin America reports the highest prevalence at 44.4%. In the U.S., the prevalence surged from 19% in 1988-1994 to 54% in 2005-2016.65,66 In Europe, there is variability in the prevalence of MASLD. A recent meta-analysis, which includes data updated until 2019, indicates an increase in prevalence to 30.9%.13 Western European nations like Germany and the U.K. displaying higher rates (25%-30%), whereas Eastern countries such as Hungary and Romania have slightly lower prevalence (around 20%).67,68,69 The trend is similarly upward in the Asia-Pacific region: China saw an increase from 25.4% in 2008-2010 to 32.3% in 2015-2018,70,71 and the latest nationwide study with 5.7 million showed that the prevalence of steatosis reached 44.39% in 2022.72 Japan experienced a rise from 20.69% in 1983 to 29.61% in 2011-2016;73 and South Korea reports a prevalence of approximately 31.5%.74 In Africa and the Middle East, data on MASLD incidence and prevalence in sub-Saharan Africa are largely missing, but this burden is expected to grow in the coming decades.75 Although data are sparse for Africa and the Middle East, the prevalence in sub-Saharan Africa is anticipated to climb in the coming decades. In the Middle East and North Africa region, high obesity and diabetes rates have significantly driven up MASLD prevalence, currently estimated at 36.5%.75,76
Notably, lean MASLD, a subtype of MASLD characterized by a BMI < 25 kg/m², shows a significantly higher prevalence in China (approximately 20%) compared to 5-10% in Western nations.77,78 This disparity is influenced by factors such as genetic variations (e.g., PNPLA3 gene variants), differences in body composition (higher percentage of body fat and visceral adipose tissue at lower BMI in Asians), dietary habits, environmental factors, and gut microbiome variations.79 Despite normal BMI, lean MASLD patients in Asian populations face similar metabolic risks and liver disease progression as their obese counterparts. The high prevalence of lean MASLD underscores the need for tailored diagnostic and screening approaches in Asian countries like China to prevent underdiagnosis, considering unique regional and ethnic characteristics.79
ALD remains a major global health concern. Roughly 2.4 billion people consume alcohol worldwide, contributing to about 2 million deaths from liver disease annually, half of which are related to cirrhosis from alcohol consumption. ALD ranks among the top 30 causes of death globally, with a death rate from alcohol-attributable cirrhosis of 7.2 per 100,000 people in 2010 (4.6 in females and 9.7 in males).80 The relationship between liver-related death rates and alcohol consumption levels varies by country. European countries have historically been the largest per capita consumers of alcohol, though consumption decreased from 12.3 to 9.8 liters annually between 2005 and 2016. Conversely, alcohol consumption has been increasing in the Western Pacific, South-East Asia, and the Americas, with future growth projected until at least 2025.81 In China, alcohol consumption has increased more rapidly over the past 30 years than in any other country.82 Countries like the U.S. are also reporting rising rates of harmful drinking. In 2019, nations severely affected by ALD included Mongolia, Kazakhstan, El Salvador, Guatemala, Greenland, Kyrgyzstan, Poland, Rwanda, Ireland, and Brazil, all of which exhibit high alcohol consumption correlating with elevated ALD incidence and mortality rates.21
During the COVID-19 pandemic, social isolation and psychological stress significantly boosted alcohol consumption in certain populations in the U.S. and Europe, potentially exacerbating ALD prevalence.83,84,85 Despite intensified public health efforts, MASLD and ALD continue to pose significant challenges due to their complex etiology involving genetic predisposition, environmental factors, and lifestyle choices such as diet and alcohol use.16,86 These diseases often progress asymptomatically, complicating early detection and treatment. The COVID-19 pandemic has further exacerbated these challenges, increasing risk factors linked to lifestyle changes and stress.87 Consequently, there is a pressing need for adaptive public health strategies that go beyond traditional interventions. This includes not only developing targeted screening and comprehensive lifestyle interventions but also exploring innovative therapeutic options and addressing social determinants of health to effectively reduce the global disease burden.
Autoimmune liver diseases
Autoimmune liver diseases, including primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and autoimmune hepatitis (AIH), form a significant part of the global spectrum of liver diseases, displaying notable regional and demographic variations in prevalence.
PBC predominantly affects middle-aged and older women, with a global prevalence around 14.6 per 100,000. The highest prevalence rates are seen in North America (approximately 21.8 per 100,000), Europe (14.6 per 100,000), and the Asia-Pacific region (9.8 per 100,000). Within continents, discrepancies exist; for instance, in Europe, the prevalence varies from 13.8 per 100,000 in Northern Europe to 10.3 per 100,000 in Western Europe, with scant data from Southern and Eastern Europe. In the Asia-Pacific region, Japan and South Korea report higher prevalence rates (10.4 and 8.5 per 100,000, respectively) compared to mainland China (5.8 per 100,000) and Taiwan district (3.7 per 100,000).88
PSC primarily affects young adult males (male-to-female ratio approximately 2:1) and shows significant regional prevalence distinctions. For instance, Northern European countries like Sweden report a high prevalence of 10.3 per 100,000, considerably greater than Southern nations like Spain (0.6 per 100,000).89 The U.K. saw a rising prevalence from 3.2 per 100,000 in 1998 to 7.4 per 100,000 in 2014.90 In North America, PSC prevalence (e.g., 13.6 per 100,000 in Olmsted County, Minnesota, U.S.) typically surpasses that in Europe. Conversely, Asian countries generally report lower rates, with Japan, South Korea, and Singapore exhibiting prevalence of 0.95, 0.45, and 0.15 per 100,000, respectively.89 Notably, around 65% of PSC patients also have inflammatory bowel disease (IBD), particularly ulcerative colitis, underscoring a significant association between PSC and intestinal immune disorders.91
AIH has a global prevalence of 17.44 per 100,000, with regional prevalence rates at 12.99 in Asia, 19.44 in Europe, and 22.80 in the Americas per 100,000 respectively.92 AIH incidence in Denmark rose from 1.37 per 100,000 in 1994 to 2.33 per 100,000 in 2014.93 In the U.K., the incidence doubled from 1.27 in 1997 to 2.56 per 100,000 in 2015, with higher latitudes correlating with increased incidence.94 Sweden saw an increase from 10.7 per 100,000 in 2003 to 17.3 per 100,000 in 2009.95 Japan reported a substantial increase in AIH prevalence from 8.1 per 100,000 in 2004 to 23.9 per 100,000 in 2016.96 While the incidence has remained relatively stable in South Korea, there has been a gradual increase in prevalence from 2009 to 2013.97 The precise etiology of AIH remains elusive, though there is speculation that environmental changes may act as triggers.88 These diseases exemplify the complex interplay of genetic, environmental, and immunological factors that characterize autoimmune pathologies affecting the liver.
Genetic and rare liver diseases
Wilson disease (WD), an autosomal recessive disorder affecting copper metabolism, leads to significant liver and neurological damage. The estimated global prevalence of WD lies between 1:30,000 and 1:40,000 but shows notable ethnic and regional variability. In Europe, WD prevalence spans from approximately 1.2 to 2.0 per 100,000. It is comparatively higher in Asia, with China reporting a rate of 5.87 per 100,000, South Korea at 2.7 per 100,000, and Japan at 1.9 per 100,000.98 A U.K. study highlighted a potentially higher-than-expected risk, suggesting a carrier rate for pathogenic mutations in WD at 1/7,026-indicative of an underestimation of WD prevalence within the general population.99 The Middle East also exhibits significant prevalence, with Iran reporting 1/31,000 and Saudi Arabia at 1/15,000. In the U.S., the prevalence is around 1:30,000 to 1:40,000, but specific subpopulations like Armenians in New York City show a higher prevalence of 1/22,000. In Brazil, the prevalence aligns closely with the global average at about 1:36,000. It is notably higher in isolated regions with high rates of consanguineous marriages, such as the Canary Islands and Sardinia, with prevalence of 1/2,600 and 1/7,000, respectively.99,100 The frequently underestimated prevalence of WD may be attributed to misdiagnosis, phenotypic diversity, inadequate sensitivity of copper metabolism tests, and low detection rates of ATP7B gene mutations.101
Alpha-1 antitrypsin deficiency (AATD) predominantly affects the lungs and liver and is another prevalent genetic condition with distinct genetic variations influencing its distribution. The most prevalent AATD genotype among people of European descent is Pi*ZZ, occurring at a rate of about 1/2,000 to 1/4,000.102 North America reports a Pi*ZZ prevalence ranging from 1/3,000 to 1/5,000, with specific populations like Newfoundland experiencing higher rates, around 1/1,100.103 Conversely, Pi*ZZ prevalence is markedly lower in Asia and Africa: Japan reports a prevalence of 1/300,000, South Korea at 1/280,000, and African Americans at 1/30,000. Notably, the prevalence of AATD in Caucasian populations is generally higher compared to other genetic liver diseases, such as AIH, PSC, and WD.104
End-stage liver diseases
Cirrhosis
Cirrhosis, representing the end stage of diverse chronic liver disorders, has experienced a global surge in incidence, from 20.7 per 100,000 people in 2000 to 23.4 per 100,000 in 2015-a 13% increase.9 The leading causes of cirrhosis include MASLD (60%), HBV (29%), HCV (9%), and ALD (2%).105
Regionally, HBV is most prevalent among cirrhosis patients in the Western Pacific (59%) and least prevalent in the Americas (5%). The highest proportion of cirrhosis due to HCV occurs in the Eastern Mediterranean (70%), while it is lowest in Africa and the Western Pacific (13% each). In terms of alcohol-related cirrhosis, Europe (16–78%) and the Americas (17–52%) report higher rates compared to Asia (0-41%). Data on MASLD as a cause of cirrhosis show that its prevalence varies, from 2% in South Korea and Brazil to 18% in Canada.8
In North America and Europe, MASLD is increasingly acknowledged as a primary cause of cirrhosis. For example, the prevalence of cirrhosis in the U.S. has increased between 1.5 to 2 fold over the past two decades, especially among younger populations due to prevalent obesity, diabetes, and metabolic syndrome.103 In Germany, MASLD-related cirrhosis cases saw a fourfold increase from 2005 to 2018.106 Similarly, in Japan, cirrhosis due to MASLD rose from 2% in 2007 to 9% in 2016107; South Korea also reports rising cirrhosis cases caused by MASLD, HCV, and alcohol.108 Despite these trends, viral hepatitis remains the dominant cause of cirrhosis in the Middle East and Africa, particularly HCV.9,109 Overall, NAFLD and ALD-related cirrhosis are becoming more common globally, although HBV and HCV infections remain the primary causes in many developing countries.
Hepatobiliary cancer
Hepatocellular carcinoma (HCC) is the most prevalent primary liver cancer globally and often develops within the context of chronic liver diseases such as cirrhosis. In 2020, the global prevalence of HCC ranged from approximately 15-30 per 100,000 with over 70% of cases in Asia.110 Mongolia reports the highest prevalence (85.6 per 100,000) in East Asia, followed by China (26.7 per 100,000), South Korea (21.8 per 100,000), and Japan (16.1 per 100,000).10,111 Southeast Asia also experiences a high HCC prevalence, with numbers like Thailand (22.2 per 100,000), Vietnam (19.4 per 100,000), and Cambodia (18.3 per 100,000). In Africa, the HCC prevalence ranges from 10-20 per 100,000, with Egypt reporting the highest at 32.2 per 100,000.112 Southern European nations like Italy (10.9 per 100,000) and Spain (8.6 per 100,000) report higher HCC rates compared to other European regions. In North America, HCC prevalence stands at about 6-8 per 100,000, while in Latin America, rates are slightly lower, ranging from 3-5 per 100,000, with Brazil and Mexico recording the highest numbers within this range. Oceania reports significantly lower HCC prevalence, as seen in Australia (3.2 per 100,000) and New Zealand (2.8 per 100,000).113,114
The global heterogeneity in HCC prevalence reflects the complex interplay of diverse etiological factors. In East and Southeast Asia, chronic HBV infection remains a major contributor, historically exacerbated by vaccination gaps and vertical transmission.111,115 Conversely, Western countries typically exhibit lower HCC rates due to effective HBV vaccination programs and improved antiviral therapy access.114,116 In Africa, particularly Egypt, high HCV prevalence is linked to elevated HCC rates, with limited healthcare access and socioeconomic challenges playing significant roles.117 In Southern Europe, cultural practices such as traditional alcohol consumption patterns also contribute to increased HCC risk.118 Moreover, the global rise in obesity and associated metabolic dysfunction represents an emerging HCC risk factor, particularly in Western regions.119 Environmental factors, including aflatoxin exposure in specific African and Asian regions, further modulate HCC risk.120 These multifaceted influences underscore the urgent need for region-specific prevention and screening strategies to effectively mitigate the global HCC burden.
Cholangiocarcinoma (CCA), though less common than HCC, has exhibited an increasing trend in the U.K. and the U.S., particularly with intrahepatic forms, while extrahepatic CCA has seen a decline.121,122 The U.S. recorded an increase in intrahepatic CCA incidence from 0.44 per 100,000 in 1973 to 1.18 per 100,000 in 2012.123 This rise is largely attributed to the increasing prevalence of obesity and metabolic syndrome in Western countries.124 In contrast, CCA incidence in Southeast Asian countries like Thailand is much higher than HCC, reaching 14.6 per 100,000.125 Factors such as liver fluke infections prevalent in Southeast Asia significantly contribute to the high regional CCA incidence. Notably, while CCA primarily affects middle-aged and older men, the rising incidence among women and younger individuals calls for further research into its evolving epidemiology.126
Clinical and pathological features of liver diseases
Clinical features
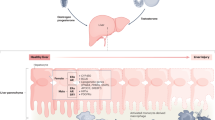
Liver diseases manifest a broad spectrum of symptoms, ranging from early nonspecific signs to advanced multisystem complications (Fig. 2A). Typically, at their onset, conditions such as acute viral hepatitis, mild DILI, early-stage chronic viral hepatitis, MASLD, and initial-phase ALD manifest with mild, nonspecific symptoms.127,128 Patients may experience minor fatigue, upper right abdominal discomfort, and a decreased appetite. Those with acute viral hepatitis might exhibit transient fever, nausea, and slight jaundice. MASLD patients often present with features of metabolic syndrome such as obesity, dyslipidemia, and hypertension.129,130 Early ALD may manifest as indigestion and abdominal discomfort. Mild DILI might lead to slight elevations in transaminases without prominent symptoms.131 Notably, many patients with early-stage liver disease are asymptomatic, being discovered incidentally during routine examinations or investigations for other reasons.

a Hepatic and extrahepatic manifestations associated with various liver diseases. b Histological progression from normal liver to hepatocellular carcinoma (HCC). Representative hematoxylin and eosin (H&E) stained sections illustrate the stages from normal liver to ballooning degeneration, alcoholic hepatitis, chronic hepatitis, and HCC. Sirius Red staining was used to visualize fibrosis and cirrhosis. Written informed consent was obtained from all patients involved. The study was approved by the Ethical Committee of West China Hospital and registered in the Chinese Clinical Trial Registry (ChiCTR2200063108). Created in BioRender. Yuan, Y. (2024) BioRender.com/o74p618
As liver diseases progress to the intermediate stage, symptoms previously mild or intermittent become more pronounced across various conditions. Patients with chronic viral hepatitis often experience ongoing fatigue, intermittent jaundice, and increasingly severe right upper quadrant pain, often accompanied by general malaise and mild hepatomegaly.35 In cases of chronic HCV, approximately 70% of patients may develop systemic complications such as mixed cryoglobulinemia and cardiovascular issues, underscoring the extensive impact of the disease.132 Concurrently, immune-mediated liver diseases demonstrate their unique progression patterns during this stage. AIH frequently manifests with nonspecific symptoms like fatigue in 85-95% of patients, often accompanied by symptoms like jaundice (67–85%) and abdominal pain (50–70%). Furthermore, 25–40% of patients display extrahepatic manifestations such as arthralgia and skin rashes, and up to 50% may suffer from concurrent autoimmune disorders such as thyroiditis.133 PBC exhibits persistent pruritus and fatigue in 65–85% of patients, with 50–60% initially asymptomatic, typically identified during routine liver function tests.134 It is often associated with other autoimmune conditions such as Sjögren’s syndrome (25%) and thyroid disorders (20%).135 PSC often exhibits pruritus and jaundice; typical symptoms include fatigue (75–80%) and right upper quadrant pain (20–40%), strongly linked to IBD, especially ulcerative colitis in 60–80% of cases, and an elevated lifetime risk of cholangiocarcinoma (10–15%) and colorectal cancer.136,137,138 In MASLD and ALD, signs of liver function abnormalities and mild coagulation disorders emerge, indicating progressive liver impairment.139,140
In the severe stages of liver diseases, patients often develop more serious and complex complications. Individuals with advanced chronic liver disease and compensated cirrhosis typically exhibit signs of portal hypertension, such as ascites, splenomegaly, and palmar erythema. These signs reflect significant changes in liver structure and function and alterations in portal systemic hemodynamics. Patients with WD often begin to exhibit significant neuropsychiatric symptoms, including movement disorders and cognitive decline at this stage, likely due to disturbances in copper metabolism affecting the central nervous system.141 Patients with AATD might concurrently suffer from respiratory symptoms such as difficulty breathing.142 Additionally, patients with late-stage liver disease commonly develop coagulopathies and thrombocytopenia, increasing the risk of bleeding. Early hepatic encephalopathy typically presents as mild cognitive impairment and disturbances in the sleep-wake cycle, early indicators of severe liver function compromise affecting the neurological system.143
The end stages of liver disease, including decompensated cirrhosis and hepatobiliary malignancies, are characterized by life-threatening complications. Patients often experience esophageal and gastric variceal bleeding, refractory ascites, and severe hepatic encephalopathy. These manifestations represent the terminal expressions of hepatic synthetic failure, altered hemodynamics, and cerebral dysfunction resulting from liver failure. Patients with HCC typically exhibit weight loss, abdominal masses, and cachexia; meanwhile, CCA patients might present as painless progressive jaundice and abdominal pain. Additionally, patients with end-stage liver disease often develop multisystem complications, including systemic coagulopathy, hepatorenal syndrome, and hepatopulmonary syndrome.144,145
In summary, the progression of liver disease constitutes a complex, multistage process that involves multiple systems and organs. Ranging from mild early symptoms to life-threatening complications in the end stages, the clinical manifestations at each stage illustrate the extent of liver damage and its systemic impact. Significantly, various types of liver diseases can progress at differing speeds and exhibit distinct clinical manifestations. This understanding is crucial for early detection of liver diseases, the assessment of disease severity, and accurate prognostication.
Pathological features
Liver histology, crucial for diagnosing various diseases, plays a pivotal role in understanding and recognizing hepatic conditions (Fig. 2B).
In the initial stages of liver disease, pathological alterations are generally mild. Acute viral hepatitis typically features mononuclear cell and lymphocyte infiltration in hepatic lobules and portal areas.146 DILI may be indicated by slight hepatocellular swelling and minimal inflammatory cell infiltration.147 MASLD and ALD initially present with steatosis without significant inflammation. These changes are typically reversible, with timely intervention preventing disease progression.19
As the disease progresses to moderate stages, pathological changes become increasingly pronounced. Chronic viral hepatitis is characterized by persistent inflammation, interface hepatitis, and varying degrees of fibrosis. In advanced MASLD and ALD, worsening steatosis is accompanied by significant inflammatory cell infiltration and hepatocyte ballooning.148 PBC is distinguished by chronic non-suppurative inflammation and interlobular bile duct destruction, while PSC presents with concentric periductal fibrosis, the classic “onion-skin” appearance.149,150 AIH is characterized by interface hepatitis with prominent plasma cell infiltration. These pathological changes reflect disease progression and potentially indicate more severe liver dysfunction.
In the severe stage of liver disease, pathological alterations become more pronounced. Advanced chronic liver disease presents with significant bridging fibrosis and early nodule formation. WD is characterized by hepatocellular copper accumulation and the presence of Mallory bodies.151 AATD manifests as PAS-positive, diastase-resistant globules in periportal hepatocytes. Early cirrhosis, characterized by fibrous septa formation and lobular architecture distortion, begins to manifest. These changes reflect severe liver structural and functional impairment, often indicating irreversible liver damage.152,153
End-stage liver disease presents the most dramatic pathological features. Liver cirrhosis is characterized by the replacement of normal liver architecture with regenerative nodules surrounded by fibrous septa, leading to severe disruption of liver structure and function. HCC shows tumor cells with varying degrees of differentiation, often with pseudoglandular structures and vascular invasion. CCA presents as adenocarcinoma with varying differentiation and prominent desmoplastic stromal reaction. Moreover, extensive hepatocellular necrosis, bile duct proliferation, and cellular atypia are observed. These end-stage changes typically signify severe liver failure, reflecting the terminal stage of the disease.150,154
While liver biopsy remains the gold standard for assessing liver pathology, its invasiveness and potential complications necessitate the development of non-invasive diagnostic methods. Advances in serological markers and imaging technologies strive to maintain diagnostic precision while reducing patient discomfort and risk, providing efficient alternatives to traditional histological examination.155
Etiology
Various factors can lead to liver injury, including viral and parasitic infections, metabolic disorders, toxic exposures (such as liver-damaging drugs), and genetic predispositions. In this section, we provide a comprehensive summary of the common causes of liver diseases.
Infection
Acute and chronic liver diseases can be caused by various viruses. HBV, an enveloped DNA virus, infects hepatocytes through bodily fluids, leading to potential CHB, cirrhosis, and HCC due to the persistence of covalently closed circular DNA (cccDNA) in hepatocytes.156 HCV can present as acute hepatitis and lead to chronic hepatitis and cirrhosis through interactions with hepatocytes via the envelope glycoprotein E2 and immune evasion mechanisms involving the core protein.23 HDV often co-infects with HBV, resulting in more severe liver diseases and increased mortality.157 HAV and HEV, transmitted feco-orally, cause self-limiting diseases, with specific mechanism interactions like gangliosides for HAV and immune response dysregulation for HEV impacting infection outcomes.35,158 Overall, viral infection serves as a common cause of various liver diseases.
Parasitic infections such as Schistosomiasis and Echinococcosis primarily occur in rural areas, leading to severe liver complications like fibrosis and portal hypertension via mechanisms like persistent immune response to parasite eggs or direct tissue infiltration by the parasite.159,160,161 Amebic liver abscesses are another consequence, originating from amoebas breaching intestinal barriers and leading to hepatic necrosis.162
Metabolic stress
MASLD represents the hepatic component of a multisystem disorder and is closely linked to the global rise in obesity, T2DM, and metabolic dysfunction.163,164 The increasing prevalence of obesity worldwide has significantly heightened the risk of MASLD development. Studies indicate that individuals with a higher body mass index are more prone to MASLD.165,166 A recent meta-analysis reported that the prevalence of T2DM among radiologically and histologically defined metabolic dysfunction-associated steatohepatitis (MASH) patients was 22.51% and 43.63%, respectively, underlining the strong association between T2DM and MASLD.75 Moreover, various manifestations of metabolic dysfunction, including insulin resistance, bile acid metabolism disorders, gut microbiota imbalances, and hyperuricemia, contribute to the pathogenesis of MASLD.167,168,169 Patients with early MASLD exhibit hepatic steatosis and steatohepatitis, with some progressing to cirrhosis and HCC.170 MASLD has emerged as a predominant chronic liver disease with the escalating prevalence of metabolic dysfunction.171
Toxic exposure
In Europe and the U.S., nonsteroidal anti-inflammatory drugs (NSAIDs), anti-infective drugs (e.g., amoxicillin-clavulanate potassium), and herbal/dietary supplements are the most common causes of DILI.43,172 However, liver injuries induced by traditional Chinese medicine, anti-tuberculosis drugs, and other anti-infective medications are more prevalent in Asia.43 The utilization of anti-cancer drugs and immunomodulators has also been linked to instances of drug-induced liver damage.173 Drug risk factors such as dose and metabolism increase the risk of liver injury with certain medications.174 Moreover, patient genetic predisposition may be another important determinant.175 Notably, DILI is becoming the main cause of ALF worldwide with increasing proportion.175 Chronic alcohol consumption leads to ALD, progressing through stages from fatty liver to cirrhosis and HCC, influenced by dosage and individual factors like gender and concurrent conditions like obesity or viral infection.19,176,177,178,179,180 Certain chemical substances in industrial production are hepatotropic poisons, which cause a susceptible period in the population. For example, exposure to chloroform and phosphorus has been associated with hepatic histological changes leading to toxic hepatitis.181,182 The use of the hepatotoxic substance carbon tetrachloride (CCl4) has become a standard method to induce murine liver fibrosis.183 Moreover, individuals occupationally exposed to chemicals like vinyl chloride and per- and polyfluoroalkyl substances are more sensitive to MASH.184,185 Of note, several fungi species could also produce mycotoxins with high hepatotoxicity, leading to necrosis of hepatocytes with various liver diseases.186
Genetic factors
Hereditary metabolic liver diseases could cause metabolic abnormalities due to the interaction between host and environmental based on genetic defects, mainly including hereditary hemochromatosis, WD, and AATD.187 There are 4 types of hereditary hemochromatosis based on gene mutations. Type 1 is a classic hereditary hemochromatosis, known as HFE-associated hemochromatosis. More than 80% of patients have a C282Y mutation or a C282Y/H63D complex heterozygous mutation.188,189 These patients show hepcidin and transferrin deficiency, which impairs the transport of iron from intracellular storage sites to plasma, resulting in iron deposition in the liver.190,191 WD results from mutations in the ATP7B gene, causing dysfunctional copper transport and accumulation.141 Excessive copper accumulation triggers a reactive oxygen species (ROS) reaction followed by hepatic inflammation and cirrhosis.192 AATD is an autosomal codominant inheritance with the mutation of the SERPINA1 gene, which encoded alpha-1 antitrypsin. Abnormal conformation of this protein is detained by the rough endoplasmic reticulum, causing cellular stress, and liver disease.193,194
Multi-level regulatory mechanisms
Liver diseases encompass a spectrum ranging from acute and chronic liver injuries to end-stage liver diseases, each characterized with distinct pathogenesis. This section outlines the multifaceted mechanisms underlying the initiation and progression of liver diseases, spanning from molecular and cellular levels to organ interactions.
Molecular mechanisms-RIG-1/MAVS signaling
Retinoic acid-inducible gene I (RIG-1) serves as a critical RNA sensor that activates the type I interferon (IFN) response crucial for antiviral defense. RIG-1 is expressed in most cell types and is primarily localized in the cytoplasm.195 Upon RNA recognition, RIG-1 undergoes conformational changes and translocates to mitochondria, where it interacts with its adaptor protein, mitochondrial antiviral-signaling protein (MAVS). MAVS then transmits signals from RIG-1 and activates downstream components such as TANK-binding kinase 1 (TBK-1) and IκB kinase-ε (IKK-ε). These kinases prompt the phosphorylation and nuclear translocation of interferon regulatory factor 3 (IRF-3) and IRF-7 as well as nuclear factor-κB (NF-κB), key transcription factors for the production of IFN and other cytokines.196 Secreted IFN activates JAK/STAT signaling within host cells to engage IFN-stimulated genes (ISGs) expression, which perform essential functions in antiviral defense and the immune response (Fig. 3).197

RIG-1/MAVS, cGAS/STING, and AMPK signaling in liver diseases. When hepatotropic viruses such as HCV and HBV infect the liver, RIG-1/MAVS and cGAS/STING signaling pathways are activated. Both pathways promote the expression of IFN and other inflammatory cytokines by the phosphorylation of IRF-3/7 and NF-κB, respectively. These cytokines perform essential roles in antiviral defense and liver inflammation. AMPK, serving as an energy sensor, regulates various cellular physiological processes. Upon exposure to excessive energy or ethanol, decreased AMPK activity and fat accumulation are observed in hepatocytes. However, activation of AMPK in the liver decreases lipogenesis and cholesterol synthesis, and cell apoptosis, while promoting fatty acid oxidation, autophagy flux, and mitochondria biogenesis. These effects help attenuate the development of MASLD and ALD. This figure was generated with Adobe Illustrator
HCV is a hepatotropic virus associated with liver inflammation, fibrosis, and HCC. The RIG-1 signaling has been demonstrated to play a vital role in HCV sensing and elimination.198 Specifically, the HCV RNA genome binds to host RIG-1 to induce IFN production in hepatocytes via RIG-1/MAVS signaling.199 This course leads to the production of ISGs like ISG15, protein kinase R (PKR), myxovirus resistance A (MxA), and 2′-5′-oligoadenylate synthetase (OAS), which are known for hindering cell cycle progression and the replication of HCV by impeding viral transcription and translation, initiating viral RNA cleavage, and modifying viral protein functions.200,201,202,203 However, Li et al. demonstrated that HCV nonstructural protein 3-4A (NS3-4A) protease cleaves MAVS protein and diminishes MAVS/IRF-3-dependent IFN and ISGs production,204 whereas NS3/4A inhibitors restore MAVS proteolysis and the IFN-dependent antiviral response.205 These data may in part explain the scarcity of endogenous IFN in some HCV-infected individuals and the persistent HCV infection, as well as the importance of RIG-1/MAVS signaling in HCV elimination.
Besides HCV, the HBV DNA is involved in RIG-1/MAVS signaling activation. Pregenomic RNA of HBV is revealed to bind with RIG-1 protein, which induces IFN and ISGs production to prevent HBV infection.206 Moreover, RIG-1 counteracts the interaction of HBV polymerase, thus suppressing viral replication and production.207 HBV covalently closed circular DNA (cccDNA), the transcriptional template for viral RNA, is indispensable for HBV persistence and chronic hepatitis. Intriguingly, Lee et al. demonstrated that RIG-1/IRF-3 signaling blocks cccDNA formation and amplification in hepatocytes.208 These findings underscore the multifunctional roles of RIG-1/MAVS signaling in anti-HBV immune response. However, HBV X protein (HBx) is shown to deactivate IFN production by deubiquitinating RIG-1 and its downstream effectors IKK-ε and IRF-3.209 Evidence also suggests that the activation of RIG-1/MAS signaling by other viruses such as HAV, HDV and HEV combats viral replication.210,211,212 In summary, RIG-1/MAVS signaling exerts pivotal roles in limiting virus replication and enhancing innate immune responses, thereby attenuating hepatic viral diseases.
Molecular mechanisms-cGAS/STING signaling
The cGAS/STING signaling pathway plays a crucial role in immune defense by detecting cytoplasmic DNA. The enzyme cyclic GMP-AMP (cGAMP) synthase (cGAS) is activated upon exposure to cytoplasmic DNA from pathogens or damaged host cells. cGAS synthesizes the second messenger cGAMP, which then binds to and activates the stimulator of interferon genes (STING). STING, localized in the ER, initiates downstream signaling to produce IFNs and other cytokines, thereby promoting a potent innate immune response.213,214
In the context of liver disease, cGAS/STING signaling has been implicated in the progression of conditions associated with viral infection and sterile inflammation (Fig. 3). Research indicates that HBV can evade immune surveillance by suppressing DNA sensor pathways, including the cGAS pathway, resulting in reduced expression of cGAS and its effectors during chronic HBV infection. Despite this viral evasion, activation of cGAS signaling can inhibit HBV replication. This is achieved by blocking the amplification of HBV cccDNA and reducing HBV RNA synthesis through the production of STING-mediated cytokines and ISGs, both in vivo and in vitro.215,216,217 Additionally, cGAS/STING signaling has been shown to restrict HCV replication in hepatocytes and is an important component in the immune response against this virus, as observed in studies involving STING knockdown models.218
The pathway is also involved in the progression of sterile inflammatory liver diseases. The release of endogenous DNA, such as mitochondrial DNA (mtDNA), during cellular damage triggers cGAS-STING signaling. This activation leads to the production of inflammatory cytokines. In cases of liver injury induced by substances such as acetaminophen and thioacetamide, hepatocyte-derived mtDNA activates cGAS-STING signaling in macrophages. This promotes an inflammatory phenotype switch in these cells, which exacerbates hepatocyte injury by promoting ferroptosis, a form of programmed cell death associated with iron.219,220,221 Additionally, mtDNA-induced cGAS-STING signaling has also been reported in mice with ALD222 and MASLD,223 whereas overexpression of RING finger protein 13 (RNF-13) in hepatocytes attenuates liver steatosis, inflammation and fibrosis by degrading the STING protein in a mouse MASLD model.224 Interestingly, the STING-NF-κB pathway in macrophages leads to metaflammation in lean MASLD mouse, which promotes lipolysis in the adipose tissue and subsequently contributes to liver lipid deposition and injury.225 The evidence shows the detrimental role of cGAS/STING signaling in the regulation of sterile liver inflammation.
Molecular mechanisms-AMPK signaling
AMP-activated protein kinase (AMPK) is a highly conserved heterotrimeric protein consisting of a catalytic α subunit and regulatory β and γ subunits. This central eukaryotic energy sensor facilitates the maintenance of physiological cellular processes.226 Upon exposure to excess energy, liver kinase B1 (LKB-1) and Ca2+/CaM-dependent protein kinase kinase β (CAMKK-β), phosphorylate the Thr172 residue on the AMPK α subunit.227 Then AMPK phosphorylates and stimulates multiple downstream substrates to regulate lipid and glucose metabolism, as well as mitochondrial function.228 Herein, we propose the primary mechanisms by which AMPK affects liver injury, especially in MASLD and ALD (Fig. 3).
AMPK inhibits lipid synthesis by deactivating acetyl-CoA carboxylases (ACC-1 and ACC-2) and HMG-CoA reductase (HMGCR), the rate-limiting enzymes in fatty acid and cholesterol synthesis, respectively.229 There is a negative correlation between AMPK and the development of MASLD and ALD, as shown by the reduced AMPK levels in these fatty liver samples.230,231 Notably, activation of AMPK by its upstream kinase LKB-1, restores hepatic lipid accumulation by downregulating lipogenesis-mediated genes, such as Srebp1c, Acc, Fas, Scd1, and Hmgcr in a high-fat diet-induced mouse model.232 SREBPs, key transcriptional factors for lipid and cholesterol synthesis, are verified to contribute to MASLD and MASH-associated HCC.233,234 Recently, studies showed that maturation and activity of SREBPs are controlled by adenosine A1 receptor (A1R) and A2R, which could be targeted to relieve MASLD.234,235 Specific agonist of A1R, 2-chloro-N6-cyclopentyladenosine (CCPA) or screened natural compound, timosaponin AIII showed promising activity in MASLD, especially MASH therapy by activating hepatic A1R.234 Therefore, hepatic A1R is a novel target for MASLD/MASH therapy with great potential through modulating SREBPs maturation and its controlled fatty acid de novo synthesis. Conversely, in vivo and in vitro studies found that hepatocytes exhibit more severe triglyceride accumulation when AMPK is depleted or blocked by its inhibitor compound C, showing the diminished protective role of AMPK against liver steatosis.236 These data indicate the pivotal role of AMPK signaling in blocking hepatic lipogenesis.
Furthermore, AMPK promotes the expression of genes related to fatty acid oxidation in an ACC-2-dependent manner. Specifically, AMPK-inhibited ACC-2 catalyzes the generation of malonyl-CoA, which inhibits the activity of carnitine palmitoyltransferase 1 (CPT-1), a rate-limiting enzyme of mitochondrial oxidation.237 Upon exposure to ethanol and lipids, decreased AMPK activity and fatty acid oxidation flux is observed in mouse hepatocytes.238,239 In contrast, metformin-driven AMPK activation rescues CPT-1 expression and diminishes lipid accumulation in rat livers impacted by chronic ethanol insult.240 Likewise, CAMKK-β-induced AMPK activation promotes fatty acid oxidation and mitochondrial biogenesis, thereby attenuating hepatic steatosis in MASLD mice.241
AMPK is essential to maintain mitochondrial homeostasis. Upon inflammatory stimuli, decreased AMPK activity together with mitochondrial dysfunction and ROS is observed in MASH models.242 Growing studies showed that ethanol-induced ROS could be repressed in an AMPK-dependent manner. Impaired mitochondrial structure and increased mtDNA were observed in ethanol-treated hepatocytes, and AMPK is verified to rescue mitochondrial biogenesis and function by increasing mitophagy and ROS removal in hepatocytes.231,243 High-fat diet-induced ROS and ER stress were inhibited by activating AMPK/NRF-2/HO-1 signaling, which attenuated hepatic lipid accumulation and inflammation.244,245,246 In addition, both in vivo and in vitro experiments demonstrated that AMPK-Caspase-6 axis relieves mitochondrial function and protect against hepatocellular apoptosis in MASH, as well as ferroptosis in ALD.247,248 Besides, AMPK plays important roles in linking metabolism to the development of liver cancer. Lower levels of AMPK are associated with poor prognosis in HCC, while activation of AMPK expression regulates metabolic reprogramming in the tumor microenvironment, improving the efficacy of tumor immunotherapy.249,250 Collectively, the above evidence elucidates the crucial roles of AMPK in liver metabolism and inflammation, which implicates AMPK might be a potential therapeutic target in liver diseases.
Molecular mechanisms-MAPK signaling
The mitogen-activated protein kinase (MAPK) signal transduction pathway is a critical mediator that orchestrates cellular proliferation, differentiation, and death. The core module of MAPK signaling is composed by three-tiered kinase cascade proteins, namely MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK) and MAPK. MAPKs encompass extracellular signal regulated kinases (ERK-1/2), p38α/β/δ/γ MAPK, and c-Jun-N-terminal kinases (JNK-1/2/3).251 MAPK signaling is activated in response to extracellular stimuli, including hormones, cytokines, growth factors. Active MAPK phosphorylates and activates downstream effectors, including transcription regulators that translocate to the nucleus to manipulate target gene expression.252 In the liver, MAPK signaling plays an important role in mediating inflammation, metabolism, and cell proliferation (Fig. 4).

MAPK, PI3K/Akt, and JAK/STAT signaling in liver injury. When liver injury occurs, activated MAPK signaling promotes gluconeogenesis but has bilateral effects on liver lipid metabolism. It enhances liver inflammation by targeting key transcription factors, including activator protein 1 (AP-1) and NF-κB, and promotes HCC and ICC by encouraging tumor cell proliferation and migration. Additionally, MAPK plays a role in antiviral defense by activating the expression of IFN and ISGs. PI3K/Akt signaling promotes liver lipid and glucose metabolism by targeting key transcription factors and enzymes, and similarly promotes liver inflammation and cancer. JAK/STAT signaling is essential for the elimination of hepatotropic viruses through the induction of IFN and ISGs. STAT proteins have bidirectional roles in liver inflammation and cancer. Specifically, STAT1 promotes inflammation, whereas STAT3 exhibits both pro-inflammatory and anti-inflammatory signals. STAT1 prevents HCC development, whereas STAT3 contributes to the liver tumorigenesis. This figure was generated with Adobe Illustrator
Multiple studies have revealed the activation of MAPK signaling in hepatocytes and macrophages during acute or chronic liver injury. Fatty acids, ethanol, and acetaminophen have been implicated in the activation of p38 MAPK, JNK and ERK signaling in hepatocytes.253,254,255 The signaling cascade triggers the activation of critical regulators of lipid synthesis, autophagy, and inflammation such as SREBP-1c, sequestosome-1 (SQSTM1/P62) and NF-κB; however, blocking JNK and p38 signaling by degradation of TAK-1, a member of the MAPKKK family, reverses fat accumulation, impaired autophagy flux, inflammation, and apoptosis in hepatocytes.253 Recently, lysosomal homeostasis and autophagic flux have been recognized as playing a beneficial role in MASLD.256 Lysosomal dysfunction can lead to impaired autophagic flux, inducing lipid droplet accumulation in hepatocytes and further activating HSCs in a hepatic steatosis model.257,258 In contrast, hepatic fat accumulation and liver fibrosis are alleviated when lysosomal dysfunction is restored.259,260 Additionally, both in vivo and in vitro studies have demonstrated that the overexpression of JNK signaling and subsequent AP-1 and NF-κB cascades in hepatocytes promotes cell proliferation and migration, thus contributing to MASLD-associated liver cancer.261 The above evidence suggests that MAPK signaling and its downstream effectors plays pivotal roles in hepatocyte survival and function.
Emerging studies suggest that MAPK signaling in hepatic macrophages acts as a key contributor to liver injury. p38 MAPK signaling in macrophages contributes to the development of nutritional steatohepatitis by promoting M1 macrophage polarization and the release of inflammatory cytokines. In contrast, macrophage p38 MAPK deficiency in mice is associated with a hepatic M2 phenotype characterized by decreased secretion of TNF-α, IL-6, and CXCL-10, which leads to reduced fat accumulation and hepatocyte apoptosis.262 Consistently, macrophage p38 MAPK-deficient mice are more resistant to drug-induced hepatotoxicity, as evidenced by decreased cytokine production and accelerated hepatocyte regeneration.263 Moreover, ERK signaling in macrophages is responsible for TGF-β production, thus triggering HSC activation in response to high-fat/high-cholesterol diet.264 Interestingly, HSC activation is directly promoted by ERK signaling. Specifically, the secretory protein ANGPTL8 from fatty hepatocytes interacts with the LILRB2 receptor on HSCs and activates ERK signaling-dependent autophagy.265 Increased autophagy flux facilitates the transdifferentiation of HSCs into a myofiblastic phenotype, ultimately contributing to liver fibrogenesis.266 These observations implicate the importance of MAPK signaling in the evolution of liver diseases.
Molecular mechanisms-PI3K/Akt signaling
The phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway represents an evolutionarily conserved signaling cascade pivotal in cellular processes such as metabolism, survival, proliferation, and cell death. The pathway consists of two core components: PI3Ks and Akts. Upon stimulation, PI3K catalyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), which serves as a second messenger to recruit and activate Akt. Activated Akt then phosphorylates numerous downstream substrates to initiate multiple pathways. In this section, we focus on PI3K/Akt signaling during liver diseases (Fig. 4).267
PI3K/Akt signaling performs bidirectional roles in response to acute liver injury and liver fibrosis.268,269 PI3K/Akt signaling impedes liver regeneration by promoting macrophage migration and fostering an inflammatory environment after partial hepatectomy.270 On the other hand, PI3K/Akt signaling is responsible for the production of hepatocyte growth factor (HGF), epidermal growth factor (EGF), and TGF-β, which are essential for hepatocyte proliferation and survival.271 In the setting of liver fibrosis, PI3K/Akt signaling in macrophages contributes to profibrotic mediators secretion, thus triggering HSC activation and ECM production.272 However, another study revealed that PI3K/Akt signaling counteracts the TGF-β/SMAD signaling-an important player in HSC activation-to balance cell survival and proliferation under chronic stimuli.273 In addition, activated Akt induces the expression of matrix metalloproteinase (MMPs), which plays an importance role in ECM breakdown.274 All these data demonstrate the dual roles of PI3K/Akt signaling in liver diseases. Considering the different stages of liver diseases, as well as the diverse cellular sources of PI3K/Akt signaling, these controversial results should be interpreted cautiously.
PI3K/Akt signaling is associated with the development of HCC through its regulation of tumor cell glycolysis, growth, and apoptosis. HCC cells exhibiting activated PI3K/Akt signaling show increased glucose uptake and lactate production, a phenomenon known as aerobic glycolysis or the Warburg effect, facilitating long-term cancer cell survival. In contrast, suppression of PI3K/Akt signaling transitions aerobic glycolysis to oxidative phosphorylation, accompanied by restored mitochondrial function, which indicates the involvement of PI3K/Akt signaling in metabolic reprogramming during HCC progression.275 In addition, inhibition of PI3K/Akt signaling elicits increased expression of caspase-3 and caspase-9, apoptotic markers, within HCC cells.276 Overall, these findings suggest the excitatory role of PI3K/Akt signaling in the evolution of HCC.
Molecular mechanisms-JAK/STAT signaling
The Janus kinase /signal transducer and activator of transcription (STAT) signaling pathway is a highly conserved pathway that performs crucial roles in cell differentiation, metabolism, growth, and immune response.277 Once extracellular signals such as cytokines, interferons, and growth factors, bind to their respective receptors, JAK proteins and downstream STAT proteins undergo phosphorylation. Activated STAT proteins translocate to the nucleus, where they bind to DNA sequences to regulate target gene expression.278,279 In this context, we discuss the dysregulation of the JAK/STAT signaling in liver diseases, particularly in autoimmune and viral hepatitis (Fig. 4).
Upregulated JAK/STAT signaling has been observed in patients with PBC, a chronic autoimmune liver disease.280 Genome-wide meta-analysis suggested a correlation between JAK/STAT signaling and PBC.281 Importantly, a JAK-1/2 inhibitor, baricitinib, has shown promising results in reducing alkaline phosphatase (ALP) levels and liver inflammation in PBC patients based on a phase II trial.282 Mechanistically, IFN-induced JAK/STAT1 signaling triggers the amplification of hepatic CD4+ T cells and CD8+ T cells, the polarization of M1 macrophages and the release of cytokines in experimental autoimmune cholangitis models, eliciting a liver immune response and inflammation.283 This finding underscores the pivotal role of JAK/STAT signaling in modulating liver autoimmunity.
In addition, JAK/STAT signaling is implicated in IFN-induced viral hepatitis. As mentioned above, secreted IFN binds to its receptor and activates ISG expression in a JAK/STAT-dependent manner.284 Essential ISGs such as PKR and OAS, which are crucial for restricting HBV replication, are induced by JAK/STAT signaling, whereas the inhibition of this pathway leads to diminished PKR and OAS expression during HBV infection.285 Similarly, in vivo and in vitro studies revealed that the JAK/STAT-dependent induction of ISG-12a plays a vital role in inhibiting HCV replication.286
In addition to ISGs, numerous mediators involved in liver inflammation and fibrosis are also the target of JAK/STAT signaling.287 The activation of JAK/STAT signaling in hepatocytes mediates the production of IL-6, CXCL-10, and iNOS, which promotes hepatocyte apoptosis, inflammatory cell infiltration and fibrogenesis in a mouse model of MASLD.288,289 In summary, JAK/STAT signaling has dual roles in liver diseases: stimulating innate and adaptive immunity while governing virus elimination within distinct disease contexts.
Molecular mechanisms-Wnt/β-catenin signaling
The Wnt/β-catenin signaling performs vital functions in embryonic process and organ development. Upon Wnt proteins bind to frizzled receptors (Fzd) and low-density lipoprotein receptor-related protein 5/6 (LRP-5/6) co-receptors, signals are transduced to β-catenin to trigger downstream events. Under physical conditions, the signaling is tightly regulated through degradation complex in the cytoplasm, where a multiprotein complex involving enzymes such as E3 ubiquitin ligases leads to the degradation of signaling proteins, maintaining an inactive state.290 Dysregulation of Wnt/β-catenin signaling is currently considered as a crucial factor in oncogenesis. The accumulation and nuclear shuttling of β-catenin result in its interaction with T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors, activating proto-oncogenes such as myelocytomatosis oncogene (c-Myc) and cyclin-D1 (CCND-1) and thus promoting cell proliferation and migration.291 Here, we summarize the crucial roles of Wnt signaling in liver cancer (Fig. 5).

TGF-β and Wnt/β-catenin canonical signaling in liver diseases. Upon liver injury, TGF-β secreted from other liver cell types binds to its receptor TGF-βRII, which subsequently recruits TGF-βRI to synergistically mediate downstream pathways: canonical SMAD-dependent pathway and non-SMAD pathways. In canonical pathway, the SMAD oligomers translocate into the nucleus, where they function as transcription factors, mediating the transcriptional activation. In non-SMAD pathway, PI3K/Akt and MAPK pathways are activated by TGF-βRs. These pathways induce expression of genes, such as α-SMA, Collagens, fibronectin, TIMP-1, LOXL-1, and Kindlin-2, leading to HSC activation, ECM production and stabilization, and cell adhesion. These processes collectively promote liver fibrosis. In a healthy liver, Wnt signaling is typically inactive due to the absence of Wnt-Wnt receptor interactions and the degradation of β-catenin by a protein complex, which includes axis inhibition protein (AXIN), adenomatous polyposis coli (APC), and E3 ubiquitin ligase. During liver oncogenic injury, Wnt proteins bind to Fzd receptor and LRP-5/6 co-receptors, activating the canonical pathway. This activation causes degradation complex to translocate to the cell membrane, preventing the degradation of β-catenin. The β-catenin then enters the nucleus, where it binds with TCF/LEF transcription factors to regulate target gene expression, such as c-Myc, cyclin-D1 and pyruvate kinase M2 (PKM-2). These genes are involved in promoting tumor cell metabolism, proliferation, migration, and metastasis in HCC and ICC. This figure was generated with Adobe Illustrator
Genetic mutations involving Wnt signaling have been reported in human HCC. Approximately 8-30% of HCC patients exhibit mutations in the β-catenin gene (CTNNB-1), which prevents β-catenin degradation and facilitates its nuclear translocation.292,293 Integrated multi-omics analyses have revealed pathologically elevated Wnt signaling in human HCC tissues.294 Notably, the interaction between Wnt-3a and Fzd-7 in human HCC cells drives tumor proliferation and migration by activating β-catenin-dependent signaling.295 Intriguingly, hepatocyte-specific overexpression and activation of β-catenin protein alone are insufficient to induce HCC.296 However, the stimulation of β-catenin in conjunction with pathological stimuli could initiate and accelerate HCC progression in mice.297 In contrast, the prevalence of tumor in the liver with Ctnnb-1 conditional knockout is 7-fold higher than that in wild type liver, indicating that the absence of β-catenin stimulates carcinogen-induced hepatocarcinogenesis.298 Mutation of hepatic Ctnnb-1 drives hepatocarcinogenesis by upregulation of pro-tumorigenic cytokines.299 It seems contradictory that the presence of the mutated β-catenin and the absence of normal β-catenin, both contribute to the development of HCC. More in-depth studies are needed to clarify the precise mechanism.
In addition to its role in HCC, Wnt/β-catenin signaling also participates in the initiation and progression of intrahepatic cholangiocarcinoma (ICC). Notably, upregulated Wnt-7b levels are observed in human ICC tumors and mouse ICC models, with evidence suggesting that macrophages are the cellular source of Wnt-7b production in vivo and in vitro.300 Pharmacological or genetic inhibition of Wnt-7b-Fzd7-β-catenin signaling has shown promise in mitigating tumor growth and metastasis.301 In summary, Wnt/β-catenin signaling contributes to the oncogenic process of liver carcinoma.
Molecular mechanisms-TGF-β signaling
Transforming growth factor-β (TGF-β) is a cytokine with three isoforms (TGF‑β1, TGF‑β2, and TGF‑β3), sharing around 80% homology in their amino acid sequences. Upon TGF-β binding, TGF-β receptor 2 (TGF-βR2) recruits and activates TGF-βR1 to synergistically mediate downstream signaling. TGF-β/TGF-βR transmits extracellular stimuli and exhibit cellular transcriptional events by two ways: canonical SMAD-dependent pathway and non-SMAD pathways.302 TGF-β signaling is well-recognized for inducing fibrosis in multiple organs, including the liver (Fig. 5).303,304
Excessive TGF-β expression is documented in both acute and chronic liver diseases across various cell types.305 In patients with diseases such as AIH and chronic hepatitis C, increased serum and hepatic levels of TGF-β are observed, correlating with disease progression.306,307 Transcriptome analysis from MASLD model reveal macrophages, LSECs, activated HSCs, and hepatocytes as sources of TGF-β production.308 Notably, macrophages are identified as the predominant cellular origin of TGF-β in the injured liver.309
HSC activation serves as a hallmark event in the initiation of liver fibrosis, with ECM deposition characterizing fibrotic progression. TGF-β, which is induced by liver injury, triggers TGFR activation in HSCs, leading to phosphorylation of downstream effectors, such as small mothers of decapentaplegic (SMAD) proteins. Activated SMAD proteins translocate into the nucleus, where they facilitate transcription of target genes by interaction with DNA-binding transcription cofactors.310 Literature supports that TGFR/SMAD in HSCs promotes expression of α smooth muscle actin (α-SMA), collagen type I and III, which are involved in HSC activation and extracellular matrix (ECM) composition, respectively.311,312 In addition, SMAD also triggers the expression of lysyl oxidase-like (LOXL) and tissue inhibitor of metalloproteinases (TIMP) proteins, both of which perform essential functions in ECM deposition and stabilization.313,314 In contrast, mice with HSC-specific inactivation of SMAD-2 have increased susceptibility to CCl4- and DDC-induced liver fibrosis.315
In addition to the canonical pathway, TGF-β also contributes to liver fibrosis through the non-SMAD pathway by interplaying with MAPK signaling and PI3K signaling. Recent work has elucidated interactions between TGF-β and p38 MAPK signaling in HSCs, driving kindlin-2 expression and subsequent immune cell adhesion, which in turn promotes HSC activation.316 In addition, TGF-β induces ADAM12 expression via the PI3K/Akt pathway in cultured human HSCs, contributing to cell adhesion and migration.317 Taken together, these data indicate the critical roles played by both SMAD and non-SMAD pathways in TGF-β-induced HSC activation and liver fibrosis.
Inter-cellular mechanisms
Cellular crosstalk is a crucial event that maintains liver homeostasis. When liver injury occurs, hepatocytes and non-parenchymal cells engage in pathological paracrine interactions. In this section, we describe the cellular crosstalk during liver diseases.
Hepatocytes
Hepatocytes, major parenchymal cells in the liver, perform diverse roles in lipid and glucose metabolism, detoxification, and protein synthesis. During disease states, hepatocytes face direct assaults from viruses or metabolites but also respond to signals from neighboring cells. The injury imposed on hepatocytes may exacerbate cellular dysfunction and subsequent death.318 Liver macrophages serve as primary reservoirs for inflammatory cytokines such as IL-1β and TNF-α, which contribute to hepatocyte death.319 It has been well-established that IL-1 drives hepatocyte inflammation and apoptosis via interacting with its receptor and downstream effectors. In contrast, hepatocyte-specific depletion of IL-1 receptor rescues hepatocyte apoptosis by blocking JNK/NF-κB signaling during acute liver injury.320 Inflammasomes released from macrophages also contribute to hepatocyte pyroptosis and liver inflammation in mouse model of MASLD.320 In addition, Wnt2 protein derived from LSECs was found to regulate cholesterol and bile acid homeostasis in hepatocytes.321 However, silencing the expression of LSEC-specific Wnt2 disturbs hepatocyte metabolic profiles in both acute and chronic liver injury, indicating the essential role of LSECs in hepatocyte function.322
Cholangiocytes
Cholangiocytes are highly specialized epithelial cells forming the bile ducts and are essential for bile acid homeostasis. In chronic liver injury, such as cholangiopathies, a pathological feature known as a ductular reaction occurs, characterized by the proliferation of reactive ductular cells.323 Ablation of β1-integrin in hepatocytes stimulates the ductular reaction, leading to cholangiocyte-derived hepatocyte regeneration during chronic liver diseases. These data indicate the potential network between hepatocytes and cholangiocytes.324 Inflammatory and fibrotic secretions from immune cells are involved in cholangiocyte activation and biliary repair, which in turn, leads to increased inflammatory cell infiltration and persistent liver impairment.325,326
LSECs
LSECs are highly differentiated endothelial cells lining the liver sinusoids. LSECs possess a unique phenotype with fenestrae, enabling substantial exchanges and cellular communications.327 Although LSECs detect extrahepatic signals and help maintain liver homeostasis through angiocrine mechanisms, their function and architecture are regulated by other cell types. Inflammatory cell populations, such as CCR2+ macrophages and CXCR1+ neutrophils, which are recruited by injured LSECs in the early phase of liver damage, could in turn compromise LSEC endocytosis capacity and cause its fenestrae impairments.328,329 Moreover, neutrophil adhesion attracts platelet recruitment, leading to the generation of sinusoidal microthrombi, which in turn induces sinusoidal dysfunction and vasoconstriction.330 This pathological cascade elevates sinusoidal pressure, exacerbating liver fibrosis and portal hypertension. HSCs are crucial players to maintain LSEC phenotype. Bone morphogenetic protein 9 (BMP-9) has been identified to control vessel homeostasis.331 Intriguingly, HSCs are the hepatic cell source of BMP-9, which emphasize the impact of HSC-derived BMP-9 on LSEC phenotype and function via targeting its receptor activin receptor-like kinase 1 (ALK-1).332 Aberrant expression of BMP-9 or ALK-1 depletion in LSECs during diseased states results in impaired angiocrine function and LSEC architecture, underlining the effect of HSCs on LSEC physiological process.333
HSCs
Quiescent hepatic stellate cells (qHSCs) represent about 5% of liver resident cells and reside in the space of Disse. Following liver damage, qHSCs convert to an activated myofibroblast phenotype characterized by proliferation, contractility, and chemotaxis.334 Fate tracing analysis has implicated that activated HSCs are the predominant source of ECM in liver diseases induced by toxic, fatty, and cholestatic insults.335,336 These activated HSCs migrate to the injured sites, where they proliferate and contribute to ECM production, thus participating in liver repair. Multiple mediators from other liver cell types contribute to HSCs activation. LSECs are responsible for the production of TGF-β and activation of HSC via the angiocrine pathway during the early stage of liver injury.337,338 Besides canonical TGF-β signaling, platelet-derived growth factor (PDGF) and IL-6 produced by capillarized LSECs also contribute to HSC activation by the JAK/STAT pathway.339 Notably, macrophages are the major producers of TGF-β, leading to subsequent HSC activation during liver injury. Loss of TGF-β results in a significant decrease in ECM deposition.340 NLRP3 inflammasome, induced by pyroptotic hepatocytes, also activates HSCs by releasing multiple cytokines in CCl4-induced liver injury.341 In summary, various liver cell types are responsible for HSC activation to induce liver fibrosis.
Macrophages
As the predominant immune cell type in the liver, macrophages play crucial roles in maintaining liver homeostasis and responding to diseases. Hepatic macrophages are composed by liver-resident Kupffer cells (KCs) and monocyte-derived macrophages (MoMFs), two heterogeneous subpopulations with distinct functions.342 KCs primarily serve as the main source of hepatic macrophages in a healthy liver, responsible for sensing injury signals and clearing cellular debris.343 Under inflammatory conditions, MoMFs infiltrate and become the major component of hepatic macrophages with the loss of KCs.344 Macrophages play essential roles in liver inflammation and fibrosis via the production of inflammatory cytokines and chemokines and the activation of inflammasomes.345 LSECs recruit MoMFs in a CCL2/CCR2-dependent manner during chronic liver injury. Specifically, by deleting LSEC-specific CCL2, infiltrating macrophage recruitment, liver inflammation, and fibrosis were reduced in CCl4-induced mice.328 In addition, transcriptome analysis and animal experiments reveal that hepatocytes are involved in the recruitment of MoMFs via the CCL2-CCR2 interaction in acute liver injury to facilitate necrotic lesion resolution.346,347 Besides, mediators secreted by hepatocytes, such as IL-17, IL-1β, and extracellular vesicles (EVs), mediate inflammatory macrophage infiltration and the development of ALD and MASLD.348,349 However, certain inflammatory macrophages switch to an anti-inflammatory LY6Clow phenotype during the progression of liver diseases, which plays an important role in ECM degradation via secretion of MMPs.350 Moreover, targeting myeloid-derived RNF-41 has been shown to promote this phenotypic switch and subsequent ECM degradation, thus leading to fibrosis resolution.351 While the data indicate the dual roles of macrophages, further investigations are needed to elucidate the precise mechanisms of macrophage phenotype switch during liver inflammation and fibrosis.
Other immune cells
The T cell-mediated adaptive immune response plays central roles in antigen-driven liver diseases such as autoimmune hepatitis and chronic viral hepatitis.352 Autoantigens in hepatocytes, such as formiminotransferase cyclodeaminase, and cytochrome P450 2D6, are presented in the naive CD4+ T-helper lymphocytes. These Th0 cells subsequently differentiate into Th1 and Th2 cells, which are responsible for macrophage and B cell infiltration, respectively. This immune response cascade attacks hepatocytes, contributing to liver injury.353,354 Similarly, the T cell response is also pivotal in the clearance of HBV and HCV. Effector T cells recognize HBV/HCV-infected hepatocytes and eliminate the virus through a combination of cytotoxic and non-cytotoxic pathways.355,356
In a healthy liver, neutrophils are typically absent, but their infiltration within liver sinusoids is noted during acute and chronic liver diseases.357 Research has highlighted that LSEC-dependent neutrophils infiltration occurs through the secretion of CXCL chemokines.358 Growing evidence has emphasized the significance of neutrophil in liver diseases, as they are thought to promote liver inflammation by releasing cytokines and chemokines, along with recruiting various other immune cells and potentially contributing to the formation of microthrombi.359,360 Collectively, these findings indicate that immune cells play multifaceted roles in the progression of liver diseases, impacting both the immune response and inflammatory processes within the liver.
Liver-organ communication
Emerging evidence has shown that pathological changes at the molecular and cellular levels may not fully account for the pathogenesis of liver diseases, implying a potential role for inter-organ communications in their development. Recently, the gut-liver-brain and adipose-liver homeostasis has gained much attention.361 In this section, we summarize the recent research on the importance of liver-organ interactions concerning metabolism, immune system, and nervous system.
Gut-liver-brain axis
Numerous preclinical and clinical studies have highlighted the role of abnormal gut microbiota and their metabolites in impairing intestinal barrier function, further contributing to liver diseases. Increased permeability is reported in mice fed a high-fat diet or with excessive ethanol intake.362,363 Gut leakage leads to the delivery of pathogens and their metabolites to the liver via portal vein. In patients with MASH, gut microbiomes predominantly exhibit Gram-positive Firmicutes. However, there is a decrease in Firmicutes and an increase in Gram-negative Proteobacteria abundance as liver fibrosis develops.364 Animal studies demonstrate that germ-free mice fed a high-fat diet show reduced liver steatosis and insulin resistance compared to wild-type mice on the same diet.365 This beneficial effect disappears following fecal microbiota transplantation (FMT) from MASLD mice to germ-free mice, resulting in increased liver triglyceride content and inflammation.366 These data indicate that dysregulated gut microbiota is essential for the progression of MASLD. Besides pathogenic bacteria, their metabolites also mediate liver diseases. For instance, short chain fatty acids (SCFAs) produced by gut bacteria have been linked to promoting hepatic de novo lipogenesis and glucose production, exacerbating the development of MASLD and ALD in mouse models.367,368 In contrast, acetate, another type of SCFA, could block the IL-6/JAK1/STAT3 signaling pathway via binding to hepatocyte-derived GPR43, and reverse the development of MASLD-associated HCC.369 This suggests the complex functions of SCFA components on liver injury. In addition, gut bacteria-derived lipopolysaccharides (LPS) influxes to the liver and activates KCs or macrophages expressing toll-like receptors (TLRs). These immune cells produce inflammatory cytokines and chemokines, thus augmenting innate immune responses and liver injuries.370,371 On the other hand, the normal enterohepatic circulation of bile acids (BAs) is important for liver and intestinal homeostasis. BA is secreted by hepatocytes and modified in the intestine for lipid digestion and absorption. Upon liver injury, changes in BA composition and level, as well as decreased BA receptor (farnesoid X receptor; FXR) was reported.372 Studies show that a decrease in intestinal FXR levels dampens the tight junctions of intestinal epithelial cells and promotes intestinal lipid absorption.373,374 Additionally, knockdown of hepatocyte-derived FXR increases hepatic triglyceride content. In contrast, activation or overexpression of FXR protects against liver injury by decreasing hepatic lipogenic gene expression, reducing intestinal lipid absorption, and promoting intestinal barrier integrity.374,375
The liver-brain axis is essential in the context of liver diseases. Upon acute or chronic injury, liver produces inflammatory cytokines and is unable to process ammonia from the intestine.376,377 These cytokines, such as TNF-α and IL-1β, impair blood-brain barrier (BBB) function and structure, and lead to neuroinflammation.378 In addition, lipid components such as ceramide and palmitate, along with peripheral insulin resistance in MASLD preclinical models, contribute to neuroinflammation and neurodegeneration.379 Moreover, ammonia crosses the damaged BBB and is absorbed by astrocytes, leading to the conversion of ammonia to glutamine and subsequently causing cerebral edema and neuronal cell death.380 Reciprocally, the central nervous system (CNS) exerts an influence on liver and intestine function. Changes in liver microenvironment are detected and transduced through hepatic vagal sensory afferent nerves to CNS, which feeds back the signal to liver vagal parasympathetic nerves.381 For instance, CNS leptin signaling has been shown to promote hepatic triglyceride export and inhibit lipogenesis via the brain-vagus-liver axis, thereby attenuating the development of MASLD in animal models, as well as in a randomized, placebo-controlled crossover trial.382,383 Chronic systemic inflammation involving liver-organ interactions, as well as bacteria translocation due to impaired intestinal barrier can further lead to acute-on-chronic liver injury and multiorgan failure.384 In summary, dysregulation of the gut-liver-brain axis partially influence the progression of liver diseases, suggesting potential therapeutic strategies targeting this axis for liver disease management.
Adipose-liver axis
Emerging studies have demonstrated the pathological crosstalk between the liver and adipose tissue during liver diseases, especially MASLD and ALD. Fat overload leads to adipocyte hypertrophy, hyperplasia, and abnormal adipokine production.385 Chronic ethanol exposure also disrupts the endocrine function of adipose tissue.386 For example, adiponectin, which promotes liver glucose use and fatty acid oxidation, is found to be decreased in MASLD and ALD; however, exogenous supplementation of adiponectin alleviates high-fat diet- and ethanol-induced liver steatosis and insulin resistance.387 Leptin, an adipocyte-derived hormone that inhibits appetite and increases fatty acid oxidation, becomes resistant in MASLD. In obese individuals, higher serum leptin levels correlate with greater severity of liver inflammation.388 In contrast, inflammatory cytokines such as IL-1β and TNF-α are produced by these diseased adipocytes. The inflammatory microenvironment recruits immune cell infiltration and further promotes adipocyte lipolysis. Systemic release of fatty acids and cytokines flows into the liver and aggravates liver steatosis and inflammation.389
Conversely, the liver also interacts with adipose tissue. Fibroblast growth factor-21 (FGF-21), an endocrine hormone mainly produced by hepatocytes, contributes to glucose uptake and adiponectin production in adipocytes.390 In obesity, the expression of FGF-21 increases with the progression of MASH, whereas its effect on adipose tissue becomes resistant, as evidenced by decreased levels of adiponectin.391 As mentioned above, a decrease in adiponectin exacerbates the development of MASLD and ALD by promoting liver steatosis and insulin resistance. In summary, pathological crosstalk between the liver and adipose tissue contributes to liver diseases.
Diagnosis, staging, prevention and therapeutic strategies
The diagnosis of liver disease usually includes the following steps: history collection, physical examination, laboratory tests, imaging examination, and histopathological examination. For several kinds of liver diseases, a liver biopsy may be required to confirm the diagnosis by pathological examination under a microscope. In the process of diagnosis, it is necessary to select targeted examination items according to the specific conditions of the patient to clarify or exclude the disease (Table 2). The accurate diagnosis of etiology and clinical staging is crucial for guiding treatment strategies and improving patient prognosis. Therefore, we have summarized the current diagnostic principles and staging plans for various liver diseases, while also providing an introduction to the corresponding treatment strategies as outlined in the guidelines.
The treatment approach for liver disease is comprehensive, encompassing etiological management, lifestyle modifications, pharmacotherapy, nutritional support, prevention and management of complications, regular monitoring, and health education. Irrespective of the underlying cause, liver transplantation may represent the sole efficacious intervention for advanced liver disease following cirrhosis or hepatic failure.
Viral hepatitis
Due to the presence of multiple types of hepatitis viruses and the possibility of acute or chronic viral infections in patients, serological testing is necessary following a thorough history collection and physical examination.392 Hepatitis virus antigen and corresponding antibody tests, along with etiological tests such as viral RNA load assessments, serve as crucial diagnostic indicators for identifying viral hepatitis in individuals presenting related symptoms.393,394 The diagnosis of viral hepatitis can be established by considering the patient’s clinical manifestations, evidence of liver function impairment in laboratory tests, and results from auxiliary imaging examinations while excluding other potential diseases that may present similar symptoms.
Vaccination is the most effective means of preventing infection with hepatitis A, B, and D viruses.395 The recombinant HBV vaccine is both safe and highly efficacious, capable of being administered as a standalone immunization or in conjunction with other antigens utilized in infant immunization programs or alongside the hepatitis A virus vaccine. The treatment approach for viral hepatitis should be tailored to each individual patient’s condition, including factors such as virus type, liver function status, presence of complications, and other relevant considerations. For patients afflicted by chronic or severe forms of hepatitis, antiviral therapy may be considered to impede progression towards cirrhosis, liver failure, and hepatocellular carcinoma. Antiviral agents like lamivudine, entecavir, and tenofovir can be employed for treating CHB while DAAs such as sofosbuvir and harvoni can be used against hepatitis C infections.396,397 Currently, nucleos(t)ide analogues have demonstrated safety along with efficacy in inhibiting HBV replication; however, they rarely achieve clearance of HBsAg necessitating long-term administration to prevent recurrence. Therefore, various classes of DAAs and immunomodulatory therapies are currently under development aiming at achieving functional cure defined as persistent undetectable HBsAg levels along with absence of detectable HBV DNA after completion of limited duration treatment.398,399 It might eventually require combination therapy involving multiple drug classes to attain this objective. Detailed information of viral hepatitis managements can be found in updated AASLD and EASL clinical practice guidelines.393,394,396,400,401,402
Acute liver injury
Ancillary examinations revealed deranged liver function tests further supporting the initial diagnosis of acute liver injury (ALI).403 Thorough medical history collection can aid in identifying potential risk factors for ALI, such as recent initiation of medications or herbal/nutritional supplements intake, exposure to possible pathogens, travel history, and vaccination status.404 The definition of mild acute liver injury typically includes an ALT level between 2 and 5 times the upper limit of normal (ULN). Moderate ALI is usually defined as an ALT level between 5 times and 15 times the ULN. Severe ALI requires meeting specific criteria, including an international normalized ratio (INR) of ≥2.0, ALT levels of ≥10 ULN, and total bilirubin (TBiL) levels of ≥3.0 mg/dL without hepatic encephalopathy.405 An increase in the INR indicates a poor prognosis for patients with severe ALI.406 Studies have demonstrated that apart from etiology, bilirubin levels, INR values, and duration of jaundice are effective predictors for poor prognosis in ALI patients with specific thresholds such as duration of jaundice >3 days, TBil>51 μmol/L, and INR > 1.7.407
The fundamental principles of ALI treatment encompass early identification and correction of reversible causes, judicious selection of medications, timely implementation of liver replacement therapy, and proactive prevention and management of complications.408 Patients with abnormal liver function who do not yet meet the criteria for ALI should be closely monitored to promptly remove pathogenic factors in order to prevent liver damage or failure.409 For patients with rapid disease progression or existing liver damage, drug therapy should be considered based on active monitoring and etiological treatment. Currently available hepatoprotective drugs can generally be categorized into agents that repair and protect the liver cell membrane, anti-inflammatory drugs, antioxidant drugs (e.g. glutathione), and cholestrogenic drugs (e.g. ursodeoxycholic acid, UDCA).410,411 Presently, there is a lack of specific medications and approaches for treating advanced ALF. Thus, emphasis should be placed on symptomatic treatment while actively preventing complications. The use of adrenocortical hormones in the management of liver failure remains controversial; comprehensive consideration must be given to etiology and patient monitoring indices before making a decision.412,413 Cytokine therapies are under investigation. For example, An open-label, dose-escalation study utilizing IL-22 agonist F-652 to treat sAH has yielded promising results as anticipated, thereby offering a potential effective treatment strategy for further reducing the case fatality rate.414,415 Guidelines related to artificial livers and liver transplantation can serve as references for their respective treatments.416,417 Detailed information of acute liver failure and acute-on-chronic liver failure managements can be found in updated AASLD and EASL clinical practice guidelines (Fig. 6).418,419,420

Comprehensive evaluation and therapeutic protocols for acute liver injury. The general diagnostic procedures for acute liver injury encompass etiological screening, identification of liver injury patterns, and assessment of severity. Based on the extent of hepatic damage, appropriate treatment modalities are employed, including causative factor elimination, supportive care administration, utilization of hepatoprotective agents, artificial liver support and liver transplantation. R = (ALT/ULN)/(ALP/ULN). This figure was generated with Adobe Illustrator
MASLD
The diagnosis of MASLD is based on three criteria: (1) imaging diagnosis of hepatic steatosis and/or liver biopsy findings of ≥ 5% hepatocyte ballooning degeneration; (2) presence of one or more metabolic syndrome score components; (3) exclusion of other causes that may contribute to hepatic steatosis.421 Ultrasound imaging is the preferred modality for diagnosing hepatic steatosis and monitoring hepatocellular carcinoma.422 Liver stiffness measurement obtained through shear wave elastography can be utilized for non-invasive assessment of hepatic steatosis and fibrosis in patients with chronic liver disease. In addition to meeting the diagnostic criteria for MASLD, the presence of ≥ 5% hepatocyte ballooning degeneration combined with lobular inflammation and/or portal inflammation can lead to a diagnosis of MASH. Given the associated risks, a liver biopsy is typically reserved for cases where MASH is suspected but cannot be confirmed by other means. Currently, clinicians make comprehensive judgments based on individual patient circumstances to select appropriate diagnostic methods.423
The management of MASLD necessitates a multidisciplinary approach, encompassing strategies such as weight and waist circumference reduction, enhancement of insulin sensitivity, prevention of metabolic syndrome and T2DM, mitigation of MASH, and reversal of fibrosis. Dietary modification and increased physical activity through health education serve as the fundamental pillars in the treatment regimen for MASLD. Greater weight loss in overweight/obese individuals confers additional benefits on metabolic cardiovascular health and liver function. A gradual weight reduction ranging from 3% to 5% within one year can reverse hepatic steatosis; a weight loss between 7% to 10% can alleviate MASH; more than 10% weight loss can lead to fibrosis regression; while a substantial decrease by 15% even improves T2DM symptoms.424 Unhealthy habits such as irregular eating patterns, soft drink consumption, smoking tobacco products, or alcohol use should be avoided alongside sedentary behavior and physical inactivity.425
Combined presence of metabolic cardiovascular risk factors and liver injury necessitates appropriate pharmacological intervention. Patients with MASLD and a BMI ≥ 28 kg/m2 may benefit from weight loss medications, while hypoglycemic drugs for weight reduction should be prioritized in the treatment of type 2 diabetes.421,426 In managing diabetic patients with MASLD, preference should be given to drugs such as metformin, pioglitazone, SGLT-2 inhibitors, GLP-1 receptor agonists, and other agents that have potential hepatoprotective effects.427 Statins are the primary choice for pharmacotherapy of arteriosclerotic lipid disorders in MASLD patients; however, caution or discontinuation is advised when using statins in individuals with severe liver diseases like decompensated cirrhosis.428 ACE inhibitors or ARBs are recommended as first-line therapy for hypertension in MASLD patients, whereas non-selective β-blockers can be used concomitantly if clinically significant portal hypertension is present.429 As an agonist of the thyroid hormone receptor-β (THR-β), resmetirom has recently gained FDA approval for the treatment of adult patients with MASH and liver fibrosis.430 For non-cirrhotic MASLD patients who meet the criteria for metabolic surgery aimed at weight loss, options such as gastric bypass surgery, sleeve gastrectomy, duodenal transposition, or adjustable gastric banding may be considered to address MASH and fibrosis.431 Liver transplantation could be an option for individuals with decompensated cirrhosis resulting from MASH complications or acute-on-chronic liver failure (ACLF) as well as those diagnosed with HCC.432 Detailed information of MASLD managements can be found in updated AASLD and EASL clinical practice guidelines (Fig. 7).421,433

Etiological diagnostic approach for steatotic liver disease. The diagnosis of steatotic liver disease necessitates the integration of medical history (hypertension, T2DM, viral hepatitis, etc.), lifestyle styles (such as alcohol consumption), physical examination (body mass index, blood pressure, etc.), laboratory tests (triglycerides, glycated hemoglobin, ALT, AST, etc.), and pathological examination. This approach is based on expert opinion of the authors and evidence from published data. This figure was generated with Adobe Illustrator
ALD
ALD is diagnosed in patients who typically have a prolonged history of alcohol consumption (more than 12 months, >2 drinks in women and >3 drinks in men per day) or recent heavy alcohol use within the past two weeks. The patient’s clinical symptoms were nonspecific and accompanied by abnormal serum liver function tests. Notably, an elevated AST/ALT ratio (>1.5), GGT levels, and mean corpuscular volume (MCV) values are characteristic of ALD.434 These indicators can significantly decrease after cessation of alcohol intake and usually return to normal within four weeks (although GGT may take longer to normalize), which aids in diagnosis.435 Imaging studies are used for diagnosis while excluding hepatitis viruses, medications, toxic liver injury, AIH, and other conditions.436 Symptomatic alcoholic hepatitis is diagnosed in the presence of jaundice and the criteria proposed by the National Institute on Alcohol Abuse and Alcoholism: last alcoholic drink within 8 weeks of onset of jaundice; serum bilirubin level greater than 3 mg/dL; AST and ALT levels less than 400 IU/L, with the AST level being greater than 50 IU/mL and an AST to ALT ratio greater than 1.5:1; and the exclusion of other potential causes of liver disease, biliary obstruction, and HCC.437
The treatment principles of ALD include abstinence and nutritional support, reducing the severity of the disease, and providing symptomatic treatment for alcoholic cirrhosis and its complications.438 Complete abstinence from alcohol enhances prognosis, mitigates liver histological injury, decreases portal vein pressure, delays fibrosis progression, and improves survival at all stages of the disease. Baclofen may be administered orally to individuals facing challenges with active abstinence.434 Moreover, patients with ALD require adequate nutritional support, including a high-protein, low-fat diet based on alcohol abstinence. Additionally, attention should be given to vitamin supplementation.439 Due to the limited expression of IL-22 receptor on epithelial cells, such as hepatocytes, IL-22 could serve as a specific target for preventing hepatocyte death and promoting hepatocyte proliferation without affecting immune cells. A multicenter trial is currently underway to investigate the use of IL-22Fc in treating ACLF, including severe alcoholic hepatitis (sAH) (CTR20212657).415 Inflammation has been extensively studied as a therapeutic target for sAH treatment due to its significant role in the pathogenesis of alcoholic liver disease. Steroid therapy has been utilized since the 1970s and emerging data suggest that it improves short-term survival in some sAH patients without impacting long-term survival.435 Treatment for anti-hepatic fibrosis should be prioritized along with actively addressing complications related to alcoholic cirrhosis such as esophageal and gastric varices rupture bleeding, spontaneous bacterial peritonitis, hepatic encephalopathy, and HCC.440 Liver transplantation may be considered for patients with severe alcoholic cirrhosis; however, many transplant centers require patients to abstain from alcohol for six months before undergoing surgery.441 Detailed information of ALD managements can be found in updated AASLD and EASL clinical practice guidelines (Fig. 7).439,442
AIH
Patients with AIH often exhibit mild to moderate elevation of ALT and AST. On the other hand, patients with PBC and PSC frequently present elevated serum ALP and GGT levels, along with increased bilirubin levels in advanced stages.134 Serum autoantibodies such as anti-nuclear antibody (ANA) and anti-smooth muscle antibody (ASMA) can be detected in AIH patients, accompanied by elevated IgG levels.443 In 90-95% of PBC patients, serum antibodies against mitochondria (AMA) and elevated IgM levels can be found.444 Ultrasound, CT scan, MRI, endoscopic retrograde cholangiopancreatography (ERCP), and magnetic resonance cholangiopancreatography (MRCP) can be utilized to exclude biliary diseases like tumors or stones affecting the hepatobiliary system.136 ERCP is considered the “gold standard” for diagnosing PSC; however, MRCP is preferred due to its non-invasive nature when diagnosing this condition initially.445,446 Liver histological biopsy serves as a means to differentiate between causes of liver injury and evaluate tissue damage.447 Liver histological biopsy serves as a means to differentiate between causes of liver injury and evaluate tissue damage.447 The Paris diagnostic criteria and the AIH simplified diagnostic system are widely utilized for diagnosing AIH and its overlap with PBC. Among these, the Paris criteria stand out as the most common and effective tool for diagnosing AIH-PBC overlap syndrome.448 In 2008, the International Autoimmune Hepatitis Group introduced a simplified diagnostic scoring system for AIH, which proves valuable in identifying patients with AIH-PBC requiring corticosteroid treatment.449
The treatment and prognosis of different autoimmune liver diseases vary significantly. Immunosuppressive therapy is the primary approach for managing AIH, with a combination of prednisolone and azathioprine being the preferred treatment for AIH patients.450 In cases of poor response, alternative immunosuppressive agents can be considered. UDCA is the first-line option for PBC treatment, while additional medications such as bate drugs, budesonide, and obeticholic acid may be added if necessary.451 Currently, there are no drugs available to alleviate liver damage caused by PSC. Therefore, the focus lies in controlling complications and monitoring liver damage. UDCA can improve liver biochemical markers, reduce liver fibrosis severity, and enhance imaging findings related to biliary tract involvement.452 Glucocorticoids are favored for inducing remission in IgG4-associated hepatobiliary diseases.453 Patients who seek early treatment for AIH generally exhibit better treatment responses and prognoses comparable to those of healthy individuals in the long term. Conversely, patients who delay seeking treatment or do not respond well to therapy have an increased risk of developing cirrhosis and liver failure. Liver transplantation remains the sole effective intervention for end-stage liver disease.454 Detailed information of AIH managements can be found in updated AASLD and EASL clinical practice guidelines.454,455
Genetic and rare liver diseases
Inherited liver diseases exhibit overlapping clinical manifestations, often requiring multiple clinical or pathological features for diagnosis.100 Genetic testing is the most crucial tool in diagnosing hereditary liver diseases. However, due to the complexity and diversity of genetic mutations in genetic liver disease, gene analysis and diagnosis remain challenging due to factors such as high cost, poor detection sensitivity, and the close relationship between heredity and acquired environment.456 Some rare or inherited liver diseases have unique clinical manifestations that aid in their diagnosis; for example, the combination of Kayser-Fleischer rings and a low serum ceruloplasmin (<0.1 g/L) level are prominent features of WD while decreased serum α1-antitrypsin levels with pulmonary damage such as emphysema suggest a1 antitrypsin deficiency.193 Notably, these features are not always reliable, and additional tests, such as high-quality imaging tests (e.g., MRI) or liver tissue biopsies, can also assist in making a definitive diagnosis.457
Several treatment options are available to alleviate symptoms and maintain optimal liver function for inherited liver disease. Dietary modifications may be necessary, such as adhering to a low-copper diet in cases of WD, and avoiding foods rich in copper like animal organs, dried fruits, and mushrooms.458 Symptomatic treatment often involves the use of medications that facilitate copper excretion or inhibit its absorption.459 Additionally, patients with liver damage can benefit from appropriate hepatoprotective therapy. For those experiencing neuropsychiatric symptoms, consultation with a neurologist is recommended for tailored management strategies. Itch relief can also be achieved through pharmacological interventions. In rare instances where hereditary liver diseases lead to ALF or decompensated cirrhosis unresponsive to conventional treatments or intolerant reactions occur, consideration should be given to liver transplantation.460 Detailed information of WD managements can be found in updated AASLD and EASL clinical practice guidelines.100,461
Liver cirrhosis
The diagnosis of liver cirrhosis should be comprehensive, taking into account clinical manifestations of hepatic hypofunction and portal hypertension, as well as imaging and endoscopy findings, and laboratory results. Liver biopsy is recommended for patients with diagnostic difficulties, while etiological screening should be conducted whenever possible.462 Typical features observed in abdominal ultrasound, CT scans, and MRI images of cirrhosis include changes in liver volume (early enlargement followed by late contraction), abnormal ratio between the left and right lobes (shrinkage of the right lobe with enlargement of the left lobe and caudate lobe), irregular or jagged liver contour, widening of liver clefts, uneven liver echo or density signal distribution, dilation of the portal vein, and collateral circulation expansion.155 Transient elastography-derived liver stiffness measurement (LSM) demonstrates a strong ability to evaluate significant liver fibrosis and cirrhosis but exhibits poor accuracy in assessing mild stages of fibrosis. Magnetic resonance elastography (MRE) offers high diagnostic accuracy along with good stability and efficiency for staging liver fibrosis because it is less influenced by factors such as obesity or ascites; however, it requires relatively more time for examination and is expensive.463 Serological indicators such as aspartate aminotransferase-platelet ratio index (APRI) and fibrosis-4 index (FIB-4) exhibit low sensitivity and specificity in diagnosing cirrhosis. Moreover, the critical value used to determine liver fibrosis/cirrhosis can also be affected by etiology among other factors.464 According to the presence of esophageal and gastric varices, hemorrhage, ascites, hepatic encephalopathy, and jaundice, cirrhosis is classified into six stages. Stage 1 does not exhibit varicose veins or any other complications; it is further divided into stages 1a and 1b based on whether the hepatic venous pressure gradient (HVPG) is ≥10 mmHg. Varicose veins appear in stage 2 but without EGVB (esophagogastric variceal bleeding) or ascites. EGVB occurs in stage 3 but without decompensation such as ascites or hepatic encephalopathy. Stage 4 includes various forms of decompensation except for EGVB, including ascites, overt hepatic encephalopathy, overt bacterial infection, and non-obstructive jaundice. Stage 5 presents two types of decompensations while stage 6 is characterized by recurrent infection, dysfunction of extrahepatic organs, ACLF, refractory ascites, persistent hepatic encephalopathy or jaundice.462,465
The most crucial treatment for cirrhosis is the removal of its underlying cause. Etiological control, particularly antiviral therapy in patients with hepatitis B/C, as well as abstinence in those with alcoholic cirrhosis, can potentially reverse liver fibrosis and cirrhosis or restore compensatory stage in decompensated cirrhosis patients.466 In cases where malnutrition complicates cirrhosis, it is recommended to consume 25–35 kcal/kg/d energy intake, 1.0–1.5 g/kg/d protein intake, increase meal frequency, add extra meals at night, and adequately supplement dietary fiber, vitamins, and trace elements. Patients with ascitic cirrhosis should moderately restrict sodium intake (85–120 mmol/d or equivalent to 5.0–6.9 g/d salt) while avoiding extreme sodium restriction (<40 mmol/d). Unless moderate to severe dilutive hyponatremia (blood sodium <125 mmol/L) is present, water intake does not generally need to be restricted in patients with ascites due to cirrhosis.467,468 Diuretics are considered the first-line treatment for ascites in cirrhotic patients; spironolactone alone or combined with furosemide or torasemide can be used. Grade 2 or 3 ascites that are unresponsive to conventional diuretics may be managed with the administration of tolvaptan.469 Massive paracentesis is a commonly employed intervention for refractory ascites, and albumin infusion should be utilized to optimize intravascular volume expansion. Teripressin represents an efficacious pharmacological option for the treatment of refractory ascites.470 Transjugular intrahepatic portosystemic shunt (TIPS) should be considered in cases where therapy for massive ascites proves ineffective.471 Liver transplantation serves as the definitive therapeutic approach for decompensated cirrhosis and warrants evaluation when patients develop esophageal variceal bleeding, refractory ascites, hepatorenal syndrome, hepato-pulmonary syndrome, recurrent hepatic encephalopathy, ACLF, or HCC.472 Detailed information of cirrhosis managements can be found in updated AASLD and EASL clinical practice guidelines.462,473,474,475,476
HCC
Traditionally, the diagnosis of HCC has been primarily based on cytology or histology. However, with advancements in staged perfusion angiography during CT and MRI cross-sectional imaging, HCC can now be reliably diagnosed radiologically in cirrhotic patients under surveillance without the need for biopsy.477 Abdominal ultrasound is currently considered the most recommended method for monitoring HCC.477,478 Although serum alpha-fetoprotein (AFP) alone lacks sensitivity and specificity to serve as an independent monitoring test, its combination with ultrasound significantly improves early detection sensitivity for HCC.479 Various integrated imaging and blood-based strategies have been proposed to enhance early detection of HCC; nevertheless, most of these approaches have only been evaluated through case-control studies and require prospective validation.480
Over the past two decades, the Barcelona Clinic Liver Cancer (BCLC) staging system has gained recognition from the majority of professional societies.481 However, managing HCC involves a complex decision-making process, and the availability of treatment options varies significantly among medical centers across different countries. Consequently, effective HCC management necessitates multidisciplinary collaboration to devise tailored strategies that cater to each patient’s unique circumstances in order to achieve optimal outcomes.482 Surgical treatment options for HCC include surgical resection and liver transplantation, both considered potentially curative treatments. However, it is important to note that nearly 70% of patients experience recurrent HCC after resection.483 Liver transplantation stands out as the most definitive treatment option for early-stage HCC since it allows removal not only of the tumor but also an unhealthy liver with limited functional capacity. A retrospective multicenter study involving 187 HCC patients revealed that 58% underwent successful downstaging followed by liver transplantation with a 5-year survival rate reaching 80%.484 Percutaneous local ablation is a potentially curative treatment modality that can be employed in patients with early HCC. The two most commonly utilized techniques are radiofrequency ablation (RFA) and microwave ablation (MWA).485 MWA demonstrates enhanced efficacy for larger tumors measuring 3-4 cm, and requires less procedural time compared to RFA.486 Transarterial chemoembolization (TACE) is a highly effective treatment modality for patients with intermediate-stage HCC. Transarterial radiation embolization (TARE) represents an alternative local regional therapy approach, which can be employed as the primary therapeutic intervention for unresectable HCC cases.487 Unlike TACE, TARE involves intratumoral brachytherapy techniques and exerts minimal embolic effects on hepatic artery distribution, making it suitable even for patients presenting portal vein thrombosis or tumor invasion (Fig. 8).488

Strategy for HCC treatment with BCLC staging system. The Barcelona Clinic Liver Cancer (BCLC) staging system categorizes hepatocellular carcinoma into five stages (0/A to D) with varying prognostic significance. The authors have provided a concise summary of the recommended first and second treatment options based on the BCLC stage. ECOG PS, Eastern Cooperative Oncology Group performance status; SBRT, stereotactic body radiation therapy. This figure was generated with Adobe Illustrator
The treatment strategy for HCC has been significantly revolutionized by the introduction of systemic pharmacological therapy worldwide.489 Sorafenib, lenvatinib, cabotinib, ramociumab, and other drugs have successively gained approval for HCC treatment.490,491,492 However, due to the heterogeneity and complexity of HCC pathogenesis, precise treatment for this disease is still under investigation. Concurrently, immune checkpoint inhibitors have emerged as a promising therapeutic option for advanced HCC.493 Various immunotherapies including checkpoint inhibitor combined targeted therapy, checkpoint inhibitor combination therapy, and non-checkpoint inhibitor immunotherapy (such as immune cell adoptive therapy) have shown significant efficacy.494,495 In conclusion, active research is still required in this field and a combination of treatment modalities may enhance therapeutic options for patients. Detailed information of HCC managements can be found in updated AASLD and EASL clinical practice guidelines.482,496
Clinical research progress
Acute liver disease-viral hepatitis
Acute viral hepatitis, resulting from viruses such as HAV-HEV, primarily benefits from prevention via inactivated vaccines.497 Currently, there is no specific antiviral for HAV, but clinical studies suggest benefits from steroids and IFN-β in improving outcomes.498 For severe acute HBV, early administration of lamivudine and entecavir has been shown to improve patient conditions and reduce progression to chronic hepatitis.499,500 Therapeutic strategies such as ledipasvir/sofosbuvir have been effective in treating HIV and HBV co-infections by shortening treatment durations.501 Meanwhile, grazoprevir combined with elbasvir shows promise in acute HCV, particularly for genotypes 1 or 4.502 For HEV, ribavirin has been effective in reducing viral load (Table 3).503 Research on treatment for acute HDV is limited and warrants further exploration. Additional studies reveal that other virus like adenovirus, cytomegalovirus (CMV), Epstein-Barr virus (EBV), and TT virus can also cause acute viral hepatitis.504,505 Rare viral hepatitis are often overlooked due to their low incidence. These less common forms underline the need for heightened clinical awareness to prevent misdiagnosis and inappropriate treatment.
Acute liver disease-DILI
Acute DILI, often caused by substances such as acetaminophen, antibiotics, or anti-inflammatory drugs, usually resolves with decreasing liver enzyme levels within days to weeks, with fewer than 10% of cases progressing to chronic liver damage.506 Acetaminophen overdose is primarily treated with N-acetylcysteine (NAC).507 Investigating additional treatments that can protect liver function during both the early and late stages of DILI is critical for minimizing long-term damage.
Acute liver disease-ALD
Acute ALD remains challenging, and therapies such as pentoxifylline and corticosteroids shown to decrease short-term, but not medium-term, mortality.508 Although corticosteroids may reduce short-term mortality, their long-term safety profile is concerning due to the risk of severe infections.509 TNF-α inhibitors, such as infliximab and etanercept, reduce inflammation but increase infection risks.510 There is emerging interest in the role of intestinal microbes in ALD, though clinical validations are still preliminary.511
Chronic liver disease-viral hepatitis
Innovations in CHB treatments include vebicorvir, a core inhibitor that has shown superior efficacy compared to traditional nucleoside reverse transcriptase inhibitors.512 Tenofovir alafenamide is used for multidrug-resistant HBV strains, improving long-term outcomes.513 PD-1 inhibitors and RNA interference therapies like ARC-520 are under investigation for their potential to enhance immune responses and reduce viral load in HBV patients.514,515 Treatments for chronic HCV have evolved with the development of DAAs, reducing concerns about VZV reactivation.516 Glecaprevir and pibrentasvir have shown improved responses in HCV patients who failed prior DAA therapies.517 Bulevirtide combined with tenofovir disoproxil fumarate offers new hope for HDV patients, although more studies are needed to confirm these findings.518,519 Overall, these advancements represent significant progress in the treatment of liver diseases, but ongoing research is crucial to optimize safety and long-term efficacy of these therapies.
Chronic liver disease-ALD
No FDA-approved medications currently exist specifically for ALD. Research indicates that alcohol consumption disrupts the intestinal microbiota, sometimes leading to an overgrowth of Candida albicans. This alteration suggests that probiotics could help mitigate ALD by modulating intestinal flora.520,521 Inflammation is a critical factor in ALD pathogenesis, hence steroids are used to manage symptoms and improve short-term survival rates, although their long-term efficacy is still not well-established.522 Corticosteroids treatments seem to be effective, which needs adequate nutritional intake throughout the treating duration.523 Efforts to target inflammation with cytokines such as IL-1 and TNF-α have been explored, but results, including attempts to combine IL-1 receptor antagonists with pentoxifylline and zinc, have not shown superior outcomes compared to corticosteroids alone.524,525 Future research is essential to develop more targeted therapies for ALD.
Chronic liver disease-MASLD
With rising global obesity rates, MASLD prevalence is expected to increase.526 In a significant development, in 2020, saroglitazar was approved by India’s Drug Administration as the first medication specifically for MASLD, demonstrating effectiveness in reducing ALT level, liver fat content, insulin resistance, and atherogenic dyslipidemia.527 Furthermore, in March 2024, Resmetirom became the first FDA-approved drug for treating non-cirrhotic NASH with moderate to advanced liver fibrosis in adults, marking a major milestone in MASLD treatment.528 Despite these advancements, there remains a substantial need for more precise non-invasive diagnostic techniques and effective treatments.
Chronic liver disease-autoimmune liver diseases
Recent advances in AIH research have improved our understanding of its causes, diagnosis, and treatment. Glucocorticoids and immunosuppressants remain treatment mainstays,529 with studies showing that combining mycophenolate mofetil (MMF) with prednisolone can lead to better outcomes and fewer side effects.530 Future directions include advancing personalized treatment strategies to enhance patient quality of life and prognosis. Cholestatic liver diseases such as PBC and PSC are primarily managed with UDCA.531 Emerging treatments, such as obeticholic acid, are undergoing evaluation for their efficacy in these diseases.532 For PSC, liver transplantation remains a definitive but severe option, underscoring the ongoing need for research into pharmacological interventions that could slow disease progression.
End-stage liver disease-cirrhosis
As a critical aspect of end-stage liver disease, cirrhosis has undergone significant advancements in both diagnosis and management. The integration of non-invasive tests like transient elastography and serum biomarkers (e.g., FibroTest) into clinical practice has greatly improved the early detection of liver fibrosis and cirrhosis, reducing reliance on invasive biopsy procedures.533 Recent studies have highlighted the potential of pegbelfermin and aldafermin in ameliorating liver fibrosis associated with MASH and compensated cirrhosis.534,535 Additionally, anti-fibrotic medications and treatments aimed at enhancing liver microcirculation are showing promising results in clinical trials. Research into the therapeutic application of mesenchymal stem cells for decompensated cirrhosis has also yielded positive outcomes, although more extensive studies are required to confirm these findings.536,537 Liver transplantation continues to be the sole curative treatment for cirrhosis, underscoring the ongoing need for the development of more effective therapies.
End-stage liver disease-liver failure
Liver failure represents the most severe manifestation of end-stage liver disease, where managing both acute and chronic forms remains a formidable challenge. Recent advancements include the development of the MKK4 inhibitor HRX215, which has been shown to promote liver regeneration and prevent liver failure.538 Extracorporeal liver support devices have also shown promise in improving patient outcomes in acute and chronic liver failure, potentially reducing mortality rates.539 Innovations in regenerative medicine, including stem cell therapies and liver bioengineering, are being explored as novel treatment avenues.540 Moreover, improvements in liver transplantation techniques and refinement of immunosuppressive treatments are crucial for enhancing patient survival and quality of life.
End-stage liver disease-HCC
There has been notable progress in clinical research on HCC, yielding several pioneering treatments. Research comparing PD-1 antibody carrelizumab with VEGFR2-targeted TKI rivoceranib has indicated better progression-free and overall survival rates in patients with unresectable HCC compared to standard treatments like sorafenib.541 In high-risk post-resection HCC, the PD-1 inhibitor sintilimab shows potential in reducing tumor recurrence.542 The LAUNCH trial revealed that combining lenvatinib with TACE significantly enhances clinical outcomes in patients with advanced HCC.543 Furthermore, a phase 1/2 trial exploring a personalized neoantigen vaccine (PTCV) combined with pembrolizumab has demonstrated promising immune responses and preliminary efficacy in advanced HCC cases.544 For patients at high risk of HCC recurrence after postoperative ablation, atezolizumab combined with bevacizumab improves recurrence-free survival.545 These emerging therapies offer new hopes and strategies in the fight against HCC, potentially transforming the therapeutic landscape.
Conclusions and perspectives
The etiology of liver diseases is continually evolving. With the global rise in obesity and T2DM, MASLD poses a growing health threat worldwide. The administration of vaccination and antiviral medications has significantly decreased the incidence of viral hepatitis in the Americas and Europe; however, the prevalence of MASLD, ALD, and DILI is rising. Despite advancements in vaccinations and antiviral medications that effectively prevent and combat viral infections, chronic hepatitis B and C remain prevalent, particularly in low-income countries lacking adequate medical resources. Additionally, the incidence of ALD is increasing, especially among younger populations. Despite heightened public health efforts, liver diseases significantly contribute to the global disease burden.546
The diagnosis of liver diseases primarily depends on liver biopsy, an invasive technique unsuitable for broad screening. The absence of reliable biomarkers for the precise diagnosis and staging of specific liver diseases poses a significant challenge.547 As such, developing novel non-invasive biomarkers and methods is crucial for the early detection of asymptomatic liver diseases. Such advancements could help identify high-risk individuals sooner, allowing for early interventions to halt disease progression.
Although there has been notable progress in understanding liver disease pathogenesis through advanced technologies, therapeutic options approved by the FDA remain limited, and existing medical interventions often provide minimal long-term survival benefits. The complexity of liver disease pathophysiology and the substantial heterogeneity in disease phenotypes mean that current mouse models do not adequately mimic the full spectrum of human liver diseases, including ALD and MASLD. Significant disparities exist between mouse models and human conditions in terms of pathophysiology and treatment outcomes, as numerous clinical trials have shown that drugs effective in mouse models fail to offer clinical benefits in humans.548 The properties of chemical absorption, distribution, metabolism, excretion, and toxicity differ between species, resulting in drug doses that are beneficial and non-toxic in mice showing insufficient efficacy or causing side effects in humans. For example, galectin-3, which was demonstrated to reduce liver inflammation and fibrosis in murine MASH models, did not translate effectively to human patients.549 Belapectin, a galectin-3 inhibitor, failed to show efficacy in MASH patients with cirrhosis and portal hypertension in a phase 2b randomized trial, possibly due to inadequate treatment duration and dosage. Pharmacokinetic analysis revealed differences in the metabolism of belapectin between mice and humans.550 This discrepancy underscores the need for the development of more standardized mammalian models, such as pigs and chimpanzees, which might bridge these gaps and improve translational success.
Moving forward, our current understanding of the pathogenesis has provided valuable insights and directed ongoing research efforts aimed at liver disease treatment. However, a comprehensive understanding of critical signaling pathways and their interactions during liver disease progression is essential to advance therapeutic strategies and improve patient outcomes.
References
Santos, A. A. et al. Spatial metabolomics and its application in the liver. Hepatology 79, 1158–1179 (2024).
Schulze, R. J. et al. The cell biology of the hepatocyte: a membrane trafficking machine. J. Cell Biol. 218, 2096–2112 (2019).
Han, H. et al. Danger signals in liver injury and restoration of homeostasis. J. Hepatol. 73, 933–951 (2020).
Devarbhavi, H. et al. Global burden of liver disease: 2023 update. J. Hepatol. 79, 516–537 (2023).
Wong, M. C. S. et al. The changing epidemiology of liver diseases in the Asia-Pacific region. Nat. Rev. Gastroenterol. Hepatol. 16, 57–73 (2019).
Vilar-Gomez, E. & Chalasani, N. Non-invasive assessment of non-alcoholic fatty liver disease: Clinical prediction rules and blood-based biomarkers. J. Hepatol. 68, 305–315 (2018).
Tapper, E. B. & Parikh, N. D. Diagnosis and management of cirrhosis and its complications: a review. JAMA 329, 1589–1602 (2023).
Ginès, P. et al. Liver cirrhosis. Lancet 398, 1359–1376 (2021).
GBD 2017 Cirrhosis Collaborators. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 5, 245–266 (2020).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Stanaway, J. D. et al. The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013. Lancet 388, 1081–1088 (2016).
Rehm, J. et al. Alcohol as a risk factor for liver cirrhosis: a systematic review and meta-analysis. Drug Alcohol Rev. 29, 437–445 (2010).
Le, M. H. et al. 2019 Global NAFLD Prevalence: a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 20, 2809–2817.e2828 (2022).
Han, J. et al. Declining disease burden of HCC in the United States, 1992-2017: a population-based analysis. Hepatology 76, 576–588 (2022).
Chen, J. et al. Sedentary lifestyle, physical activity, and gastrointestinal diseases: evidence from Mendelian randomization analysis. EBioMedicine 103, 105110 (2024).
Ivancovsky Wajcman, D. et al. A narrative review of lifestyle management guidelines for metabolic dysfunction-associated steatotic liver disease. Hepatology https://doi.org/10.1097/hep.0000000000001058 (2024).
Wong, V. W., Ekstedt, M., Wong, G. L. & Hagström, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. 79, 842–852 (2023).
Sheron, N. Alcohol and liver disease in Europe-Simple measures have the potential to prevent tens of thousands of premature deaths. J. Hepatol. 64, 957–967 (2016).
Seitz, H. K. et al. Alcoholic liver disease. Nat. Rev. Dis. Prim. 4, 16 (2018).
Askgaard, G. et al. Population-based study of alcohol-related liver disease in england in 2001-2018: influence of socioeconomic position. Am. J. Gastroenterol. 119, 1337–1345 (2024).
Younossi, Z. M., Wong, G., Anstee, Q. M. & Henry, L. The global burden of liver disease. Clin. Gastroenterol. Hepatol. 21, 1978–1991 (2023).
Jiang, H., Room, R. & Hao, W. Alcohol and related health issues in China: action needed. Lancet Glob. Health 3, e190–e191 (2015).
Manns, M. P. & Maasoumy, B. Breakthroughs in hepatitis C research: from discovery to cure. Nat. Rev. Gastroenterol. Hepatol. 19, 533–550 (2022).
Trickey, A. et al. The effectiveness of low dead space syringes for reducing the risk of hepatitis C virus acquisition among people who inject drugs: findings from a National Survey in England, Wales, and Northern Ireland. Clin. Infect. Dis. 75, 1073–1077 (2022).
Falade-Nwulia, O. et al. Oral direct-acting agent therapy for hepatitis C virus infection: a systematic review. Ann. Intern Med 166, 637–648 (2017).
Polaris Observatory HCV Collaborators. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol. 2, 161–176 (2017).
Lemon, S. M., Ott, J. J., Van Damme, P. & Shouval, D. Type A viral hepatitis: a summary and update on the molecular virology, epidemiology, pathogenesis and prevention. J. Hepatol. 68, 167–184 (2017).
Hakim, M. S. et al. The global burden of hepatitis E outbreaks: a systematic review. Liver Int. 37, 19–31 (2017).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Barouki, R. et al. The exposome and liver disease—how environmental factors affect liver health. J. Hepatol. 79, 492–505 (2023).
Rani, J., Dhull, S. B., Rose, P. K. & Kidwai, M. K. Drug-induced liver injury and anti-hepatotoxic effect of herbal compounds: a metabolic mechanism perspective. Phytomedicine 122, 155142 (2024).
Organization, W. H. WHO Position Paper on Hepatitis A Vaccines https://www.who.int/publications/i/item/who-wer9740-493-512 (2022).
Jacobsen, K. H. Globalization and the changing epidemiology of hepatitis A virus. Cold Spring Harb. Perspect. Med. 8 (2018).
Jacobsen, K. H. & Wiersma, S. T. Hepatitis A virus seroprevalence by age and world region, 1990 and 2005. Vaccine 28, 6653–6657 (2010).
Van Damme, P. et al. Hepatitis A virus infection. Nat. Rev. Dis. Prim. 9, 51 (2023).
Migueres, M., Lhomme, S. & Izopet, J. Hepatitis A: epidemiology, high-risk groups, prevention and research on antiviral treatment. Viruses 13 (2021).
Khuroo, M. S., Khuroo, M. S. & Khuroo, N. S. Transmission of Hepatitis E virus in developing countries. Viruses 8 (2016).
Zeng, D. Y. et al. Global burden of acute viral hepatitis and its association with socioeconomic development status, 1990-2019. J. Hepatol. 75, 547–556 (2021).
Ricci, A. et al. Public health risks associated with hepatitis E virus (HEV) as a food-borne pathogen. EFSA J. 15, e04886 (2017).
Thom, K. et al. Hepatitis E virus (HEV) in Scotland: evidence of recent increase in viral circulation in humans. Euro Surveill. 23 (2018).
Dalton, H. R. & Izopet, J. Transmission and epidemiology of Hepatitis E virus genotype 3 and 4 infections. Cold Spring Harb. Perspect. Med. 8 (2018).
Mahrt, H. et al. Continuous decline of hepatitis E virus seroprevalence in southern Germany despite increasing notifications, 2003-2015. Emerg. Microbes Infect. 7, 133 (2018).
Shen, T. et al. Incidence and etiology of drug-induced liver injury in Mainland China. Gastroenterology 156, 2230–2241.e2211 (2019).
Hussaini, S. H., O’Brien, C. S., Despott, E. J. & Dalton, H. R. Antibiotic therapy: a major cause of drug-induced jaundice in southwest England. Eur. J. Gastroenterol. Hepatol. 19, 15–20 (2007).
de Abajo, F. J., Montero, D., Madurga, M. & García Rodríguez, L. A. Acute and clinically relevant drug-induced liver injury: a population based case-control study. Br. J. Clin. Pharm. 58, 71–80 (2004).
Sgro, C. et al. Incidence of drug-induced hepatic injuries: a French population-based study. Hepatology 36, 451–455 (2002).
Björnsson, E. S. et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 144, 1419–1425 (2013).
Vega, M. et al. The incidence of drug- and herbal and dietary supplement-induced liver injury: preliminary findings from gastroenterologist-based surveillance in the population of the state of Delaware. Drug Saf. 40, 783–787 (2017).
Suk, K. T. et al. A prospective nationwide study of drug-induced liver injury in Korea. Am. J. Gastroenterol. 107, 1380–1387 (2012).
Organization, W. H. Estimated Mortality Rate from Cirrhosis and Other Chronic Liver Diseases https://www.who.int/data/gho/indicator-metadata-registry/imr-details/1179 (2019).
GBD 2019 Hepatitis B Collaborators. Global, regional, and national burden of hepatitis B, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol Hepatol. 7, 796–829 (2022).
Indolfi, G. et al. Hepatitis B virus infection in children and adolescents. Lancet Gastroenterol. Hepatol. 4, 466–476 (2019).
Polaris Observatory Collaborators. Global prevalence, cascade of care, and prophylaxis coverage of hepatitis B in 2022: a modelling study. Lancet Gastroenterol Hepatol. 8, 879–907 (2023).
Cooke, G. S. et al. Accelerating the elimination of viral hepatitis: a Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 4, 135–184 (2019).
Rochwerg, B. et al. High flow nasal cannula compared with conventional oxygen therapy for acute hypoxemic respiratory failure: a systematic review and meta-analysis. Intensive Care Med 45, 563–572 (2019).
Sarin, S. K. et al. Liver diseases in the Asia-Pacific region: a Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 5, 167–228 (2020).
Pimpin, L. et al. Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 69, 718–735 (2018).
Asrani, S. K., Devarbhavi, H., Eaton, J. & Kamath, P. S. Burden of liver diseases in the world. J. Hepatol. 70, 151–171 (2019).
Rizzetto, M., Hamid, S. & Negro, F. The changing context of hepatitis D. J. Hepatol. 74, 1200–1211 (2021).
Chen, H. Y. et al. Prevalence and burden of hepatitis D virus infection in the global population: a systematic review and meta-analysis. Gut 68, 512–521 (2019).
Stockdale, A. J. et al. The global prevalence of hepatitis D virus infection: systematic review and meta-analysis. J. Hepatol. 73, 523–532 (2020).
Polaris Observatory HCV Collaborators. Global change in hepatitis C virus prevalence and cascade of care between 2015 and 2020: a modelling study. Lancet Gastroenterol Hepatol. 7, 396-415 (2022).
Mohd Hanafiah, K., Groeger, J., Flaxman, A. D. & Wiersma, S. T. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57, 1333–1342 (2013).
Younossi, Z. M. et al. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology 77, 1335–1347 (2023).
Younossi, Z. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20 (2018).
Yip, T. C. et al. Geographical similarity and differences in the burden and genetic predisposition of NAFLD. Hepatology 77, 1404–1427 (2023).
Riazi, K. et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 7, 851–861 (2022).
Armstrong, M. J. et al. Presence and severity of non-alcoholic fatty liver disease in a large prospective primary care cohort. J. Hepatol. 56, 234–240 (2012).
Tarnoki, A. D. et al. Heritability of non-alcoholic fatty liver disease and association with abnormal vascular parameters: a twin study. Liver Int. 32, 1287–1293 (2012).
Zhou, F. et al. Unexpected rapid increase in the burden of NAFLD in China from 2008 to 2018: a systematic review and meta-analysis. Hepatology 70, 1119–1133 (2019).
Xiao, J. et al. Global liver disease burdens and research trends: analysis from a Chinese perspective. J. Hepatol. 71, 212–221 (2019).
Man, S. et al. Prevalence of liver steatosis and fibrosis in the general population and various high-risk populations: a nationwide study with 5.7 million adults in China. Gastroenterology 165, 1025–1040 (2023).
Ito, T. et al. The epidemiology of NAFLD and lean NAFLD in Japan: a meta-analysis with individual and forecasting analysis, 1995-2040. Hepatol. Int. 15, 366–379 (2021).
Park, J. et al. NASH/liver fibrosis prevalence and incidence of nonliver comorbidities among people with NAFLD and incidence of NAFLD by metabolic comorbidities: lessons from South Korea. Dig. Dis. 39, 634–645 (2021).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016).
Ahmed, M. H. et al. Non-alcoholic fatty liver disease in africa and middle east: an attempt to predict the present and future implications on the healthcare system. Gastroenterol. Res 10, 271–279 (2017).
Long, M. T., Noureddin, M. & Lim, J. K. AGA clinical practice update: diagnosis and management of nonalcoholic fatty liver disease in lean individuals: expert review. Gastroenterology 163, 764–774.e761 (2022).
Tang, A. et al. Comparative burden of metabolic dysfunction in lean NAFLD vs non-lean NAFLD—a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 21, 1750–1760.e1712 (2023).
Eslam, M. et al. Metabolic (dysfunction)-associated fatty liver disease in individuals of normal weight. Nat. Rev. Gastroenterol. Hepatol. 19, 638–651 (2022).
Rehm, J., Samokhvalov, A. V. & Shield, K. D. Global burden of alcoholic liver diseases. J. Hepatol. 59, 160–168 (2013).
WHO. Global Status Report on Alcohol and Health 2018 450 (WHO, 2018).
Xiao, J. et al. Epidemiological realities of alcoholic liver disease: global burden, research trends, and therapeutic promise. Gene Expr. 20, 105–118 (2020).
Kim, J. U. et al. Effect of COVID-19 lockdown on alcohol consumption in patients with pre-existing alcohol use disorder. Lancet Gastroenterol. Hepatol. 5, 886–887 (2020).
Kilian, C. et al. Changes in alcohol use during the COVID-19 pandemic in Europe: a meta-analysis of observational studies. Drug Alcohol Rev. 41, 918–931 (2022).
Zhong, P. et al. COVID-19-associated gastrointestinal and liver injury: clinical features and potential mechanisms. Signal Transduct. Target Ther. 5, 256 (2020).
Lazarus, J. V. et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat. Rev. Gastroenterol. Hepatol. 19, 60–78 (2022).
Meyers, J. L. et al. COVID-19 pandemic stressors are associated with reported increases in frequency of drunkenness among individuals with a history of alcohol use disorder. Transl. Psychiatry 13, 311 (2023).
Lv, T. et al. Regional variation and temporal trend of primary biliary cholangitis epidemiology: a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 36, 1423–1434 (2021).
Tanaka, A. & Takikawa, H. Geoepidemiology of primary sclerosing cholangitis: a critical review. J. Autoimmun. 46, 35–40 (2013).
Liang, H. et al. Incidence, prevalence, and natural history of primary sclerosing cholangitis in the United Kingdom. Medicine (Baltimore) 96, e7116 (2017).
Boonstra, K. et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 58, 2045–2055 (2013).
Lv, T. et al. Systematic review and meta-analysis on the incidence and prevalence of autoimmune hepatitis in Asian, European, and American population. J. Gastroenterol. Hepatol. 34, 1676–1684 (2019).
Dinse, G. E. et al. Increasing prevalence of antinuclear antibodies in the United States. Arthritis Rheumatol. 72, 1026–1035 (2020).
Webb, G. J., Ryan, R. P., Marshall, T. P. & Hirschfield, G. M. The epidemiology of UK autoimmune liver disease varies with geographic latitude. Clin. Gastroenterol. Hepatol. 19, 2587–2596 (2021).
Danielsson Borssén, Å. et al. Epidemiology and causes of death in a Swedish cohort of patients with autoimmune hepatitis. Scand. J. Gastroenterol. 52, 1022–1028 (2017).
Tanaka, A. et al. Increase trend in the prevalence and male-to-female ratio of primary biliary cholangitis, autoimmune hepatitis, and primary sclerosing cholangitis in Japan. Hepatol. Res 49, 881–889 (2019).
Kim, B. H. et al. Population-based prevalence, incidence, and disease burden of autoimmune hepatitis in South Korea. PLoS ONE 12, e0182391 (2017).
Xie, J. J. & Wu, Z. Y. Wilson’s disease in China. Neurosci. Bull. 33, 323–330 (2017).
Lo, C. & Bandmann, O. Epidemiology and introduction to the clinical presentation of Wilson disease. Handb. Clin. Neurol. 142, 7–17 (2017).
European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 56, 671-685 (2012).
Coffey, A. J. et al. A genetic study of Wilson’s disease in the United Kingdom. Brain 136, 1476–1487 (2013).
Hamesch, K. et al. Liver fibrosis and metabolic alterations in adults with alpha-1-antitrypsin deficiency caused by the Pi*ZZ mutation. Gastroenterology 157, 705–719.e718 (2019).
Flemming, J. A. et al. Incidence of cirrhosis in young birth cohorts in Canada from 1997 to 2016: a retrospective population-based study. Lancet Gastroenterol. Hepatol. 4, 217–226 (2019).
Ala, A. et al. Wilson’s disease. Lancet 369, 397–408 (2007).
Moon, A. M., Singal, A. G. & Tapper, E. B. Contemporary epidemiology of chronic liver disease and cirrhosis. Clin. Gastroenterol. Hepatol. 18, 2650–2666 (2020).
Gu, W. et al. Trends and the course of liver cirrhosis and its complications in Germany: nationwide population-based study (2005 to 2018). Lancet Reg. Health Eur. 12, 100240 (2022).
Enomoto, H. et al. Transition in the etiology of liver cirrhosis in Japan: a nationwide survey. J. Gastroenterol. 55, 353–362 (2020).
Jang, W. Y. et al. Changes in characteristics of patients with liver cirrhosis visiting a tertiary hospital over 15 years: a retrospective multi-center Study in Korea. J. Korean Med. Sci. 35, e233 (2020).
Vento, S., Dzudzor, B., Cainelli, F. & Tachi, K. Liver cirrhosis in sub-Saharan Africa: neglected, yet important. Lancet Glob. Health 6, e1060–e1061 (2018).
Zhang, C. H. et al. Changing epidemiology of hepatocellular carcinoma in Asia. Liver Int. 42, 2029–2041 (2022).
Singal, A. G., Lampertico, P. & Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 72, 250–261 (2020).
Konyn, P., Ahmed, A. & Kim, D. Current epidemiology in hepatocellular carcinoma. Expert Rev. Gastroenterol. Hepatol. 15, 1295–1307 (2021).
Rich, N. E., Yopp, A. C., Singal, A. G. & Murphy, C. C. Hepatocellular carcinoma incidence is decreasing among younger adults in the United States. Clin. Gastroenterol. Hepatol. 18, 242–248.e245 (2020).
Toh, M. R. et al. Global epidemiology and genetics of hepatocellular carcinoma. Gastroenterology 164, 766–782 (2023).
Fanning, G. C., Zoulim, F., Hou, J. & Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat. Rev. Drug Discov. 18, 827–844 (2019).
Suarez, A. A. R. & Zoulim, F. Opportunities and challenges for hepatitis B cure. eGastroenterology 1, e100021 (2023).
Lemoine, M. & Thursz, M. R. Battlefield against hepatitis B infection and HCC in Africa. J. Hepatol. 66, 645–654 (2017).
Mathurin, P. & Bataller, R. Trends in the management and burden of alcoholic liver disease. J. Hepatol. 62, S38–S46 (2015).
Danpanichkul, P. et al. Incidence of liver cancer in young adults according to the Global Burden of Disease database 2019. Hepatology 80, 828–843 (2024).
Meijer, N. et al. The aflatoxin situation in Africa: Systematic literature review. Compr. Rev. Food Sci. Food Saf. 20, 2286–2304 (2021).
Bertuccio, P. et al. A comparison of trends in mortality from primary liver cancer and intrahepatic cholangiocarcinoma in Europe. Ann. Oncol. 24, 1667–1674 (2013).
Beal, E. W. et al. Cohort contributions to trends in the incidence and mortality of intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 7, 270–276 (2018).
Saha, S. K., Zhu, A. X., Fuchs, C. S. & Brooks, G. A. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 21, 594–599 (2016).
Florio, A. A. et al. Global trends in intrahepatic and extrahepatic cholangiocarcinoma incidence from 1993 to 2012. Cancer 126, 2666–2678 (2020).
Khan, S. A., Tavolari, S. & Brandi, G. Cholangiocarcinoma: epidemiology and risk factors. Liver Int. 39, 19–31 (2019).
Ouyang, G. et al. The global, regional, and national burden of gallbladder and biliary tract cancer and its attributable risk factors in 195 countries and territories, 1990 to 2017: a systematic analysis for the Global Burden of Disease Study 2017. Cancer 127, 2238–2250 (2021).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines on nutrition in chronic liver disease. J. Hepatol. 70, 172–193 (2019).
Hansen, L. et al. Symptom classes in decompensated liver disease. Clin. Gastroenterol. Hepatol. 20, 2551–2557.e2551 (2022).
Kaya, E. & Yilmaz, Y. Metabolic-associated Fatty Liver Disease (MAFLD): a multi-systemic disease beyond the liver. J. Clin. Transl. Hepatol. 10, 329–338 (2022).
Hernandez-Tejero, M., Clemente-Sanchez, A. & Bataller, R. Spectrum, screening, and diagnosis of alcohol-related liver disease. J. Clin. Exp. Hepatol. 13, 75–87 (2023).
Ortega-Alonso, A., Stephens, C., Lucena, M. I. & Andrade, R. J. Case characterization, clinical features and risk factors in drug-induced liver injury. Int. J. Mol. Sci. 17 (2016).
Mazzaro, C. et al. A review on extrahepatic manifestations of chronic Hepatitis C virus infection and the impact of direct-acting antiviral therapy. Viruses. 13 (2021).
Terziroli Beretta-Piccoli, B., Mieli-Vergani, G. & Vergani, D. Autoimmmune hepatitis. Cell Mol. Immunol. 19, 158–176 (2022).
Younossi, Z. M. et al. Diagnosis and management of primary biliary cholangitis. Am. J. Gastroenterol. 114, 48–63 (2019).
Li, H. et al. Plasma lipidomics of primary biliary cholangitis and its comparison with Sjögren’s syndrome. Front. Immunol. 14, 1124443 (2023).
Barberio, B. et al. Prevalence of primary sclerosing cholangitis in patients with inflammatory bowel disease: a systematic review and meta-analysis. Gastroenterology 161, 1865–1877 (2021).
Assis, D. N. & Bowlus, C. L. Recent advances in the management of primary sclerosing cholangitis. Clin. Gastroenterol. Hepatol. 21, 2065–2075 (2023).
van Munster, K. N., Bergquist, A. & Ponsioen, C. Y. Inflammatory bowel disease and primary sclerosing cholangitis: one disease or two? J. Hepatol. 80, 155–168 (2024).
Eslam, M., Sanyal, A. J. & George, J. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014.e1991 (2020).
Gómez-Medina, C., Melo, L., Martí-Aguado, D. & Bataller, R. Subclinical versus advanced forms of alcohol-related liver disease: Need for early detection. Clin. Mol. Hepatol. 29, 1–15 (2023).
Członkowska, A. et al. Wilson disease. Nat. Rev. Dis. Prim. 4, 21 (2018).
Wu, T. et al. Liver disease progression in patients with alpha-1 antitrypsin deficiency and protease inhibitor ZZ genotype with or without lung disease. Aliment Pharm. Ther. 58, 1075–1085 (2023).
D’Amico, G., Bernardi, M. & Angeli, P. Towards a new definition of decompensated cirrhosis. J. Hepatol. 76, 202–207 (2022).
Rose, C. F. et al. Hepatic encephalopathy: novel insights into classification, pathophysiology and therapy. J. Hepatol. 73, 1526–1547 (2020).
Llovet, J. M. et al. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: pathogenesis and treatment. Nat. Rev. Gastroenterol. Hepatol. 20, 487–503 (2023).
Clouston, A. D. et al. Severe acute liver disease in adults: contemporary role of histopathology. Histopathology 85, 549–561 (2024).
Ahmad, J. et al. Value of liver biopsy in the diagnosis of drug-induced liver injury. J. Hepatol. 76, 1070–1078 (2022).
Brown, G. T. & Kleiner, D. E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 65, 1080–1086 (2016).
Portmann, B. & Zen, Y. Inflammatory disease of the bile ducts-cholangiopathies: liver biopsy challenge and clinicopathological correlation. Histopathology 60, 236–248 (2012).
Floreani, A., Gabbia, D. & De Martin, S. Current perspectives on the molecular and clinical relationships between primary biliary cholangitis and hepatocellular carcinoma. Int. J. Mol. Sci. 25 (2024).
de Boer, Y. S. et al. Assessment of the histopathological key features in autoimmune hepatitis. Histopathology 66, 351–362 (2015).
Cardoso, A. C., Figueiredo-Mendes, C., Villela-Nogueira, C. A. & Marcellin, P. Staging fibrosis in chronic viral hepatitis. Viruses. 14 (2022).
Loomba, R. et al. Review article: new developments in biomarkers and clinical drug development in alpha-1 antitrypsin deficiency-related liver disease. Aliment Pharm. Ther. 59, 1183–1195 (2024).
Leone, V. et al. Liver Inflammation and Hepatobiliary Cancers. Trends Cancer 7, 606–623 (2021).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines on non-invasive tests for evaluation of liver disease severity and prognosis—2021 update. J. Hepatol. 75, 659–689 (2021).
Yuen, M. F. et al. Hepatitis B virus infection. Nat. Rev. Dis. Prim. 4, 18035 (2018).
Negro, F. & Lok, A. S. Hepatitis D: a review. Jama 330, 2376–2387 (2023).
Ma, Z., de Man, R. A., Kamar, N. & Pan, Q. Chronic hepatitis E: advancing research and patient care. J. Hepatol. 77, 1109–1123 (2022).
Colley, D. G., Bustinduy, A. L., Secor, W. E. & King, C. H. Human schistosomiasis. Lancet 383, 2253–2264 (2014).
McManus, D. P., Zhang, W., Li, J. & Bartley, P. B. Echinococcosis. Lancet 362, 1295–1304 (2003).
Zhang, C. et al. Involvement of TIGIT in natural killer cell exhaustion and immune escape in patients and mouse model with liver Echinococcus multilocularis Infection. Hepatology 74, 3376–3393 (2021).
Morán, P. et al. Amoebiasis: advances in diagnosis, treatment, immunology features and the interaction with the intestinal ecosystem. Int. J. Mol. Sci. 24 (2023).
Eslam, M. et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J. Hepatol. 73, 202–209 (2020).
Miao, L. et al. Current status and future trends of the global burden of MASLD. Trends Endocrinol. Metab. 35, 697–707 (2024).
Nasereldin, D. S. et al. Association of metabolic health phenotypes, obesity, and hepatocellular carcinoma risk. Dig. Liver Dis. 54, 964–972 (2022).
Kwon, Y. M. et al. Association of nonalcoholic fatty liver disease with components of metabolic syndrome according to body mass index in Korean adults. Am. J. Gastroenterol. 107, 1852–1858 (2012).
Arab, J. P. et al. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 65, 350–362 (2017).
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M. & Sanyal, A. J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922 (2018).
Tilg, H., Adolph, T. E., Dudek, M. & Knolle, P. Non-alcoholic fatty liver disease: the interplay between metabolism, microbes and immunity. Nat. Metab. 3, 1596–1607 (2021).
Feng, G. et al. Recompensation in cirrhosis: unravelling the evolving natural history of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 21, 46–56 (2024).
Xu, X. et al. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target Ther. 7, 287 (2022).
Chalasani, N. et al. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology 148, 1340–1352.e1347 (2015).
Li, X., Tang, J. & Mao, Y. Incidence and risk factors of drug-induced liver injury. Liver Int. 42, 1999–2014 (2022).
Andrade, R. J. et al. Drug-induced liver injury. Nat. Rev. Dis. Prim. 5, 58 (2019).
Stephens, C., Lucena, M. I. & Andrade, R. J. Genetic risk factors in the development of idiosyncratic drug-induced liver injury. Expert Opin. Drug Metab. Toxicol. 17, 153–169 (2021).
Mackowiak, B., Fu, Y., Maccioni, L. & Gao, B. Alcohol-associated liver disease. J. Clin. Invest. 134 (2024).
Kamper-Jørgensen, M., Grønbaek, M., Tolstrup, J. & Becker, U. Alcohol and cirrhosis: dose-response or threshold effect? J. Hepatol. 41, 25–30 (2004).
Inan-Eroglu, E. et al. Joint associations of adiposity and alcohol consumption with liver disease-related morbidity and mortality risk: findings from the UK Biobank. Eur. J. Clin. Nutr. 76, 74–83 (2022).
Sinn, D. H. et al. Alcohol intake and mortality in patients with chronic viral hepatitis: a nationwide cohort study. Am. J. Gastroenterol. 116, 329–335 (2021).
Chang, B. et al. Prevalence and prediction of hepatocellular carcinoma in alcohol-associated liver disease: a retrospective study of 136 571 patients with chronic liver diseases. eGastroenterology 2, e100036 (2024).
Panetta, M., Brightmore, A. & Waring, W. S. Delayed onset of liver injury after intentional chloroform overdose: a case report and literature review. Acute Med. 18, 192–196 (2019).
Santos, O. et al. Acute liver failure due to white phosphorus ingestion. Ann. Hepatol. 8, 162–165 (2009).
Scholten, D., Trebicka, J., Liedtke, C. & Weiskirchen, R. The carbon tetrachloride model in mice. Lab Anim. 49, 4–11 (2015).
Cave, M. et al. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology 51, 474–481 (2010).
Wu, Y. et al. Association between per- and poly-fluoroalkyl substances and nonalcoholic fatty liver disease: a nested case-control study in northwest China. Environ. Pollut. 350, 123937 (2024).
Liu, Y., Galani Yamdeu, J. H., Gong, Y. Y. & Orfila, C. A review of postharvest approaches to reduce fungal and mycotoxin contamination of foods. Compr. Rev. Food Sci. Food Saf. 19, 1521–1560 (2020).
Zabaleta, N., Hommel, M., Salas, D. & Gonzalez-Aseguinolaza, G. Genetic-based approaches to inherited metabolic liver diseases. Hum. Gene Ther. 30, 1190–1203 (2019).
Pietrangelo, A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 139, 393–408 (2010). 408.e391-392.
Feder, J. N. et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 13, 399–408 (1996).
Nemeth, E. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 (2004).
Aschemeyer, S. et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 131, 899–910 (2018).
Chen, L., Min, J. & Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target Ther. 7, 378 (2022).
Greene, C. M. et al. α1-Antitrypsin deficiency. Nat. Rev. Dis. Prim. 2, 16051 (2016).
Lomas, D. A., Hurst, J. R. & Gooptu, B. Update on alpha-1 antitrypsin deficiency: new therapies. J. Hepatol. 65, 413–424 (2016).
Rehwinkel, J. & Gack, M. U. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551 (2020).
Onomoto, K., Onoguchi, K. & Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell Mol. Immunol. 18, 539–555 (2021).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Żeromski, J. et al. Pattern recognition receptors: significance of expression in the liver. Arch. Immunol. Ther. Exp. (Warsz.) 68, 29 (2020).
Li, Y. et al. Activation of endogenous type I IFN signaling contributes to persistent HCV infection. Rev. Med. Virol. 24, 332–342 (2014).
Asahina, Y. et al. Association of gene expression involving innate immunity and genetic variation in interleukin 28B with antiviral response. Hepatology 55, 20–29 (2012).
Dixit, U. et al. Staufen1 promotes HCV replication by inhibiting protein kinase R and transporting viral RNA to the site of translation and replication in the cells. Nucleic Acids Res. 44, 5271–5287 (2016).
Shi, X. et al. MxA is a positive regulator of type I IFN signaling in HCV infection. J. Med Virol. 89, 2173–2180 (2017).
Li, C. Z. et al. Polymorphism of OAS-1 determines liver fibrosis progression in hepatitis C by reduced ability to inhibit viral replication. Liver Int. 29, 1413–1421 (2009).
Li, X. D. et al. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl Acad. Sci. USA 102, 17717–17722 (2005).
Liang, Y. et al. Antiviral suppression vs restoration of RIG-I signaling by hepatitis C protease and polymerase inhibitors. Gastroenterology 135, 1710–1718.e1712 (2008).
Song, H. et al. TRIM25 inhibits HBV replication by promoting HBx degradation and the RIG-I-mediated pgRNA recognition. Chin. Med. J. (Engl.) 136, 799–806 (2023).
Sato, S. et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 (2015).
Lee, S. et al. Suppression of hepatitis B virus through therapeutic activation of RIG-I and IRF3 signaling in hepatocytes. iScience 24, 101969 (2021).
Jiang, J. & Tang, H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 1, 1106–1117 (2010).
Giersch, K. et al. Strain-specific responsiveness of hepatitis D virus to interferon-alpha treatment. JHEP Rep. 5, 100673 (2023).
Devhare, P. B., Desai, S. & Lole, K. S. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci. Rep. 6, 26827 (2016).
Sung, P. S. et al. CXCL10 is produced in hepatitis A virus-infected cells in an IRF3-dependent but IFN-independent manner. Sci. Rep. 7, 6387 (2017).
Sun, L. et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Chen, Q., Sun, L. & Chen, Z. J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016).
Kuipery, A., Gehring, A. J. & Isogawa, M. Mechanisms of HBV immune evasion. Antivir. Res. 179, 104816 (2020).
He, J. et al. Inhibition of hepatitis B virus replication by activation of the cGAS-STING pathway. J. Gen. Virol. 97, 3368–3378 (2016).
Dansako, H. et al. The cyclic GMP-AMP synthetase-STING signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J. 283, 144–156 (2016).
Ding, Q. et al. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 59, 52–58 (2013).
Chen, L. et al. Loss of Sam50 in hepatocytes induces cardiolipin-dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology 76, 1389–1408 (2022).
Liu, Z. et al. XBP1 deficiency promotes hepatocyte pyroptosis by impairing mitophagy to activate mtDNA-cGAS-STING signaling in macrophages during acute liver injury. Redox Biol. 52, 102305 (2022).
Li, Y. et al. Ginsenoside Rd inhibited ferroptosis to alleviate CCl(4)-induced acute liver injury in mice via cGAS/STING Pathway. Am. J. Chin. Med. 51, 91–105 (2023).
Ma, X. et al. Loss of hepatic DRP1 exacerbates alcoholic hepatitis by inducing megamitochondria and mitochondrial maladaptation. Hepatology 77, 159–175 (2023).
Lin, Z. et al. RING finger protein 13 protects against nonalcoholic steatohepatitis by targeting STING-relayed signaling pathways. Nat. Commun. 14, 6635 (2023).
Liu, K. et al. Lipotoxicity-induced STING1 activation stimulates MTORC1 and restricts hepatic lipophagy. Autophagy 18, 860–876 (2022).
Huang, X. et al. Macrophage SCAP contributes to metaflammation and lean NAFLD by activating STING-NF-κB signaling pathway. Cell Mol. Gastroenterol. Hepatol. 14, 1–26 (2022).
Steinberg, G. R. & Hardie, D. G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 24, 255–272 (2023).
Xu, X. et al. Nuclear UHRF1 is a gate-keeper of cellular AMPK activity and function. Cell Res. 32, 54–71 (2022).
Herzig, S. & Shaw, R. J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135 (2018).
Göransson, O., Kopietz, F. & Rider, M. H. Metabolic control by AMPK in white adipose tissue. Trends Endocrinol. Metab. 34, 704–717 (2023).
Zhao, L. et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 69, 705–717 (2018).
Lu, X. et al. AMPK protects against alcohol-induced liver injury through UQCRC2 to up-regulate mitophagy. Autophagy 17, 3622–3643 (2021).
Castaño, D. et al. Cardiotrophin-1 eliminates hepatic steatosis in obese mice by mechanisms involving AMPK activation. J. Hepatol. 60, 1017–1025 (2014).
Cai, Y. et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology 68, 48–61 (2018).
Allard, B. et al. Adenosine A2A receptor is a tumor suppressor of NASH-associated hepatocellular carcinoma. Cell Rep. Med. 4, 101188 (2023).
Zhu, W. et al. Activation of hepatic adenosine A1 receptor ameliorates MASH via inhibiting SREBPs maturation. Cell Rep. Med. 5, 101477 (2024).
Wan, J. et al. Gastrodin improves nonalcoholic fatty liver disease through activation of the adenosine monophosphate-activated protein kinase signaling pathway. Hepatology 74, 3074–3090 (2021).
O’Neill, H. M. et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia 57, 1693–1702 (2014).
Shearn, C. T. et al. Identification of 5’ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J. Biol. Chem. 289, 15449–15462 (2014).
Handa, P. et al. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology 60, 133–145 (2014).
Zhu, Z. et al. Involvement of insulin resistance in the protective effect of metformin against alcoholic liver injury. Alcohol Clin. Exp. Res 38, 1510–1519 (2014).
Chen, M. et al. Cdo1-Camkk2-AMPK axis confers the protective effects of exercise against NAFLD in mice. Nat. Commun. 14, 8391 (2023).
Qiu, B. et al. MKP1 promotes nonalcoholic steatohepatitis by suppressing AMPK activity through LKB1 nuclear retention. Nat. Commun. 14, 5405 (2023).
You, Y. et al. SNX10 mediates alcohol-induced liver injury and steatosis by regulating the activation of chaperone-mediated autophagy. J. Hepatol. 69, 129–141 (2018).
Shen, B. et al. Aucubin inhibited lipid accumulation and oxidative stress via Nrf2/HO-1 and AMPK signalling pathways. J. Cell Mol. Med. 23, 4063–4075 (2019).
Zhang, Y. et al. Exercise and Metformin intervention prevents lipotoxicity-induced hepatocyte apoptosis by alleviating oxidative and ER stress and activating the AMPK/Nrf2/HO-1 signaling pathway in db/db mice. Oxid. Med Cell Longev. 2022, 2297268 (2022).
Peng, W. et al. Zanthoxylum bungeanum amides ameliorates nonalcoholic fatty liver via regulating gut microbiota and activating AMPK/Nrf2 signaling. J. Ethnopharmacol. 318, 116848 (2024).
Zhao, P. et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 367, 652–660 (2020).
You, Y. et al. FNDC3B protects steatosis and ferroptosis via the AMPK pathway in alcoholic fatty liver disease. Free Radic. Biol. Med 193, 808–819 (2022).
Zheng, L. et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 19, 5372–5380 (2013).
Cheng, L. et al. Hepatic mitochondrial NAD + transporter SLC25A47 activates AMPKα mediating lipid metabolism and tumorigenesis. Hepatology 78, 1828–1842 (2023).
Canovas, B. & Nebreda, A. R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 22, 346–366 (2021).
Bahar, M. E., Kim, H. J. & Kim, D. R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct. Target Ther. 8, 455 (2023).
Wang, L. et al. Tripartite motif 16 ameliorates nonalcoholic steatohepatitis by promoting the degradation of phospho-TAK1. Cell Metab. 33, 1372–1388.e1377 (2021).
Che, Z. et al. Melatonin alleviates alcoholic liver disease via EGFR-BRG1-TERT axis regulation. Acta Pharm. Sin. B 13, 100–112 (2023).
Fang, Z. et al. Narirutin activates TFEB (transcription factor EB) to protect against Acetaminophen-induced liver injury by targeting PPP3/calcineurin. Autophagy 19, 2240–2256 (2023).
Kim, H. J., Han, Y. H., Kim, J. Y. & Lee, M. O. RORα Enhances Lysosomal Acidification and Autophagic Flux in the Hepatocytes. Hepatol. Commun. 5, 2121–2138 (2021).
Inami, Y. et al. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys. Res Commun. 412, 618–625 (2011).
Lee, A. Y. et al. Dihydroceramide is a key metabolite that regulates autophagy and promotes fibrosis in hepatic steatosis model. Biochem. Biophys. Res Commun. 494, 460–469 (2017).
Zeng, J. et al. Restoration of lysosomal acidification rescues autophagy and metabolic dysfunction in non-alcoholic fatty liver disease. Nat. Commun. 14, 2573 (2023).
Lo, C. H. et al. Acidic nanoparticles restore lysosomal acidification and rescue metabolic dysfunction in pancreatic β-cells under lipotoxic conditions. ACS Nano 18, 15452–15467 (2024).
Xu, W. et al. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J. Hepatol. 67, 310–320 (2017).
Zhang, X. et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J. Hepatol. 71, 163–174 (2019).
Liu, J. et al. Deficiency of p38α in macrophage ameliorates d-galactosamine/TNF-α-induced acute liver injury in mice. FEBS J. 284, 4200–4215 (2017).
Cai, B. et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 31, 406–421.e407 (2020).
Zhang, Z. et al. ANGPTL8 accelerates liver fibrosis mediated by HFD-induced inflammatory activity via LILRB2/ERK signaling pathways. J. Adv. Res. 47, 41–56 (2023).
Thoen, L. F. et al. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 55, 1353–1360 (2011).
He, Y. et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target Ther. 6, 425 (2021).
Jung, K. et al. Farnesoid X receptor activation impairs liver progenitor cell-mediated liver regeneration via the PTEN-PI3K-AKT-mTOR axis in zebrafish. Hepatology 74, 397–410 (2021).
Zhang, Z. et al. Exosomes derived from human adipose mesenchymal stem cells ameliorate hepatic fibrosis by inhibiting PI3K/Akt/mTOR pathway and remodeling choline metabolism. J. Nanobiotechnol. 21, 29 (2023).
Jackson, L. N. et al. PI3K/Akt activation is critical for early hepatic regeneration after partial hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1401–1410 (2008).
Nechemia-Arbely, Y. et al. Early hepatocyte DNA synthetic response posthepatectomy is modulated by IL-6 trans-signaling and PI3K/AKT activation. J. Hepatol. 54, 922–929 (2011).
Ye, Q. et al. Deficiency of gluconeogenic enzyme PCK1 promotes metabolic-associated fatty liver disease through PI3K/AKT/PDGF axis activation in male mice. Nat. Commun. 14, 1402 (2023).
Zhang, L., Zhou, F. & ten Dijke, P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 38, 612–620 (2013).
Yuan, M. et al. DC-SIGN-LEF1/TCF1-miR-185 feedback loop promotes colorectal cancer invasion and metastasis. Cell Death Differ. 27, 379–395 (2020).
Zhao, C., Wang, B., Liu, E. & Zhang, Z. Loss of PTEN expression is associated with PI3K pathway-dependent metabolic reprogramming in hepatocellular carcinoma. Cell Commun. Signal 18, 131 (2020).
Luo, Y. et al. PI3K/AKT1 signaling pathway mediates sinomenine-induced hepatocellular carcinoma cells apoptosis: an in vitro and in vivo study. Biol. Pharm. Bull. 45, 614–624 (2022).
Philips, R. L. et al. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 185, 3857–3876 (2022).
Xue, C. et al. Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer. Signal Transduct. Target Ther. 8, 204 (2023).
Rocca, S. et al. Targeting few to help hundreds: JAK, MAPK and ROCK pathways as druggable targets in atypical chronic myeloid leukemia. Mol. Cancer 17, 40 (2018).
Asuri, S. et al. Primary biliary cholangitis in British Columbia First Nations: clinical features and discovery of novel genetic susceptibility loci. Liver Int 38, 940–948 (2018).
Cordell, H. J. et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat. Commun. 6, 8019 (2015).
Gordon, S. C. et al. Baricitinib and primary biliary cholangitis. J. Transl. Autoimmun. 4, 100107 (2021).
Shao, T. et al. Treatment with a JAK1/2 inhibitor ameliorates murine autoimmune cholangitis induced by IFN overexpression. Cell Mol. Immunol. 19, 1130–1140 (2022).
Schoggins, J. W. Interferon-stimulated genes: what do they all do? Annu Rev. Virol. 6, 567–584 (2019).
Makjaroen, J. et al. Comprehensive proteomics identification of IFN-λ3-regulated antiviral proteins in HBV-transfected cells. Mol. Cell Proteom. 17, 2197–2215 (2018).
Chen, Y. et al. ISG12a inhibits HCV replication and potentiates the anti-HCV activity of IFN-α through activation of the Jak/STAT signaling pathway independent of autophagy and apoptosis. Virus Res 227, 231–239 (2017).
Zhao, J., Qi, Y. F. & Yu, Y. R. STAT3: a key regulator in liver fibrosis. Ann. Hepatol. 21, 100224 (2021).
Wang, F. et al. Activated natural killer cell promotes nonalcoholic steatohepatitis through mediating JAK/STAT pathway. Cell Mol. Gastroenterol. Hepatol. 13, 257–274 (2022).
Grohmann, M. et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 175, 1289–1306.e1220 (2018).
Liu, J. et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target Ther. 7, 3 (2022).
Pez, F. et al. Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. J. Hepatol. 59, 1107–1117 (2013).
Rebouissou, S. et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 64, 2047–2061 (2016).
Laurent-Puig, P. et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 120, 1763–1773 (2001).
Sun, Y. et al. Integrated multi-omics profiling to dissect the spatiotemporal evolution of metastatic hepatocellular carcinoma. Cancer Cell 42, 135–156.e117 (2024).
Kim, M. et al. Functional interaction between Wnt3 and Frizzled-7 leads to activation of the Wnt/beta-catenin signaling pathway in hepatocellular carcinoma cells. J. Hepatol. 48, 780–791 (2008).
Cadoret, A. et al. Hepatomegaly in transgenic mice expressing an oncogenic form of beta-catenin. Cancer Res. 61, 3245–3249 (2001).
Nejak-Bowen, K. N. et al. Accelerated liver regeneration and hepatocarcinogenesis in mice overexpressing serine-45 mutant beta-catenin. Hepatology 51, 1603–1613 (2010).
Rignall, B., Braeuning, A., Buchmann, A. & Schwarz, M. Tumor formation in liver of conditional β-catenin-deficient mice exposed to a diethylnitrosamine/phenobarbital tumor promotion regimen. Carcinogenesis 32, 52–57 (2011).
Liang, Y. et al. β-catenin deficiency in hepatocytes aggravates hepatocarcinogenesis driven by oncogenic β-catenin and MET. Hepatology 67, 1807–1822 (2018).
Boulter, L. et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Invest. 125, 1269–1285 (2015).
Chen, Q. et al. Circular RNA ACTN4 promotes intrahepatic cholangiocarcinoma progression by recruiting YBX1 to initiate FZD7 transcription. J. Hepatol. 76, 135–147 (2022).
Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012).
Deng, Z. et al. TGF-β signaling in health, disease, and therapeutics. Signal Transduct. Target Ther. 9, 61 (2024).
Henderson, N. C. et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 19, 1617–1624 (2013).
Tahashi, Y. et al. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 35, 49–61 (2002).
Gorham, J. D. Transforming growth factor-beta1, Th1 responses, and autoimmune liver disease. Transfusion 45, 51s–59s (2005).
Chida, T. et al. Critical role of CREBH-mediated induction of transforming growth factor β2 by hepatitis C virus infection in fibrogenic responses in hepatic stellate cells. Hepatology 66, 1430–1443 (2017).
He, W. et al. Identifying a distinct fibrosis subset of NAFLD via molecular profiling and the involvement of profibrotic macrophages. J. Transl. Med. 21, 448 (2023).
Lurje, I., Gaisa, N. T., Weiskirchen, R. & Tacke, F. Mechanisms of organ fibrosis: emerging concepts and implications for novel treatment strategies. Mol. Asp. Med. 92, 101191 (2023).
Meng, L. et al. NPRC deletion attenuates cardiac fibrosis in diabetic mice by activating PKA/PKG and inhibiting TGF-β1/Smad pathways. Sci. Adv. 9, eadd4222 (2023).
Sharma, A. et al. Lipopolysaccharide reverses hepatic stellate cell activation through modulation of cMyb, small mothers against decapentaplegic, and CCAAT/enhancer-binding protein C/EBP transcription factors. Hepatology 72, 1800–1818 (2020).
Song, Y. et al. Tyrosine kinase receptor B attenuates liver fibrosis by inhibiting TGF-β/SMAD signaling. Hepatology 78, 1433–1447 (2023).
Russo, I. et al. Protective effects of activated myofibroblasts in the pressure-overloaded myocardium are mediated through smad-dependent activation of a matrix-preserving program. Circ. Res. 124, 1214–1227 (2019).
Busnadiego, O. et al. LOXL4 is induced by transforming growth factor β1 through Smad and JunB/Fra2 and contributes to vascular matrix remodeling. Mol. Cell Biol. 33, 2388–2401 (2013).
Zhang, J. et al. Sirt6 alleviated liver fibrosis by deacetylating conserved lysine 54 on Smad2 in hepatic stellate cells. Hepatology 73, 1140–1157 (2021).
Yu, J. et al. Kindlin-2 regulates hepatic stellate cells activation and liver fibrogenesis. Cell Death Discov. 4, 34 (2018).
Le Pabic, H. et al. Involvement of the serine/threonine p70S6 kinase in TGF-beta1-induced ADAM12 expression in cultured human hepatic stellate cells. J. Hepatol. 43, 1038–1044 (2005).
Gong, J., Tu, W., Liu, J. & Tian, D. Hepatocytes: a key role in liver inflammation. Front Immunol. 13, 1083780 (2022).
Loft, A. et al. A macrophage-hepatocyte glucocorticoid receptor axis coordinates fasting ketogenesis. Cell Metab. 34, 473–486.e479 (2022).
Gehrke, N. et al. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J. Hepatol. 68, 986–995 (2018).
Dong, X. et al. Ursodesoxycholic acid alleviates liver fibrosis via proregeneration by activation of the ID1-WNT2/HGF signaling pathway. Clin. Transl. Med. 11, e296 (2021).
Kaffe, E. et al. Humanized mouse liver reveals endothelial control of essential hepatic metabolic functions. Cell 186, 3793–3809.e3726 (2023).
Banales, J. M. et al. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 16, 269–281 (2019).
Raven, A. et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 547, 350–354 (2017).
Pinto, C., Giordano, D. M., Maroni, L. & Marzioni, M. Role of inflammation and proinflammatory cytokines in cholangiocyte pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1270–1278 (2018).
Strazzabosco, M. et al. Pathophysiologic implications of innate immunity and autoinflammation in the biliary epithelium. Biochim Biophys. Acta Mol. Basis Dis. 1864, 1374–1379 (2018).
Gao, J. et al. Angiocrine signaling in sinusoidal homeostasis and liver diseases. J. Hepatol. 81, 543–561 (2024).
Gao, J. et al. Endothelial p300 promotes portal hypertension and hepatic fibrosis through C-C motif chemokine ligand 2-mediated angiocrine signaling. Hepatology 73, 2468–2483 (2021).
Greuter, T. et al. Mechanotransduction-induced glycolysis epigenetically regulates a CXCL1-dominant angiocrine signaling program in liver sinusoidal endothelial cells in vitro and in vivo. J. Hepatol. 77, 723–734 (2022).
Hilscher, M. B. et al. Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology 157, 193–209.e199 (2019).
Desroches-Castan, A. et al. Bone morphogenetic protein 9 is a paracrine factor controlling liver sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis. Hepatology 70, 1392–1408 (2019).
Gaitantzi, H. et al. BMP-9 Modulates the Hepatic Responses to LPS. Cells 9 (2020).
Schmid, C. D. et al. ALK1 controls hepatic vessel formation, angiodiversity, and angiocrine functions in hereditary hemorrhagic telangiectasia of the liver. Hepatology 77, 1211–1227 (2023).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Mederacke, I. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 4, 2823 (2013).
Iwaisako, K. et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl Acad. Sci. USA 111, E3297–E3305 (2014).
Poisson, J. et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J. Hepatol. 66, 212–227 (2017).
Colucci, S. et al. Liver sinusoidal endothelial cells suppress bone morphogenetic protein 2 production in response to TGFβ pathway activation. Hepatology 74, 2186–2200 (2021).
Meyer, J. et al. Platelet interactions with liver sinusoidal endothelial cells and hepatic stellate cells lead to hepatocyte proliferation. Cells 9 (2020).
Wang, Q. et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J. Hepatol. 77, 312–325 (2022).
Xiao, Y. et al. STING mediates hepatocyte pyroptosis in liver fibrosis by epigenetically activating the NLRP3 inflammasome. Redox Biol. 62, 102691 (2023).
Wen, Y., Lambrecht, J., Ju, C. & Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 18, 45–56 (2021).
Blériot, C. et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity 54, 2101–2116.e2106 (2021).
Tran, S. et al. Impaired Kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity 53, 627–640.e625 (2020).
Krenkel, O. et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 69, 551–563 (2020).
Sun, X. et al. Transcriptional switch of hepatocytes initiates macrophage recruitment and T-cell suppression in endotoxemia. J. Hepatol. 77, 436–452 (2022).
Feng, D. et al. Monocyte-derived macrophages orchestrate multiple cell-type interactions to repair necrotic liver lesions in disease models. J. Clin. Invest. 133 (2023).
Ma, H. Y. et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 72, 946–959 (2020).
Xiong, X. et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e645 (2019).
Ramachandran, P. et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl Acad. Sci. USA 109, E3186–E3195 (2012).
Moreno-Lanceta, A. et al. RNF41 orchestrates macrophage-driven fibrosis resolution and hepatic regeneration. Sci. Transl. Med. 15, eabq6225 (2023).
Ficht, X. & Iannacone, M. Immune surveillance of the liver by T cells. Sci. Immunol. 5 (2020).
Mieli-Vergani, G. et al. Autoimmune hepatitis. Nat. Rev. Dis. Prim. 4, 18017 (2018).
Hardtke-Wolenski, M. et al. The influence of genetic predisposition and autoimmune hepatitis inducing antigens in disease development. J. Autoimmun. 78, 39–45 (2017).
Iannacone, M. & Guidotti, L. G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 22, 19–32 (2022).
Binder, B. & Thimme, R. CD4+ T cell responses in human viral infection: lessons from hepatitis C. J. Clin. Invest 130, 595–597 (2020).
Liu, K., Wang, F. S. & Xu, R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cell Mol. Immunol. 18, 38–44 (2021).
Liu, M. et al. Super enhancer regulation of cytokine-induced chemokine production in alcoholic hepatitis. Nat. Commun. 12, 4560 (2021).
Huang, H. et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 62, 600–614 (2015).
Ariño, S. et al. Ductular reaction-associated neutrophils promote biliary epithelium proliferation in chronic liver disease. J. Hepatol. 79, 1025–1036 (2023).
Yan, M. et al. Gut liver brain axis in diseases: the implications for therapeutic interventions. Signal Transduct. Target Ther. 8, 443 (2023).
Mouries, J. et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 71, 1216–1228 (2019).
Shao, T. et al. Intestinal HIF-1α deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J. Hepatol. 69, 886–895 (2018).
Loomba, R. et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 25, 1054–1062.e1055 (2017).
Rabot, S. et al. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 24, 4948–4959 (2010).
Le Roy, T. et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794 (2013).
Steensels, S. et al. Acyl-coenzyme a thioesterase 9 traffics mitochondrial short-chain fatty acids toward de novo lipogenesis and glucose production in the liver. Hepatology 72, 857–872 (2020).
Niu, Y. et al. Blautia coccoides is a newly identified bacterium increased by leucine deprivation and has a novel function in improving metabolic disorders. Adv. Sci. (Weinh.) 11, e2309255 (2024).
Song, Q. et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J. Hepatol. 79, 1352–1365 (2023).
Park, B. S. et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 (2009).
Carpino, G. et al. Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology 72, 470–485 (2020).
Wang, S. et al. Hyperoside attenuates non-alcoholic fatty liver disease in rats via cholesterol metabolism and bile acid metabolism. J. Adv. Res. 34, 109–122 (2021).
Modica, S. et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 142, 355–365.e351-354 (2012).
Clifford, B. L. et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 33, 1671–1684.e1674 (2021).
Liu, S. et al. Melatonin mitigates aflatoxin B1-induced liver injury via modulation of gut microbiota/intestinal FXR/liver TLR4 signaling axis in mice. J. Pineal Res. 73, e12812 (2022).
Cheon, S. Y. & Song, J. The association between hepatic encephalopathy and diabetic encephalopathy: the brain-liver axis. Int. J. Mol. Sci. 22 (2021).
Görg, B. et al. O-GlcNAcylation-dependent upregulation of HO1 triggers ammonia-induced oxidative stress and senescence in hepatic encephalopathy. J. Hepatol. 71, 930–941 (2019).
Lama, A. et al. Palmitoylethanolamide dampens neuroinflammation and anxiety-like behavior in obese mice. Brain Behav. Immun. 102, 110–123 (2022).
Asimakidou, E., Saipuljumri, E. N., Lo, C. H. & Zeng, J. Role of metabolic dysfunction and inflammation along the liver-brain axis in animal models with obesity-induced neurodegeneration. Neural Regen. Res. 20, 1069–1076 (2025).
Fallahzadeh, M. A. & Rahimi, R. S. Hepatic encephalopathy: current and emerging treatment modalities. Clin. Gastroenterol. Hepatol. 20, S9–s19 (2022).
Patterson, T. T. et al. Complex feed-forward and feedback mechanisms underlie the relationship between traumatic brain injury and the gut-microbiota-brain axis. Shock 52, 318–325 (2019).
Hackl, M. T. et al. Brain leptin reduces liver lipids by increasing hepatic triglyceride secretion and lowering lipogenesis. Nat. Commun. 10, 2717 (2019).
Metz, M. et al. Leptin increases hepatic triglyceride export via a vagal mechanism in humans. Cell Metab. 34, 1719–1731.e1715 (2022).
Saeidinejad, M. et al. Novel therapeutic approaches in treatment of acute-on-chronic liver failure. Semin. Liver Dis. 43, 429–445 (2023).
Duwaerts, C. C. & Maher, J. J. Macronutrients and the adipose-liver axis in obesity and fatty liver. Cell Mol. Gastroenterol. Hepatol. 7, 749–761 (2019).
Wu, X. et al. Recent advances in understanding of pathogenesis of alcohol-associated liver disease. Annu. Rev. Pathol. 18, 411–438 (2023).
Xu, A. et al. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Invest. 112, 91–100 (2003).
Polyzos, S. A. et al. Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia 59, 30–43 (2016).
Lee, E., Korf, H. & Vidal-Puig, A. An adipocentric perspective on the development and progression of non-alcoholic fatty liver disease. J. Hepatol. 78, 1048–1062 (2023).
Zarei, M. et al. Targeting FGF21 for the treatment of nonalcoholic steatohepatitis. Trends Pharm. Sci. 41, 199–208 (2020).
Holland, W. L. et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 17, 790–797 (2013).
Xiao, G., Yang, J. & Yan, L. Comparison of diagnostic accuracy of aspartate aminotransferase to platelet ratio index and fibrosis-4 index for detecting liver fibrosis in adult patients with chronic hepatitis B virus infection: a systemic review and meta-analysis. Hepatology 61, 292–302 (2015).
Terrault, N. A. et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 67, 1560–1599 (2018).
Ghany, M. G. & Morgan, T. R. Hepatitis C Guidance 2019 Update: American Association for the Study of Liver Diseases-Infectious Diseases Society of America Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Hepatology 71, 686–721 (2020).
Muhoza, P. et al. Routine vaccination coverage—worldwide, 2020. MMWR Morb. Mortal. Wkly. Rep. 70, 1495–1500 (2021).
European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 67, 370–398 (2017).
Tseng, C. H. et al. Hepatocellular carcinoma incidence with tenofovir versus entecavir in chronic hepatitis B: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 5, 1039–1052 (2020).
Martinez, M. G. et al. Covalently closed circular DNA: The ultimate therapeutic target for curing HBV infections. J. Hepatol. 75, 706–717 (2021).
Yip, T. C. et al. HBsAg seroclearance further reduces hepatocellular carcinoma risk after complete viral suppression with nucleos(t)ide analogues. J. Hepatol. 70, 361–370 (2019).
European Association for the Study of the Liver. EASL recommendations on treatment of hepatitis C: Final update of the series(☆). J. Hepatol. 73, 1170–1218 (2020).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines on hepatitis delta virus. J.Hepatol. 79, 433–460 (2023).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines on hepatitis E virus infection. J. Hepatol. 68, 1256–1271 (2018).
Koch, D. G. et al. The Natural History of Severe Acute Liver Injury. Am. J. Gastroenterol. 112, 1389–1396 (2017).
Palmer, M. et al. Consensus guidelines: best practices for detection, assessment and management of suspected acute drug-induced liver injury occurring during clinical trials in adults with chronic cholestatic liver disease. Aliment Pharm. Ther. 51, 90–109 (2020).
Kim, W. R., Flamm, S. L., Di Bisceglie, A. M. & Bodenheimer, H. C. Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology 47, 1363–1370 (2008).
Stravitz, R. T. et al. Bleeding complications in acute liver failure. Hepatology 67, 1931–1942 (2018).
Björnsson, H. K. & Björnsson, E. S. Drug-induced liver injury: Pathogenesis, epidemiology, clinical features, and practical management. Eur. J. Intern. Med. 97, 26–31 (2022).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 70, 1222–1261 (2019).
Stravitz, R. T. et al. Future directions in acute liver failure. Hepatology 78, 1266–1289 (2023).
Fontana, R. J. et al. AASLD practice guidance on drug, herbal, and dietary supplement-induced liver injury. Hepatology 77, 1036–1065 (2023).
Li, M. et al. Pharmacotherapies for drug-induced liver injury: a current literature review. Front Pharm. 12, 806249 (2021).
Moosa, M. S. et al. A randomized controlled trial of intravenous n-acetylcysteine in the management of anti-tuberculosis drug-induced liver injury. Clin. Infect. Dis. 73, e3377–e3383 (2021).
Flamm, S. L., Yang, Y. X., Singh, S. & Falck-Ytter, Y. T. American gastroenterological association institute guidelines for the diagnosis and management of acute liver failure. Gastroenterology 152, 644–647 (2017).
Hwang, S. et al. Novel treatment of acute and acute-on-chronic liver failure: Interleukin-22. Liver Int. https://doi.org/10.1111/liv.15619 (2023).
Arab, J. P. et al. An open-label, dose-escalation study to assess the safety and efficacy of IL-22 agonist F-652 in patients with alcohol-associated hepatitis. Hepatology 72, 441–453 (2020).
Saliba, F. et al. Artificial liver support in patients with liver failure: a modified DELPHI consensus of international experts. Intensive Care Med. 48, 1352–1367 (2022).
Artru, F., Goldberg, D. & Kamath, P. S. Should patients with acute-on-chronic liver failure grade 3 receive higher priority for liver transplantation? J. Hepatol. 78, 1118–1123 (2023).
Karvellas, C. J. et al. AASLD Practice Guidance on Acute-on-chronic liver failure and the management of critically ill patients with cirrhosis. Hepatology 79, 1463–1502 (2024).
Wendon, J. et al. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure. J. Hepatol. 66, 1047–1081 (2017).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines on acute-on-chronic liver failure. J. Hepatol. 79, 461–491 (2023).
Rinella, M. E. et al. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology 77, 1797–1835 (2023).
Eslam, M. et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 14, 889–919 (2020).
Cusi, K. et al. American Association of Clinical Endocrinology Clinical Practice Guideline for the Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Primary Care and Endocrinology Clinical Settings: Co-Sponsored by the American Association for the Study of Liver Diseases (AASLD). Endocr. Pr. 28, 528–562 (2022).
Younossi, Z. M., Zelber-Sagi, S., Henry, L. & Gerber, L. H. Lifestyle interventions in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 20, 708–722 (2023).
Zhang, X. et al. Unhealthy lifestyle habits and physical inactivity among Asian patients with non-alcoholic fatty liver disease. Liver Int. 40, 2719–2731 (2020).
Park, M. J., Kim, H., Kim, M. G. & Kim, K. Comparison of glucagon-like peptide-1 receptor agonists and thiazolidinediones on treating nonalcoholic fatty liver disease: a network meta-analysis. Clin. Mol. Hepatol. 29, 693–704 (2023).
Wang, Z. et al. Response to pioglitazone in non-alcoholic fatty liver disease patients with vs. without type 2 diabetes: a meta-analysis of randomized controlled trials. Front. Endocrinol. (Lausanne) 14, 1111430 (2023).
Pose, E. et al. Safety of two different doses of simvastatin plus rifaximin in decompensated cirrhosis (LIVERHOPE-SAFETY): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Gastroenterol. Hepatol. 5, 31–41 (2020).
Ramandi, A. et al. Polypill protects MAFLD patients from cardiovascular events and mortality: a prospective trial. Hepatol. Int. 17, 882–888 (2023).
Harrison, S. A. et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 394, 2012–2024 (2019).
Wang, G. et al. Impacts of bariatric surgery on adverse liver outcomes: a systematic review and meta-analysis. Surg. Obes. Relat. Dis. 19, 717–726 (2023).
Younossi, Z. M. et al. The changing epidemiology of adult liver transplantation in the United States in 2013-2022: the dominance of metabolic dysfunction-associated steatotic liver disease and alcohol-associated liver disease. Hepatol Commun. 8 (2024).
European Association for the Study of the Liver. EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J. Hepatol. 81, 492–542 (2024).
Singal, A. K. & Mathurin, P. Diagnosis and treatment of alcohol-associated liver disease: a review. JAMA 326, 165–176 (2021).
Bataller, R., Arab, J. P. & Shah, V. H. Alcohol-associated hepatitis. N. Engl. J. Med. 387, 2436–2448 (2022).
Nahon, P. et al. Assessment of liver fibrosis using transient elastography in patients with alcoholic liver disease. J. Hepatol. 49, 1062–1068 (2008).
Crabb, D. W. et al. Standard definitions and common data elements for clinical trials in patients with alcoholic hepatitis: recommendation from the NIAAA Alcoholic Hepatitis Consortia. Gastroenterology 150, 785–790 (2016).
European Association for the Study of the Liver. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 64, 1388–1402 (2016).
Crabb, D. W. et al. Diagnosis and treatment of alcohol-associated liver diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology 71, 306–333 (2020).
Vannier, A. G. L. et al. Incidence and progression of alcohol-associated liver disease after medical therapy for alcohol use disorder. JAMA Netw. Open 5, e2213014 (2022).
Mathurin, P. & Lucey, M. R. Liver transplantation in patients with alcohol-related liver disease: current status and future directions. Lancet Gastroenterol. Hepatol. 5, 507–514 (2020).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of alcohol-related liver disease. J. Hepatol. 69, 154–181 (2018).
Wang, Q. X. et al. Clinical and histological features of autoantibody-negative autoimmune hepatitis in Chinese patients: a single center experience. J. Dig. Dis. 14, 175–180 (2013).
Dahlqvist, G. et al. Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology 65, 152–163 (2017).
Aabakken, L. et al. Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline. Endoscopy 49, 588–608 (2017).
Kovač, J. D. & Weber, M. A. Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis: an Update on MR Imaging Findings with Recent Developments. J. Gastrointestin Liver Dis. 25, 517–524 (2016).
Gurung, A. et al. Histologic features of autoimmune hepatitis: a critical appraisal. Hum. Pathol. 82, 51–60 (2018).
Purohit, T. & Cappell, M. S. Primary biliary cirrhosis: pathophysiology, clinical presentation and therapy. World J. Hepatol. 7, 926–941 (2015).
Hennes, E. M. et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 48, 169–176 (2008).
Wang, Z. et al. The management of autoimmune hepatitis patients with decompensated cirrhosis: real-world experience and a comprehensive review. Clin. Rev. Allergy Immunol. 52, 424–435 (2017).
Harms, M. H. et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J. Hepatol. 71, 357–365 (2019).
Deneau, M. R. et al. Oral vancomycin, ursodeoxycholic acid, or no therapy for pediatric primary sclerosing cholangitis: a matched analysis. Hepatology 73, 1061–1073 (2021).
Minaga, K., Watanabe, T., Chung, H. & Kudo, M. Autoimmune hepatitis and IgG4-related disease. World J. Gastroenterol. 25, 2308–2314 (2019).
Mack, C. L. et al. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines From the American Association for the Study of Liver Diseases. Hepatology 72, 671–722 (2020).
Clinical, E. A. S. L. Practice guidelines: autoimmune hepatitis. J. Hepatol. 63, 971–1004 (2015).
Konkwo, C., Chowdhury, S. & Vilarinho, S. Genetics of liver disease in adults. Hepatol. Commun. 8 (2024).
Tarnacka, B., Szeszkowski, W., Golebiowski, M. & Czlonkowska, A. MR spectroscopy in monitoring the treatment of Wilson’s disease patients. Mov. Disord. 23, 1560–1566 (2008).
Brewer, G. J. et al. Oral zinc therapy for Wilson’s disease. Ann. Intern. Med. 99, 314–319 (1983).
Wiggelinkhuizen, M., Tilanus, M. E., Bollen, C. W. & Houwen, R. H. Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharm. Ther. 29, 947–958 (2009).
Schilsky, M. L. Wilson disease: diagnosis, treatment, and follow-up. Clin. Liver Dis. 21, 755–767 (2017).
Schilsky, M. L. et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: Executive summary of the 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology 77, 1428–1455 (2023).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 69, 406–460 (2018).
Pennisi, G. et al. Noninvasive assessment of liver disease severity in patients with nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes. Hepatology 78, 195–211 (2023).
Newsome, P. N. et al. FibroScan-AST (FAST) score for the non-invasive identification of patients with non-alcoholic steatohepatitis with significant activity and fibrosis: a prospective derivation and global validation study. Lancet Gastroenterol. Hepatol. 5, 362–373 (2020).
Garcia-Tsao, G., Abraldes, J. G., Berzigotti, A. & Bosch, J. Portal hypertensive bleeding in cirrhosis: Risk stratification, diagnosis, and management: 2016 practice guidance by the American Association for the study of liver diseases. Hepatology 65, 310–335 (2017).
Marcellin, P. et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 381, 468–475 (2013).
Cai, X. et al. Hydronidone for the treatment of liver fibrosis related to chronic hepatitis B: a phase 2 randomized controlled trial. Clin. Gastroenterol. Hepatol. 21, 1893–1901.e1897 (2023).
Brusilovskaya, K., Königshofer, P., Schwabl, P. & Reiberger, T. Vascular targets for the treatment of portal hypertension. Semin Liver Dis. 39, 483–501 (2019).
Biggins, S. W. et al. Diagnosis, evaluation, and management of ascites, spontaneous bacterial peritonitis and hepatorenal syndrome: 2021 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 74, 1014–1048 (2021).
Bai, Z. et al. Role of terlipressin in cirrhotic patients with ascites and without hepatorenal syndrome: a systematic review of current evidence. Can. J. Gastroenterol. Hepatol. 2020, 5106958 (2020).
de Franchis, R. et al. Baveno VII—renewing consensus in portal hypertension. J. Hepatol. 76, 959–974 (2022).
Lim, C. et al. Auxiliary liver transplantation for cirrhosis: from APOLT to RAPID: a scoping review. Ann. Surg. 275, 551–559 (2022).
Rogal, S. S. et al. AASLD Practice Guidance: Palliative care and symptom-based management in decompensated cirrhosis. Hepatology 76, 819–853 (2022).
Lai, J. C. et al. Malnutrition, frailty, and sarcopenia in patients with cirrhosis: 2021 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 74, 1611–1644 (2021).
Kaplan, D. E. et al. AASLD Practice Guidance on risk stratification and management of portal hypertension and varices in cirrhosis. Hepatology 79, 1180–1211 (2024).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines on prevention and management of bleeding and thrombosis in patients with cirrhosis. J. Hepatol. 76, 1151–1184 (2022).
Marrero, J. A. et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 68, 723–750 (2018).
Tan, C. H., Low, S. C. & Thng, C. H. APASL and AASLD consensus guidelines on imaging diagnosis of hepatocellular carcinoma: a review. Int J. Hepatol. 2011, 519783 (2011).
Chang, T. S. et al. Alpha-fetoprotein measurement benefits hepatocellular carcinoma surveillance in patients with cirrhosis. Am. J. Gastroenterol. 110, 836–844 (2015).
Singal, A. G. et al. International Liver Cancer Association (ILCA) white paper on biomarker development for hepatocellular carcinoma. Gastroenterology 160, 2572–2584 (2021).
Reig, M. et al. BCLC strategy for prognosis prediction and treatment recommendation: the 2022 update. J. Hepatol. 76, 681–693 (2022).
Singal, A. G. et al. AASLD Practice Guidance on prevention, diagnosis, and treatment of hepatocellular carcinoma. Hepatology 78, 1922–1965 (2023).
Tabrizian, P. et al. Ten-year outcomes of liver transplant and downstaging for hepatocellular carcinoma. JAMA Surg. 157, 779–788 (2022).
Mehta, N. et al. Excellent outcomes of liver transplantation following down-staging of hepatocellular carcinoma to within milan criteria: a multicenter study. Clin. Gastroenterol. Hepatol. 16, 955–964 (2018).
Lencioni, R. et al. Early-stage hepatocellular carcinoma in patients with cirrhosis: long-term results of percutaneous image-guided radiofrequency ablation. Radiology 234, 961–967 (2005).
Vietti Violi, N. et al. Efficacy of microwave ablation versus radiofrequency ablation for the treatment of hepatocellular carcinoma in patients with chronic liver disease: a randomised controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 3, 317–325 (2018).
Salem, R. et al. Y90 radioembolization significantly prolongs time to progression compared with chemoembolization in patients with hepatocellular carcinoma. Gastroenterology 151, 1155–1163.e1152 (2016).
Kulik, L. M. et al. Safety and efficacy of 90Y radiotherapy for hepatocellular carcinoma with and without portal vein thrombosis. Hepatology 47, 71–81 (2008).
Yang, J. D. et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16, 589–604 (2019).
Kudo, M. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018).
Abou-Alfa, G. K. et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 379, 54–63 (2018).
Zhu, A. X. et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 282–296 (2019).
Llovet, J. M. et al. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 19, 151–172 (2022).
Haber, P. K. et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002-2020). Gastroenterology 161, 879–898 (2021).
Llovet, J. M. Exploring a new pathway for biomarker-based approval of immunotherapies. Nat. Rev. Clin. Oncol. 20, 279–280 (2023).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J.Hepatol. 69, 182–236 (2018).
Elbahrawy, A. et al. Recent advances in protective vaccines against hepatitis viruses: a narrative review. Viruses 15 (2023).
Zakaria, H. M. et al. Steroid therapy in children with fulminant hepatitis A. J. Viral Hepat. 25, 853–859 (2018).
Wiegand, J. et al. Treatment of severe, nonfulminant acute hepatitis B with lamivudine vs placebo: a prospective randomized double-blinded multicentre trial. J. Viral Hepat. 21, 744–750 (2014).
Streinu-Cercel, A., Sandulescu, O., Stefan, M. & Streinu-Cercel, A. Treatment with lamivudine and entecavir in severe acute hepatitis B. Indian J. Med. Microbiol 34, 166–172 (2016).
Naggie, S. et al. Ledipasvir/sofosbuvir for 8 weeks to treat acute hepatitis C virus infections in men with human immunodeficiency virus infections: sofosbuvir-containing regimens without interferon for treatment of acute HCV in HIV-1 infected individuals. Clin. Infect. Dis. 69, 514–522 (2019).
Boerekamps, A. et al. Treatment of acute hepatitis C genotypes 1 and 4 with 8 weeks of grazoprevir plus elbasvir (DAHHS2): an open-label, multicentre, single-arm, phase 3b trial. Lancet Gastroenterol. Hepatol. 4, 269–277 (2019).
Pischke, S. et al. Ribavirin treatment of acute and chronic hepatitis E: a single-centre experience. Liver Int. 33, 722–726 (2013).
Da Cunha, T. & Wu, G. Y. Cytomegalovirus hepatitis in immunocompetent and immunocompromised hosts. J. Clin. Transl. Hepatol. 9, 106–115 (2021).
Williams, R. & Riordan, S. M. Acute liver failure: established and putative hepatitis viruses and therapeutic implications. J. Gastroenterol. Hepatol. 15, G17–G25 (2000).
Kumachev, A. & Wu, P. E. Drug-induced liver injury. CMAJ 193, E310 (2021).
Dear, J. W. et al. A metabolomic analysis of thiol response for standard and modified N-acetyl cysteine treatment regimens in patients with acetaminophen overdose. Clin. Transl. Sci. 14, 1476–1489 (2021).
Singh, S. et al. Comparative effectiveness of pharmacological interventions for severe alcoholic hepatitis: a systematic review and network meta-analysis. Gastroenterology 149, 958–970.e912 (2015).
Sehrawat, T. S., Liu, M. & Shah, V. H. The knowns and unknowns of treatment for alcoholic hepatitis. Lancet Gastroenterol. Hepatol. 5, 494–506 (2020).
Naveau, S. et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 39, 1390–1397 (2004).
Sarin, S. K., Pande, A. & Schnabl, B. Microbiome as a therapeutic target in alcohol-related liver disease. J. Hepatol. 70, 260–272 (2019).
Yuen, M. F. et al. Safety and efficacy of vebicorvir in virologically suppressed patients with chronic hepatitis B virus infection. J. Hepatol. 77, 642–652 (2022).
Byun, K. S. et al. Tenofovir alafenamide for drug-resistant hepatitis b: a randomized trial for switching from tenofovir disoproxil fumarate. Clin. Gastroenterol. Hepatol. 20, 427–437.e425 (2022).
Salimzadeh, L. et al. PD-1 blockade partially recovers dysfunctional virus-specific B cells in chronic hepatitis B infection. J. Clin. Invest 128, 4573–4587 (2018).
Yuen, M. F. et al. RNA interference therapy with ARC-520 results in prolonged hepatitis B surface antigen response in patients with chronic hepatitis B infection. Hepatology 72, 19–31 (2020).
Rohde, M. D. et al. No association between DAA treatment for HCV infection and herpes zoster infection in analysis of data from 37 clinical trials. Clin. Gastroenterol. Hepatol. 19, 1670–1678 (2021).
Poordad, F. et al. Glecaprevir and pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment. Hepatology 66, 389–397 (2017).
Wedemeyer, H. et al. Safety and efficacy of bulevirtide in combination with tenofovir disoproxil fumarate in patients with hepatitis B virus and hepatitis D virus coinfection (MYR202): a multicentre, randomised, parallel-group, open-label, phase 2 trial. Lancet Infect. Dis. 23, 117–129 (2023).
Wedemeyer, H. et al. Peginterferon alfa-2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT-II): a randomised, placebo controlled, phase 2 trial. Lancet Infect. Dis. 19, 275–286 (2019).
Lang, S. & Schnabl, B. Microbiota and fatty liver disease-the known, the unknown, and the future. Cell Host Microbe 28, 233–244 (2020).
Chu, H. et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J. Hepatol. 72, 391–400 (2020).
Louvet, A. et al. Corticosteroids reduce risk of death within 28 days for patients with severe alcoholic hepatitis, compared with pentoxifylline or placebo-a meta-analysis of individual data from controlled trials. Gastroenterology 155, 458–468.e458 (2018).
Moreno, C. et al. Intensive enteral nutrition is ineffective for patients with severe alcoholic hepatitis treated with corticosteroids. Gastroenterology 150, 903–910.e908 (2016).
Szabo, G. et al. IL-1 receptor antagonist plus pentoxifylline and zinc for severe alcohol-associated hepatitis. Hepatology 76, 1058–1068 (2022).
He, Y. et al. Immunopathobiology and therapeutic targets related to cytokines in liver diseases. Cell Mol. Immunol. 18, 18–37 (2021).
Teng, M. L. et al. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 29, S32–s42 (2023).
Gawrieh, S. et al. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology 74, 1809–1824 (2021).
Keam, S. J. Resmetirom: first Approval. Drugs 84, 729–735 (2024).
Díaz-González, Á. et al. Budesonide as first-line treatment in patients with autoimmune hepatitis seems inferior to standard predniso(lo)ne administration. Hepatology 77, 1095–1105 (2023).
Snijders, R. et al. An open-label randomised-controlled trial of azathioprine vs. mycophenolate mofetil for the induction of remission in treatment-naive autoimmune hepatitis. J. Hepatol. 80, 576–585 (2024).
Tonin, F. & Arends, I. Latest development in the synthesis of ursodeoxycholic acid (UDCA): a critical review. Beilstein J. Org. Chem. 14, 470–483 (2018).
Kowdley, K. V. et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 73, 94–101 (2020).
Rinella, M. E. et al. Non-invasive evaluation of response to obeticholic acid in patients with NASH: Results from the REGENERATE study. J. Hepatol. 76, 536–548 (2022).
Abdelmalek, M. F. et al. Pegbelfermin in patients with nonalcoholic steatohepatitis and compensated cirrhosis (FALCON 2): a randomized phase 2b study. Clin. Gastroenterol. Hepatol. 22, 113–123.e119 (2024).
Rinella, M. E. et al. A randomized, double-blind, placebo-controlled trial of aldafermin in patients with NASH and compensated cirrhosis. Hepatology 79, 674–689 (2024).
Shi, M. et al. Mesenchymal stem cell therapy in decompensated liver cirrhosis: a long-term follow-up analysis of the randomized controlled clinical trial. Hepatol. Int. 15, 1431–1441 (2021).
Wang, H. et al. Meta-analysis on last ten years of clinical injection of bone marrow-derived and umbilical cord MSC to reverse cirrhosis or rescue patients with acute-on-chronic liver failure. Stem Cell Res. Ther. 14, 267 (2023).
Zwirner, S. et al. First-in-class MKK4 inhibitors enhance liver regeneration and prevent liver failure. Cell 187, 1666–1684.e1626 (2024).
Alshamsi, F. et al. Extracorporeal liver support in patients with liver failure: a systematic review and meta-analysis of randomized trials. Intensive Care Med. 46, 1–16 (2020).
Jin, Y. et al. Stem cell-derived hepatocyte therapy using versatile biomimetic nanozyme incorporated nanofiber-reinforced decellularized extracellular matrix hydrogels for the treatment of acute liver failure. Bioact. Mater. 28, 112–131 (2023).
Qin, S. et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international phase 3 study. Lancet 402, 1133–1146 (2023).
Wang, K. et al. Adjuvant sintilimab in resected high-risk hepatocellular carcinoma: a randomized, controlled, phase 2 trial. Nat. Med 30, 708–715 (2024).
Peng, Z. et al. Lenvatinib combined with transarterial chemoembolization as first-line treatment for advanced hepatocellular carcinoma: a phase III, randomized clinical trial (LAUNCH). J. Clin. Oncol. 41, 117–127 (2023).
Yarchoan, M. et al. Personalized neoantigen vaccine and pembrolizumab in advanced hepatocellular carcinoma: a phase 1/2 trial. Nat. Med. 30, 1044–1053 (2024).
Qin, S. et al. Atezolizumab plus bevacizumab versus active surveillance in patients with resected or ablated high-risk hepatocellular carcinoma (IMbrave050): a randomised, open-label, multicentre, phase 3 trial. Lancet 402, 1835–1847 (2023).
Díaz, L. A. et al. Impact of public health policies on alcohol-associated liver disease in Latin America: an ecological multinational study. Hepatology 74, 2478–2490 (2021).
Wong, V. W. et al. Noninvasive biomarkers in NAFLD and NASH—current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 15, 461–478 (2018).
Harrison, S. A. et al. Challenges and opportunities in NASH drug development. Nat. Med 29, 562–573 (2023).
Henderson, N. C. et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl Acad. Sci. USA 103, 5060–5065 (2006).
Chalasani, N. et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology 158, 1334–1345.e1335 (2020).
Havelaar, A. H. et al. World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med. 12, e1001923 (2015).
Gozlan, Y. et al. Ongoing hepatitis A among men who have sex with men (MSM) linked to outbreaks in Europe in Tel Aviv area, Israel, December 2016–June 2017. Euro Surveill. 22 (2017).
Hofmeister, M. G. et al. Hepatitis A person-to-person outbreaks: epidemiology, morbidity burden, and factors associated with hospitalization-multiple states, 2016-2019. J. Infect. Dis. 223, 426–434 (2021).
Foster, M. A. et al. Widespread Hepatitis A outbreaks associated with person-to-person transmission—United States, 2016-2020. MMWR Morb. Mortal. Wkly. Rep. 71, 1229–1234 (2022).
Zaaijer, H. L. No artifact, hepatitis E is emerging. Hepatology 62, 654 (2015).
Schweitzer, A. et al. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386, 1546–1555 (2015).
Eaton, J. E. et al. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology 145, 521–536 (2013).
Bambha, K. et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology 125, 1364–1369 (2003).
Sandahl, T. D. et al. The prevalence of Wilson’s disease: an update. Hepatology 71, 722–732 (2020).
Beyzaei, Z., Mehrzadeh, A., Hashemi, N. & Geramizadeh, B. The mutation spectrum and ethnic distribution of Wilson disease, a review. Mol. Genet. Metab. Rep. 38, 101034 (2024).
Ovchinnikova, E. V., Garbuz, M. M., Ovchinnikova, A. A. & Kumeiko, V. V. Epidemiology of Wilson’s disease and pathogenic variants of the ATP7B gene leading to diversified protein disfunctions. Int. J. Mol. Sci. 25 (2024).
Blanco, I. et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int. J. Chron. Obstruct Pulmon. Dis. 12, 561–569 (2017).
Beste, L. A. et al. Trends in burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US veterans, 2001-2013. Gastroenterology 149, 1471–1482.e1475 (2015).
Nakanishi, T. et al. The undiagnosed disease burden associated with alpha-1 antitrypsin deficiency genotypes. Eur. Respir. J. 56 (2020).
Ashenhurst, J. R. et al. Prevalence of alpha-1 antitrypsin deficiency, self-reported behavior change, and health care engagement among direct-to-consumer recipients of a personalized genetic risk report. Chest 161, 373–381 (2022).
Hasegawa, K. et al. Clinical practice guidelines for hepatocellular carcinoma: The Japan Society of Hepatology 2021 version (5th JSH-HCC Guidelines). Hepatol. Res. 53, 383–390 (2023).
Giannitrapani, L. et al. The changing epidemiology of hepatocellular carcinoma : experience of a single center. Biomed. Res. Int. 2020, 5309307 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82122009, 82225008, 82322011, 82300711, and U23A20401), Science and Technology Projects in Guangzhou (202201020066), and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2021-I2M-5-003).
Author information
Authors and Affiliations
Contributions
H.W., E.D., and J.X. conceptualized and wrote the outline of the manuscript. C.G., Y.Y., H.S., J.G. X.K., Z.C., and Y.G. performed the literature search and wrote the manuscript draft. J.G., H.W., and J.X. reviewed and edited the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gan, C., Yuan, Y., Shen, H. et al. Liver diseases: epidemiology, causes, trends and predictions. Sig Transduct Target Ther 10, 33 (2025). https://doi.org/10.1038/s41392-024-02072-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-02072-z
