
Abstract
Neurodegenerative diseases (e.g., Alzheimer’s, Parkinson’s, Huntington’s disease, and Amyotrophic Lateral Sclerosis) are major health threats for the aging population and their prevalences continue to rise with the increasing of life expectancy. Although progress has been made, there is still a lack of effective cures to date, and an in-depth understanding of the molecular and cellular mechanisms of these neurodegenerative diseases is imperative for drug development. Protein phosphorylation, regulated by protein kinases and protein phosphatases, participates in most cellular events, whereas aberrant phosphorylation manifests as a main cause of diseases. As evidenced by pharmacological and pathological studies, protein kinases are proven to be promising therapeutic targets for various diseases, such as cancers, central nervous system disorders, and cardiovascular diseases. The mechanisms of protein phosphatases in pathophysiology have been extensively reviewed, but a systematic summary of the role of protein kinases in the nervous system is lacking. Here, we focus on the involvement of protein kinases in neurodegenerative diseases, by summarizing the current knowledge on the major kinases and related regulatory signal transduction pathways implicated in diseases. We further discuss the role and complexity of kinase–kinase networks in the pathogenesis of neurodegenerative diseases, illustrate the advances of clinical applications of protein kinase inhibitors or novel kinase-targeted therapeutic strategies (such as antisense oligonucleotides and gene therapy) for effective prevention and early intervention.
Similar content being viewed by others
Introduction
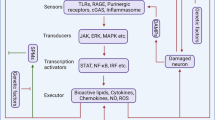
Kinases catalyze the transfer of the γ-phosphate group of ATP to the specific substrate,1 making the specific amino acid of the substrate phosphorylated (Fig. 1). Considering the type of substrates, kinases can be divided into protein kinases, lipid kinases, carbohydrate kinases, and other kinases (including Riboflavin kinase and Thymidine kinase, etc.), the most important group of which is protein kinase.2 The first protein kinase that was biochemically characterized in 1955 is phosphorylase kinase, which catalyzes the ATP-dependent phosphorylation of the specific phosphorylase.1 Since then, 518 human protein kinases to date (including 478 human eukaryotic protein kinases and 40 atypical protein kinases) and more than 900 genes encoding proteins with kinase activity have been identified.3,4 These protein kinases are classified into Ser/Thr kinases (385 members), tyrosine kinases (TK, 90 members), and tyrosine kinase-like kinases (TKL, 43 members) based on the targeted phosphate group of substrate residue.3 Protein kinases play indispensable roles in human diseases, especially in cancer and neurodegenerative diseases, and have been widely considered as drug targets for precision therapy.5 Small-molecule kinase inhibitors such as imatinib (inhibitor for TK, including BCR-Abl) and kinase-directed biological molecules such as margetuximab (a monoclonal antibody drug targeting human epidermal growth factor receptor 2 (HER2)-TK) have already been approved clinically for cancer therapy worldwide.1 However, despite these advancements, no kinase-related therapies targeting neurodegeneration have yet been approved.

Schematic diagram of the general working mechanism of kinases. Kinases catalyze the transfer of the γ-phosphate group of ATP to the specific substrate, making the specific amino acid of the substrate phosphorylated. This figure was created with BioRender.com
Neurodegenerative diseases, mainly affecting the brain, are a general term for a series of diseases caused by the progressive loss of structure or function of neurons, mainly including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and Amyotrophic Lateral Sclerosis (ALS). A shared characteristic of these diseases is the deposition of misfolded protein aggregates owing to the increased resistance of degradation of specific mutant proteins or the excessive accumulation of wild-type proteins. Aging is a major risk factor shared in neurodegenerative diseases. During brain aging, the metabolic regulation of neurons, neuronal development, and the immune microenvironment change, leading to a dysregulated molecular network around neurons, thereby promoting cognitive dysfunction.6 As the population ages, the rate of neurodegenerative diseases is increasing. For example, more than 55 million people currently live with dementia, and it is estimated that by 2050, ~150 million people globally will be affected by dementia, which will bring an economic burden of nearly $10 trillion7 and immeasurable losses to patients and their families.
However, these neurodegenerative diseases are currently incurable, and the treatments can only relieve symptoms and delay the progress of the diseases. Strategies under development for neurodegenerative diseases mainly include small molecules, antisense oligonucleotides (ASOs), gene therapy, and cell therapy. Immunotherapies targeting β-amyloid (Aβ) (Aducanumab,8 Lecanemab,9 Donanemab,10 all of which have been approved), tau (BIIB080,11 NI0752,12 JNJ-63733657,12,13 ACI-3513), and α-synuclein (α-syn) (Cinpanemab,14 Prasinezumab,15 Lu AF8242216) reduce internalization and diffusion of extracellular protein aggregates into neighboring cells in AD and PD.17 Drugs that maximize the quality of life for ALS patients have been approved, including, Relyvrio,18 Riluzole,19 and Eladarone.19 In addition to drug treatment, ASO therapies targeting several ALS causative genes (SOD1, C9orf72, ATXN-2, FUS), including ISIS333611,20 Tofersen (approved),21 BIIB078,22 and BIIB10523 are entered in clinical studies, and gene therapy drugs using adeno-associated virus as a vector are also under development. Nevertheless, there is still an unmet need for drugs or new potential strategies that can reverse neurodegeneration.
Protein phosphorylation is a crucial type of post-translational modification in neurodegeneration that has been most extensively investigated. The process of adding phosphate groups to the substrate, is catalyzed by kinases, while phosphatases are enzymes that remove phosphate groups. The addition or removal of phosphate groups results in the gaining or losing function of the substrate, thereby positively or negatively regulating the subsequent pathophysiological processes. In pathological processes, such as tangle formation in AD, residues in the proline-rich and microtubule-binding regions of tau protein are highly susceptible to phosphorylation modification. Phosphorylation of tau at Thr231 and Ser262 results in decreased affinity for microtubules.24 Abnormal hyperphosphorylation of tau increases its self-aggregation, leading to its mislocalization in neurons and impairment of synaptic functions.25,26 However, during physiological processes, such as axon formation, phosphorylations of tau at Ser199/202 and Thr205 sites are essential for axon formation, as evidenced by that about 80% of tau is phosphorylated at these sites in the cell body and proximal axons, while about 20% of tau is phosphorylated in the growth cones to regulate axonogenesis.27 In PD, hyperphosphorylation of α-syn at Ser129 leads to its misfolding and aggregation, forming pathological Lewy-bodies and Lewy-neurites.28 On the other hand, the dynamic changes of phosphorylation and dephosphorylation at Ser129 of α-syn at physiological levels are essential for fine-tuning of neuronal synaptic transmission.28 Furthermore, hyperphosphorylation of 43 kDa transactive response DNA-binding protein (TDP-43) protein at Ser409/410 leads to its mislocalization and aggregation in the neuronal cytoplasm in ALS.29 These observations together have highlighted the great potential to target abnormal protein phosphorylation to treat neurodegenerative diseases.
Protein kinases, as protein phosphorylation writer, are thereby crucial for neuronal homeostasis in neurodegenerative diseases as supported by numerous evidence. For example, the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (PKB/AKT)/mechanistic target of rapamycin (mTOR) pathway and metabolic central regulator-AMP activated kinases (AMPKs) are involved in neural development by regulating cell growth, proliferation, survival, and metabolism.30,31 Dysregulation of the apoptotic signal-regulating kinase 1 (ASK1)/p38 mitogen-activated protein kinase (MAPK) pathway and the abnormal activation of key signaling molecule-Ca2+/calmodulin (CaM)-dependent protein kinase II (CAMKII) in synaptic plasticity contributes to AD progression by impairing the long-term potentiation, triggering inflammation and cell apoptosis.32 Phosphorylation of α-syn at the Ser129 site is regulated by multiple kinases such as casein kinase II (CKII) and polo-like kinase (PLK),33,34 leading to abnormal aggregation of α-syn in Lewy-bodies of PD patients. Furthermore, an imbalance in the receptor-interacting Ser/Thr-protein kinase-1 (RIPK1),35 receptor of activated protein kinase C1,36 and leucine-rich repeat kinase 2 (LRRK2)37 kinase signaling cascades exacerbates the course of both PD and ALS.
Here, we summarize the extensive evidence linking protein kinases with neurodegenerative diseases, and the current progress of kinase inhibitors in clinical trials targeting neurodegenerative diseases. We also discussed the challenges and future directions of kinase-targeted therapeutic strategies for clinical applications. Taken together, understanding the mechanisms of how protein kinases participate in neurodegeneration still holds great promise and substantial opportunities for future kinase-directed drug development.
Definition and classification of protein kinases
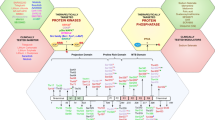
There are 478 typical (containing a eukaryotic protein kinase domain) kinases and 40 atypical kinases (exhibiting kinase activity without the eukaryotic protein kinase domain) identified to date. Based on the sequence similarity of protein kinase domains, the typical kinases can be further classified into eight groups,38,39 including the tyrosine kinases (TK) group, tyrosine kinase-like kinases (TKL) group, containing cyclin-dependent kinase (CDK), MAPK, glycogen synthase kinase (GSK), cell division cycle (CDC)-like kinase (CLK) families) (CMGC) group, homologs of yeast Sterile 7, Sterile 11, Sterile 20 kinases (STE) group, containing cAMP-dependent protein kinase (PKA), cGMP-dependent protein kinase (PKG), protein kinase C (PKC) families (AGC) group, calmodulin-dependent protein kinases (CAMK) group, casein kinase 1 (CK1) group (a small group of Ser/Thr protein kinases), and the other group. Additionally, 32 of 40 atypical protein kinases have also been identified as atypical protein kinases group (containing activity of bc1 complex kinase, alpha-protein kinase, bromodomain protein kinase, pyruvate dehydrogenase kinase, phosphoinositide-3-kinase-related kinase, right open kinase, transcription intermediary factor 1 kinase, etc), where the remaining atypical protein kinases have been further classified as protein kinase-like since they share the same structural fold as eukaryotic protein kinase.40 The kinase classification38 is illustrated in Fig. 2 and discussed in detail below.

Kinase classification map. Based on the similarity of kinase domain sequences, 518 human protein kinases were divided into nine groups: tyrosine kinase group (TK group), tyrosine kinase-like group (TKL group), homologs of yeast Sterile 7, Sterile 11, Sterile 20 kinases group (STE group), containing PKA, PKG, PKC families group (AGC group), calcium/calmodulin-dependent protein kinase group (CAMK group), containing CDK, MAPK, GSK3, CLK families group (CMGC group), casein kinase 1 group (CK1 group), other group, atypical protein kinase group. Proteins of pathological significance for neurodegenerative diseases were highlighted in the figure using different colors as indicated in the figure. Figures generated using KinMap (http://kinhub.org/kinmap/index.html), and illustrations reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com)
TK group
Tyrosine kinases catalyze the transfer of phosphate groups from ATP to the tyrosine residues of substrates. In the TK group, 58 kinases are receptor tyrosine kinases (RTKs) that can be divided into 20 families, composed of transmembrane receptors carrying tyrosine kinase domains in their intracellular segments; the other 32 kinases are non-receptor tyrosine kinases (non-RTKs) that can be divided into 10 families and they are consistently located in the cytoplasm.41,42
Growth factor receptors such as platelet-derived growth factor receptor (PDGFR), epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGFR), and fibroblast growth factor receptor (FGFR), along with tyrosine kinase receptor B (TrkB) and insulin-like growth factor 1 (IGF-1R), all fall within the category of RTKs. Activation of these receptors by corresponding cytokines, growth factors, and hormones induces auto-phosphorylation of intracellular auto-receptor tyrosine residues, leading to further amplification of the kinase activity and exposure of the docking sites for tyrosine phosphorylation which allows its recognition by cytoplasmic proteins containing Src homology 2 domain (SH2) or phosphotyrosine-binding (PTB) domains, and eventually triggering the activation of various downstream signaling cascades43 (Fig. 3). PI3K/AKT and Ras/Raf extracellular regulated protein kinases 1/2 (ERK1/2) pathways, typical signaling cascades activated by RTKs, are responsible for the regulation of cell cycle, cell survival, and cell proliferation. For instance, brain-derived neurotrophic factor (BDNF) improves the neuronal survival, plasticity, and function by activating the TrkB-PI3K/AKT signaling,44 and activation of IGF1-MEK/ERK and IGF1-PI3K/AKT signal transduction cascades modulates neurogenesis in the brain.45,46

Typical RTKs-activated signaling cascades. Activation of the receptors (including growth factor receptors such as platelet-derived growth factor receptor (PDGFR), epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGFR), and fibroblast growth factor receptor (FGFR), as well as tyrosine kinase receptor B (TrkB) and insulin-like growth factor 1 (IGF-1R)) by the corresponding cytokines, growth factors, and hormones induces autophosphorylation of their receptor tyrosine residues within the cell, thereby further amplifying the kinase activity, exposing the tyrosine phosphorylation docking sites, allowing them to be recognized by cytoplasmic proteins with Src homology 2 domain (SH2) or phosphotyrosine-binding (PTB) domains. Activated RTKs are able to recruit various signaling molecules and initiate downstream pathways. One major pathway involves the activation of phosphatidylinositol-3 kinase (PI3K), which converts PIP2 to PIP3, thereby activating AKT. Another pathway involves phospholipase C gamma (PLCγ), which, upon phosphorylation, stimulates protein kinase C (PKC) activity and mobilizes intracellular calcium (Ca2+). Concurrently, the RTK signaling cascade engages the Ras-Raf-MEK-ERK pathway via GRB2. Collectively, these pathways regulate diverse cellular processes, including cell cycle progression, proliferation, differentiation, survival, migration, metabolism, and other key functions. This figure was created with BioRender.com
Typical non-RTKs consist of the Src family (including Src, Fyn, Lyn, Lck), the Abelson tyrosine kinase (Abl) family (including Abl1, Abl2), and the Janus kinase (JAK) family (including JAK1, JAK2, JAK3, TYK2). Structurally, non-RTKs have a variable number of protein domains (e.g., SH2 or SH3 domains responsible for binding to other signaling molecules) in addition to the conserved kinase domains.41,42 Representative diagram of the non-RTK signal transduction pathway is depicted in Fig. 4. Src and Fyn are ubiquitously expressed in various tissues, while Lyn and Lck expressions are more tissue-specific in the nerves, liver, adipose, and lymphoid tissue. Functionally, Src family kinases often act as signaling mediators, for example, Src phosphorylates and upregulates N-methyl-D-aspartate receptors (NMDAR) functioning at postsynapses, resulting in an aberrant influx of Ca2+ that eventually leads to neuronal death in the brain.47,48,49,50 Fyn regulates excitatory or inhibitory neurotransmission by interacting with various effector proteins such as CDK5, tau, Dab1, mGluR1, and GluN2B, and Fyn-related modulations are tightly associated with learning and memory processes (reviewed elsewhere51). Fyn is highly similar to other Src members, and the typical motifs of Fyn consist of SH4, SH3, SH2, and SH1 domains, with the catalytic SH1 domain showing TK activity highly conserved among Src family members.51,52 The phosphorylation status of Tyr420 and Tyr531 in Fyn, by tyrosine kinases and phosphatase, are critical for the precise regulation of its kinase activity. Both Csk-mediated phosphorylation of Fyn at Tyr531 and striatal enriched phosphatase (STEP) mediated dephosphorylation of Fyn at Tyr420 lead to Fyn inactivation.53,54 Conversely, the dephosphorylated Tyr531 epitope allows the opening of the inactive conformation, enabling the exposure of SH2 and SH3 domains for protein interaction, followed by full activation of the catalytic loop in the SH1 domain.

Typical non-RTKs activated signaling cascades. In addition to the conserved kinase domain, non-RTKs also have a variable number of protein domains (e.g., SH2 or SH3 domains responsible for binding to other signaling molecules). Typical non-RTKs consist of the Src family (including Src, Fyn, Lyn, Lck), the Abelson tyrosine kinase (Abl) family (including Abl1, Abl2), and the Janus kinase (JAK) family (including JAK1, JAK2, JAK3, TYK2). The Src-mediated Ras-Raf-MEK pathway leads to transcriptional regulation in the nucleus, impacting cellular functions. Simultaneously, Fyn activates cyclin-dependent kinase 5 (CDK5), which modifies tau protein to facilitate microtubule remodeling. The activation of the JAK-STAT pathway allows STATs to translocate to the nucleus and directly regulate gene transcription. The regulation of non-RTK signal transduction pathways is closely related to synaptic function remodeling, neuronal excitability regulation, immune regulation, cell proliferation, etc. This figure was created with BioRender.com
Abl localizes in neuronal axons of the central nervous system (CNS) and regulates the axonal growth. In Drosophila melanogaster, an impaired signaling network mediated by Abl mutation interrupts the dynamics of actin cytoskeleton in the neuronal growth cones.55 c-Abl has been shown to phosphorylate tau protein at Tyr394, and both Fyn and c-Abl are critical regulators in the neurodegenerations involving tau lesions.56 The JAK phosphorylates its substrate, signal transducer and activator of transcription (STAT), causing STAT to dimerize, which then translocates into the nucleus and triggers the expression of target genes, thereby promoting cytokine-mediated cellular activation. The failure of this signaling pathway may disrupt the normal immune responses and induce pathological effects in diseases.57,58,59,60 Different from JAK3 which is expressed exclusively in the bone marrow and lymphatic system as well as endothelial cells and vascular smooth muscle cells, other JAK family members are expressed in almost all tissues.
TKL group
TKL generally lack the TK-specific motifs of the TK group, but they share similar amino acid sequences with TKs, such as the catalytic motif (His-Arg-Asp) in the kinase domain, and act as a Ser/Thr kinase in biochemical processes.61 LRRK2 belongs to the TKL group with GTPase and Ser/Thr kinase activities.62 Multi-domains in LRRK2 contain an armadillo repeat motif (ARM), an ankyrin repeat (ANK), a leucine-rich repeat (LRR), a Ras-of-complex (ROC)-C-terminal of Roc (COR) domain, a Ser/Thr kinase (KIN) domain and a WD40 domain. The ROC-COR and KIN domain are responsible for the GTPase activity and kinase activity, respectively. Mutations at specific sites within these domains may alter cellular processes such as vesicle trafficking, cytoskeletal dynamics, lysosomal function, and microglial responses.37 LRRK2 forms a monomer or dimer under physiological conditions, and generates a filamentous structure through microtubule-dependent aggregation in a pathological state. Recently, the cryo-EM structure of full-length human LRRK2 shows that the monomer adopts an elongated conformation similar to the letter ‘J’.63 The interaction of LRRK2 and Dlp1/ dynamin-related protein 1 regulates mitochondrial dynamics through the kinase activity of LRRK2 in neurons,64 and its activation promotes mitochondrial fragmentation in microglia.65,66
RIPK1, a key mediator of apoptotic and necrotic cell death and inflammatory pathways, belongs to the TLK group. Structurally, RIPK1 consists of an N-terminal kinase domain, an intermediate domain, and a C-terminal death domain (DD). The intermediate domain contains a receptor-interacting protein homotypic interaction motif (RHIM), which mediates the formation of amyloid proteins, and is involved in the interaction of RIPK1 with other RHIM-containing proteins such as RIPK3, TIR-domain-containing adaptor-inducing IFNβ, and Z-DNA binding protein 1.67 The C-terminal DD interacts with other proteins containing C-terminal DD (such as tumor necrosis factor (TNF)-α receptor 1 and FAS-associated death domain protein) to mediate their homodimerization, thereby promoting the activation of the N-terminal kinase domain.68 In the TNF signaling-mediated apoptosis and necroptosis pathway, RIPK1 assembles with TNFR1-associated death domain protein, TNFR-associated factor 2, and multiple E3 ubiquitin ligases (cellular inhibitor of apoptosis protein 1/2, linear ubiquitin chain assembly complex) into a large receptor-bound signaling complex I, mediating the first step of TNF signaling.69 When downstream nuclear factor-kappaB (NF-κB) activation is inhibited, a cytoplasmic complex called complex IIa can be formed, which mediates caspase-8 activation, RIPK1 cleavage, and RIPK1-independent apoptosis. While caspase-8 activation is blocked, RIPK1 C-terminal DD dimerization leads to RIPK1 activation and the formation of complex IIb (including FAS-associated death domain protein, caspase-8, RIPK1, RIPK3, and mixed lineage kinase domain-like, of which RIPK1 activity is required for the formation of complex IIb).70 Complex II is a downstream mediator after the first step of TNF signaling. The transmission of its downstream signal is regulated by the activity of caspase-8 and RIPK3, leading to apoptosis, necroptosis, and increased expression of inflammatory genes.70 Deletion and kinase-inactivating knock-in mutations of the RIPK3 (including D138N, K45A, K584R, D161N) exhibit resistance to inflammatory and neurodegenerative processes.68,71,72,73,74
Dual leucine zipper kinase (DLK) is a Ser/Thr protein kinase in the TLK group. The number of amino acids that make up mouse and human DLK is 888 and 859, respectively. DLK has four characteristic domains: kinase domain, leucine zipper domain, glycine-serine-proline rich domain, and glycine-proline rich domain.75 The human DLK kinase domain consists of 127-375 amino acid residues, and its activity is regulated by the dimerization of DLK mediated by the leucine zipper chain domain.76 DLK binds to the c-Jun N-terminal kinase (JNK) interacting protein (JIP) through its N-terminal region, activating transcription factors downstream of the mitogen-activated protein kinase kinase 7/JNK signaling pathway (including STAT3, transcription activator 2, and myocyte-specific enhancer factor 2A, etc.), causing axon regeneration to respond to cell death signals caused by axon damage.77,78 DLK maintains the establishment of axonal bundles originating from pyramidal neurons in the cerebral neocortex, and mice deficient in DLK exhibit defects in axonal growth and neuronal migration.79
Interleukin-1 receptor associated kinase-M, specifically expressed in microglia,80 downregulates the toll-like receptor 4-myeloid differentiation primary response gene 88 signaling, which leads to the differentiation of microglia to a neuroprotective and anti-inflammatory M-phenotype, ultimately preventing the pathogenesis of experimental autoimmune encephalitis.81,82 Furthermore, two transmembrane Ser/Thr kinase receptors, Transforming growth factor-β receptor 1 and 2, transmit transforming growth factor-β signaling to intracellular mediators and contribute to neurogenesis, dentin regeneration, and carcinogenesis.83,84,85,86
CMGC group
Major kinases in the CMGC group include CDK, MAPK, glycogen synthase kinase 3 (GSK3), and CLK. Among them, CDKs and MAPKs are two of the largest and most well-studied CMGC kinases.87
CDKs are originally identified to regulate the cell cycle. In mammals, the CDK family can be divided into 2 categories, functionally as cell cycle-related CDKs (e.g., CDK1, CDK4, and CDK5), and transcriptional CDKs (CDK7, CDK8, CDK9, CDK11, and CDK20). Within the first category, CDK1, CDK2, and CDK4/6 are located in the nucleus and binds with CycA/E, CycA/B, and CycD, respectively, to regulate the transformation of cell cycle stages (Fig. 5). However, CDK5 located in the cytoplasm, is mainly active in post-mitotic neurons,88 and participates in neuronal differentiation, migration, synaptic function, and memory consolidation. CDK5 affects synaptic plasticity and memory formation by directly phosphorylating relevant substrates and interacting proteins. For instance, postsynaptic density protein-95 (PSD95), NMDAR, dopamine and adenosine 3’5’-monophosphate-regulated phospho-protein 32 kDa, and dopamine D2 receptors are all substrates of CDK5 in the postsynaptic compartment.89,90,91,92 CDK5 can also regulate protein phosphatases PP1, TrkB (BDNF receptor), and PKA (reviewed elsewhere.93). Furthermore, CDK5 regulates the expression of receptor tyrosine-protein kinase erbB-3 and postsynaptic acetylcholine receptor by phosphorylating STAT3 at Ser727, thereby negatively regulating the formation of neuromuscular synapses.94,95 Under physiological conditions, CDK5 is also shown to maintain survival signals by regulating PI3K/AKT activity, and CDK5/p35 blocks neuronal apoptosis by inducing Bcl-2 expression through ERK activiation.96

CDK family and functions. In mammals, the CDK family can be divided into two categories according to their functions: cell cycle-related CDKs (such as CDK1, CDK4, and CDK5) and transcription-related CDKs (CDK7, CDK8, CDK9, CDK11, and CDK20). In the first category, CDK1, CDK2, and CDK4/6 are located in the cell nucleus and combine with CycA/E, CycA/B, and CycD, respectively, to regulate the transformation of different cell cycle stages; while CDK5 is located in the cytoplasm in cells, is mainly active in post-mitotic neurons, and participates in neuronal differentiation, migration, synaptic function, and memory consolidation. Unlike classical CDKs, CDK5 is not activated by cyclins. Instead, it is primarily activated by its neuron-specific cofactor, p35, a regulatory protein that binds to CDK5 and induces a conformational change, enabling its catalytic activity, while cleavage of p35 to p25 under pathological conditions such as oxidative stress, calcium dysregulation, or neurotoxic insults, results in the overactivation of CDK5, driving neurotoxic processes. This figure was created with BioRender.com
All transcriptional CDKs, including CDK7, CDK8, CDK9, CDK11, and CDK20, are located in the nucleus. Among them, CDK7 and CDK9 bind to CycH and Cyc7 respectively to directly phosphorylate the C-terminal domain of RNA polymerase II, thereby regulating the transcription process involving CDK8-mediated complex. CDK11 binds to CycL to control transcription by modulating the phosphorylation of hormone receptors and associated regulators or splicing factors.97 In a recent study, CDK20 has been reported to regulate the Wnt and Keap1-Nrf2 signaling pathways to facilitate cell proliferation.98
MAPKs regulate diverse cellular processes and are broadly involved in cell fate determinations across all eukaryotic phyla. MAPK family members, including ERK, JNK, and p38 MAPK, regulate cell proliferation, differentiation, and apoptosis.99 The JNK signaling pathway regulates axonal regeneration, nervous system development, and neuronal degeneration after acute injury or in chronic neurodegenerative diseases.100 Specifically, in the nervous system, negative regulation of MAPK by enhanced activity of MAP kinase phosphatase 1 (MKP-1, also known as dual-specificity phosphatase 1) is neuroprotective,101 and inhibition of p38α/β-MAPK activity reduces the number of degenerated neurons in the brain with improved cognitive function.102
GSK3 comprises two isoforms α and β, which share about 85% amino acid sequence homology in humans, and these two isoforms adopt similar secondary structures.103 GSK3α is only significantly expressed in the cortex, hippocampus, striatum, and cerebellum,104 whereas GSK3β is uniformly expressed in all brain regions. In physiological processes, GSK3α is localized in the cytoplasm, while truncated GSK3α lacking the N-terminal region accumulates in the nucleus. In contrast, GSK3β is more likely to be localized in the nucleus, especially in the context of cell proliferation and apoptosis.105 Functionally, GSK3α is generally associated with lifespan, mental state, behavior, and lipid metabolism, while GSK3β plays a crucial role in promoting neural development, the formation of neuronal polarity, and the maintenance of brain structure and function.105 Hence, the expression of GSK3α/GSK3β is strictly regulated in the spatiotemporal sequence. As a protein kinase, GSK3 can phosphorylate almost all downstream proteins with the S/T-X-X-X-S/T(P) motif, and is also dynamically regulated by multiple kinases including AKT, PKA, and PKC.106 Studies have confirmed that excessive activity of GSK3 is strongly correlated with neurodegenerative diseases such as AD, as described in detail below.
CLK family, relatively less studied, includes dual-specificity Tyr-regulated kinases (Dyrks) and Ser-Arg protein kinases,87 of which Dyrk1A functions in both of the cytoplasm and nucleus, interacts with histone acetyltransferase p300/CBP, and contributes to mental retardation and microcephaly.107 A global proteomic analysis of the human CMGC kinome complex provides extensive insights into resources and approaches for the analysis of CMGC kinases and human diseases,87 with information detail also available in the IntAct database (accession number: IM-17935, http://www.ebi.ac.uk/intact/).
STE group
STE kinase group consists of three main families, Sterile 7 (Ste7, also known as MAP2K), Sterile 11 (Ste11, also known as MAP3K), and Sterile 20 (Ste20, also known as MAP4K). After being activated sequentially, they activate the MAPK family (Fig. 6). The MAP2Ks directly phosphorylate MAPKs. In a typical MAPK cascade, MAP3K activates MAP2K by phosphorylating two conserved Ser/Thr residues in the activation loop, while MAP4K acts on MAP3K.108

MAP3K-MAP2K-MAPK signaling pathway. In a typical MAPK cascade, MAP3K activates MAP2K (also known as MKK, MEK) by phosphorylating two conserved Ser/Thr residues in the activation loop, and the MAP2Ks directly phosphorylate MAPKs. The activation of MAPK cascades is initiated by various extracellular stimuli, including growth factors, G-protein-coupled receptor (GPCR) signaling, stress, and cytokines. Activated Ras recruits and activates Raf, which phosphorylates and activates downstream MEK1/2. MEK1/2 then phosphorylates ERK1/2, which translocates to the nucleus and regulates gene transcription. Stress signals (e.g., reactive oxygen species or osmotic stress) and cytokines activate distinct MAPKKKs, leading to the phosphorylation of different MAPKKs. MKK4/7 phosphorylates and activates JNK/p38 MAPK, while MKK3/6 phosphorylates p38 MAPK. The hierarchical organization of the MAPK pathways ensures signal specificity, playing a critical role in cell apoptosis, cell survival, and other cellular events. This figure was created with BioRender.com
In neurons, MAP4Ks serve as critical regulators of the DLK/JNK signaling by phosphorylating DLK, an axonal stress-responsive MAP3K, followed by translocation of JNK-dependent c-Jun signaling complex to the nucleus in response to stress.100 However, JNK is not the primary target for MAP4Ks in immune cells, and the latter regulates immune responses, related signaling, and inflammation activation through other targets.109 Indeed, activation of MAP3K/DLK leads to rapid cell death, whereas activation of MAP3K/leucine-zipper-bearing kinase (LZK) causes slow degeneration in cerebellar Purkinje cells in mouse models with conditional knockout or overexpression of DLK or LZK, and these two MAP3Ks independently induce JNK activation and caspase-mediated apoptosis. Therefore, precise control of DLK and LZK activation is essential for neuronal survival.110 Compounds targeting the MAP4K family exert neuroprotective effects, as suggested by that an exceptionally potent, blood-brain barrier (BBB)-penetrant and metabolically stable compound, prostetin/12k, has been screened out and demonstrates the potential to treat ALS.111
AGC group
In the human genome, more than 63 protein kinases share the AGC group features, including 20 Ser/Thr protein kinases such as PKA, 3-phosphoinositide-dependent protein kinase 1 (PDK1), AKT, and Rho-associated coiled-coil containing kinase (ROCK), which can be divided into 14 families. Two other families, aurora kinase and PLK are most closely related to the AGC group.112,113
PKA is composed of four subunits, two regulatory subunits (types I and II) that bind to two catalytic subunits (α and β) to modulate a variety of cAMP-dependent cellular responses, such as pro-survival gene transcription, neuronal differentiation, and synaptic plasticity.114,115 The PKA catalytic subunit domain consists of a small N-terminal lobe containing a 5-strand β-sheet and an αC helix, and a large C-terminal lobe mainly composed of α-helix (AGC kinases usually have a second helix αB adjacent to the αC helix).116,117 Between the two lobes, a connected deep pocket serves as the ATP binding site,118 which is one of the major targets for drug development.113 These structural conformations represent a common model for understanding the structures of the entire superfamily. Moreover, AGC kinases have two regulatory phosphorylation sites (hydrophobic motif and turn motif/zipper phosphorylation site, respectively) in addition to the activation loop that is shared by the diverse kinase groups.113
As a superfamily widely distributed, kinases in the AGC group broadly regulate physiological processes.119,120,121,122 Knockin of PDK1K465E/K465 in mouse neurons causes inadequate phosphorylation of Thr308 of AKT (PDK1 substrate), incomplete phosphorylation and inactivation of proline-rich AKT substrate of 40 kDa and tuberous sclerosis complex 2, and reduced activation of the mechanistic target of rapamycin complex 1 (mTORC1), followed by declined protein synthesis of brain-specific kinase and insufficient neuronal differentiation, giving rise to reduced brain size in mouse.123 Mutations in the docking site of AKT-independent PDK1 substrate cause microcephaly and abnormal brain morphogenesis in the developing mouse brain, leading to cognitive impairment and disruptive behavior in adult mice.124 Furthermore, in cultured primary rat hippocampal neurons, increased ROCK2 induces dendritic spine loss via the serine and threonine kinase LIM domain kinase 1 whereas administration of SR7826, an inhibitor of LIM domain kinase 1, rescues ROCK2-mediated dendritic spines loss and morphological distortion.125
CAMK group
CaMKs are critical Ca2+ sensors that convert glutamatergic activation into synaptic plasticity during the formation of learning and memory.126 CaMKII is one of the Ca2+/CaM-regulated kinases and is evolutionarily closer to the phosphorylase kinase, whose intrinsic regulatory δ-subunit is recognized to be CaM.127,128 Originally, CaMKI-IV were named according to the elution order of brain extract separated with a fractionating column.129,130 Later studies indicated that this nomenclature was not rational, i.e., CaMKI/IV and CaMKII were members of related sister groups, but CaMKIII (known as eukaryotic elongation factor 2 kinase, eEF2) is now classified as atypical kinase instead of CaMKs.3,130 Thus, although the role of CaM regulation seems to be the shared feature of the CaMK family, not all CaM-regulated kinases are CaMKs. For example, death-associated protein kinase (DAPK) 1 and 2 (DAPK2, also known as DRP-1) can be activated by CaM, but DAPK3 (also known as zipper-interacting protein kinase, ZIPK) cannot respond to CaM due to a lack of CaM binding site.130,131 In addition, the CaMK subfamily that lacks CaM regulation also includes AMPKs and 90 kDa ribosomal S6 kinases.130 As a key regulator of cellular energy homeostasis, AMPK regulates a diverse range of physiopathological processes, especially in the homeostasis of mitochondrial function and autophagy.132,133,134 ATP/Ca2+ regulates liver kinase B1 phosphorylates AMPKα subunit at Thr172 and activates the main metabolic AMPK signaling, thereby exhibiting a protective effect on neural energy metabolism and autophagic degradation.135,136
As a result of the enriched presence of CaM in the synapses, the influx of Ca2+ through NMDA receptors leads to the formation of Ca2+/CaM complexes that activate CaMKs, causing induction of persistent synaptic plasticity via calcium signaling.126,137,138 CaMKII is profoundly abundant in the brain and has a remarkable biochemical profile as a multifunctional kinase.126,139 Among the 12 subunits of CaMKII, αCaMKII, and βCaMKII are the most abundant subunits in the brain, and the former is exclusively expressed in glutamatergic neurons, while the latter is present in both excitatory and inhibitory neurons.140 Upon Ca2+/CaM binding, attachment of αCaMKII to F-actin is attenuated, which dissociates CaMKII from F-actin and modulates actin polymerization to form synaptic morphologies.126,141 The αCaMKII-dependent CaMKII function is illustrated by that autophosphorylated αCaMKII at Thr286 prolongs the activity of CaMKII at synapses after Ca2+ stimulation, leading to the transport of glutamate receptors to the postsynaptic density and subsequent enhanced synaptic transmission.138,142 Therefore, Ca2+/CaMKII contributes to synaptic transmission and is required for long-term potentiation (LTP) maintenance.143 Furthermore, as a Ca2+/CaM-dependent Ser/Thr kinase, DAPK1 overexpression and phosphorylation at Thr231, Ser262, and Ser396 sites are implicated in various neurological disorders, such as AD,144,145 PD,146 and stroke.147
CK1 group
CK1 is so named because of its ability to phosphorylate milk protein casein in vitro, and the CK1 group includes the CK1 isoform, vaccinia-related kinase, and tau tubulin kinase 1/2 members.3,148,149 To date, seven mammalian CK1 isoforms have been grouped, including α, α-like, γ1, γ2, γ3, δ, and ε, due to the high homology among their N-terminal kinase domains.150 The α, α-like, δ, and ε isoforms have higher sequence similarity in the kinase domain than the γ isoform.151,152 Furthermore, CK1 isoform distribution is specific in organs and cells.153,154 For example, both CK1δ and CK1ε are mainly expressed in the brain.155 Activation of these two isoforms is regulated by the inhibitory autophosphorylation in the C-terminal region,156 and is associated with brain activities such as circadian rhythm,157 dopamine signaling,158 and neurotransmission.159 CK1 phosphorylates β-catenin at Ser45 and primes subsequent sequential phosphorylation at Thr41, Ser37, and Ser33 by GSK3.160,161,162 CK1 isoforms regulate the Wnt pathway via antagonistic roles in the signaling cascade,150 as well as p53 signaling,163 Hippo signaling,164,165 and Hedgehog signaling.166,167 Reduced phosphorylation of LRRK2 mediated by CK1 triggers the degradation of LRRK2, thereby disrupting the LRRK2 homeostasis.168 Both tau tubulin kinase (TTBK) 1 and TTBK2 belong to the CK1 superfamily, and can phosphorylate microtubule-associated proteins at 10 different residues to regulate neuronal function.3,169,170
Other groups
Within this group, nearly all members are Ser/Thr kinases with distinct sequence homology, and almost all are involved in cell division. This group consists of 30 families (such as Aurora, PLK, cell division cycle 7, never in mitosis gene A (NIMA)-related kinase (NEK), CaM-dependent protein kinase kinase (CAMKK), IkappaB kinase (IKK), TBC1-domain containing kinase) and 2 subfamilies (including general control nonrepressible 2 and pancreatic eIF2alpha kinase), and many of which involved in neuronal processes will be discussed below.
The NEK family consists of 11 kinases named NEK1 to NEK11. Among them, NEK2/6/7/9 promotes the establishment of a microtubule-based mitotic spindle, while NEK1/10/11 is related to DNA damage response.171 All 11 human NEKs contain a His-Arg-Asp (HRD) motif within the catalytic domain as well as sites for activation modification at Ser or Thr residues within the activation loop. Compared with the conserved catalytic domain, the C-terminal regions of the 11 NEK species differ greatly in length, sequence, and domain organization.172 The diversities in these domains or motifs may explain the selectivity of NEKs during the cell cycle progression and differentiation processes.
The IKK family includes typical and atypical IKK kinases. The typical IKK includes IKKα, IKKβ, and IKKγ (also known as NEMO), and atypical IKK includes IKKε and TANK-binding kinase 1 (TBK1). The typical IKKα-IKKβ-IKKγ complex serves as the signal integration center for NF-κBactivation and consists of two Ser/Thr kinases (IKKα and IKKβ) and the regulatory subunit IKKγ. There is about 50% high sequence homology between IKKα and IKKβ, both of which contain an N-terminal kinase domain, dimerization domain, and C-terminal IKKγ binding domain (NBD). The kinase activity of IKKα mainly depends on the phosphorylation of Lys44/Ser176/Ser180, while IKKβ activation mainly depends on the phosphorylation of Ser177/181.173 The complex catalyzes the phosphorylation of inhibitor-κB and p65 proteins and other substrates, and mediates downstream immune and inflammatory responses, cell proliferation and differentiation, autophagy and apoptosis physiological processes.174 Dysregulation of these physiological functions is associated with a variety of diseases (such as cancer, neurodegenerative diseases, and heart disease).
The structure of atypical IKK, TBK1, includes an N-terminal kinase domain (KD), ubiquitin-like domain (ULD), α-helical scaffold dimerization domain (SDD) and C-terminal adapter binding domain (CTD). The phosphorylation site of TBK1 is Ser172 on the KD activation loop,175,176 and TBK1 controls selective autophagy of damaged mitochondria by phosphorylating sequestosome 1 (p62) and optineurin (OPTN) autophagy receptors, while mutations in TBK1 lead to impairment of the selective autophagy pathway in protein aggregation.177 In the innate immune system, activation of TBK1 promotes the release of type I interferon (IFN) through the nuclear translocation of phosphorylated IFN regulatory factors 3 and 7.178 TBK1 is also involved in the mitosis of cancer cells to regulate cell survival by mediating PLK1 phosphorylation.179
CAMKK is the upstream kinase of CAMK and is responsible for CAMK activation. CAMKK belongs to the other group since its sequence differs from the CAMK family, however, both CAMKKα and CAMKKβ are highly homologous and compatible. They catalyze the phosphorylation of Thr177 of CAMKI and Thr196 of CAMKIV, activating CAMKI and CAMKII to regulate cell energy metabolism, proliferation, differentiation, and survival.180
Kinases in neurodegenerative diseases
AD
Dementia is an age-related, progressive, and irreversible neurodegenerative disorder, and is characterized by cognitive and memory impairment, compromised executive function, and difficulties in performing daily activities.181,182 AD is the most common form of dementia in the elderly, with more than 50 million people suffering from AD or AD-related dementia worldwide.181 To date, the U.S. Food and Drug Administration (FDA) has approved a variety of prescription medications for the relief of AD symptoms, including acetylcholinesterase (AchE) inhibitors (donepezil, galantamine, rivastigmine),183 NMDAR antagonists (memantine),184 and anti-Aβ antibodies (Aduhelm, Leqembi) that were recently approved through accelerated approval in 2021185 and 2023.9 In addition, Donanemab, developed by Eli Lilly, is also a monoclonal antibody that binds to Aβ subtype N3pG.186 and is approved by the FDA in 2024. Although these drugs may relieve the cognitive and behavioral symptoms in AD patients, they do not cure the disease. Therefore, an in-depth investigation of AD pathogenesis will be of great significance for targeted drug development.
Pathological features of AD include neurofibrillary tangles (NFTs, composed by tau aggregation), extracellular senile plaques (SPs, composed by Aβ aggregation), gliosis, and dystrophic neurites, accompanied by cerebrovascular amyloidosis, neuronal loss, metal dysregulation, and synaptic alterations.187,188,189,190,191,192,193,194 Aβ is generated by the amyloidogenic processing of amyloid precursor protein (APP).195 The cleavage of APP involves three proteolytic secretases including α-secretase (ADAM9, ADAM10, ADAM17), β-secretase (BACE1/2), and γ-secretase (composed of at least four core components including presenilins1 and 2, nicastrin, anterior pharynx defective 1, and presenilin enhancer 2).195 In the amyloidogenic pathway, APP is initially cleaved by BACE1 to release the sAPPβ ectodomain, and the 99 amino acids C-terminus of APP (C99) is further cleaved at various sites by γ-secretase,195 resulting in Aβ peptides in different length (including Aβ37-43), which tend to accumulate in the AD brain. Aβ40 and Aβ42 are the two major Aβ species, but neurotoxic Aβ42 is the main component of amyloid plaques.196 In addition to Aβ plaque neuropathology, several other neurochemical abnormalities, including elemental signatures of iron, copper, zinc, and selenium, have been widely validated in AD brains.197
Tau undergoes a series of post-translational modifications including hyperphosphorylation, acetylation, carboxy-terminal truncation, O-GlcNAcylation, and N-glycosylation before NFTs are formed.198,199 In AD, early hyperphosphorylation of tau disrupts the association between tau and the microtubules, prompts the mislocalization of tau from axons to the somatodendritic compartment, resulting in increasing levels of tau in the somatic domain where p-tau422 epitope turns positive,198,200 whereas other epitopes such as p-tau396 only become more prominent later in the disease.201 Various kinases such as CDK5 and CDK5 activator 1, truncated form of a CDK regulator p25,202 GSK3β, and phosphorylated JNK are upregulated in brain tissue from AD patients203,204; hyperactive GSK3β induces inflammation through NF-κB, impairs axonal transport, and promotes apoptosis,198 and these findings together suggest that increased activity of kinases may be responsible for tau pathology.
Major kinases in AD
We here summarized the major kinases that are involved in AD and the kinase regulatory network (Figs. 2 and 7). These kinases include GSK3β, CDK5, CK1, PKA, p38 MAPK, Fyn, TTBK1, and AMPK. The details of each kinase involved in AD pathogenesis will be discussed below.

Schematic description of kinase signaling pathways in Alzheimer’s disease. GSK3β, as one of the main kinases involved in tau phosphorylation, adds a phosphate group to the Thr231 site on tau. This process triggers tau oligomerization, NFTs formation, and participates in the regulation of the Nrf2-ARE pathway by phosphorylating Nrf2 Ser334-338 residues, thereby reducing the antioxidant capacity. The CDK5/p35 complex plays a critical role in maintaining synaptic function by modulating STAT3, synaptic components including PSD95 and DARPP32, ErbB3, BDNF/TrkB, or other regulators, while in AD, the CDK5/p35 is cleaved by Ca2+, Aβ, and calpain-1, giving rise to the abnormally hyperactive CDK5/p25 variant, promoting the pathway of cellular apoptosis, reentry into the cell cycle, and mitochondrial dysfunction. The phosphorylation of p38 MAPK exacerbates oxidative stress, decreases synaptic plasticity, and increases the release of inflammatory factors. Meanwhile, overactivated Fyn contributes to the phosphorylation of APP at Tyr682, leading to increased generation of intracellular Aβ. Fyn phosphorylation at Tyr416 causes cellular toxicity and imbalances in neural network function by modulating NMDAR, Pyk2, and eEF2. TTBK1 activates CDK5 and triggers downstream signaling, promoting NMDAR internalization and imbalanced degradation of the neural network. On the other hand, it triggers the phosphorylation of tau protein at Ser422 via calpain-1, exacerbating tau aggregation. In the progression of AD, the reduced activity of AMPK increases the phosphorylation level of mTOR. This, in turn, hinders autophagy processes while concurrently enhancing Aβ generation. The downregulation of PKA expression in AD pathology leads to decreased activation of both SIRT1 and CREB, increasing Aβ production and synaptic plasticity vulnerability. CK1 abnormalities not only regulate the transmission of their inherent signals but also wield regulatory influence over the downstream signaling pathways of crucial kinases such as GSK3β and CDK5. This intricate interplay between kinases forms an interconnected regulatory network that functions in AD. This figure was created with BioRender.com
GSK3β
As a ubiquitously expressed Ser/Thr kinase, GSK3β activity in the peripheral blood of AD patients is positively correlated with the degree of dementia.205 GSK3β mainly affects the pathology of AD, including Aβ formation, tau pathology, neuronal survival and apoptosis, oxidative stress, and neuroinflammation. During Aβ production, GSK3β promotes the phosphorylation of APP and aggravates the β-cleavage of APP,206,207 and also inhibits APP autophagic degradation by reducing lysosomal biogenesis, thereby increasing Aβ levels in AD animal and cell models.105 In NFT formation, GSK3β acts as one of the major tau kinases by specifically phosphorylating tau at Thr231, which accelerates tau dissociation from the microtubules, promoting tau oligomerization and NFT formation.208 GSK3β interacts with other kinases such as CDK5 to amplify the hyperphosphorylation of tau. Overactivation of GSK3β inhibits the activity of protein phosphatase 2A, resulting in the blockage of tau dephosphorylation and subsequent synaptic dysfunction.209,210 The impairment of the Wnt/β-catenin signaling pathway further aggravates the elevated GSK3β activity and promotes tau hyperphosphorylation, leading to abnormal neuronal structure and dysfunction in PS1 knockout fibroblasts from and mutant PS1 transgenic mice.211,212,213
GSK3β also inhibits BDNF-induced TrkB receptor endocytosis by directly phosphorylating mixed-lineage kinase 3 or phosphorylating dynamin1, impairing the activation of AKT signaling downstream of BDNF-TrkB, thereby reducing neuronal survival and promoting neuronal apoptosis both in vitro and in vivo.214,215 Additionally, reduced PI3K/AKT activation in AD patient brains216 increases GSK3β activity, which further phosphorylates Nrf2 at Ser sites within residues 334-338, leading to enhanced degradation of Nrf2, accumulated oxidative stress, and cognitive deterioration.217 GSK3β is also involved in AD-related neuroinflammation by mediating the phosphorylation of CCAAT/enhancer-binding protein δ to upregulate MCP-1, MMP3, and MMP1 expression in astrocytes, promoting microglia reactivity.218 Consistently, GSK3β transgenic mice exhibit hyperphosphorylated tau, increased Aβ accumulation, reactive gliosis, neuronal death, enhanced oxidative stress, and cognitive deficits, all of which can be reversed by GSK3β inhibitors.217,219,220 These results highlight GSK3β as a promising therapeutic target against AD.
CDK5
In AD, abnormally elevated CDK5 activity enhances the phosphorylation of its substrates such as APP, tau, and neurofilaments.93 Mechanistically, CDK5 is activated by its neuron-specific and membrane-localized activators p35 and p39, which are cleaved by calpain into p25 and p29, respectively. Upon increased Ca2+ concentrations, CDK5/p25 binding is more stable, leading to the hyperphosphorylation of multiple substrates of CDK5, thus promoting AD progression.93,221 Increased phosphorylation of APP (p-Thr668) by CDK5 and excessive Aβ accumulation were observed in p25 transgenic mice222; whereas Aβ aggregation in turn contributes to abnormal CDK5 activity, forming a toxic feedback loop that aggravates AD progression.223,224,225
There is abundant evidence that CDK5 upregulates GSK3β activity and promotes phosphorylation of tau and other substrates, and c-Abl tyrosine kinase is responsible for CDK5 activation in this process.226,227 CDK5/p25 enhances p53 activity by phosphorylating p53, and promoting p53-Bax induced neuronal apoptosis,93,228 a process that also involves CDK5/p25-regulated JNK3 pathway and regulation of MEF-2, a direct target of CDK5 in the nucleus.229,230,231 Dysfunction of CDK5 also causes mitochondrial dysfunction and ROS accumulation, promoting neuronal apoptosis.232,233
On the other hand, CDK5/p25 phosphorylates phosphatases such as cell division cycle 25A (cdc25A), cdc25B, and cdc25C, resulting in the upregulation of CDK1/2/4 and subsequent re-entry of the cell cycle, which ultimately leads to cell cycle-related neuronal death and degeneration.234,235 It was identified that the ubiquitination and degradation of p35 are regulated by hexokinase 2. The abnormal decrease of hexokinase 2 is conducive to the cleavage of p35 into p25, which leads to the overactivation of CDK5 and interferes with the degradation of β-catenin induced by GSK3β, thus inhibiting the activation of cell cycle machinery.236 In addition, CDK5/p25 interacts with and phosphorylates BM88 (also known as cell-cycle exit and neuronal differentiation 1), thereby promoting the degradation of BM88 and upregulating dynamin-related protein 1, leading to mitochondrial dysfunction and neuronal death in the brains of 5xFAD mice.237 Abnormal CDK5 also was reported to mediate Bcl2-associated athanogene-3 loss, leading to neuronal synaptic dysfunction in AD pathology.238
CK1
Overexpression of CK1ε in N2a cells stably expressing APP (N2A-APP695 cells) results in increased production of Aβ, and CK1 inhibitors block β-secretase cleavage of APP without affecting the Notch cleavage.153 Furthermore, in response to inflammation, CK1 is delivered from astrocytes to neurons through extracellular vesicles, which promotes translation and amyloidogenic processing of APP.239 Inhibition of the interaction between CK1δ and APP695 mitigates the pathogenic metabolism of APP.240 Expression of CK1δ is increased in the AD brain and correlated with tau pathology.241 Overexpressed CK1δ in N2a cells promoted the cytoplasmic aggregation of TDP-43 by phosphorylating Ser379, Ser403/404 and Ser409/410 residues of TDP-43, enhancing the instability and inhibiting the inclusion of exon 10 in tau mRNA.242 In addition, analysis of tau441 phosphorylation mutants showed that Ser68/Thr71, Ser214, and Ser289 of tau are specific substrates for CK1δ in vitro.243
Besides being directly involved in AD pathology, CK1 also acts as a priming kinase of other key AD-related kinases such as GSK3β.244 Pre-phosphorylation and activation of GSK3β by CK1 leads to elevated Aβ and tau hyperphosphorylation thereby aggravating AD pathology. CK1 also acts upstream of CDK5 and regulates its activity and downstream signals.245,246 Consequently, abnormal CK1 expression affects AD pathogenesis via direct CK1-associated signalings as well as other critical kinases in AD. Since current CK1 kinase inhibitors mainly take effect by targeting the ATP-binding sites, which are highly conserved among CK1 isoforms, these inhibitors are of less specificity and targeted drug design remains challenging for AD therapy.
PKA
In AD, calcium dysregulation leads to increased degradation of PKA subunits through excessive activation of calpain, which reduces PKA activity and subsequently downregulates the phosphorylation of transcription factor cAMP response element binding protein (CREB) and BDNF expression.247,248,249 Moreover, Aβ42-treatment interferes with BDNF-induced activation of other pro-survival Ser/Thr kinases such as PI3K/Akt and ERK.250
Additionally, overexpression of BACE1 interacts with cAMP at the transmembrane domain of BACE1, and downregulates cAMP levels, PKA activation, and CREB phosphorylation, thereby leading to cognitive impairment due to compromised cAMP/PKA/CREB signaling pathway in AD.251 Sirtuin 1 (SIRT1) exerts its deacetylase activity after phosphorylated by activated cAMP/PKA signaling,252 and upregulates ADAM10 and downregulates BACE1 to prevent Aβ production in APP-overexpressing cells and animal models.253 Taken together, increasing PKA activity to regulate downstream CREB and SIRT1 signaling may be of benefit for AD.
p38 MAPK
Phosphorylation of p38 MAPK is mediated by the MAP3K-MAP2K signaling cascade.254 Composed of α, β, γ, δ isoforms, p38 MAPKs are activated after dual phosphorylation of Tyr182 and Thr180 residues,254,255 resulting in adaptive responses through phosphorylation of p38-dependent kinases or transcription factors. In postmortem brain tissue of AD patients, intense phosphorylation of p38 MAPK was associated with Aβ-dependent inflammatory response and NFT formation, and the activation was shown to occur at an early stage in AD.254,256 In both glial cells and neurons, p38 MAPK-mediated signal transduction contributes to AD progression. For example, stimulation of microglia by Aβ fragments activates p38 MAPK signaling to promote the secretion of pro-inflammatory cytokines interleukin-1β and TNF-α, leading to chronic inflammation.257,258 Subsequently, interleukin-1β activates the p38 MAPK signaling pathway in neurons and astrocytes, resulting in the production of pro-inflammatory mediators such as nitric oxide (NO) and TNF-α in astrocytes.259,260 In neurons, activated p38 MAPK phosphorylates Nrf2, facilitates the interaction between Nrf2 and Keap1, and inhibits the nuclear translocation of Nrf2, leading to a decrease in ARE-dependent transcription of antioxidant enzymes.217,261
Furthermore, p38 MAPK activation in neurons alters synaptic plasticity and promotes tau phosphorylation.262,263,264 Several studies have shown that deficiency of p38α-MAPK in all myeloid cells prevents AD progression by increasing microglial clearance of Aβ and reducing pathological hyperphosphorylated tau.265,266,267 However, unlike p38α-MAPK, p38γ-MAPK is specifically localized to the post-synaptic density in neurons and mediates the phosphorylation of postsynaptic tau protein at Thr205.268,269 Phosphorylation of the Thr205 site promotes the removal of tau from the PSD95/NMDAR complex.269,270 The dissolution of the interaction between PSD95 and tau further uncouples tau from downstream factors such as Fyn and ERK, thereby inhibiting toxic signals downstream of NMDAR, such as Aβ excitotoxicity.269,271 These results suggest that p38 MAPK activation is significantly related to defective Aβ clearance, Aβ-induced neuroexcitotoxicity, and tau phosphorylation. However, specific subtypes may need to be considered when targeting p38 MAPK to alleviate AD pathology.
Fyn
Fyn is a member of the non-receptor tyrosine Src family. In AD pathogenesis, overactivation of Fyn promotes the phosphorylation of APP at Tyr682 and tau at Tyr18, leading to increased production of extracellular Aβ and intracellular NFTs formed by tau, respectively.272 Extracellular Aβ oligomer binding to cellular prion protein stimulates Fyn phosphorylation at Tyr420 and activates Fyn, triggering pathological signaling cascades and excitotoxicity caused by phosphorylation of NR2B/NMDAR and persistent activation of both eEF2 and protein tyrosine kinase 2β downstream of Fyn.273,274 In addition, hyperactivation of Fyn not only participates in tau pathology through direct interaction and phosphorylation, but also indirectly through the activation of p35/CDK5.275,276
Fyn is recruited to the postsynaptic NMDAR complex in a tau-dependent manner and mediates Aβ excitotoxicity in postsynaptic toxic signaling.269,271,277 In this recruitment pathway, tau interacts with Fyn through the proline-X-X-proline motif in its proline-rich region.271,278 Subsequently, the tau/Fyn complex interacts with the key scaffolding protein PSD95 to mediate excitotoxicity induced by Aβ or excess glutamate levels.271,279 On the contrary, phosphorylation of NR2B by Fyn at the Y1472 site can enhance the binding of PSD95 to NMDAR, thereby increasing the level of tau in the NMDAR complex.269,280 Therefore, Aβ-Fyn-tau closely contributes to AD pathogenesis, and Fyn inhibition (using the Src family kinase inhibitor, AZD0530) has been demonstrated to rescue spatial memory deficits and synaptic density loss in APP/PS1 mice, and rescue abnormalities in tau phosphorylation and deposition in 3xTg-AD mice,271,277,281,282 suggestive of Fyn as an attractive target for the treatment of AD.
TTBK1
TTBK1, a CNS-specific kinase that mediates tau phosphorylation and aggregation,170 is significantly upregulated in the frontal cortex of AD patients,283 and genetic variation in the TTBK1 gene (single nucleotide polymorphism of rs2651206) is associated with late-onset AD in two cohorts of patients in China and Spain.284,285 TTBK1 directly phosphorylates tau at Ser422, an epitope present before NFT formation.286 Consistently, brain-permeable TTBK1 inhibitors are capable of reducing tau phosphorylation and alleviating AD pathology in both the mouse isoflurane-induced hypothermia model and rat developmental model.287
In addition, elevated levels of CDK5 co-activators p35 and p25, as well as increased calpain-1 activity and p35/CDK5 activity were found in TTBK1 transgenic mice,149 suggesting that TTBK1 activates CDK5 to trigger downstream signaling cascades. Both p35/CDK5 and calpain-1 (via proteinase activity288) closely regulate the turnover of NMDAR subunit NR2B on the membrane surface, and affect hippocampal spatial learning and LTP.289,290 Therefore, enhanced TTBK1 activity is assumed to upregulate p35/CDK5 and calpain-1 activity, which causes cognitive impairment via endocytosis and degradation of NMDAR. Moreover, p25 generated from the cleavage of p35 by calpain-1 acts as a more potential activator of CDK5 and subsequently forms the p25/CDK5 complex, resulting in augmented activity compared to the p35/CDK5 complex.149,291 All these events further disrupt the homeostasis of NMDAR and the balance of neural networks.
AMPK
AMPK belongs to the CAMK family, but lacks CaM regulation because it does not directly interact with AMPK due to the lack of CaM binding site. AMPK alleviates tau phosphorylation by reducing the activity of GSK3β, and activation of the AMPK signaling pathway induces SIRT1 activity to deacetylate tau followed by degradation of misfolded tau.292,293 However, in AD, disturbance of energy metabolism moderates AMPK activity in the brain, which on one hand increases mTOR phosphorylation and inhibits autophagy294,295,296,297; on the other hand reduces SIRT1 activation, resulting in augmented Aβ production and tau phosphorylation.293,298 Aβ42 oligomers can trigger synaptic loss through CAMKK2/AMPK-dependent modulation of mitochondrial fission and mitophagy in APPSwe/Swe-knockin human embryonic stem cell lines.299 Thus, AMPK, as a master regulator of energy-related signaling pathways, extensively participates in neurodegeneration.
PD
PD, a chronic and progressive movement disorder, is the second most common neurodegenerative disease.300,301 The number of worldwide PD patients increased from ~2.5 million in 1990 to 6.1 million in 2016, and the incidence is predicted to rise to 13 million by 2040.302,303 The primary clinical symptom of PD is motor impairment, in addition to non-motor deficits including cognitive decline,304,305 depression, and pain, which greatly affect the quality of life of patients.306 The complexity of PD pathophysiology involves several functional abnormalities, such as mitochondrial, lysosomal, and synaptic dysfunctions, all of which may synergistically cause selective loss of dopaminergic neurons in the substantia nigra.307,308
The accumulation of α-syn in Lewy bodies and Lewy neurites, characterized by crowded organelles and lipid membranes, is the pathological hallmark of PD.309 Currently, inhibition of misfolded α-syn diffusion or promoting its clearance has been validated to mitigate the progression of PD.310 α-syn propagates from neuron to neuron and induces normal α-syn aggregation,311 a process that is closely related to the post-translational modifications (PTMs) of α-syn protein. Phosphorylation of α-syn occurs at 39, 87, 125, 129, 133, and 136 sites, and involves kinases such as CK, PLK, and G protein-coupled receptor kinase (GRK) families.312,313 Among all the phosphorylation sites, the pathological phosphorylation at Ser129 (pSer129) accounts for ~90% of the deposition in Lewy bodies.314,315,316,317 In vitro, α-syn phosphorylation at Ser129 leads to a higher tendency to form fibrils.318 Likewise, pSer129 α-syn induced by GRK5 facilitates α-syn aggregation in cells co-transfected with α-syn and GRK5.319
Selective loss of neurons in the SNpc is a typical pathological feature of PD comparable to the neuronal loss of the dorsal tier during normal aging.320,321 Although PD pathology involves the entire brain, specific types of neurons, especially the dopaminergic neurons of SNpc, are the most vulnerable to damage in PD. Given that the axons of SNpc dopaminergic neurons forming a huge branch network contain a higher density of mitochondria and therefore display a higher rate of oxidative phosphorylation than other neurons, these neurons will inevitably be more affected if mitochondrial dysfunction occurs.322,323 It was also found that iron and dopamine levels in the SNpc are significantly higher than in the adjacent ventral tegmental region, which is less susceptible to neuronal loss in PD.
The current first-line treatment for PD is L-3.4-dihydroxyphenylalanine (L-DOPA) supplementation, which is only a symptomatic treatment. Since the loss of dopaminergic neurons continues and the survival neurons may be exhausted by the treatment, L-DOPA works within 20 to 30 min of dosing, with maximum effects reaching about 1.5 h. Therefore, therapies for PD are expected to prevent ongoing neuronal death, and understanding the kinases involved in the pathomechanisms of PD is of particular significance.
Major kinases in PD
Approximately 3–5% of PD is attributed to monogenic PD. Mutations of SNCA (encoding α-syn), LRRK2 (encoding LRRK2), PRKN (encoding Parkin), PINK1 (encoding PINK1), and GBA genes have been identified as causal mutations of PD. For example, mutations in the SNCA cause familial PD in an autosomal dominant manner and increase the risk of sporadic PD.324,325 PD patients with SNCA mutations exhibit earlier onset age, more rapid progression of motor symptoms, and significant cognitive impairment.326
Kinases regulate α-syn during disease progression. While other causal genes encode kinases, such as LRRK2 and PINK1, and their genetic mutations directly increase susceptibility to PD. In addition, other kinase signaling pathways including MAPK and AKT are also affected in PD. Activation of JNK or p38 MAPK in PD promotes the apoptosis of neurons,327 while activation of ERK1/2 MAPK and PI3K/AKT pathways supports cell survival.328 Here, we summarize the major PD risk genes highly associated with kinases and elucidate their multifaceted contributions to PD-related disturbance of kinase signaling pathways (Figs. 2 and 8).

Schematic description of kinase signaling pathways in Parkinson’s disease. PINK1 and LRRK2 are involved in mitochondrial and lysosomal function, and their mutations lead to mitochondrial dysfunction and autophagy defects. These mutations induce typical α-syn aggregation in the form of Lewy bodies and neurites, as well as neuronal loss. The accumulation of PINK1 in the mitochondrial membrane and subsequent recruitment of Parkin can trigger the initiation of mitophagy. LRRK2 mutations failed to phosphorylate AKT and promote cell survival via inhibition of FOXO1, whereas phosphorylated JNK and p38 MAPK, promoting cell death. Activated ASK1 can also phosphorylate MAPKK and subsequent MAPK to form a cascade amplification of signaling, promoting cell death by regulating the activation of JNK and p38. Phosphorylation of α-syn occurs at several sites involving kinases such as CK, PLK, c-Abl, and GRK, contributing to its aggregation and the formation of Lewy bodies, which alters the activity of numerous kinases and triggers neuroinflammation and increased ROS. This figure was created with BioRender.com
LRRK2
The LRRK2 gene, encoding the LRRK2, is one of the most commonly mutated genes in familial PD, and its mutations are also found in sporadic PD.37 LRRK2 kinase activity in dopaminergic neurons and microglia in the SNpc is increased in patients with idiopathic PD (iPD), suggesting the involvement of LRRK2 in iPD.329 Patients with LRRK2 mutations are very similar to iPD in clinical features and treatment responses.330,331 G2019S, the most common LRRK2 mutation in PD, accounts for 4% of familial PD cases and 1% of sporadic PD cases worldwide.37 The mutation induces typical α-syn aggregation in Lewy bodies and neurites, as well as neuronal loss in specific brain regions.
LRRK2 belongs to the TKL group and is a member of the leucine-rich repeat kinase family, with a Ser/Thr kinase domain, a GTPase functional enzyme domain, and protein-protein interaction domains such as armadillo, ankyrin, and WD40. LRRK2 is mainly present in the cytoplasm, but also in the intracellular membrane and microtubules under certain conditions.332 It primarily serves as a kinase for Rab GTPases, which are the main regulators of membrane transport, autophagy, and lysosomal degradation.333 Impairments of endolysosomal function and autophagy have been observed in PD involving LRRK2 mutation.334,335,336 As LRRK2 regulates the balance between membrane repair and organelle replacement to maintain endolysosomal homeostasis, pathogenic mutations lead to membrane damage followed by disruption of endolysosomal homeostasis.337 In addition, wild-type LRRK2 can be degraded by chaperon-mediated autophagy in lysosomes, and mutations such as G2019S block such degradation pathway.335
The mechanisms relating to LRRK2 mutations in PD pathogenesis are generally associated with the kinase activity of LRRK2, inhibition of which has displayed therapeutic effects in models of PD in vitro and in vivo.338,339,340 For instance, a novel LRRK2 kinase inhibitor, FL090, ameliorates lysosomal dysfunction and loss of dopaminergic neurons in the SNpc by upregulating microtubule-associated protein 1B in both genetic and pharmacological PD animal models.341 In addition, reducing LRRK2 expression with anti-sense oligonucleotide has recently shown promising results in ameliorating α-syn inclusion formation in preclinical trials.342 Therefore, a variety of LRRK2 kinase inhibitors, including WXWH0226 (https://synapse.patsnap.com/), ARV-102 (https://www.arvinas.com/), DNL201,343 and DNL151,344 are now being tested in clinical trials for PD.345
PINK1 and Parkin
Quality control such as the structure and function of mitochondria is achieved by homeostasis of degradation-related enzymes, mitophagy, mitochondria-derived vesicles, and other manners.346 Mitochondrial dysfunction is considered a key event in both familial and sporadic PD,347 and has been widely observed in autopsy of patients with PD.348,349 Reduced mitophagy is one of the forms of mitochondrial defects in PD patients and serves as a possible mechanism of pathogenesis, largely due to the loss-of-function of the key mitophagy regulators PINK1 and Parkin.350 The cytoplasmic protein Parkin, acting with the ubiquitin kinase PINK1 (a Ser/Thr protein kinase), mediates the mitochondria-related autophagy process, namely mitophagy.351,352 When mitochondrial damage is perceived, PINK1/Parkin is the first to activate the mitochondrial quality control pathways.353 In brief, PINK1 phosphorylates ubiquitin on Ser65, and stabilizes the active conformation of Parkin, allowing the charged E2 ligases to bind, therefore the E3 ubiquitin ligase activity of Parkin is enhanced.354 The accumulation of PINK1 on the mitochondrial membrane and subsequent recruitment of Parkin triggers the initiation of mitophagy.355
PINK1 and PRKN mutations are the main causes of autosomal recessive PD, and dysfunctions of these proteins also contribute to early-onset PD.356 As for clinical manifestations, although the onset is relatively early, it shows slow progression, and cognition is rarely affected in patients with PINK1 or PRKN mutations.357,358 The G309D and L347P mutations of PINK1 result in a significantly reduced PINK1 activity,359,360 leading to the loss of its anti-apoptotic function by suppressing the release of cytochrome c from mitochondria.361 The accumulation of pathogenic substrates, such as aminoacyl-tRNA synthetase complex interacting multifunctional protein-2 (AIMP2), occurs as a consequence of PRKN mutations and Parkin inactivation.362 AIMP2 present in Lewy bodies causes DNA damage and cell death by activating poly (ADP-ribose) polymerase-1.363 The non-RTK c-Abl phosphorylates Parkin at Tyr143, resulting in decreased Parkin activity. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced c-Abl activation leads to the Parkin inactivation, which in turn causes the accumulation of AIMP2 and neuronal death.364 Furthermore, c-Abl was activated and Parkin was phosphorylated at Tyr143 in postmortem human brain tissue from PD patients.364 Therefore, c-Abl may serve as a pathogenic kinase in PD by inhibiting the Parkin activity.
GAK
Genome-wide association analysis of patients with familial PD revealed an association between single nucleotide polymorphisms at the GAK locus and PD susceptibility,365 which was supported by followed-up meta-analyses.366,367,368,369 Cyclin G-associated kinase (GAK) encoded by the GAK gene is a Ser/Thr kinase comprised of a kinase domain in the N-terminus and a clathrin binding domain.370 Neuron-specific knockout of GAK leads to cell loss in neonatal mice due to deficient proliferation of neural progenitors,371 and GAK inhibition is associated with neurodevelopmental disorders.372
GAK activity affects PRKN-independent mitophagy by altering the mitochondrial network and lysosome morphology, shedding light on the regulation of mitophagy.373 Downregulation of auxilin, the homolog of GAK in Drosophila, leads to impaired climbing ability, shortened lifespan, and dopaminergic neuron loss.374 In vitro experiments demonstrated that knockdown of GAK in cells overexpressing α-syn increased α-syn level and resulted in cytotoxicity,375 suggesting that GAK may play a protective role in PD.
AKT
Mutations in several PD-associated genes cause abnormalities in the AKT signaling pathway which maintains fundamental functions in dopaminergic neurons.376 In an in vitro model of PD, 1-Methyl-4-phenylpyridinium (MPP+) treatment has been found to inactivate AKT.377 PINK1 regulates the activation of insulin-dependent AKT signaling pathway,378 possibly by phosphorylating FK506 binding protein 5, and rescues the damage of mitochondrial complex I induced by MPP+.379,380 Consistently, the MPTP-induced inactivation of AKT was recovered by overexpression of Parkin.381 LRRK2 phosphorylates AKT and promotes cell survival via inhibition of forkhead box protein O1. G2019S and R1441C mutants of LRRK2 reduce the phosphorylation of AKT, and subsequent blockade of pro-cell survival by AKT inhibition may contribute to neuronal death in PD.382 DJ-1, a homodimeric protein, acts as an oxidative stress sensor in stressed conditions.383 Loss-of-function mutations of DJ-1 are associated with autosomal recessive PD.384,385 A cytoprotective role of DJ-1 has been proposed involving the activation of cell survival-related ERK1/2 and PI3K/AKT pathways.386,387 Therefore, restoring the function of AKT and activating the AKT signaling pathway may show protective benefits in PD.
JNK/p38
MAPK family members, belonging to the CMGC group, are dysregulated in PD, and among them, JNK and p38 MAPK are activated in various PD models.388,389,390,391,392 JNK2 and JNK3, but not JNK1, induce upregulation of cyclooxygenase-2 expression and activate c-Jun-induced death of dopaminergic neurons in the MPTP-induced PD model.393 The activation of c-Jun and up-regulated expression of cyclooxygenase-2 were also observed in dopaminergic neurons of PD patients.394 Toxins such as Rotenone and MPTP can directly or indirectly activate the p38 MAPK pathway, which in turn results in ROS accumulation.395 In α-syn-A53T transgenic mice, increased p38 MAPK activity leads to direct phosphorylation of Parkin and subsequent mitochondrial dysfunction.391 Additionally, α-syn activates toll-like receptor 4-dependent p38 MAPK and triggers autophagy impairment in microglia and induction of neuroinflammation.392
Interestingly, JNK and p38 MAPK can be indirectly phosphorylated by LRRK2 via MAP2K, possibly because the LRRK2 kinase domain has a high homology with the MAP3K family members.396,397 Moreover, PD-associated LRRK2-G2019S mutation has been found to exhibit augmented phosphotransferase activity for MAP2K.396 Several drugs, such as SKF-86002,398 SB203580,399 and SB202190,400 inhibiting JNK/p38 MAPK and their upstream pathways have been tested in several models of PD. However, one such inhibitor CEP-1347 failed to reach the clinical endpoint in a clinical trial.401 This indicates that treatment with broad effects needs to be used with caution when applied to human patients.
ASK1
ASK1, a member of the MAP3K family, phosphorylates and activates MAP2K and subsequent MAPK to amplify the downstream signaling cascades. ASK1 itself takes part in physiological processes as well as cell death, by regulating the activation of JNK and p38.402 In the MPTP model, ASK1-MAPK was activated possibly due to TNF-dependent thioredoxin-1 oxidation,403 and knockout of ASK1 rescued MPTP-induced motor deficits, dopaminergic neuron loss, and neuroinflammation.404 ASK1 inhibitor, JNK3-N-Tat alleviated mitochondrial damages and inhibited the apoptosis of dopaminergic neurons in MPP+-treated primary cortical cells as well as MPTP mouse model.405,406 In addition, Apelin-36, a neuromodulatory peptide, protects neurons from apoptosis in the MPTP mouse model by inhibiting the ASK1/JNK/caspase-3 pathway.407
ASK1 activation was also found in α-syn-overexpressing cells and transgenic animal models, suggestive of the involvement of ASK1 in the cascade effects of α-syn.408 In support, ASK1 deletion rescues the behavioral deficits induced by intrastriatal injections of α-syn pre-formed fibrils and accumulation of phosphorylated α-syn in the striatum and cortex.409 ASK1 also activates cell death-related pathways after phosphorylated by LRRK2 at Thr832,410 and abolished activation of ASK1 mediated by DJ-1 shows protective benefits in oxidative stress-induced cell death.411 However, inhibitors of ASK1 (including Selonsertib,412 ASK1-IN-2,413, and GS-444217414) have not been yet tested in clinical PD.
HD
HD is a monogenic autosomal dominant genetic disease caused by mutations in the huntingtin (HTT) gene, the abnormal copy of which will have a 50% of probability being passed to the offsprings.415 At the molecular level, tandem repeats of triplet CAG are abnormally expanded in the encoding region of HTT gene, resulting in an insertion of polyglutamine (polyQ) polypeptide in the N-terminal domain of the mutant HTT (mHTT) protein which drives the death of the brain cells.416,417,418 HD symptoms usually appear between the ages of 30 and 50, and are influenced by familial inheritance. For example, the age of onset may be earlier in families with associated chromosomal abnormalities and about 8% of cases show onset before the age of 20.419,420 Early symptoms manifest as mild emotional or intellectual impairment, with only an uncoordinated and unsteady gait. As the disease progresses, incoordination of body movements becomes more pronounced, and progressively and eventually worsens until movement becomes difficult, speech becomes impossible, and decline of mental abilities develops into dementia.421 The incidence of HD is similar between sexes, and is about 12 per 100,000 people in the European population, but the incidence in Africa and Asia is approximately one-tenth of that in European.422
In HD, mHTT protein is defective in its normal physiological functions, instead, misfolded mHTT fragments form a variety of intracellular aggregation structures with toxicity to adversely impair cell functions.423 HTT protein undergoes various types of PTM, including phosphorylation, acetylation, palmitoylation, and ubiquitination. These modifications modulate the toxicity of mHTT, leading to potential clinical implications. Growing evidence from cellular and animal models shows that phosphorylation or pseudophosphorylation of Ser13, Ser16, Ser434, or Ser536, in HTT reduces the toxicity of polyQ-mHTT, and levels of phosphorylation are weakened by polyQ expansion,424,425,426 suggesting that phosphorylation state of HTT tightly links to toxicity in HD pathogenesis. Phosphorylations of HTT at Ser13 and Ser16 have been most studied, and are found to regulate the structure, aggregation, and subcellular localization of HTT.425
Major kinases in HD
Kinases are involved in the pathophysiology of HD through multiple regulatory processes including integration of glutamate transmission and BDNF signaling, neuroimmune regulation, resistance to mHTT toxicity, neuronal apoptosis, cellular energy metabolism, and autophagy pathways. Phosphorylation of HTT at Ser1/Ser16 by IKKβ increases the clearance of mHTT by proteasomes and lysosomes thereby reducing mHTT-mediated cytotoxicity.427 Similarly, phosphorylation of HTT at Ser434 by CDK5 ameliorates HTT aggregates-stimulated cell toxicity and cell death.88 Interestingly, autophagic clearance of mHTT and suppressed accumulation of mHTT are found to be facilitated by TBK1-medicated HTT phosphorylation.428 In addition, kinases such as AKT, ERK, and JNK are also phosphor-activated to mediate the toxicity of mHTT.429,430,431 Here we summarize the major kinases and their regulatory mechanisms in HD pathogenesis (Figs. 2 and 9).

Schematic description of kinase signaling pathways in Huntington’s disease. mHTT promotes neuronal apoptosis and inflammatory responses by inhibiting ERK and activating the p38, JNK, and IKKβ signaling pathways. It establishes connections with HTT via the MEK/ERK and AKT signaling pathways, regulating neuronal autophagy, neuroprotection, and glucose uptake pathways. In addition to causing DNA damage by itself, mHTT’s aggregation also leads to pancreatic β-cells damage and activation of CDK5 through its interaction with IRS-2. The aberrant activation of CDK5 reduces microtubule stability, which, in turn, contributes to the exacerbation of mHTT pathology. In addition, the activation of the IKKβ signaling pathway by AKT promotes neuroinflammatory responses, while the inhibition of the JNK signaling cascade by AKT hinders cell apoptosis. The regulatory network formed by the mutual activation and inhibition of kinases plays distinct roles in different stages of the HD pathological process. This figure was created with BioRender.com
MEK/ERK
Studies have concluded that MEK (a member of the MAP2K family in the STE group) /ERK (a member of the MAPK family in the CMGC group) acts as a double-edged sword in HD. On the one hand, MEK/ERK signaling is considered to integrate glutamate transmission and BDNF signaling in HD.415 In R6/1 and R6/2 mouse models expressing exon 1 of HTT, wide variations in ERK activation related to age, brain regions, and cell types are identified.432,433 Mice expressing full-length HTT tend to exhibit suppressed ERK activity and downstream BDNF expression.434 ERK deficiency in the mHTT model triggers striatal degeneration and increases glutamate susceptibility.435,436 Therefore, reduced ERK activation leads to lessened trophic support by BDNF, and upregulated expression of glutamate transporters (reviewed elsewhere415), leading to subsequent neuronal excitotoxicity and apoptosis.
On the other hand, MEK/ERK activation is believed to be neuroprotective in HD.430,432 The reduction of nutrients obtained by striatal neurons will in turn activate ERK to increase compensatory responses.430 Increased MEK activity promotes the activation of ERK/CREB, reduces the caspase-3 activity, increases the phosphorylation of HTT and autophagic clearance of mHTT, and ultimately supports neuronal survival.415,436,437
IKK
IKK is a member of the other group of typical protein kinases and is a complex composed of two catalytic subunits IKKα and IKKβ, and a regulatory subunit IKKγ.438 The human IKK family has four members, the IKKα, IKKβ, TBK1, and IKKε. IKK directly interacts with mHTT,439 and chronic elevation of IKK caused by DNA damage and NF-κB pathway activation is associated with neurodegeneration in HD.440,441,442 Clinical data indicate that HD patients exhibit high levels of inflammatory factors resulting from the chronic increase of IKK/NF-κB activity in the CNS and serum specimen/samples before symptom onset, with the immune cells maintained in a sensitive state.438,440,443 Similar effects of IKK/NF-κB signaling involving the immune environment were observed in the HD cell model and R6/2 mouse brains,439,444 and increased IKK in HdhQ150 mice was observed in the striatum.445
Specifically, IKKβ phosphorylates HTT at Ser13/Ser16, which activates the scavenging of HTT, and knockout of IKKβ further deteriorates deficits in motor behavior of R6/1 HD mice, accompanied by striatal neurodegeneration and microglia activation.446 Hence, IKKβ may protect the striatum from neuronal degeneration and slow HD progression through multiple mechanisms.427,446 However, since IKK is regulated by various factors such as AKT, TNF-α, and interleukin-1β, and both the activation and inhibition of IKKβ have been shown to exert neuroprotective effects on HTT aggregation,447,448,449 the confirmative role of IKKβ in HD needs to be further elucidated.
CDK5
CDK5 belongs to the CDK family, the activity of which is abnormally elevated in AD pathology, whereas in HD, reduced levels of CDK5 and p35 have been detected in human HD patients and brains of HD mouse model.426,450 In the mouse model, although CDK5 reduces the HTT aggregation by phosphorylating HTT at Ser434 and reduces its cleavage by caspases,88 phosphorylation of HTT at Ser434 by CDK5 was repressed.426 In addition, DNA damage-triggered CDK5 activation causes phosphorylation of HTT at Ser1181 and Ser1201, thereby protecting striatal neurons from mHTT-induced toxicity.88 Mitigated CDK5 activity stabilizes microtubules and facilitates the formation of mHTT inclusion bodies in HD.451 Conversely, CDK5 activation has been shown to dissociate microtubules in primary cortical neurons, thereby weakening mHTT aggregation.451,452,453 However, enhanced CDK5 activation in StHdhQ111 cells promotes cell death, possibly through dysregulation of glutamatergic and dopaminergic signaling.454 Therefore, single-targeting CDK5 to increase its activity may produce certain toxic side effects during treatment for HD.
JNK/p38
As a member of the MAPK family, JNK activates the transcription factor c-Jun, one of the activator protein-1 transcription complexes, to regulate the expression of pro-apoptotic genes.455,456 JNK also phosphorylates and stabilizes p53 thereby enhancing the pro-apoptotic activity of p53.457 Knockout of CNS-specific JNK3 exerts resistance to excitotoxicity and increases PI3K/AKT activity, implying that JNK3 is antagonistic to AKT signaling.458,459 So far, upregulated JNK signaling can be observed in most animal models of HD, except the StHdhQ111 knockin model.415 Activation of MAP2K4 and MAP3K1, upstream activators of JNK, accelerates the formation of mHTT inclusion bodies and augments the resultant toxicity in HD transgenic models.460,461 Negative regulatory mutations of MAP3K1 and JNK-interacting protein 1 reduced dopamine and adenosine 3’5’-monophosphate-regulated phosphoprotein 32 kDa, but did not alter c-Jun expression and mHTT inclusion formation in a rat lentiviral model of HD.462 Furthermore, ASK1 activated by mHTT acts as an upstream kinase of JNK and p38 to indirectly promote apoptosis.463,464 Therefore, inhibition of JNK activation protects neurons from mHTT-induced toxicity and apoptosis.
In HD patients and mouse models, increased p38 activity is found to correlate with neuronal death.465,466 Phosphatase MAPK phosphatase 1 inactivates p38 by dephosphorylating p38 in the Thr-Gly-Tyr motif, and the decreased MAPK phosphatase 1 activity therefore accounts for enhanced p38 activity.467 Pharmacological stimulation of MAPK phosphatase 1 reduced p38 activity and protected cultured cells and mHTT-injected mice from neurotoxic outcomes resulting from mHTT expression.101 In HD mice, increased activation of p38 is partially responsible for striatal degeneration, and p38 inhibitor SB-239063 successfully protects neurons from degeneration,468,469 which may involve blockade of neuronal apoptotic signals and glial cell-mediated neuroinflammation.
AKT
Activation of AKT signaling in all HD cells, mouse models, and patients’ brains has been suggested to be neuroprotective.415,470,471 Phosphorylation of mHTT by AKT is essential for IGF-mediated neuroprotection,429 while Mn2+-induced reduction of p-IGFR/IR-dependent AKT phosphorylation leads to reduced glucose uptake in HD cells.472 In addition, insulin and IGF-1 signaling pathway improves mitochondrial function and mitigates reactive oxygen species production in a PI3K/AKT-dependent manner in HD knockin striatal cells,473 while enhancing autophagy by modulating PI3K/AKT/mTOR signaling improves HD-like lesions in rats.474 In the study of the relationship between HD and the pathogenesis of diabetes, it was shown that in the mouse pancreatic insulinoma cell line NIT-1 (160Q cells) expressing the N-terminal mHTT containing 160 polyglutamine, insulin receptor substrate 2 was recruited to the mHTT aggregates in HD β cells, leading to the inhibition of the PI3K/AKT/forkhead box protein O1 pathway and causing damage to pancreatic β cells.475 In addition, 11-week-old R6/2 mice showed smaller pancreatic islets, loss of insulin, glucagon, and somatostatin expression, and reduced activation and expression of PKA, AKT, ERK1/2, and STAT3 in the pancreas.476 These studies suggest that diabetes or abnormal glucose and lipid metabolism may be involved in the development of HD. mHTT regulates both pro-survival and pro-apoptotic signals via CDK5/AKT modulation.415 In brief, mHTT activates CDK5/AKT signaling to promote HTT phosphorylation for cell survival; on the other hand, the same process may inhibit JNK/caspase-3 signaling, accelerating cell apoptosis. The indication of this line of events depends on the diversities of research models and experimental conditions.
ALS
ALS is a rare but fatal neurodegenerative disease characterized by progressive degeneration and loss of motor neurons. Familial ALS (fALS) accounts for 10–15% of all cases,477 and more than 40 ALS-related genes have been identified, including C9orf72, TARDBP, SOD1, and FUS genes.478 Variations in these four genes account for ~60% of fALS and 10% of sporadic ALS.479 In addition to genetic factors, environmental elements such as exposure to chemicals, metal contaminations, or radiation, can increase the risk of ALS through mechanisms such as augmented neuronal oxidative stress and mitochondrial damage.480 These factors also, by affecting protein phosphorylation, drive disease progression and selective degeneration of motor neurons.
Mutations in the TDP-43-encoding gene TARDBP account for approximately 4% of fALS patients.481 Interestingly, the TDP-43 pathology appears in the vast majority of ALS patients.482 Normally, TDP-43 is predominantly localized in the nucleus. However, pathological aggregates of TDP-43 in the cytoplasm are present in ALS pathology.483 Various PTM of TDP-43 such as ubiquitination,484 acetylation,485 and phosphorylation,486 phosphorylation on multiple sites (Ser409, Ser410, Ser379, Ser403, and Ser404) have been observed in experimental and clinical ALS specimens, and other potential phosphorylation sites in TDP-43 are continuously discovered.487 With the aggravation of TDP-43 pathology, the cognitive decline appears more and more rapid.488 Possible causes of TDP-43 toxicity involve loss of normal physiological activity, gain-of-function mutation induced by pathological aggregation, or combined effects.489
Mutations in FUS cause ALS in an autosomal dominant manner, and most FUS mutations lead to mislocalization of FUS (fused in sarcomas) from the nucleus to the cytoplasm as inclusions.490 FUS is a DNA/RNA binding protein mainly expressed and localized in the nucleus, and participates in physiological processes such as DNA repair and RNA metabolism.491 Transgenic mice overexpressing wild-type human FUS, as well as expressing ALS-associated FUS mutants or FUS lacking a nuclear localization signal greatly triggered the formation of cytoplasmic FUS inclusions, and exhibited ALS-like motor impairments.492,493,494 In both mouse and human brain tissue, FUS has been observed to bind to over 5500 pre-mRNAs.495 Knockdown of FUS results in decreased expression of mRNAs containing long introns, many of which encode proteins involved in synaptic function, such as neurexin 3 and neuroligin 1.495 Hence, the contribution of FUS in ALS may be primarily due to the loss-of-function of normal FUS in the nucleus and the gain-of-function of toxic FUS secondary to the mislocalized FUS aggregation in the cytoplasm.
To date, more than 200 SOD1 variations have been disclosed in ALS (https://alsod.iop.kcl.ac.uk/), occurring in 14.8% of European fALS patients and 30% of Asian fALS patients.496 Cu/Zn superoxide dismutase 1 (SOD1), encoded by SOD1, is an antioxidant enzyme that regulates mitochondrial and cytosolic superoxide levels. Mutant SOD1 forms unstable depositions in the cytoplasm, and then greater aggregates and inclusions,497 which impair axonal transport.480 The spinal motor neurons in SOD1G93A transgenic mice showed mitochondrial membrane potential loss, respiratory chain activity disruption, calcium homeostasis disruption, and mitochondrial vacuolation.498 However, for a long time, the loss-of-function hypothesis was not recognized due to the lack of correlations between enzyme activity of SOD1 and aggressiveness of clinical phenotypes in ALS patients,499 as well as the absence of obvious phenotypes in SOD1 null mice in early studies.500 Nonetheless, a comprehensive review501 on SOD1-ALS patients revealed that almost all mutations lead to a substantial reduction in SOD1 enzyme activity, and significant deleterious effects in the nervous system were evident in SOD1 knockout mice, supporting the contribution of loss-of-function mutation of SOD1 to ALS pathogenesis.
Defects in mitochondrial morphology, functions, and dynamics were consistently observed in ALS patients and mouse models. C9orf72 is a mitochondrial inner membrane-associated protein stabilizing mitochondrial complex I assembly,502 and haploinsufficiency and loss-of-function mutation of C9orf72 lead to decreased activity of neuronal mitochondrial complex I.502,503 Given the discovery of C9orf72 hexanucleotide repeat expansions as a critical cause of ALS,504,505 studies have ever since revealed the multifaceted effects of C9orf72. In addition to the detrimental effects on mitochondrial functions mentioned above, mutated C9orf72 also impairs a variety of intracellular processes such as RNA metabolism and autophagy. C9orf72 haploinsufficiency also negatively affects vesicle trafficking and inhibits the initiation of autophagy.506 Inhibition of autophagy impedes the clearance of misfolded and aggregated proteins, such as TDP-43 and FUS aggregates in ALS, thereby exacerbating cellular damage.
To date, Rilutek and Radicava have been approved by the FDA for ALS treatment to slow the progression of symptoms and improve the quality of life, although they are unable to reverse the existing damages caused by ALS.507 Continuing endeavors into the pathophysiology of ALS have led to several potential mechanism-based therapies that are currently being tested in clinical trials.
Major kinases in ALS
Protein kinase-encoding genes such as TBK1 and NEK1 were recently discovered as ALS causal genes, as well as that several other kinases are widely involved in ALS pathogenesis (Figs. 2 and 10). In general, they take part in the phosphorylation of causative proteins in ALS such as TDP-43, leading to altered functions and abnormal aggregation of these proteins. Furthermore, dysregulation of the kinase signaling network may directly contribute to the death of motor neurons via complex interactions.

Schematic description of kinase signaling pathways in Amyotrophic lateral sclerosis. ALS genes encoding protein kinases, such as TBK1 and NEK1, affect the proteostasis process, autophagy, and DNA damage. TBK1 phosphorylates OPTN and p62, leading to autophagy clearance and thus ensuring efficient degradation of ubiquitinated mitochondria. TBK1 inactivates RIPK1 and loss of TBK1 boosts RIPK1 activation and promotes cell death. NEK1 mutations disable DNA damage response and its deficiency reduced the phosphorylation of VPS26B, leading to disruption of endosomal transport and further dysfunction of mitochondria and lysosome. Several kinases are involved in the phosphorylation of TDP-43 and others. The decrease of AKT activity in ALS leads to the upregulated GSK3β activity, contributing to TDP-43-induced axonal degeneration. c-Abl kinase mediates the accumulation of toxic FUS by phosphorylating FUS. Proteins abnormally aggregated alter the kinase signaling networks, eventually leading to cell death of motor neurons. This figure was created with BioRender.com
TBK1
Whole-exome sequencing data of 2869 ALS patients and 6405 controls have identified TBK1 as an ALS-associated gene,508 further supported by another independent study showing that the haploinsufficiency of TBK1 caused ALS.509 Thereafter, the pathogenic role of ALS-TBK1 mutations has been validated,510,511,512 e.g., nonsense, frameshift, and missense mutant forms were found in sporadic and fALS patients.513
TBK1 is a IKK family protein, composed of a Ser/Thr kinase domain with two lobes, a ubiquitin-like domain, and two coiled-coil domains.514 Activation of TBK1 involves a multi-step mechanism whereby Lys30 and Lys401 are first poly-ubiquitinated, followed by phosphorylation of Ser172, resulting in a conformational change to allow substrates to bind.176 Regulated by adaptor proteins such as NAK-associated protein 1, TANK, and Sintbad, TBK1 participates in physiological activities such as induction of interferons and autophagy regulation.515 Activated TBK1 modulates autophagy by phosphorylating autophagic adaptor proteins and membrane components in autophagosome such as LC3 and C9orf72-binding partner SMCR8.516,517 In addition, other autophagy receptors OPTN, nuclear dot protein 52, Tax1 (human T cell leukemia virus type I) binding protein 1, and p62, are all TBK1 substrates, and phosphorylation of these receptors leads to autophagy clearance and degradation of ubiquitinated mitochondria, a process of which is of great importance considering that both TBK1 and OPTN are genetically linked to ALS.518
ALS-associated TBK1-S172A mutation blocks the efficient formation of autophagosomes and damages autophagosomal membranes, suggesting that impairment of mitophagy may be a key pathophysiological mechanism for ALS.517,519 In the early disease stage of SOD1G93A mice, the heterozygous deletion of TBK1 impairs autophagy in motor neurons and precedes disease onset.520 However, in the late disease stage, heterozygous TBK1 deletion shows opposite effects of significantly attenuated microglial neuroinflammation and prolonged survival.520 Similarly, TBK1 G217R and R228H mutations show decreased kinase activity, thereby accelerating the onset of disease in SOD1G93A mice, whilst extending their lifespan.177 Given these observations, TBK1 may exert a multifaceted role in SOD1-related ALS.
NEK1
Another ALS-associated gene NEK1 has also been screened out from a whole-exome analysis of familial and sporadic ALS patients, and NEK1 variations usually result in a loss-of-function outcome for the NEK1-encoding kinase NEK1.521,522,523,524 Comparing hiPSC-derived motor neurons carrying a NEK1 mutation and neurons carrying a C9orf72 mutation with wild-type neurons, neurons with NEK1 mutation showed the most severe DNA damage, implying that NEK1 mutations disrupt DNA damage response in ALS pathology.525 In addition, NEK1 deficiency reduces the phosphorylation of VPS26B and causes disruption of endosomal transport and consequent dysfunction of mitochondria and lysosome.526
DLK
As a member of the mixed-lineage kinase, DLK acts upstream of MAPK signaling and activates JNK and p38 via MAPK-kinase 4/7.527 DLK in the CNS facilitates the establishment of neural circuits during development528 and acts as an axonal damage sensor in diseased states.527,529 DLK/JNK activity is profoundly elevated, and pharmacological inhibition or genetic knockout of DLK reduces the neuronal loss and axonal degeneration in ALS mouse models.530 Moreover, overexpression of activating transcription factor 3 post-DLK deletion significantly protects motor neurons from death and axonal degeneration in ALS mice, highlighting the feasibility of combination therapy in ALS.531
RIPK1
RIPK1 mainly regulates cell death and activates inflammatory pathways in ALS patients.71 TBK1 deficiency in mice causes strong activation of RIPK1 and embryonic death, which can be rescued by RIPK1 inactivation. Mechanistically, TBK1 inactivates RIPK1 by phosphorylating RIPK1 at Thr189, and loss of TBK1 boosts RIPK1 activation and aggravates late-onset ALS/frontotemporal dementia-like pathology,532,533 providing insights on intermolecular regulation between TBK1 and RIPK1 in ALS. In addition, defects in glucose uptake caused by NEK1 depletion stimulate RIPK1 activation, while inhibition of RIPK1 restores BBB damage, neuroinflammation, and accumulation of misfolded proteins, suggesting ALS may be treated by blocking RIPK1 activation.534
Mutations in another gene OPTN are associated with both familial and sporadic ALS.71 Expression of OPTN encoded by OPTN attenuates RIPK1-dependent cell death by modulating the turnover of RIPK1, and loss of OPTN leads to progressive axonal degeneration through RIPK1-dependent necrosis.71,535 RIPK1/BRM-associated factor complex was recently reported to promote the transcription of pro-inflammatory genes in ALS patients as a key regulator of chromatin remodeling.536 These results highlight the importance of RIPK1 in ALS, and consequently, RIPK1 inhibitors have entered clinical trials as a new class of drugs for the treatment of ALS.537
AMPK
AMPK coordinates cellular energy homeostasis by promoting catabolism and reducing ATP consumption. In ALS, AMPK activity is dysregulated, and abnormal activation of AMPK has been observed in motor neurons of ALS patients.538 As ALS progresses, patients may lose weight and body fat, or even exhibit malnutrition, stongly suggesting a metabolic dysfunction and an energy imbalance.539 Consistently, in the SOD1G93A mouse model, AMPK activity is increased in motor neurons, likely attributed to a severe reduction in glucose uptake and a significant decline in ATP production.540,541 Activation of AMPK disrupts the distribution of human antigen R, a well-characterized mRNA stabilizer mainly located in the nucleus, through the phosphorylation of importin,538 which leads to an imbalance in RNA metabolism, presenting an alternative mechanism for the pathogenesis of ALS.
Nevertheless, controversial results have been reported on AMPK activation for ALS treatment. Several studies have indicated that the inhibition of AMPK activity may offer protection against ALS. Reducing aak-2, an ortholog of AMPKα2 catalytic subunit, improved the motor behavior in SOD1-G85R mutant nematodes.542 Disease progression of TDP-43 mutant mice can also be delayed by down-regulating AMPKα1 or inhibition of AMPK activity using cAMP analogs.543 In another study, the activation of AMPK by latrepirdine delayed the onset of motor symptoms and expanded the lifespan of SOD1G93A mice.544 The effect of AMPK activation or inhibition on ALS may depend on the specific model used and the time window for pharmacological intervention, and future studies are still desired to determine whether the modulation of AMPK activity can represent a viable therapeutic avenue for ALS patients.
PI3K-AKT-GSK3β pathway
As one of the major anti-apoptotic pathways, the PI3K/AKT signaling is suppressed in the spinal cord of an ALS mouse model545; consequently, downstream proteins and their phosphorylation are broadly affected including GSK3β. AKT phosphorylates GSK3β at Ser9 thereby down-regulates GSK3β activity.546 Consequently, it is expected that the reduced AKT activity in ALS enhances GSK3β activity. Consistently, enhanced GSK3β activity was present in both animal models and patients with ALS,547,548 and GSK3β inactivation showed neuroprotective effects in ALS pathogenesis, partly through amelioration of phenotypic defects caused by expression of TDP-43, SOD1, or FUS mutants.549 GSK3β is also involved in TDP-43-induced axonal degeneration, since the loss of GSK3β/Shaggy inhibits TDP-43Q331K-mediated degeneration of axon and neuromuscular junction in Drosophila.550 A small molecule chemical, kenpaullone, was screened out and found to significantly prolong the survival of embryonic stem cell-derived motor neurons from SOD1 mice, partly due to its inhibitory effects on GSK3 activity.551
ROCK
Both isoforms of ROCK (ROCK1 and ROCK2) are Ser/Thr kinases.552 ROCK is catalytically inactive in its natural state as the kinase domain is automatically blocked by the C-terminal region.553 Expression of ROCK in adult mouse brains demonstrates that glial cells express ROCK1 while neurons express ROCK2,554 and ROCK2 levels are significantly increased in the skeletal muscle of patients with ALS.555 Upregulated ROCK activity activates phosphatase and tensin homolog and decreases AKT activity in ALS mice model556; ROCK inhibitor Fasudil greatly prolonged the survival of ALS mice, accompanied by reduced phosphatase and tensin homolog phosphorylation and restored AKT activity, in addition to increase the number of protective microglia in the spinal cord and decreased secretion of pro-inflammatory cytokines and chemokines in the early stage of the disease (day 100).557 Considering this drug is clinically available, it may be a promising treatment for ALS trials.
c-Abl
c-Abl is a member of the non-RTKs family with DNA binding domain sequence, nuclear export sequence, and nuclear localization signal. c-Abl participates in neurite extension and dendritic spine stability directly through its F-actin-binding region or indirectly through phosphorylation of actin-binding proteins (such as the small GTPase RhoA/Rac1 and CDK5),558,559 and it has recently emerged as a potential target for neurodegenerative diseases.560 Increased phosphorylation of Src/c-Abl was found in post-mortem spinal cord tissues of ALS patients, and inhibition of Src/c-Abl by pharmacological or genetic manipulations promotes the survival of motor neurons in various ALS models.561,562 Phosphorylation of pathological ALS proteins such as TDP-43 and FUS also involves c-Abl,563,564 e.g., c-Abl-mediated phosphorylation of TDP-43 at Tyr43 leads to its cytoplasmic accumulation and triggers neuronal cell death.563 Similarly, c-Abl kinase stimulates the cytoplasmic mislocalization and accumulation of toxic FUS by phosphorylating FUS at Tyr526.564
Kinases that phosphorylate TDP-43
TDP-43 is recognized as a major target of ALS, and the several kinases responsible for TDP-43 phosphorylation have been identified. In TDP-43cKO mice, IKKβ promoted the degradation of cytoplasmic TDP-43 by proteasomes in hippocampal neurons via phosphorylating Thr8 and Ser92 at the N-terminus of TDP-43.565 IKKβ also significantly reduced the expression level and toxicity of pathogenic TDP-43-encoding gene TARDBP mutations (A321V and K181E) in N2a.565 Negative regulation of the cAMP/PKA signaling pathway by the phosphodiesterase dunce and inhibitory subunit PKA-R2 promotes the aggregation and mislocalization of the Drosophila TDP-43 ortholog TBPH in the cell bodies of motor neurons, leading to motor defects and shortened lifespan.566 Overexpression of the PKA target CrebA rescued TBPH mislocalization in the TBPH-overexpressing ALS Drosophila model.566 Therefore, activation of cAMP/PKA may be a strategy to improve the molecular and functional effects of pathological TDP-43, although this study was only in Drosophila.
CK1 phosphorylates multiple Ser residues in the glycine-rich region of the C-terminus of TDP-43 (including Ser379, Ser403/404, and Ser409/410), suggesting that CK1 may be involved in the pathological phosphorylation of TDP-43 in vivo.486,567 CDC7 strongly phosphorylates Ser409/410 of TDP-43, promoting neurotoxicity in TDP-43 transgenic animals.568 TTBK1/2 also strongly phosphorylates TDP-43 at Ser409/410, leading to synergistic exacerbation of behavioral abnormalities and increased pathological protein phosphorylation in TDP-43/TTBK1 transgenic Caenorhabditis elegans.569 In addition, elevated TTBK1/2 protein levels were found in the postmortem frontal cortex of patients with frontotemporal lobar degeneration, and TTBK1/2 colocalized with TDP-43 inclusions in ALS spinal cord.570 These studies suggest that targeting kinases may be an effective strategy for treating TDP-43 proteinopathy.
Kinases in other neurodegenerative diseases
Kinases also are considered crucial in other neurodegenerative diseases, such as spinocerebellar ataxias (SCAs) and chronic traumatic encephalopathy (CTE).
Autosomal dominant SCA is a chronic progressive neurological disease, in which SCA type 14 is affected by point mutations in PRKCG (encoding PKCγ),571 a Ser/Thr protein kinase. Specifically, mutations in PRKCG, such as D115Y, enhance the basal activity of the kinase by compromising its autoinhibition, thereby promoting SCA type 14 disease progression.572 A study on SCA type 17 has shown that activation of MAPK/ERK occurs in the cerebellum of SCA type 17 mice, and that increased gliosis due to ERK activation may lead to neural apoptosis, suggesting that Purkinje cell loss may lag behind ERK activation in the SCA type 17 mouse model.573 In SCA type 11, TTBK2 inhibits disease progression by initiating ciliogenesis in vivo, and dominant truncating mutations in human TTBK2 cause SCA type 11.574 The NMDA receptor NR2D subunit interacts with the SH3 domain of c-Abl, inhibiting the autophosphorylation activity of c-Abl, thereby reducing the loss of cerebellar neuronal function, suggesting that changes in c-Abl activity are associated with the development of cerebellar ataxia.575 Therefore, abnormal activation of kinases and mutations in specific sites of their encoding genes may lead to diverse types of SCAs.
CTE is a progressive neurodegenerative disease associated with repeated traumatic brain injury (TBI) that causes symptoms of cognitive impairment, behavioral changes, mood disturbances, and movement disorders.576 The pathology of CTE mainly includes the accumulation of hyperphosphorylated tau in neurons around blood vessels, and a unique molecular structural configuration of p-tau fibrils that is distinct from changes observed in aging, AD, or any other tauopathies.577 DAPK1 was found to induce cis-p-tau conformational changes and neurodegeneration by phosphorylating Pin1 (a unique prolyl isomerase known to inhibit the conformational state of p-tau) at Ser71 in the mouse brain of CTE model.578 In CTE brains, overactivation of GSK3β and CDK5 increased abnormal phosphorylation of tau, further leading to synaptic defects, a process that could be reversed by inhibition of GSK3β and CDK5.579 In the mouse model of TBI, the activity of ASK1-K716R was significantly reduced. Specifically increasing the activity of ASK1-K716R may maintain the integrity of the BBB, decrease the number of inflammatory microglia/macrophages, and white matter damage, and improve the nerve conduction of nerve fibers after TBI by suppressing the kinase activity of ASK1/JNK and p38.580 Activation of JAK2/STAT3 promotes the expression of interleukin-2 receptor γ, interleukin-4 receptor α in astrocytes, thereby increasing the inflammatory response in the cortex and hippocampus of adult male rats.581 Furthermore, axonemal dynein light intermediate polypeptide 1 was shown to promote neurodegeneration after TBI by preventing the clearance of phosphorylated tau via inhibition of autophagosome-lysosome fusion.582
Clinical trials targeting kinases in neurodegenerative diseases
As summarized above, protein kinases are crucial for disease pathogenesis. In AD, the regulatory roles of kinases are predominantly manifested in tau phosphorylation, Aβ metabolism, and neuronal survival activities. In PD, kinases regulate α-syn phosphorylation, mitochondrial dynamics, autophagy pathways, and formation/plasticity of neuronal synapses. In HD, the occurrence is facilitated by PKC kinase which phosphorylates HTT and alters its structure and function, resulting in further aggregation and damage to the neurons. As one of the hotspots in ALS-related research, kinases participate in ALS pathogenesis by regulating the phosphorylation and aggregation/metabolism of pathological protein TDP-43 and other causative proteins in ALS. In other diseases such as SCAs and TBI, PRKCG gene mutations and abnormal activation of MAPK/ERK, c-Abl, and TTBK2 are involved in the pathogenesis of SCAs, while abnormal activation of DAPK1, GSK3β, CDK5, JAK2 and decreased activity of ASK1-K716R promote neuropathy after TBI. Collectively, these findings support the notion that drug development targeting protein kinases may provide new directions and prospects for neurodegenerative diseases. Several clinical trials with kinase-targeted drugs are completed or currently underway (Table 1 and Table 2).
AD
p38α MAPK inhibitors
MW01-18-150SRM (MW150), an isoform-selective inhibitor of p38α MAPK, effectively rescues hippocampal-dependent associative and spatial memory deficits in two AD mouse models.583 Subsequently, in the APP/PS1 knockin mouse model, effective MW150 administration selectively downregulates neuroinflammatory responses associated with pathological progression, without altering the physiological function of microglia.584 Selectively inhibiting stress-activated p38α MAPK with MW150 attenuates entorhinal cortex dysfunction associated with neuroinflammation early in AD progression.585 Dysregulated LTP in 2-month-old AD mice was restored by MW150 treatment, with efficacy comparable to that of widely used multi-kinase inhibitor SB203580,585 avoiding the limitations of using isoform-unspecific inhibitor drugs of p38 MAPK. These data have provided preliminary clinical results supporting p38α MAPK-directed drug development, and a phase II clinical trial of MW150 is underway (ClinicalTrials.gov identifier: NCT05194163).
VX-745 (also known as neflamapimod) is a novel selective p38α kinase inhibitor with excellent pharmacokinetic characteristics and in vivo activity in inflammation models.586 The main difference between VX-745 and MW150 is that VX-745 exhibits potent, BBB permeable, and highly selective inhibition of p38α, and has no inhibition of p38γ. Phase IIa clinical evidence in patients with early AD suggested that 6-12 weeks of VX-745 treatment significantly improves episodic memory, supporting its potential use in human AD.587 Oral intake of VX-745 in early AD patients attenuated Aβ production and improved episodic memory.588 These two clinical trials investigated the effects of VX-745 treatment (40 mg or 150 mg twice daily) for episodic memory improvement and Aβ production in AD patients for 6 to 12 weeks, which are relatively short period of time for AD trials. Therefore, a phase II clinical study of the multi-center, randomized, double-blind, placebo-controlled trial of VX-745 was conducted between December 29, 2017, and June 17, 2019, and it concluded that 24 weeks of VX-745 (40 mg, twice daily) treatment did not improve episodic memory in patients with mild AD compared to placebo, although the drug resulted in significant reductions of CSF total tau and p-tau 181 levels (ClinicalTrials.gov identifier: NCT03761849). The follow-up pharmacokinetic-pharmacodynamic analysis suggested that further studies of longer-term and higher doses of VX-745 treatment are needed to assess the effects on AD progression.589
GSK3 inhibitors
NP031112 (Tideglusib), a non-ATP competitive inhibitor of GSK3β, is a thiazolidinedione compound with anti-inflammatory and neuroprotective effects.590,591 In rats, injection of NP031112 into the hippocampus significantly reduced inflammation and hippocampal damages induced by kainic acid, with neuroprotective effects involving the activation of peroxisome proliferator-activated receptor-γ.592 Considering the potential beneficial effect of GSK3β inhibition for AD, a pilot human study was conducted in 2013 on 30 patients with mild to moderate AD, indicating a positive benefit in the NP031112 treated group compared to the placebo in multifaceted evaluations including Mini-Mental State Examination, AD Assessment Scale-Cognitive subscale, Geriatric Depression Scale, and Global Clinical Assessment, providing critical safety and efficacy evaluation for the use of NP031112 in AD. However, due to the small sample size and dose escalation method, although there are positive trends in clinical indications (including Mini-Mental State Examination, AD Assessment Scale-Cognitive subscale, Vocabulary Fluency, Geriatric Depression Scale, and final Global Clinical Assessment), it lacked statistical significance to support or reject the benefits of NP031112 in AD.593 A second study conducted in 2014 on 306 AD patients who received 26 weeks of NP031112 or placebo treatment reported that short-term (26 weeks) NP031112 treatment was safe and well-tolerated, but did not offer clinical benefit.594 Additionally, no increase in AchE activity was observed with NP031112 in the CSF of AD patients, which is disappointing because AChE co-localizes with hyperphosphorylated tau in NFTs, and inhibition of GSK3β is expected to preserve the AchE activity.595
Lithium is another GSK3β inhibitor that has demonstrated promising effects in animal models of AD. In PDAPP (APP-V717F) transgenic mice, lithium treatment reduces plaque load by suppressing APP processing and eliminating GSK3β-mediated Aβ synthesis in the brain.596,597 In addition, oral treatment with lithium carbonate can prevent NFT formation but does not improve motor or working memory deficits in aged animals.598 Lithium carbonate also prevents object recognition memory impairment caused by Aβ25-35 in rats.599 Moreover, chronic lithium therapy in 3xTg-AD mice showed dose-dependent improvement in brain inflammation and oxidative stress.600 Low doses of novel lithium salicylate proline ion co-crystal, lithium carbonate, and lithium salicylate prevented spatial cognitive decline and depressive-like behavior in APPswe/PS1dE9 mice, while lithium salicylate proline ion co-crystal also rescued hippocampus-dependent associative memory decline and irritability.601 Consistently, long-term lithium therapy in AD patients is associated with a lower incidence of AD and increased BDNF activity, suggesting a role of lithium in preventing early-stage AD patients at risk from deterioration.602 A study using [18F]FDG-PET imaging shows that long-term lithium therapy can reduce glucose metabolism in the cerebellum and hippocampus of non-demented elderly individuals.603 However, a multicenter, short-term (10-week) lithium treatment clinical trial in 71 patients with mild AD did not find an effect of lithium treatment on GSK-3 activity or CSF-based biomarker concentrations (Controlled-Trials.com identifier: ISRCTN72046462).604 Despite the long-term lithium therapy may have beneficial effects in AD animal models or patients, there are also studies demonstrating that lithium-induced Aβ elevation may not be related to GSK3β inhibition,605,606 and the neuronal protection provided by lithium under tau pathology is independent of GSK3β.606,607 It was also pointed out that lithium treatment also reduces tau in cell culture and mouse brain, and chronic treatment may have the side-effect of tau-dependent iron accumulation demonstrated by MRI imaging analysis.608 Therefore, targetting a multi-functional kinase such as GSK3β may not be the primary choice of drug target for AD.
Tyrosine kinase inhibitors
Two prominent tyrosine kinase inhibitors, masitinib, and nilotinib, have been developed to alleviate AD symptoms. Masitinib specifically targets mast cells and microglia activity in the neuroimmune system, and may improve AD symptoms by preventing the cell cycle re-entry, improving neuronal plasticity, inhibiting tau phosphorylation, and modulating NMDA receptors by blocking Fyn kinase activity and activation of mast cells and microglia.609 In non-neuronal cells, nilotinib treatment also effectively stimulates the activation of microglia and the proliferation of astrocytes, promotes Aβ clearance, and modulates immune responses in early-stage AD.610
A previous phase II study (ClinicalTrials.gov identifier: NCT00976118) demonstrated that masitinib considerably improves cognitive decline in patients with mild to moderate AD, offering guidance to larger clinical trials.611 Another completed phase IIb/III study (ClinicalTrials.gov identifier: NCT01872598) showed that masitinib (4.5 mg/kg/day) as an adjunct to cholinesterase inhibitors memantine may benefit patients with mild-to-moderate AD.612 In a completed phase II trial (ClinicalTrials.gov identifier: NCT02947893), compared to the placebo group, the nilotinib group exhibits significantly decreased Aβ load in the frontal lobe of patients with mild-to-moderate dementia due to AD, Aβ40, and Aβ42 in the CSF were reduced at 6 and 12 months after treatment, respectively. Additionally, hippocampal volume was larger and pTau-181 levels in CSF were lower in the nilotinib group than that in the placebo group.613 These trials provide strong prospects for the upcoming phase III clinical trial to evaluate the effectiveness and safety of masitinib and nilotinib in early-stage AD.
AZD0530, an inhibitor of Src family kinases including Fyn, is developed to treat mild to moderate AD. In P301S transgenic mice, chronic AZD0530 treatment prevents spatial memory and passive avoidance learning deficits, accompanied by reduced accumulation of phosphorylated tau in the hippocampus, decreased glial activation, and restoration of presynaptic markers indicative of notable neuroprotection.614 A phase Ib multicenter, randomized, double-blind, placebo-controlled trial (ClinicalTrials.gov identifier: NCT01864655) lasted 4 weeks with 24 subjects and was conducted using escalating doses (50 mg, 100 mg, 125 mg per day) of AZD0530, and demonstrated that an oral dose of 100-125 mg achieved significant CNS penetration in patients with mild to moderate AD, with good safety and tolerability. Subsequently, a larger phase IIa clinical trial was conducted to evaluate whether AZD0530 treatment could slow down the decline in the cerebral metabolic rate of glucose (CMRgl), as well as its safety and tolerability. However, no significant effects on relative CMRgl in AD-related regions of interest were observed. Secondary volumetric magnetic resonance imaging analysis showed no therapeutic protective effects of AZD0530 on total brain volume or ventricular volume, but did indicate a trend of slowing down the reduction of hippocampal volume and olfactory thickness. Therefore, targeting the Fyn kinase may be further evaluated as a therapeutic strategy for AD.
Other kinase modulators
Other kinase modulators have been investigated for AD treatment, with targets including PKC activators, Dyrk1A modulators, and JAK1/2 inhibitors. Bryostatin 1 activates PKCε by binding to the C1A and C1B domains of both conventional and novel PKC subtypes, which leads to increased levels of BDNF and PSD95, reversed synaptic loss, and accelerated synaptic maturation in AD mouse models.615 Additionally, bryostatin 1 enhanced synaptogenesis in cortical neurons while reducing dendritic spine density in a PKC-dependent manner.616 Elevated levels of PKCε during the first 24 weeks in one expanded access patient trial (ClinicalTrials.gov identifier: NCT02221947) were closely associated with cognitive benefits indicated by Mini-Mental State Examination and ADCS-ADL psychometrics.617 These findings offer preliminary support for bryostatin 1 as a potential therapeutic drug for AD.
Epigallocatechin-3-gallate (EGCG) is the predominant catechin present in green tea and has been shown to attenuate the excessive expression of Dyrk1A and APP in the brains of Down syndrome (DS) mouse model.618 Clinical trials have demonstrated that a 12-month intervention with EGCG and cognitive training yields significant improvements in visual recognition memory, inhibitory control, and adaptive behavior compared to placebo-alone and cognitive training-alone groups in DS patients.619 Given the high-degree association between DS and AD, subsequent preclinical investigations using the combination of EGCG with docosahexaenoic acid and α-lipoic acid have demonstrated potent anti-inflammatory and neuroprotective effects in a mouse model of AD, highlighting a protective role of EGCG from a daily diet in reducing the risk of AD.620 Nevertheless, it should be noted that EGCG is also an antioxidant and thus the effects may not necessarily be related to its kinase activity.
JAK1/2 inhibitor baricitinib, an anti-inflammatory drug used to treat rheumatoid arthritis,621 rescues inflammatory biomarkers and attenuates cell death in a dose-dependent manner in vitro.622,623 A recent phase I/II clinical trial (ClinicalTrials.gov identifier: NCT05189106) targeting mild cognitive impairment, AD, and ALS has commenced to investigate the penetration of baricitinib into CSF at daily doses of 2 mg or 4 mg and the effects on inflammatory biomarkers in the CSF of patients at risk of AD or with ALS. It will be concluded by June 2025.
PD, SCAs, and HD
There are fewer kinase inhibitors entering phase II-III stages of clinical trials for PD, and completed and ongoing ones primarily assess c-Abl tyrosine kinase inhibitors such as nilotinib and vodobatinib (K0706). However, in clinical practice, nilotinib at the maximal permissible dose for 1 to 6 months has limited penetration into the CNS, resulting in minimal improvement in symptoms and no improvement in cognitive impairment in PD patients.624 In contrast, a more specific c-Abl inhibitor, K0706, has shown higher affinity with c-Abl in vitro than nilotinib. When orally administered to healthy volunteers and PD patients for 7 and 14 days, respectively, vodobatinib indicated better CNS penetration in PD patients than nilotinib, and more importantly, a significantly higher inhibitory potential on c-Abl is observed with vodobatinib in the brain, which was also indicated by the CSF pharmacokinetic curve of drugs, showing the same conclusion of better neuroprotective effects of vodobatinib.624 A phase II clinical trial evaluating the safety and efficacy of K0706 in early-stage PD patients has commenced the recruitment phase (ClinicalTrials.gov identifier: NCT03655236). Currently, a phase II clinical trial evaluating the efficacy and safety of nilotinib for 1 year has been completed in patients with autosomal dominant spinocerebellar ataxia (ClinicalTrials.gov identifier: NCT03932669). The data from this study suggest that nilotinib may improve the severity of ataxia in patients with autosomal dominant spinocerebellar ataxia, and serum protein markers (including leucine-rich α-2-glycoprotein, vitamin D binding protein, and C4b binding protein β and α chain) may be clues to predict response to nilotinib.625
Similarly, limited kinase-targeted drugs are tested clinically for HD. Since the low levels of BDNF in the brain and CSF of HD patients may account for the disruption of the cortical-striatal circuit and disease progression, kinase-related drugs responsible for BDNF regulation or improving vesicular transport or expression levels of BDNF may be a viable approach.626,627 In line with this, a phase II clinical trial (ClinicalTrials.gov identifier: NCT00095355) initiated in 2004 has investigated the effects of lithium carbonate and sodium valproate (primarily used for mood disorders and epileptic seizures) on BDNF expression in CSF of HD patients, and whether TKs and ERK signaling pathways are involved, although the final report was not released after the study was completed in 2005. The protein expression and function of CREB and BDNF are abnormal in the brains of HD patients. Phosphodiesterase inhibition restored the protein levels of CREB and BDNF in the brain of the R6/2 transgenic mouse model, thereby reducing the degeneration of the striatum and cortex.628 Therefore, the primary focus of HD-related clinical trials later turned to the efficacy and safety of phosphodiesterase inhibitors. For example, a positron emission tomography clinical trial of rolipram, a selective phosphodiesterase 4 inhibitor, began in 2011 (ClinicalTrials.gov identifier: NCT01602900), but the results have not yet been published.
ALS and CTE
ROCK inhibitor
Fasudil, which effectively targets ROCK, is proven to be a potential drug for the treatment of ALS. In 2013, oral doses of fasudil at 30 or 100 mg/kg were administered to SOD1G93A mice, and fasudil reduced ROCK activity and tensin homolog expression, increased p-AKT levels, and thereby prevented the death of motor neurons and mitigated the disease progression.556 Intriguingly, the release of pro-inflammatory factors tumor necrosis factor-α, interleukin-6, and chemokines such as C-C motif ligand 2, C-C motif ligand 3, and C-C motif ligand 5 was consistently decreased by fasudil in the spinal cord of SOD1G93A mice. Fasudil also facilitated the process of neuromuscular junction by remodeling the motor axons of the sciatic nerve. It notably improved the motor behavior of male SOD1G93A mice with good tolerance, likely due to the higher ROCK activity present in the male mice at the advanced stage of ALS receiving better drug response.629 Taken together, although firstly approved for vasospasm, fasudil exerts great therapeutic potential in treating ALS.
The first clinical trial (ClinicalTrials.gov identifier: NCT01935518), initiated in September 2013 aiming to assess the safety and effectiveness of fasudil in ALS patients, was concluded without releasing the results. In addition, a phase IIa clinical trial, known as the ROCK-ALS trial (initiated recruitment in 2019 from 16 centers across Germany, France, and Switzerland. ClinicalTrials.gov identifier: NCT03792490), was proposed to evaluate the safety, tolerance, and efficacy of fasudil as an intervention in early-stage ALS patients, encompassing a total of 120 individuals suspected or diagnosed with ALS within 6-18 months of experiencing muscle weakness, and the results of this study have shown that fasudil is well tolerated and safe in patients with ALS.630,631 In March 2020, the compassionate use of fasudil was reported in three ALS patients, a 66-year-old male and two females aged 62 and 68, indicating well tolerance.632 Overall, fasudil has shown promising effects in ALS animal models, and the clinical benefits have been demonstrated in trials.
RIPK1 inhibitors
Since RIPK1 is essential in cell death decisions, including necroptosis and RIPK1-dependent apoptosis, its inhibitor has been tested in several disease models including ALS. Inhibition of RIPK1 prevents oligodendrocyte degeneration that precedes the onset of motor dysfunction in SODG93A transgenic mice.71 GSK2982772, a type III inhibitor of RIPK1, is being developed for the treatment of chronic inflammatory diseases. A phase I safety trial of oral safety, tolerability, pharmacokinetics, and exploratory pharmacodynamics of GSK2982772 has been completed in healthy male volunteers. The results showed that single and repeated doses of oral GSK2982772 are safe and well tolerated633 (ClinicalTrials.gov identifier: NCT02302404). Another brain-penetrant small molecule inhibitor of RIPK1 is DNL747, also known as SAR443060 (reducing RIPK1 phosphorylation at Ser166), which has also completed phase I safety, tolerability, pharmacokinetics, and pharmacodynamics trials in AD or ALS patients (ClinicalTrials.gov identifier: NCT03757325, NCT03757351). The results showed that DNL747 was safe and well tolerated in AD and ALS patients, exhibited good blood-brain barrier permeability after oral administration, and had good peripheral target binding ability,634 which brings potential therapeutic prospects for AD, ALS, and other neurodegenerative diseases. In addition, DNL788 (also known as SAR443820), as a potent, selective, brain-penetrating RIPK1 small molecule inhibitor, showed good safety, pharmacokinetics, and strong interaction with the target in a phase I clinical trial conducted in healthy adult participants (ClinicalTrials.gov identifier: NCT05795907). Subsequently, DNL788 entered phase II clinical trials for the treatment of ALS in 2022 (ClinicalTrials.gov identifier: NCT05237284). Unfortunately, Denali recently stated in a document submitted to the U.S. Securities and Exchange Commission that the phase II Himalaya trial of DNL788 failed to reach the primary endpoint of the ALS-Functional Rating Scale (https://alsnewstoday.com/news/denali-therapy-candidate-fails-slowing-als-progression-trial/), where the official clinical trial data has not yet been released. Therefore, the clinical drug research targeting RIPK1 for the treatment of ALS still encounters significant challenges.
Other kinase inhibitors
Other potential kinase inhibitors tested for ALS include TK inhibitors (such as JAK inhibitors) and MEK inhibitors that downregulate the MAPK/ERK pathway. Highly selective TK inhibitor masitinib exhibits considerable preventive effects against CNS neuroinflammation in ALS, stroke, and AD. Neuronal death and rapid progression of paralysis involve abnormal proliferation of spinal cord microglial cells and activation of astrocytes in the SOD1G93A rat, while oral administration of masitinib after the onset of paralysis reduced these pathologies. Noteworthily, masitinib treatment initiated 7 days after the onset of paralysis extended post-paralysis survival by 40%.635 In vitro, masitinib also effectively inhibits colony-stimulating factor-induced proliferation, cell migration, and expression of inflammatory mediators in cultured spinal cord microglial cells.635,636 These preclinical findings provide compelling biological evidence supporting the use of masitinib for ALS-related neuroinflammation.
A randomized phase III clinical trial AB10015 (ClinicalTrials.gov identifier: NCT02588677) has investigated masitinib as an adjunct therapy to riluzole in ALS patients. In this trial, 394 patients were assigned in a 1:1:1 ratio to receive either riluzole (100 mg/day) plus placebo, or masitinib at doses of 4.5 or 3.0 mg/kg/day. A higher dose of masitinib was well-tolerated and effectively decelerated the rate of functional decline in ALS patients, as assessed by the ALS-Functional Rating Scale-Revised.637 Subsequently, an evaluation of long-term overall survival data was conducted for all participants in the AB10015 study, suggesting that initiating oral administration of masitinib (4.5 mg/kg/day) before severe impairment of neuromuscular function can prolong patient survival by over 2 years compared to that of the placebo.638 A confirmatory phase III clinical trial to validate the efficacy and safety of masitinib versus placebo in combination with riluzole for ALS treatment was initiated in 2021 (ClinicalTrials.gov identifier: NCT03127267) and is currently in the recruitment phase.
JAK inhibitors proven to be potent anti-inflammatory drugs include tofacitinib (inhibiting JAK1/2/3), baricitinib (inhibiting JAK1/2), and upadacitinib (selectively inhibiting JAK1), all of which have received FDA approval for treating rheumatoid arthritis. However, the clinical investigation of JAK inhibitors for the treatment of neurodegenerative diseases is still in the early stages. A phase I/II clinical trial has been initiated to assess whether baricitinib (at doses of 2 or 4 mg/day) could achieve therapeutic concentration in the CSF and inhibit type I interferon-related inflammatory biomarkers in ALS, AD, and mild cognitive impairment patients, with an estimated completion in June 2025 (ClinicalTrials.gov identifier: NCT05189106). In addition to ALS and AD, a multicenter randomized controlled phase II clinical trial studying the therapeutic effect of baricitinib (given daily at the dosage of 4 mg) on patients with moderate to severe traumatic intracerebral hemorrhage/cerebral contusion is also being planned (the current status is not yet recruiting), and is expected to be completed in December 2025 (ClinicalTrials.gov identifier: NCT06065046).
Trametinib (GSK1120212, SNR1611) is a selective MEK1/2 inhibitor approved for the clinical treatment of metastatic melanoma with BRAF V600E or V600K mutations.639,640 A phase I/II clinical trial evaluating the safety, tolerability, and efficacy of trametinib in ALS patients was initiated in 2020 (ClinicalTrials.gov identifier: NCT04326283), aiming to assess whether suppressing the MAPK/ERK pathway by trametinib is of benefit for ALS, and the trial is currently recruiting participants.
Concluding remarks
Protein kinases play diverse roles in signal pathways engaged in cellular metabolism, cell growth, and neuroinflammation, positioning them as central targets for treating neurodegenerative diseases. However, the intricate network of protein kinase-protein interactions and mechanisms of action have limited the thorough understanding of their functions, inhibitors, and activators in diseases. Extensive research has been focusing on unraveling the unique roles and regulatory functions of different protein kinases in specific neurodegenerative diseases.
The inter-regulatory network constructed by diverse protein kinases such as GSK3β, CDK5, CK1, PKA, p38 MAPK, Fyn, TTBK1, AMPK, and others serves as an imperative hub to guide the pathogenesis and progression of AD. In the AD brain, abnormal protein hydrolysis of p35-CDK5 by Aβ, Ca2+, and calpain-1 leads to the generation of p25-CDK5, which in turn activates the cell cycle re-entry, mitochondrial dysfunction, and apoptosis. Elevated CK1 activity leads to the excessive activation of GSK3β and CDK5, and the former further triggers Fyn activation, exacerbating neuronal excitotoxicity and neural network function, and ultimately contributing to neurodegeneration in AD. Additionally, AMPK, by inhibiting mTOR activity, promotes autophagy and reduces Aβ toxicity. The reduced PKA activity in AD results in decreased expression of CREB/BDNF and SIRT1, giving rise to enhanced Aβ production and impaired synaptic plasticity and memory function. Furthermore, increased activity of p38 MAPK is mainly responsible for the neuroinflammation and oxidative stress in AD. Therefore, the predominant focus is being put on p38α MAPK, GSK3, and tyrosine kinases in the therapeutic landscape for AD, and the majority of these therapies are still in phase I/II clinical trials.
mHTT pathogenesis is notably accelerated by a signaling network that engages AKT, ERK, p38, JNK, CDK5, and IKKβ. Conversely, these kinase signaling cascades are inevitably influenced by the action of abnormal mHTT. For instance, mHTT aggregation inhibits the upstream PI3K/AKT or the ERK/CREB signaling, which are proven to be neuroprotective. Alternatively, mHTT activates the IKKβ/NF-κB pathway and promotes inflammatory signaling, in addition to p38 activation-induced cell apoptosis. Furthermore, MEK/ERK activation by mHTT enhances HTT phosphorylation, thereby disrupting the transport and uptake of neurotransmitter glutamate. While fALS only accounts for a very small percentage of ALS, the majority of sporadic ALS cases with indefinable causes are the principal challenges for clinical treatment. Etiological studies of ALS have highlighted the importance of a homeostatic environment regulated by kinase networks. Clinical trials targeting ROCK, RIPK1, and MEK in ALS are currently in phase I/II, and tyrosine kinase inhibitors have reached phase III. However, due to the complexity of protein kinase signaling networks, modifying the activity of one kinase in purpose may yield broad outcomes from various respects in distinct neurodegenerative diseases, possibly resulting from interferences of the dynamics of its interacting kinases, therefore disruption of physiological processes by such inhibitor may also result in potential side effects.641
It is worth noting that most neurodegenerative diseases are related to family genetic inheritance. Although gene expression is related to the environment, genes determine the phenotype to a large extent. Therefore, variations in kinase genes may be another important reason that affects kinase function, leads to disease and affects pathological progression. In AD cohort studies, it has been confirmed that a single nucleotide polymorphism (rs2651206) located in the TTBK1 gene may promote the occurrence of sporadic late-onset AD in the Chinese Han population.285 SNPs in nucleotides within the functional domain of LRRK2 have been identified that cause common and sporadic forms of PD, including G2019S (the most common and inherited in an autosomal dominant manner), I2020T, N1437H, R1441C, R1441G, I1441H, and Y1699C, these variants all lead to increased LRRK2 kinase activity.63 On the contrary, LRRK2 also has protective variants in PD (such as R1398H and N551K) that reduce LRRK2 activity,642 which suggests that kinase gene mutations have two sides in disease progression and maintaining wild-type LRRK2 kinase activity is not necessarily optimal in PD treatment. NEK1 variants include frameshift variants (p.Glu929Asnfs*12) and missense variants (p.Val713Met, p.Ser909Cys and p.Arg1073Cys), all of which reduce the protein level of NEK1 in fibroblasts from ALS patients, leading to a partial loss of NEK1 function and promoting the progression of ALS.643 TBK1 site mutation (R228H) causes neurodegeneration by reducing TBK1 activity, thereby increasing the course of frontotemporal dementia-ALS.644 Gene sequence analysis of population cohort data showed that missense mutations (Gln127Arg) in the PRKCG gene may cause the occurrence of SCA14645 (disease-related variations in genes can be found at https://rddc.tsinghua-gd.org/). Similarly, mutations in non-kinase genes such as TARDBP, mHTT, and OPTN may also promote/reduce kinase activity by regulating abnormal protein expression, thereby unbalancing protein homeostasis and promoting the occurrence of neurodegenerative diseases. Therefore, when performing gene therapy or ASO therapy on kinase genes, it may be necessary to consider the physiological function of the kinase.
In summary, kinase signaling networks play a double-edged sword role in the progression of neurodegenerative diseases and are closely linked to neurodegenerative lesions advance. When developing kinase-targeted drugs and formulating dosing strategies, in addition to physiological barriers in the CNS like BBB and the blood-CSF barrier, factors such as the drug delivery window, chronic inflammation, and acute immune activation may also need to be considered. While the link between kinase regulatory networks and neurodegenerative diseases remains incompletely understood, researchers are making steady progress in developing better drugs targeting protein kinases and more efficient delivery techniques directed to the CNS. Targeted kinase therapy for neurodegenerative diseases continues to hold potential and challenges simultaneously.
References
Roskoski, R. Jr A historical overview of protein kinases and their targeted small molecule inhibitors. Pharm. Res. 100, 1–23 (2015).
Attwood, M. M. et al. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat. Rev. Drug Discov. 20, 839–861 (2021).
Manning, G. et al. The protein kinase complement of the human genome. Science 298, 1912–1934 (2002).
Zhang, H. et al. A subcellular map of the human kinome. Elife. 10, e64943 (2021).
Klaeger, S. et al. The target landscape of clinical kinase drugs. Science 358, eaan4368 (2017).
Liu, W. S. et al. Plasma proteomics identify biomarkers and undulating changes of brain aging. Nat Aging 5, 99–112 (2024).
Nandi, A. et al. Global and regional projections of the economic burden of Alzheimer’s disease and related dementias from 2019 to 2050: a value of statistical life approach. EClinicalMedicine 51, 101580 (2022).
Budd Haeberlein, S. et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 9, 197–210 (2022).
van Dyck, C. H. et al. Lecanemab in early Alzheimer’s Disease. N. Engl. J. Med. 388, 9–21 (2023).
Sims, J. R. et al. Donanemab in early symptomatic Alzheimer Disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 330, 512–527 (2023).
Mummery, C. J. et al. Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nat. Med. 29, 1437–1447 (2023).
Bejanin, A. & Villain, N. Posterior cortical atrophy: new insights into treatments and biomarkers for Alzheimer’s disease. Lancet Neurol. 23, 127–128 (2024).
Congdon, E. E. et al. Tau-targeting therapies for Alzheimer disease: current status and future directions. Nat. Rev. Neurol. 19, 715–736 (2023).
Lang, A. E. et al. Trial of cinpanemab in early Parkinson’s Disease. N. Engl. J. Med. 387, 408–420 (2022).
Pagano, G. et al. Trial of prasinezumab in early-stage Parkinson’s Disease. N. Engl. J. Med. 387, 421–432 (2022).
Buur, L. et al. Randomized phase I trial of the alpha-synuclein antibody Lu AF82422. Mov. Disord. 39, 936–944 (2024).
Sengupta, U. & Kayed, R. Amyloid beta, Tau, and alpha-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 214, 102270 (2022).
Paganoni, S. et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 383, 919–930 (2020).
Jaiswal, M. K. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 39, 733–748 (2019).
Miller, T. M. et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442 (2013).
Blair, H. A. Tofersen: first approval. Drugs 83, 1039–1043 (2023).
van den Berg, L. H. et al. Safety, tolerability, and pharmacokinetics of antisense oligonucleotide BIIB078 in adults with C9orf72-associated amyotrophic lateral sclerosis: a phase 1, randomised, double blinded, placebo-controlled, multiple ascending dose study. Lancet Neurol. 23, 901–912 (2024).
Kim, G. et al. Genome-wide CRISPR screen reveals v-ATPase as a drug target to lower levels of ALS protein ataxin-2. Cell Rep. 41, 111508 (2022).
Sengupta, A. et al. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 357, 299–309 (1998).
Parra Bravo, C., Naguib, S. A. & Gan, L. Cellular and pathological functions of tau. Nat. Rev. Mol. Cell Biol. 25, 845–864 (2024).
Mangiafico, S. P. et al. Tau suppresses microtubule-regulated pancreatic insulin secretion. Mol. Psychiatr. 28, 3982–3993 (2023).
Johnson, G. V. & Stoothoff, W. H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 117, 5721–5729 (2004).
Ramalingam, N. & Dettmer, U. alpha-Synuclein serine129 phosphorylation - the physiology of pathology. Mol. Neurodegener. 18, 84 (2023).
Neumann, M. et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 117, 137–149 (2009).
Heras-Sandoval, D., Perez-Rojas, J. M., Hernandez-Damian, J. & Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 26, 2694–2701 (2014).
Muraleedharan, R. & Dasgupta, B. AMPK in the brain: its roles in glucose and neural metabolism. FEBS J. 289, 2247–2262 (2022).
Ding, C., Wu, Y., Dabas, H. & Hammarlund, M. Activation of the CaMKII-Sarm1-ASK1-p38 MAP kinase pathway protects against axon degeneration caused by loss of mitochondria. Elife. 11 (2022).
Hara, S. et al. Serine 129 phosphorylation of membrane-associated alpha-synuclein modulates dopamine transporter function in a G protein-coupled receptor kinase-dependent manner. Mol. Biol. Cell 24, 1649–1660 (2013). S1641-1643.
Bergeron, M. et al. In vivo modulation of polo-like kinases supports a key role for PLK2 in Ser129 alpha-synuclein phosphorylation in mouse brain. Neuroscience 256, 72–82 (2014).
Degterev, A., Ofengeim, D. & Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl Acad. Sci. USA 116, 9714–9722 (2019).
Zhao, B. et al. Targeting RACK1 to alleviate TDP-43 and FUS proteinopathy-mediated suppression of protein translation and neurodegeneration. Acta Neuropathol. Com. 11, 200 (2023).
Tolosa, E., Vila, M., Klein, C. & Rascol, O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107 (2020).
Eid, S. et al. KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinforma. 18, 16 (2017).
Wang, B. et al. An overview of kinase downregulators and recent advances in discovery approaches. Signal Transduct. Tar. 6, 423 (2021).
Kanev, G. K. et al. The landscape of atypical and eukaryotic protein kinases. Trends Pharm. Sci. 40, 818–832 (2019).
Gomez-Puerta, J. A. & Mocsai, A. Tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Curr. Top. Med. Chem. 13, 760–773 (2013).
Robinson, D. R., Wu, Y. M. & Lin, S. F. The protein tyrosine kinase family of the human genome. Oncogene 19, 5548–5557 (2000).
Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010).
Qi, D. et al. HO-1 attenuates hippocampal neurons injury via the activation of BDNF-TrkB-PI3K/Akt signaling pathway in stroke. Brain Res. 1577, 69–76 (2014).
Yuan, H. et al. The regulatory mechanism of neurogenesis by IGF-1 in adult mice. Mol. Neurobiol. 51, 512–522 (2015).
Shen, J. et al. Neurovascular coupling in the dentate gyrus regulates adult hippocampal neurogenesis. Neuron 103, 878–890 e873 (2019).
Liang, S. et al. Neuroprotective profile of novel SRC kinase inhibitors in rodent models of cerebral ischemia. J. Pharm. Exp. Ther. 331, 827–835 (2009).
Lennmyr, F. et al. Src family kinase-inhibitor PP2 reduces focal ischemic brain injury. Acta Neurol. Scand. 110, 175–179 (2004).
Tuo, Q. Z. et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct. Target Ther. 7, 59 (2022).
Tuo, Q. Z. & Lei, P. Ferroptosis in ischemic stroke: animal models and mechanisms. Zool. Res. 45, 1235–1248 (2024).
Matrone, C., Petrillo, F., Nasso, R. & Ferretti, G. Fyn tyrosine kinase as harmonizing factor in neuronal functions and dysfunctions. Int. J. Mol. Sci. 21, 4444 (2020).
Brown, M. T. & Cooper, J. A. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1287, 121–149 (1996).
Nada, S. et al. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell 73, 1125–1135 (1993).
Nguyen, T. H., Liu, J. & Lombroso, P. J. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 277, 24274–24279 (2002).
Liebl, E. C. et al. Dosage-sensitive, reciprocal genetic interactions between the Abl tyrosine kinase and the putative GEF trio reveal trio’s role in axon pathfinding. Neuron 26, 107–118 (2000).
Lebouvier, T. et al. The microtubule-associated protein tau is also phosphorylated on tyrosine. J. Alzheimers Dis. 18, 1–9 (2009).
Hatami, M. et al. STAT5a and STAT6 gene expression levels in multiple sclerosis patients. Cytokine 106, 108–113 (2018).
Brooks, A. J. et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 344, 1249783 (2014).
Hebenstreit, D., Horejs-Hoeck, J. & Duschl, A. JAK/STAT-dependent gene regulation by cytokines. Drug N. Perspect. 18, 243–249 (2005).
Luoqian, J. et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell Mol. Immunol. 19, 913–924 (2022).
Shen, D. et al. Genome-wide and functional analyses of tyrosine kinase-like family genes reveal potential roles in development and virulence in mosquito pathogen Pythium guiyangense. Fungal Genet. Biol. 130, 11–18 (2019).
Galatsis, P. Leucine-rich repeat kinase 2 inhibitors: a patent review (2014-2016). Expert Opin. Ther. Pat. 27, 667–676 (2017).
Myasnikov, A. et al. Structural analysis of the full-length human LRRK2. Cell 184, 3519–3527 e3510 (2021).
Wang, X. et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 21, 1931–1944 (2012).
Seol, W., Nam, D. & Son, I. Rab GTPases as physiological substrates of LRRK2 Kinase. Exp. Neurobiol. 28, 134–145 (2019).
Ho, D. H. et al. LRRK2 kinase activity induces mitochondrial fission in microglia via Drp1 and modulates neuroinflammation. Exp. Neurobiol. 27, 171–180 (2018).
Liu, J. et al. The structure of mouse RIPK1 RHIM-containing domain as a homo-amyloid and in RIPK1/RIPK3 complex. Nat. Commun. 15, 6975 (2024).
Meng, H. et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc. Natl. Acad. Sci. USA 115, E2001–E2009 (2018).
Ofengeim, D. & Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14, 727–736 (2013).
Yuan, J., Amin, P. & Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2019).
Ito, Y. et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608 (2016).
Ofengeim, D. et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 114, E8788–E8797 (2017).
Lule, S. et al. Genetic inhibition of receptor interacting protein kinase-1 reduces cell death and improves functional outcome after intracerebral hemorrhage in mice. Stroke 48, 2549–2556 (2017).
Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014).
Holzman, L. B., Merritt, S. E. & Fan, G. Identification, molecular cloning, and characterization of dual leucine zipper bearing kinase. A novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. J. Biol. Chem. 269, 30808–30817 (1994).
Ferraris, D., Yang, Z. & Welsbie, D. Dual leucine zipper kinase as a therapeutic target for neurodegenerative conditions. Future Med. Chem. 5, 1923–1934 (2013).
Shin, J. E. et al. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74, 1015–1022 (2012).
Welsbie, D. S. et al. Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron 94, 1142–1154 e1146 (2017).
Hirai, S. et al. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J. Neurosci. 26, 11992–12002 (2006).
Hu, X. et al. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 11, 56–64 (2015).
Liu, B. et al. Interleukin-1 receptor-associated kinase (IRAK)-M -mediated type 2 microglia polarization ameliorates the severity of experimental autoimmune encephalomyelitis (EAE). J. Autoimmun. 102, 77–88 (2019).
Miranda-Hernandez, S. & Baxter, A. G. Role of toll-like receptors in multiple sclerosis. Am. J. Clin. Exp. Immunol. 2, 75–93 (2013).
Akhurst, R. J. & Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811 (2012).
Wu, X. et al. Photoactivation of TGFbeta/SMAD signaling pathway ameliorates adult hippocampal neurogenesis in Alzheimer’s disease model. Stem Cell Res. Ther. 12, 345 (2021).
Arany, P. R. et al. Photoactivation of endogenous latent transforming growth factor-beta1 directs dental stem cell differentiation for regeneration. Sci. Transl. Med. 6, 238ra269 (2014).
Tebben, A. J. et al. Crystal structures of apo and inhibitor-bound TGFbetaR2 kinase domain: insights into TGFbetaR isoform selectivity. Acta Crystallogr. D Struct. Biol. 72, 658–674 (2016).
Varjosalo, M. et al. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 3, 1306–1320 (2013).
Cheung, Z. H. & Ip, N. Y. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 22, 169–175 (2012).
Bianchetta, M. J., Lam, T. T., Jones, S. N. & Morabito, M. A. Cyclin-dependent kinase 5 regulates PSD-95 ubiquitination in neurons. J. Neurosci. 31, 12029–12035 (2011).
Plattner, F. et al. Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron 81, 1070–1083 (2014).
Zhong, P. et al. Cyclin-dependent kinase 5 in the ventral tegmental area regulates depression-related behaviors. J. Neurosci. 34, 6352–6366 (2014).
Lebel, M. et al. Dopamine D1 receptor activation induces tau phosphorylation via cdk5 and GSK3 signaling pathways. Neuropharmacology 57, 392–402 (2009).
Liu, S. L. et al. The role of Cdk5 in Alzheimer’s disease. Mol. Neurobiol. 53, 4328–4342 (2016).
Fu, A. K. et al. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc. Natl. Acad. Sci. USA 101, 6728–6733 (2004).
Lin, W. et al. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron 46, 569–579 (2005).
Wang, C. X. et al. Cyclin-dependent kinase-5 prevents neuronal apoptosis through ERK-mediated upregulation of Bcl-2. Cell Death Differ. 13, 1203–1212 (2006).
Malumbres, M. Cyclin-dependent kinases. Genome Biol. 15, 122 (2014).
Chivukula, S. & Malkhed, V. The role of CDK20 protein in carcinogenesis. Curr. Drug Targets 24, 790–796 (2023).
Kyriakis, J. M. & Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737 (2012).
Larhammar, M. et al. The Ste20 family kinases MAP4K4, MINK1, and TNIK converge to regulate stress-induced JNK signaling in neurons. J. Neurosci. 37, 11074–11084 (2017).
Taylor, D. M. et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J. Neurosci. 33, 2313–2325 (2013).
Gee, M. S. et al. A selective p38alpha/beta MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimers Res. Ther. 12, 45 (2020).
Shaw, P. C. et al. Isolation and chromosomal mapping of human glycogen synthase kinase-3 alpha and -3 beta encoding genes. Genome 41, 720–727 (1998).
Yao, H. B., Shaw, P. C., Wong, C. C. & Wan, D. C. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 23, 291–297 (2002).
Zhao, J. et al. GSK3: a potential target and pending issues for treatment of Alzheimer’s disease. CNS Neurosci. Ther. 30, e14818 (2024).
Wang, L., Li, J. & Di, L. J. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 42, 946–982 (2022).
Li, S. et al. DYRK1A interacts with histone acetyl transferase p300 and CBP and localizes to enhancers. Nucleic Acids Res. 46, 11202–11213 (2018).
Hao, N. et al. Combined computational and experimental analysis reveals mitogen-activated protein kinase-mediated feedback phosphorylation as a mechanism for signaling specificity. Mol. Biol. Cell 23, 3899–3910 (2012).
Chuang, H. C., Wang, X. & Tan, T. H. MAP4K Family Kinases in Immunity and Inflammation. Adv. Immunol. 129, 277–314 (2016).
Li, Y. et al. Activation of MAP3K DLK and LZK in Purkinje cells causes rapid and slow degeneration depending on signaling strength. Elife. 10, e63509 (2021).
Bos, P. H. et al. Development of MAP4 kinase inhibitors as motor neuron-protecting agents. Cell Chem. Biol. 26, 1703–1715.e1737 (2019).
Arencibia, J. M. et al. AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim. Biophys. Acta 1834, 1302–1321 (2013).
Leroux, A. E., Schulze, J. O. & Biondi, R. M. AGC kinases, mechanisms of regulation and innovative drug development. Semin Cancer Biol. 48, 1–17 (2018).
Brandon, E. P., Idzerda, R. L. & McKnight, G. S. PKA isoforms, neural pathways, and behaviour: making the connection. Curr. Opin. Neurobiol. 7, 397–403 (1997).
Dagda, R. K. & Das Banerjee, T. Role of protein kinase A in regulating mitochondrial function and neuronal development: implications to neurodegenerative diseases. Rev. Neurosci. 26, 359–370 (2015).
Knighton, D. R. et al. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 407–414 (1991).
Taylor, S. S. et al. PKA: lessons learned after twenty years. Biochim Biophys. Acta 1834, 1271–1278 (2013).
Zheng, J. et al. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 32, 2154–2161 (1993).
Pearce, L. R., Komander, D. & Alessi, D. R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 (2010).
Hergovich, A. Regulation and functions of mammalian LATS/NDR kinases: looking beyond canonical Hippo signalling. Cell Biosci. 3, 32 (2013).
Turnham, R. E. & Scott, J. D. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene 577, 101–108 (2016).
Pretre, V. & Wicki, A. Inhibition of Akt and other AGC kinases: a target for clinical cancer therapy? Semin. Cancer Biol. 48, 70–77 (2018).
Zurashvili, T. et al. Interaction of PDK1 with phosphoinositides is essential for neuronal differentiation but dispensable for neuronal survival. Mol. Cell Biol. 33, 1027–1040 (2013).
Cordon-Barris, L. et al. Mutation of the 3-phosphoinositide-dependent protein kinase 1 (PDK1) substrate-docking site in the developing brain causes microcephaly with abnormal brain morphogenesis independently of Akt, leading to impaired cognition and disruptive behaviors. Mol. Cell Biol. 36, 2967–2982 (2016).
Henderson, B. W. et al. Pharmacologic inhibition of LIMK1 provides dendritic spine resilience against beta-amyloid. Sci Signal. 12, eaaw9318 (2019).
Muller, C. P. et al. CaM kinases: from memories to addiction. Trends Pharm. Sci. 37, 153–166 (2016).
Krebs, E. G. & Fischer, E. H. The phosphorylase b to a converting enzyme of rabbit skeletal muscle. Biochim. Biophys. Acta 20, 150–157 (1956).
Krebs, E. G., Kent, A. B. & Fischer, E. H. The muscle phosphorylase b kinase reaction. J. Biol. Chem. 231, 73–83 (1958).
Yamauchi, T. & Fujisawa, H. Evidence for three distinct forms of calmodulin-dependent protein kinases from rat brain. FEBS Lett. 116, 141–144 (1980).
Bayer, K. U. & Schulman, H. CaM Kinase: still Inspiring at 40. Neuron 103, 380–394 (2019).
Zhang, L., Luo, B., Lu, Y. & Chen, Y. Targeting death-associated protein kinases for treatment of human diseases: recent advances and future directions. J. Med. Chem. 66, 1112–1136 (2023).
Egan, D., Kim, J., Shaw, R. J. & Guan, K. L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644 (2011).
Lin, S. C. & Hardie, D. G. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 27, 299–313 (2018).
Vingtdeux, V., Davies, P., Dickson, D. W. & Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 121, 337–349 (2011).
Woods, A. et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 (2003).
Wang, X., Zimmermann, H. R. & Ma, T. Therapeutic potential of AMP-activated protein kinase in Alzheimer’s disease. J. Alzheimers Dis. 68, 33–38 (2019).
Martin, S. J., Grimwood, P. D. & Morris, R. G. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu. Rev. Neurosci. 23, 649–711 (2000).
Lisman, J., Yasuda, R. & Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182 (2012).
Hanson, P. I. & Schulman, H. Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J. Biol. Chem. 267, 17216–17224 (1992).
Lamsa, K., Irvine, E. E., Giese, K. P. & Kullmann, D. M. NMDA receptor-dependent long-term potentiation in mouse hippocampal interneurons shows a unique dependence on Ca(2+)/calmodulin-dependent kinases. J. Physiol. 584, 885–894 (2007).
Yasuda, R., Hayashi, Y. & Hell, J. W. CaMKII: a central molecular organizer of synaptic plasticity, learning and memory. Nat. Rev. Neurosci. 23, 666–682 (2022).
Harda, Z. et al. Autophosphorylation of alphaCaMKII affects social interactions in mice. Genes Brain Behav. 17, e12457 (2018).
Tao, W. et al. Synaptic memory requires CaMKII. Elife. 10, e60360 (2021).
Kim, B. M. et al. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis. 5, e1237 (2014).
Zhang, T., Kim, B. M. & Lee, T. H. Death-associated protein kinase 1 as a therapeutic target for Alzheimer’s disease. Transl. Neurodegener. 13, 4 (2024).
Su, Y. et al. MicroRNA-26a/death-associated protein kinase 1 signaling induces synucleinopathy and dopaminergic neuron degeneration in Parkinson’s disease. Biol. Psychiatry 85, 769–781 (2019).
Pei, L. et al. DAPK1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J. Neurosci. 34, 6546–6556 (2014).
Venerando, A., Ruzzene, M. & Pinna, L. A. Casein kinase: the triple meaning of a misnomer. Biochem J. 460, 141–156 (2014).
Ikezu, S. & Ikezu, T. Tau-tubulin kinase. Front Mol. Neurosci. 7, 33 (2014).
Fulcher, L. J. & Sapkota, G. P. Functions and regulation of the serine/threonine protein kinase CK1 family: moving beyond promiscuity. Biochem. J. 477, 4603–4621 (2020).
Schittek, B. & Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 13, 231 (2014).
Knippschild, U. et al. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal 17, 675–689 (2005).
Flajolet, M. et al. Regulation of Alzheimer’s disease amyloid-beta formation by casein kinase I. Proc. Natl. Acad. Sci. USA 104, 4159–4164 (2007).
Sunkari, Y. K., Meijer, L. & Flajolet, M. The protein kinase CK1: inhibition, activation, and possible allosteric modulation. Front. Mol. Biosci. 9, 916232 (2022).
Gross, S. D. & Anderson, R. A. Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell Signal 10, 699–711 (1998).
Gietzen, K. F. & Virshup, D. M. Identification of inhibitory autophosphorylation sites in casein kinase I epsilon. J. Biol. Chem. 274, 32063–32070 (1999).
Singh, T. J., Grundke-Iqbal, I. & Iqbal, K. Phosphorylation of tau protein by casein kinase-1 converts it to an abnormal Alzheimer-like state. J. Neurochem. 64, 1420–1423 (1995).
Desdouits, F., Siciliano, J. C., Greengard, P. & Girault, J. A. Dopamine- and cAMP-regulated phosphoprotein DARPP-32: phosphorylation of Ser-137 by casein kinase I inhibits dephosphorylation of Thr-34 by calcineurin. Proc. Natl. Acad. Sci. USA 92, 2682–2685 (1995).
Kloss, B., Rothenfluh, A., Young, M. W. & Saez, L. Phosphorylation of period is influenced by cycling physical associations of double-time, period, and timeless in the Drosophila clock. Neuron 30, 699–706 (2001).
Marin, O. et al. A noncanonical sequence phosphorylated by casein kinase 1 in beta-catenin may play a role in casein kinase 1 targeting of important signaling proteins. Proc. Natl. Acad. Sci. USA 100, 10193–10200 (2003).
Liu, C. et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 (2002).
Amit, S. et al. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 16, 1066–1076 (2002).
Winter, M. et al. Protein kinase CK1delta phosphorylates key sites in the acidic domain of murine double-minute clone 2 protein (MDM2) that regulate p53 turnover. Biochemistry 43, 16356–16364 (2004).
Xu, F. et al. Mammalian sterile 20-like kinase 1/2 inhibits the Wnt/beta-catenin signalling pathway by directly binding casein kinase 1epsilon. Biochem. J. 458, 159–169 (2014).
Liu, C. Y. et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J. Biol. Chem. 285, 37159–37169 (2010).
Denef, N., Neubuser, D., Perez, L. & Cohen, S. M. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102, 521–531 (2000).
Chen, Y. et al. Sonic Hedgehog-dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 9, e1001083 (2011).
De Wit, T., Baekelandt, V. & Lobbestael, E. Inhibition of LRRK2 or casein kinase 1 results in LRRK2 protein destabilization. Mol. Neurobiol. 56, 5273–5286 (2019).
Sergeant, N. et al. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev. Proteom. 5, 207–224 (2008).
Sato, S., Cerny, R. L., Buescher, J. L. & Ikezu, T. Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J. Neurochem. 98, 1573–1584 (2006).
Fry, A. M., O’Regan, L., Sabir, S. R. & Bayliss, R. Cell cycle regulation by the NEK family of protein kinases. J. Cell Sci. 125, 4423–4433 (2012).
Johnson, L. N., Noble, M. E. & Owen, D. J. Active and inactive protein kinases: structural basis for regulation. Cell 85, 149–158 (1996).
Hinz, M. & Scheidereit, C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 15, 46–61 (2014).
Courtois, G. & Gilmore, T. D. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene 25, 6831–6843 (2006).
Xiang, S. et al. TANK-binding kinase 1 (TBK1): An emerging therapeutic target for drug discovery. Drug Discov. Today 26, 2445–2455 (2021).
Tu, D. et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 3, 747–758 (2013).
Gerbino, V. et al. The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 106, 789–805.e785 (2020).
Sharma, S. et al. Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151 (2003).
Kim, J. Y. et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl. Acad. Sci. USA 110, 12414–12419 (2013).
Tokumitsu, H. & Sakagami, H. Molecular mechanisms underlying Ca(2+)/calmodulin-dependent protein kinase kinase signal transduction. Int. J. Mol. Sci. 23, 11025 (2022).
Guo, T. et al. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 15, 40 (2020).
Tarawneh, R. & Holtzman, D. M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. CSH Perspect. Med. 2, a006148 (2012).
Walczak-Nowicka, L. J. & Herbet, M. Acetylcholinesterase inhibitors in the treatment of neurodegenerative diseases and the role of acetylcholinesterase in their pathogenesis. Int. J. Mol. Sci. 22, 9290 (2021).
Song, X. et al. Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature 556, 515–519 (2018).
Tolar, M. et al. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near-term approval. Alzheimers Res Ther. 12, 95 (2020).
Mintun, M. A. et al. Donanemab in early Alzheimer’s Disease. N. Engl. J. Med. 384, 1691–1704 (2021).
Tonnies, E. & Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 57, 1105–1121 (2017).
Iqbal, K., Liu, F. & Gong, C. X. Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 12, 15–27 (2016).
Greenberg, S. M. et al. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat. Rev. Neurol. 16, 30–42 (2020).
Sharoar, M. G. et al. Accumulation of saposin in dystrophic neurites is linked to impaired lysosomal functions in Alzheimer’s disease brains. Mol. Neurodegener. 16, 45 (2021).
Lei, P. et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295 (2012).
Jiao, L. et al. Iron metabolism mediates microglia susceptibility in ferroptosis. Front. Cell Neurosci. 16, 995084 (2022).
Kwapong, W. R. et al. Choriocapillaris reduction accurately discriminates against early-onset Alzheimer’s disease. Alzheimers Dement. 20, 4185–4198 (2024).
Yan, H. F., Tuo, Q. Z. & Lei, P. Cell density impacts the susceptibility to ferroptosis by modulating IRP1-mediated iron homeostasis. J. Neurochem. 168, 1359–1373 (2024).
Zhang, Y. W., Thompson, R., Zhang, H. & Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 4, 3 (2011).
Iwatsubo, T. et al. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13, 45–53 (1994).
Lei, P., Ayton, S. & Bush, A. I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 296, 100105 (2021).
Congdon, E. E. & Sigurdsson, E. M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415 (2018).
Lei, P. & Ayton, S. TRIMming the tangles. Sci. Bull. 68, 2507–2509 (2023).
Lei, P. et al. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 9, 29 (2014).
Augustinack, J. C., Schneider, A., Mandelkow, E. M. & Hyman, B. T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 103, 26–35 (2002).
Shukla, V., Skuntz, S. & Pant, H. C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 43, 655–662 (2012).
Planel, E., Sun, X. & Takashima, A. Role of GSK-3? In Alzheimer’s disease pathology. Drug Dev. Res. 56, 491–510 (2002).
Zhu, X. et al. Activation and redistribution of c-jun N-terminal kinase/stress-activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 76, 435–441 (2001).
Platenik, J. et al. GSK3beta, CREB, and BDNF in peripheral blood of patients with Alzheimer’s disease and depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 50, 83–93 (2014).
Triaca, V. et al. NGF controls APP cleavage by downregulating APP phosphorylation at Thr668: relevance for Alzheimer’s disease. Aging Cell 15, 661–672 (2016).
Rockenstein, E. et al. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 27, 1981–1991 (2007).
Lauretti, E., Dincer, O. & Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys. Acta Mol. Cell Res. 1867, 118664 (2020).
Qian, W. et al. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J. Alzheimers Dis. 19, 1221–1229 (2010).
Manoharan, S. D. et al. Could protein phosphatase 2A and glycogen synthase kinase-3 beta be targeted by natural compounds to ameliorate Alzheimer’s pathologies? Brain Res. 1829, 148793 (2024).
Baki, L. et al. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 23, 2586–2596 (2004).
Hadi, F., Akrami, H., Shahpasand, K. & Fattahi, M. R. Wnt signalling pathway and tau phosphorylation: a comprehensive study on known connections. Cell Biochem. Funct. 38, 686–694 (2020).
Zhang, Z. et al. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395, 698–702 (1998).
Mishra, R. et al. Glycogen synthase kinase-3beta induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J. Biol. Chem. 282, 30393–30405 (2007).
Liu, X. H. et al. Blocking GSK3beta-mediated dynamin1 phosphorylation enhances BDNF-dependent TrkB endocytosis and the protective effects of BDNF in neuronal and mouse models of Alzheimer’s disease. Neurobiol. Dis. 74, 377–391 (2015).
Mercado-Gomez, O. et al. Inhibition of Wnt and PI3K signaling modulates GSK-3beta activity and induces morphological changes in cortical neurons: role of tau phosphorylation. Neurochem. Res. 33, 1599–1609 (2008).
Fao, L., Mota, S. I. & Rego, A. C. Shaping the Nrf2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing Res. Rev. 54, 100942 (2019).
Ko, C. Y. et al. Glycogen synthase kinase-3beta-mediated CCAAT/enhancer-binding protein delta phosphorylation in astrocytes promotes migration and activation of microglia/macrophages. Neurobiol. Aging 35, 24–34 (2014).
Hernandez, F., Lucas, J. J. & Avila, J. GSK3 and tau: two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 33, S141–S144 (2013).
Ly, P. T. et al. Inhibition of GSK3 beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 123, 224–235 (2013).
Zempel, H., Thies, E., Mandelkow, E. & Mandelkow, E. M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950 (2010).
Cruz, J. C. et al. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40, 471–483 (2003).
Lopes, J. P., Oliveira, C. R. & Agostinho, P. Neurodegeneration in an Abeta-induced model of Alzheimer’s disease: the role of Cdk5. Aging Cell 9, 64–77 (2010).
Matrone, C. et al. Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc. Natl. Acad. Sci. USA 106, 11358–11363 (2009).
Lau, K. F. et al. Cyclin-dependent kinase-5/p35 phosphorylates Presenilin 1 to regulate carboxy-terminal fragment stability. Mol. Cell Neurosci. 20, 13–20 (2002).
Jayapalan, S. & Natarajan, J. The role of CDK5 and GSK3B kinases in hyperphosphorylation of microtubule-associated protein tau (MAPT) in Alzheimer’s disease. Bioinformation 9, 1023–1030 (2013).
Cancino, G. I. et al. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol. Aging 32, 1249–1261 (2011).
Tian, B., Yang, Q. & Mao, Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat. Cell Biol. 11, 211–218 (2009).
Sun, K. H., Lee, H. G., Smith, M. A. & Shah, K. Direct and indirect roles of cyclin-dependent kinase 5 as an upstream regulator in the c-Jun NH2-terminal kinase cascade: relevance to neurotoxic insults in Alzheimer’s disease. Mol. Biol. Cell 20, 4611–4619 (2009).
Li, B. S. et al. Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J. 21, 324–333 (2002).
Gong, X. et al. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 38, 33–46 (2003).
Sun, K. H., de Pablo, Y., Vincent, F. & Shah, K. Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J. Neurochem. 107, 265–278 (2008).
Park, J. et al. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem. 132, 687–702 (2015).
Chang, K. H., Vincent, F. & Shah, K. Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J. Cell Sci. 125, 5124–5137 (2012).
Kim, D. et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60, 803–817 (2008).
Chow, H. M. et al. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 22, 1806–1819 (2019).
Xie, W. et al. CEND1 deficiency induces mitochondrial dysfunction and cognitive impairment in Alzheimer’s disease. Cell Death Differ. 29, 2417–2428 (2022).
Zhou, J. et al. Cyclin-Dependent Kinase 5-Dependent BAG3 Degradation Modulates Synaptic Protein Turnover. Biol. Psychiatry 87, 756–769 (2020).
Li, Z. et al. Astrocytes deliver CK1 to neurons via extracellular vesicles in response to inflammation promoting the translation and amyloidogenic processing of APP. J. Extracell. Vesicles 10, e12035 (2020).
Roth, A. et al. CK1delta-derived peptides as novel tools inhibiting the interactions between CK1delta and APP695 to modulate the pathogenic metabolism of APP. Int. J .Mol. Sci. 22, 6423 (2021).
Kuret, J. et al. Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer’s disease brain. J. Neurochem. 69, 2506–2515 (1997).
Yang, M. et al. Casein Kinase 1delta phosphorylates TDP-43 and suppresses its function in tau mRNA processing. J. Alzheimers Dis. 91, 1527–1539 (2023).
Roth, A. et al. Comprehensive characterization of CK1delta-mediated tau phosphorylation in Alzheimer’s Disease. Front. Mol. Biosci. 9, 872171 (2022).
Meijer, L., Flajolet, M. & Greengard, P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharm. Sci. 25, 471–480 (2004).
Liu, F. et al. Regulation of cyclin-dependent kinase 5 and casein kinase 1 by metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA 98, 11062–11068 (2001).
Walter, J. et al. Phosphorylation regulates intracellular trafficking of beta-secretase. J. Biol. Chem. 276, 14634–14641 (2001).
Tong, L., Thornton, P. L., Balazs, R. & Cotman, C. W. Beta -amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival Is not compromised. J. Biol. Chem. 276, 17301–17306 (2001).
Meng, C., He, Z. & Xing, D. Low-level laser therapy rescues dendrite atrophy via upregulating BDNF expression: implications for Alzheimer’s disease. J. Neurosci. 33, 13505–13517 (2013).
Liang, Z. et al. Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer’s disease brain. J. Neurochem. 103, 2462–2470 (2007).
Tong, L., Balazs, R., Thornton, P. L. & Cotman, C. W. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J. Neurosci. 24, 6799–6809 (2004).
Chen, Y. et al. Alzheimer’s beta-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of beta-amyloid. J. Neurosci. 32, 11390–11395 (2012).
Herskovits, A. Z. & Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 81, 471–483 (2014).
Zhang, Z. et al. Activation of PKA/SIRT1 signaling pathway by photobiomodulation therapy reduces Abeta levels in Alzheimer’s disease models. Aging Cell 19, e13054 (2020).
Munoz, L. & Ammit, A. J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 58, 561–568 (2010).
Lee, J. C. et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 (1994).
Sun, A. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 183, 394–405 (2003).
Kim, S. H., Smith, C. J. & Van Eldik, L. J. Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1 beta production. Neurobiol. Aging 25, 431–439 (2004).
Decourt, B., Lahiri, D. K. & Sabbagh, M. N. Targeting tumor necrosis factor-alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 14, 412–425 (2017).
Lee, Y. B., Schrader, J. W. & Kim, S. U. p38 map kinase regulates TNF-alpha production in human astrocytes and microglia by multiple mechanisms. Cytokine 12, 874–880 (2000).
Hua, L. L. et al. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J. Neuroimmunol. 126, 180–189 (2002).
Keum, Y. S. et al. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 66, 8804–8813 (2006).
Kelleher, I. et al. Kinase activities increase during the development of tauopathy in htau mice. J. Neurochem. 103, 2256–2267 (2007).
Griffin, R. et al. The age-related attenuation in long-term potentiation is associated with microglial activation. J. Neurochem. 99, 1263–1272 (2006).
Bains, J. S. & Oliet, S. H. Glia: they make your memories stick! Trends Neurosci. 30, 417–424 (2007).
Luo, Q. et al. p38alpha-MAPK-deficient myeloid cells ameliorate symptoms and pathology of APP-transgenic Alzheimer’s disease mice. Aging Cell 21, e13679 (2022).
Schnoder, L. et al. Neuronal deficiency of p38alpha-MAPK ameliorates symptoms and pathology of APP or Tau-transgenic Alzheimer’s mouse models. FASEB J. 34, 9628–9649 (2020).
Schnoder, L. et al. Deficiency of neuronal p38alpha MAPK attenuates amyloid pathology in Alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J. Biol. Chem. 291, 2067–2079 (2016).
Ittner, A. et al. Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer’s mice. Science 354, 904–908 (2016).
Ittner, A. & Ittner, L. M. Dendritic tau in Alzheimer’s Disease. Neuron 99, 13–27 (2018).
Ittner, A. et al. Reduction of advanced tau-mediated memory deficits by the MAP kinase p38gamma. Acta Neuropathol. 140, 279–294 (2020).
Ittner, L. M. et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397 (2010).
Matrone, C., Iannuzzi, F. & Annunziato, L. The Y682ENPTY687 motif of APP: Progress and insights toward a targeted therapy for Alzheimer’s disease patients. Ageing Res. Rev. 52, 120–128 (2019).
Um, J. W. et al. Alzheimer’s amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235 (2012).
Nygaard, H. B. Targeting Fyn kinase in Alzheimer’s Disease. Biol. Psychiatry 83, 369–376 (2018).
Hernandez, P., Lee, G., Sjoberg, M. & Maccioni, R. B. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25–35): involvement of lipid rafts. J. Alzheimers Dis. 16, 149–156 (2009).
Lee, G. et al. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J. Neurosci. 24, 2304–2312 (2004).
Roberson, E. D. et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 31, 700–711 (2011).
Lau, D. H. et al. Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer’s disease. Acta Neuropathol. Commun. 4, 49 (2016).
Lopes, S. et al. Tau protein is essential for stress-induced brain pathology. Proc. Natl. Acad. Sci. USA 113, E3755–E3763 (2016).
Rong, Y. et al. Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J. Neurochem. 79, 382–390 (2001).
Kaufman, A. C. et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer’s mice. Ann. Neurol. 77, 953–971 (2015).
Li, C. & Gotz, J. Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J. 36, 3120–3138 (2017).
Sato, S. et al. Spatial learning impairment, enhanced CDK5/p35 activity, and downregulation of NMDA receptor expression in transgenic mice expressing tau-tubulin kinase 1. J. Neurosci. 28, 14511–14521 (2008).
Vazquez-Higuera, J. L. et al. Genetic variations in tau-tubulin kinase-1 are linked to Alzheimer’s disease in a Spanish case-control cohort. Neurobiol. Aging 32, 550.e555–559 (2011).
Yu, N. N. et al. Tau-tubulin kinase-1 gene variants are associated with Alzheimer’s disease in Han Chinese. Neurosci. Lett. 491, 83–86 (2011).
Lund, H. et al. Tau-tubulin kinase 1 expression, phosphorylation and co-localization with phospho-Ser422 tau in the Alzheimer’s disease brain. Brain Pathol. 23, 378–389 (2013).
Halkina, T. et al. Discovery of potent and brain-penetrant tau tubulin kinase 1 (TTBK1) inhibitors that lower tau phosphorylation in vivo. J. Med. Chem. 64, 6358–6380 (2021).
Simpkins, K. L. et al. Selective activation induced cleavage of the NR2B subunit by calpain. J. Neurosci. 23, 11322–11331 (2003).
Zhang, S. et al. Cdk5 regulates the phosphorylation of tyrosine 1472 NR2B and the surface expression of NMDA receptors. J. Neurosci. 28, 415–424 (2008).
Hawasli, A. H. et al. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat. Neurosci. 10, 880–886 (2007).
Wang, Q. et al. Scopolamine causes delirium-like brain network dysfunction and reversible cognitive impairment without neuronal loss. Zool. Res. 44, 712–724 (2023).
Assefa, B. T., Tafere, G. G., Wondafrash, D. Z. & Gidey, M. T. The bewildering effect of AMPK activators in Alzheimer’s Disease: review of the current evidence. Biomed. Res. Int. 2020, 9895121 (2020).
Ibrahim, W. W., Kamel, A. S., Wahid, A. & Abdelkader, N. F. Dapagliflozin as an autophagic enhancer via LKB1/AMPK/SIRT1 pathway in ovariectomized/D-galactose Alzheimer’s rat model. Inflammopharmacology 30, 2505–2520 (2022).
Schubert, D. Glucose metabolism and Alzheimer’s disease. Ageing Res. Rev. 4, 240–257 (2005).
Brown, G. C., Murphy, M. P., Jornayvaz, F. R. & Shulman, G. I. Regulation of mitochondrial biogenesis. Essays Biochem. 47, 69–84 (2010).
Ling, D. & Salvaterra, P. M. Brain aging and Abeta(1)(-)(4)(2) neurotoxicity converge via deterioration in autophagy-lysosomal system: a conditional Drosophila model linking Alzheimer’s neurodegeneration with aging. Acta Neuropathol. 121, 183–191 (2011).
Caccamo, A. et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120 (2010).
Cai, Z. et al. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromology 14, 1–14 (2012).
Lee, A. et al. Abeta42 oligomers trigger synaptic loss through CAMKK2-AMPK-dependent effectors coordinating mitochondrial fission and mitophagy. Nat. Commun. 13, 4444 (2022).
Berg, D. et al. Changing the research criteria for the diagnosis of Parkinson’s disease: obstacles and opportunities. Lancet Neurol. 12, 514–524 (2013).
Tang, F. et al. Inhibition of ACSL4 alleviates parkinsonism phenotypes by reduction of lipid reactive oxygen species. Neurotherapeutics 20, 1154–1166 (2023).
Jankovic, J. & Tan, E. K. Parkinson’s disease: etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 91, 795–808 (2020).
Collaborators, G. B. D. N. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 459–480 (2019).
Aarsland, D. et al. Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Prim. 7, 47 (2021).
Guo, Y. J. et al. Brain regions susceptible to alpha-synuclein spreading. Mol. Psychiatry 27, 758–770 (2022).
Santos Garcia, D. et al. Non-motor symptoms burden, mood, and gait problems are the most significant factors contributing to a poor quality of life in non-demented Parkinson’s disease patients: Results from the COPPADIS Study Cohort. Parkinsonism Relat. Disord. 66, 151–157 (2019).
Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. Lancet 397, 2284–2303 (2021).
Chen, K., Guo, Y.-J., Lei, P. & Finkelstein, D. I. Can alpha-synuclein be both the cause and a consequence of Parkinson’s disease? Ageing Neurodegener. Dis. 3, 10 (2023).
Shahmoradian, S. H. et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109 (2019).
Jankovic, J. et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson's Disease: a randomized clinical trial. JAMA Neurol. 75, 1206–1214 (2018).
Dunning, C. J., George, S. & Brundin, P. What’s to like about the prion-like hypothesis for the spreading of aggregated alpha-synuclein in Parkinson's Disease? Prion 7, 92–97 (2013).
He, S. et al. Effects of alpha-synuclein-associated post-translational modifications in Parkinson’s Disease. ACS Chem. Neurosci. 12, 1061–1071 (2021).
Dzamko, N., Zhou, J., Huang, Y. & Halliday, G. M. Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis. Front. Mol. Neurosci. 7, 57 (2014).
Chen, L. & Feany, M. B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson's disease. Nat. Neurosci. 8, 657–663 (2005).
Febbraro, F. et al. Ser129D mutant alpha-synuclein induces earlier motor dysfunction while S129A results in distinctive pathology in a rat model of Parkinson’s disease. Neurobiol. Dis. 56, 47–58 (2013).
McFarland, N. R. et al. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson's disease. J. Neuropathol. Exp. Neurol. 68, 515–524 (2009).
Sato, H., Kato, T. & Arawaka, S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: a review of in vivo models. Rev. Neurosci. 24, 115–123 (2013).
Fujiwara, H. et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164 (2002).
Arawaka, S. et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. J. Neurosci. 26, 9227–9238 (2006).
Jellinger, K. A. Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov. Disord. 27, 8–30 (2012).
Fearnley, J. M. & Lees, A. J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114, 2283–2301 (1991).
Pacelli, C. et al. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 25, 2349–2360 (2015).
Matsuda, W. et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453 (2009).
Polymeropoulos, M. H. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997).
Pihlstrom, L. & Toft, M. Genetic variability in SNCA and Parkinson’s disease. Neurogenetics 12, 283–293 (2011).
Trinh, J. et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDS gene systematic review. Mov. Disord. 33, 1857–1870 (2018).
Jha, S. K. et al. p38 MAPK and PI3K/AKT signalling cascades in Parkinson’s Disease. Int. J. Mol. Cell Med. 4, 67–86 (2015).
Morrison, R. S. et al. Neuronal survival and cell death signaling pathways. Adv. Exp. Med. Biol. 513, 41–86 (2002).
Di Maio, R. et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 10, eaar5429 (2018).
Aasly, J. O. et al. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann. Neurol. 57, 762–765 (2005).
Marras, C. et al. Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology 77, 325–333 (2011).
Bonet-Ponce, L. et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv. 6, eabb2454 (2020).
Steger, M. et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 5, e12813 (2016).
Vazquez-Velez, G. E. & Zoghbi, H. Y. Parkinson’s disease genetics and pathophysiology. Annu. Rev. Neurosci. 44, 87–108 (2021).
Orenstein, S. J. et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406 (2013).
Nguyen, M. & Krainc, D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc. Natl. Acad. Sci. USA 115, 5576–5581 (2018).
Herbst, S. et al. LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 39, e104494 (2020).
West, A. B. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp. Neurol. 298, 236–245 (2017).
Chan, S. L. & Tan, E. K. Targeting LRRK2 in Parkinson’s disease: an update on recent developments. Expert Opin. Ther. Targets 21, 601–610 (2017).
Shihabuddin, L. S. et al. New frontiers in Parkinson’s Disease: from genetics to the clinic. J. Neurosci. 38, 9375–9382 (2018).
Chen, K. et al. Leucine-rich repeat kinase 2 (LRRK2) inhibition upregulates microtubule-associated protein 1B to ameliorate lysosomal dysfunction and parkinsonism. MedComm 4, e429 (2023).
Zhao, H. T. et al. LRRK2 antisense oligonucleotides ameliorate alpha-synuclein inclusion formation in a Parkinson’s Disease mouse model. Mol. Ther. Nucleic Acids 8, 508–519 (2017).
Jennings, D. et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci. Transl. Med. 14, eabj2658 (2022).
Jennings, D. et al. LRRK2 inhibition by BIIB122 in healthy participants and patients with Parkinson’s disease. Mov. Disord. 38, 386–398 (2023).
Cao, R. et al. Recent advances in targeting leucine-rich repeat kinase 2 as a potential strategy for the treatment of Parkinson’s disease. Bioorg. Chem. 141, 106906 (2023).
Misgeld, T. & Schwarz, T. L. Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666 (2017).
Dawson, T. M. & Dawson, V. L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302, 819–822 (2003).
Schapira, A. H. et al. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269 (1989).
Giannoccaro, M. P., La Morgia, C., Rizzo, G. & Carelli, V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. 32, 346–363 (2017).
Clark, E. H., Vazquez de la Torre, A., Hoshikawa, T. & Briston, T. Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 296, 100209 (2021).
Vives-Bauza, C. et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 107, 378–383 (2010).
Rakovic, A. et al. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 26, 1428–1441 (2019).
Harper, J. W., Ordureau, A. & Heo, J. M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108 (2018).
Ge, P., Dawson, V. L. & Dawson, T. M. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 15, 20 (2020).
Matsuda, N. & Tanaka, K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy 6, 952–954 (2010).
Kasten, M. et al. Genotype-phenotype relations for the Parkinson’s Disease genes parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741 (2018).
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998).
Valente, E. M. et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160 (2004).
Beilina, A. et al. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA 102, 5703–5708 (2005).
Sim, C. H. et al. C-terminal truncation and Parkinson’s disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum. Mol. Genet.15, 3251–3262 (2006).
Wang, H. L. et al. PINK1 mutants associated with recessive Parkinson’s disease are defective in inhibiting mitochondrial release of cytochrome C. Neurobiol. Dis. 28, 216–226 (2007).
Ko, H. S. et al. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J. Neurosci. 25, 7968–7978 (2005).
Lee, Y. et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 16, 1392–1400 (2013).
Ko, H. S. et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits Parkin’s ubiquitination and protective function. Proc. Natl. Acad. Sci. USA 107, 16691–16696 (2010).
Pankratz, N. et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 124, 593–605 (2009).
Rhodes, S. L. et al. Replication of GWAS associations for GAK and MAPT in Parkinson’s disease. Ann. Hum. Genet. 75, 195–200 (2011).
Li, N. N. et al. GWAS-linked GAK locus in Parkinson’s disease in Han Chinese and meta-analysis. Hum. Genet. 131, 1089–1093 (2012).
Li, H., Zhang, C. & Ji, Y. Association of GAK rs1564282 With Susceptibility to Parkinson’s Disease in Chinese populations. Front. Genet. 12, 777942 (2021).
Shulman, J. M. et al. Association of Parkinson disease risk loci with mild parkinsonian signs in older persons. JAMA Neurol. 71, 429–435 (2014).
Eisenberg, E. & Greene, L. E. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic 8, 640–646 (2007).
Lee, D. W. et al. Essential role of cyclin-G-associated kinase (Auxilin-2) in developing and mature mice. Mol. Biol. Cell 19, 2766–2776 (2008).
Egawa, J. et al. The cyclin G-associated kinase (GAK) inhibitor SGC-GAK-1 inhibits neurite outgrowth and synapse formation. Mol. Brain 15, 68 (2022).
Munson, M. J. et al. GAK and PRKCD are positive regulators of PRKN-independent mitophagy. Nat. Commun. 12, 6101 (2021).
Song, L. et al. Auxilin underlies progressive locomotor deficits and dopaminergic neuron loss in a drosophila model of Parkinson’s Disease. Cell Rep. 18, 1132–1143 (2017).
Dumitriu, A. et al. Cyclin-G-associated kinase modifies alpha-synuclein expression levels and toxicity in Parkinson’s disease: results from the GenePD study. Hum. Mol. Genet. 20, 1478–1487 (2011).
Ali, M. Z. & Dholaniya, P. S. Oxidative phosphorylation mediated pathogenesis of Parkinson’s disease and its implication via Akt signaling. Neurochem. Int. 157, 105344 (2022).
Koutsilieri, E., Chan, W. W., Reinitzer, D. & Rausch, W. D. Functional changes in cocultures of mesencephalon and striatal neurons from embryonic C57/BL6 mice due to low concentrations of 1-methyl-4-phenylpyridinium (MPP+). J. Neural Transm. Gen. Sect. 94, 189–197 (1993).
Akundi, R. S., Zhi, L. & Bueler, H. PINK1 enhances insulin-like growth factor-1-dependent Akt signaling and protection against apoptosis. Neurobiol. Dis. 45, 469–478 (2012).
Boonying, W. et al. Pink1 regulates FKBP5 interaction with AKT/PHLPP and protects neurons from neurotoxin stress induced by MPP. J. Neurochem. 150, 312–329 (2019).
Morais, V. A. et al. Parkinson’s disease mutations in PINK1 result in decreased complex I activity and deficient synaptic function. EMBO Mol. Med. 1, 99–111 (2009).
Yasuda, T. et al. Parkin-mediated protection of dopaminergic neurons in a chronic MPTP-minipump mouse model of Parkinson's disease. J. Neuropathol. Exp. Neurol. 70, 686–697 (2011).
Chuang, C. L., Lu, Y. N., Wang, H. C. & Chang, H. Y. Genetic dissection reveals that Akt is the critical kinase downstream of LRRK2 to phosphorylate and inhibit FOXO1, and promotes neuron survival. Hum. Mol. Genet 23, 5649–5658 (2014).
Neves, M., Graos, M., Anjo, S. I. & Manadas, B. Modulation of signaling pathways by DJ-1: An updated overview. Redox Biol. 51, 102283 (2022).
van Duijn, C. M. et al. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 69, 629–634 (2001).
Oh, S. E. & Mouradian, M. M. Cytoprotective mechanisms of DJ-1 against oxidative stress through modulating ERK1/2 and ASK1 signal transduction. Redox Biol. 14, 211–217 (2018).
Takahashi-Niki, K. et al. Epidermal growth factor-dependent activation of the extracellular signal-regulated kinase pathway by DJ-1 protein through its direct binding to c-Raf protein. J. Biol. Chem. 290, 17838–17847 (2015).
Kim, R. H. et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 7, 263–273 (2005).
Xia, X. G. et al. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 98, 10433–10438 (2001).
Pan, J. et al. Expression of FasL and its interaction with Fas are mediated by c-Jun N-terminal kinase (JNK) pathway in 6-OHDA-induced rat model of Parkinson disease. Neurosci. Lett. 428, 82–87 (2007).
Saporito, M. S., Brown, E. M., Miller, M. S. & Carswell, S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J. Pharm. Exp. Ther. 288, 421–427 (1999).
Chen, J. et al. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson’s disease. Cell Death Dis. 9, 700 (2018).
Tu, H. Y. et al. alpha-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 20, e13522 (2021).
Hunot, S. et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 101, 665–670 (2004).
Teismann, P. et al. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 100, 5473–5478 (2003).
Gao, F., Chen, D., Hu, Q. & Wang, G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS One 8, e72046 (2013).
Gloeckner, C. J., Schumacher, A., Boldt, K. & Ueffing, M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J. Neurochem. 109, 959–968 (2009).
Hsu, C. H., Chan, D. & Wolozin, B. LRRK2 and the stress response: interaction with MKKs and JNK-interacting proteins. Neurodegener. Dis. 7, 68–75 (2010).
Iba, M. et al. Inhibition of p38alpha MAPK restores neuronal p38gamma MAPK and ameliorates synaptic degeneration in a mouse model of DLB/PD. Sci. Transl. Med. 15, eabq6089 (2023).
Chen, J. et al. p38-TFEB pathways promote microglia activation through inhibiting CMA-mediated NLRP3 degradation in Parkinson’s disease. J. Neuroinflamm. 18, 295 (2021).
Zawada, W. M. et al. Inhibitors of p38 MAP kinase increase the survival of transplanted dopamine neurons. Brain Res. 891, 185–196 (2001).
Parkinson Study Group, P. I. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 69, 1480–1490 (2007).
Ichijo, H. et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275, 90–94 (1997).
Ray, A. et al. MPTP activates ASK1-p38 MAPK signaling pathway through TNF-dependent Trx1 oxidation in Parkinsonism mouse model. Free Radic. Biol. Med. 87, 312–325 (2015).
Lee, K. W. et al. Apoptosis signal-regulating kinase 1 mediates MPTP toxicity and regulates glial activation. PLoS One 7, e29935 (2012).
Ortner, E. & Moelling, K. Heteromeric complex formation of ASK2 and ASK1 regulates stress-induced signaling. Biochem. Biophys. Res. Commun. 362, 454–459 (2007).
Mansour, H. M., Mohamed, A. F., El-Khatib, A. S. & Khattab, M. M. Kinases control of regulated cell death revealing druggable targets for Parkinson’s disease. Ageing Res. Rev. 85, 101841 (2023).
Zhu, J. et al. Apelin-36 mediates neuroprotective effects by regulating oxidative stress, autophagy and apoptosis in MPTP-induced Parkinson’s disease model mice. Brain Res. 1726, 146493 (2020).
Lee, K. W. et al. Apoptosis signal-regulating kinase 1 modulates the phenotype of alpha-synuclein transgenic mice. Neurobiol. Aging 36, 519–526 (2015).
Zhang, J. et al. Apoptosis signal-regulating kinase 1 deletion mitigates alpha-synuclein pre-formed fibril propagation in mice. Neurobiol. Aging 85, 49–57 (2020).
Yoon, J. H. et al. LRRK2 functions as a scaffolding kinase of ASK1-mediated neuronal cell death. Biochim. Biophys. Acta Mol. Cell Res. 1864, 2356–2368 (2017).
Junn, E. et al. Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proc. Natl. Acad. Sci. USA 102, 9691–9696 (2005).
Chertow, G. M. et al. Effects of selonsertib in patients with diabetic kidney disease. J. Am. Soc. Nephrol. 30, 1980–1990 (2019).
Hou, S. et al. Structure-based discovery of 1H-indole-2-carboxamide derivatives as potent ASK1 inhibitors for potential treatment of ulcerative colitis. Eur. J. Med. Chem. 211, 113114 (2021).
Liles, J. T. et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J. Clin. Investig. 128, 4485–4500 (2018).
Bowles, K. R. & Jones, L. Kinase signalling in Huntington’s disease. J. Huntingt. Dis. 3, 89–123 (2014).
Andrew, S. E. et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 4, 398–403 (1993).
Squitieri, F. et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain 126, 946–955 (2003).
Tabrizi, S. J., Flower, M. D., Ross, C. A. & Wild, E. J. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 16, 529–546 (2020).
Roos, R. A. Huntington’s disease: a clinical review. Orphanet J. Rare Dis. 5, 40 (2010).
Leavitt, B. R., Kordasiewicz, H. B. & Schobel, S. A. Huntingtin-lowering therapies for Huntington disease: a review of the evidence of potential benefits and risks. JAMA Neurol. 77, 764–772 (2020).
Caron, N. S., Dorsey, E. R. & Hayden, M. R. Therapeutic approaches to Huntington disease: from the bench to the clinic. Nat. Rev. Drug Discov. 17, 729–750 (2018).
Evans, S. J. et al. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 84, 1156–1160 (2013).
Arrasate, M. & Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11 (2012).
Schilling, B. et al. Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J. Biol. Chem. 281, 23686–23697 (2006).
Gu, X. et al. Serines 13 and 16 are critical determinants of full-length human mutant Huntingtin-induced disease pathogenesis in HD mice. Neuron 64, 828–840 (2009).
Luo, S., Vacher, C., Davies, J. E. & Rubinsztein, D. C. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J. Cell Biol. 169, 647–656 (2005).
Thompson, L. M. et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187, 1083–1099 (2009).
Hegde, R. N. et al. TBK1 phosphorylates mutant Huntingtin and suppresses its aggregation and toxicity in Huntington’s disease models. EMBO J. 39, e104671 (2020).
Humbert, S. et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2, 831–837 (2002).
Apostol, B. L. et al. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum. Mol. Genet. 15, 273–285 (2006).
Merienne, K. et al. Polyglutamine expansion induces a protein-damaging stress connecting heat shock protein 70 to the JNK pathway. J. Biol. Chem. 278, 16957–16967 (2003).
Saavedra, A. et al. Striatal-enriched protein tyrosine phosphatase expression and activity in Huntington’s disease: a STEP in the resistance to excitotoxicity. J. Neurosci. 31, 8150–8162 (2011).
Lievens, J. C., Woodman, B., Mahal, A. & Bates, G. P. Abnormal phosphorylation of synapsin I predicts a neuronal transmission impairment in the R6/2 Huntington’s disease transgenic mice. Mol. Cell Neurosci. 20, 638–648 (2002).
Liot, G. et al. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 33, 6298–6309 (2013).
Bodai, L. & Marsh, J. L. A novel target for Huntington’s disease: ERK at the crossroads of signaling. The ERK signaling pathway is implicated in Huntington’s disease and its upregulation ameliorates pathology. Bioessays 34, 142–148 (2012).
Roze, E. et al. Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 22, 1083–1093 (2008).
Wang, P. et al. The KDEL receptor induces autophagy to promote the clearance of neurodegenerative disease-related proteins. Neuroscience 190, 43–55 (2011).
Hacker, H. & Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13 (2006).
Khoshnan, A. et al. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci. 24, 7999–8008 (2004).
Trager, U. et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain 137, 819–833 (2014).
Marcora, E. & Kennedy, M. B. The Huntington’s disease mutation impairs Huntingtin’s role in the transport of NF-kappaB from the synapse to the nucleus. Hum. Mol. Genet 19, 4373–4384 (2010).
Singh, S. & Singh, T. G. Role of nuclear factor Kappa B (NF-kappaB) signalling in neurodegenerative diseases: an mechanistic approach. Curr. Neuropharmacol. 18, 918–935 (2020).
Bjorkqvist, M. et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 205, 1869–1877 (2008).
Zuccato, C., Valenza, M. & Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 90, 905–981 (2010).
Landles, C. et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 285, 8808–8823 (2010).
Ochaba, J. et al. IKKbeta slows Huntington’s disease progression in R6/1 mice. Proc. Natl Acad. Sci. USA 116, 10952–10961 (2019).
Kane, L. P., Shapiro, V. S., Stokoe, D. & Weiss, A. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. 9, 601–604 (1999).
Meng, F., Liu, L., Chin, P. C. & D’Mello, S. R. Akt is a downstream target of NF-kappa B. J. Biol. Chem. 277, 29674–29680 (2002).
Hsiao, H. Y. et al. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet. 22, 1826–1842 (2013).
Anne, S. L., Saudou, F. & Humbert, S. Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 27, 7318–7328 (2007).
Kaminosono, S. et al. Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J. Neurosci. 28, 8747–8755 (2008).
Muchowski, P. J., Ning, K., D’Souza-Schorey, C. & Fields, S. Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc. Natl Acad. Sci. USA 99, 727–732 (2002).
Taylor, J. P. et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 12, 749–757 (2003).
Paoletti, P. et al. Dopaminergic and glutamatergic signaling crosstalk in Huntington’s disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J. Neurosci. 28, 10090–10101 (2008).
Pulverer, B. J. et al. Phosphorylation of c-jun mediated by MAP kinases. Nature 353, 670–674 (1991).
Weston, C. R. & Davis, R. J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 (2007).
Jebelli, J. D., Hooper, C., Garden, G. A. & Pocock, J. M. Emerging roles of p53 in glial cell function in health and disease. Glia 60, 515–525 (2012).
Junyent, F. et al. Gene expression profile in JNK3 null mice: a novel specific activation of the PI3K/AKT pathway. J. Neurochem. 117, 244–252 (2011).
Song, G., Ouyang, G. & Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 9, 59–71 (2005).
Meriin, A. B. et al. Intracellular aggregation of polypeptides with expanded polyglutamine domain is stimulated by stress-activated kinase MEKK1. J. Cell Biol. 153, 851–864 (2001).
Liu, Y. F. Expression of polyglutamine-expanded Huntingtin activates the SEK1-JNK pathway and induces apoptosis in a hippocampal neuronal cell line. J. Biol. Chem. 273, 28873–28877 (1998).
Perrin, V. et al. Implication of the JNK pathway in a rat model of Huntington’s disease. Exp. Neurol. 215, 191–200 (2009).
Guo, X. et al. ASK1 in neurodegeneration. Adv. Biol. Regul. 66, 63–71 (2017).
Nishitoh, H. et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355 (2002).
Fan, J. et al. P38 MAPK is involved in enhanced NMDA receptor-dependent excitotoxicity in YAC transgenic mouse model of Huntington disease. Neurobiol. Dis. 45, 999–1009 (2012).
Huang, Z. N. et al. Inhibition of p38 mitogen-activated protein kinase ameliorates HAP40 depletion-induced toxicity and proteasomal defect in Huntington’s Disease model. Mol. Neurobiol. 58, 2704–2723 (2021).
Patterson, K. I., Brummer, T., O’Brien, P. M. & Daly, R. J. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem. J. 418, 475–489 (2009).
Gladding, C. M. et al. Alterations in STriatal-enriched protein tyrosine phosphatase expression, activation, and downstream signaling in early and late stages of the YAC128 Huntington’s disease mouse model. J. Neurochem. 130, 145–159 (2014).
Legos, J. J. et al. The selective p38 inhibitor SB-239063 protects primary neurons from mild to moderate excitotoxic injury. Eur. J. Pharm. 447, 37–42 (2002).
Gines, S. et al. Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-D-aspartate receptor activation in Huntington’s disease knock-in striatal cells. J. Biol. Chem. 278, 50514–50522 (2003).
Warby, S. C. et al. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum. Mol. Genet. 14, 1569–1577 (2005).
Bryan, M. R. et al. Manganese acts upon Insulin/IGF receptors to phosphorylate AKT and increase glucose uptake in Huntington’s disease cells. Mol. Neurobiol. 57, 1570–1593 (2020).
Ribeiro, M. et al. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 74, 129–144 (2014).
Senousy, M. A., Hanafy, M. E., Shehata, N. & Rizk, S. M. Erythropoietin and Bacillus calmette-guerin vaccination mitigate 3-nitropropionic acid-induced Huntington-like disease in rats by modulating the PI3K/Akt/mTOR/P70S6K pathway and enhancing the autophagy. ACS Chem. Neurosci. 13, 721–732 (2022).
Li, L. et al. Mutant Huntingtin impairs pancreatic beta-cells by recruiting IRS-2 and disturbing the PI3K/AKT/FoxO1 signaling pathway in Huntington’s disease. J. Mol. Neurosci. 71, 2646–2658 (2021).
Somvanshi, R. K., Jhajj, A., Heer, M. & Kumar, U. Characterization of somatostatin receptors and associated signaling pathways in pancreas of R6/2 transgenic mice. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 359–373 (2018).
Goutman, S. A. et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 21, 465–479 (2022).
Chia, R., Chio, A. & Traynor, B. J. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102 (2018).
Renton, A. E., Chio, A. & Traynor, B. J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 (2014).
Zarei, S. et al. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 6, 171 (2015).
Renton, A. E., Chiò, A. & Traynor, B. J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 (2014).
Johnson, B. S. et al. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339 (2009).
Peters, O. M., Ghasemi, M. & Brown, R. H. Jr Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 125, 2548 (2015).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
Cohen, T. J. et al. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 6, 5845 (2015).
Hasegawa, M. et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70 (2008).
Taylor, L. M., McMillan, P. J., Kraemer, B. C. & Liachko, N. F. Tau tubulin kinases in proteinopathy. FEBS J. 286, 2434–2446 (2019).
Wilson, R. S. et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol. 70, 1418–1424 (2013).
Lee, E. B., Lee, V. M. & Trojanowski, J. Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50 (2011).
Deng, H., Gao, K. & Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 10, 337–348 (2014).
Kim, G. et al. ALS genetics: gains, losses, and implications for future therapies. Neuron 108, 822–842 (2020).
Mitchell, J. C. et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 125, 273–288 (2013).
Sharma, A. et al. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 7, 10465 (2016).
Shiihashi, G. et al. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain 139, 2380–2394 (2016).
Lagier-Tourenne, C. et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497 (2012).
Zou, Z. Y. et al. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 88, 540–549 (2017).
Lindberg, M. J. et al. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc. Natl. Acad. Sci. USA 102, 9754–9759 (2005).
Shi, P. et al. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1802, 45–51 (2010).
Ratovitski, T. et al. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum. Mol. Genet. 8, 1451–1460 (1999).
Reaume, A. G. et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13, 43–47 (1996).
Saccon, R. A., Bunton-Stasyshyn, R. K., Fisher, E. M. & Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358 (2013).
Wang, T. et al. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 33, 531–546.e539 (2021).
Sakae, N. et al. Poly-GR dipeptide repeat polymers correlate with neurodegeneration and clinicopathological subtypes in C9ORF72-related brain disease. Acta Neuropathol. Commun. 6, 63 (2018).
Renton, A. E. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Beckers, J., Tharkeshwar, A. K. & Van Damme, P. C9orf72 ALS-FTD: recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 17, 3306–3322 (2021).
Mejzini, R. et al. ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 13, 1310 (2019).
Cirulli, E. T. et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441 (2015).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636 (2015).
Williams, K. L. et al. Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiol. Aging 36, 3334 e3331–3334 e3335 (2015).
Tsai, P. C. et al. Mutational analysis of TBK1 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 40, 191.e111–116 (2016).
Oakes, J. A., Davies, M. C. & Collins, M. O. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10, 5 (2017).
Freischmidt, A. et al. Association of mutations in TBK1 with sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. JAMA Neurol. 74, 110–113 (2017).
Larabi, A. et al. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 3, 734–746 (2013).
Goncalves, A. et al. Functional dissection of the TBK1 molecular network. PLoS One 6, e23971 (2011).
Herhaus, L. et al. TBK1-mediated phosphorylation of LC3C and GABARAP-L2 controls autophagosome shedding by ATG4 protease. EMBO Rep. 21, e48317 (2020).
Sullivan, P. M. et al. The ALS/FTLD-associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4, 51 (2016).
Matsumoto, G., Shimogori, T., Hattori, N. & Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 24, 4429–4442 (2015).
Moore, A. S. & Holzbaur, E. L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 113, E3349–E3358 (2016).
Brenner, D. et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J. Exp. Med. 216, 267–278 (2019).
Brenner, D. et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, e28 (2016).
Kenna, K. P. et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037 (2016).
Nguyen, H. P. et al. NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol. Aging 61, e1–255.e7 (2018).
Riva, N. et al. NEK1 Variants in a cohort of Italian patients with amyotrophic lateral sclerosis. Front. Neurosci. 16, 833051 (2022).
Higelin, J. et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res 30, 150–162 (2018).
Wang, H. B. et al. NEK1-mediated retromer trafficking promotes blood-brain barrier integrity by regulating glucose metabolism and RIPK1 activation. Nat Commun. 12, 4826 (2021).
Asghari Adib, E., Smithson, L. J. & Collins, C. A. An axonal stress response pathway: degenerative and regenerative signaling by DLK. Curr. Opin. Neurobiol. 53, 110–119 (2018).
Ghosh, A. S. et al. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol. 194, 751–764 (2011).
Xiong, H. et al. Neural circuit changes in neurological disorders: evidence from in vivo two-photon imaging. Ageing Res Rev. 87, 101933 (2023).
Le Pichon, C. E. et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 9, eaag0394 (2017).
Wlaschin, J. J. et al. Promoting regeneration while blocking cell death preserves motor neuron function in a model of ALS. Brain 146, 2016–2028 (2022).
Xu, D. et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e1419 (2018).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Wang, H. et al. NEK1-mediated retromer trafficking promotes blood-brain barrier integrity by regulating glucose metabolism and RIPK1 activation. Nat. Commun. 12, 4826 (2021).
Du, B. et al. Iron promotes both ferroptosis and necroptosis in the early stage of reperfusion in ischemic stroke. Genes Dis. 11, 101262 (2024).
Li, W. et al. Nuclear RIPK1 promotes chromatin remodeling to mediate inflammatory response. Cell Res. 32, 621–637 (2022).
Mifflin, L., Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 19, 553–571 (2020).
Liu, Y. J., Lee, L. M., Lai, H. L. & Chern, Y. Aberrant activation of AMP-activated protein kinase contributes to the abnormal distribution of HuR in amyotrophic lateral sclerosis. FEBS Lett. 589, 432–439 (2015).
Scarmeas, N. et al. Premorbid weight, body mass, and varsity athletics in ALS. Neurology 59, 773–775 (2002).
Browne, S. E. et al. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 22, 599–610 (2006).
Sui, Y. et al. Adenosine monophosphate-activated protein kinase activation enhances embryonic neural stem cell apoptosis in a mouse model of amyotrophic lateral sclerosis. Neural Regen. Res. 9, 1770–1778 (2014).
Lim, M. A. et al. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 32, 1123–1141 (2012).
Liu, Y. J. et al. Activation of AMP-activated protein kinase alpha1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 787–801 (2015).
Coughlan, K. S., Mitchem, M. R., Hogg, M. C. & Prehn, J. H. Preconditioning” with latrepirdine, an adenosine 5’-monophosphate-activated protein kinase activator, delays amyotrophic lateral sclerosis progression in SOD1(G93A) mice. Neurobiol. Aging 36, 1140–1150 (2015).
Warita, H. et al. Early decrease of survival signal-related proteins in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Apoptosis 6, 345–352 (2001).
Manning, B. D. & Toker, A. AKT/PKB signaling: navigating the network. Cell 169, 381–405 (2017).
Hu, J. H. et al. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 85, 432–442 (2003).
Yang, W., Leystra-Lantz, C. & Strong, M. J. Upregulation of GSK3beta expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci). Brain Res. 1196, 131–139 (2008).
Choi, H. J. et al. Recent advances on the role of GSK3beta in the pathogenesis of amyotrophic lateral sclerosis. Brain Sci. 10, 675 (2020).
Sreedharan, J., Neukomm, L. J., Brown, R. H. Jr. & Freeman, M. R. Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr. Biol. 25, 2130–2136 (2015).
Yang, Y. M. et al. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12, 713–726 (2013).
Nakagawa, O. et al. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 392, 189–193 (1996).
Jacobs, M. et al. The structure of dimeric ROCK I reveals the mechanism for ligand selectivity. J. Biol. Chem. 281, 260–268 (2006).
Iizuka, M. et al. Distinct distribution and localization of Rho-kinase in mouse epithelial, muscle and neural tissues. Cell Struct. Funct. 37, 155–175 (2012).
Conti, A. et al. Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim Biophys. Acta 1842, 99–106 (2014).
Takata, M. et al. Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br. J. Pharm. 170, 341–351 (2013).
Tonges, L. et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 62, 217–232 (2014).
Preyer, M., Shu, C. W. & Wang, J. Y. J. Delayed activation of Bax by DNA damage in embryonic stem cells with knock-in mutations of the AbI nuclear localization signals. Cell Death Differ. 14, 1139–1148 (2007).
Gutierrez, D. A. et al. c-Abl kinase at the crossroads of healthy synaptic remodeling and synaptic dysfunction in neurodegenerative diseases. Neural Regen. Res. 18, 237–243 (2023).
Schlatterer, S. D., Acker, C. M. & Davies, P. c-Abl in neurodegenerative disease. J. Mol. Neurosci. 45, 445–452 (2011).
Katsumata, R. et al. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS One 7, e46185 (2012).
Imamura, K. et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 9, eaaf3962 (2017).
Lee, S. B. et al. c-Abl regulates the pathological deposition of TDP-43 via tyrosine 43 phosphorylation. Cells. 11, 3972 (2022).
Motaln, H. et al. Abl kinase-mediated FUS Tyr526 phosphorylation alters nucleocytoplasmic FUS localization in FTLD-FUS. Brain 146, 4088-4104 (2023).
Iguchi, Y. et al. IkappaB kinase phosphorylates cytoplasmic TDP-43 and promotes its proteasome degradation. J. Cell Biol. 223, e202302048 (2024).
Ho, D. M. et al. cAMP/PKA signaling regulates TDP-43 aggregation and mislocalization. Proc. Natl. Acad. Sci. USA 121, e2400732121 (2024).
Kametani, F. et al. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 382, 405–409 (2009).
Liachko, N. F. et al. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann. Neurol. 74, 39–52 (2013).
Taylor, L. M. et al. Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol. Neurodegener. 13, 7 (2018).
Liachko, N. F. et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet 10, e1004803 (2014).
Pilo, C. A. et al. Mutations in protein kinase C gamma promote spinocerebellar ataxia type 14 by impairing kinase autoinhibition. Sci. Signal 15, eabk1147 (2022).
Klebe, S. et al. New mutations in protein kinase C gamma associated with spinocerebellar ataxia type 14. Ann. Neurol. 58, 720–729 (2005).
Lin, C. W., Fan, C. H., Chang, Y. C. & Hsieh-Li, H. M. ERK activation precedes Purkinje cell loss in mice with Spinocerebellar ataxia type 17. Neurosci. Lett. 738, 135337 (2020).
Goetz, S. C., Liem, K. F. Jr. & Anderson, K. V. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 151, 847–858 (2012).
Glover, R. T. et al. Interaction of the N-methyl-D-aspartic acid receptor NR2D subunit with the c-Abl tyrosine kinase. J. Biol. Chem. 275, 12725–12729 (2000).
Smith, D. H., Johnson, V. E., Trojanowski, J. Q. & Stewart, W. Chronic traumatic encephalopathy—confusion and controversies. Nat. Rev. Neurol. 15, 179–183 (2019).
McKee, A. C. et al. Chronic traumatic encephalopathy (CTE): criteria for neuropathological diagnosis and relationship to repetitive head impacts. Acta Neuropathol. 145, 371–394 (2023).
Kim, N. et al. Inhibition of death-associated protein kinase 1 attenuates cis P-tau and neurodegeneration in traumatic brain injury. Prog. Neurobiol. 203, 102072 (2021).
Braun, N. J., Yao, K. R., Alford, P. W. & Liao, D. Mechanical injuries of neurons induce tau mislocalization to dendritic spines and tau-dependent synaptic dysfunction. Proc. Natl. Acad. Sci. USA 117, 29069–29079 (2020).
Meng, S. et al. ASK1-K716R reduces neuroinflammation and white matter injury via preserving blood-brain barrier integrity after traumatic brain injury. J. Neuroinflamm. 20, 244 (2023).
Oliva, A. A. Jr. et al. STAT3 signaling after traumatic brain injury. J. Neurochem. 120, 710–720 (2012).
Ding, X. et al. DNALI1 promotes neurodegeneration after traumatic brain injury via inhibition of autophagosome-lysosome fusion. Adv. Sci. 11, e2306399 (2024).
Roy, S. M. et al. Targeting human central nervous system protein kinases: an isoform selective p38alphaMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem. Neurosci. 6, 666–680 (2015).
Zhou, Z. et al. Retention of normal glia function by an isoform-selective protein kinase inhibitor drug candidate that modulates cytokine production and cognitive outcomes. J. Neuroinflamm. 14, 75 (2017).
Rutigliano, G. et al. An isoform-selective p38alpha mitogen-activated protein kinase inhibitor rescues early entorhinal cortex dysfunctions in a mouse model of Alzheimer’s disease. Neurobiol. Aging 70, 86–91 (2018).
Duffy, J. P. et al. The discovery of VX-745: a novel and selective p38alpha kinase inhibitor. ACS Med. Chem. Lett. 2, 758–763 (2011).
Alam, J., Blackburn, K. & Patrick, D. Neflamapimod: clinical phase 2b-ready oral small molecule inhibitor of p38alpha to reverse synaptic dysfunction in early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 4, 273–278 (2017).
Scheltens, P. et al. An exploratory clinical study of p38alpha kinase inhibition in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 5, 464–473 (2018).
Prins, N. D. et al. A phase 2 double-blind placebo-controlled 24-week treatment clinical study of the p38 alpha kinase inhibitor neflamapimod in mild Alzheimer’s disease. Alzheimers Res. Ther. 13, 106 (2021).
Martinez, A. et al. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 45, 1292–1299 (2002).
Martinez, A., Castro, A., Dorronsoro, I. & Alonso, M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med. Res. Rev. 22, 373–384 (2002).
Luna-Medina, R. et al. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: potential therapeutic role in brain disorders. J. Neurosci. 27, 5766–5776 (2007).
del Ser, T. et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J. Alzheimers Dis. 33, 205–215 (2013).
Lovestone, S. et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88 (2015).
Cortes-Gomez, M. A. et al. Tau phosphorylation by glycogen synthase kinase 3beta modulates enzyme acetylcholinesterase expression. J. Neurochem. 157, 2091–2105 (2021).
Su, Y. et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 43, 6899–6908 (2004).
Du, B., Chen, K., Wang, W. & Lei, P. Targeting metals in Alzheimer’s disease: an update. J. Alzheimers Dis. 101, S141–S154 (2024).
Leroy, K. et al. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J. Alzheimers Dis. 19, 705–719 (2010).
Alvarez-Ruiz, Y. & Carrillo-Mora, P. Amyloid beta 25-35 impairs reconsolidation of object recognition memory in rats and this effect is prevented by lithium carbonate. Neurosci. Lett. 548, 79–83 (2013).
Cardillo, G. M. et al. Chronic lithium treatment increases telomere length in parietal cortex and hippocampus of triple-transgenic Alzheimer’s disease mice. J. Alzheimers Dis. 63, 93–101 (2018).
Habib, A. et al. Comparing the effect of the novel ionic cocrystal of lithium salicylate proline (LISPRO) with lithium carbonate and lithium salicylate on memory and behavior in female APPswe/PS1dE9 Alzheimer’s mice. J. Neurosci. Res. 97, 1066–1080 (2019).
Yeh, H. L. & Tsai, S. J. Lithium may be useful in the prevention of Alzheimer’s disease in individuals at risk of presenile familial Alzheimer’s disease. Med. Hypotheses 71, 948–951 (2008).
Forlenza, O. V. et al. Long-term lithium treatment reduces glucose metabolism in the cerebellum and hippocampus of nondemented older adults: an [(1)(8)F]FDG-PET study. ACS Chem. Neurosci. 5, 484–489 (2014).
Hampel, H. et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931 (2009).
Feyt, C. et al. Lithium chloride increases the production of amyloid-beta peptide independently from its inhibition of glycogen synthase kinase 3. J. Biol. Chem. 280, 33220–33227 (2005).
Lei, P., Ayton, S., Bush, A. I. & Adlard, P. A. GSK-3 in neurodegenerative diseases. Int. J. Alzheimers Dis. 2011, 189246 (2011).
Nakashima, H. et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 110, 547–556 (2005).
Lei, P. et al. Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol. Psychiatr. 22, 396–406 (2017).
Folch, J. et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 15, 587–596 (2015).
Lonskaya, I. et al. Nilotinib and bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in Alzheimer’s disease models. Neuroscience 304, 316–327 (2015).
Piette, F. et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: a randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 3, 16 (2011).
Dubois, B. et al. Masitinib for mild-to-moderate Alzheimer’s disease: results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimers Res. Ther. 15, 39 (2023).
Turner, R. S. et al. Nilotinib effects on safety, tolerability, and biomarkers in Alzheimer’s disease. Ann. Neurol. 88, 183–194 (2020).
Tang, S. J. et al. Fyn kinase inhibition reduces protein aggregation, increases synapse density and improves memory in transgenic and traumatic Tauopathy. Acta Neuropathol. Commun. 8, 96 (2020).
Dinda, B. et al. Therapeutic potentials of plant iridoids in Alzheimer’s and Parkinson’s diseases: a review. Eur. J. Med. Chem. 169, 185–199 (2019).
Ly, C. et al. Bryostatin 1 promotes synaptogenesis and reduces dendritic spine density in cortical cultures through a PKC-dependent mechanism. ACS Chem. Neurosci. 11, 1545–1554 (2020).
Nelson, T. J. et al. Bryostatin effects on cognitive function and PKCvarepsilon in Alzheimer’s disease phase IIa and expanded access trials. J. Alzheimers Dis. 58, 521–535 (2017).
Costa, A. C. & Scott-McKean, J. J. Prospects for improving brain function in individuals with Down syndrome. CNS Drugs 27, 679–702 (2013).
de la Torre, R. et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 15, 801–810 (2016).
Sharman, M. J. et al. Assessment of diets containing curcumin, epigallocatechin-3-gallate, docosahexaenoic acid and alpha-lipoic acid on amyloid load and inflammation in a male transgenic mouse model of Alzheimer’s disease: are combinations more effective? Neurobiol. Dis. 124, 505–519 (2019).
Fridman, J. S. et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J. Immunol. 184, 5298–5307 (2010).
Simpson, E. L. et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet 396, 255–266 (2020).
Samuel, C., Cornman, H., Kambala, A. & Kwatra, S. G. A review on the safety of using JAK inhibitors in dermatology: clinical and laboratory monitoring. Dermatol. Ther. 13, 729–749 (2023).
Walsh, R. R. et al. Plasma and cerebrospinal fluid pharmacokinetics of vodobatinib, a neuroprotective c-Abl tyrosine kinase inhibitor for the treatment of Parkinson’s disease. Parkinsonism Relat. Disord. 108, 105281 (2023).
Lee, W. J. et al. Nilotinib treatment outcomes in autosomal dominant spinocerebellar ataxia over one year. Sci. Rep. 14, 16303 (2024).
Virlogeux, A. et al. Increasing brain palmitoylation rescues behavior and neuropathology in Huntington disease mice. Sci. Adv. 7, eabb0799 (2021).
Einat, H. et al. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J. Neurosci. 23, 7311–7316 (2003).
Fusco, F. R. & Paldino, E. Role of phosphodiesterases in Huntington’s disease. Adv. Neurobiol. 17, 285–304 (2017).
Gunther, R. et al. Rho kinase inhibition with Fasudil in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis-symptomatic treatment potential after disease onset. Front. Pharm. 8, 17 (2017).
Lingor, P. et al. ROCK-ALS: protocol for a randomized, placebo-controlled, double-blind phase IIa Trial of safety, tolerability and efficacy of the Rho Kinase (ROCK) inhibitor Fasudil in amyotrophic lateral sclerosis. Front. Neurol. 10, 293 (2019).
Koch, J. C. et al. Safety, tolerability, and efficacy of fasudil in amyotrophic lateral sclerosis (ROCK-ALS): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 23, 1133–1146 (2024).
Koch, J. C. et al. Compassionate use of the ROCK inhibitor Fasudil in three patients with amyotrophic lateral sclerosis. Front. Neurol. 11, 173 (2020).
Weisel, K. et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 5, e00365 (2017).
Vissers, M. et al. Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: randomized, placebo-controlled, double-blind phase I/Ib studies in healthy subjects and patients. Clin. Transl. Sci. 15, 2010–2023 (2022).
Trias, E. et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 13, 177 (2016).
Ketabforoush, A. et al. Masitinib: the promising actor in the next season of the Amyotrophic Lateral Sclerosis treatment series. Biomed. Pharmacother. 160, 114378 (2023).
Mora, J. S. et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph. Lat. Scl FR 21, 5–14 (2020).
Mora, J. S. et al. Long-term survival analysis of masitinib in amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 14, 17562864211030365(2021).
Zeiser, R., Andrlova, H. & Meiss, F. Trametinib (GSK1120212). Recent Results Cancer Res. 211, 91–100 (2018).
Sun, C. et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 (2014).
Nance, E., Pun, S. H., Saigal, R. & Sellers, D. L. Drug delivery to the central nervous system. Nat. Rev. Mater. 7, 314–331 (2021).
Ng, A. S. L. et al. Case-control analysis of leucine-rich repeat kinase 2 protective variants in Alzheimer’s disease. Neurobiol. Aging 64, 157.e157–159 (2018).
Lattante, S. et al. Novel variants and cellular studies on patients’ primary fibroblasts support a role for NEK1 missense variants in ALS pathogenesis. Hum. Mol. Genet. 30, 65–71 (2021).
Shao, W. et al. Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration. Science 378, 94–99 (2022).
Yabe, I. et al. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch. Neurol. 60, 1749–1751 (2003).
Martinez-Gonzalez, L. et al. Tideglusib, a non-ATP competitive inhibitor of GSK-3beta as a drug candidate for the treatment of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 22, 8975 (2021).
Acknowledgements
This work was supported by the Noncommunicable Chronic Diseases-National Science and Technology Major Project (2023ZD0507200), National Natural Science Foundation of China (32070961), Natural Science Foundation of Sichuan Province (2024NSFSC1643), Sichuan University Postdoctoral Interdisciplinary Innovation Fund (JCXK2206), and Postdoctoral Research Program of West China Hospital, Sichuan University (2023HXBH126).
Author information
Authors and Affiliations
Contributions
P.L. conceived the review; X.W., Z.Y., J.Z., H.G., and Z.S. searched for literature and wrote the draft; X.W. and Z.Y. drew the figures; P.L. and C.L. edited the manuscript. P.L., C.L., and X.W. provided funding support. All authors have read and approved the article and agree with publication in this journal.
Corresponding authors
Ethics declarations
Competing interests
P.L. serves on the editorial board of Signal Transduction and Targeted Therapy but was not involved in the handling of the manuscript. The other authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, X., Yang, Z., Zou, J. et al. Protein kinases in neurodegenerative diseases: current understandings and implications for drug discovery. Sig Transduct Target Ther 10, 146 (2025). https://doi.org/10.1038/s41392-025-02179-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-025-02179-x
This article is cited by
-
Toward a Unified Framework in Molecular Neurobiology of Alzheimer’s Disease: Revisiting the Pathophysiological Hypotheses
Molecular Neurobiology (2026)
-
Phosphoproteomics unveils the signaling dynamics in neuronal cells stimulated with insulin and insulin-like growth factors
Cell Communication and Signaling (2025)
