
Abstract
Background
The habenula is an epithalamic brain structure that acts as a neuroanatomical hub connecting the limbic forebrain to the major monoamine centres. Abnormal habenula activity is increasingly implicated in depression, with a surge in publications on this topic in the last 5 years. Direct activation of the habenula is sufficient to induce a depressive phenotype in rodents, suggesting a causative role in depression. However, the molecular basis of habenula dysfunction in depression remains elusive and it is unclear how the preclinical advancements translate to the clinical field.
Methods
A systematic literature search was conducted following the PRISMA guidelines. The two search terms depress* and habenula* were applied across Scopus, Web of Science and PubMed databases. Studies eligible for inclusion must have examined the habenula in clinical cases of depression or preclinical models of depression and compared their measures to an appropriate control.
Results
Preclinical studies (n = 63) measured markers of habenula activity (n = 16) and neuronal firing (n = 22), largely implicating habenula hyperactivity in depression. Neurotransmission was briefly explored (n = 15), suggesting imbalances within excitatory and inhibitory habenula signalling. Additional preclinical studies reported neuroconnectivity (n = 1), inflammatory (n = 3), genomic (n = 3) and circadian rhythm (n = 3) abnormalities. Seven preclinical studies (11%) included both males and females. From these, 5 studies (71%) reported a significant difference between the sexes in at least one habenula measure taken. Clinical studies (n = 24) reported abnormalities in habenula connectivity (n = 15), volume (n = 6) and molecular markers (n = 3). Clinical studies generally included male and female subjects (n = 16), however, few of these studies examined sex as a biological variable (n = 6).
Conclusions
Both preclinical and clinical evidence suggest the habenula is disrupted in depression. However, there are opportunities for sex-specific analyses across both areas. Preclinical evidence consistently suggests habenula hyperactivity as a primary driver for the development of depressive symptoms. Clinical studies support gross habenula abnormalities such as altered activation, connectivity, and volume, with emerging evidence of blood brain barrier dysfunction, however, progress is limited by a lack of detailed molecular analyses and limited imaging resolution.

Similar content being viewed by others
Introduction
Depression is a highly prevalent and debilitating mood disorder, affecting over 300 million individuals and is the leading cause of disability worldwide [1]. Depression is characterised by a long disease course, high suicide rates and symptoms that include depressed mood, an inability to feel pleasure (anhedonia), sleep disturbances, impaired cognition and lethargy [1, 2]. Women are disproportionately affected and account for approximately two-thirds of the clinical population [3]. Despite the tremendous contribution to the global burden of disease, current pharmacological treatment options for depression are associated with a wide range of side effects [4, 5], have a slow onset of action and limited efficacy leading to a high incidence of treatment resistance [6, 7]. Due to the complex and heterogeneous nature of depression, the underlying molecular mechanisms remain unclear. Novel therapeutic targets and approaches are needed to reduce the social, economic, and psychological burden associated with the symptoms of depression and provide reprieve for those living with the illness.
Recently an evolutionary conserved, epithalamic brain structure known as the habenula (Hb) has garnered substantial interest in the context of mood disorders, particularly, depression. The past 5 years has seen a surge in publications (Fig. 1) as researchers seek to understand how the anatomically tiny, yet functionally vital, Hb goes awry in depression. The Hb is comprised of two divisions, the medial (MHb) and lateral Hb (LHb) [8]. The LHb, colloquially termed the “anti-reward” or “disappointment” centre of the brain, is involved in the regulation of motivational states [9], stress adaptation [10], sleep [11], and the encoding of negative stimuli [12]. Our understanding of the molecular and cellular properties of the Hb is predominately derived from rodent studies [13,14,15]. Primarily comprised of glutamatergic neurons, the LHb receives inputs from the basal ganglia and limbic forebrain and projects onto GABAergic interneurons located in the ventral tegmental area (VTA) and rostromedial tegmental nucleus, inhibiting the major monoamine centres including the dopaminergic VTA and serotonergic dorsal raphe nucleus [13]. Greater neuronal activity within the LHb leads to an inhibition within each of these regions, making it a key point of convergence modulating monoamine transmission [13]. The LHb connection to several brain regions involved in emotional regulation, stress and reward suggests it may act as a central driver influencing multiple aspects of depression [13]. Like the LHb, the MHb contains a large population of glutamatergic neurons, however in addition, it is characterised by both cholinergic and substance P neuronal populations [16, 17]. Interestingly, acetylcholine is co-released with glutamate in cholinergic neurons in the MHb [18]. The MHb projects onto the interpeduncular nucleus [13] and has been linked to depression through its downstream regulatory effects on the dopaminergic and serotonergic systems via the interpeduncular-dorsal raphe nucleus and interpeduncular-VTA circuitry [19, 20]. In addition, the MHb is involved in the regulation of fear and anxiety [21] and has been heavily implicated in nicotine addiction [22]. Preclinical research has suggested a possible role for the MHb in depression [23,24,25]; however, studies of the MHb are challenged by the small size of the subregion. Accordingly, there has been less investigation into the MHb, and the clinical significance of these findings is unclear. The human Hb exhibits similar neuroanatomical connectivity to rodents; however proportionately the LHb is significantly larger than the MHb, comprising up to 95% of the total Hb volume, comparatively, the rodent LHb makes up ~60% of the total Hb volume [26, 27]. The relative size difference may suggest a more complex modulation within the human LHb, highlighting the importance of clinical research.

The number of publications containing the terms “habenula” and ”depression” published from 1979 to 2024 in the PubMed database. *Data taken from Jan–May 2024.
The majority of literature reporting on the Hb is centred on preclinical investigations. In rodents, direct activation of the LHb is sufficient to induce a depressive-like phenotype [28, 29]. Further, inhibition of the LHb via optogenetics and local administration of rapid-acting anti-depressants (ketamine) can reverse these behavioural deficits [30,31,32]. Emerging research has identified sexual dimorphisms in the Hb circuitry [33, 34]. Recently, female mice were reported to exhibit a higher number of excitatory inputs to the LHb, specifically from the lateral septum and hypothalamus; conversely, males displayed more inhibitory inputs, specifically from the medial septum to the LHb [34]. Despite this, the literature exploring whether there are sex differences in the Hb in clinical depression or preclinical models of depression is sparse.
Despite the recent surge in interest in the Hb, the molecular, cellular and circuit properties of this region remain elusive in humans, and it is unclear if the remarkable developments observed in preclinical models translate to the human brain. Additionally, it’s important to highlight a variety of approaches are often used to induce depressive-like behaviours in preclinical models; these range from environmental insults to selective breeding and genetic manipulation [35]. As a result, the question of whether Hb dysfunction is consistently observed across each of these models remains unanswered. Therefore, in this article, we present a systematic review of studies examining the habenula in clinical cases of depression and preclinical models of relevance to depression, to synthesize the current landscape of this growing field. We aim to determine the consistency of findings and identify opportunities to advance research in this area, exploring its clinical impact and implications for future treatment approaches focussing on the Hb.
Methods
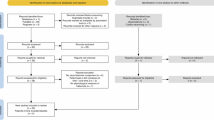
The systematic literature review was conducted adhering to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 guidelines [36] (see Supplementary Table S1). Articles retained and excluded were recorded in a four-phase flow diagram following the PRISMA statement requirements (Fig. 2) [37].

PRISMA Flow Diagram identifying the studies meeting the inclusion criteria for systematic review.
Search strategy
A literature search was completed using the following electronic databases; PubMed, Scopus, and Web of Science. All publication dates until May 2024 were included in the literature search. The two search terms depress* and habenula* were applied to each of the relevant databases in the field of title, abstract, and/or keyword. For example, the search strategy used for PubMed was (depress* AND habenula*).
Eligibility criteria
Studies eligible for inclusion in this systematic review must have been full-text, primary, research articles published in English and examined changes in the Hb in clinical cases of depression (living or post-mortem) or preclinical models relevant to depression. Any measure of the habenula was eligible for review including structural, functional, cellular, molecular, and electrophysiological measures. Preclinical articles were excluded if they examined differences in models of depression following the administration of an external treatment or following external manipulation of the Hb, without including a baseline measure in the model compared to control. Clinical studies were excluded if they did not have a non-psychiatric control group or examined depression amongst other diagnoses, where depression was not an independent factor.
Data selection
In adherence to the PRISMA guidelines, articles obtained from the initial electronic database search were screened by title and abstract by one author (SC). Duplicates were then removed using the reference management software Zotero (Version 6.0.18, Roy Rosenzweig Centre for History and New Media, Virginia, United States). Relevant articles were assessed by full-text based on the inclusion and exclusion criteria to determine their eligibility for review. Where there was ambiguity surrounding the articles, all authors discussed until consensus was reached. The reference list of eligible studies was also screened to identify additional studies.
Data extraction and analysis
The following data was extracted from articles that adhered to the inclusion criteria and were determined eligible for review: author, publication date, sample size, age, sex (and for clinical studies analysis of sex as a biological variable), the region of habenula examined (medial/lateral), strain/species and preclinical model used (for preclinical studies only), medication usage (for clinical studies only), methods used to obtain habenula measures, and the key findings of the research. Data extraction was recorded using Microsoft Excel (Version 16.68, Microsoft, Washington, United States) and Zotero (Version 6.0.18, Roy Rosenzweig Centre for History and New Media, Virginia, United States) was used to collate references. The current review includes both animal and human studies; accordingly, no risk of bias assessment was performed due to the heterogeneity of study designs and the lack of standardised reporting requirements. In addition, due to the lack of empirically validated risk of bias tools in animal studies, methodical quality assessment was not justified [38].
Results
The primary database search yielded a total of 1836 articles from PubMed (528 items), Scopus (703 items) and Web of Science (605 items) (Fig. 2). One additional record was identified through the screening of reference lists. Following the exclusion of duplicates, a total of 738 articles were screened by title and abstract for eligibility, 612 articles were excluded as they were not primary research articles, or they did not assess alterations in habenula measures in the clinically depressed population or preclinical models relevant to depression. 126 articles passed the initial screening process and were assessed by full-text review for eligibility. A further 31 articles were excluded because they did not examine baseline changes in preclinical models of depression prior to external manipulation of the Hb, three articles were excluded as they did not independently examine depression within the psychiatric cohort (grouped with schizophrenia, bipolar or other disorders) and 11 articles were excluded due to the lack of an adequate control group. A total of 81 articles that adhered to the eligibility criteria were included in the qualitative synthesis (n = 63 preclinical studies; n = 24 clinical studies, note: n = 6 articles included multiple study designs and were categorized under different subheadings accordingly).
The majority of the preclinical studies examined changes in markers of habenula activity (n = 16), neuronal firing (n = 22) and neurotransmission (n = 15) (Table 1). There were an additional 10 studies that assessed other Hb measures related to volume and connectivity (n = 1), inflammation (n = 3), genomic changes (n = 3) and circadian rhythm (n = 3). Of the 63 preclinical studies, 7 experimental designs (11%) included both male and female animals. From these, 5 studies (71%) reported a significant difference between the sexes in at least one measure taken. Most preclinical investigations were conducted on adult animals (n = 47), with some inclusion of infants (n = 2), juveniles (n = 6) and late adolescents (n = 8). In addition, the majority of preclinical studies specifically analysed the LHb, with only 13 preclinical studies measuring changes in the MHb (Table 1).
Clinical studies included for review (n = 20 living; n = 4 post-mortem) measured changes in Hb functional connectivity (n = 15), volumetric differences (n = 6) and molecular alterations (n = 3) (Table 2). Of these 24 clinical studies, 21 examined the whole Hb complex and did not differentiate the medial and lateral subdivisions. Clinical studies generally included male and female subjects (n = 22), however, few of these studies examined sex as a biological variable (n = 6). Instead, sex was typically examined as a covariate and male and female data were pooled together (n = 16). In addition, all clinical studies were conducted in adults with the exception of 1 study that examined adolescents.
Alterations in the habenula across preclinical models of depression
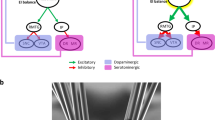
Depressive-like states in preclinical models are commonly induced via exposure to aversive stress. Despite varying in type, duration, and life stage the stressor occurs, these environmental models typically induce behavioural changes analogous to the depressive phenotypes observed in the clinical population. In the current review, the majority of preclinical studies employed an environmental stress paradigm (n = 51). Within this group, over 37 different stress protocols were used. Genetic models of depression largely relied on the selective breeding of stress-susceptible rodents (n = 7) or insertion/knockout of depressive-related genes (n = 1) to induce behavioural changes. Chemical-induced models of depression depended on the use of an external treatment and generally provided a more mechanistic approach to understanding the disorder. The initiation of depressive-like states via liposaccharide treatment (inflammatory agent) was the most common chemical-induced model included for review (n = 3). Across the preclinical models, evidence suggests LHb hyperactivity, caused by a combination of altered glutamatergic and GABAergic signalling, astrocyte dysfunction and neuroinflammation (Fig. 3).

Excitatory changes are shown in green, inhibitory changes are shown in red and the endocannabinoid system, a neural system influencing both inhibitory and excitatory systems, is shown in orange. Circles in grey illustrate the key efferent projections of the Hb of relevance to depression. AEA anandamide, AMPA α-amino-3-hydroxy-5-methyl-4-isoxazoldpropionic acid, ßCaMKII ß calmodulin-dependent kinase II, DNA deoxyribonucleic acid, eNOS endothelial nitric oxide synthase, Glu glutamate, GABA gamma aminobutyric acid, IL-4 interleukin-4, IL-6 interleukin-6, IL-1ß interleukin-1ß, iNOS inducible nitric oxide synthase, Kir4.1 inward rectifying potassium 4.1 channel, LHb lateral habenula, nNOS neuronal nitric oxide, NMDA N-methyl-D-aspartate, TNF-α tumour necrosis factor α, 2-AG 2- arachidonoylglycerol → activation, inhibition.
Markers of habenula neuronal and metabolic activity
Across preclinical models of depression, markers of neuronal and metabolic activity were consistently upregulated in the habenula (n = 16). The neuronal activation marker, c-Fos, was increased in the LHb following exposure to acute restraint stress [39, 40], chronic restraint stress [41], chronic inescapable shock stress [42], chronic unpredictable stress [31, 43] and social defeat stress [44,45,46]. Only two of these studies examined c-Fos expression in male and female rodents; both studies observed sex differences in the Hb [39, 40]. While both males and females displayed an elevation in c-Fos expression following exposure to chronic restraint stress, the increase was significantly greater in the LHb of stressed females compared to stressed males [39]. Conversely, following acute stress exposure, Sood et al. [40]. reported a significantly higher c-Fos expression in immobilisation stressed males compared to females, suggesting sex differences warrant further investigation.
A total of 4 studies reported that Hb neuronal activity did not significantly differ between control and depressive-like rodents; however, each of these studies supported a general trend towards greater Hb activity within the stressed cohort [25, 46,47,48]. One study found that c-Fos cell count was unchanged in the MHb and LHb of rats exposed to chronic restraint stress [47]. Despite this, an increase in c-Fos staining intensity was observed in animals susceptible to stress compared to those deemed resilient [47]. Exposure to social defeat stress in balb/c mice also did not alter c-Fos activity (24 h post stress) [46]. However, the stress did lead to a delayed increase in c-Fos activity in the LHb 2 weeks following the initial stress exposure; a similar trend was observed within the MHb [46]. Similarly, c-Fos was not changed in the MHb following exposure to social isolation stress; however, a positive trend was noted [25].
The enzyme, cytochrome oxidase, is an indicator of brain metabolic capacity and greater expression has been shown to correspond to a greater degree of neuronal activity [49,50,51]. In congenitally learned helpless rats, an increase in cytochrome oxidase was noted in both the MHb and LHb when compared to non-helpless controls [23]. Greater Hb cytochrome oxidase expression was also reported in a model of chronic unpredictable stress [52, 53]. However, a study conducted by Spivey et al. reported that early life stress, involving either exposure to early handling or maternal separation, did not alter the cytochrome oxidase activity in the whole Hb complex [48], suggesting developmental stress may have less effect on Hb metabolic activity. Despite these findings, female rats recorded greater baseline cytochrome oxidase activity in the Hb compared to males, irrespective of stress or control group, further supporting sex differences in Hb activity [48].
Habenula neuronal firing
The most consistent finding across preclinical models of depression was aberrant neuronal firing of the Hb, specifically hyperactivity within the LHb. LHb hyperactivity was reported across 11 experimental designs that utilised 14 models of depression. LHb hyperactivity was consistently found following exposure to chronic (n = 7), acute (n = 1) and early life stress paradigms (n = 3) [32, 54,55,56,57,58,59,60]. Similarly, this phenomenon was observed in the genetic congenitally learned helplessness model (n = 2) [32, 61] and an inflammatory model of depression [54]. The dysregulated Hb neuronal activity may be specific to a subtype of burst neurons; greater spontaneous bursting activity in the LHb was reported in a model of chronic stress [32]. Moreover, general baseline Hb neuronal activity was greater in the Hb of Wistar Kyoto rats (endogenous treatment-resistant model) when compared to the non-depressive-like hypertensive rat [62].
Exposure to stress is theorised to lead to increased sensitivity within the cells of the LHb, thereby leading to more frequent firing. This is supported by work showing that a lower rheobase (minimum frequency required for an action potential) was recorded in excitatory neurons projecting from the m-PFC to the LHb in both a chronic stress paradigm and a genetic knockout (itpr2 knockout) model [63]. LHb outputs, specifically to the dopaminergic VTA, have a similar increase in excitatory postsynaptic currents [57] and overall firing rate [64] in models of acute learned helplessness, congenitally learned helplessness [57] and chronic unpredictable mild stress [64]. More recently however Cerniauskas et al. [65]. showed that chronic stress-induced LHb-VTA hyperactivity was specific to animals that showed greater immobility in the tail suspension test (TST), but not those that showed anhedonic behaviour in the absence of immobility, suggesting LHb hyperactivity may be specific to a learned helplessness or passive coping phenotype [65]. Similarly, early life stress via maternal deprivation, which induced only a mild depressive phenotype with a modest reduction in sucrose preference and no change in mobility (OFT), grooming behaviour (splash test) or time spent in open borders (OFT), was associated with LHb hypoactivation, again suggesting LHb hyperactivity may be specific or more pronounced in specific behavioural adaptations [66].
Excitatory/inhibitory neurotransmission balance
Alterations in Hb neurotransmission have been briefly characterised in models of relevance to depression, largely focusing on aspects of excitatory and inhibitory neurotransmission. To date, studies have largely focussed on glutamatergic dysfunction (n = 6), the primary neurotransmitter of the LHb. Stress-induced models consistently potentiate LHb glutamatergic transmission, shifting the excitatory/inhibitory (E:I) balance towards excitation [55, 67]. Both stress-induced and genetic models of depression have reported significant elevations in ßCaMKII (which enhances glutamate activity) in the LHb [68, 69]. In line with the role of ßCaMKII to enhance glutamatergic synaptic transmission via AMPAR phosphorylation and insertion into the synapse, the post-synaptic expression of calcium-permeable AMPARs was increased in the LHb following chronic stress exposure [65]. More recently, upregulation of NMDA receptor-mediating signalling has been suggested as an additional driving mechanism that may underlie the heightened LHb excitability [70]. Following exposure to early life stress, juvenile rats displayed increased expression of the NMDA 2A and 2B receptor subunits in the LHb [70]. Moreover, stressed rats showed a reduction in the glutamate transporter, GLT-1, in the LHb that may disrupt glutamate clearance and further exacerbate excitatory neurotransmission [70].
Dysfunction within upstream regulators of glutamatergic transmission has also been reported in preclinical models of depression (n = 2) [71, 72]. P11 is a multifunctional protein that acts to promote glutamatergic (and serotonergic) signalling [71]. Seo et al. [71] reported an increase in p11+ cells, p11 protein and mRNA expression within the LHb following exposure to chronic stress. The increase in p11 was accompanied by a long-lasting increase in neuronal excitability within the LHb [71]. Furthermore, DNA hypomethylation, via a reduction in the DNA methylation marker, DNMT1, has been reported in the LHb following chronic stress [72]. Interestingly, the authors reported that inducing local LHb DNA hypomethylation led to an increase in glutamatergic excitatory genes (ßCaMKII, GluR1) suggesting DNA hypomethylation may be a possible upstream mechanism contributing to hyperactivity within the region [72].
Disrupted inhibitory signalling has previously been reported in environmental stress models (n = 2), with mice susceptible to stress following chronic social defeat showing lower GABAR B1 and B2 expression in the LHb [44]. In contrast, the MHb displayed no change in GABAR B1 and B2 following chronic social defeat irrespective of whether the mice were resilient or susceptible to that stress, further highlighting the functional differences between the medial and lateral subdivisions [44]. In addition, the inhibitory somatostatin 2 receptor, was increased in the MHb following chronic mild stress exposure (the LHb was not examined) [24].
Dysfunctional endocannabinoid signalling has been reported in one study design. The endocannabinoid system is a neuromodulatory system that regulates both glutamatergic and GABAergic signalling [73]. Exposure to acute, chronic, and social defeat stress increased the endocannabinoid, 2-arachidonoylglycerol, which was correlated to greater LHb neuronal activity, an effect that was independent of sex [73]. While stress exposure had no significant effect on anandamide levels, control females recorded higher baseline anandamide concentrations compared to males [73].
Other molecular changes in the habenula
The inflammatory hypothesis of depression has gained momentum in recent years and inducing inflammation is an established preclinical model to study depression [74]. Interestingly, following systematic administration of the inflammatory agent, lipopolysaccharide, Yang et al. [74]. reported that depressive-like behaviours were accompanied by a significant reduction in the volume of the whole Hb complex. When accounting for the different divisions it was determined this change was predominantly driven by changes within the MHb not LHb [74]. Inflammatory changes are also described as a secondary response to stress exposure and two studies have reported an elevation in inflammatory cytokines within the LHb following chronic stress [75, 76]. Specifically, an increase in TNF-α, IL-1ß and IL-6 was noted in a model of chronic unpredictable stress and was accompanied by greater activation within the inflammatory nuclear factor NF-kB/NOD-like receptor pyrin domain-containing 3 (NLRP3) signalling pathway [76]. In addition, mice that were susceptible to chronic social defeat stress (i.e. exhibited depressive-like behaviours) displayed an increase in the pro-inflammatory markers TNF-a, IL-1ß and IL-6 in the LHb, compared to mice deemed resilient to stress and non-stressed control mice [75]. Both stress-exposed groups (susceptible and resilient) displayed greater activation within the matrix metalloproteinase (MMP) 12-MMP2 pathway compared to controls, however; changes were most robust in the stress-susceptible group. An increase in Pcsk5 (MMP upstream regulator that facilitates microglia motility) and the increase in monocyte mobilisation was greater in the stress susceptible group compared to stress resilient [75]. Ito et al. suggest that greater cytokine release may lead to a remodelling of the extracellular matrix providing a possible explanation for these findings [75]. More recently, Chen et al. 2023 reported that rats exposed to chronic stress exhibited reduced levels of the anti-inflammatory cytokine, IL-4 in the LHb [77]. In addition, the stressed rats showed differential expression of nitric oxide synthase isoforms (NOS) [77].
Using RNA sequencing, a comprehensive genomic profile of the Hb in rats exposed to chronic stress revealed the differential expression of 379 genes [78]. Of these, stress exposure particularly affected cholinergic synapse-related genes which are typically associated with neuronal signalling in the MHb. Further, a downregulation of cholinergic genes (CHAT, VACHT, CHT, CHRNA3, CHRNB3, CHRNB4) in the MHb was noted in the same model using qPCR [79]. In addition, the downregulation of several serotonergic receptor genes was reported in stressed rats compared to non-stressed controls [78]. Serotonergic dysfunction has also been reported in the Hb of the Wistar Kyoto rat, an endogenous model of treatment-resistant depression [80], with downregulation of the serotonergic receptor genes Htr7 and Htr2a in both the MHb and LHb and increased expression of Htr4 in the LHb but not MHb [80]. Similarly, a reduction in 5-HTR7 receptor expression was isolated to the LHb [80]. In line with these findings, reduced aromatic L-amino acid decarboxylase mRNA, a precursor to dopamine and serotonin synthesis, was reported in the LHb in rats following chronic stress [81]. More recently, changes in small nucleolar RNA (soRNA) have been identified in the LHb of BALB/c mice exposed to chronic stress [82]. Increased expression of Snora69 specifically, suggests upstream dysregulation may be driving downstream adaptions at the level of gene and protein [82].
Disruptions in circadian rhythm have previously been linked to depression in both clinical and preclinical investigations (reviewed in 96). The Hb is situated near the pineal gland, a prominent regulator of the circadian cycle. Consequently, Hb dysfunction has also been examined in the context of sleep, specifically clock genes that contribute to the homeostatic aspect of sleep regulation (n = 2) [83, 84]. Christiansen et al. [83] reported a reduction in the Per2 clock gene in the LHb of rats exposed to chronic stress. The downregulation of Per2 has previously been noted in other brain regions that are hypoactive in depression and is suggested to contribute to depressive symptoms at night [83]. Similarly, in a clomipramine-induced (20 mg/kg subcutaneous) model of relevance to depression, Per2 mRNA was significantly reduced in the LHb compared to controls, in a time-dependent manner [84]. The endogenous circadian rhythm is closely influenced by fluctuations in body temperature [85]. Thermosensitive genes (cirbp and rbm3), that are expressed in response to changes in temperature, were altered in the LHb of mice deemed susceptible to social defeat stress immediately following stress, however; these changes were restored to control levels 10 days post stress [85].
Alteration in habenula measures in clinical depression
Clinical research is limited by the accessibility of the Hb, leading studies to predominately rely on non-invasive neuroimaging methods. The majority of the literature examined basal changes in Hb connectivity in cases of clinical depression (n = 12) or during task-based conditions (n = 3). Volumetric and morphological changes in the Hb were also assessed (n = 6). A further three studies conducted post-mortem molecular analyses of the LHb. Subtypes of depression examined included major depressive disorder (MDD) (n = 16), treatment-resistant depression (TRD) (n = 3), persistent depressive disorder (n = 1), late-life depression (n = 2) and subclinical depression (n = 1). In addition, as expected, subjects with depression in nearly all included studies had some history of anti-depressant drug use (n = 17).
Resting-state fMRI
Functional connectivity is correlated to greater cerebral blood flow and is suggested to be indicative of increased metabolic and subsequent neuronal activity [86]. Reduced functional connectivity has been reported between the Hb (hemisphere not specified) and the right anterior cingulate in individuals with MDD [87]. Further reductions in functional connectivity were observed between the right Hb and bilateral inferior temporal gyrus in a cohort of late-life depression [88]. Interestingly within this cohort, the reduction in Hb connectivity was associated with the severity of depressive symptoms and cognitive impairment [88]. Conversely, a study that examined dynamic functional connectivity, a technique that works off the assumption that connectivity between regions can fluctuate at various time points within a scan, reported an increase in connectivity between the right Hb and left orbitofrontal cortex in late-life depression [89]. A study by Yang et al. [90] further supports the correlations between greater functional connectivity of the Hb and disease severity. Compared to controls and those with subclinical depression, individuals with a diagnosis of MDD showed greater functional connectivity between the posterior parietal thalamus (region containing Hb) and the inferior temporal gyrus. Individuals with depression also showed greater functional connectivity between the left Hb and the inferior temporal and superior frontal gyrus but reduced connectivity to the calcarine sulcus [91] and cerebellum [92]; no change was recorded in the right Hb. Interestingly, Barreiros et al. [93] identified that patients with treatment-resistant depression exhibited greater functional connectivity between the left Hb and precuneus cortex. Conversely, individuals deemed responsive to traditional anti-depressants showed hypoconnectivity between the left Hb and precuneus cortex when compared to controls [93].
A study conducted by Amirir et al. [94] reported that a greater Hb node degree (indicative of a higher number of connections) was characteristic of TRD. The greater node degree was specific to the left hemisphere and more pronounced in females compared to males within the same diagnosis group [94]. Expanding on these initial findings, dynamic causal modelling was conducted to measure effective connectivity. Effective connectivity, unlike functional connectivity, detects a causal relationship between nodes (i.e. excitatory or inhibitory) [95]. Using this technique, it was found that people with TRD exhibited greater excitatory effective connectivity between the left subcallosal cingulate and left Hb but greater inhibitory effective connectivity between the right Hb and regions including the ventral caudate and nucleus accumbens [95]. Together, these findings suggest Hb dysregulation extends to both changes in excitation and inhibition and is influenced by hemisphere, which may, in part, explain the large variability within results. In addition, in a subclinical depression cohort, the left Hb node degree was not different compared to non-psychiatric controls, again suggesting depression severity may influence Hb connectivity [96].
Reward learning task fMRI
Three studies have reported abnormalities in Hb activity in depressed individuals during reward-task-based fMRI. Considering the Hb signals negative stimuli, it is expected that Hb activity would increase in response to a negative outcome. Indeed, both left and right Hb activation has been shown to increase in healthy controls when exposed to a negative outcome [97, 98]. However, those with a diagnosis of MDD recorded either no or attenuated change in Hb activation following exposure to the same negative stimuli [97, 98]. Similarly, in an error prediction learning task, adolescents with MDD exhibited a reduction in negative related signalling in the Hb when compared to controls [99]. Moreover, the reduction in loss-related prediction error correlated with anhedonia symptom severity [99]. While the reduction in Hb activation appears to contradict the preclinical research, it does support a perturbed habenula response in MDD.
Volumetric changes
Habenula volumetric studies (n = 6) have produced inconsistent results, and comparison is difficult due to the various methods used, which include a combination of ante- and post-mortem analyses [100,101,102,103,104,105]. It is important to note when considering these results, that volumetric analysis of the habenula is challenging due to its extremely small size, making it susceptible to measurement bias which may contribute to the significant variance in results.
Ranft et al. reported a decrease in whole Hb volume in post-mortem depression cases (n = 14) compared to controls (n = 13) [103]. This was accompanied by a reduction in cell number and cell area in the right Hb; a similar though insignificant trend was noted in the left Hb [103]. A neuroimaging study with a larger sample (n = 28; M = 13, F = 15) reported a similar reduction in total Hb volume, however; these findings only pertained to females with a diagnosis of MDD [104]. Interestingly, while Carceller-Sindreu et al. reported no overall difference in whole Hb volume in MDD, the study found an increase in Hb white matter volume in female first-episode MDD patients compared to female treatment-resistant MDD patients [101]. These results did not extend to the male MDD cohort or the Hb grey matter [101]. Two studies have reported a positive correlation between Hb volume and symptom severity, one noting this effect was independent of sex [102, 105]. However, more recently, a study comprising a significantly larger cohort (n = 234, M = 124, F = 110) that subcategorised patients with mild, moderate or severe depression reported a significant relationship between smaller Hb volumes and greater disease severity in females only [100]. A similar, though non-significant trend was noted in the male cohort [100]. Taken together, these studies highlight the importance of considering sex in studies of the Hb in depression.
Post-mortem molecular changes
One post-mortem study comprehensively investigated differences in gene expression profiles between male MDD subjects who died by suicide and non-psychiatric controls [106]. The study found a total of 251 differentially expressed genes in the Hb of MDD patients compared to controls. These genes primarily affected endothelial cell-enriched genes encoding cell-cell junction complexes (suggesting blood-brain barrier dysfunction) and plasma membrane-associated proteins and their upstream regulators [106]. In an additional study conducted by the same research group, individuals with MDD who died by suicide showed a reduction in cholinergic genes (CHT, CHRNB3) in the Hb post-mortem [79]. Recently, Lin et al. investigated the non-coding small nucleolar RNA (soRNA) profile of the LHb. The study identified 4 soRNAs that were significantly upregulated in the LHb of individuals that died with depression, two of which were validated by qPCR (SNOR69 and SNOR54) [82]. This supports preclinical findings by the same author, suggesting upstream RNA modification may be an important driver of molecular dysfunction in the habenula in depression [82].
Discussion
Overall, the developing body of evidence consistently points to aberrant Hb neuronal firing and dysregulated Hb neurotransmission, particularly excitatory/inhibitory balance, in preclinical models of depression. The hyperactivity observed within preclinical models translates to greater functional connectivity observed within a subgroup of Hb projections in the clinical population [87, 89, 107]. The small number of clinical studies that have examined molecular changes in the Hb, shed light on previously unexplored mechanisms in preclinical models of depression including endothelial dysfunction and compromised blood brain barrier integrity. A summary of the proposed molecular mechanisms underlying Hb dysfunction in preclinical models of depression and the clinical population is illustrated in Fig. 4.

DNA hypomethylation leads to an increased expression of glutamatergic excitatory genes. Greater ßCaMKII gene and subsequent protein expression then results in an increased surface expression of AMPAR. Together with increased NMDA subunit expression and p11 protein expression, these molecular changes potentiate glutamatergic signalling. GABAergic transmission is attenuated via reduced expression of GABAßR1/2 and increased activity of the PP2A enzyme which leads to GABA-GIRK complex internalisation. Increased expression of the endocannabinoid, 2-AG, acts via the CB1 receptor to inhibit GABA release. Greater expression of the Astrocytic Kir4.1 potassium transporter, reduces the resting membrane potential leading the neuron in a state more primed to fire and compromised BBB integrity leads to greater cytokine infiltration. A comparison of the a human and b rodent Hb and the respective boundaries of the MHb and LHb is shown in the bottom left. AMPA α-amino-3-hydroxy-5-methyl-4-isoxazoldpropionic acid, ßCaMKII ß calmodulin-dependent kinase II, BBB blood brain barrier, CB1 cannabinoid type 1 receptor, Glu glutamate, GABA gamma aminobutyric acid, Kir4.1 inward rectifying potassium 4.1 channel, LHb lateral habenula, MHb medial habenula, NMDA N-methyl-D-aspartate receptor, PP2A protein phosphatase 2A, VHb ventral habenula, 2-AG 2- arachidonoylglycerol.
LHb: an emerging model of altered excitatory/inhibitory balance in preclinical models of depression
A clear consensus emerges from the preclinical research linking aberrant Hb neuronal firing to the development of depressive-like behaviours. Enhanced glutamatergic transmission combined with attenuated inhibitory signalling may lead to a state of abnormal activation, specifically within the LHb. While investigations in the MHb are in their infancy, the findings of reduced acetylcholine [79] and increased somatostatin [24] in the MHb in preclinical models of depression, suggest a possible reduction in excitatory activity in this division, particularly MHb-IPN synaptic transmission. However, further research of this small subdivision is necessary. Subsequent advancements in optogenetics have allowed researchers to determine whether LHb hyperactivity plays a causative role in the development of depressive-like behaviours. Indeed, in healthy rodents, direct activation of the LHb is sufficient to induce a depressive-like phenotype, suggesting LHb hyperactivity may be a primary pathology in depression [28, 67]. Preclinical evidence suggests molecular substrates involved in glutamatergic transmission in the Hb may be a valuable target to suppress neuronal firing [55, 68,69,70, 108]. Overexpression of the enzyme, ßCaMKII, which promotes glutamatergic transmission, in the LHb induces symptoms of behavioural despair and anhedonia whereas blockade of ßCaMKII ameliorates these depressive-like behaviours [69]. Moreover, the antidepressants imipramine and ketamine downregulate ßCaMKII levels in the LHb [68, 69].
Enhanced glutamatergic transmission may be the primary driver of LHb hyperactivity; however, dampened GABAergic signalling seems to further offset the Hb excitatory/inhibitory balance [44, 55, 109]. Expression of GABA(B) receptors is downregulated in mice susceptible to stress [44]. Moreover, GABA(B)-GIRK complex internalisation within the LHb has been implicated in the development of depressive behaviours [56]. Interestingly, treatment with both a GABA(B) agonist and antagonist produced anti-depressant like effects [44]. The localisation of GABA(B) receptors in the Hb is unclear, and it may be that they are located on both the pre- and post-synaptic terminals [44]. An important consideration of these treatments is first identifying the location of each receptor and whether the drugs differ in their affinity for the differentially located subtypes [44]. The endocannabinoid system is an additional neural system responsible for regulating glutamatergic and GABAergic transmission [73]. Results reported by Berger et al. suggest an increase in endocannabinoids, a mechanism intended to aid in neuronal homeostasis, further accentuates neural imbalances in the LHb [73]. Rather than suppressing neuronal excitability, as previously noted in cortical and hippocampal brain regions [110], an increase in the endocannabinoid, 2-AG, further facilitates neuronal activation in the LHb [73]. The paradoxical results may be explained via the endocannabinoid’s preferential suppression of GABA, via its potentiation of glutamate or via differing actions on the pre- and post-synaptic terminals [111]. Understanding how this applies in the human brain, including the localisation of cannabinoid receptors on glutamatergic or GABAergic terminals in the Hb, will be an important step forward.
An additional driving regulator of LHb bursting activity involves glial-neuron interactions. The potassium channel, Kir4.1, expressed on astrocytes regulates the degree of membrane polarisation [112]. Overexpression of astrocytic Kir4.1 in the LHb induces neuronal bursting that is reversed via ablation of Kir4.1 [112]. Moreover, preclinical models of depression show greater expression of Kir4.1 in the LHb [112]. Upregulation of Kir4.1 leads to an increased clearance of extracellular K+ [112]. The lower levels of extracellular K+ then induce membrane hyperpolarisation which may explain the abnormal firing pattern [112] Maladaptive astrocyte functioning and subsequent disrupted glutamate clearance have been associated with depressive states and may contribute to abnormal Hb activity (reviewed in ref. [113]). Moreover, astrocytes play a vital role in neuroimmune response and are capable of both mitigating and promoting inflammatory signalling [114]. While astrocytes are known to be abundant in the healthy Hb, changes in their expression are yet to be examined in the context of depression [115].
Neuroinflammation is an emerging phenotype evident in depression. In the LHb, chronic stress exposure has been shown to lead to an increase in pro-inflammatory cytokines [75, 76]. Both inducible NOS (iNOS) and neuronal NOS (nNOS), typically associated with inflammation and neuronal activity respectively, were increased in the LHb in a model of chronic stress [77]. The elevated levels of nitric oxide produced from nNOS and iNOS may lead to excitotoxic/neurotoxic effects in the LHb [77]. Conversely, endothelial NOS (eNOS) levels were decreased in the LHb of stressed rats. The reduction in eNOS-positive cells suggests a compromised endothelial function, which may lead to impaired vasodilation and subsequent reduced anti-inflammatory responses within the LHb [77]. Local injection of TNF- α into the LHb was sufficient to induce depressive-like symptoms in rodents [76]. Moreover, administration of the anti-inflammatory agents aspirin or a NF-kB inhibitor restored these behavioural deficits [76]. Interestingly, these findings did not translate when administered locally within the DRN or paraventricular nucleus, suggesting a specific role for the LHb in the inflammatory response [76]. Lesions to the LHb also reduced the expression of proinflammatory cytokines induced by chronic stress, improved the hippocampal anti-inflammatory response via activation of the PI3K/mTOR pathway and reduced the expression of apoptosis-related proteins [76]. Given the proximity of the Hb to the third ventricle and previous reports of altered blood brain barrier permeability in depression, the region may be particularly vulnerable to cytokine infiltration and be a unique target in the therapeutic action of anti-inflammatory treatments [106].
An important consideration in preclinical research is individual variation between models. For instance, neural responses in the LHb have previously been found to differ between acute and chronic stress exposure [81]. Chronic stress leads to sustained activation of RMTg GABAergic neurons, whereas acute stress inhibits these neurons [81]. These findings may indicate dynamic neural adaptation within the LHb in response to different types of stress [81]. Chronic stress is more likely to induce a strong depressive-like phenotype in rodents due its sustained effects and may serve as a more validated model of relevance to depression. While the findings in this SLR show many consistencies across the preclinical models, particularly related to LHb excitation, it is important to consider that variations in molecular findings may be influenced by the preclinical model of depression employed or specific stress protocol. However, these variations might provide insights into the potential nuances and underlying mechanisms of different forms of depression, which is considered a heterogenous disorder in the human/clinical scenario [116].
Clinical insights
Clinical studies support a perturbed Hb response in individuals with depression; the limited evidence points to a general pattern of hyperconnectivity and volumetric changes in depression. However, it is unclear how Hb functionality may vary across sex, age, depression severity and subtype or how external medications may influence its activity in humans. Additionally, clinical studies typically utilised neuroimaging approaches such as fMRI, which rely on accurate volumetric analysis of the region of interest. However, this analysis may be complicated by the minute size of the Hb. Therefore, alternative neuroimaging methods should be prioritised moving forward, including 7-T scanning and DTI [117]. 7-T imaging has been proven to achieve a sufficient spatial resolution of the Hb capable of differentiating lateral and medial divisions [118]. Additionally, DTI offers advantages over volumetric analysis as it can provide detailed microstructural information by measuring the diffusion of water molecules along white matter tracts, which is particularly valuable for small and complex regions like the habenula [107, 118]. This technique allows for the mapping of the brain region’s intricate fiber pathways rather than total volume, enhancing our understanding of the functional and structural connectivity of the Hb (reviewed in [119]). In addition, Ultra-High Resolution Positron Emission Tomography (UHR PET) imaging offers a novel complementary approach for detailed investigations of metabolic and molecular processes in small brain regions like the Hb [18, 120]. Traditional PET imaging provides a functional understanding of alterations in neurotransmitters and receptor binding and density, but its limited spatial resolution makes it less suitable for Hb research (Reviewed in refs. [121, 122]). While UHR PET shows great promise, its application specifically to the Hb is still emerging and to date, there are no studies that have utilised UHR PET exclusively for Hb imaging. Nonetheless, its potential to provide high-resolution functional imaging makes it a valuable tool that when used in conjunction with 7-T MRI and DTI, could offer a comprehensive understanding of Hb dysfunction in depression.
Only three studies have conducted molecular (gene expression) analysis of the Hb in people with depression and two were limited to males [79, 106]. The intriguing results suggest abnormal Hb activity may be a downstream consequence of endothelial cell dysfunction and altered blood brain barrier permeability [106]. However, further research is required to understand how these findings integrate into the current hypothesis around Hb dysfunction in depression and determine if the findings are replicable in the female brain. In particular, it will be important to determine whether the evidence of excitatory/inhibitory imbalance observed in preclinical studies, is replicated in the human brain.
Considering the variety of networks projecting from the Hb, it is possible that some circuitry may be hyperactive and others hypoactive in depressive states. For instance, while overall Hb activation was consistently elevated in depression, specific connections to the cerebellum and calcarine sulcus were found to be reduced compared to controls [91, 92]. Interestingly, a post-mortem human study identified individuals who died with depression displayed a reduction in total Hb volume and cell count [103]. While both male and female subjects were included in experiments, sex was not analysed as a biological variable [103]. The pooling of sex is an important limitation considering more recent investigations reported Hb volume reductions only pertained to females with a diagnosis of depression [104]. Of important consideration, reductions in Hb volume were limited to depression and did not extend to the post-mortem brain of schizophrenia patients [103]. Considering schizophrenia often includes depressive symptoms, an interesting avenue for future research involves investigating whether Hb abnormalities are unique to primary depression or occur when depression is secondary to a differential diagnosis such as schizophrenia, dementia, stroke, or Parkinson’s disease.
Therapeutic potential
People with depression often do not experience an alleviation of symptoms until weeks following the initiation of SSRI treatment [6]. This delayed therapeutic response aligns with the time required to restore Hb activity in preclinical models [25, 67]. In the clinical population, following the administration of single-dose ketamine, subjects with treatment-resistant depression exhibited a significant reduction in LHb glucose metabolism compared to baseline, suggesting an attenuating effect on LHb activity [123]. Furthermore, recent preclinical work has shown that systemic ketamine administration in depressive-like mice, blocks NMDA receptors specifically in the LHb, and that knockout of NMDA receptors in this region eliminated the antidepressant effect of ketamine [124]. These studies highlight ketamine’s ability to normalise Hb hyperactivity and suggest direct targeting of the Hb may be a promising avenue for future research.
Clinical studies have also suggested alterations within the Hb complex may be indicative of treatment response. In treatment-resistant psychiatric patients (diagnosis of depression, anorexia nervosa or bipolar disorder) receiving subcallosal cingulate deep brain stimulation, individuals unresponsive to treatment had a reduction in total Hb volume at 12 months follow up [125]. Conversely, patients who exhibited a therapeutic response displayed an overall increase in Hb volume 12 months post treatment [125]. In addition, high-frequency deep brain stimulation of the LHb led to complete remission in a female living with treatment-resistant depression [126]. Interestingly, depressive symptoms returned within days following the ceasing of treatment but resolved following the reinstatement of LHb stimulation for 12 weeks [126]. It is unclear how deep brain stimulation impacts LHb connectivity and neurotransmission; however, deciphering the underlying mechanisms may help identify the molecular changes in the Hb leading to the antidepressant effect. Furthermore, as previously highlighted, local administration of agents that modulate excitatory (ßCaMKII blockade, NMDA antagonist), inhibitory (GABA-B agonist/antagonist) and inflammatory (NF-kB inhibitor, aspirin) processes in the Hb has yielded notable therapeutic outcomes. It has even been shown that non-invasive light therapy, which has antidepressant potential in clinical populations (reviewed in [127]) and preclinical studies [128], mediates its effect via reducing CaMKIIα excitation in the LHb, via a retina-lateral geniculate nuclei-LHb circuit, further reinforcing the potential of novel approaches to reduce LHb excitation in depression. Examining the molecular composition and cellular architecture of the human Hb may reveal unique treatment targets in this crucial brain region.
Considerations, limitations, and future directions
A major limitation of the current preclinical and clinical literature is the lack of consideration of biological sex. The few studies that conducted molecular analyses on the female Hb demonstrate the results often differ from males. For instance, in rodents while both sexes exhibit LHb hyperactivation, the changes appear to be more pronounced in females following chronic but not acute stress exposure [39, 40]. Similarly, females displayed greater basal LHb activation compared to males, suggesting they may be particularly vulnerable to changes in Hb firing activity [39, 64]. Considering the LHb signals disappointment and aversion, it is theorised that the greater excitation within female mice may bias the animals towards negative emotional states and impair their resilience to stress [106, 129]. Whether these molecular sexual dimorphisms contribute to the clinical sex differences observed in affective disorders is yet to be explored. To date, molecular characterisation of the human Hb is predominately derived from males and has exclusively been conducted at the transcriptomic level [82, 106]. Furthermore, the current literature lacks a fundamental understanding as to whether the cellular, anatomical, and functional composition of the Hb is synonymous between sexes and what the clinical implications of any differences may be.
Despite emerging evidence highlighting the importance of neurodevelopment in depression, the current literature largely focused on the mature, adult Hb. Depression is influenced by both early life environmental factors and genetic predispositions [130]. Stressful early life experiences have been shown to disrupt LHb maturation, leading to long-lasting behavioural changes in adulthood [131]. Additionally, specific developmental Hb neuronal clusters (subset of Hb neurons with a similar molecular signature) have been linked to known risk genes (Rbfox1 and Lhx2) for MDD [132], hinting that changes in the development of the LHb may contribute to the development of MDD. Despite these findings, our understanding surrounding the organisation and development of Hb neurocircuitry remains incomplete [132]. It is also unclear when Hb dysregulation occurs during depression onset and how Hb function changes over the course of the disease. At the other end of the spectrum, there is a lack of evidence on Hb functionality in ageing. A critical step forward will be to determine how the properties and function of the Hb alter in the context of the ageing brain, and whether these changes are protective or increase susceptibility to Hb dysregulation and depression in later life.
The small size of the medial and lateral Hb has been a major limitation in clinical investigations; typical imaging techniques lack the spatial resolution to differentiate the nuclei from one another [133]. Consequently, the precise roles and molecular profile of the medial and lateral subdivisions remain elusive in humans. Considering current neuroimaging studies are unable to isolate the MHb and the preclinical research has predominately focussed on the LHb, the specific role of the MHb has largely been neglected. This review highlights a need for further consideration of the individualised functions of each subdivision, specifically in the human brain, moving towards advanced neuroimaging methods and considering post-mortem molecular analyses.
The invasive nature of preclinical research has allowed for comprehensive analysis of Hb neuronal signatures including neurotransmitter populations, neuroconnectivity and cytoarchitecture within rodents [115, 134]. However, the morphologic properties of the human Hb have previously been reported to differ from rodents indicating potential molecular differences [4, 27]. An essential step in developing the clinical literature is to determine the comparative physiology and cellular organisation (neuronal and non-neuronal cell types, neurotransmitter populations, afferent/efferent projections) of the human Hb. Subsequent investigations should then consider how the Hb may change across human disease states including the various subtypes of depression (bipolar depression, post-stroke depression etc.) and what the implication of these changes may be.
Despite advancements in preclinical research on pharmacological treatments for depression, there remains a critical unmet need in the clinical population. While a large array of antidepressant drugs is available, these medications broadly exhibit similar mechanisms of action (except for the newer emerging therapeutics such as ketamine) and are often prescribed as first-line treatments without considering individual patient differences [4]. This non-specific approach makes predicting treatment responses challenging. By understanding the neurobiological underpinnings of depression within the heterogeneous patient population, more targeted and effective interventions can be developed. Although preclinical findings consistently report Hb hyperactivity, clinical results vary widely, likely due to the diverse patient populations studied which include TRD, MDD and late-life depression. Defining appropriate patient subgroups based on Hb neurobiology may help prescribe more effective personalised treatment approaches, whether that is pharmacological, neuromodulatory (e.g. TMS, DBS), environmental (e.g. nutrition/exercise interventions, light therapy) or a combination of these. Through this approach, we may be able to take advantage of the substantial number of drugs currently available (and non-drug alternatives) and avoid the substantial cost and time investment associated with drug development. On the other hand, understanding the role of the Hb in the neurobiology of depression may reveal novel therapeutic targets that enable the development of new antidepressant drugs with a different mechanism of action to the existing pharmaceutical options that justifies the challenges that pharmaceutical companies face in drug development for mental health disorders.
Conclusion
Overall, the preclinical studies presented in this systematic review support LHb hyperactivity as a primary driver for the development of depressive symptoms. The molecular mechanisms driving LHb hyperactivation have been explored and centre on altered inhibitory/excitatory balance, abnormal glial interactions, and neuroinflammation. Moreover, when successful in eliciting a therapeutic response, serval antidepressant treatments including SSRIs, deep brain stimulation, and ketamine intersect in their ability to restore and normalise Hb connectivity, suggesting a role for the Hb as a site for therapeutic agents. However, clinical findings suggest the complexity of depression may go beyond the simplicity of an overactive Hb. Evidence from human studies does support gross Hb abnormalities such as altered activation, connectivity, and volume as a clinical feature of depression; however, it highlights the need for considerations including hemispheric differences, the specific roles of the medial and lateral subdivisions, and greater molecular characterisation of the human Hb in depression. Furthermore, considerations need to be given to the influence of sex and age across preclinical and clinical contexts. Bridging these gaps in the research is essential to enhance our understanding of how Hb dysregulation mediates the core symptoms of depression and shed light on the development of novel therapeutic approaches.
References
World Health Organisation. Depression and other common mental health disorders: Global health estimates. Geneva: World Health Organisation; 2017. 24. https://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
Salk RH, Hyde JS, Abramson LY. Gender differences in depression in representative national samples: Meta-analyses of diagnoses and symptoms. Psychol Bull. 2017;143:783–822.
Habert J, Katzman MA, Oluboka OJ, McIntyre RS, McIntosh D, MacQueen GM, et al. Functional recovery in major depressive disorder: focus on early optimized treatment. Prim Care Companion CNS Disord. 2016;18.
Read J, Gee A, Diggle J, Butler H. The interpersonal adverse effects reported by 1008 users of antidepressants; and the incremental impact of polypharmacy. Psychiatry Res. 2017;256:423–7.
Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354:1243–52.
Kraus C, Kadriu B, Lanzenberger R, Zarate CA, Kasper S. Prognosis and improved outcomes in major depression: a review. Transl Psychiatry. 2019;9:127.
Namboodiri VMK, Rodriguez-Romaguera J, Stuber GD. The habenula. Curr Biol. 2016;26:R873–7.
Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447:1111–5.
Mathis V, Cosquer B, Barbelivien A, Herbeaux K, Bothorel B, Sage-Ciocca D, et al. The lateral habenula interacts with the hypothalamo-pituitary adrenal axis response upon stressful cognitive demand in rats. Behav Brain Res. 2018;341:63–70.
Aizawa H, Yanagihara S, Kobayashi M, Niisato K, Takekawa T, Harukuni R, et al. The synchronous activity of lateral habenular neurons is essential for regulating hippocampal theta oscillation. J Neurosci. 2013;33:8909–21.
Wang D, Li Y, Feng Q, Guo Q, Zhou J, Luo M. Learning shapes the aversion and reward responses of lateral habenula neurons. Costa RM, editor. eLife. 2017;6:e23045.
Aizawa H, Kobayashi M, Tanaka S, Fukai T, Okamoto H. Molecular characterization of the subnuclei in rat habenula. J Comp Neurol. 2012;520:4051–66.
Meye FJ, Lecca S, Valentinova K, Mameli M. Synaptic and cellular profile of neurons in the lateral habenula. Front Hum Neurosci. 2013;7:860.
Yang N, Anapindi KDB, Rubakhin SS, Wei P, Yu Q, Li L, et al. Neuropeptidomics of the Rat Habenular Nuclei. J Proteome Res. 2018;17:1463–73.
Contestabile A, Villani L, Fasolo A, Franzoni MF, Gribaudo L, Øktedalen O, et al. Topography of cholinergic and substance P pathways in the habenulo-interpeduncular system of the rat. An immunocytochemical and microchemical approach. Neuroscience. 1987;21:253–70.
Wagner F, Bernard R, Derst C, French L, Veh RW. Microarray analysis of transcripts with elevated expressions in the rat medial or lateral habenula suggest fast GABAergic excitation in the medial habenula and habenular involvement in the regulation of feeding and energy balance. Brain Struct Funct. 2016;221:4663–89.
Gaudin É, Toussaint M, Thibaudeau C, Paillé M, Fontaine R, Lecomte R. Performance simulation of an ultra-high resolution brain PET scanner using 1.2-mm pixel detectors. IEEE Trans Radiat Plasma Med Sci. 2019;3:334–42.
Hayakawa T, Seki M, Zyo K. Studies on the efferent projections of the interpeduncular complex in cats. Okajimas Folia Anat Jpn. 1981;58:1–15.
Groenewegen HJ, Ahlenius S, Haber SN, Kowall NW, Nauta WJH. Cytoarchitecture, fiber connections, and some histochemical aspects of the interpeduncular nucleus in the rat. J Comp Neurol. 1986;249:65–102.
Mathuru AS, Jesuthasan S. The medial habenula as a regulator of anxiety in adult zebrafish. Front Neural Circuits. 2013;7:99.
Frahm S, Slimak MA, Ferrarese L, Santos-Torres J, Antolin-Fontes B, Auer S, et al. Aversion to nicotine is regulated by the balanced activity of β4 and α5 nicotinic receptor subunits in the medial habenula. Neuron. 2011;70:522–35.
Shumake J, Edwards E, Gonzalez-Lima F. Opposite metabolic changes in the habenula and ventral tegmental area of a genetic model of helpless behavior. Brain Res. 2003;963:274–81.
Faron-Górecka A, Kuśmider M, Solich J, Kolasa M, Pabian P, Gruca P, et al. Regulation of somatostatin receptor 2 in the context of antidepressant treatment response in chronic mild stress in rat. Psychopharmacology. 2018;235:2137–49.
Stanisavljević A, Perić I, Gass P, Inta D, Lang UE, Borgwardt S, et al. Fluoxetine modulates neuronal activity in stress-related limbic areas of adult rats subjected to the chronic social isolation. Brain Res Bull. 2020;163:95–108.
Aizawa H, Amo R, Okamoto H. Phylogeny and ontogeny of the habenular structure. Front. Neurosci. 2011;5:138.
Díaz E, Bravo D, Rojas X, Concha ML. Morphologic and immunohistochemical organization of the human habenular complex. J Comp Neurol. 2011;519:3727–47.
Stamatakis AM, Stuber GD. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci. 2012;15:1105–7.
Knowland D, Lim BK. Circuit-based frameworks of depressive behaviors: the role of reward circuitry and beyond. Pharmacol Biochem Behav. 2018;174:42–52.
Tchenio A, Lecca S, Valentinova K, Mameli M. Limiting habenular hyperactivity ameliorates maternal separation-driven depressive-like symptoms. Nat Commun. 2017;8:1135.
Kingir E, Sevinc C, Unal G. Chronic oral ketamine prevents anhedonia and alters neuronal activation in the lateral habenula and nucleus accumbens in rats under chronic unpredictable mild stress. Neuropharmacology. 2023;228:109468.
Yang Y, Cui Y, Sang K, Dong Y, Ni Z, Ma S, et al. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature. 2018;554:317–22.
Hitti FL, Parker D, Yang AI, Brem S, Verma R. Laterality and sex differences of human lateral habenula afferent and efferent fiber tracts. Front Neurosci. 2022;16:837624.
Liu X, Huang H, Zhang Y, Wang L, Wang F. Sexual dimorphism of inputs to the lateral habenula in mice. Neurosci Bull. 2022;38:1439–56.
Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. Behav. Neurosci. 2011;7:121–47.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
Krauth D, Woodruff TJ, Bero L. Instruments for assessing risk of bias and other methodological criteria of published animal studies: a systematic review. Environ Health Perspect. 2013;121:985–92.
Kim W, Chung C. Brain-wide cellular mapping of acute stress-induced activation in male and female mice. FASEB J. 2021;35:e22041.
Sood A, Chaudhari K, Vaidya VA. Acute stress evokes sexually dimorphic, stressor-specific patterns of neural activation across multiple limbic brain regions in adult rats. Stress Amst Neth. 2018;21:136–50.
Kim Y, Morath B, Hu C, Byrne LK, Sutor SL, Frye MA, et al. Antidepressant actions of lateral habenula deep brain stimulation differentially correlate with CaMKII/GSK3/AMPK signaling locally and in the infralimbic cortex. Behav Brain Res. 2016;306:170–7.
Andalman AS, Burns VM, Lovett-Barron M, Broxton M, Poole B, Yang SJ, et al. Neuronal dynamics regulating brain and behavioral state transitions. Cell. 2019;177:970–985.e20.
Greenwood BN, Foley TE, Burhans D, Maier SF, Fleshner M. The consequences of uncontrollable stress are sensitive to duration of prior wheel running. Brain Res. 2005;1033:164–78.
Li ZL, Wang Y, Zou HW, Jing XY, Liu YJ, Li LF. GABA(B) receptors within the lateral habenula modulate stress resilience and vulnerability in mice. Physiol Behav. 2021;230:113311.
Nakajo H, Tsuboi T, Okamoto H. The behavioral paradigm to induce repeated social defeats in zebrafish. Neurosci Res. 2020;161:24–32.
Okamura H, Yasugaki S, Suzuki-Abe H, Arai Y, Sakurai K, Yanagisawa M, et al. Long-term effects of repeated social defeat stress on brain activity during social interaction in BALB/c mice. eNeuro. 2022;9:ENEURO.0068–22.2022.
Febbraro F, Svenningsen K, Tran TP, Wiborg O. Neuronal substrates underlying stress resilience and susceptibility in rats. PLoS ONE. 2017;12:e0179434.
Spivey JM, Padilla E, Shumake JD, Gonzalez-Lima F. Effects of maternal separation, early handling, and gonadal sex on regional metabolic capacity of the preweanling rat brain. Brain Res. 2011;1367:198–206.
Hevner RF, Liu S, Wong-Riley MTT. A metabolic map of cytochrome oxidase in the rat brain: histochemical, densitometric and biochemical studies. Neuroscience. 1995;65:313–42.
Millard SJ, Weston-Green K, Newell KA. The Wistar-Kyoto rat model of endogenous depression: a tool for exploring treatment resistance with an urgent need to focus on sex differences. Prog Neuropsychopharmacol Biol Psychiatry. 2020;101:109908.
Daut RA, Fonken LK. Circadian regulation of depression: a role for serotonin. Front Neuroendocrinol. 2019;54:100746.
Begega A, Cuesta Lopez I, Cuesta Izquierdo M, Jove CI, Moreno-Fernández RD, López M. Reorganization of brain networks as a substrate of resilience: an analysis of cytochrome c oxidase activity in rats. Neuroscience. 2023;516:75–90.
Xu C, Sun Y, Cai X, You T, Zhao H, Li Y, et al. Medial habenula-interpeduncular nucleus circuit contributes to anhedonia-like behavior in a rat model of depression. Front Behav Neurosci. 2018;12:238.
Guan YF, Huang GB, Xu MD, Gao F, Lin S, Huang J, et al. Anti-depression effects of ketogenic diet are mediated via the restoration of microglial activation and neuronal excitability in the lateral habenula. Brain Behav Immun. 2020;88:748–62.
Langlois LD, Berman RY, Shepard RD, Simmons SC, Tsuda MC, Gouty S, et al. Potentiation of glutamatergic synaptic transmission onto lateral habenula neurons following early life stress and intravenous morphine self-administration in rats. Addict Biol. 2022;27:e13064.
Lecca S, Pelosi A, Tchenio A, Moutkine I, Lujan R, Hervé D, et al. Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat Med. 2016;22:254–61.
Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, et al. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470:535–9.
Shepard RD, Nugent FS. Targeting endocannabinoid signaling in the lateral habenula as an intervention to prevent mental illnesses following early life stress: a perspective. Front Synaptic Neurosci. 2021;13:689518.
Simmons SC, Shepard RD, Gouty S, Langlois LD, Flerlage WJ, Cox BM, et al. Early life stress dysregulates kappa opioid receptor signaling within the lateral habenula. Neurobiol Stress. 2020;13:100267.
Zhang Y, Ma L, Zhang X, Yue L, Wang J, Zheng J, et al. Deep brain stimulation in the lateral habenula reverses local neuronal hyperactivity and ameliorates depression-like behaviors in rats. Neurobiol Dis. 2023;180:106069.
Klein ME, Chandra J, Sheriff S, Malinow R. Opioid system is necessary but not sufficient for antidepressive actions of ketamine in rodents. Proc Natl Acad Sci USA. 2020;117:2656–62.
Zubcevic J, Watkins J, Perez PD, Colon-Perez LM, Long MT, Febo M, et al. MEMRI reveals altered activity in brain regions associated with anxiety, locomotion, and cardiovascular reactivity on the elevated plus maze in the WKY vs SHR rats. Brain Imaging Behav. 2018;12:1318–31.
Lin S, Huang L, Luo ZC, Li X, Jin SY, Du ZJ, et al. The ATP level in the medial prefrontal cortex regulates depressive-like behavior via the medial prefrontal cortex-lateral Habenula pathway. Biol Psychiatry. 2022;92:179–92.
Zhang S, Zhang H, Ku SM, Juarez B, Morel C, Tzavaras N, et al. Sex differences in the neuroadaptations of reward-related circuits in response to subchronic variable stress. Neuroscience. 2018;376:108–16.
Cerniauskas I, Winterer J, de Jong JW, Lukacsovich D, Yang H, Khan F, et al. Chronic stress induces activity, synaptic, and transcriptional remodeling of the lateral habenula associated with deficits in motivated behaviors. Neuron. 2019;104:899–915.e8.
Webster JF, Beerens S, Wozny C. Effects of early life stress and subsequent re-exposure to stresson neuronal activity in the lateral habenula. Neuropsychopharmacology. 2023;48:745–53.
Knowland D, Lilascharoen V, Pacia CP, Shin S, Wang EHJ, Lim BK. Distinct ventral pallidal neural populations mediate separate symptoms of depression. Cell. 2017;170:284–297.e18.
Zhang M, Lyu D, Wang F, Shi S, Wang M, Yang W, et al. Ketamine may exert rapid antidepressant effects through modulation of neuroplasticity, autophagy, and ferroptosis in the habenular nucleus. Neuroscience. 2022;506:29–37.
Li K, Zhou T, Liao L, Yang Z, Wong C, Henn F, et al. βCaMKII in lateral habenula mediates core symptoms of depression. Science. 2013;341:1016–20.
Kang M, Chung JM, Noh J, Kim J. The mineralocorticoid receptor and extra-synaptic NMDA receptor in the lateral habenula involve in the vulnerability to early life stress in the maternal separation model. Neurobiol Stress. 2023;27:100570.
Seo JS, Zhong P, Liu A, Yan Z, Greengard P. Elevation of p11 in lateral habenula mediates depression-like behavior. Mol Psychiatry. 2018;23:1113–9.
Shen XF, Yuan HB, Wang GQ, Xue H, Liu YF, Zhang CX. Role of DNA hypomethylation in lateral habenular nucleus in the development of depressive-like behavior in rats. J Affect Disord. 2019;252:373–81
Berger AL, Henricks AM, Lugo JM, Wright HR, Warrick CR, Sticht MA, et al. The lateral habenula directs coping styles under conditions of stress via recruitment of the endocannabinoid system. Biol Psychiatry. 2018;84:611–23.
Yang E, Kim JY, Yang SH, Lee E, Sun W, Lee HW, et al. Three-dimensional analysis of mouse habenula subnuclei reveals reduced volume and gene expression in the lipopolysaccharide-mediated depression model. Exp Neurobiol. 2019;28:709–19.
Ito H, Nozaki K, Sakimura K, Abe M, Yamawaki S, Aizawa H. Activation of proprotein convertase in the mouse habenula causes depressive-like behaviors through remodeling of extracellular matrix. Neuropsychopharmacology. 2021;46:442–54.
Wang Y, Qu P, Sun Y, Li Z, Liu L, Yang L. Association between increased inflammatory cytokine expression in the lateral habenular nucleus and depressive-like behavior induced by unpredictable chronic stress in rats. Exp Neurol. 2022;349:113964.
Chen W, Chen Y, Aslam MS, Shen J, Tong T, Yan S, et al. The effect of acupuncture on lateral habenular nucleus and intestinal microflora in depression model rats. Behav Brain Res. 2023;455:114627.
Yoo H, Kim HJ, Yang SH, Son GH, Gim JA, Lee HW, et al. Gene expression profiling of the habenula in rats exposed to chronic restraint stress. Mol Cells. 2022;45:306–16.
Han S, Yang SH, Kim JY, Mo S, Yang E, Song KM, et al. Down-regulation of cholinergic signaling in the habenula induces anhedonia-like behavior. Sci Rep. 2017;7:900.
Korlatowicz A, Pabian P, Solich J, Kolasa M, Latocha K, Dziedzicka-Wasylewska M, et al. Habenula as a possible target for treatment-resistant depression phenotype in Wistar Kyoto rats. Mol Neurobiol. 2023;60:643–54.
Yang SH, Yang E, Lee J, Kim JY, Yoo H, Park HS. et al. Neural mechanism of acute stress regulation by trace aminergic signalling in the lateral habenula in male mice. Nat Commun. 2023;14:2435.
Lin R, Mitsuhashi H, Fiori LM, Denniston R, Ibrahim EC, Belzung C, et al. SNORA69 is up-regulated in the lateral habenula of individuals with major depressive disorder. Sci Rep. 2024;14:8258.
Christiansen S, Bouzinova E, Fahrenkrug J, Wiborg O. Altered expression pattern of clock genes in a rat model of depression. Int J Neuropsychopharmacol. 2016;19:pyw061.
Li Y, Li G, Li J, Cai X, Sun Y, Zhang B, et al. Depression-like behavior is associated with lower Per2mRNA expression in the lateral habenula of rats. Genes Brain Behav. 2021;20:e12702.
Haniffa S, Narain P, Hughes MA, Petkovic A, Susic M, Mlambo V, et al. Chronic social stress blunts corebody temperature and molecular rhythms of Rbm3 and Cirbp in mouse lateral habenula. Open Biol. 2023;13:220380.
Guo B, Zhou F, Li M, Gore JC, Ding Z. Correlated functional connectivity and glucose metabolism in brain white matter revealed by simultaneous MRI/positron emission tomography. Magn Reson Med. 2022;87:1507–14.
Wu Y, Chu Z, Chen X, Zhu Y, Xu X, Shen Z. Functional connectivity between the habenula and posterior default mode network contributes to the response of the duloxetine effect in major depressive disorder. Neuroreport. 2024;35:380–6.
Su T, Chen B, Yang M, Wang Q, Zhou H, Zhang M, et al. Disrupted functional connectivity of the habenula links psychomotor retardation and deficit of verbal fluency and working memory in late-life depression. CNS Neurosci Ther. 2024;30:e14490.
Chen B, Su T, Yang M, Wang Q, Zhou H, Tan G, et al. Static and dynamic functional connectivity of the habenula in late-life depression patient with suicidal ideation. J Affect Disord. 2024;356:499–506.
Yang L, Jin C, Qi S, Teng Y, Li C, Yao Y, et al. Maladaptive avoidance learning in the orbitofrontal cortex in adolescents with major depression. BMC Psychiatry. 2022;22:588.
Zhu Z, Wang S, Lee TMC, Zhang R. Habenula functional connectivity variability increases with disease severity in individuals with major depression. J Affect Disord. 2023;333:216–24.
Jung JY, Cho SE, Kim N, Kang CK, Kang SG. Decreased resting-state functional connectivity of the habenula-cerebellar in a major depressive disorder. Front Psychiatry. 2022;13:925823.
Barreiros AR, Breukelaar I, Mayur P, Andepalli J, Tomimatsu Y, Funayama K, et al. Abnormal habenulafunctional connectivity characterizes treatment-resistant depression. NeuroImage Clin. 2022;34:102990.
Amiri S, Arbabi M, Kazemi K, Parvaresh-Rizi M, Mirbagheri MM. Characterization of brain functional connectivity in treatment-resistant depression. Prog Neuropsychopharmacol Biol Psychiatry. 2021;111:110346.
Amiri S, Arbabi M, Rahimi M, Parvaresh-Rizi M, Mirbagheri MM. Effective connectivity between deep brain stimulation targets in individuals with treatment-resistant depression. Brain Commun. 2023;5:fcad256.
Zhu Y, Qi S, Zhang B, He D, Teng Y, Hu J, et al. Connectome-based biomarkers predict subclinical depression and identify abnormal brain connections with the lateral habenula and thalamus. Front Psychiatry. 2019;10:371.
Furman DJ, Gotlib IH. Habenula responses to potential and actual loss in major depression: preliminary evidence for lateralized dysfunction. Soc Cogn Affect Neurosci. 2016;11:843–51.
Lawson RP, Nord CL, Seymour B, Thomas DL, Dayan P, Pilling S, et al. Disrupted habenula function in major depressionwill. Mol Psychiatry. 2017;22:202–8.
Willinger D, Karipidis II, Neuer S, Emery S, Rauch C, Häberling I, et al. Maladaptive avoidance learning in the orbitofrontal cortex in adolescents with major depression. Biol Psychiatry Cogn Neurosci Neuroimaging. 2022;7:293–301.
Kyuragi Y, Oishi N, Hatakoshi M, Hirano J, Noda T, Yoshihara Y, et al. Segmentation and volume estimation of the habenula using deep learning in patients with depression. Biol Psychiatry Glob Open Sci. 2024;4:100314.
Carceller-Sindreu M, de Diego-Adeliño J, Serra-Blasco M, Vives-Gilabert Y, Martí;n-Blanco A, Puigdemont D, et al. Volumetric MRI study of the habenula in first episode, recurrent and chronic major depression. Eur Neuropsychopharmacol. 2015;25:2015–21.
Liu WH, Valton V, Wang LZ, Zhu YH, Roiser JP. Association between habenula dysfunction and motivational symptoms in unmedicated major depressive disorder. Soc Cogn Affect Neurosci. 2017;12:1520–33.
Ranft K, Dobrowolny H, Krell D, Bielau H, Bogerts B, Bernstein HG. Evidence for structural abnormalities of the human habenular complex in affective disorders but not in schizophrenia. Psychol Med. 2010;40:557–67.
Savitz JB, Rauch SL, Drevets WC. Habenula volume in bipolar disorder and major depressive disorder: a high-resolution magnetic resonance imaging study. Biopsych. 2011;69:336–43.
Schmidt FM, Schindler S, Adamidis M, Strauß M, Tränkner A, Trampel R, et al. Habenula volume increases with disease severity in unmedicated major depressive disorder as revealed by 7T MRI. Eur Arch Psychiatry Clin Neurosci. 2017;267:107–15.
Kim HJ, Yoo H, Kim JY, Yang SH, Lee HW, Lee HJ, et al. Postmortem gene expression profiles in the habenulae of suicides: implication of endothelial dysfunction in the neurovascular system. Mol Brain. 2022;15:48.
Cho S-E, Kim N, Na K-S, Kang C-K, Kang S-G. Thalamo-habenular connection differences between patients with major depressive disorder and normal controls. Front Psychiatry. 2021;12:699416.
Kang M, Noh J, Chung JM. NMDA receptor-dependent long-term depression in the lateral habenula: implications in physiology and depression. Sci Rep. 2020;10:17921.
Wang D, Li A, Dong K, Li H, Guo Y, Zhang X, et al. Lateral hypothalamus orexinergic inputs to lateral habenula modulate maladaptation after social defeat stress. Neurobiol Stress. 2021;14:100298.
Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81.
Winters ND, Kondev V, Loomba N, Delpire E, Grueter BA, Patel S. Opposing retrograde and astrocyte-dependent endocannabinoid signaling mechanisms regulate lateral habenula synaptic transmission. Cell Rep. 2023;42:112159.
Cui Y, Yang Y, Ni Z, Dong Y, Cai G, Foncelle A, et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature. 2018;554:323–7.
Zhou X, Xiao Q, Xie L, Yang F, Wang L, Tu J. Astrocyte, a promising target for mood disorder interventions. Front Mol Neurosci. 2019;12:136.
Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte crosstalk in CNS inflammation. Neuron. 2020;108:608–22.
Hashikawa Y, Hashikawa K, Rossi MA, Basiri ML, Liu Y, Johnston NL, et al. Transcriptional and spatial resolution of cell types in the mammalian habenula. Neuron. 2020;106:743–758.e5.
Nguyen TD, Harder A, Xiong Y, Kowalec K, Hägg S, Cai N, et al. Genetic heterogeneity and subtypes of major depression. Mol Psychiatry. 2022;27:1667–75.
Roiser JP, Levy J, Fromm SJ, Nugent AC, Talagala SL, Hasler G, et al. The effects of tryptophan depletion on neural responses to emotional words in remitted depression. Biol Psychiatry. 2009;66:441–50.
Strotmann B, Kögler C, Bazin PL, Weiss M, Villringer A, Turner R. Mapping of the internal structure of human habenula with ex vivo MRI at 7T. Front Hum Neurosci. 2013;7:878.
Tohyama S, Walker MR, Sammartino F, Krishna V, Hodaie M. The utility of diffusion tensor imaging in neuromodulation: moving beyond conventional magnetic resonance imaging. Neuromodulation Technol Neural Interface. 2020;23:427–35.
Doyon V, Sarrhini O, Loignon-Houle F, Auger É, Toussaint M, Thibaudeau C, et al. First PET investigation of the human brain at 2 µL resolution with the ultra-high-resolution (UHR) scanner. J Nucl Med. 2023;64:P726.
Singh SB, Tiwari A, Katta MR, Kafle R, Ayubcha C, Patel KH, et al. The utility of PET imaging in depression. Front Psychiatry. 2024;25:1322118.
Frahm S, Antolin-Fontes B, Görlich A, Zander JF, Ahnert-Hilger G, Ibañez-Tallon I. An essential role of acetylcholine-glutamate synergy at habenular synapses in nicotine dependence. Nelson SB, editor. eLife. 2015;4:e11396.
Carlson PJ, Diazgranados N, Nugent AC, Ibrahim L, Luckenbaugh DA, Brutsche N, et al. Neural correlates of rapid antidepressant response to ketamine in treatment-resistant unipolar depression: a preliminary positron emission tomography study. Biol Psychiatry. 2013;73:1213–21.
Chen M, Ma S, Liu H, Dong Y, Tang J, Ni Z, et al. Brain region–specific action of ketamine as a rapid antidepressant. Science. 2024;385:eado7010.
Elias GJB, Germann J, Loh A, Boutet A, Pancholi A, Beyn ME, et al. Habenular involvement in response to subcallosal cingulate deep brain stimulation for depression. Front Psychiatry. 2022;13:810777.
Sartorius A, Henn FA. Deep brain stimulation of the lateral habenula in treatment resistant major depression. Med Hypotheses. 2007;69:1305–8.
Menegaz de Almeida A, Aquino de Moraes FC, Cavalcanti Souza ME, Cavalcanti Orestes Cardoso JH, Tamashiro F, Miranda C, et al. Bright light therapy for nonseasonal depressive disorders: a systematic review and meta-analysis. JAMA Psychiatry. 2024:2871.
Huang L, Xi Y, Peng Y, Yang Y, Huang X, Fu Y, et al. A visual circuit related to habenula underlies the antidepressive effects of light therapy. Neuron. 2019;102:128–142.e8.
Browne CA, Hammack R, Lucki I. Dysregulation of the lateral habenula in major depressive disorder. Front Synaptic Neurosci. 2018;10:46
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Nakamura T, Kurosaki K, Kanemoto M, Sasahara M, Ichijo H. Early-life experiences altered the maturation of the lateral habenula in mouse models, resulting in behavioural disorders in adulthood. J Psychiatry Neurosci Jpn. 2021;46:E480–9.
van de Haar LL, Riga D, Boer JE, Garritsen O, Adolfs Y, Sieburgh TE, et al. Molecular signatures and cellular diversity during mouse habenula development. Cell Rep. 2022;40:111029.
Wang M, Chen X, Hu Y, Zhou Y, Wang C, Zheng W, et al. Functional connectivity between the habenula and default mode network and its association with the antidepressant effect of ketamine. Depress Anxiety. 2022;39:352–62.
Hu H, Cui Y, Yang Y. Circuits and functions of the lateral habenula in health and in disease. Nat Rev Neurosci. 2020;21:277–95.
Caldecott-Hazard S, Mazziotta J, Phelps M. Cerebral correlates of depressed behavior in rats, visualized using 14C-2-deoxyglucose autoradiography. J Neurosci Off J Soc Neurosci. 1988;8:1951–61.
Gordon N, Goelman G. Understanding alterations in serotonin connectivity in a rat model of depression within the monoamine-deficiency and the hippocampal-neurogenesis frameworks. Behav Brain Res. 2016;296:141–8.
Mirrione MM, Schulz D, Lapidus KAB, Zhang S, Goodman W, Henn FA. Increased metabolic activity in the septum and habenula during stress is linked to subsequent expression of learned helplessness behavior. Front Hum Neurosci. 2014;8:29.
Park H, Rhee J, Lee S, Chung C. Selectively impaired endocannabinoid-dependent long-term depression in the lateral habenula in an animal model of depression. Cell Rep. 2017;20:289–96.
Svenningsen K, Venø MT, Henningsen K, Mallien AS, Jensen L, Christensen T, et al. MicroRNA profiling in the medial and lateral habenula of rats exposed to the learned helplessness paradigm: candidate biomarkers for susceptibility and resilience to inescapable shock. PLoS ONE. 2016;11:e0160318.
Tong T, Chen Y, Hao C, Shen J, Chen W, Cheng W, et al. The effects of acupuncture on depression by regulating BDNF-related balance via lateral habenular nucleus BDNF/TrkB/CREB signaling pathway in rats. Behav Brain Res. 2023;451:114509.
Zhang H, Li K, Chen HS, Gao SQ, Xia ZX, Zhang JT, et al. Dorsal raphe projection inhibits the excitatory inputs on lateral habenula and alleviates depressive behaviors in rats. Brain Struct Funct. 2018;223:2243–58.
Acknowledgements
This study has been conducted with the support of the Australian Government Research Training Program Scholarship awarded to S.C.
Author information
Authors and Affiliations
Contributions
SC, KWG, and KAN conceived this study. SC performed the literature search and data extraction. KWG and KAN assisted with the data selection. SC wrote the first draft of the manuscript. SC, KWG, and KAN revised the manuscript and all authors approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cameron, S., Weston-Green, K. & Newell, K.A. The disappointment centre of the brain gets exciting: a systematic review of habenula dysfunction in depression. Transl Psychiatry 14, 499 (2024). https://doi.org/10.1038/s41398-024-03199-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-03199-x
This article is cited by
-
Altered habenular and whole brain functional connectivity in early Parkinson’s disease using 7 T MRI
npj Parkinson's Disease (2025)
-
The Clinical Impact of Habenular Dysfunction on Depression and Suicidality: A Literature Review and Discussion on the Implications for Psychiatric Practice
Current Behavioral Neuroscience Reports (2025)
