
Abstract
Growth and differentiation factor 15 (GDF15) is a significant player in cellular stress and energy homeostasis. GDF15 is elevated in cancer cachexia, chemotherapy-induced anorexia, hyperemesis gravidarum, and mitochondrial disorders. Here we analyze GDF15 in anorexia nervosa (AN), a psychiatric disorder characterized by low weight and persistent restriction of food intake. While no significant difference in plasma GDF15 concentration was seen across the three included groups; active AN, recovered AN, and healthy controls, a subgroup of study participants with high GDF15 plasma was noted to a significantly higher extent in the AN groups. Sparse partial least squares discriminant analysis (sPLS-DA) identified six markers related to inflammatory processes or cellular stress from a set of 74 markers that distinguished AN with high GDF15 from the rest, with fibroblast growth factor 21 (FGF21) being the most important contributor. Moreover, FGF21 plasma concentration was significantly higher in the group with high GDF15, suggesting an involvement of mitochondrial dysfunction. In fact, mitochondrial polygenic risk score (PRS) was significantly associated with AN risk in a large AN case-control cohort. In line with this, we also report elevated liver expression of GDF15 in the anx/anx mouse displaying anorexia associated with mitochondrial dysfunction. We conclude that mitochondrial dysfunction should be further explored in AN. Clinical trials of GDF15 immunoneutralization in patients with AN and high levels of GDF15 are worthy of consideration.
Similar content being viewed by others


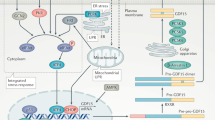
Introduction
Anorexia, i.e., low appetite and/or food intake, accompanies several conditions including cachexia of cancer and inflammatory disorders, chemotherapy-induced anorexia, nausea and vomiting of pregnancy, hyperemesis gravidarum, and the psychiatric disorder anorexia nervosa (AN). The potent appetite-inhibiting cytokine growth and differentiation factor 15 (GDF15), previously known as macrophage inhibitory cytokine-1 (MIC-1) [1], has been reported as elevated in all these conditions [1,2,3,4], although to our knowledge, only two studies with small sample sizes (n = 16/20) have explored GDF15 in AN [5, 6]. This cytokine, originally identified as a product of activated macrophages, is now well established as a cellular and nutritional stress-induced hormone that acts on the hindbrain to co-ordinate an “illness response” including anorexia, nausea, vomiting, physical inactivity, and the activation of neuroendocrine stress responses [3, 7]. It is increased in obesity [8, 9] and overfeeding appears to activate a stress response in the liver, the major source for systemic elevations of this hormone [10]. Nevertheless, animals and humans lacking GDF15 are not obese [11], while overexpressing GDF15 in mice results in reduced body weight [12]. Additionally, data indicate that GDF15 causes anorexia by inducing nausea and/or by engaging emetic neurocircuitry in the area postrema and nucleus tractus solitarius [1]. The receptor for GDF15, glial cell-derived neurotrophic factor receptor alpha-like (GFRAL), is exclusively expressed in these two brain regions [13,14,15]. Activation of GFRAL reduces food intake and body weight in animal models [12]. Inhibition of GDF15 in mice and humans reverses cancer cachexia [16, 17], including reducing anorexia and emesis [4], restoring muscle function as well as physical performance [18]. Lastly, intense exercise has been shown to increase circulating GDF15 in humans [19, 20].
The two small studies that previously explored GDF15 in females with active AN reported increased serum levels [5, 6]. Two months of nutritional therapy leading to partial weight recovery significantly decreased GDF15 in AN, even if the concentrations remained significantly higher in AN compared with healthy controls [5]. AN is a severe mental disorder with a high mortality rate, characterized by persistent restriction of food intake, fear of gaining weight, and concerns about body weight and shape [21]. The twin-based heritability of AN is estimated to be 50–60% and genome-wide association studies (GWAS) have to date identified eight variants in the genome significantly associated with AN risk [22]. A puzzling part of AN is a seemingly paradoxical response to negative energy balance, wherein patients actively strive to consistently expend more energy than they consume, resulting in continued loss of body weight and low energy stores. Loss of weight promotes changes in hormonal and other signals from adipose tissue (e.g., reduced leptin) and gut to instruct the brain to increase appetite and reduce energy expenditure, thus restoring energy stores and ensuring survival [23]. However, individuals with AN reside in persistent severe starvation, emaciation, and negative energy balance often for many years, and frequently revert to that state even after therapeutic weight restoration as if their biology defends their low weight state. The definition of the molecular underpinnings of anorexia, i.e., the loss or absence of appetite, might therefore define drug targets supporting renourishment and weight gain in AN.
The anx/anx mouse is a genetically spontaneously arisen model mimicking central aspects of AN; starvation and underweight [24]. The mouse eats less compared with its healthy littermates despite unrestricted access to food, subsequently becomes emaciated, and dies prematurely around three weeks of age. A range of deviations in hypothalamic neuropeptidergic and neurotransmitter systems have been documented in the anx/anx mouse [25]. The mouse exhibits a dysfunction in complex I of the oxidative phosphorylation system of the mitochondria present before the anorectic phenotype develops [26]. A small study in humans also described a similar mitochondrial dysfunction in leukocytes from patients with AN [27]. Interestingly, GDF15 is reported to be elevated in other animal models of mitochondrial dysfunction [28, 29], and human mitochondrial disorders [30] and has therefore been proposed as a biomarker to screen for mitochondrial disorders [31].
Based on our hypothesis that elevated plasma GDF15 in AN disrupts energy homeostatic regulation, because of mitochondrial dysfunction, we evaluated plasma levels of GDF15 in the to date largest cohort of women with active (n = 70) or recovered from AN (n = 89) and normal-weight women with no histories of eating disorders (n = 72). We explored potential contributors to the difference of GDF15 in the two AN groups by feature selection using a large set of circulating proteins many of but not all of which are involved in inflammation. Given the genetic basis of AN [22, 32,33,34], we further evaluate the association between mitochondrial function polygenic risk score (PRS), a personalized genetic risk score based on single nucleotide polymorphisms (SNPs) within the gene regions associated with mitochondrial function, and the risk of AN in a larger AN case-control cohort. Furthermore, we analyze correlations between plasma GDF15 and the adipose-derived hormone leptin and perform stratified analyses of typical AN characteristic such as purging behaviors. Lastly, we evaluated expression of GDF15 and an associated marker in the liver of the anorectic anx/anx mouse with established mitochondrial dysfunction.
Materials and methods
Study participants and design
The study participants were identified from the Swedish sample of the Anorexia Nervosa Genetics Initiative (ANGI-SE, including in total 4118 AN cases and 4035 controls). Details on the recruitment procedure for ANGI-SE have been described previously [35].
For the subcohort that provided plasma, the AN group comprised women at least 18 years of age at recruitment, meeting DSM-IV criteria for AN [36] except for amenorrhea, and a minimum of one year since AN onset (n = 70). For those recovered from AN, the inclusion criteria were a history of DSM-IV AN followed by weight restoration (BMI > 20 kg/m2) and no eating disorder behaviors for at least a year (AN-REC, n = 89). The age-matched normal-weight controls reported no history of disordered-eating behavior (CTRL, n = 72).
Exploratory analyses of the two AN groups were done investigating subtypes: defined by episode(s) of binge eating with loss of control as AN with binge eating (AN-B, n = 26, AN-REC-B n = 59) compared to the complete absence of such episodes as AN without binge eating (AN-noB, n = 37, AN-REC-noB, n = 24); episodes of laxative use (AN-LAX, n = 11, AN-REC-LAX, n = 18) compared to without laxative use (AN-noLAX, n = 58, AN-REC-noLAX, n = 69); documented self-induced vomiting (AN-VOM, n = 29, AN-REC-VOM, n = 46) compared to without self-induced vomiting (AN-noVOM, n = 40, AN-REC-noVOM, n = 41); reported episodes with use of diuretics (AN-DIU, n = 6, AN-REC-DIU, n = 5) compared to without diuretics use (AN-noDIU, n = 43, AN-REC-noDIU, n = 55); and reported compensatory exercise (AN-EXE, n = 41, AN-REC-EXE, n = 54) compared to without compensatory exercise (AN-noEXE, n = 5, AN-REC-noEXE, n = 7). See Table 1 for detailed characteristics of the study participants. The ANGI-SE study was approved by the Regional Ethics Review Board in Stockholm. All participants gave written informed consent. When applicable the investigators were blinded to group allocation when conducting the analyses.
Blood sampling
Blood samples were collected in EDTA tubes at a hospital near the participant’s home address, sent to Karolinska Institutet Biobank with overnight mail, and processed upon arrival. After centrifugation, plasma samples were stored at −80 °C. All samples were exposed to two freeze-thaw cycles prior to analyzing GDF15 concentrations.
Plasma GDF15 concentrations
We measured GDF15 in plasma by quantitative sandwich enzyme immunoassay (n = 6 plates, R&D Systems Quantikine® ELISA assay, Minneapolis, MN). In brief, plasma samples were diluted fourfold. Samples and standards were pipetted in duplicates into the wells and any GDF15 present was bound by the immobilized antibody on the bottom of the wells. After adding horseradish peroxidase (HRP)-conjugated polyclonal antibody recognizing GDF15 followed by a substrate solution, color developed in proportion to the amount of GDF15 bound in the initial step. The absorbance was measured on a plate reader (Spectramax Plus, Molecular Devices, San Jose, CA).
A very common histidine to aspartate variant at position 202 of the GDF15 pro-peptide in humans substantially affects its measurement by the R&D assay [37]. We here corrected the concentration of GDF15 based on this variant as described by Karusheva et al. [37], utilizing the genetic data collected in ANGI-SE [35]. All statistical analyses were conducted on the genotype-corrected concentrations.
Plasma leptin
We measured leptin in plasma by an in-house quantitative sandwich time-resolved fluorescence (DELFIA®) immunoassay (n = 7 plates) using antibodies and standards from R&D Systems, Minneapolis, MN, and Europium-labelled Streptavidin, buffers and enhancement solution from Revvity, Waltham, MA). In brief, the microtiter plate was coated with a monoclonal anti-leptin capture antibody. Standards, controls, and samples were added to the plate in duplicate. After incubation and washing, a biotinylated polyclonal anti-leptin detection antibody was added to the plate. After incubation and washing, Europium-labelled Streptavidin was added. After another wash Enhancement Solution was added to the wells which allowed the fluorescence to be generated in the wells when illuminated. The intensity of the time-resolved fluorescence is directly proportional to the concentration of leptin in the standards, controls, and samples. The leptin analysis was undertaken on the AutoDELFIA® analyzer (Revvity, Waltham, MA).
We analyzed the correlation between plasma leptin and plasma GDF15.
Plasma markers related to inflammatory processes and cellular stress
In a recent paper from our group exploring the Olink Proteomics inflammation panel (Uppsala, Sweden) utilizing the same cohort as here, we quantified the plasma concentrations of 74 immune activation markers [38]. The log2-transformed normalized expression data of these markers, together with leptin and GDF15, were used to select the most discriminative features that distinguished individuals with high GDF15 (>800 pg/ml) from the remaining individuals (<800 pg/ml) by sparse partial least squares discriminant analysis (sPLS-DA).
Mitochondrial polygenic risk score
We constructed a PRS of mitochondrial genes for each individual in ANGI-SE, which aggregates genetic risk for each individual from single nucleotide polymorphisms (SNPs) located within the regions of mitochondrial genes, using PRS-continued shrinkage (CS) [39]. Mitochondrial genes (n = 1136) were obtained from MitoCarta 3.0 database [40]. MitoCarta is an inventory of human and mouse genes with strong evidence for mitochondrial function. Genotype quality control of the ANGI-SE sample has been described in detail previously [22, 41] which resulted in 4054 AN cases and 3922 controls. AN GWAS summary statistics [22] without Swedish participants were used as the discovery dataset providing effect sizes and standard errors for PRS-CS [39] to get posterior SNP effect size estimates. We included common non-ambiguous SNPs within the listed regions with genes related to mitochondrial function (Supplementary Table 1) with high imputation quality (INFO ≥ 0.8) and minor allele frequency (MAF) ≥ 0.01 in the PRS calculation, resulting in a subset of 2475 SNPs. The 1000 Genomes Project Phase 3 EUR reference was used as linkage disequilibrium reference panel. After applying the posterior SNP effect size estimates from PRS-CS across chromosomes to get individual PRS via PLINK 1.9 [42], these scores were then standardized in R (version 4.3.2) [43] for further regression analyses.
Animals
Experiments involving animals followed the procedures approved by the ethical committee (Stockholms norra djurförsöksetiska nämnd) and were designed to minimize suffering. An intercross was set up using heterozygous anx breeding pairs (B6C3Fe–a/a–anx A/+a) acquired from the Jackson Laboratory (Bar Harbor, ME). All mice were genotyped using simple sequence length polymorphism markers mapped to the subchromosomal region, where the anx mutation is located. Phenotypic characterization was based on body weight. The mice were housed in ventilated cages at 25 °C in an animal room with a 12 h light-dark cycle (lights on at 7:00 AM) and with unrestricted access to the mother’s milk. Pups were sacrificed by decapitation between postnatal days (P) 19–21, with the day of delivery considered P1. The liver was rapidly dissected and frozen in ice-cold isopentane. We included seven liver samples from anx/anx and six from healthy wild-type siblings (+/+ or anx/+), all females. All analyses were conducted blinded of genotype.
Quantitative polymerase chain reaction (qPCR)
Liver tissues were homogenized with ZR BashingBead Lysis Tubes (S6012–50, Zymo Research, Irvine, USA) in Trizol Reagent (15596018, Invitrogen, Carlsbad, USA). Total RNA was extracted using Direct-zol™ RNA Miniprep (R2050, Zymo Research) according to the manufacturer’s instructions. DNase-treated RNA was reverse transcribed to cDNA using SuperScript™ III First-Strand Synthesis System (18080051, Invitrogen). Reactions were performed in three replicates with primers for GDF15 (5′-AACCCCTGGTCTGGGGATAC-3′ (forward); 5′-CATGTCGCTTGTGTCCTTTCAG-3′(reverse)), FGF21 (5′-CCTTGAAGCCAGGGGTCATT-3′ (forward); 5′-GGATCAAAGTGAGGCGATCCA-3′(reverse)), and GAPDH (5′-ACCCTTAAGAGGGATGCTGC-3′ (forward); 5′-CCCAATACGGCCAAATCCGT-3′(reverse)) using iTaq™ Universal SYBR® Green PCR kit (1725124, Bio-Rad, Hercules, USA) on a QuantStudio™ 6 Real-Time PCR Instrument (ThermoFisher, Waltham, USA). The PCR consists of 95 °C for 10 min, 95 °C 15 s −> 60 °C for 60 s for 40 cycles, and 72 °C for 5 min. The measured GDF15 transcript abundance was normalized to GAPDH using the delta-delta Ct method.
Statistical analyses
Group differences in plasma concentrations of GDF15 and leptin in AN, AN-REC, and CTRL were analyzed using the Kruskal-Wallis test since the concentrations were not normally distributed. This was followed by post-hoc Dunn’s test with Bonferroni correction to evaluate pairwise comparisons. ANCOVA was used to control for the effect of BMI when comparing the leptin levels of AN and AN-REC, followed by Tukey’s test. Associations among GDF15, body mass index (BMI), leptin, and immune activation markers were assessed using Spearman correlation. Principal component analysis (PCA) was used to identify multi-dimensional outliers in the immune activation markers dataset. sPLS-DA was then used to discriminate the groups of individuals with high GDF15 (>800 pg/ml) and the rest of the individuals (<800 pg/ml). A five-fold, ten-repeat cross-validation procedure was used to select the number of components and variables of the sPLS-DA model, and balanced error rate to evaluate model performance. The sPLS-DA results were supported by other feature selection techniques such as Lasso regression (data not shown). Group differences in plasma concentrations of GDF15 in AN-B vs. AN-noB, AN-REC-B vs. AN-REC-noB, AN-LAX vs. AN-noLAX, AN-REC-LAX vs. AN-REC-noLAX, AN-VOM vs. AN-noVOM, AN-REC-VOM vs AN-REC-noVOM, AN-DIU vs AN-noDIU, AN-REC-DIU vs AN-REC-noDIU, AN-EXE vs AN-noEXE, AN-REC-EXE vs AN-REC-noEXE were tested using the nonparametric Mann-Whitney U test, and effect sizes were measured by Cohen’s d. To evaluate the associations between high plasma GDF15 and AN diagnosis and behaviors, the Chi-square test was used, whereas Fisher’s exact test was used when at least one cell of the contingency tables contained values below 5. Phi Coefficient (φ) and odds ratio (OR) were reported as effect size estimates for the Chi-square and Fisher’s exact test, respectively. Logistics regressions were conducted to examine the associations between AN risk and mitochondrial function PRS, including the first ten genetic principal components (PCs) as covariates. The ANGI-SE cohort was then divided into quartiles based on the mitochondrial PRS level (i.e., low, medium-low, medium-high, and high), with the lowest quartile serving as the reference group. The risk of AN was then compared across these mitochondrial PRS quartiles using logistic regressions, adjusting for the first ten genetic PCs. Finally, linear regression was conducted to test the association between GDF15 and FGF21 level and mitochondrial PRS. All analyses were carried out using R programming language version 4.3.2. Graphs were made using the R package ggplot2 [44]. sPLS-DA was carried out by R package mixOmics (v6.26.0) [45].
Results
The demographic and clinical characteristics of the cohort are summarized in Table 1. BMI at sampling and minimum BMI during AN were significantly lower, whereas the duration of amenorrhea was significantly longer in AN vs. AN-REC. The other characteristics were not significantly different between the two patient groups. Note that BMI at sampling was not significantly different between AN-REC and CTRL.
Plasma GDF15
Analysis of the genotype-corrected plasma concentration of GDF15 revealed no significant differences across groups (Fig. 1A). However, a subgroup of individuals had high concentrations of GDF15 (>800 pg/ml), with a significantly larger proportion found in the AN (n = 9) and AN-REC groups (n = 10) compared to CTRL (n = 2) (p = 0.038). Plasma GDF15 levels were negatively correlated with BMI in the AN group (p = 0.0084), while a positive correlation was found in the AN-REC group (p = 0.0058), (Fig. 1B). Inter-assay variability ranged between 13–15%.

A Plasma GDF15 in anorexia nervosa (AN), recovered anorexia nervosa (AN-REC), and healthy controls (CTRL). The median is shown as a straight line and the box denotes the interquartile range. B Correlations between plasma GDF15 and body mass index (BMI) in AN (−0.32, p = 0.0084, 95% CI [−0.53, −0.079]), AN-REC (0.29, p = 0.0058, 95% CI [0.082, 0.48]), CTRL (−0.15, p = 0.21, 95% CI [−0.38, 0.09]), and all groups combined (−0.031, p = 0.64, 95% CI [−0.16, 0.10]). The colored lines correspond to the correlation for all groups, and for the AN, AN-REC, and CTRL groups separately. The shaded area around each linear fit line represents a 95% confidence interval (CI).
Plasma leptin and GDF15
The plasma concentration of leptin was significantly reduced in AN and AN-REC compared with healthy controls, despite no difference in mean BMI at the time of sampling between the latter two groups. The plasma concentration of leptin in AN-REC was significantly higher than in the AN group (p < 0.001) (Fig. 2A) even after controlling for BMI (p = 0.01). Plasma leptin concentration was significantly correlated with BMI in all groups (Fig. 2B). Furthermore, plasma GDF15 was positively correlated with leptin concentration only in the AN-REC group (p = 0.028) (Fig. 2C).

A Plasma leptin in anorexia nervosa (AN), recovered anorexia nervosa (ANREC), and healthy controls (CTRL). The median is shown as a straight line and the box denotes the interquartile range. B Correlations between plasma leptin and BMI in AN (0.28, p = 0.021, 95% CI [0.037, 0.49]), AN-REC (0.59, p = 1.94e–09, 95% CI [0.43, 0.71]), CTRL (0.55, p = 5.54e–07, 95% CI [0.36, 0.70]), and all groups combined (0.81, p 2.20e–16, 95% CI [0.75, 0.85]). C Correlations between plasma GDF15 and leptin in AN (−0.053, p = 0.67, 95% CI [−0.30, 0.20]), AN-REC (0.24, p = 0.028, 95% CI [0.020, 0.43]), CTRL (−0.16, p = 0.18, 95% CI [−0.39, 0.08]), and all groups combined (−0.013, p = 0.85, 95% CI [−0.15, 0.12]). The colored lines correspond to the correlation for all groups, and for the AN, AN-REC, and CTRL groups separately. The shaded area around each linear fit line represents a 95% confidence interval.
Plasma markers of inflammatory processes or cellular stress and GDF15
A preliminary unsupervised PCA identified no major multi-dimensional outliers but also showed no separation between the group with high GDF15 concentration and the others (Supplementary Fig. 1). sPLS-DA revealed discrimination between the profiles of markers related to inflammatory processes or cellular stress of the two groups (Fig. 3A). The tuned sPLS-DA model (balanced error rate = 0.21), yielded by component and variable selection, consisted of two components, of which the first one best separated the group with high GDF15 from the rest by explaining 13% of the variance in GDF15 status. Six proteins constituted the first component, of which FGF21 was the major contributor (Fig. 3B). Furthermore, differential protein expression analysis identified that FGF21 was significantly higher in the high GDF15 group vs. the rest and was the only protein with a log2 fold change larger than 1.5 (Fig. 3C). Plasma FGF21 was significantly higher in the group of individuals with a high concentration of GDF15 in plasma (>800 pg/ml), compared with the rest of the individuals (p = 0.001). Group differences of other plasma markers identified by sPLS-DA and their correlation estimates with plasma GDF15 concentration are included in Supplementary Fig. 2.

A Sparse partial least squares discriminant analysis (sPLS-DA) sample plot of the 74 immune markers based on high GDF15 and normal GDF15 group with 95% confidence ellipses. Variances retained by the first two components are reported. B The loading plot represents the absolute values of the loading vectors for markers selected on Component 1 of the sPLS-DA model. C Volcano plot showing fold difference between high GDF15 group and normal GDF15 group. The x-axis represents fold change (log2), and the y-axis represents p-value (−log10). The data point representing FGF-21 is labeled (p = 0.0047). CDCP1 CUB domain-containing protein 1, CSF1 colony stimulating factor 1, FGF21 fibroblast growth factor 21, GDF15 growth and differentiation factor 15, HGF hepatocyte growth factor, TGF-a Transforming Growth Factor alpha.
As a proxy for recent physical exercise, we also reevaluated plasma levels of IL6 reported in our previous publication [38] and detected no significant difference between individuals with high GDF15 and the rest of the individuals. We also saw no significant correlation between plasma concentrations of IL-6 and GDF15 (Supplementary Fig. 2K, L).
Mitochondrial PRS
Based on GDF15 and FGF21 in combination being used as markers of mitochondrial disorders [31] we evaluated PRS of mitochondrial function. Each standard deviation increase in the mitochondrial PRS was associated with 1.05 times greater odds of AN risk (95% CI [1.00, 1.09], p = 0.045). Individuals in the highest quartile of mitochondrial PRS had 1.13 times higher odds of AN compared to those in the lowest quartile (95% CI [1.00, 1.28]). There was a trend that the risk of AN increased across the mitochondrial PRS quartiles (Fig. 4), though larger sample sizes are needed to confirm these findings. However, no significant correlation was found between PRS scores and GDF15 level (p = 0.75) as well as FGF21 level (p = 0.29).

Odds Ratio (OR) with 95% confidence interval (95% CI) for anorexia nervosa (AN) by mitochondrial polygenic risk score (PRS) quartiles in ANGI-SE. The risk of AN is estimated using OR with 95% CI, with the lowest PRS quartile (1) as the reference group. The mitochondrial polygenic risk score (PRS) quartiles are defined as follows: 1 = low, 2 = mid-low, 3 = mid-high, and 4 = high.
GDF15 and FGF21 expression in anx/anx liver
The relative expression of GDF15 in the liver of the anorectic anx/anx mouse was significantly higher compared with their wild-type siblings (Fig. 5A), while FGF21 was significantly lower (Fig. 5B).

A Relative expression levels of GDF15 and B FGF21 in anx/anx and wild-type female mice determined by qPCR. The bar charts present values as mean ± SD.
Plasma GDF15 and eating disorder characteristics
To further analyze GDF15 in relation to AN, we performed an exploratory analysis of plasma levels and eating disorder characteristics. Nominally significantly higher plasma concentrations of GDF15 were detected in the group of active AN patients who reported laxative use (p = 0.010, d = 0.91), self-induced vomiting (p = 0.017, d = 0.75), or compensatory exercise (p = 0.002, d = 0.74) compared with patients who reported no history of these behaviors (Supplementary Fig. 3). No significant differences were found in the group of AN-REC comparing those with and those without these behaviors, nor were any differences seen between those with vs without reported binge-eating behavior, or with vs without diuretic use in AN or AN-REC.
Supplementary Table 2 summarizes the clinical and eating disorder characteristics in the groups with elevated (>800 pg/ml) vs normal plasma GDF15(<800 pg/ml). Supplementary Table 3 shows the associations with these traits of the same two groups. Reported use of diuretics was nominally significantly associated with high plasma GDF15. Only two individuals in the full cohort, both within the group with high GDF15, were diagnosed with type 2 diabetes and cancer, thus no analysis of a potential association with these diagnoses was possible.
Discussion
In contrast to smaller studies [5, 6], we did not detect any significant differences in plasma GDF15 concentration across the three groups; AN, AN-REC, and healthy controls. However, we found that a subgroup of women of a significantly larger number in the AN groups, have increased plasma GDF15 concentrations. We also observed a significant positive correlation between GDF15 and BMI, as well as with plasma leptin, in the AN-REC group. In the active AN group we only see a significant negative correlation between GDF15 and BMI, but not leptin, which may be explained by the near-zero concentrations of plasma leptin in this group. We speculate that the opposing correlations between GDF15 levels in plasma and BMI in AN (negative) vs AN-REC (positive) may be related to the history of starvation in the latter, since no correlation is seen in the control group with similar BMIs. When combining this plasma GDF15 and leptin data with data on 74 markers related to inflammatory processes or cellular stress from a previously published report from our group [38] in a sPLS-DA, we identified six markers that distinguished individuals with high plasma GDF15 from the rest of the individuals in the AN groups. Of these, FGF21 was defined as the most important contributor (see Supplementary Text). Plasma FGF21 was significantly higher only in the group of individuals with high GDF15. As mentioned above, FGF21 and GDF15 are elevated in animals [28, 29] as well as humans with mitochondrial dysfunction [30], and have been proposed to be used in combination as biomarkers to screen for pediatric mitochondrial disorders [31]. It is established that complex I dysfunction within the liver seen in e.g., Cockayne syndrome, gives rise to high concentrations of circulating GDF15 and FGF21 resulting in suppressed food intake, which is reversed by blocking GDF15 alone [46]. Thus, we speculate that mitochondrial dysfunction may be a factor in explaining the elevated plasma GDF15 seen in individuals predominantly belonging to the patient groups. In line with this, PRS calculated with SNPs within genes associated with mitochondrial function show a significant association with AN status in the full ANGI-SE cohort, suggesting a potential role of mitochondrial genes in AN risk and highlighting possible shared genetics between mitochondrial dysfunction and AN. The lack of correlations between the PRS scores and plasma levels of GDF15 and FGF21 were expected based on the in the context of PRS calculations small sample size used here. We also show significantly higher GDF15 expression in the liver from the anx/anx mouse compared to their wild-type siblings, whereas the liver expression of FGF21 was significantly lower in the anx/anx mouse, which was not in line with our hypothesis. We were however unable to measure the two proteins in anx/anx plasma, due to the small blood volume of these young and emaciated mice and thus cannot with certainty say that the plasma levels of FGF21 correspond to the level of expression of FGF21 in liver. The anorectic mouse model also displays dysfunction in complex I of the mitochondrial oxidative phosphorylation system [26], which has similarly been reported in a small cohort of patients with AN [27]. This indicates that further studies on mitochondrial dysfunction in AN should be prioritized.
FGF21 is another important energy metabolic regulator [47]. Animal studies show that the injection of FGF21 leads to elevated energy expenditure and adiponectin secretion from adipose tissue [48]. By simple diffusion, FGF21 can cross the blood-brain barrier and bind to its receptor expressed throughout the brain, particularly in the hypothalamus [49], which is the central control of feeding and energy expenditure. For example, FGF21 targets the lateral hypothalamus and acts on GABAergic neurons, which in turn stimulates thermogenesis and energy expenditure that leads to reduced weight gain [50]. FGF21 also activates glutamatergic neurons in the ventromedial hypothalamus to reduce sugar intake [51]. Moreover, subcutaneous administration of FGF21 increases the expression of the orexigenic neuropeptide AgRP and NPY in the arcuate nucleus of the hypothalamus [52]. In the context of AN, contradictory results on FGF21 levels in patients compared to controls have been reported. In a cohort of 11 patients and 12 controls, Pouneh et al. [53] reported significantly higher FGF21 in active AN patients, while Ivana et al. [54] showed reduced plasma FGF21 in AN patients in a cohort with 17 patients and controls. In Nilsson et al. we detected no difference in plasma FGF21 in active AN (n = 113), recovered AN (n = 113) and controls (n = 114) [36]. This despite that FGF21 expression is reported to be strongly induced by fasting [55] and protein restriction [56], and regulates energy homeostasis during starvation [55]. Based on this we were again surprised to see FGF21 expression not upregulated, but rather downregulated, in the anx/anx mouse liver.
Of note, lower plasma leptin levels were observed not only in AN but also in AN-REC compared to healthy controls, despite the latter two groups having similar mean BMI. Previous data on leptin in individuals recovered from AN have been mixed, while some studies have reported that serum leptin rises with weight recovery [57, 58] and even reaches values above those observed in controls matched for BMI [59], others report normal leptin in both cerebrospinal fluid and serum with long term recovery [60]. Thus, the potential role of leptin in AN recovery remains to be established.
As with leptin, GDF15 is related to physical activity. Although low leptin is speculated to play a role in the increased physical activity of AN [61], GDF15 is reported to markedly increase with intense exercise in humans as well as mice [19, 20], and we here detected increased plasma GDF15 in the group with active AN reporting compensatory exercise. However, we lack information on physical activity directly prior to sampling. As a proxy for recent intense exercise [62], we compared plasma concentration of the exercise-induced myokine IL6 in the group of individuals with high vs. normal plasma GDF15 but saw no differences. Thus, this indicates that the increased GDF15 seen in the group of individuals here is unlikely to be related to intense exercise prior to sampling.
In stratified analyses, we found that individuals with active AN who report compensatory behaviors, i.e., laxative use, self-induced vomiting, and as already mentioned compensatory exercise, have nominally significantly higher plasma GDF15 than individuals with active AN reporting no such behaviors. Purging behaviors were not associated with higher plasma GDF15 in AN-REC.
This is the to date largest study evaluating GDF15 levels in AN. The inclusion of samples from individuals recovered from AN, data from anorectic animals as well as genetic data gives strength to the study. A limitation of this study is that we did not have information about the state of the participants, in particular fasted vs fed, when sampling was done. However, little variation in GDF15 concentration has been reported with meals [63], fasting, and refeeding [64]. One study reported peak plasma GDF15 concentration after 48 h of severe caloric restriction [63], while other studies have shown no change in plasma GDF15 following 8 weeks or 6 months of low-calorie dietary plans, despite a total body weight loss of 11 and 13.5%, respectively [65, 66]. We also did not have information on the time of the day of sampling. But even if circulating levels of GDF15 vary in a diurnal pattern with 10% plus or minus [64], this should not account for the high plasma concentrations seen for some individuals in this study. With the exception of cancer and diabetic diagnoses, we were limited by having had no information on other factors/conditions that could influence GDF15 levels in plasma e.g., smoking [67] and medications. Lastly, due to the small blood volume of the young and anorectic anx/anx mouse we were restricted to measuring GDF15 expression in liver rather than GDF15 protein in plasma.
To conclude, we observed no differences in plasma GDF15 across the AN, AN-REC, or CTRL groups, but identified a subgroup of individuals almost exclusively within the two patient groups who have high plasma GDF15 concentrations. This leads us to hypothesize that, if our results are replicated, neutralizing GDF15 may have the potential to support appetite and aid in normalizing food intake in some individuals with AN. The group of individuals with high plasma GDF15 also had higher concentrations of FGF21 in plasma. FGF21 was the main distinguishing contributor of the group of individuals with high GDF15, which may suggest mitochondrial dysfunction in this group of individuals with current or past AN. Associations between mitochondrial PRS and AN risk further support the potential shared genetic basis between AN and mitochondrial dysfunction. In line with this, we report markedly increased GDF15 expression, while FGF21 surprisingly is reduced, in the liver from the anorectic anx/anx mouse previously reported to display mitochondrial dysfunction. Thus, our findings support the continued evaluation of mitochondrial function in AN, and clinical trials of GDF15 immunoneutralization in patients with AN and high levels of GDF15 are worth consideration.
Data availability
Data supporting the findings of this study can be made available from the corresponding authors upon request.
References
Borner T, Shaulson ED, Ghidewon MY, Barnett AB, Horn CC, Doyle RP, et al. GDF15 induces anorexia through nausea and emesis. Cell Metab. 2020;31:351–62.e355.
Petry CJ, Ong KK, Burling KA, Barker P, Goodburn SF, Perry JRB, et al. Associations of vomiting and antiemetic use in pregnancy with levels of circulating GDF15 early in the second trimester: a nested case-control study. Wellcome Open Res. 2018;3:123.
Tsai VWW, Husaini Y, Sainsbury A, Brown DA, Breit SN. The MIC-1/GDF15-GFRAL pathway in energy homeostasis: implications for obesity, cachexia, and other associated diseases. Cell Metab. 2018;28:353–68.
Breen DM, Kim H, Bennett D, Calle RA, Collins S, Esquejo RM, et al. GDF-15 neutralization alleviates platinum-based chemotherapy-induced emesis, anorexia, and weight loss in mice and nonhuman primates. Cell Metab. 2020;32:938–50.e936.
Dostalova I, Kavalkova P, Papezova H, Domluvilova D, Zikan V, Haluzik M. Association of macrophage inhibitory cytokine-1 with nutritional status, body composition and bone mineral density in patients with anorexia nervosa: the influence of partial realimentation. Nutr Metab. 2010;7:34.
Karczewska-Kupczewska M, Kowalska I, Nikolajuk A, Adamska A, Otziomek E, Gorska M, et al. Hyperinsulinemia acutely increases serum macrophage inhibitory cytokine-1 concentration in anorexia nervosa and obesity. Clin Endocrinol. 2012;76:46–50.
Lockhart SM, Saudek V, O’Rahilly S. GDF15: a hormone conveying somatic distress to the brain. Endocr Rev. 2020;41:bnaa007.
Xiong Y, Walker K, Min X, Hale C, Tran T, Komorowski R, et al. Long-acting MIC-1/GDF15 molecules to treat obesity: evidence from mice to monkeys. Sci Transl Med. 2017;9:eaan8732.
Kempf T, Guba-Quint A, Torgerson J, Magnone MC, Haefliger C, Bobadilla M, et al. Growth differentiation factor 15 predicts future insulin resistance and impaired glucose control in obese nondiabetic individuals: results from the XENDOS trial. Eur J Endocrinol. 2012;167:671–8.
Patel S, Haider A, Alvarez-Guaita A, Bidault G, El-Sayed Moustafa JS, Guiu-Jurado E, et al. Combined genetic deletion of GDF15 and FGF21 has modest effects on body weight, hepatic steatosis and insulin resistance in high fat fed mice. Mol Metab. 2022;65:101589.
Gurtan AM, Khalid S, Koch C, Khan MZ, Lamarche LB, Splawski I, et al. Identification and characterization of human GDF15 knockouts. Nat Metab. 2024;6:1913–21.
Macia L, Tsai VW, Nguyen AD, Johnen H, Kuffner T, Shi YC, et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) decreases food intake, body weight and improves glucose tolerance in mice on normal & obesogenic diets. PLoS ONE. 2012;7:e34868.
Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med. 2017;23:1150–7.
Hsu JY, Crawley S, Chen M, Ayupova DA, Lindhout DA, Higbee J, et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature. 2017;550:255–9.
Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med. 2017;23:1215–9.
Suriben R, Chen M, Higbee J, Oeffinger J, Ventura R, Li B, et al. Antibody-mediated inhibition of GDF15-GFRAL activity reverses cancer cachexia in mice. Nat Med. 2020;26:1264–70.
Groarke JD, Crawford J, Collins SM, Lubaczewski S, Roeland EJ, Naito T, et al. Ponsegromab for the treatment of cancer cachexia. N Engl J Med. 2024;391:2291–303.
Kim-Muller JY, Song L, LaCarubba Paulhus B, Pashos E, Li X, Rinaldi A, et al. GDF15 neutralization restores muscle function and physical performance in a mouse model of cancer cachexia. Cell Rep. 2023;42:111947.
Klein AB, Nicolaisen TS, Ortenblad N, Gejl KD, Jensen R, Fritzen AM, et al. Pharmacological but not physiological GDF15 suppresses feeding and the motivation to exercise. Nat Commun. 2021;12:1041.
Kleinert M, Clemmensen C, Sjoberg KA, Carl CS, Jeppesen JF, Wojtaszewski JFP, et al. Exercise increases circulating GDF15 in humans. Mol Metab. 2018;9:187–91.
Association AP. Diagnostic and statistical manual of mental disorders (DSM-5). American Psychiatric Publishing, Washington, DC; 2013.
Watson HJ, Yilmaz Z, Thornton LM, Hubel C, Coleman JRI, Gaspar HA, et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat Genet. 2019;51:1207–14.
Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci. 2014;15:367–78.
Maltais LJ, Lane PW, Beamer WG. Anorexia, a recessive mutation causing starvation in preweanling mice. J Hered. 1984;75:468–72.
Nilsson IAK. The anx/anx mouse - a valuable resource in anorexia nervosa research. Front Neurosci. 2019;13:59.
Lindfors C, Nilsson IA, Garcia-Roves PM, Zuberi AR, Karimi M, Donahue LR, et al. Hypothalamic mitochondrial dysfunction associated with anorexia in the anx/anx mouse. Proc Natl Acad Sci USA. 2011;108:18108–13.
Victor VM, Rovira-Llopis S, Saiz-Alarcon V, Sanguesa MC, Rojo-Bofill L, Banuls C, et al. Altered mitochondrial function and oxidative stress in leukocytes of anorexia nervosa patients. PLoS ONE. 2014;9:e106463.
Forsstrom S, Jackson CB, Carroll CJ, Kuronen M, Pirinen E, Pradhan S, et al. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 2019;30:1040–54.e1047.
Lehtonen JM, Forsstrom S, Bottani E, Viscomi C, Baris OR, Isoniemi H, et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology. 2016;87:2290–9.
Suomalainen A, Elo JM, Pietilainen KH, Hakonen AH, Sevastianova K, Korpela M, et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10:806–18.
Li Y, Li S, Qiu Y, Zhou M, Chen M, Hu Y, et al. Circulating FGF21 and GDF15 as biomarkers for screening, diagnosis, and severity assessment of primary mitochondrial disorders in children. Front Pediatr. 2022;10:851534.
Bulik CM, Sullivan PF, Wade TD, Kendler KS. Twin studies of eating disorders: a review. Int J Eat Disord. 2000;27:1–20.
Wade TD, Bulik CM, Neale M, Kendler KS. Anorexia nervosa and major depression: shared genetic and environmental risk factors. Am J Psychiatry. 2000;157:469–71.
Bulik CM, Sullivan PF, Tozzi F, Furberg H, Lichtenstein P, Pedersen NL. Prevalence, heritability, and prospective risk factors for anorexia nervosa. Arch Gen Psychiatry. 2006;63:305–12.
Thornton LM, Munn-Chernoff MA, Baker JH, Jureus A, Parker R, Henders AK, et al. The anorexia nervosa genetics initiative (ANGI): overview and methods. Contemp Clin Trials. 2018;74:61–69.
Association AP. Diagnostic and statistical manual of mental disorders. 4th ed. American Psychiatric Publishing, Washington, DC; 1994.
Karusheva Y, Ratcliff M, Morseburg A, Barker P, Melvin A, Sattar N, et al. The common H202D variant in GDF-15 does not affect its bioactivity but can significantly interfere with measurement of its circulating levels. J Appl Lab Med. 2022;7:1388–1400.
Nilsson IAK, Millischer V, Goteson A, Hubel C, Thornton LM, Bulik CM, et al. Aberrant inflammatory profile in acute but not recovered anorexia nervosa. Brain Behav Immun. 2020;88:718–24.
Ge T, Chen CY, Ni Y, Feng YA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10:1776.
Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021;49:D1541–D1547.
Zhang R, Kuja-Halkola R, Borg S, Leppa V, Thornton LM, Birgegard A, et al. The impact of genetic risk for schizophrenia on eating disorder clinical presentations. Transl Psychiatry. 2023;13:366.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. 2021.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016.
Rohart F, Gautier B, Singh A, Le Cao KA. mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol. 2017;13:e1005752.
Mulderrig L, Garaycoechea JI, Tuong ZK, Millington CL, Dingler FA, Ferdinand JR, et al. Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response. Nature. 2021;600:158–63.
Chau MD, Gao J, Yang Q, Wu Z, Gromada J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci USA. 2010;107:12553–8.
Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17:779–89.
Hsuchou H, Pan W, Kastin AJ. The fasting polypeptide FGF21 can enter brain from blood. Peptides. 2007;28:2382–6.
Pena-Leon V, Folgueira C, Barja-Fernandez S, Perez-Lois R, Da Silva Lima N, Martin M, et al. Prolonged breastfeeding protects from obesity by hypothalamic action of hepatic FGF21. Nat Metab. 2022;4:901–17.
Jensen-Cody SO, Flippo KH, Claflin KE, Yavuz Y, Sapouckey SA, Walters GC, et al. FGF21 signals to glutamatergic neurons in the ventromedial hypothalamus to suppress carbohydrate intake. Cell Metab. 2020;32:273–86.e276.
Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–27.
Fazeli PK, Misra M, Goldstein M, Miller KK, Klibanski A. Fibroblast growth factor-21 may mediate growth hormone resistance in anorexia nervosa. J Clin Endocrinol Metab. 2010;95:369–74.
Dostalova I, Kavalkova P, Haluzikova D, Lacinova Z, Mraz M, Papezova H, et al. Plasma concentrations of fibroblast growth factors 19 and 21 in patients with anorexia nervosa. J Clin Endocrinol Metab. 2008;93:3627–32.
Fazeli PK, Lun M, Kim SM, Bredella MA, Wright S, Zhang Y, et al. FGF21 and the late adaptive response to starvation in humans. J Clin Invest. 2015;125:4601–11.
Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest. 2014;124:3913–22.
Audi L, Mantzoros CS, Vidal-Puig A, Vargas D, Gussinye M, Carrascosa A. Leptin in relation to resumption of menses in women with anorexia nervosa. Mol Psychiatry. 1998;3:544–7.
Casanueva FF, Dieguez C, Popovic V, Peino R, Considine RV, Caro JF. Serum immunoreactive leptin concentrations in patients with anorexia nervosa before and after partial weight recovery. Biochem Mol Med. 1997;60:116–20.
Hebebrand J, Blum WF, Barth N, Coners H, Englaro P, Juul A, et al. Leptin levels in patients with anorexia nervosa are reduced in the acute stage and elevated upon short-term weight restoration. Mol Psychiatry. 1997;2:330–4.
Gendall KA, Kaye WH, Altemus M, McConaha CW, La Via MC. Leptin, neuropeptide Y, and peptide YY in long-term recovered eating disorder patients. Biol Psychiatry. 1999;46:292–9.
Holtkamp K, Herpertz-Dahlmann B, Hebebrand K, Mika C, Kratzsch J, Hebebrand J. Physical activity and restlessness correlate with leptin levels in patients with adolescent anorexia nervosa. Biol Psychiatry. 2006;60:311–3.
Steensberg A. The role of IL-6 in exercise-induced immune changes and metabolism. Exerc Immunol Rev. 2003;9:40–47.
Patel S, Alvarez-Guaita A, Melvin A, Rimmington D, Dattilo A, Miedzybrodzka EL, et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab. 2019;29:707–18.e708.
Tsai VW, Macia L, Feinle-Bisset C, Manandhar R, Astrup A, Raben A, et al. Serum levels of human MIC-1/GDF15 vary in a diurnal pattern, do not display a profile suggestive of a satiety factor and are related to BMI. PLoS ONE. 2015;10:e0133362.
Imbert A, Vialaneix N, Marquis J, Vion J, Charpagne A, Metairon S, et al. Network analyses reveal negative link between changes in adipose tissue GDF15 and BMI during dietary-induced weight loss. J Clin Endocrinol Metab. 2022;107:e130–42.
Thom G, Dombrowski SU, Brosnahan N, Algindan YY, Rosario Lopez-Gonzalez M, Roditi G, et al. The role of appetite-related hormones, adaptive thermogenesis, perceived hunger and stress in long-term weight-loss maintenance: a mixed-methods study. Eur J Clin Nutr. 2020;74:622–32.
Wada H, Suzuki M, Matsuda M, Ajiro Y, Shinozaki T, Sakagami S, et al. Impact of smoking status on growth differentiation factor 15 and mortality in patients with suspected or known coronary artery disease: The ANOX study. J Am Heart Assoc. 2020;9:e018217.
Acknowledgements
We are truly grateful to all participants contributing to this research. We thank the collection team that worked to recruit them as well as data managers Stina Borg and Bozenna Iliadou. The Anorexia Nervosa Genetics Initiative (ANGI) was an initiative of the Klarman Family Foundation. We also wish to acknowledge Laura Thornton for guidance on the interpretation of the data in the ANGI survey, the National Quality Registry for Eating Disorders (Riksät) for helping with recruiting patients, the BBMRI.se and KI Biobank at Karolinska Institutet for their professional biobank service. RZ is supported by the Swedish Research Council (Vetenskapsrådet, grant no. 2022-00242). ZY is supported by NIMH (R01MH136156), Independent Research Fund Denmark (3166-00063B) and Lundbeck Foundation Ascending Investigator (R434-2023-269). VG is supported by the CAMH Discovery Grant, WomenMind Seed Fund, Larry and Judy Tanebaun Foundation. ML is supported by grants from the Swedish Research Council (2022-01643), Hjärnfonden/the Swedish Brain Foundation (FO2022-0217), and the Swedish Government under the LUA/ALF agreement (ALFGBG-965444). SOR was supported by the Medical research Council of the UIK and by the Cambridge NIHR Biomedical Research Centre. CMB is supported by NIMH (R56MH129437; R01MH120170; R01MH124871; R01MH119084; R01MH118278; R01 MH124871) and the Swedish Research Council (Vetenskapsrådet, award: 538-2013-8864). Finally, IAKN acknowledge generous financial support from Sten and Birgitta Westerberg, Ulf Lundahls Minnesfond via the Swedish Brain Foundation (FO2019 -0122), the China Sweden Council Scholarship, and funds from Karolinska Institutet.
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
JX designed and performed experiments analyzed data and drafted the manuscript. IAKN formed the hypothesis, designed the study, handled animal breeding and experiments, and drafted the manuscript. RZ, ZY & VG performed PRS analysis. VM, CT & SM performed experiments. MS and CL designed analyses. ML designed and conducted the collection of plasma samples. CB collected plasma samples and designed the study. SOR designed experiments and generated hypotheses. PB designed and performed experiments. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
CMB is an author and royalty recipient from Pearson Education Inc. (unrelated to the content of the paper).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, J., Zhang, R., Millischer, V. et al. Elevated plasma GDF15 combined with FGF21 suggests mitochondrial dysfunction in a subgroup of anorexia nervosa patients. Transl Psychiatry 15, 215 (2025). https://doi.org/10.1038/s41398-025-03425-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03425-0
