
Abstract
The relationship between fatty acids and Alzheimer’s Disease (AD) risk has been an area of growing interest but remains insufficiently understood. This study aimed to develop and validate a fatty acid score (FAS) derived from blood fatty acid levels and explore its association with AD risk. We analyzed 148,308 UK Biobank participants (age 37–73; mean 55.96 years) with a mean follow-up of 12.3 years (maximum 16), and 1193 ADNI subjects (age 55–90; mean 73.50 years) with a mean follow-up of 4.2 years (maximum 8). Lasso regression was used to construct the FAS based on UKB, and Cox regression and linear regression was employed to assess the relationships of FAS with AD risk, cognition, hippocampal volume, and/or cerebrospinal fluid markers in both cohorts. Stratified effects by APOE ε4 status were examined. Causal mediation, proteomic, and bioinformatic analyses were performed to reveal potential mechanisms. Higher FAS was associated with increased AD risk in both cohorts (UKB: HR = 1.298, 95% CI 1.183–1.423, P < 0.001; ADNI: HR = 1.413, 95% CI 1.105–1.808, P = 0.006). In UKB, higher FAS was linked to reduced hippocampal volume (P < 0.001), and in ADNI, it was associated with faster hippocampal atrophy (P = 0.002) and cognitive decline (P < 0.001). These associations were stronger in APOE ε4 carriers. Hippocampal volume partly mediated the link between FAS and cognitive decline. Proteomic analyses demonstrated that the protein expression levels of Adhesion G protein-coupled receptor G1 (ADGRG1), Chitinase-3-like protein 1 (CHI3L1), RNA-binding FOX-1 homolog 3 (RBFOX3), and Growth differentiation factor 15 (GDF15) could mediate the effect of FAS on AD risk. The enriched pathways include cytokine activity, neurotrophic signaling, and pathways related to nervous system development. Blood levels of fatty acid could aid in AD prediction, but further research is needed to confirm causality.
Similar content being viewed by others

Introduction
Alzheimer’s Disease (AD) is the leading cause of dementia globally and places significant burdens on societies [1]. It is crucial to prevent and postpone AD occurrence. Compared to the biomarker framework based on cerebrospinal fluid (CSF) or PET imaging, the blood-based biomarker exhibited multiple advantages including accessible reproducibility, non-invasiveness, ease of measurement, and cost-effectiveness [2]. Identifying reliable blood signals can facilitate risk stratification and personalized intervention in early stage of AD. Lines of studies have shown that multiple circulating proteins are associated with risk of dementia [3] and AD [4,5,6,7,8]. Moreover, peripheral blood proteins may drive AD onset by modulating neuroinflammation, metabolism, and the extracellular matrix [9,10,11].
Recently, some specific blood fatty acids were reported to modulate the levels of peripheral blood proteins associated with AD pathology [12, 13], making them potential biomarkers for predicting AD. We and other researchers have found that higher levels of omega-3 polyunsaturated fatty acids (PUFAs) were associated with a reduced risk of cognitive decline [14] and AD [15, 16], whereas higher levels of saturated fatty acids (SFAs) could increase the risk of AD [17]. However, the associations of blood fatty acids with cognitive function [18] or AD [19] remained inconclusive. These inconsistencies may be attributed to variations in sample size and confounding by APOE genotype [20,21,22]. In addition, the diverse types of blood fatty acids and their interactions will also complicate the associations [20]. Therefore, a strategy of weighting and incorporating different components of blood fatty acids could better estimate the contributions to AD.
On the other hand, the mechanisms by which blood fatty acids were associated with AD were still unclear. Previous evidence showed that disrupted fatty acid metabolism and imbalance in proportions were critically implicated in AD pathogenesis [20, 23, 24]. High-fat diet increased hippocampal SFAs and decreased PUFAs [25], while this intake triggered inflammation, endoplasmic reticulum stress, and apoptotic signaling in the murine hippocampus [26]. Additionally, the omega-6/omega-3 fatty acid ratio imbalance influenced amyloid pathology in the hippocampus and cortex of transgenic mice [27]. It was reasonably hypothesized that neurodegeneration and neuroinflammation could be the underlying mechanisms, but other mediating pathways could exist.
In the present study, we aimed to (1) develop a blood FAS associated with incident AD risk using a UK cohort and validate this association in an independent North America cohort, (2) explore the relationships of the FAS with cognition, hippocampus, and cerebrospinal fluid (CSF) AD biomarkers, (3) investigate the effect of hippocampus in mediating the relationship between FAS and cognition, and (4) examine the potential biological mechanisms by which FAS was associated with AD.
Methods
Participants
The UK Biobank (UKB) is a prospective cohort study of around 500,000 individuals aged 37–73 years recruited between 2006 and 2010. UKB participants completed comprehensive assessments including questionnaires, interviews, biological sampling, and physical measurements at 22 centers across the UK [28]. Baseline data covered socio-demographics, lifestyle, diet, and medical history, with follow-up via electronic health records. Ethical approval was obtained (Ref. 11/NW/0382), and all participants provided informed consent.
Serving as the validation cohort, ADNI is an established multi-center cohort in North America (adni.loni.usc.edu). ADNI is launched to test clinical, imaging, genetic, and biochemical biomarkers of AD. The participants, aged 55–90 years, were enrolled after approval from the institutional review boards of all participating centers, with written informed consent obtained from all participants or their authorized representatives in accordance with the 1975 Declaration of Helsinki. Detailed information can be found at http://www.adniinfo.org.
For both cohorts, we excluded participants who were diagnosed with dementia or major neuropsychiatric disorders at baseline, or were lost to follow-up, or had incomplete covariate or fatty acid data. Any participant with a missing value for variables of interest was excluded.
Assessment of plasma fatty acids
In UKB, plasma fatty acids levels (absolute concentrations and percentages of specific component to total fatty acids) were measured by nuclear magnetic resonance (NMR) in blood samples [29]. In ADNI, serum fatty acids were analyzed using Nightingale Health’s NMR metabolomics platform [17]. For both cohorts, 17 fatty acid indicators were included: total fatty acids (TFA), saturated fatty acids (SFA), monounsaturated fatty acids (MUFA), polyunsaturated fatty acid (PUFA), omega-3, omega-6, docosahexaenoic acid (DHA), and linoleic acid (LA), and various ratios including SFA to TFA ratio, MUFA to TFA ratio, PUFA to TFA ratio, omega-3 to TFA ratio, omega-6 to TFA ratio, DHA to TFA ratio, LA to TFA ratio, PUFA to MUFA ratio, omega-6 to omega-3 ratio.
Diagnosis of AD dementia
In UKB, dementia diagnoses were ascertained using the International Classification of Diseases (ICD) coding system from hospital records and death registers. The Alzheimer’s disease codes included F00, F00.0, F00.1, F00.2, F00.9, G30, G30.0, G30.1, G30.8, and G30.9. The follow-up duration was calculated as the shorter interval between the initial evaluation and the diagnosis of dementia, death, last follow-up, or loss to follow-up [30, 31]. In ADNI, neuropsychological testing and diagnostic criteria details can be found on the ADNI website (http://adni.loni.usc.edu/methods). In brief, AD patients had an MMSE score of 20–26, a CDR-SB score of 0.5 or 1, and met the NINDS-ADRDA criteria for probable AD. Cognitive diagnoses were recorded annually, with progressors identified by changes from NC to AD dementia or MCI to AD dementia [22].
Cognitive assessments
In UKB, the Numeric Memory Test (NMT) was selected as the primary cognitive outcome. The NMT collects comprehensive data, including the number and value of digits remembered, response accuracy, response times, and overall test completion status [32]. Higher scores in NMT indicated better cognitive performance. In ADNI, cognitive functions were evaluated using multiple scales, including global cognition by the cognitive section of the Alzheimer’s Disease Assessment Scale (ADAS) and specific memory functions (MEM) by extracting relevant neuropsychological batteries to identify relevant items [33, 34].
Hippocampal volume measurement
The volume of hippocampus in both UKB and ADNI were obtained from magnetic resonance imaging (MRI). T1-weighted and T2-FLAIR structural images were acquired in a straight sagittal orientation and centrally processed to extract hippocampal volumes. Detailed descriptions of image processing for both UKB and ADNI cohorts are available in other publications [35, 36].
Measurements of CSF biomarkers
Data on CSF biomarkers were accessible only in ADNI cohort. The detailed protocols for CSF procedures have been documented in [37]. Briefly, concentrations of CSF Aβ1–42, p-tau181, and total tau proteins (pg/mL) were measured using electrochemiluminescence immunoassays (Elecsys; Roche Diagnostics) on a fully automated Elecsys cobas e 601 instruments, as detailed in the UPENNBIOMK9.csv file [38].
Blood proteomics
In UKB, blood samples were collected in EDTA tubes, centrifuged at 2500 g for 10 min at 4 °C, and the plasma was aliquoted and stored at −80 °C. Proteomic assays were conducted on approximately 55,000 plasma samples using dual-barcoded antibody technology on the Olink platform [39]. A total of 2923 types of proteins were retained for current analyses.
Covariate measurements
In UKB, the covariates included age, gender, educational attainment level, APOE ε4, Townsend deprivation index, depression, anxiety, hypertension, diabetes mellitus, hyperlipidemia, smoking, alcohol, stroke, obesity, and cancer. In ADNI, the covariates included age, gender, education, APOE ε4, diagnosis, depression, anxiety, hypertension, diabetes, smoking, stroke, obesity, and cancer. APOE ε4 carrier status was determined by genetic information (rs7412 and rs429358).
Statistical analyses
R software version 4.3.1 was used for statistical analyses. A two-sided p < 0.05 was considered as statistical significance. Baseline characteristics were summarized as mean (standard deviation [SD]) for normally distributed continuous variables, median (interquartile range [IQR]) for non-normally distributed continuous variables, and number (percentage) for categorical variables. The population was divided into high-risk and low-risk groups based on the highest quartile of blood fatty acid score (FAS). Comparisons between groups were performed using the Mann-Whitney U test for non-normally distributed continuous variables, the t-test for normally distributed continuous variables, and the chi-square test for categorical variables.
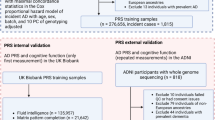
The research design and flowchart are shown in Fig. 1. Firstly, Lasso regression and Cox proportional hazards regression were employed to identify significant variables associated with risk of AD based on UKB cohort. The identified variables were then used to construct risk scores using Cox regression which met the proportional hazards assumption. The proportional hazards assumption for the Cox regression model was assessed using Schoenfeld residuals. If the assumption was violated, interaction terms with time were incorporated. Subsequently, the FAS for each participant was calculated based on the coefficients of the feature variables. Specifically, variable selection was performed using the Lasso regression model. Twenty-fold cross-validation was conducted to identify the optimal penalty parameter (lambda). Virtual vertical lines were drawn at lambda1.1SE and lambda.min, with the model at lambda.min set to 0.000 being selected as the best diagnostic model. Predictors with non-zero coefficients were considered relevant and included in further analysis to develop a more concise multi-factor Cox regression prediction model. A nomogram of the multi-factor Cox regression prediction model was then constructed using the R package ‘rms’ to visualize the AD risk related to FAS. The population was then stratified into high-risk and low-risk groups according to the highest quartile of FAS. The restricted cubic spline analysis was used to explore the linear or nonlinear relationship between FAS and AD risk, validating the appropriateness of the group classifications. Next, multivariable Cox proportional hazard regression models were used to investigate the relationship of FAS with the risk of AD in UKB and ADNI. The “survival”, “ggplot2”, “pub”, “gritty” and “survminer” packages were used for these analyses.

Seventeen blood fatty acid indicators were initially considered. Cox proportional hazard models were used to identify indicators closely associated with the risk of Alzheimer’s disease (AD). After Lasso regression (n = 7) and considering contribution degree in the nomogram (n = 2), a total of 8 indicators were finally included to construct the fatty acid risk score. The association of risk score with AD risk, cognition, hippocampus, and/or cerebrospinal fluid AD markers were tested in both UKB and ADNI cohorts. Mediation effects and interaction analyses by APOE ε4 were performed. Finally, proteomic analyses were conducted to elucidate the biological mechanisms of the interaction. “*” See e-Fig. 1 for details. FAS Fatty acid score.
Next, multiple linear regression models were employed to explore the associations of FAS with cognition and hippocampal volume in UKB and ADNI. All dependent variables were checked for normal distribution. When dependent variables exhibited skewed distributions, Box-Cox transformation was applied to make the data meet or approximate normality (Kolmogorov-Smirnov test p-value > 0.01). Residuals were visually assessed for linearity, homoscedasticity, and normality. To further validate the cross-sectional findings, the linear mixed-effects (LME) models were used to depict the longitudinal relationship of FAS with cognitive decline, hippocampal atrophy, and changing rates of CSF biomarkers in ADNI. The LME models had random intercepts and slopes for time and an unstructured covariance matrix for the random effects, and included the interaction between time (continuous) and the dependent variable as predictors. Regression diagnostics were performed, and outliers were excluded to confirm that all models satisfied the required assumptions: residuals following a normal distribution and no signs of heteroscedasticity. In all the analyses mentioned above, covariate adjustments were made across three models: Model I (no variables were adjusted), Model II (UKB: adjusted for age, gender, education, APOE ε4, and Townsend index; ADNI: adjusted for age, gender, education, APOE ε4, and baseline cognitive diagnosis [mild cognitive impairment vs. normal cognition]), and Model III (UKB: covariates from Model II plus depression, anxiety, hypertension, diabetes mellitus, hyperlipidemia, smoking, alcohol use, stroke, obesity, and cancer; ADNI: covariates from Model II plus depression, anxiety, hypertension, diabetes, smoking, stroke, obesity, and cancer). The “lm”, “name”, “ggplot2” and “car” packages were used to conduct the above analyses.
Mediation analyses were performed to investigate the mediating role of hippocampus in the relationship of FAS with cognitive function scores in UKB and ADNI. To strengthen the robustness of our findings, we calculated the longitudinal changing rates of hippocampal atrophy and cognitive decline based on LME models and performed mediation analysis on these rates in ADNI. Covariates were consistent with those used in Model II. Mediation analyses were executed using the “Mediation” package, employing nonparametric bootstrapping with 10,000 iterations to estimate direct effects (DE), indirect effects (IE), the proportion mediated, and associated P-values.
Moreover, interaction and stratified analyses by APOE ε4 status were performed to investigate whether the associations of FAS with AD risk, hippocampus, cognition, and CSF biomarkers was influenced by APOE ε4 status. As for the longitudinal interaction analyses, LME models were employed due to their suitability for managing unbalanced and censored data, as well as continuous time variables. Fixed effects included the primary effects of FAS, APOE ε4 status, and follow-up duration (time), in addition to interaction terms such as FAS × APOE ε4, time × FAS, time × APOE ε4, and the three-way interaction of time × FAS × APOE ε4. The overall significance of the three-way interaction was assessed using a likelihood ratio test by contrasting the full model with a nested model that excluded the three-way interaction term. Diagnostic evaluations of the regression models were conducted, with outliers removed to ensure that the assumptions of normality in residuals and homoscedasticity were met. Group differences were statistically examined by comparing model coefficients using the Wald test, executed via the ‘aod’ package.
Lastly, proteomic and bioinformatic analyses were conducted to explore the potential biological mechanisms through which FAS was associated with AD risk. Cox proportional hazards adjusting for covariates in Model II and logistic regression models adjusting for age and gender were used to identified differentially expressed proteins associated with AD risk and FAS, respectively. Bonferroni correction was applied to define statistical significance (P < 1.71 × 10−5, number of proteins tested = 2923). Mediation analyses were further used to assess the roles of the overlapped proteins in mediating the relationships of FAS with AD risk. Subsequently, functional enrichment analyses targeting these tagged proteins were performed using the STRING database (http://string-db.org). The Benjamini-Hochberg (BH) procedure was employed for multiple testing corrections. The False Discovery Rate (FDR) value indicates the significance of the enrichment. Finally, we selected the top 10 pathways with the lowest FDR values in each category for bubble mapping. In addition, we included the FDR correction as sensitivity analysis. These results are reported in the supplementary materials.
Results
Construction and validation of fatty acid risk score
In UKB cohort (N = 148,308; median age 57 years; maximum follow-up = 16 years), participants with high FAS were older, were more likely to be female, had lower education levels, and had higher rates of obesity, hypertension, diabetes, smoking, and stroke (P < 0.001). In ADNI cohort (N = 1193; median age 74 years; maximum follow-up 8 years), participants with high FAS were more often female (P = 0.004). (Table 1).
Based on LASSO regression and cross-validation, a total of ten components were chosen, including MUFA, omega-6, omega-3, DHA, LA, the SFA to TFA ratio, the MUFA to TFA ratio, the omega-3 to TFA ratio, the LA to TFA ratio, and the omega-6 to omega-3 ratio. After excluding MUFA to TFA ratio and omega-3 which had negligible contributions in the nomogram, eight components were finally retained to construct the FAS (e-Fig. 1). When FAS was treated as continuous variable, higher FAS was significantly associated with an increased risk of AD (hazard ratio [HR] = 1.089, 95% confidence interval [CI] = 1.055–1.125, P < 0.001) in UKB cohort (Model II). The association still reached borderline significance in Model III (HR = 1.033, 95% CI: 0.998–1.070, P = 0.064). In ADNI, higher FAS was significantly associated with an increased risk of AD in both Model II (HR = 1.051, 95% CI: 1.018–1.085, P = 0.002) and Model III (HR = 1.047, 95% CI: 1.014–1.081, P = 0.005) (e-Table 1). Restricted cubic spline analysis supported the validity of the FAS high- and low-risk group stratification (e-Fig. 2). When FAS was treated as categorical variable, higher FAS was associated with higher risk of AD in Model II (HR = 1.298, 95% CI: 1.183–1.423, P < 0.001, Fig. 2A) and Model III. The associations were validated in ADNI (HR = 1.413, 95% CI 1.105–1.808, P = 0.006, Fig. 2B). The proportional hazards assumption for the Cox regression models in the UKB and ADNI cohort is presented in e-Fig. 3 and detailed in e-Table 2. No significant interaction by APOE ε4 status was found in both cohorts (e-Table 1). The stratified analysis by APOE ε4 status in two cohorts revealed that the association between FAS and AD risk remained significant in the APOE ε4 carrier group, but not in the non-carrier group (e-Table 3).

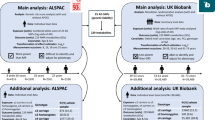
Higher fatty acid score was associated with elevated risk of AD in UKB A and ADNI B.
Higher FAS was associated with lower levels and faster decline of memory function
The characteristics of participants (N = 13,035 for UKB and N = 1193 for ADNI) included for analyses were given in the e-Table 4. The cross-sectional analyses showed that higher FAS was associated with worse numeric memory test performance (Model I, β = −0.084, P < 0.001) in UKB. The association reached borderline significance in Model II (β = −0.036, P = 0.083, Fig. 3A). In ADNI, individuals with higher FAS exhibited worse cognition, as indicated by a higher level of ADAS (Model II, β = 0.122, P = 0.026, Fig. 3B) and lower scores of MEM (Model II, β = −0.101, P = 0.012, Fig. 3C). The associations remained significant after controlling for more covariates (e-Table 5). Longitudinal analyses showed that higher FAS was associated with faster cognitive decline in memory (Model II, β = −0.034, P < 0.001, Fig. 3D). The association remained significant in Model III. Stratified analyses by APOE ε4 status showed that the association was significant only in the APOE ε4 carrier group (β = −0.058, P < 0.001, e-Table 6). No association was found for changing rates of general function (Model II, β = 0.003, P = 0.812, e-Table 7). No significant was found for interaction by APOE ε4 status (e-Table 8).

In UKB, numeric memory test scores were lower in the high-risk group, with marginal statistical significance A. In ADNI, ADAS B was higher and MEM C was lower in the high-risk group, with a faster rate of MEM decline D. Hippocampal volume was lower in the high-risk group in both UKB E and ADNI F, with a faster rate of decline in ADNI G. In ADNI, Aβ levels showed no difference H, while tau I and p-tau J were higher in the high-risk group. In mediation analyses, no significant mediation was found in UKB K, but in ADNI, hippocampal volume mediated effects on MEM L and ADAS M. Longitudinal analysis in ADNI showed hippocampal changes mediated MEM decline N, but not ADAS O. AD Alzheimer’s disease, ADAS Alzheimer’s Disease Assessment Scale, MEM memory function, Numeric Test numeric memory test, IE indirect effect.
Higher FAS was associated with lower volume and faster atrophy of hippocampus
The characteristics of participants (N = 22,626 for UKB and N = 977 for ADNI) for analyses were given in the e-Table 9. Higher FAS was associated with lower volume of hippocampus at baseline (Model II, UKB: β = −0.075, P = 5.83 × 10−7, Fig. 3E; ADNI: β = −0.119, P = 0.048, Fig. 3F). Longitudinal analyses of ADNI data showed that individuals with higher FAS exhibited faster rates of hippocampal atrophy (Model II, β = −0.024, P = 0.002, Fig. 3G). The associations remained significant in Model III (e-Table 7). No significant interaction by APOE ε4 status was found in the longitudinal analysis (e-Table 8). However, subgroup analyses in ADNI showed that FAS was associated with hippocampus atrophy only in APOE ε4 carrier group (β = −0.038, P = 0.003, e-Table 6).
Higher FAS was associated with higher levels of CSF tau proteins
The characteristics of ADNI participants for CSF biomarker analyses were given in the e-Table 10. Individuals with higher FAS were older (P = 0.021), more often female (P = 0.007), and had a higher prevalence of APOE ε4 (P = 0.035). Among 870 participants with baseline data of CSF biomarker, 65.4% completed at least two follow-up evaluations over a maximum of 6 years. Higher FAS was linked to higher levels of CSF t-tau (Model II, β = 0.183, P = 0.015, Fig. 3I) and p-tau (Model II, β = 0.197, P = 0.008, Fig. 3J). The associations remained unchanged after controlling for more covariates (e-Table 5). No significant correlation was found between FAS and CSF Aβ1–42 at baseline (P = 0.302, Fig. 3H). Though no longitudinal relationships were uncovered of FAS with CSF AD markers (e-Table 7), the likelihood ratio test indicated that the three-way interaction of FAS × APOE ε4 × time accounted for a significant amount of variance in CSF Aβ1–42 (Model II, P = 0.017, e-Fig. 4A), t-tau protein (Model II, P = 0.013, e-Fig. 4B), and p-tau protein (Model II, P = 0.032, e-Fig. 4C). Specifically, compared to other groups, a greater rate of CSF t-tau or p-tau increase (β = 0.04, P = 0.032) was observed in the APOE ε4 carriers with higher FAS group (e-Table 6).
The association of FAS with cognition was mediated by hippocampus volume
In UKB, potential mediating effects of hippocampus were found on the relationship of FAS with numeric test scores (P = 0.048, Fig. 3K). In ADNI, hippocampus volume mediated the relationship of FAS with MEM (P = 0.048, proportion = 19.2%, Fig. 3L) and ADAS (P = 0.038, proportion = 21.1%, Fig. 3M) scores. Furthermore, the relationship of FAS with changing rates of MEM (P = 0.008, proportion = 38.6%, Fig. 3N) but not ADAS (P = 0.016, proportion = 81.7%, Fig. 3O) was mediated by the atrophy rate of hippocampus.
Proteomic and bioinformatics analyses to reveal biological pathways
After Bonferroni correction (P < 1.71 × 10−5), we uncovered 16 proteins associated with AD risk (Fig. 4A) and 968 proteins associated with higher FAS (Fig. 4B). The Venn diagram illustrated four overlapping proteins that were positively correlated with both AD risk and higher FAS, including ADGRG1, CHI3L1, GDF15, and RBFOX3 (Fig. 4C). We found that the relationships of FAS with AD risk were mediated by these four proteins (proportion ranging from 7.7–19.2%, P < 2 × 10−16, Fig. 4D). Because no significant protein-protein interaction (PPI) enrichment was found (P = 0.053), we separately explored the underpinning pathways for each protein. RBFOX3 is markedly involved in nervous system development, with significant emphasis on brain regions like the dentate gyrus, subventricular zone, and hippocampus (Fig. 5A). ADGRG1 is enriched in key pathways, including dopamine receptor signaling and G protein-coupled receptor (GPCR) signaling pathways (Fig. 5B). GDF15 is primarily involved in pathways like the glial cell-derived neurotrophic factor receptor signaling pathway and transforming growth factor beta receptor signaling pathway (Fig. 5C). The CHI3L1 bubble chart highlights its pivotal role in immune response, cytokine signaling, and inflammatory pathways, including cytokine and inflammatory responses, cytokine-cytokine receptor interactions, and regulation of leukocyte activation (Fig. 5D). FDR-corrected proteomics and bioinformatics results are presented in e-Fig. 5 and e-Table 11.

Volcano plots of proteins associated with AD incidence A and FAS B. The Venn diagram illustrated the differential proteins among the four groups. Group A: proteins positively associated with AD incidence; Group B: proteins negatively associated with AD incidence; Group C: proteins negatively associated with higher FAS; Group D: proteins positively associated with higher FAS. Four overlapping proteins (adgrg1, chi3l1, gdf15, and rbfox3) were shared between Groups A and D, indicating significant relevance to both AD incidence and higher FAS. C. Mediation analyses indicated that the relationship between FAS and AD risk was mediated by screening differential proteins D. FAS Fatty acid score, IE indirect effect.

The enrichment bubble diagram showed functional and pathway analysis of four proteins, displaying the top 10 terms with the lowest p-values. The y-axis represented enriched pathways, the x-axis showed the gene ratio, the bubble color indicated the p-value, and the size reflected the number of enriched genes. KEGG, other pathways, and GO enrichment analyses were shown for enriched pathways associated with RBFOX3 A, ADGRG1 B, GDF15 C, and CHI3L1 D.
Discussion
In this study, we 1) developed and validated a blood fatty acid score associated with AD risk, 2) confirmed the relationships of the scores with cognitive decline, hippocampus, and CSF AD markers, especially among APOE ε4 carriers, 3) found that hippocampal neurodegeneration could mediate the effects of FAS on cognition, and 4) uncovered several pathways, such as inflammatory pathways and neurogenesis, that were potentially responsible for bridging the relationship between blood fatty acids and AD occurrence. Overall, these findings underscored critical roles of blood fatty acid metabolic homeostasis in contributing to AD development.
Several fatty acid components included for the FAS calculation were also previously related to AD risk, such as omega-3 fatty acids family and SFAs [16, 40]. Elevated omega-3 levels were considered to mitigate the risk of dementia [15, 41], whereas SFAs were associated with cognitive decline and an increased risk of AD [40, 42]. While these results underscored the roles of omega-3 fatty acids and SFAs in AD risk, integrating these key fatty acids into a comprehensive risk score offered a more holistic approach. By integrating these key fatty acids into a comprehensive risk score, we for the first time offered a practical tool for clinicians to assist in risk assessment. In future trials, this scoring system might be used to evaluate the efficacy of dietary interventions or fatty acid supplementation in AD prevention.
As indicated by the proteomic analyses, the mechanisms by which blood fatty acids were involved in AD occurrence included multiple processes, such as modulation of neuroinflammatory pathways, promotion of synaptic plasticity, and neurogenesis [24, 43, 44]. CHI3L1 was found as a critical differential protein linking fatty acids to AD. CHI3L1 was primarily expressed in reactive astrocytes and microglia, acting as a key driver of neuroinflammation in AD pathogenesis [45]. In genetically modified mice models, high‐fat diet feeding markedly increased CHI3L1 mRNA and protein expression in white adipose tissue and lung [46]. Mechanistically, SFAs might activate the TLR4–MyD88–IKK axis, leading to NF-κB [47,48,49] induction and subsequent upregulation of CHI3L1 [50]. CHI3L1 then engages IL-13Rα2 and RAGE, triggering MAPK/ERK and PI3K–Akt signaling cascades [45] that exacerbate neuroinflammation and amyloid-β–associated inflammatory responses, thereby aggravating AD pathology. In contrast, unsaturated fatty acids (UFAs) activate PPARγ [51] and Nrf2 [52] pathways, suppress NF-κB signaling, and downregulate CHI3L1 expression [53]. Therefore, we hypothesize that modulation of CHI3L1 by distinct fatty acid species could regulate neuroinflammatory processes and thus influence AD progression. The disruption of SFA/UFA balance could amplify CHI3L1-mediated inflammation through NF-κB pathways, immune activation, and cytokine release. Future animal studies are needed to validate these hypotheses. Similarly, GDF15 is induced in response to cellular stress, mitochondrial dysfunction, and inflammation to maintain cellular and tissue homeostasis [54]. Saturated fatty acids promote GDF15 expression and secretion in human macrophages through inducing ER stress and activating the PERK/eIF2/CHOP signaling pathway [55]. Subsequently, GDF15 alleviates neuroinflammation and energy metabolism by inhibiting the TAK1/IKK/NF-κB cascade [56] and modulating GFRAL-mediated central metabolic routes [57], thereby indirectly mitigating amyloid-β and tau pathologies. It could be reasonably postulated that neuroinflammation could at least partially underpin the association of blood fatty acid with AD.
ADGRG1 is an adhesion G protein-coupled receptor closely associated with developmental processes [58]. Although no studies have directly examined the relationship between fatty acids and ADGRG1. Other adhesion GPCRs such as GPR116 and GPR97 have been implicated in lipid homeostasis and high-fat diet–induced metabolic changes [59, 60], suggesting potential roles for ADGRG1 in fatty acid regulation. We hypothesize that ADGRG1 may act as a novel lipid-regulatory receptor. Its intracellular G protein–coupled domains activate downstream cAMP and RhoA signaling networks [61], influencing metabolic homeostasis. Notably, beyond its potential role in lipid metabolism, ADGRG1 has also been implicated in immune regulation. ADGRG1 has been shown to regulate the cytotoxicity of natural killer (NK) cells [58]. As sentinels of the immune system, NK cells are pivotal in the early coordination of local inflammatory responses and have profound implications for the onset and progression of neuroinflammation in aging and AD-related neurodegenerative disease [62]. Fatty acids may engage ADGRG1 through ligand binding or co-regulatory mechanisms, thereby modulating NK cell–mediated inflammatory responses to drive Alzheimer’s disease pathogenesis. Elucidating this axis could inform novel therapeutic strategies targeting both lipid metabolism and immune regulation in AD.
In addition, our mediation analyses revealed that hippocampal volume mediated the relationship between fatty acids and cognition, especially memory function. This aligned with previous research linking fatty acids to hippocampus. High-fat diets were reported to trigger neuroinflammation in the hippocampus, promoting its atrophy and cognitive decline [25, 63]. Elevated blood levels of omega-3 were instead associated with less hippocampal atrophy, better cognitive performance, and reduced risk of cognitive decline in the elderly [64]. These results emphasized the modulation effects of fatty acid profile on neurodegeneration in hippocampus. The possible mechanisms included neuroinflammation, synaptic activity, and neurogenesis [65]. Interestingly, our bioinformatics analyses identified RBFOX3 as a key bridging protein, which was enriched in pathways critical for hippocampal neurogenesis and synaptic plasticity in subventricular zone and dentate gyrus. These regions are vital for neuronal differentiation, synaptic remodeling, and cognition [66]. RBFOX3 is crucial for neurogenesis and synaptogenesis, with knockout models showing deficits in synaptic plasticity and cognition [67]. RBFOX proteins also supported neuronal maturation and axon assembly, which were critical for neuron integration into circuits [68]. We thus inferred that one key pathway by which blood fatty acids influenced cognition was by regulating RBFOX3 and enhancing synaptic plasticity. Regrettably, there have been no studies confirming that fatty acids or their metabolites can regulate the expression of RBFOX3. Future studies should investigate the impact of fatty acids on RBFOX3 expression. Gene knockout techniques could be employed to investigate whether the effects of fatty acids on synaptic function are mediated through RBFOX3, in both in vivo and in vitro models.
The interaction of fatty acids such as omega-3 with APOE ε4 gene has been a controversial topic in the field. We previously reported the cognitive and pathological benefits of omega-3 supplementation were depending on presence of APOE ε4 [22], while others reported greater benefits in individuals with lower genetic risk [69, 70]. In the present study, we found that the associations of FAS with AD were more pronounced among APOE ε4 carriers, reinforcing the idea that APOE ε4 may amplify the effects of fatty acids on AD [14, 22]. Two mechanisms could help explain the interaction effect. Firstly, APOE ε4 is associated with reduced delivery of DHA to the brain [21], which may limit the protective effects of beneficial fatty acids. Secondly, dietary fatty acids have been shown to modulate microglial states, thereby influencing neuroinflammation and contributing to AD-related neuropathology [71]. Similarly, APOE ε4 may cause overactivation of microglia and the release of pro-inflammatory cytokines, leading to neuronal damage and tau hyperphosphorylation [72]. The combined effects of imbalanced fatty acid ratios and APOE ε4 may amplify neuroinflammation, thereby exacerbating the progression of AD. However, this APOE ε4 × FAS interaction should be interpreted cautiously, considering several important limitations: First, as an observational study, causal inference is difficult; the interaction may reflect shared upstream determinants rather than a true biological synergism. Second, residual confounding by unmeasured factors—such as dietary patterns, or other genetic variants—could influence both fatty acid profiles and AD risk. Finally, statistical interaction does not necessarily imply mechanistic interaction, and replication in independent cohorts with more detailed phenotyping will be necessary to confirm these findings.
Our study demonstrated several strengths. First, we constructed a risk score by rigorously screening blood fatty acids profile associated with AD risk. This comprehensive approach allows for a thorough evaluation of the interactions among various fatty acids. Second, the association of FAS with AD was validated across independent cohorts from two different continents, enhancing the generalizability and reliability of our findings. Third, multiple methods were further used to explore the potential mechanisms. There were several limitations. First, plasma fatty acids can fluctuate with dietary intake, which may not accurately represent the accumulating levels of fatty acid during the follow-up. Second, the associations or the pathways were based on the observational study but not equal to the causal relationship. Future experiments were warranted to test the hypothesis in the future. Third, the absence of CSF data and ATN‐based AD confirmation in UKB remains a key limitation of our study. Future studies should leverage multicenter cohorts with multimodal data (CSF, PET, and plasma biomarkers) to confirm these findings. Fourth, differences in ethnicity between ADNI and UKB may limit generalizability. Future studies should include multi-ethnic cohorts and adjust for ancestry to minimize confounding.
Conclusion
Overall, our study developed and validated a blood fatty acid score associated with incident AD risk in two dependent cohorts. The potential underpinning mechanisms could be hippocampal neurodegeneration, neuroinflammation, neurogenesis, and synaptic plasticity. Future researches are needed to further validate this risk score in more community-based populations as well as in the clinic settings.
Data availability
All data are available upon reasonable request or can be obtained from the UKB (https://biobank.ctsu.ox.ac.uk/) and Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu).
References
Patterson C. World Alzheimer Report 2018: The state of the art of dementia research: New frontiers. London: Alzheimer’s Disease International; 2018. Available from: https://www.alzint.org/u/WorldAlzheimerReport2018.pdf
Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018;136:821–53. https://doi.org/10.1007/s00401-018-1932-x
Hampel H, O’Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14:639–52. https://doi.org/10.1038/s41582-018-0079-7
Blennow K. A review of fluid biomarkers for Alzheimer’s disease: moving from CSF to blood. Neurol Ther. 2017;6:15–24. https://doi.org/10.1007/s40120-017-0073-9
Kim KY, Shin KY, Chang KA. GFAP as a potential biomarker for Alzheimer’s disease: a systematic review and meta-analysis. Cells. 2023;12:1309 https://doi.org/10.3390/cells12091309
Guo Y, You J, Zhang Y, Liu WS, Huang YY, Zhang YR, et al. Plasma proteomic profiles predict future dementia in healthy adults. Nat Aging. 2024;4:247–60. https://doi.org/10.1038/s43587-023-00565-0
Walker KA, Chen J, Shi L, Yang Y, Fornage M, Zhou L, et al. Proteomics analysis of plasma from middle-aged adults identifies protein markers of dementia risk in later life. Sci Transl Med. 2023;15:eadf5681 https://doi.org/10.1126/scitranslmed.adf5681
Pase MP, Himali JJ, Puerta R, Beiser AS, Gonzales MM, Satizabal CL, et al. Association of plasma YKL-40 With MRI, CSF, and cognitive markers of brain health and dementia. Neurology. 2024;102:e208075 https://doi.org/10.1212/wnl.0000000000208075
Mwale PF, Hsieh CT, Yen TL, Jan JS, Taliyan R, Yang CH, et al. Chitinase-3-like-1: a multifaceted player in neuroinflammation and degenerative pathologies with therapeutic implications. Mol Neurodegener. 2025;20:7 https://doi.org/10.1186/s13024-025-00801-8
Dhauria M, Mondal R, Deb S, Shome G, Chowdhury D, Sarkar S, et al. Blood-based biomarkers in Alzheimer’s disease: advancing non-invasive diagnostics and prognostics. Int J Mol Sci. 2024;25:10911 https://doi.org/10.3390/ijms252010911
Ehtewish H, Mesleh A, Ponirakis G, De la Fuente A, Parray A, Bensmail I, et al. Blood-based proteomic profiling identifies potential biomarker candidates and pathogenic pathways in dementia. Int J Mol Sci. 2023;24:8117 https://doi.org/10.3390/ijms24098117
Scrimgeour AG, Condlin ML, Loban A, DeMar JC. Omega-3 fatty acids and vitamin D decrease plasma T-tau, GFAP, and UCH-L1 in experimental traumatic brain injury. Front Nutr. 2021;8:685220 https://doi.org/10.3389/fnut.2021.685220
Lv X, Zhao Q, Liu Q, Ji Q, Huang X, Zhou L, et al. Serum fatty acid profiles and neurofilament light chain levels in the general population. J Nutr. 2024;154:3070–8. https://doi.org/10.1016/j.tjnut.2024.07.007
Wei BZ, Li L, Dong CW, Tan CC, Xu W. The relationship of Omega-3 fatty acids with dementia and cognitive decline: evidence from prospective cohort studies of supplementation, dietary intake, and blood markers. Am J Clin Nutr. 2023;117:1096–109. https://doi.org/10.1016/j.ajcnut.2023.04.001
He Y, Huang SY, Wang HF, Zhang W, Deng YT, Zhang YR, et al. Circulating polyunsaturated fatty acids, fish oil supplementation, and risk of incident dementia: a prospective cohort study of 440,750 participants. GeroScience. 2023;45:1997–2009. https://doi.org/10.1007/s11357-023-00778-6
Thomas A, Baillet M, Proust-Lima C, Féart C, Foubert-Samier A, Helmer C, et al. Blood polyunsaturated omega-3 fatty acids, brain atrophy, cognitive decline, and dementia risk. Alzheimers Dement. 2020. https://doi.org/10.1002/alz.12195.
Fan L, Borenstein AR, Wang S, Nho K, Zhu X, Wen W, et al. Associations of circulating saturated long-chain fatty acids with risk of mild cognitive impairment and Alzheimer’s disease in the Alzheimer’s disease neuroimaging initiative (ADNI) cohort. EBioMedicine. 2023;97:104818 https://doi.org/10.1016/j.ebiom.2023.104818
Ammann EM, Pottala JV, Harris WS, Espeland MA, Wallace R, Denburg NL, et al. ω-3 fatty acids and domain-specific cognitive aging: secondary analyses of data from WHISCA. Neurology. 2013;81:1484–91. https://doi.org/10.1212/WNL.0b013e3182a9584c
Lin PY, Cheng C, Satyanarayanan SK, Chiu LT, Chien YC, Chuu CP, et al. Omega-3 fatty acids and blood-based biomarkers in Alzheimer’s disease and mild cognitive impairment: a randomized placebo-controlled trial. Brain Behav Immun. 2022;99:289–98. https://doi.org/10.1016/j.bbi.2021.10.014
Hosseini M, Poljak A, Braidy N, Crawford J, Sachdev P. Blood fatty acids in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review. Ageing Res Rev. 2020;60:101043 https://doi.org/10.1016/j.arr.2020.101043
Dakterzada F, Jové M, Cantero JL, Mota-Martorell N, Pamplona R, Piñoll-Ripoll G. The shift in the fatty acid composition of the circulating lipidome in Alzheimer’s disease. Alzheimers Dement. 2024;20:3322–33. https://doi.org/10.1002/alz.13792
Li L, Xu W, Tan CC, Cao XP, Wei BZ, Dong CW, et al. A gene-environment interplay between omega-3 supplementation and APOE ε4 provides insights for Alzheimer’s disease precise prevention amongst high-genetic-risk population. Eur J Neurol. 2022;29:422–31. https://doi.org/10.1111/ene.15160
Mett J. The impact of medium chain and polyunsaturated ω-3-fatty acids on amyloid-β deposition, oxidative stress and metabolic dysfunction associated with Alzheimer’s disease. Antioxidants. 2021;10:1991 https://doi.org/10.3390/antiox10121991
Bogie JFJ, Haidar M, Kooij G, Hendriks JJA. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv Drug Deliv Rev. 2020;159:198–213. https://doi.org/10.1016/j.addr.2020.01.004
Butler MJ, Cole RM, Deems NP, Belury MA, Barrientos RM. Fatty food, fatty acids, and microglial priming in the adult and aged hippocampus and amygdala. Brain Behav Immun. 2020;89:145–58. https://doi.org/10.1016/j.bbi.2020.06.010
Nakandakari S, Muñoz VR, Kuga GK, Gaspar RC, Sant’Ana MR, Pavan ICB, et al. Short-term high-fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav Immun. 2019;79:284–93. https://doi.org/10.1016/j.bbi.2019.02.016
Ordóñez-Gutiérrez L, Fábrias G, Casas J, Wandosell F. Diets with higher ω-6/ω-3 ratios show differences in ceramides and fatty acid levels accompanied by increased amyloid-beta in the brains of male APP/PS1 transgenic mice. Int J Mol Sci. 2021;22:10907 https://doi.org/10.3390/ijms222010907
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wi de range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779 https://doi.org/10.1371/journal.pmed.1001779
Würtz P, Kangas AJ, Soininen P, Lawlor DA, Davey Smith G, Ala-Korpela M. Quantitative serum nuclear magnetic resonance metabolomics in large-scale epidemiology: a primer on -omic technologies. Am J Epidemiol. 2017;186:1084–96. https://doi.org/10.1093/aje/kwx016
Li Y, Liu X, Zhuang P, Zhang L, Wu Y, Wu S, et al. Fish oil supplementation and risk of dementia among diabetic patients: a prospective study of 16,061 older patients. J Nutr Health Aging. 2024;28:100176 https://doi.org/10.1016/j.jnha.2024.100176
Huang Y, Deng Y, Zhang P, Lin J, Guo D, Yang L, et al. Associations of fish oil supplementation with incident dementia: evidence from the UK biobank cohort study. Front Neurosci. 2022;16:910977 https://doi.org/10.3389/fnins.2022.910977
Wang HF, Zhang W, Rolls ET, Li Y, Wang L, Ma YH, et al. Hearing impairment is associated with cognitive decline, brain atrophy and tau pathology. EBioMedicine. 2022;86:104336 https://doi.org/10.1016/j.ebiom.2022.104336
Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, et al. Development and assessment of a composite score for memory in the Alzheimer’s disease neuroimaging initiative (ADNI). Brain Imaging Behav. 2012;6:502–16. https://doi.org/10.1007/s11682-012-9186-z
Gibbons LE, Carle AC, Mackin RS, Harvey D, Mukherjee S, Insel P, et al. A composite score for executive functioning, validated in Alzheimer’s disease neuroimaging initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 2012;6:517–27. https://doi.org/10.1007/s11682-012-9176-1
Jack CR Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–91. https://doi.org/10.1002/jmri.21049
Alfaro-Almagro F, Jenkinson M, Bangerter NK, Andersson JLR, Griffanti L, Douaud G, et al. Image processing and quality control for the first 10,000 brain imaging datasets from UK biobank. Neuroimage. 2018;166:400–24. https://doi.org/10.1016/j.neuroimage.2017.10.034
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. https://doi.org/10.1002/ana.21610
Hansson O, Seibyl J, Stomrud E, Zetterberg H, Trojanowski JQ, Bittner T, et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470–81. https://doi.org/10.1016/j.jalz.2018.01.010
Sun BB, Chiou J, Traylor M, Benner C, Hsu YH, Richardson TG, et al. Plasma proteomic associations with genetics and health in the UK biobank. Nature. 2023;622:329–38. https://doi.org/10.1038/s41586-023-06592-6
Koch M, Furtado JD, DeKosky ST, Fitzpatrick AL, Lopez OL, Kuller LH, et al. Case-cohort study of plasma phospholipid fatty acid profiles, cognitive function, and risk of dementia: a secondary analysis in the Ginkgo evaluation of memory study. Am J Clin Nutr. 2021;114:154–62. https://doi.org/10.1093/ajcn/nqab087
Power R, Nolan JM, Prado-Cabrero A, Roche W, Coen R, Power T, et al. Omega-3 fatty acid, carotenoid and vitamin E supplementation improves working memory in older adults: a randomised clinical trial. Clin Nutr. 2022;41:405–14. https://doi.org/10.1016/j.clnu.2021.12.004
Heude B, Ducimetière P, Berr C. Cognitive decline and fatty acid composition of erythrocyte membranes–the EVA study. Am J Clin Nutr. 2003;77:803–8. https://doi.org/10.1093/ajcn/77.4.803
Layé S, Nadjar A, Joffre C, Bazinet RP. Anti-inflammatory effects of omega-3 fatty acids in the brain: physiological mechanisms and relevance to pharmacology. Pharmacol Rev. 2018;70:12–38. https://doi.org/10.1124/pr.117.014092
Peineau S, Rabiant K, Pierrefiche O, Potier B. Synaptic plasticity modulation by circulating peptides and metaplasticity: involvement in Alzheimer’s disease. Pharmacol Res. 2018;130:385–401. https://doi.org/10.1016/j.phrs.2018.01.018
Connolly K, Lehoux M, O’Rourke R, Assetta B, Erdemir GA, Elias JA, et al. Potential role of chitinase-3-like protein 1 (CHI3L1/YKL-40) in neurodegeneration and Alzheimer’s disease. Alzheimers Dement. 2023;19:9–24. https://doi.org/10.1002/alz.12612
Ahangari F, Sood A, Ma B, Takyar S, Schuyler M, Qualls C, et al. Chitinase 3-like-1 regulates both visceral fat accumulation and asthma-like Th2 inflammation. Am J Respir Crit Care Med. 2015;191:746–57. https://doi.org/10.1164/rccm.201405-0796OC
Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–9. https://doi.org/10.1074/jbc.M011695200
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. https://doi.org/10.1172/jci28898
Vesga-Jiménez DJ, Martin C, Barreto GE, Aristizábal-Pachón AF, Pinzón A, González J. Fatty acids: an insight into the pathogenesis of neurodegenerative diseases and therapeutic potential. Int J Mol Sci. 2022;23:2577 https://doi.org/10.3390/ijms23052577
Yu JE, Yeo IJ, Han SB, Yun J, Kim B, Yong YJ, et al. Significance of chitinase-3-like protein 1 in the pathogenesis of inflammatory diseases and cancer. Exp Mol Med. 2024;56:1–18. https://doi.org/10.1038/s12276-023-01131-9
Li H, Ruan XZ, Powis SH, Fernando R, Mon WY, Wheeler DC, et al. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-gamma-dependent mechanism. Kidney Int. 2005;67:867–74. https://doi.org/10.1111/j.1523-1755.2005.00151.x
Khoo NKH, Li L, Salvatore SR, Schopfer FJ, Freeman BA. Electrophilic fatty acid nitroalkenes regulate Nrf2 and NF-κB signaling: a medicinal chemistry investigation of structure-function relationships. Sci Rep. 2018;8:2295 https://doi.org/10.1038/s41598-018-20460-8
Hübner K, Karwelat D, Pietsch E, Beinborn I, Winterberg S, Bedenbender K, et al. NF-κB-mediated inhibition of microRNA-149-5p regulates Chitinase-3-like 1 expression in human airway epithelial cells. Cell Signal. 2020;67:109498 https://doi.org/10.1016/j.cellsig.2019.109498
Rochette L, Dogon G, Zeller M, Cottin Y, Vergely C. GDF15 and cardiac cells: current concepts and new insights. Int J Mol Sci. 2021;22:8889 https://doi.org/10.3390/ijms22168889
L’Homme L, Sermikli BP, Staels B, Piette J, Legrand-Poels S, Dombrowicz D. Saturated fatty acids promote GDF15 expression in human macrophages through the PERK/eIF2/CHOP signaling pathway. Nutrients. 2020;12:3771 https://doi.org/10.3390/nu12123771
Ratnam NM, Peterson JM, Talbert EE, Ladner KJ, Rajasekera PV, Schmidt CR, et al. NF-κB regulates GDF-15 to suppress macrophage surveillance during early tumor development. J Clin Invest. 2017;127:3796–809. https://doi.org/10.1172/jci91561
Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjær SB, et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med. 2017;23:1158–66. https://doi.org/10.1038/nm.4394
Chang GW, Hsiao CC, Peng YM, Vieira Braga FA, Kragten NA, Remmerswaal EB, et al. The adhesion g protein-coupled receptor GPR56/ADGRG1 is an inhibitory receptor on human NK cells. Cell Rep. 2016;15:1757–70. https://doi.org/10.1016/j.celrep.2016.04.053
Kaczmarek I, Suchý T, Prömel S, Schöneberg T, Liebscher I, Thor D. The relevance of adhesion G protein-coupled receptors in metabolic functions. Biol Chem. 2022;403:195–209. https://doi.org/10.1515/hsz-2021-0146
Liessmann F, von Bredow L, Meiler J, Liebscher I. Targeting adhesion G protein-coupled receptors. Current status and future perspectives. Structure. 2024;32:2188–205. https://doi.org/10.1016/j.str.2024.10.022
Wu J, Wang Z, Cai M, Wang X, Lo B, Li Q, et al. GPR56 promotes diabetic kidney disease through eNOS regulation in glomerular endothelial cells. Diabetes. 2023;72:1652–63. https://doi.org/10.2337/db23-0124
Qi C, Liu Q. Natural killer cells in aging and age-related diseases. Neurobiol Dis. 2023;183:106156 https://doi.org/10.1016/j.nbd.2023.106156
Yao X, Yang C, Jia X, Yu Z, Wang C, Zhao J, et al. High-fat diet consumption promotes adolescent neurobehavioral abnormalities and hippocampal structural alterations via microglial overactivation accompanied by an elevated serum free fatty acid concentration. Brain Behav Immun. 2024;119:236–50. https://doi.org/10.1016/j.bbi.2024.04.005
Chen C, Xun P, Kaufman JD, Hayden KM, Espeland MA, Whitsel EA, et al. Erythrocyte omega-3 index, ambient fine particle exposure, and brain aging. Neurology. 2020;95:e995–e1007. https://doi.org/10.1212/wnl.0000000000010074
Crupi R, Marino A, Cuzzocrea S. n-3 fatty acids: role in neurogenesis and neuroplasticity. Curr Med Chem. 2013;20:2953–63. https://doi.org/10.2174/09298673113209990140
Lopez-Rojas J, Kreutz MR. Mature granule cells of the dentate gyrus–passive bystanders or principal performers in hippocampal function? Neurosci Biobehav Rev. 2016;64:167–74. https://doi.org/10.1016/j.neubiorev.2016.02.021
Lin YS, Wang HY, Huang DF, Hsieh PF, Lin MY, Chou CH, et al. Neuronal splicing regulator RBFOX3 (NeuN) regulates adult hippocampal neurogenesis and synaptogenesis. PLoS ONE. 2016;11:e0164164 https://doi.org/10.1371/journal.pone.0164164
Jacko M, Weyn-Vanhentenryck SM, Smerdon JW, Yan R, Feng H, Williams DJ, et al. Rbfox splicing factors promote neuronal maturation and axon initial segment assembly. Neuron. 2018;97:853–68.e856 https://doi.org/10.1016/j.neuron.2018.01.020
Huang TL, Zandi PP, Tucker KL, Fitzpatrick AL, Kuller LH, Fried LP, et al. Benefits of fatty fish on dementia risk are stronger for those without APOE epsilon4. Neurology. 2005;65:1409–14. https://doi.org/10.1212/01.wnl.0000183148.34197.2e
Daiello LA, Gongvatana A, Dunsiger S, Cohen RA, Ott BR. Association of fish oil supplement use with preservation of brain volume and cognitive function. Alzheimers Dement. 2015;11:226–35. https://doi.org/10.1016/j.jalz.2014.02.005
Desale SE, Chinnathambi S. Role of dietary fatty acids in microglial polarization in Alzheimer’s disease. J Neuroinflammation. 2020;17:93 https://doi.org/10.1186/s12974-020-01742-3
Parhizkar S, Holtzman DM. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin Immunol. 2022;59:101594 https://doi.org/10.1016/j.smim.2022.101594
Acknowledgements
This research has been conducted using the UK Biobank resource under application number 108930. The authors also thank contributors, including the staff at Alzheimer’s Disease Centers who collected samples used in this study, patients, and their families whose help and participation made this work possible. Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Funding
This study was supported by grants from the Taishan Scholar Project (NO.tsqn202211375 and NO.tsqn202312391).
Author information
Authors and Affiliations
Consortia
Contributions
Dr. Wen-Zheng Liu and Dr. Liang-Yu Huang: analysis of the data, drafting and revision of the manuscript, and prepared all the figures. Prof. Lan Tan, Prof. Chen-Chen Tan, Prof. Song Chi, and Dr Ya-Hui Ma: revision of the manuscript. Prof. Wei Xu: conceptualization and design of the study, analysis of the data, drafting and revision of the manuscript. Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The UKB and ADNI were approved by the institutional review boards of all participating centers, and written informed consent was obtained from all participants or authorized representatives according to the 1975 Declaration of Helsinki. All methods in this study were performed in accordance with the relevant guidelines and regulations, and the UK Biobank has approval from the North West Multi-centre Research Ethics Committee as a Research Tissue Bank (RTB) approval. Researchers with approved access operate under the RTB approval and do not require separate ethical approval. The ADNI was approved by the Institutional Review Boards (IRB) all participating centers.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
*Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, WZ., Huang, LY., Chi, S. et al. The blood lipidome fatty acid profile predicts the disease risk and clinical phenotypes of Alzheimer’s disease: associations from two prospective cohort studies. Transl Psychiatry 15, 373 (2025). https://doi.org/10.1038/s41398-025-03526-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-025-03526-w
