
Abstract
The human brain has high energy demands and tightly regulated mechanisms ensure its activity-dependent energy supply. Glucose hypometabolism is associated with brain aging and has also been linked to neurodegenerative diseases such as Alzheimer’s disease (AD). The apolipoprotein E4 (APOE4) allele is the strongest genetic risk factor for AD while APOE2 reduces the risk and APOE3 has been referred to as risk neutral allele. APOE is a major lipid carrier in the brain and is not only involved in the build-up of the two AD hallmark pathologies, β-amyloid (Aβ) plaques and neurofibrillary tangles, but also in several other (patho-)physiological processes including immune response, neuronal growth, synaptic plasticity and energy metabolism. Although there has been recent progress in understanding APOE biology, the exact mechanisms of how APOE (especially APOE4) affects brain energy metabolism are still largely unclear. This review highlights the recent evidence of how APOE isoforms differentially affect the bioenergetic homeostasis of the brain, thereby affecting AD etiology and pathophysiology, and identifies critical questions and emerging topics that require further investigation.
Similar content being viewed by others


Background
The human brain is a highly metabolically active organ, requiring substantial energy to sustain its functions. Although the brain accounts for only 2% of body weight, it consumes 20% of the body’s total energy [1]. Alterations in bioenergetic processes and brain energy metabolism are commonly observed during normal aging. However, they are also implicated in various brain diseases.
Alzheimer’s disease (AD) is the most prevalent age-related neurodegenerative disorder. It is characterized by memory deficit and progressive cognitive decline, with significant impairment in activities of daily living. Currently, over 55 million individuals worldwide are affected by dementia and nearly 10 million new cases emerge annually. Dementia is already the seventh leading cause of death and one of the major causes of disability and dependency among the elderly [2, 3].
Histopathologically, AD is characterized by severe neurodegeneration, synapse loss, extracellular depositions of aggregated the β-amyloid peptide (Aβ) as well as intracellular accumulation of hyperphosphorylated tau protein [4]. Advanced brain imaging tools and cerebrospinal fluid (CSF) biomarkers show that pathophysiological processes precede clinical symptoms by 20–30 years. AD can be classified into two forms, familial and sporadic. Familial AD is caused by distinct mutations with 100% penetrance but accounts only for 1–5% of all AD cases [5]. For sporadic AD, several genetic and non-genetic risk factors have been identified so far. Among the numerous genes associated with sporadic AD, as identified through genome-wide association studies (GWAS), the presence of the APOE4 allele represents the strongest genetic risk factor.
This review will draw the link between APOE4 and aberrant energy metabolism in AD and discuss how different APOE isoforms modulate glycolytic and mitochondrial metabolism in neurons and astrocytes and the implications for AD pathophysiology.
APOE
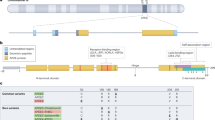
Three major common allelic variants of human APOE have been identified: APOE2, E3 and E4. APOE4 increases the risk of developing AD by 2.5–4-fold in heterozygous and 12–15-fold in homozygous carriers [6,7,8,9,10,11]. APOE4 homozygosity was recently even suggested to represent a distinct genetic form of AD [12]. In contrast, APOE2 is known to lower the AD risk [13,14,15]. In Caucasians, the prevalence of the APOE3/4 and APOE4/4 genotype in non-demented subjects is 21.3% or 1.8% respectively, while AD subjects show higher allelic frequencies, i.e. 41.1% (APOE3/4) or 14.8% (APOE4/4) (Fig. 1A). However, the correlation strength between APOE4 and AD also depends on the ethnic origin and is higher in Caucasians than in African Americans or Hispanics [14].

A APOE allele frequencies in ND controls and AD subjects and odds ratios for developing AD in a Caucasian population [14]. green: genotypes with odds ratio <1; red: odds ratio >1. B Structure of the three major APOE isoforms. ND non-demented.
Encoded by the APOE gene on chromosome 19, APOE is a 299-amino acid (34 kDa) secretory glycoprotein. Its N-terminal domain (1–167) includes the receptor binding region (136–150) and its C-terminal domain (206–299) contains the lipid binding site (244–272), both connected by a hinge region (167–206) (Fig. 1B) [16, 17]. The three human APOE isoforms differ in two amino acid residues at position 112 and 158, resulting in APOE2 (Cys112, Cys158), APOE3 (Cys112, Arg158) and APOE4 (Arg112, Arg158) [17, 18].
After secretion, APOE associates with lipoproteins that transport cholesterol and other lipids and mediates their uptake by target cells via receptor-mediated endocytosis. APOE is a ligand for the low density lipoprotein (LDL) receptor family, including the low density lipoprotein receptor (LDLR), APOE receptor 2 (ApoER2), very low density lipoprotein receptor (VLDLR), LDL-related protein 1 (LRP1) and LDL-related protein 1b (LRP1b) [19]. The single amino acid changes lead to different binding affinities. APOE2 preferentially binds to HDL receptors but poorly to LDL receptors while APOE4 has high affinity to triglyceride-rich VLDLs causing downregulation of LDL receptors [20].
The blood brain barrier (BBB) limits the transport of APOE leading to two different pools of APOE, one peripheral pool and one cerebral pool. In peripheral tissues, APOE is primarily produced by hepatocytes, macrophages and adipocytes [21, 22]. This review focuses on APOE in the brain, which is mainly generated and secreted by astrocytes, but to some extent by microglia and neurons [23]. APOE belongs to the family of apolipoproteins, which combine lipids to form lipoproteins that are responsible for packaging and transporting cholesterol and other phospholipids. In brain tissue, APOE has many physiological functions such as cellular lipid transport, and is involved in neuronal growth, synaptic plasticity, transcriptional regulation, energy metabolism and immune response [24,25,26,27].
While the APOE effect on several AD-related pathologies, such as Aβ generation and clearance [28, 29] or tau-induced neurodegeneration, neuroinflammation and BBB disruption [30], has already been extensively studied (for details see [31, 32]), the knowledge on the role of different APOE isoforms on brain energy metabolism is limited. This review highlights the role of APOE in cerebral energy metabolism and how this might contribute to etiology and pathophysiology of AD.
Metabolic changes in Alzheimer’s disease
Using various techniques such as magnetic resonance imaging (MRI) and positron emission tomography (PET), it is possible to identify brain atrophy and vascular injuries as well as changes in specific biomarkers. By applying these methods in combination with specific tracers, typical amyloid depositions and neurofibrillary tangles (NFTs) can reliably be visualized in mild cognitive impairment (MCI) subjects, a prodromal stage of AD, and early-stage AD patients [33,34,35,36]. Amyloid PET imaging showed that APOE4 carriers have a higher amyloid burden and a faster Aβ deposition rate compared to non-carriers, suggesting APOE4 increases the risk for brain amyloidosis [37,38,39,40,41]. The APOE genotype is important to consider when interpreting amyloid PET results since the presence of at least one APOE4 allele can predict a positive amyloid PET image in amnestic MCI patients [36, 42]. A gene-dose effect of APOE genotype was observed with APOE4 carriers showing higher rates of cerebral amyloid while APOE2 carriers displayed no increase, which was in agreement with the observed decreased CSF Aβ42 levels in APOE4 carriers and increased CSF Aβ42 levels in APOE2 carriers [43]. Furthermore, APOE4 carriers showed higher tau burden in the lateral, and medial temporal, cingulate, and insula cortices compared to non-carriers [37].
While Aβ depositions and tau accumulation are the major hallmarks of AD, abnormalities in cerebral glucose metabolism in specific brain areas forecast the diagnosis of AD based on histopathological changes and can even be correlated with the disease progression [44, 45]. Cerebral glucose metabolism can be detected by [18F]fluorodeoxyglucose (FDG)-PET, an imaging technique based on the uptake of a radiolabeled glucose analogue. Thereby, reduced glucose metabolism (hypometabolism) was detected in AD brains, especially in the temporoparietal cortex including the precuneus and posterior cingulate cortex (Fig. 2) [46,47,48]. A negative correlation was observed between amyloid depositions and glucose metabolism bilaterally in the temporal and parietal lobes, brain areas that showed glucose hypometabolism in AD subjects [49]. Adding FDG-PET as a clinical tool improves the accuracy and confidence of diagnosing AD, indicating that glucose hypometabolism plays an essential role in AD pathogenesis [46, 50]. Additionally, FDG-PET is useful for staging the disease and was found to be more reliable in predicting the conversion of MCI to AD than MRI or CSF biomarkers [51,52,53]. FDG-PET even provides a higher predictive value compared to the amyloid PET, however, the combination of both methods provides the most accurate prediction of the conversion from MCI to AD [54, 55].

FDG-PET (upper row,) β-amyloid-PET (second row), tau-PET (third row) and structural MRI images in the respective horizontal (left), sagittal (middle) and coronal (right) view from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. AD subjects show reduced FDG-PET intensity, increased amyloid- and tau-PET signals as well as brain atrophy in MRI. HCS: Female, 67 y, MMSE 30, APOE3/3, amyloid tracer: [18F]florbetapir, tau tracer: [18F]flortaucepir; AD subject: Male, 75 y, MMSE 20, APOE3/4, amyloid tracer: [18F]florbetaben, tau tracer: [18F]flortaucepir; Colorbars: Standard visual display with comparable reference tissue color level.
Brain energy metabolism
Insulin signaling
Type 2 diabetes mellitus (T2DM) is another risk factor for cognitive decline and late-onset AD. It is a metabolic disorder characterized by insulin resistance, impaired insulin signaling and glucose intolerance. The link between dementia including AD and diabetes has been shown [56,57,58,59,60].
Insulin is a peptide hormone which is secreted from beta cells located in the pancreas as a response to increased glucose levels in the blood. The insulin receptor (IR) can be found in various regions and most cell types of the developing and adult brain. It is still controversial whether insulin can also be produced in the brain [61]. By passing the BBB, peripheral-produced insulin can enter the central nervous system (CNS) where it regulates food intake but also affects cognitive functions such as memory. On the cellular level, insulin modulates multiple essential mechanisms [62]. In neurons, insulin regulates synaptic plasticity by enhancing neurite outgrowth, regulating the expression of NMDA, AMPA and GABA receptors and inhibiting apoptosis to promote neuronal survival. Even though IRs are more concentrated in neurons, they are also expressed in 20–40% of glia cells which provide support for neuronal cells in the brain. In astrocytes, IRs are involved in brain glucose sensing and in the entry of glucose and insulin into the brain [63].
Activation of the insulin signaling pathway starts with insulin binding to the alpha subunit of the IR located at the plasma membrane (Fig. 3) [64] Binding of insulin causes a conformational change and autophosphorylation of the intracellular β subunit of the IR leading to the endocytosis of the insulin-IR complex. At the same time, autophosphorylation of the IR enables growth factor binding protein 2 (GRB2) to bind to the Src homology 2 (SH2) domain of the phosphorylated tyrosine residues of the IR. This activates the downstream cascade of RAS, RAF and MEK1 kinase and finally ERK1 and ERK2, two MAP kinase isoforms. Next to the activation of the MAP kinase pathway, insulin also induces the AKT/PI3 kinase pathway. Phosphorylated IR substrate proteins interact with phosphatidylinositol 3-kinase (PI3K), which then catalyzes the formation of phosphatidylinositol (3,4)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) [64]. As a result, 3-phosphoinositide-dependent protein kinase 1 (PDK1) is recruited which phosphorylates and activates AKT. AKT is a key regulator in the insulin signaling pathway and regulates several downstream proteins, such as the transcription factor FOXO1, glycogen synthase kinase 3 (GSK3) as well as mTOR and Forkhead transcription factors [65, 66]. While the MAP kinase pathway promotes mitogenic effects, the AKT/PI3 kinase pathway is involved in metabolic effects such as glucose uptake, glycogen synthesis and gluconeogenesis [67].

The figure illustrates the insulin signaling pathway and and the various sites of action through which APOE exerts its influence. LRP1 LDL-related protein 1, PIP2 phosphatidylinositol (3,4)-bisphosphate, PIP3 phosphatidylinositol (3,4,5)-trisphosphate, PDK1 3- phosphoinositide-dependent protein kinase 1, PI3K phosphatidylinositol 3-kinase, IR Insulin receptor, SHC Src homology collagen, SOS Son of Sevenless, GRB2 growth factor receptor-bound protein 2, MEK mitogen-activated protein kinase kinase, ERK extracellular signal-regulated kinase.
APOE influences insulin signaling at different levels. Sex as well as APOE genotype-based differences were observed in insulin BBB pharmacokinetics with APOE4 mice displaying higher insulin binding to brain vasculature than APOE3 mice [68]. Sex and APOE4 genotype were also found to influence CSF insulin levels. Women and APOE4 non-carriers had higher levels of CSF insulin which was associated with worse cognitive performance and higher CSF levels of (p-)tau indicating cerebral insulin resistance and impaired cerebral insulin signaling [69]. However, research have been mainly focusing on APOE3 and E4 carriers, leaving open questions about the effect of APOE2 on insulin signaling. High fat diet (HFD)-induced insulin resistance in female homozygous human APOE3 and APOE4 targeted replacement mice led to alterations in purine metabolism, glutamate metabolism and the pentose phosphate pathway (PPP). Further, cognitive deficits in hippocampal-dependent spatial learning and memory after HFD were enhanced in APOE4 mice [70]. APOE4, but not APOE3, also impaired insulin-stimulated mitochondrial respiration and glycolysis [71]. Protein expression of insulin signaling pathways and mitochondrial electron transport chain was changed in APOE4 mice [72]. LRP1, a major APOE receptor, interacts with the IR β-subunit in the brain. Deficiency of LRP1 resulted in impaired insulin signaling via reduced levels of IRβ in the presence but also absence of insulin and consequently also reduced levels of phosphorylated AKT (p-AKT). Additionally, in the absence of insulin the levels of LRP1 were downregulated. Treatment with insulin resulted in higher presentation of LRP1 at the cell surface [73]. LRP1 also regulates hepatic insulin response and somatic inactivation of LRP1 has been linked to insulin resistance [74].
Further, APOE can modulate insulin signaling directly. Recombinant APOE binds to IRs, with APOE4 showing a higher affinity than APOE3 resulting in less insulin-IR interaction with 50% reduction in the presence of APOE4 and 25% less insulin binding in the presence of APOE3. Interestingly, binding between APOE4 and IR increased with the age of the mice [71]. APOE4 suppresses activation of insulin signaling by blocking the interaction between insulin and IR. Thereby, IR internalization and signaling activation is downregulated. Further, APOE4 impairs IR trafficking by trapping it in the endosomes which leads to downregulation of IRs at the cell surface [71]. Bioinformatic analysis revealed insulin signaling pathway key molecules being affected in AD patients of which the six kinases, leucine-rich repeat kinase 2 (LRRK2), glycogen synthase kinase 3 beta (GSK3B), AKT serine/threonine kinase 1 (AKT1), epidermal growth factor receptor (EGFR), mitogen-activated protein kinase 1 (MAPK1), and the non-receptor tyrosine kinase FYN, played central roles [56]. In addition to the interaction with IRs, APOE can also activate the AKT pathway by direct binding to AKT. Interestingly, AKT is phosphorylated in an APOE isoform dependent manner [75, 76]. APOE can induce AKT phosphorylation via two pathways. In the first, APOE binds to heparan sulfate proteoglycan/ LDL receptor-related protein complex. This pathway is only activated by APOE3 and APOE4 but not by APOE2. The second and later activated pathway is mediated by calcium and also APOE genotype dependent [76]. Insulin treatment leads to activation of AKT pathway and higher phosphorylation of tau (at Ser202/ Thr205) in APOE4 mice compared to APOE3 mice, indicating that APOE4 potentiates insulin-dependent activation of AKT pathway and tau hyperphosphorylation in the brain as observed in APOE4 carriers [77]. APOE4 reduces brain insulin signaling by lowering insulin receptor substrate 1 (IRS1) at S636/639 and AKT phosphorylation at T308 which was linked to lower brain glucose in fasting human APOE4 targeted replacement mice and with increasing age reduction in IRS2 expression, IRS1 phosphorylation at Y608, AKT phosphorylation at S473 and MAPK activation [78].
APOE has been shown to activate the PI3K/ AKT pathway to modulate food intake [79]. APOE4, bound to VLDL, inhibited the apoptotic pathway normally activated by HDL by reducing the phosphorylation of AKT through inhibition of PIP3. APOE4-VLDL inhibited PIP3 via SHIP2, known as risk factor for T2DM [80]. PDK1 mutant mice with reduced ability of AKT activation displayed enhanced membrane localization and activity of α-secretase and reduced neuronal resistance against neurotoxicity indicating that the AKT pathway also plays an important role in Aβ pathology. At the same time, PDK1 knock-in mice showed protection of neurons against ER stressors [81]. Pre-treatments with APOE2 and APOE3 reduced stress-induced neuronal apoptosis by regulating the AKT/ FOXO3a/ BIM signaling pathway, while no neuroprotective effect was found with APOE4 with higher concentrations of APOE4 showing a rather toxic effect [75]. These studies indicate that insulin signaling plays a crucial role in AD pathology revealing key elements such as the AKT pathway. However, more research is needed to decipher the exact mechanisms and how APOE isoforms influence key elements in these signaling cascades.
Glycolysis
The human brain is a highly metabolically active organ that requires a constant delivery of fuels and oxygen to maintain its function. While the adult brain can utilize lactate and ketone bodies during special situations like exercise and fasting, its primary substrate is glucose [82]. This is taken up into the cytosol by highly efficient glucose transporters (GLUTs). In the cytosol, glucose is either stored as glycogen or further metabolized via glycolysis, the stepwise conversion to pyruvate. The presence of oxygen stimulates pyruvate dehydrogenase (PDH) to convert pyruvate to acetyl coenzyme A (Acetyl-CoA) in the mitochondria. Acetyl-CoA subsequently enters the tricarboxylic acid (TCA) cycle, also known as Krebs cycle, which finally produces NADH that feeds the electron transport chain (ETC) for oxidative phosphorylation (OXPHOS) to produce energy in the form of ATP. Under low oxygen conditions, cells depend on anaerobic glycolysis, in which pyruvate is converted to lactate by lactate dehydrogenase (LDH). This reaction is coupled to the conversion of NADH+H+ into NAD+ and is reversible, allowing cells to either produce or consume lactate dependent on their metabolic needs [83].
Glucose hypometabolism has been demonstrated as a common phenotype in MCI and AD, especially in APOE4 carriers. The metabolism of glucose is one of the processes that becomes dysfunctional at an early preclinical stage and correlates to cognitive impairment [84]. Elevated glucose concentration in the brain and reduced glycolytic flux are related to the severity of AD pathology and the establishment of clinical symptoms [85]. In recent years, the role of glycolysis in AD pathology has become more and more prominent. Analysis of glycolytic metabolites in the CSF of AD subjects showed that 5 of 11 metabolites, including phosphoenolpyruvate (PEP), 2-phosphoglycerate (2PG), 3-phosphoglycerate (3PG), pyruvate and dihydroxyacetone phosphate (DHAP), were significantly different from cognitively healthy non-AD controls. Except for pyruvate, a positive correlation with Aβ1-42 and Aβ1-42/Aβ1-40 was found [86]. Aβ resistant nerve cells have been shown to upregulate PDK1 expression and LDHA activity and subsequently lactate production a phenomenon called Warburg effect by which cells favor aerobic glycolysis over oxidative phosphorylation for energy production, even in the presence of oxygen, impacting metabolic efficiency [87]. This indicates that the Warburg effect is a contributing factor to support neuronal survival in the AD brain [88].
Different brain cell types display distinct metabolic phenotypes. Glucose can be taken up directly by neurons and used for oxidative phosphorylation. While neurons generally show high oxidative metabolism, astrocytes rather display glycolytic activity. Thus, glucose can be taken up by astrocytes and converted to lactate. According to the astrocyte-to-neuron lactate shuttle (ANLS) hypothesis, astrocytes are the primary producers of lactate which is then shuttled to neurons via monocarboxylate transporters (MCTs) and metabolized [89].
Glycolytic alterations in neurons
The impact of Aβ on glycolysis and the relation between enhanced neuronal glycolysis and Aβ protection has been shown [85, 88, 90,91,92]. However, APOE genotypes have major impacts on neuronal glycolysis as well (Fig. 4). Lactate production was shown to be higher in differentiated N2a-APOE2-expressing cells and lower in N2a-APOE4-expressing cells compared to N2a-APOE3-expressing cells [93]. Neuro 2A (N2a) is a mouse neuroblastoma cell line with neural precursor cell properties. An APOE4-induced glycolytic phenotype became more severe in aged N2a cells and older primary astrocytes, while N2a-APOE2-expressing cells displayed more robust glycolytic activity [94, 95].

The figure illustrates the impact of APOE on metabolic pathways in astrocytes and neurons. The corresponding publications are cited in the figure. ACoA acetyl co-enzyme A, TCA tricarboxylic acid, GLUT1, GLUT3 glucose transporters GLUT1 and GLUT3; MCT2, MCT4 monocarboxylate transporters MCT2 and MCT4, PPP pentose phosphate pathway, PYR pyruvate, LD lipid droplets, β-OX beta oxidation.
When combining APOE2- and APOE4-expressing cells, an increase in APOE protein levels was observed together with enhanced glycolytic function based on elevated basal glycolytic rate and maximum glycolytic capacity. This effect was only found with APOE2 and not APOE3 indicating a neuroprotective function for APOE2 [95]. ATP can be released by astrocytes to mediate neuronal activity and ATP levels were shown to be higher in N2a-APOE2-expressing cells compared to N2a-APOE3-expressing cells supporting the hypothesis that APOE2 provides better neuronal support [93].
APOE4-associated deficits in glycolysis were compensated by enhancing the activity of peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1), a key regulator of energy metabolism, which might be due to an increase hexokinase 1 (Hk1) expression [93]. Hexokinases are the initial enzymes of glycolysis, converting glucose to glucose 6-phosphate (G-6-P). Hk1 is mainly associated with mitochondria exclusively promoting glycolysis, whereas Hk2 translocate between mitochondria and cytosol mediating glycolysis when bound to mitochondria and glycogen synthesis when located in the cytosol [96]. APOE4 leads to reduced Hk1 and Hk2 protein expression and impaired functional activity as well as the upregulation of two other key rate-limiting enzymes of the glycolysis, phosphofructokinase (PFK) and pyruvate kinase M1 (PKM1) and deficient glycolytic activity. Gene expression profiles of Hk1 and Hk2 were significantly downregulated in APOE4 brains, similar to Slc2a4 which encodes a protein that functions as an insulin-regulated facilitative glucose transporter. In contrast, an increased Hk protein expression and enzymatic activity as well as basal and maximum glycolytic flux and ATP production was observed in APOE2 transgenic mice [93]. Additionally, APOE2-expressing cells show downregulation of PFK and PKM1. Interestingly, when combining APOE2- and APOE4-expressing N2A cells, APOE2 mitigates the APOE4-induced hexokinase impairments [95].
Alterations in glycolysis by APOE4 were observed in an insulin-dependent manner [71]. Increased glycolytic activity was associated with elevated energy-related metabolite D-Fructose 6-phosphate and TCA cycle metabolites malate, citrate and isocitrate in APOE4 mice compared to APOE3 [97]. Insulin treatment leads to an increase in glycolytic rate and capacity in APOE -/- cells and treatment with APOE4 inhibited this increase in glycolysis, whereas treatment with APOE3 had no effect on insulin-induced glycolysis [71]. However, not only APOE but also LRP1 can interact directly with the insulin receptor and thereby regulate insulin signaling and glucose uptake. Higher glucose levels suppress LRP1 expression, and the absence of insulin downregulates levels of LRP1. LRP1 downregulation in neurons leads to reduced levels of GLUT3 and GLUT4 and consequently in reduced glucose uptake. In mice, LRP1 deficiency causes glucose intolerance in the brain and hyperglycemia was observed to suppress LRP1 expression, finally causing insulin resistance and glucose intolerance [73]. Interestingly, LRP1 is a major GLUT4 storage vesicle cargo protein necessary for functional GLUT4 storage vesicle trafficking. LRP1 interacts with the luminal domains to constituents of the vesicle including GLUT4 and with the cytoplasmic tail to AKT substrate of 160 kDa (AS160), an insulin-signaling pathway target. LRP1 deficiency reduces GLUT4 expression and decreases insulin-dependent glucose uptake [98]. Even though APOE isoforms did not cause changes in the expression levels of major glucose transporters of the brain, GLUT1, GLUT3 and GLUT4 [95], APOE can indirectly alter glucose uptake in an isoform-dependent manner.
As an alternative energy source, ketone bodies can provide energy in times of starvation. Ketone bodies include β-hydroxybutyrate (BHB), acetoacetate (ACA) and acetone can cross the BBB and ketones are rapidly metabolized by neurons. They can bypass glucose metabolism by entering the TCA cycle in the mitochondria for NADH synthesis and subsequently oxidation of the ETC and ATP synthesis [99]. In contrast to non-APOE4 carriers, elevated ketone body levels do not show cognitive benefits in APOE4 carriers indicating that APOE4 might impact the transport of ketone bodies executed by MCTs or the metabolism of ketone bodies in the mitochondria [100, 101]. Interestingly, cells expressing APOE2 or APOE4 both demonstrated a robust profile in uptake and metabolism of ketone bodies compared to APOE3 which showed less efficient ketone body utilization [93].
Glycolytic alterations in astrocytes
Neurons and astrocytes are tightly metabolically coupled and therefore dysfunction in astrocyte physiology has a great impact on the vitality of neurons [102].
Earlier studies showed that the presence of the E4 allele is associated with altered glucose metabolism [103, 104]. Astrocytes, known to be the major source of APOE production in the brain, displayed the highest glycolysis gene expression [105]. APOE4 astrocytes showed deficits in glucose uptake capability but increased glycolytic activity and decreased oxygen consumption compared to APOE3 [105, 106]. An APOE-directed global metabolic shift at the cost of glucose metabolism towards lipid oxidation and enhanced thermogenesis was observed in humanized APOE4 mice [107].
APOE2 astrocytes showed increased glucose uptake, higher maximum glycolytic capacity and glycolytic reserve compared to APOE3 cells [93]. APOE2 astrocytes also had a higher glycolytic flux whereas APOE4 astrocytes cycled more glucose through the PPP, a major regulator of redox homeostasis and biosynthesis, compared to APOE3 [106]. Treatment with Aβ led to an increase in astrocytic glycolytic activity, independent of the APOE isoform, however APOE4 astrocytes had a stronger increase in glycolytic ATP production compared to APOE3 astrocytes with no differences in the glycolytic output [108].
The production of lactate during glycolysis also dependent on the APOE isoform. Lactate synthesis from glucose as well as the total amount of NADH was higher in APOE4 compared to APOE3 and APOE2 [105, 106]. APOE4 was shown to increase LDH enzyme activity, whereas pyruvate entry into the TCA cycle was decreased and PDH enzyme activity was not altered indicating a shift towards aerobic glycolysis [105]. Compared to APOE2 and APOE3, APOE4 lead to an increase in the two main enzymes that control the entry of pyruvate into the TCA cycle, pyruvate carboxylase (PC) and PDH [106]. Interestingly, this shift towards aerobic glycolysis was also observed in humans. Decreased energy expenditure driven by a lower rate of oxygen consumption and increased lactate plasma levels was found in female APOE4 carriers compared to E4 non-carriers [105].
Mitochondrial function
Mitochondria are essential for energy metabolism but also crucial for producing reactive oxygen species (ROS) to activate transcription factors and the release of cytochrome c to induce cell death [109]. Mitochondrial dynamics are regulated by the balance of continuous fission and fusion events and maintain mitochondrial shape, distribution, size and function [110].
Mitochondria are highly dynamic organelles that produce most of the chemical energy to cover the bioenergetic needs of the cell. In the mitochondrial cristae, a series of metabolic reactions and processes convert energy that is stored in macronutrients to the universal energy donor ATP. Enzymatic reactions in the TCA cycle, also known as Krebs or citric acid cycle, produce FADH2 and NADH that are required for the transfer of electrons in the ETC. This is composed of four major protein complexes (I-IV) that generate a charge and proton gradient across the inner mitochondrial membrane (IMM) which drives the ATP synthesis in complex V, known as ATP synthase. This process requires oxygen as a final electron acceptor and is called OXPHOS [109, 111].
Mitochondrial dysfunction presents a risk factor for neurodegenerative diseases including AD. The close link between early mitochondrial dysfunction and AD pathology has been demonstrated (for details see [112]) and research to untangle the role of mitochondria in AD pathology is still ongoing. Recently, the discovery of new mitochondrial components such as SHMOOSE, a small reading frame encoded microprotein, or a mitochondrial protein called cell-cycle exit and neuronal differentiation 1 (CEND1) that have been linked to AD pathology has been reported [113, 114].
Early mitochondrial dysfunction can be observed in AD patients [115]. In AD patient brains, the activity of the mitochondrial enzyme α-ketoglutarate dehydrogenase complex (KGDHC) correlates better with the clinical dementia rating than amyloid plaques or NFTs. This was especially found for APOE4 carriers, indicating that mitochondrial dysfunction might play an even more important role in subjects carrying the APOE4 allele [116]. Before the onset of any cognitive changes and amyloid or NFT pathology, APOE4 carriers display mitochondrial dysfunction especially in brain areas affected in AD supporting the hypothesis that mitochondrial dysfunction plays an essential role in early development of AD pathology [117].
Further, APOE4 carriers show downregulation of proteins responsible for mitochondrial biogenesis compared to non-APOE4 carriers. PGC-1α and NAD-dependent deacetylase sirtuin-3 (SIRT3) as well as the dynamic proteins mitofusin 1 (MFN1), mitofusin 2 (MFN2), and dynamin like protein 1 (DLP1) were all decreased in APOE4 carriers. These proteomic changes could be observed in AD compared to healthy control post-mortem brain tissue but not in MCI. APOE4 carriers display decreased levels of antioxidative stress response proteins such as superoxide dismutase 2 (SOD2) and forkhead-box protein O3a (Foxo3a) compared to non-APOE4 carriers.
Mitochondrial alterations in neurons
Mitochondrial dysfunction is closely connected with AD pathology which has been primarily studied in neurons. ETC gene- and protein expression were altered in AD patients [118] and AD patient-derived neuronal cells [119]. Mitochondrial fission protein dynamin related protein 1 (DRP1) was shown to interact with Aβ, a process that increases with disease progression. Partial deficiency of DRP1 reduced Aβ production, mitochondrial dysfunction, maintained mitochondrial dynamics and increased mitochondrial biogenesis as well as synaptic activity in APP mice indicating that inhibition of DRP1-Aβ interactions protects Aβ-induced mitochondrial dysfunctions [120].
Furthermore, PGC-1α and PPAR-γ, both crucial factors in mitochondrial biogenesis, are implicated in Aβ generation and aggregation [121,122,123]. For example, PGC-1α, a co-factor for transcription factors including PPAR-γ, regulates the transcription of the β-APP cleaving enzyme BACE1. Both PGC-1α and PPAR-γ regulate mitochondrial dynamics and their levels were decreased in AD patients. Agonist-induced activation of PPAR-γ resulted in mitochondrial protection against oxidative stress by modulating mitochondrial fusion and fission events in hippocampal neurons [124].
The impact of APOE on neuronal energy metabolism has already been shown. APOE4 expression reduced protein expression of all subunits of mitochondrial respiratory complexes in neurons cultured from APOE transgenic mouse brains and N2A cells [125]. APOE4 neurons showed reduced expression of mitochondrial biogenesis regulators, PGC1α and PGC1β, as well as reduced maximal mitochondrial respiration and lower OXPHOS capacity. APOE4 neurons had smaller and more dispersed mitochondria in the cytoplasm compared to APOE3 neurons [126]. Enzymatic activity of complex IV was also shown to be decreased in APOE4 N2A cells. Interestingly, these reductions were only observed in neurons and not in astrocytes, indicating that APOE4 might have a different effect on neuronal mitochondria than on astrocytic mitochondria [125]. Mitochondrial dysfunction was not affected in mutant N2a-APOE4-expressing cells that lack the intramolecular interaction between the amino-terminal domain (Arg-61) and the carboxyl-terminal domain (Glu-255). Levels of complex IV were even restored in N2a cells with the treatment of a small molecule that disrupts the domain interaction, indicating that this domain interaction within APOE4 plays a crucial role in mediating mitochondrial dysfunction [125]. ETC complexes can be targeted directly by C-terminal-truncated APOE4 fragments and overexpressing APOE4 (1–272) fragment affects their activities resulting in altered mitochondrial function in neuronal cells [127].
APOE4 causes impairments in insulin signaling and a link between brain insulin signaling and mitochondrial function is evident. RNA sequencing analysis on endothelial monolayers showed that mitochondrial function is one of insulin-responsive pathways in the BBB endothelium [66]. Treatment with insulin increased basal and maximal mitochondrial respiration, glycolysis rate and glycolytic capacity in APOE-/- neurons and neurons treated with recombinant APOE3, while recombinant APOE4 inhibited insulin’s ability to increase mitochondrial respiration and glycolysis. This was also observed for lipidated APOE4 secreted from astrocytes, indicating that APOE4 but not APOE3 suppresses insulin-stimulated mitochondrial respiration and glycolysis in neurons [71]. Changes in proteins involved in insulin signaling and mitochondrial ETC were also observed in APOE4 mice. Most prominent differences were found in mitochondrial signaling including higher expression of complexes I, II and III which stands in contrast to other studies reporting reduced expression of all subunits of mitochondrial respiratory complexes in neurons cultured from APOE transgenic mouse brains as discussed above. Further, increased expression of pErk1/2 but decreased pGSK3β levels in APOE transgenic mice were observed while the pAKT/AKT ratio was not affected [72].
A study based on fMRI, electrophysiology and metabolomics showed that APOE4 disrupts mitochondrial function by causing a hyperactivity phenotype in the brains of aged mice, especially in the entorhinal cortex. This neuronal hypermetabolism was supported by the APOE4-induced increase of mitochondrial metabolites that are involved in cellular energy metabolism including the TCA cycle metabolites malate, citrate and isocitrate as well as carnitine, an important metabolite for the transport of FAs into the mitochondrial matrix for energy usage via β-oxidation. Additionally, APOE4 caused an upregulation of ATP and upregulation of several metabolites that are involved in mitochondria-related ammonia regulation such as leucine, inosine monophosphate and succinoadenosine [97]. This is in agreement with increased ATP production and OXPHOS observed in APOE4 iPSC-derived neurons, compared to APOE2 and APOE3 [128].
Mitochondrial alterations in astrocytes
Mitochondrial impairments in neurons and astrocytes intricately converge within the complex pathogenesis of AD, highlighting the interconnected nature of these cellular processes in both cell types in AD. APOE4-driven astrocyte TCA cycle dysfunction in AD may impair neurotransmitter recycling, potentially contributing to synaptic dysfunction and neurodegeneration [129].
Dysfunctions in ETC have been shown to induce a 49-fold upregulation of APOE transcript and protein as well as APOE secretion in diverse cells including iPSC-derived human astrocytes. Further, a correlation between APOE expression and ETC subunits was shown in the 5xFAD mouse model, indicating a direct link between APOE expression and mitochondrial function [130].
Mitochondria in APOE4 astrocytes were more fragmented and formed less tubule structures. The fission protein DRP1 was upregulated, but no change in levels of the fusion protein OPA1 was observed. When adding APOE3 astrocytes to APOE3 neuronal cultures, mitochondrial function and aerobic glycolysis was increased. Adding APOE4 astrocytes however caused a shift towards glucose metabolism which was demonstrated by a higher basal respiration on glucose, a higher glycolytic rate, increased expression of multiple OXPHOS subunit complexes, higher levels of ATP and lower AMPK activation [126]. These studies indicate that APOE4 induces mitochondrial fission and a shift towards increased basal glucose metabolism.
Elevated function of mitochondria-associated ER membranes (MAM), a subdomain of the ER involved in lipid and cholesterol homeostasis, have been shown in AD patients. Fibroblasts treated with APOE4-conditioned astrocyte medium displayed an increase in ER-mitochondrial communication and MAM function compared fibroblasts treated with APOE3-containing medium, indicating that mitochondrial dysfunction affects multiple cellular communication pathways. Interestingly, this effect was only observed with lipoprotein-enriched but not unlipidated APOE protein [131].
APOE is also implicated in mitochondrial fatty acid (FA) metabolism. Active neurons produce toxic FAs but have a low capacity to store or consume them. Thus, they release these FAs, which can be taken up and metabolized by astrocytes. Lindner and colleagues showed that APOE4 astrocytes with ectopic accumulation of fat, a stress-associated condition, had a reduced capacity to clear potentially neurotoxic FAs because APOE4 reroutes them to secretion [132].
Increased accumulation of damaged mitochondria in the lysosome was linked with lysosomal dysfunction which we earlier showed to be affected in APOE4 astrocytes [24]. APOE4 causes cholesterol accumulation which hinders the lysosomal-dependent clearance of damaged mitochondria resulting in a shift in energy production with increased glycolysis but impaired mitochondrial function in human iPSC-derived APOE4 astrocytes [133]. The main type of mitochondrial damage in APOE4 transgenic mice was mitochondria-derived lysosomes associated with lipofuscin [134]. Mitochondrial function and cell survival could be improved by reducing DRP1/FIS1-mediated mitochondrial damage in human cardiomyocytes. Treatment with P110, a selective inhibitor for FIS1-mediated DRP1 recruitment to the mitochondria, enhanced mitochondrial structure [135].
Mitochondrial function is closely related with fatty acid metabolism, LD lipolysis, autophagy and glycolysis which could be especially observed in astrocytes. However, defects in mitochondrial metabolism also affect neuronal functions including synaptic function, oxidative stress, and finally cognitive function. APOE4 disrupts mitochondrial structure and function in the brain by targeting several mechanisms making it even more challenging for single target treatment approaches. A limitation of most of these studies, however, is the focus on APOE3 and E4 genotypes, leaving open the questions on how APOE2 affects mitochondrial function.
Hyper- and hypometabolism: a causal dilemma
Increased brain metabolism (hypermetabolism) has been observed in the brain of individuals with MCI [136, 137]. Longitudinal FDG PET imaging in AD patients revealed hypermetabolism 25 years before symptoms onset. Remarkably, hypermetabolism was detected in brain regions that were hypometabolic at later stages of the disease [138]. However, the role of glucose metabolism in disease pathophysiology and whether it is cause or consequence, remains unclear.
Interestingly, APOE4 has been linked to increased brain metabolism and activity, especially in young individuals. Functional MRI analysis in 20–35-year-old healthy individuals showed greater task-related activation in APOE4 carriers relative to non-carriers in hippocampal regions [139]. Further, young APOE4 carriers exhibited greater hippocampal activity relative to non-carriers while accomplishing the same encoding task [140]. This indicates that APOE4 influences brain activity and metabolism decades before the onset of clinical symptoms or pathological hallmarks. This is supported by studies in APOE4 knock-in mice showing brain hypermetabolism in the absence of neurodegeneration [97, 141].
While this appears to contradict the reports of a hypometabolism in AD patients, it may be explained by an age- and pathology-dependent APOE effect on the brain. Increased metabolic activity in young brains of APOE4 carriers may drive the classical AD pathology and contribute to the accumulation of β-amyloid aggregates and phospho-tau [97] leading to neuroinflammation, synapse loss and neurodegeneration. This may subsequently result in a hypoactive state once brain pathology has reached a certain threshold (Fig. 5). Natural aging may further support this metabolic switch. This hypothesis is further supported by a recent study showing that APP knock-in mice display mitochondrial hypermetabolism before onset of pathology. Upon increasing pathologies, such as neuroinflammation, synapse loss and amyloid plaque accumulation, the brain shifted to a state of hypometabolism [142]. A time-dependent metabolic shift was also shown for cultured human astrocytes. Upon exposure to Aβ, astrocytes initially enhanced mitochondrial fusion to stabilize their energy production and increased glycolytic ATP production. This however, changed with prolonged Aβ-mediated stress, and cells showed impaired mitochondrial function and a shift towards fatty acid β oxidation to maintain constant ATP levels [90]. We hypothesize that this metabolic shift is further exacerbated by the presence of APOE4, leading to the typical brain hypometabolism observed in AD patients.

In young, healthy APOE4 carriers, cellular energy metabolism is elevated, characterized by increased glucose uptake, enhanced glycolysis and elevated lactate release, alongside high mitochondrial respiration. However, over time and with increasing AD pathologies, this state may shift towards decreased energy metabolism, as cells become unable to sustain these processes. This decline may be further exacerbated by the presence of APOE4, further contributing to disease progression. MCT monocarboxylate transporters, MPC mitochondrial pyruvate carrier 2, Glu glucose, GLUT glucose transporter, TCA tricarboxylic acid cycle, Lac lactate.
Nevertheless, an APOE4-dependent metabolic switch during aging, from an early hypermetabolism to a disease-associated hypometabolism, needs to be confirmed by further research and requires more attention in order to better understand the APOE4-induced transition from a healthy to a diseased brain.
Conclusions
This review provides key insights into the critical role of energy metabolism in AD pathology, with a focus on the role of APOE, particularly APOE4, in metabolic dysfunction. It highlights how APOE4 impacts glucose metabolism, mitochondrial function and insulin signaling, all of which are central to AD pathophysiology. While most studies suggest hypometabolism linked to APOE4, especially in AD patients, others report increased metabolic activity. Primary discrepancies observed among studies are likely due to variations in experimental models and differences in the timing of analyses such as the age of human subjects, highlighting the need for further investigation.
We propose an APOE4-dependent metabolic switch, suggesting that early compensatory hypermetabolism transitions into hypometabolism as the disease progresses. It will be interesting to see whether this effect involves all neural cell types to a similar extent or whether it is cell type-specific. This shift could be a key factor in understanding disease progression and may offer valuable implications for early diagnosis and treatment strategies targeting metabolic dysfunctions in AD. APOE4’s role in energy metabolism is particularly significant, but more research is needed to fully elucidate the molecular mechanisms involved and to explore the effects of APOE2 and other recently identified protective APOE variants.
In conclusion, this review highlights the importance of further research into the metabolic aspects of AD, as understanding these processes could lead to more effective diagnostic approaches and treatments aimed at slowing or preventing disease progression.
References
Padamsey Z, Rochefort NL. Paying the brain’s energy bill. Curr Opin Neurobiol. 2023;78:102668.
World Health Organization (WHO). Key facts Dementia. 2025 https://www.who.int/news-room/fact-sheets/detail/dementia.
Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17:327–406.
de Leeuw SM, Tackenberg C. Alzheimer’s in a dish – induced pluripotent stem cell-based disease modeling. Transl Neurodegener. 2019;8:21.
Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P, Vargas-Rodríguez I, Cadena-Suárez AR, Sánchez-Garibay C, et al. Alzheimer’s disease: an updated overview of its genetics. Int J Mol Sci. 2023;24:3754.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3.
Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010;19:3295–301.
Grupe A, Abraham R, Li Y, Rowland C, Hollingworth P, Morgan A, et al. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum Mol Genet. 2007;16:865–73.
Michaelson DM. APOE ε4: the most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement. 2014;10:861–8.
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–81.
Fortea J, Pegueroles J, Alcolea D, Belbin O, Dols-Icardo O, Vaqué-Alcázar L, et al. APOE4 homozygosity represents a distinct genetic form of Alzheimer’s disease. Nat Med. 2024;30:1284–91.
Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA. 1997;278:1349–56.
Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11:667.
Chen Y, Strickland MR, Soranno A, Holtzman DM. Apolipoprotein E: structural insights and links to Alzheimer disease pathogenesis. Neuron. 2021;109:205–21.
Rall SC, Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 1982;257:4171–8.
Weisgraber KH, Rall SC, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem. 1981;256:9077–83.
Rebeck GW, LaDu MJ, Estus S, Bu G, Weeber EJ. The generation and function of soluble apoE receptors in the CNS. Mol Neurodegener. 2006;1:15.
Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med. 2016;94:739–46.
Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci USA. 1985;82:203–7.
Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–94.
Blumenfeld J, Yip O, Kim MJ, Huang Y. Cell type-specific roles of APOE4 in Alzheimer disease. Nat Rev Neurosci. 2024;25:91–110.
de Leeuw SM, Kirschner AWT, Lindner K, Rust R, Budny V, Wolski WE, et al. APOE2, E3, and E4 differentially modulate cellular homeostasis, cholesterol metabolism, and inflammatory response in isogenic iPSC-derived astrocytes. Stem Cell Rep. 2022;17:110–26.
Fernandez CG, Hamby ME, McReynolds ML, Ray WJ. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front Aging Neurosci. 2019;11:14.
Kim J, Yoon H, Basak J, Kim J. Apolipoprotein E in synaptic plasticity and Alzheimer’s disease: potential cellular and molecular mechanisms. Mol Cells. 2014;37:767–76.
Mahley RW, Rall SC. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37.
Huang YWA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell. 2017;168:427–41.e21.
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57.
Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20:68–80.
Chai AB, Lam HHJ, Kockx M, Gelissen IC. Apolipoprotein E isoform-dependent effects on the processing of Alzheimer’s amyloid-β. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866:158980.
Fernández-Calle R, Konings SC, Frontiñán-Rubio J, García-Revilla J, Camprubí-Ferrer L, Svensson M, et al. APOE in the bullseye of neurodegenerative diseases: impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol Neurodegener. 2022;17:62.
Alongi P, Sardina DS, Coppola R, Scalisi S, Puglisi V, Arnone A, et al. 18F-Florbetaben PET/CT to assess Alzheimer’s disease: a new analysis method for regional amyloid quantification. J Neuroimaging. 2019;29:383–93.
Ciarmiello A, Tartaglione A, Giovannini E, Riondato M, Giovacchini G, Ferrando O, et al. Amyloid burden identifies neuropsychological phenotypes at increased risk of progression to Alzheimer’s disease in mild cognitive impairment patients. Eur J Nucl Med Mol Imaging. 2019;46:288–96.
Miki T, Shimada H, Kim JS, Yamamoto Y, Sugino M, Kowa H, et al. Brain uptake and safety of flutemetamol F 18 injection in Japanese subjects with probable Alzheimer’s disease, subjects with amnestic mild cognitive impairment and healthy volunteers. Ann Nucl Med. 2017;31:260–72.
Higashi T, Nishii R, Kagawa S, Kishibe Y, Takahashi M, Okina T, et al. 18F-FPYBF-2, a new F-18-labelled amyloid imaging PET tracer: first experience in 61 volunteers and 55 patients with dementia. Ann Nucl Med. 2018;32:206–16.
Baek MS, Cho H, Lee HS, Lee JH, Ryu YH, Lyoo CH. Effect of APOE ε4 genotype on amyloid-β and tau accumulation in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:140.
Bouter C, Vogelgsang J, Wiltfang J. Comparison between amyloid-PET and CSF amyloid-β biomarkers in a clinical cohort with memory deficits. Clin Chim Acta. 2019;492:62–8.
Drzezga A, Grimmer T, Henriksen G, Mühlau M, Perneczky R, Miederer I, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72:1487–94.
Jack CR, Wiste HJ, Weigand SD, Rocca WA, Knopman DS, Mielke MM, et al. Age-specific population frequencies of cerebral β-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: a cross-sectional study. Lancet Neurol. 2014;13:997–1005.
Toledo JB, Habes M, Sotiras A, Bjerke M, Fan Y, Weiner MW, et al. APOE effect on amyloid-β PET spatial distribution, deposition rate, and cut-points. J Alzheimers Dis. 2019;69:783–93.
Risacher SL, Kim S, Shen L, Nho K, Foroud T, Green RC, et al. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E-MCI). Front Aging Neurosci. 2013;5:11.
Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31.
Mosconi L, Tsui WH, Herholz K, Pupi A, Drzezga A, Lucignani G, et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J Nucl Med. 2008;49:390–8.
Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36:811–22.
Foster NL, Heidebrink JL, Clark CM, Jagust WJ, Arnold SE, Barbas NR, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130:2616–35.
Mosconi L, Tsui WH, Pupi A, De Santi S, Drzezga A, Minoshima S, et al. 18)F-FDG PET database of longitudinally confirmed healthy elderly individuals improves detection of mild cognitive impairment and Alzheimer’s disease. J Nucl Med. 2007;48:1129–34.
Rabinovici GD. Late-onset Alzheimer disease. Continuum. 2019;25:14–33.
Chiaravalloti A, Castellano AE, Ricci M, Barbagallo G, Sannino P, Ursini F, et al. Coupled imaging with [18F]FBB and [18F]FDG in AD subjects show a selective association between amyloid burden and cortical dysfunction in the brain. Mol Imaging Biol. 2018;20:659–66.
Jagust W, Reed B, Mungas D, Ellis W, Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–7.
Caminiti SP, Ballarini T, Sala A, Cerami C, Presotto L, Santangelo R, et al. FDG-PET and CSF biomarker accuracy in prediction of conversion to different dementias in a large multicentre MCI cohort. NeuroImage Clin. 2018;18:167–77.
Stonnington CM, Chen Y, Savage CR, Lee W, Bauer RJ, Sharieff S, et al. Predicting imminent progression to clinically significant memory decline using volumetric MRI and FDG PET. J Alzheimers Dis. 2018;63:603–15.
Yuan Y, Gu ZX, Wei WS. Fluorodeoxyglucose-positron-emission tomography, single-photon emission tomography, and structural MR imaging for prediction of rapid conversion to Alzheimer disease in patients with mild cognitive impairment: a meta-analysis. AJNR Am J Neuroradiol. 2009;30:404–10.
Blazhenets G, Ma Y, Sörensen A, Schiller F, Rücker G, Eidelberg D, et al. Predictive Value of 18F-Florbetapir and 18F-FDG PET for conversion from mild cognitive impairment to Alzheimer dementia. J Nucl Med. 2020;61:597–603.
Frings L, Hellwig S, Bormann T, Spehl TS, Buchert R, Meyer PT. Amyloid load but not regional glucose metabolism predicts conversion to Alzheimer’s dementia in a memory clinic population. Eur J Nucl Med Mol Imaging. 2018;45:1442–8.
Alves SR, da Cruz e Silva C, Rosa IM, Henriques AG, da Cruz e Silva OAB. A Bioinformatics approach toward unravelling the synaptic molecular crosstalk between Alzheimer’s disease and diabetes. J Alzheimers Dis. 2022;86:1917–33.
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5:64–74.
Grünblatt E, Homolak J, Babic Perhoc A, Davor V, Knezovic A, Osmanovic Barilar J, et al. From attention-deficit hyperactivity disorder to sporadic Alzheimer’s disease-Wnt/mTOR pathways hypothesis. Front Neurosci. 2023;17:1104985.
Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6:551–9.
Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–7.
Dakic T, Jevdjovic T, Lakic I, Ruzicic A, Jasnic N, Djurasevic S, et al. The expression of insulin in the central nervous system: what have we learned so far? Int J Mol Sci. 2023;24:6586.
Stockhorst U, de Fries D, Steingrueber HJ, Scherbaum WA. Insulin and the CNS: effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol Behav. 2004;83:47–54.
García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell. 2016;166:867–80.
Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98:2133–223.
Scherer T, Sakamoto K, Buettner C. Brain insulin signalling in metabolic homeostasis and disease. Nat Rev Endocrinol. 2021;17:468–83.
Wang Z, Tang X, Swaminathan SK, Kandimalla KK, Kalari KR. Mapping the dynamics of insulin-responsive pathways in the blood-brain barrier endothelium using time-series transcriptomics data. NPJ Syst Biol Appl. 2022;8:29.
Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer. 2009;16:1103–23.
Rhea EM, Banks WA. Role of the blood-brain barrier in central nervous system insulin resistance. Front Neurosci. 2019;13:521.
Geijselaers SLC, Aalten P, Ramakers IHGB, De Deyn PP, Heijboer AC, Koek HL, et al. Association of cerebrospinal fluid (CSF) insulin with cognitive performance and CSF biomarkers of Alzheimer’s disease. J Alzheimers Dis. 2018;61:309–20.
Johnson LA, Torres ERS, Impey S, Stevens JF, Raber J. Apolipoprotein E4 and insulin resistance interact to impair cognition and alter the epigenome and metabolome. Sci Rep. 2017;7:43701.
Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, Knight JA, et al. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron. 2017;96:115–29.e5.
He K, Nie L, Zhou Q, Rahman SU, Liu J, Yang X, et al. Proteomic profiles of the early mitochondrial changes in APP/PS1 and ApoE4 transgenic mice models of Alzheimer’s disease. J Proteome Res. 2019;18:2632–42.
Liu CC, Hu J, Tsai CW, Yue M, Melrose HL, Kanekiyo T, et al. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J Neurosci. 2015;35:5851–9.
Ding Y, Xian X, Holland WL, Tsai S, Herz J. Low-density lipoprotein receptor-related protein-1 protects against hepatic insulin resistance and hepatic steatosis. EBioMedicine. 2016;7:135–45.
Gao H, Zheng W, Li C, Xu H. Isoform-specific effects of apolipoprotein e on hydrogen peroxide-induced apoptosis in human induced pluripotent stem cell (iPSC)-derived cortical neurons. Int J Mol Sci. 2021;22:11582.
Laffont I, Takahashi M, Shibukawa Y, Honke K, Shuvaev VV, Siest G, et al. Apolipoprotein E activates Akt pathway in neuro-2a in an isoform-specific manner. Biochem Biophys Res Commun. 2002;292:83–7.
Traversy MT, Vandal M, Tremblay C, Tournissac M, Giguère-Rancourt A, Bennett AD, et al. Altered cerebral insulin response in transgenic mice expressing the epsilon-4 allele of the human apolipoprotein E gene. Psychoneuroendocrinology. 2017;77:203–10.
Ong QR, Chan ES, Lim ML, Cole GM, Wong BS. Reduced phosphorylation of brain insulin receptor substrate and Akt proteins in apolipoprotein-E4 targeted replacement mice. Sci Rep. 2014;4:3754.
Shen L, Wang DQH, Tso P, Jandacek RJ, Woods SC, Liu M. Apolipoprotein E reduces food intake via PI3K/Akt signaling pathway in the hypothalamus. Physiol Behav. 2011;105:124–8.
DeKroon R, Robinette JB, Hjelmeland AB, Wiggins E, Blackwell M, Mihovilovic M, et al. APOE4-VLDL inhibits the HDL-activated phosphatidylinositol 3-kinase/Akt Pathway via the phosphoinositol phosphatase SHIP2. Circ Res. 2006;99:829–36.
Yang S, Pascual-Guiral S, Ponce R, Giménez-Llort L, Baltrons MA, Arancio O, et al. Reducing the levels of Akt activation by PDK1 knock-in mutation protects neuronal cultures against synthetic amyloid-beta peptides. Front Aging Neurosci. 2017;9:435.
Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–97.
Ruminot I, Schmälzle J, Leyton B, Barros LF, Deitmer JW. Tight coupling of astrocyte energy metabolism to synaptic activity revealed by genetically encoded FRET nanosensors in hippocampal tissue. J Cereb Blood Flow Metab. 2019;39:513–23.
Berlanga-Acosta J, Guillén-Nieto G, Rodríguez-Rodríguez N, Bringas-Vega ML, García-del-Barco-Herrera D, Berlanga-Saez JO, et al. Insulin resistance at the crossroad of Alzheimer disease pathology: a review. Front Endocrinol. 2020;11:560375.
An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 2018;14:318–29.
Bergau N, Maul S, Rujescu D, Simm A, Navarrete Santos A. Reduction of glycolysis intermediate concentrations in the cerebrospinal fluid of Alzheimer’s disease patients. Front Neurosci. 2019;13:871.
Barros LF, Ruminot I, San Martín A, Lerchundi R, Fernández-Moncada I, Baeza-Lehnert F. Aerobic glycolysis in the brain: warburg and crabtree contra pasteur. Neurochem Res. 2021;46:15–22.
Newington JT, Pitts A, Chien A, Arseneault R, Schubert D, Cumming RC. Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS ONE. 2011;6:e19191.
Figley CR, Stroman PW. The role(s) of astrocytes and astrocyte activity in neurometabolism, neurovascular coupling, and the production of functional neuroimaging signals. Eur J Neurosci. 2011;33:577–88.
Zyśk M, Beretta C, Naia L, Dakhel A, Påvénius L, Brismar H, et al. Amyloid-β accumulation in human astrocytes induces mitochondrial disruption and changed energy metabolism. J Neuroinflammation. 2023;20:43.
Santangelo R, Giuffrida ML, Satriano C, Tomasello MF, Zimbone S, Copani A. β-amyloid monomers drive up neuronal aerobic glycolysis in response to energy stressors. Aging. 2021;13:18033–50.
Soucek T, Cumming R, Dargusch R, Maher P, Schubert D. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron. 2003;39:43–56.
Wu L, Zhang X, Zhao L. Human ApoE isoforms differentially modulate brain glucose and ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J Neurosci. 2018;38:6665–81.
Fang W, Xiao N, Zeng G, Bi D, Dai X, Mi X, et al. APOE4 genotype exacerbates the depression-like behavior of mice during aging through ATP decline. Transl Psychiatry. 2021;11:507.
Zhang X, Wu L, Swerdlow RH, Zhao L. Opposing effects of ApoE2 and ApoE4 on glycolytic metabolism in neuronal aging supports a warburg neuroprotective cascade against Alzheimer’s disease. Cells. 2023;12:410.
John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE. 2011;6:e17674.
Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY, et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun. 2017;8:1464.
Jedrychowski MP, Gartner CA, Gygi SP, Zhou L, Herz J, Kandror KV, et al. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J Biol Chem. 2010;285:104–14.
Costantini LC, Barr LJ, Vogel JL, Henderson ST. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008;9:S16.
Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94:1–14.
Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, et al. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol Aging. 2004;25:311–4.
Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–38.
Hanson AJ, Bayer JL, Baker LD, Cholerton B, VanFossen B, Trittschuh E, et al. Differential effects of meal challenges on cognition, metabolism, and biomarkers for apolipoprotein E ɛ4 carriers and adults with mild cognitive impairment. J Alzheimers Dis. 2015;48:205–18.
Hanson AJ, Banks WA, Hernandez Saucedo H, Craft S. Apolipoprotein E genotype and sex influence glucose tolerance in older adults: a Cross-Sectional Study. Dement Geriatr Cogn Dis Extra. 2016;6:78–89.
Farmer BC, Williams HC, Devanney NA, Piron MA, Nation GK, Carter DJ, et al. APOΕ4 lowers energy expenditure in females and impairs glucose oxidation by increasing flux through aerobic glycolysis. Mol Neurodegener. 2021;16:62.
Williams HC, Farmer BC, Piron MA, Walsh AE, Bruntz RC, Gentry MS, et al. APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol Dis. 2020;136:104742.
Arbones-Mainar JM, Johnson LA, Torres-Perez E, Garcia AE, Perez-Diaz S, Raber J, et al. Metabolic shifts toward fatty-acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int J Obes. 2016;40:1574–81.
Fleeman RM, Kuhn MK, Chan DC, Proctor EA. Apolipoprotein E ε4 modulates astrocyte neuronal support functions in the presence of amyloid-β. J Neurochem. 2023;165:536–49.
Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102.
Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21:204–24.
Rich PR, Maréchal A. The mitochondrial respiratory chain. Essays Biochem. 2010;47:1–23.
Mary A, Eysert F, Checler F, Chami M. Mitophagy in Alzheimer’s disease: molecular defects and therapeutic approaches. Mol Psychiatry. 2023;28:202–16.
Miller B, Kim SJ, Mehta HH, Cao K, Kumagai H, Thumaty N, et al. Mitochondrial DNA variation in Alzheimer’s disease reveals a unique microprotein called SHMOOSE. Mol Psychiatry. 2023;28:1813–26.
Xie W, Guo D, Li J, Yue L, Kang Q, Chen G, et al. CEND1 deficiency induces mitochondrial dysfunction and cognitive impairment in Alzheimer’s disease. Cell Death Differ. 2022;29:2417–28.
Terada T, Obi T, Bunai T, Matsudaira T, Yoshikawa E, Ando I, et al. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology. 2020;94:e1592–604.
Gibson GE, Haroutunian V, Zhang H, Park LC, Shi Q, Lesser M, et al. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann Neurol. 2000;48:297–303.
Valla J, Yaari R, Wolf AB, Kusne Y, Beach TG, Roher AE, et al. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE ε4 allele, the major late-onset Alzheimer’s susceptibility gene. J Alzheimers Dis. 2010;22:307–13.
Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci USA. 2008;105:4441–6.
Birnbaum JH, Wanner D, Gietl AF, Saake A, Hock C, Nitsch RM, et al. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-beta and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018;27:121–30.
Manczak M, Kandimalla R, Fry D, Sesaki H, Reddy PH. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet. 2016;25:5148–66.
Corona JC, Duchen MR. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic Biol Med. 2016;100:153–63.
Johri A, Chandra A, Flint Beal M. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic Biol Med. 2013;62:37–46.
Katsouri L, Lim YM, Blondrath K, Eleftheriadou I, Lombardero L, Birch AM, et al. PPARγ-coactivator-1α gene transfer reduces neuronal loss and amyloid-β generation by reducing β-secretase in an Alzheimer’s disease model. Proc Natl Acad Sci USA. 2016;113:12292–7.
Zolezzi JM, Silva-Alvarez C, Ordenes D, Godoy JA, Carvajal FJ, Santos MJ, et al. Peroxisome proliferator-activated receptor (PPAR) γ and PPARα agonists modulate mitochondrial fusion-fission dynamics: relevance to reactive oxygen species (ROS)-related neurodegenerative disorders? PLoS ONE. 2013;8:e64019.
Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, et al. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem. 2011;286:5215–21.
Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021;34:108572.
Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener. 2009;4:35.
Budny V, Knöpfli Y, Meier D, Zürcher K, Bodenmann C, Peter SL, et al. APOE4 increases energy metabolism in APOE-Isogenic iPSC-derived neurons. Cells. 2024;13:1207.
Andersen JV, Schousboe A, Verkhratsky A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022;217:102331.
Wynne ME, Ogunbona O, Lane AR, Gokhale A, Zlatic SA, Xu C, et al. APOE expression and secretion are modulated by mitochondrial dysfunction. eLife. 2023;12:e85779.
Tambini MD, Pera M, Kanter E, Yang H, Guardia-Laguarta C, Holtzman D, et al. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016;17:27–36.
Lindner K, Beckenbauer K, van Ek LC, Titeca K, de Leeuw SM, Awwad K, et al. Isoform- and cell-state-specific lipidation of ApoE in astrocytes. Cell Rep. 2022;38:110435.
Lee H, Cho S, Kim MJ, Park YJ, Cho E, Jo YS, et al. ApoE4-dependent lysosomal cholesterol accumulation impairs mitochondrial homeostasis and oxidative phosphorylation in human astrocytes. Cell Rep. 2023;42:113183.
Shenk JC, Liu J, Fischbach K, Xu K, Puchowicz M, Obrenovich ME, et al. The effect of acetyl-L-carnitine and R-alpha-lipoic acid treatment in ApoE4 mouse as a model of human Alzheimer’s disease. J Neurol Sci. 2009;283:199–206.
Joshi AU, Ebert AE, Haileselassie B, Mochly-Rosen D. Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington’s disease. J Mol Cell Cardiol. 2019;127:125–33.
Ashraf A, Fan Z, Brooks DJ, Edison P. Cortical hypermetabolism in MCI subjects: a compensatory mechanism? Eur J Nucl Med Mol Imaging. 2015;42:447–58.
Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, et al. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci. 2009;29:14770–8.
Benzinger TLS, Blazey T, Jack CR, Koeppe RA, Su Y, Xiong C, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci USA. 2013;110:E4502–4509.
Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci USA. 2009;106:7209–14.
Evans S, Dowell NG, Tabet N, King SL, Hutton SB, Rusted JM. Disrupted neural activity patterns to novelty and effort in young adult APOE-e4 carriers performing a subsequent memory task. Brain Behav. 2017;7:e00612.
Venzi M, Tóth M, Häggkvist J, Bogstedt A, Rachalski A, Mattsson A, et al. Differential effect of APOE alleles on brain glucose metabolism in targeted replacement mice: an [18F]FDG-μPET study. J Alzheimers Dis Rep. 2017;1:169–80.
Naia L, Shimozawa M, Bereczki E, Li X, Liu J, Jiang R, et al. Mitochondrial hypermetabolism precedes impaired autophagy and synaptic disorganization in App knock-in Alzheimer mouse models. Mol Psychiatry. 2023;28:3966–81.
Funding
The authors acknowledge the following funding. Vanessa Budny and Christian Tackenberg: Dr. Wilhelm Hurka Foundation and Hartmann Müller Foundation (2711); Iván Ruminot: Fondecyt 1230682; L Felipe Barros: Fondecyt 1230145.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Budny, V., Ruminot, I., Wybitul, M. et al. Fueling the brain - the role of apolipoprotein E in brain energy metabolism and its implications for Alzheimer’s disease. Transl Psychiatry 15, 316 (2025). https://doi.org/10.1038/s41398-025-03550-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03550-w
