
Abstract
Genomic Copy Number variants (CNVs) increase risk for neurodevelopmental disorders (NDDs) and affect cognition, but their impact on sleep remains understudied despite the well-established link between sleep disturbances, NDDs, and cognition. We investigated the relationship between CNVs, sleep traits, cognitive ability, and executive function in 498,852 individuals from an unselected population in the UK Biobank. We replicated the U-shape relationship between measures of cognitive ability and sleep duration. The effects of CNVs on sleep duration were evident at the genome-wide level; CNV-burden analyses showed that overall, CNVs with an increasing number of intolerant genes were associated with increased or decreased sleep duration in a U-shape pattern (p < 2e−16), but did not increase risk of insomnia. Sleep duration only marginally mediated the robust association between CNVs and poorer cognitive performance, suggesting that sleep and cognitive phenotypes may result from pleiotropic effects of CNVs with minimal causal relationship.
Similar content being viewed by others

Introduction
Genomic Copy Number Variants (CNVs) are deletions and duplications over 1000 base pairs.
CNVs are well-established biological factors strongly linked to susceptibility to Neurodevelopmental disorders (NDD) and are associated with negative effects on cognition, even in individuals from unselected populations who do not meet criteria for an NDD diagnosis [1]. Cognitive ability is one of the most commonly assessed quantitative traits in pediatric clinics, as it helps predict outcomes and adaptive skills in children with neurodevelopmental symptoms [2]. Many studies have shown that the effect size of CNVs on cognitive ability is related to their effect size on liability for NDDs [1]. As such, measuring effect size on cognitive ability provides a nuanced insight into the severity of genetic variants associated with NDD risk in general population cohorts where the prevalence of NDD is low.
Measures of intolerance to haploinsufficiency (i.e., genetic variants that inactivate protein-coding genes) of genes encompassed in CNVs have emerged as the best metrics to predict the effect size of multigenic CNVs (genome-wide) on cognition [3], risk for NDDs and psychiatric conditions such as autism, schizophrenia, Major Depressive Disorder (MDD), bipolar, and Obsessive-Compulsive Disorder (OCD) [4,5,6].
Despite the fact that sleep disturbances are some of the most common comorbidities across NDDs (i.e., up to 86% of individuals with NDDs are reported to experience sleep problems [7]), they remain understudied in individuals who carry genetic risk for NDDs.
Indeed, sleep disturbances have long been linked to cognitive difficulties across the developmental lifespan in individuals with and without a diagnosis of NDD or psychiatric condition [8,9,10,11,12,13]. It has been posited that disturbed sleep may contribute to exacerbating cognitive impairments in NDD groups, in particular executive functioning (EF) [14], which enables goal-directed thought and behavior. There is a large body of research showing that executive functions are especially vulnerable to sleep disruption [15, 16]. Sleep has also been proposed as a modifiable lifestyle factor to improve EF specifically [17]. In recent years, support for an inverted U-shape association between sleep duration and cognition has emerged [18,19,20,21]. Such that individuals who have shorter or longer sleep compared to the average duration in their cohort, experience poorer cognitive performance. Studies documenting that sleep problems are associated with genetic NDDs [22] are limited to specific well-known recurrent CNVs, like 16p11.2, 22q11.2, 15q11.2–13.1 [23,24,25]. Mouse models have also demonstrated evidence for such relationships, including sleep alterations in 16p11.2 deletions mouse models and circadian rhythm disruptions in 17p11.2 deletion mouse models [26]. However, the relationship between rare large effect genetic risk for NDDs, cognitive deficits, and sleep traits is still understudied.
Knowledge Gap: The effects of NDD-associated CNVs on sleep traits have not been investigated genome-wide. Understanding whether sleep disturbances may mediate some of the well-known effects of CNVs on cognitive abilities would provide insights into the mechanisms underlying genetic risk for neurodevelopmental disorders.
Our overarching aim is to investigate the relationship between sleep and cognition and the effects of CNVs on both traits in an unselected population.
Specifically, we aim to : (i) Examine the relationship between self-reported and actigraphy-derived measures of sleep with cognition and executive functions in the UK Biobank (UKBB); (ii) Investigate if recurrent CNVs associated with cognition and NDD risk are associated with sleep quantity and quality as well as lower cognition in the UKBB; (iii) Investigate if sleep traits mediate the relationship between CNVs and cognitive ability.
To reach these goals, we investigated all CNVs genome-wide in 458,436 individuals from an unselected population (UKBB) with measures of general cognitive ability, executive functioning, self-reported, and accelerometer-derived measures of sleep. Measures of general cognitive ability and executive functioning were used to evaluate whether associations remain robust across cognitive domains. As such, this large sample provided an opportunity to disentangle the relationship between genes, sleep and cognition.
Methods
Participants
Data was analyzed from the UK Biobank study, which has been extensively described elsewhere [27]. Over 500,000 individuals from the United Kingdom aged 37–74 were recruited between 2006–2010 (initial assessment visit). Information was also obtained from additional assessment visits between 2012–2020, along with online follow-up questionnaires.
Cognitive measures
General cognitive ability
We computed a g-factor separately for clinic-based and online assessments using principal component analysis (PCA), when the complete set of cognitive tasks for each modality was available. The clinic g-factor was computed using five cognitive tasks, based on previous research [28], selecting the earliest available assessment measures for each participant: Fluid intelligence score (20016), Pairs matching (399), Numeric memory (4282), Prospective memory (20018) and Reaction time (20023). The online g-factor was derived from the following tasks at the first assessment only: Fluid intelligence score (20191), Trail making #2 (20157), Symbol digit substitution (20159), Pairs matching (20132), Numeric memory (20240).
Each g-factor was normalized separately for age and sex. To maximize participant inclusion, we used the most robust available cognitive measure, following this priority: clinic g-factor, online g-factor, clinic Fluid Intelligence, and online Fluid Intelligence; if a g-factor could not be computed (due to incomplete cognitive data).
This composite cognitive score was used as a proxy for general cognitive ability, as described elsewhere [29] (eFig. 1).
The trail making task
The cognitive shifting component of executive functions was measured using the Trail Making Task. Participants were asked to link consecutive numbers (numeric version A), then link letters and numbers sequentially (alphanumeric version B). The difference in completion time between version B and A was computed to account for motor ability. Z-score directions were reversed to indicate lower scores correspond with worst performance. Performance scores were obtained from initial visit assessments (A:6348; B: 6350) and pooled with online follow-up scores (A:20156; B:20157) if initial scores were not available.
Tower rearranging task
Planning abilities were measured using accuracy on the Tower Rearranging Task available only for the initial assessment visit (21004). For 18 rounds participants were shown three pegs (“towers”) with three differently colored hoops, then asked to indicate how many moves were required to re-arrange the hoops to a specified reference location. Participants’ score based on the number of correct responses (maximum 18).
All cognitive scores described were transformed into z scores, adjusting for age and sex.
Sleep measures
Self-report questionnaires
We analyzed two sleep traits self-reported during initial assessment:
-
(a)
Sleep duration (1160); the number of hours spent sleeping over a 24 hr period (including naps) responses were made in 1-hr increments. Extreme responses of less than 3 h and more than 12 h were excluded as recommended by previous studies [30, 31]
-
(b)
Probable insomnia (1200); participants responding “sometimes” or “usually” to “Do you have trouble falling asleep at night, or do you wake up in the middle of the night?”, were coded as having probable insomnia. Participants responding “never/rarely” were coded as controls, while participants indicating “prefer not to answer” were removed. Due to the way the question was phrased, responses of ‘usually’ or ‘sometimes’ do not specify the number of days participants experience these symptoms. Hence, they could both indicate probable insomnia. This coding approach has also been used in a previous study [32].
Accelerometer
A subset of participants were invited to wear a triaxial accelerometer device (Axivity AX3) between 3 to 10 years after the initial study assessment. Individuals were excluded based on UKBB quality checks previously described [31, 33], we further excluded individuals wearing the device for <5 days and those with extreme short (<3 h) or long (>12 h) mean sleep duration [30, 31]. Detailed descriptions of raw accelerometer data is available elsewhere [33]. Converted and processed raw data using the R package GGIR was retrieved from UKBB return 1862.
Two Accelerometer derived sleep traits were analyzed:
-
(a)
Sleep duration; calculated as sleep episodes within the Sleep Period Time (SPT) lasting more than 5 min with no detected change >°5 associated with the z-axis of the accelerometer. Duration for all sleep episodes were summed and averaged across nights available.
-
(b)
Sleep efficiency; calculated as a ratio of sleep duration divided by the time elapsed between the beginning of the first inactivity bout and end of the last inactivity bout (SPT window). The ratio represents the mean of all nights available.
Medications
Participants self-reporting medication (20003) use that interfere or likely interfere with sleep and circadian rhythms [30] were excluded in sensitivity analysis (eTable 1).
Genetic information
Genetic information was retrieved from the UKBB. Genotyping collection, quality control and imputation procedures have been described in detail [34]. Genome wide-analyses controlled for ancestry. Ancestry was computed by the UKBB using a PCA yielding 10 distinct ancestral groups (22009).
CNV calling
CNVs were called using PennCNV [35] and QuantiSNP [36], and filtered based on previously published methods [3, 37] available online (https://github.com/JacquemontLab/MIND-GENESPARALLELCNV). The final CNV calls can be requested from UKBB [Return ID: 3104]. The following parameters were used for both algorithms: number of probes for CNV ≥ 3, CNV size ≥1Kb, likelihood scores ≥15. CNVs detected by both algorithms were combined (CNVision [38]) to minimize the number of potential false discoveries. We defined all CNVs with less than 2 copies as deletions and all CNVs with more than 2 copies as duplications. After this merging step, an in-house algorithm based on CNV was applied to concatenate adjacent CNVs of the same type into a single event, based on the following criteria: a) gap between CNVs ≤150 kb; b) size of the CNVs ≥1000 bp; and c) number of probes ≥3.
Array filtering
We used the following quality-control criteria: call rate ≥95%; log R ratio-standard deviation <0.35; B allele frequency-standard deviation <0.08; |waviness factor| <0.05, and CNVs <50 observations per array. From a total of 488,288 people with genotypic data, 28,240 were excluded for failing one of these filters. Finally, we excluded participants carrying at least one CNV ≥ 10 Mb (N = 431, a widely used QC threshold for CNVs [3, 37, 39]) because such large CNVs are rarely observed in general population cohorts and are almost always present as mosaics and/or somatic CNVs that can not be analyzed together with germline CNVs.
CNV filtering
Only autosomal CNVs were considered for this study. CNVs with the following criteria were selected for analyses: likelihood score ≥30 (for at least one of both detection algorithms), size ≥50 kb, unambiguous type (deletions and duplications) and overlap with segmental duplications, HLA regions (chr6:32400000–32700000, hg19) or centromeric regions <50% (regions defined from UCSC hg19). In addition, we applied an in-house algorithm based on a machine learning method to detect additional artifact CNVs (DigCNV). This algorithm combines the consensus of three methods (Random forest, bagging of KNN and SVM) and uses 9 CNV features (Array criteria: log R ratio-standard deviation, B allele frequency-standard deviation, wave frequency; Localization CNV criteria: % of CNV overlap with centromeric regions and with segmental duplications; CNV criteria: density of SNPs (numbers of SNPs / size of CNV), likelihood score / number of SNPs, % algorithms overlapping, percentage of shared sequence found by the both algorithms), type of CNV). The model was trained on 66% and tested on 33% of a manually curated reference set of 34,156 CNVs (31,746 true CNVs and 2410 artifacts from 6 cohorts [29]), validated using log R and BAF plots by two experts (a third in case of disagreement). DigCNV achieved an AUC of 0.95, sensitivity 0.95, and specificity 0.85.
Annotation of CNVs
CNVs were annotated using Gencode V19 annotation (the reference release for hg19 Human genome release) with ENSEMBL gene (https://grch37.ensembl.org/index.html). Each gene was annotated with “Loss-of-function observed/expected upper bound fraction” (LOEUF) [40]. We used the LOEUF score to measure intolerance to haploinsufficiency. As described by gnomAD, LOEUF is a continuous variable, ranging from 0 to 2, which provides insight across the full spectrum of Loss of Function constraint. Values below 0.35 are suggestive to be highly intolerant to haploinsufficiency.
We defined recurrent CNVs as those previously associated with neurodevelopmental or neuropsychiatric conditions (eTable 2) [41,42,43,44,45]. CNVs were identified based on >50% reciprocal overlap with a known recurrent CNV, disruption of specific gene(s), and/or their size. All recurrent CNVs with a likelihood score below 150 were visually inspected, and those identified as false positives (<1%) were excluded from the analyses.
Data analysis
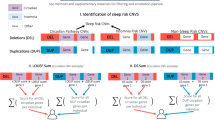
An overview of the different analysis methodologies is illustrated in Fig. 1.

Methodology.
All analyses were performed with R 4.3.1
Associations between sleep and cognition
The relationship between cognitive variables (general cognitive ability, trail making and tower rearranging task) and sleep duration (self-report and accelerometer derived) was first plotted with a General Additive Model (GAM) smooth line using the R ‘gam’ package. To confirm plotted relationships and to test a priori inverted U-shape relationships published, linear and quadratic models were run for each cognitive variable, with sleep duration as the independent variable. Likelihood ratio test (ANOVA for nested models) was applied to compare significant differences between models, using higher adjusted R-squared values and sleep duration p-values to select the optimal model. Sleep duration associated with the highest cognitive performance (x-coordinate of a parabola’s vertex) was calculated as: \(-\frac{B}{2A}\). A refers to the estimated coefficient for the squared sleep duration variable and B the duration coefficient for the linear term. Separate general linear models were used to test the relationship between sleep efficiency and probable insomnia, with each cognitive variable. For all models, age at which sleep traits were reported was controlled for, along with the 10 principal components. Subsequent sensitivity analyses included: restricting comparisons between sleep and cognitive variables reported at the same initial assessment and removing individuals taking medication and above 65 years.
Effects of individual CNVs on sleep and cognition
Separate linear regression models were applied to investigate associations between independent CNVs of interest with each cognitive and sleep dependent variable, with the exception of insomnia, for which a logistic regression was used. In all models, carrier status for each individual CNV was the independent variable, with carriers as the reference group. All models controlled for age, sex and the 10 first principal components. In order to correct for multiple testing, FDR correction was applied within each phenotype for deletions and duplications.
Effects of haploinsufficiency on sleep and cognition
Based on LOEUF categories
As LOEUF is a continuous measure of constraint, we defined consecutive categories based on gnomAD thresholds [40] (LOEUF < 0.35 for highly intolerant genes and LOEUF < 1 for moderately constrained genes). We therefore added additional categories within the highly intolerant range to better capture differences within highly intolerant genes. This approach was motivated by prior work showing a gradient of cognitive effects even within the LOEUF < 0.35 range [29]. The effect size of genes deleted and duplicated were stratified by score of intolerance to Loss of Function variants. For each individual, the number of genes deleted or duplicated was assessed in four categories defined across the range of LOEUF values: highly intolerant genes (LOEUF < 0.2), intolerant genes (0.2 ≤ LOEUF < 0.35), moderately intolerant genes (0.35 ≤ LOEUF < 1), and tolerant genes (1 ≤ LOEUF ≤ 2).
Regressions were performed as follows, separately for deletions and duplications:
Where Σ(genesi inside the windowj)DEL/DUP is the number of genes deleted (DEL) or duplicated (DUP) for the individual i inside the LOEUF category j. β0, β1, β2, β3 and β4 are the vectors of coefficients for fixed effects. Linear regressions were applied for all sleep and cognitive measures, with the exception of probable insomnia (logistic regression).
Based on aggregate LOEUF Values
We computed the \(\varSigma \frac{1}{{LOEUF}}\) separately for all genes included in deletions or duplications to investigate the relationship between genetic factors and sleep duration measures. We applied the same steps, as previously for investigating U-shape association between sleep and cognition : (i) fitting a General Additive Model (GAM) smooth line, (ii) run linear and quadratic models (iii) Compare linear and quadratic models using likelihood ratio test.
Separate linear models were used to test the relationship between genetic factors and other sleep/cognitive variables. All models were controlled for age, sex and the 10 principal components.
Mediation analyses
To investigate if sleep traits mediate the relationship between increased haploinsufficiency measured by LOEUF (independent variable) and cognitive ability (dependent variable), indirect effects were tested using the R ‘mediation’ package. Bootstrap procedures were applied, with indirect effects computed from 1000 imputations. Models were computed separately for \(\varSigma \frac{1}{{LOEUF}}\) in deletions and duplications. The “Prop.mediated” output shows how the mediator (sleep trait) influences the relationship between LOEUF and cognitive factors.
Results
Associations between sleep and cognition in the UKBB
Participant characteristics can be found in Table 1. Measures of sleep duration examined using GAM smooth line exhibited an inverted U-shaped relationship with cognition (eFigs. 2, 3), hence we fitted a quadratic model. The latter demonstrated a significant relationship between sleep duration, general cognitive ability, as well as executive functioning (Fig. 2). The only non-significant association was between accelerometer-derived sleep duration and the Tower Rearranging task, which had the smallest sample size. (eTable 3). Quadratic trends demonstrated that any deviation from the mean sleep duration (7 to 7.5 h) was associated with worse cognitive performance. A Likelihood Ratio Test, showed that the quadratic model outperformed linear models (eTable 3). Moreover, this quadratic trend remained when restricting the analysis to self-reported sleep duration and cognitive variables administered at the same time-point and when removing individuals above 65 years of age, as well as those taking any psychiatric or sleep medication (eTables 1, 3). In contrast with sleep duration, sleep efficiency (derived from accelerometer data) was not associated with general cognitive ability or executive functioning measured by the tower rearranging task, and a weak association was only observed with trail making task (β = 0.2, p = 4.84−04, eTable 4). These observations remained unchanged when removing individuals with medication and above 65 years (eTable 4).

Fitted curves of the Quadratic model associating cognition and sleep duration (dark line). The light line represents the GAM model. The dotted vertical line indicates the vertex of the parabola. a Relationship between general cognition aggregated across visits and sleep duration (self-reported on the left and accelerometer on the right). b Relationship between executive functioning measured by the trail-making task administered in person and online and self-reported sleep duration (self-reported on the left and accelerometer on the right). c Relationship between executive functioning measured by the tower rearranging task administered in person and self-reported sleep duration (Note: this relationship was not significant with accelerometer-derived sleep duration). Blue = General cognitive ability, Green = Trail making task, Purple = Tower task. X-axis: durations are in hours (first line) and z-scored duration (second line). Y-axis: z-scored cognitive ability.
The reporting of insomnia traits was also associated with lower measures of general cognitive ability (β = −0.04, p = 6.31−22, eTable 4). This effect size was unchanged when the analysis was restricted to probable insomnia and cognitive assessments collected at the same time point (β = −0.08, p = 4.69−12, eTable 4) and when removing individuals taking medication as well as the elderly (β = −0.04, p = 2.31−13, eTable 4). Executive functioning measured by the trail making task and tower rearranging task (eTable 4) showed weak to no association with self-reported insomnia.
Associations between recurrent CNVs, sleep and cognition
A total of 14,295 individuals carrying a CNV at one of 29 genomic loci associated with cognitive ability and risk for NDD were identified in the UKBB (eTable 2). Estimates were computed only for CNVs with more than 10 carriers for a given measure. As a result, some phenotypes (accelerometer-derived) did not have enough carriers at specific loci to carry out the analysis. As expected, several CNVs (n = 11) negatively affected general cognitive ability with mild to large effect sizes, except for a positive effect associated with deletions at the ZNF92 locus (β = 0.08, p = 8.30−03, Fig. 3, eTable 5) previously reported to have a protective effect on schizophrenia [43].

Forest plots depicting the associations between specific recurrent CNV (red = deletion; blue = duplications) and 7 cognitive and sleep measures. Estimates are computed only for CNVs with more than 10 carriers for a given measure. As a result, some phenotypes (accelerometer-derived) have fewer estimates depicted in the figure. Only CNVs with a significant association (FDR < 0.05) with at least one trait are shown. The Y-axis represents the cognitive and sleep traits. The X-axis represents the estimate of the linear models associating CNVs and traits. Abbreviation: TMT trail making task.
We also identified 6 significant associations with executive functioning measured by the Trail Making Task, including a large negative effect for deletions at the VPS13B locus (β = −1.85, p = 2.73−16, Fig. 3, eTable 5). Among all tested CNVs, FDR-significant effects on self-reported sleep duration were observed only for 10q11.21q11.23 deletions, and a significant association with probable insomnia was identified for the 15q13.3 duplication (Fig. 3, eTable 5).
Genome-wide effects of deletions and duplications on sleep and cognition in the UKBB
The analysis above was limited to recurrent CNVs observed in more than 10 individuals. It is likely, however, that these effects may be observed for a much broader spectrum of CNVs burden (genome-wide). We therefore used a previously published burden method to measure the average effects of CNVs weighted by intolerance to haploinsufficiency (measured by LOEUF). This method has been used to associate CNV burden with phenotypes, including cognitive ability and risk for neurodevelopmental disorders. As previously reported, in other cohorts and the UKBB [3, 4, 37], there was a strong association between the weighted burden of CNVs (measured by the \(\varSigma \frac{1}{{LOEUF}}\)) and general cognitive ability (β = −0.04, p = 1.11−40 for deletions; β = −0.09, p = 3.51−31 for duplications, eTable 6).
Using GAM, we observed a U-shaped trend between sleep duration (self-reported) and the severity of CNVs measured by the \(\varSigma \frac{1}{{LOEUF}}\). We therefore tested this association using a linear and quadratic model. The quadratic model showed a significant association for deletions and duplications (Fig. 4a, b, eTable 7). In contrast, the linear model showed no association and was outperformed (ANOVA : p = 3.56−24 for deletions ; p = 1.97−20 for duplications) by the quadratic model for deletions and duplications (Fig. 4a, b, eTable 7). Sensitivity analysis revealed that the quadratic relationship was not influenced by medication, age, or genotyping batches. (eTable 7). Overall, this suggests that individuals carrying CNVs of increasing severity (higher intolerance to haploinsufficiency) exhibit increasingly shorter or longer sleep duration. There was only a weak relationship between CNVs burden (measured by the \(\varSigma \frac{1}{{LOEUF}}\)) and probable insomnia for deletions and duplications (eTable 6). No association were detected for accelerometer-derived sleep measures (eTables 6, 7).

a, b represent the fitted curves of the quadratic model associating CNVs measured by their sum of 1/LOEUF (for (a) deletions and (b) duplications) with self-reported sleep duration (dark line). The light line represents the GAM model. The dotted vertical line indicates the vertex of the parabola. c, d illustrates the results of the mediation analysis between the sum of LOEUF (for deleted and duplicated genes), sleep duration, and cognitive ability (*p-value < 2e−16). The Average Causal Mediation Effect effect was 0.005 × −0.129 = −0.0007 (p < 2e-16) for deletions and 0.002 × −0.146 = −0.0003 (p < 2e-16) for duplications. The proportion mediated was 3.8% for deletions and 4.5% for duplications.
To further characterize the relationship between CNV burden and sleep duration as well as cognition, we stratified the coding genome into 4 categories with increasing intolerance to haploinsufficiency as measured by LOEUF (highly intolerant <0.2, intolerant = [0.2;0.35[, mildly-intolerant = [0.35;1[, and tolerant ≥1). The three categories of increasingly intolerant genes were associated with increasingly negative effects on general cognitive ability. CNVs encompassing intolerant and highly intolerant genes were associated with increased deviation from the average self-reported sleep duration (eFig. 4). Genes within the intolerant category were associated with worst performance on both executive functioning tasks, but overall results were less robust, likely due to smaller sample sizes (eFig. 4). These results remained unchanged after removing individuals taking medication and above 65 years (eFig. 4).
To better understand the relationships between CNVs (based on \(\varSigma \frac{1}{{LOEUF}}\)), general cognitive ability and sleep duration (self-reported), we performed a mediation analysis using a linear model and the absolute measure of sleep duration (Fig. 4c, d). The analyses suggested that the genome-wide effects of duplications and deletions on cognitive ability were only very mildly mediated (4% for deletions and 5% for duplications) by sleep duration (eTable 8). In other words, there is a genetic impact on general cognitive ability and sleep duration, but the effects on these 2 traits are mostly independent. Results remained when removing individuals taking medication and >65 years (eFig. 5).
Discussion
This study represents the largest investigation of rare CNVs, sleep traits and cognition. We confirm the inverted U-shaped relationship between sleep duration and cognitive abilities as previously documented [18,19,20,21]. While the power to study the association between individual recurrent CNVs and sleep phenotypes was limited, the genome-wide CNV-burden (weighted by LOEUF) was strongly associated with worse cognitive performance and sleep duration. Interestingly, the latter association was in a U-shape fashion and, therefore, only visible using a quadratic model or the absolute values of sleep duration. In contrast, CNVs showed no or weak association with sleep efficiency and insomnia. Although the genome-wide burden of CNVs was associated with both sleep duration and general cognitive performance, the association with these two traits were largely independent, as shown by the mediation analysis.
The U shape relationship between sleep duration, biological factors, and other phenotypes
We replicated the inverted U-shape relationship between sleep duration and cognitive measures.
Our results echo the findings in other studies that have reported a U-shape relationship between sleep duration and health [46, 47], psychiatric symptoms [48, 49], and mortality [50]. Sleep duration is often investigated as a linear effect, which may overlook U-shape relationships and may explain some of the conflicting results such as those reported between ASD and sleep duration [14]. Quadratic U-shape relationships have also been established between sleep duration and intermediate brain phenotypes. Brain volume of 46 cortical and subcortical regions were associated with deviation from the average sleep duration in a U-shape fashion [20]. The same study also showed that markers of cerebrovascular burden (i.e., white matter hyperintensities) show the same quadratic association with sleep duration. Interestingly, we also identified a U-shape relationship between genome-wide CNV burden and sleep duration, which could not have been identified without adding a quadratic term. The mechanisms underlying the U-shaped association in which both short and long sleep duration are associated with worse cognitive performance remain to be elucidated. One key question raised by previous studies is whether short and long sleep duration might be risk factors, early markers, or a result of cognitive deficits [19]. Our mediation analysis suggests that the association between CNVs and cognitive performance is not mediated by sleep duration. We therefore posit that deviation from average sleep duration may be an additional phenotype (rather than a causal factor) of biological factors (including CNVs) contributing to cognitive deficits and risk for psychiatric conditions.
CNVs increasing risk for NDDs and decreasing cognition only mildly affect sleep traits
Our findings on the effects of individual recurrent CNVs on cognition are in line with previous research in UKBB [51]. As expected, we only detected negative associations with cognition for recurrent CNVs. The only exception was the positive effect on general cognitive ability for the deletions of ZNF92, which have previously been associated with a protective effect for schizophrenia [43]. Further studies are required to replicate this unusual positive association.
Overall, the association between CNVs and cognition are more robust for general cognitive ability than for specific tasks, such as executive functioning. This was true for recurrent CNVs and the genome-wide burden analysis. However, results suggest that some genomic loci may show preferential effects on executive functions, such as VPS13B.
While our power was limited to detect the effect of recurrent CNVs on sleep phenotypes, the genome-wide CNV burden analysis showed a clear association with sleep duration.
Overall, the associations between sleep duration and CNV-burden is consistent with the polygenic nature of this trait [52] and suggest that, when deleted or duplicated, many genomic loci are related to variations in sleep duration. Our burden analysis (weighted by LOEUF) as well as our sensitivity analysis, stratifying genes based on their constraint score, shows that intolerant genes are more likely to alter sleep duration and cognitive abilities but this association is much stronger for cognitive ability. This may indicate that, as opposed to cognitive ability, sleep may be linked to a more restricted set of biological functions, such as those identified through GWAS of common variants [52, 53]. Indeed, LOEUF values reflect genetic fitness, which may only be weakly related to mechanisms involved in sleep traits.
CNVs affecting intolerant genes are not associated with insomnia
We did not detect any robust association between insomnia and recurrent CNVs or with the genome-wide burden of CNVs (weighted by LOEUF). This may be due to the fact that heritability estimates for insomnia (22–25%) [54] are much lower than those estimated for cognitive ability [55] or NDDs with well-known associations with CNVs (e.g., autism spectrum disorder [56]).
Recent large GWAS [52] of insomnia have reported many associated loci, and a strong genetic correlation with depression and anxiety. The latter may in part explain the lack of association between insomnia and CNVs, which show only weak association with anxiety and depression [57, 58]. This lack of association may also be due to the differing impacts of rare versus common variants for insomnia traits or a recruitment bias in the UKBB. In contrast, CNVs showed a clear association with sleep duration. This is consistent with the fact that the strongest genetic correlation between long or short sleep duration was previously observed for educational attainment [30], and the latter is also strongly associated with CNVs [51, 59].
Limitations
Given the nature of the UKBB protocol, not all sleep and cognitive variables were collected at concurrent time-points. Of note, our results were unchanged when restricting analyses to sleep and cognitive measures collected at the same time-point. Previous findings also demonstrate that cognitive performance remains stable during follow-up in the UKBB [60]. Although sleep duration was mostly self-reported, when available, an accelerometer confirmed these reports. Insomnia was assessed using self-reported data, due to the insufficient number of ICD-10 codes for sleep disorders in the UKBB to perform robust statistical analysis.
Similar to previous studies in UK biobank and other cohorts, we do not attempt to infer any causal relationships between sleep, cognitive or other phenotypic measures.
Conclusion
Our study showed that the genome-wide burden of CNVs across a very large proportion is associated with decreased cognitive abilities and sleep duration in a U-shape fashion. While several previous studies have suggested that changing sleep duration may improve cognitive outcomes, our mediation analysis may suggest that both sleep duration and cognitive ability are two phenotypes with little causal relationship and may result from pleiotropic effects of genetic and non genetic factors. These results add to the emerging complexity of the quadratic relationship between sleep duration and multiple health and behavior outcomes.
Code availability
The scripts used for CNV calling and annotation are publicly available on GitHub: 1. CNV calling and annotation: https://github.com/JacquemontLab/MIND-GENESPARALLELCNV. 2. CNV filtering and validation: https://github.com/labjacquemont/DigCNV. All scripts and summary statistics for the analyses presented in this manuscript are available on Zenodo : https://doi.org/10.5281/zenodo.15757500.
References
Jacquemont S, Huguet G, Klein M, Chawner SJRA, Donald KA, Van Den Bree MBM, et al. Genes To Mental Health (G2MH): a framework to map the combined effects of rare and common variants on dimensions of cognition and psychopathology. Am J Psychiatry. 2022;179:189–203.
Deary IJ. Intelligence. Annu Rev Psychol. 2012;63:453–82.
Huguet G, Schramm C, Douard E, Tamer P, Main A, Monin P, et al. Genome-wide analysis of gene dosage in 24,092 individuals estimates that 10,000 genes modulate cognitive ability. Mol Psychiatry. 2021. https://doi.org/10.1038/s41380-020-00985-z.
Alexander-Bloch A, Huguet G, Schultz LM, Huffnagle N, Jacquemont S, Seidlitz J, et al. Copy number variant risk scores associated with cognition, psychopathology, and brain structure in youths in the philadelphia neurodevelopmental cohort. JAMA Psychiatry. 2022;79:699.
Wainberg M, Merico D, Huguet G, Zarrei M, Jacquemont S, Scherer SW, et al. Deletion of loss-of-function–intolerant genes and risk of 5 psychiatric disorders. JAMA Psychiatry. 2022;79:78.
Douard E, Zeribi A, Schramm C, Tamer P, Loum MA, Nowak S, et al. Effect sizes of deletions and duplications on autism risk across the genome. Am J Psychiatry. 2021;178:87–98.
Shelton AR, Malow B. Neurodevelopmental disorders commonly presenting with sleep disturbances. Neurotherapeutics. 2021;18:156–69.
Tham E, Schneider N, Broekman B. Infant sleep and its relation with cognition and growth: a narrative review. Nat Sci Sleep. 2017;9:135–49.
Carruthers SP, Brunetti G, Rossell SL. Sleep disturbances and cognitive impairment in schizophrenia spectrum disorders: a systematic review and narrative synthesis. Sleep Med. 2021;84:8–19.
Roussis S, Richdale AL, Katz T, Malow BA, Barbaro J, Sadka N. Behaviour, cognition, and autism symptoms and their relationship with sleep problem severity in young children with autism spectrum disorder. Res Autism Spectr Disord. 2021;83:101743.
Scullin MK, Bliwise DL. Sleep, cognition, and normal aging: integrating a half century of multidisciplinary research. Perspect Psychol Sci. 2015;10:97–137.
Xu W, Tan C-C, Zou J-J, Cao X-P, Tan L. Sleep problems and risk of all-cause cognitive decline or dementia: an updated systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2020;91:236–44.
Cheng W, Rolls E, Gong W, Du J, Zhang J, Zhang X-Y, et al. Sleep duration, brain structure, and psychiatric and cognitive problems in children. Mol Psychiatry. 2021;26:3992–4003.
Tesfaye R, Wright N, Zaidman-Zait A, Bedford R, Zwaigenbaum L, Kerns CM, et al. Investigating longitudinal associations between parent reported sleep in early childhood and teacher reported executive functioning in school-aged children with autism. Sleep. 2021;44:zsab122.
Tucker AM, Whitney P, Belenky G, Hinson JM, Van Dongen HPA. Effects of sleep deprivation on dissociated components of executive functioning. Sleep. 2010;33:47–57.
Turnbull K, Reid GJ, Morton JB. Behavioral sleep problems and their potential impact on developing executive function in children. Sleep. 2013;36:1077–84.
Sen A, Tai XY. Sleep duration and executive function in adults. Curr Neurol Neurosci Rep. 2023;23:801–13.
Wild CJ, Nichols ES, Battista ME, Stojanoski B, Owen AM. Dissociable effects of self-reported daily sleep duration on high-level cognitive abilities. Sleep. 2018;41:zsy182.
Leng Y, Yaffe K. Sleep duration and cognitive aging—beyond a U-shaped association. JAMA Netw Open. 2020;3:e2014008.
Tai XY, Chen C, Manohar S, Husain M. Impact of sleep duration on executive function and brain structure. Commun Biol. 2022;5:201.
Ma Y, Liang L, Zheng F, Shi L, Zhong B, Xie W. Association between sleep duration and cognitive decline. JAMA Netw Open. 2020;3:e2013573.
Agar G, Brown C, Sutherland D, Coulborn S, Oliver C, Richards C. Sleep disorders in rare genetic syndromes: a meta-analysis of prevalence and profile. Mol Autism. 2021;12:18.
Moulding HA, Bartsch U, Hall J, Jones MW, Linden DE, Owen MJ, et al. Sleep problems and associations with psychopathology and cognition in young people with 22q11.2 deletion syndrome (22q11.2DS). Psychol Med. 2020;50:1191–202.
Kamara D, De Boeck P, Lecavalier L, Neuhaus E, Beauchaine TP. Characterizing sleep problems in 16p11.2 deletion and duplication. J Autism Dev Disord. 2023;53:1462–75.
Saravanapandian V, Nadkarni D, Hsu S-H, Hussain SA, Maski K, Golshani P, et al. Abnormal sleep physiology in children with 15q11.2–13.1 duplication (Dup15q) syndrome. Mol Autism. 2021;12:54.
Wintler T, Schoch H, Frank MG, Peixoto L. Sleep, brain development, and autism spectrum disorders: insights from animal models. J Neurosci Res. 2020;98:1137–49.
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med. 2015;12:e1001779.
Lyall DM, Cullen B, Allerhand M, Smith DJ, Mackay D, Evans J, et al. Cognitive test scores in UK Biobank: data reduction in 480,416 participants and longitudinal stability in 20,346 participants. PLOS ONE. 2016;11:e0154222.
Huguet G, Renne T, Poulain C, Dubuc A, Kumar K, Kazem S, et al. Effects of gene dosage on cognitive ability: a function-based association study across brain and non-brain processes. Cell Genomics. 2024;4:100721.
Dashti HS, Jones SE, Wood AR, Lane JM, Van Hees VT, Wang H, et al. Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nat Commun. 2019;10:1100.
Jones SE, Van Hees VT, Mazzotti DR, Marques-Vidal P, Sabia S, Van Der Spek A, et al. Genetic studies of accelerometer-based sleep measures yield new insights into human sleep behaviour. Nat Commun. 2019;10:1585.
Lane JM, Jones SE, Dashti HS, Wood AR, Aragam KG, van Hees VT, et al. Biological and clinical insights from genetics of insomnia symptoms. Nat Genet. 2019;51:387–93.
Doherty A, Jackson D, Hammerla N, Plötz T, Olivier P, Granat MH, et al. Large scale population assessment of physical activity using wrist worn accelerometers: the UK Biobank Study. PLOS ONE. 2017;12:e0169649.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–74.
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, et al. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–25.
Huguet G, Schramm C, Douard E, Jiang L, Labbe A, Tihy F, et al. Measuring and estimating the effect sizes of copy number variants on general intelligence in community-based samples. JAMA Psychiatry. 2018;75:447.
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–85.
Auwerx C, Lepamets M, Sadler MC, Patxot M, Stojanov M, Baud D, et al. The individual and global impact of copy-number variants on complex human traits. Am J Hum Genet. 2022;109:647–68.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Coe BP, Witherspoon K, Rosenfeld JA, Van Bon BWM, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46:1063–71.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180:568–584.e23.
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35.
Sanders SJ, Sahin M, Hostyk J, Thurm A, Jacquemont S, Avillach P, et al. A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat Med. 2019;25:1477–87.
Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505:361–6.
Ohkuma T, Fujii H, Iwase M, Ogata-Kaizu S, Ide H, Kikuchi Y, et al. U-shaped association of sleep duration with metabolic syndrome and insulin resistance in patients with type 2 diabetes: The Fukuoka Diabetes Registry. Metabolism. 2014;63:484–91.
Li X, Lin L, Lv L, Pang X, Du S, Zhang W, et al. U-shaped relationships between sleep duration and metabolic syndrome and metabolic syndrome components in males: a prospective cohort study. Sleep Med. 2015;16:949–54.
Liu B-P, Wang X-T, Liu Z-Z, Wang Z-Y, An D, Wei Y-X, et al. Depressive symptoms are associated with short and long sleep duration: a Longitudinal Study of Chinese adolescents. J Affect Disord. 2020;263:267–73.
Li Y, Sahakian BJ, Kang J, Langley C, Zhang W, Xie C, et al. The brain structure and genetic mechanisms underlying the nonlinear association between sleep duration, cognition and mental health. Nat Aging. 2022;2:425–37.
Cappuccio FP, D’Elia L, Strazzullo P, Miller MA. Sleep duration and all-cause mortality: a systematic review and meta-analysis of prospective studies. Sleep. 2010;33:585–92.
Kendall KM, Bracher-Smith M, Fitzpatrick H, Lynham A, Rees E, Escott-Price V, et al. Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br J Psychiatry. 2019;214:297–304.
The 23andMe Research Team, Jansen PR, Watanabe K, Stringer S, Skene N, Bryois J, et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat Genet. 2019;51:394–403.
HUNT All In Sleep, Lane JM, Jones SE, Dashti HS, Wood AR, Aragam KG, et al. Biological and clinical insights from genetics of insomnia symptoms. Nat Genet. 2019;51:387–93.
Lind MJ, Aggen SH, Kirkpatrick RM, Kendler KS, Amstadter AB. A Longitudinal Twin Study of insomnia symptoms in adults. Sleep. 2015;38:1423–30.
Plomin R, von Stumm S. The new genetics of intelligence. Nat Rev Genet. 2018;19:148–59.
Huguet, G, Benabou, M & Bourgeron, T The genetics of autism spectrum disorders. in A time for metabolism and hormones (eds. Sassone-Corsi, P & Christen, Y) (Cham: Springer International Publishing; 2016) pp 101-29. https://doi.org/10.1007/978-3-319-27069-2_11.
Mollon J, Schultz LM, Huguet G, Knowles EEM, Mathias SR, Rodrigue A, et al. Impact of copy number variants and polygenic risk scores on psychopathology in the UK Biobank. Biol Psychiatry. 2023;94:591–600.
Kendall KM, Rees E, Bracher-Smith M, Legge S, Riglin L, Zammit S, et al. Association of rare copy number variants with risk of depression. JAMA Psychiatry. 2019;76:818–25.
Männik K, Mägi R, Macé A, Cole B, Guyatt A, Shihab HA, et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA. 2015;313:2044–54.
Fawns-Ritchie C, Deary IJ. Reliability and validity of the UK Biobank cognitive tests. PLOS ONE. 2020;15:e0231627.
Acknowledgements
Icons in Fig. 1 were designed by Freepik.
Funding
This research was enabled by support provided by Calcul Quebec (http://www.calculquebec.ca) and Compute Canada (http://www.computecanada.ca). Sebastien Jacquemont is a recipient of a Canada Research Chair in neurodevelopmental disorders, and a chair from the Jeanne et Jean Louis Levesque Foundation. This work is supported by a grant from the CIHR grant 159734 (Sebastien Jacquemont). Funding for the project was provided by the Wellcome Trust. This work was also supported by an NIH award U01 MH119690 granted to Laura Almasy, Sebastien Jacquemont and David Glahn and U01 MH119739. “This research has been conducted using the UK Biobank Resource under Application Number 40980”. The Cardiff Copy Number Variant cohort (UKBB) was supported by the Wellcome Trust Strategic Award DEFINE and the National Centre for Mental Health with funds from Health and Care Research Wales (code 100202/Z/12/Z). The funder had no role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
Conceptualization: RT, CP, GH, ME, SJ; Data curation: RT, CP, M J-L, ZS, GH; Formal Analysis: RT, CP; Funding acquisition: DC G, LA, SJ, ME; Methodology: RT, CP, M J-L, ZS, GH; Writing – review & editing: CP, RT, GH, ED, M J-L, ZS, AL, DC G, LA, ME, SJ.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflicts of interest.
Ethics statement
The study design has been previously detailed, and it received approval from the Sainte Justine Hospital Ethics Committee (2021-2812). All listed authors have approved the manuscript and have agreed to its publication. All participants provided informed consent. For further details, please refer to the UK Biobank website at www.ukbiobank.ac.uk. We confirm that all methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Poulain, C., Tesfaye, R., Douard, E. et al. The interplay between genomic copy number variants, sleep, and cognition in the general population. Transl Psychiatry 15, 351 (2025). https://doi.org/10.1038/s41398-025-03579-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03579-x
