
Abstract
A significant body of evidence suggests that mitochondrial dysfunction plays a key role in the development and progression of Alzheimer’s disease (AD). However, the absence of peripheral biomarkers for mitochondrial dysfunction limits its clinical applicability. Mitochondrial DNA (mtDNA) copy number, a proxy for mitochondrial function, has shown promise in detecting early stages of AD and predicting AD risk in cerebrospinal fluid (CSF) and blood, respectively. Surprisingly, recent studies have identified mtDNA molecules in human saliva, but their relationship with AD remains unexplored. Here, we investigated potential associations between salivary mtDNA copy number and cortical amyloid-β (Aβ) load measured with PET, and blood AD markers measured with ultrasensitive single molecule array (SIMOA) assays, in cognitively normal older adults. We found that salivary mtDNA copy number was positively correlated with cortical Aβ burden and plasma levels of tau phosphorylated at threonine 181 (pTau-181), and negatively correlated with general cognitive ability. It is worth noting that salivary mtDNA was not significantly associated with other blood-based AD biomarkers, including Aβ1-40, Aβ1-42, neurofilament-light chain (NfL), or glial fibrillary acidic protein (GFAP). Additionally, plasma pTau-181 levels moderated the association between salivary mtDNA and Aβ accumulation in the inferior temporal lobe, while Aβ load in the occipital cortex mediated the association between plasma pTau-181 and salivary mtDNA. Together, these findings represent the first evidence linking salivary mtDNA to well-established AD biomarkers in normal aging, suggesting that salivary mtDNA may serve as a potential non-invasive biomarker for identifying individuals at risk for developing AD in the general population.
Similar content being viewed by others


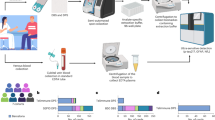
Introduction
Mitochondria are cellular organelles that play a pivotal role in energy production and oxidative balance in most eukaryotic cells. Beyond their metabolic functions, they serve as major regulatory centers involved in programmed cell death, immune response modulation, macromolecule synthesis, calcium homeostasis, and both intracellular and endocrine signaling pathways [1]. Mitochondrial function and mitophagy declines with age, leading to alterations in cellular physiology that may contribute to the development of aging-related chronic diseases [2].
Mitochondria contain their own DNA (mtDNA), which encodes proteins critical for mitochondrial function. Recent studies have identified mtDNA in different body fluids, either as unprotected circulating cell-free mtDNA (cf-mtDNA) or encapsulated in extracellular vesicles [3,4,5,6,7]. Notably, these forms of mtDNA appear to be resistant to nuclease degradation. While the cellular origins and physiological roles of cf-mtDNA remain unclear, latest findings suggest its potential as a biomarker for Alzheimer’s disease (AD) in CSF [8, 9], as well as its predictive value for AD-related risk in blood [10].
Saliva is attracting considerable attention as a suitable biofluid for identifying biomarkers of complex systemic diseases, due to its easy accessibility, minimally invasive collection procedures and low patient compliance demands. Furthermore, saliva-based analyses offer a potentially cost-effective strategy for screening of large-scale population and longitudinal follow-up studies [11]. Although mtDNA has been previously detected in human saliva [12,13,14,15,16,17,18], research examining its potential as a surrogate biomarker for AD in cognitively normal older adults is lacking. In this study, we first explored whether variations in salivary mtDNA levels were associated with changes in cognitive function. Next, we investigated potential associations between salivary mtDNA levels and cortical Aβ load measured with PET, as well as its relationship with a panel of blood-based AD biomarkers measured with ultrasensitive SIMOA assays. Finally, we assessed whether these variables acted as moderators or mediators in the observed associations.
Materials and methods
Participants
Ninety-one cognitively normal older adults participated in the study. To rule out the presence of mild cognitive impairment or dementia, each participant underwent a complete neurological evaluation. Individuals with conditions such as stroke, epilepsy, head trauma, neurodevelopmental disorders, intellectual disability, alcohol abuse, hydrocephalus, or intracranial tumors, were excluded from participation.
Inclusion criteria were: i) normal cognitive performance on cognitive tests, based on age- and education-adjusted reference values; ii) Clinical Dementia Rating score of 0, indicating no evidence of dementia; iii) autonomy in daily life; and iv) a score lower than 5 on the short form of the Geriatric Depression Scale, indicating no symptoms of depression.
All participants provided their informed consent before taking part in the study. The protocol was approved by the Ethical Committee for Clinical Research of the Junta de Andalucía, following the ethical standards outlined in the Declaration of Helsinki.
Neuropsychological assessment
Participants completed a comprehensive neuropsychological assessment. The assessment included the Spanish version of the memory binding test [19], the face-name associative memory test [20], the D2 attention test, as well as the letter-number sequencing and digit span subtests from the Wechsler Adult Intelligence Scale-III. Additionally, the spatial span subtest of the Wechsler Memory Scale-III was administered together with the short form of the Boston Naming Test, and semantic and phonological fluency tasks based on the “Animal” and letter “P” naming categories. Processing speed was assessed using the Symbol Digit Modalities Test. To evaluate executive function, both forms of the Trail Making Test (A and B) were administered, along with the Tower of London test, which measures planning and problem-solving abilities.
Saliva collection
Unstimulated whole saliva samples were collected into 50 ml sterile polypropylene conical tubes, which had been previously treated with a 2% sodium azide solution. Immediately after collection, samples were placed on ice and subjected to an initial centrifugation at 162 x g (1000 rpm) for 10 min at 4 °C. The supernatant was then centrifuged again at 2287 x g (4500 rpm) for 15 min at 4 °C. Following centrifugation, a protease inhibitor cocktail (cOmplete™ Ultra Tablets Mini, Roche, Basel, Switzerland) was added at a concentration of 1X. The resulting sample was aliquoted into 1.5 ml polypropylene tubes and stored at −80 °C until analysis.
Measurement of mtDNA copy number and nuclear genomes in saliva
Quantification of mtDNA and nuclear DNA (nDNA) was carried out directly in saliva samples without performing nucleic acid extraction following a modified protocol [21]. Saliva was mixed with the 100ST DNA/RNA/protein solubilization reagent (DirectQuant) in a 4:1 ratio (v:v) and heated at 90 °C for 3 min with continuous shaking at 750 rpm. The resulting solubilized saliva was then diluted 1:20 using double-distilled water for mtDNA quantification. The mtDNA was amplified using the primer pair mt92-CYTB, which targets the region from base pairs 15427 to 15518 of the mtDNA reference sequence NC_012920.1 (Forward, 5’-AGACGCCCTCGGCTTACTTC-3’; Reverse, 5’-GGGTATAATTGTCTGGGTCGCC-3’). For nDNA measurement, the TBP1-73 primer pair, targeting the TBP1 gene, was used (Forward, 5’-CACCACAGCTCTTCCACTCA-3’; Reverse, 5’-GGGGAGGGATACAGTGGAGT-3’). The digital PCR (dPCR) reaction for both mtDNA and nDNA quantification was prepared in a 20 µl final volume, consisting of 5 µl of saliva (diluted for mtDNA or undiluted for nDNA), 10 µl of 2X QX200™ dPCR™ EvaGreen Supermix (Bio-Rad, #1864033), 1 µl of either 2.7 µM mt92-CYTB primers (135 nM final concentration) or 1 µl of 1.9 µM TBP1-73 primers (95 nM final concentration), 1 µl of restriction enzyme AluI (#FD0014, Thermo Scientific) for mtDNA analysis, or 0.5 µl of SaqAI (#FD2174, Thermo Scientific) and 0.5 µl of BsuRI (#FD0154, Thermo Scientific) for nDNA. The final reaction volume was adjusted to 20 µl with double-distilled water. Enzyme digestion was performed at 37 °C for 15 min before partitioning the reaction mixture into droplets using the Bio-Rad QX200™ Droplet Reader.
The dPCR amplification conditions were as follows: initial denaturation at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 30 s, and annealing at 60 °C for 1 min, with a final extension at 4 °C for 5 min, 90 °C for 5 min, and cooling at 4 °C for 10 min. The quantification of the amplified PCR products was performed with the Bio-Rad QX200™ Droplet Reader. The number of mtDNA copies per sample was calculated by the formula: (mtDNA copies per µl saliva) / (nuclear DNA copies per µl saliva / 2).
Blood biomarkers of AD
Blood samples were collected after an overnight fast, immediately processed, and stored at −80 °C until analysis. Plasma levels of Aβ1-40, Aβ1-42, and pTau-181 along with serum levels of NfL and GFAP, were measured in duplicate using SIMOA assays (Quanterix, MA, USA) on a SR-X analyzer according to the manufacturer’s instructions. The average of the two measurements (pg/ml) was used for statistical analysis. To ensure measurement accuracy and consistency, samples with coefficients of variation greater than 20% were repeated.
Acquisition and preprocessing of MRI scans
Structural brain images were acquired on a Philips Ingenia 3.0 T CX MRI scanner equipped with a 32-channel receive-only radio-frequency (RF) head coil (Philips, Best, Netherlands). A high-resolution 3D T1-weighted magnetization prepared rapid gradient echo (MPRAGE) sequence was obtained in the sagittal plane with no inter-slice gaps using the following parameters: repetition time/echo time = 2600 ms/4.7 ms, flip angle = 9°, acquisition matrix = 384 ×384, and isotropic voxel resolution of 0.65 mm³.
T1-weighted (T1w) images were processed using Freesurfer version 6.0 (https://surfer.nmr.mgh.harvard.edu/), which involved a series of preprocessing steps: brain extraction, automated segmentation of tissue types, and the creation of white matter (WM) and pial surfaces. Additional corrections for surface topology and inflation, as well as co-registration and spherical projection of cortical surfaces, were applied to establish a surface-based coordinate system [22]. Manual adjustments were made by a trained technician on a slice-by-slice basis to correct misalignments in the pial surface and inaccuracies in WM segmentation. Visual inspection was performed at each stage to ensure quality control and accuracy of the processing pipeline.
Acquisition and preprocessing of amyloid PET scans
Participants received an injection of 300 MBq of 18F-Florbetaben (FBB, NeuraCeq™, Curium Pharma) 90 min prior to imaging with a Philips Gemini 16 PET/CT scanner (Philips, Best, Netherlands). A dynamic 20-min FBB-PET scan was conducted, consisting of four 5-min frames. Each frame was reviewed for excessive motion artifacts. As no significant head movements were observed, the four frames were averaged to generate a single static FBB brain image for subsequent quantitative analysis. Amyloid PET images were corrected for radioactive decay, dead time, attenuation, and scatter, and were then reconstructed with an iterative algorithm, yielding an isotropic voxel resolution of 2 mm³.
Partial volume correction (PVC) of the FBB-PET images was performed with the PetSurfer tool (https://surfer.nmr.mgh.harvard.edu/fswiki/PetSurfer), employing a region-based voxel-wise method derived from the geometric transfer matrix [23]. This method assumes a uniform 6 mm point spread function. Initial alignment of the cerebral FBB images to T1w scans was followed by transformation of the PVC-corrected FBB cortical images into a standardized uptake value ratio (SUVR) format, with the mean PVC uptake in the cerebellar grey matter serving as the reference region. The resulting cortical-to-cerebellum SUVR images were then mapped to individual cortical surfaces and smoothed with non-linear spherical wavelet-based denoising techniques [24]. The global Aβ load was computed by obtaining a composite FBB measure for each participant [25].
Statistical analysis
Scores of cognitive tests were transformed into Z-scores for each participant. Z-scores were reversed if a higher score indicates a worse test performance. General cognitive ability was estimated in principal component analysis using the g-factor score, which was derived from the first unrotated component with the highest eigenvalue. Higher values in the g-factor score indicated better cognitive performance. The analysis was performed with R Statistical Software v3.0.1 (R Foundation for Statistical Computing, Vienna, Austria), applying the prcomp function.
Before conducting statistical analyses, we applied a Box-Cox transformation to dependent variables in order to address non-normality and reduce heteroscedasticity [26]. Multiple linear regression models were used to assess the relationships between salivary mtDNA and variables such as age, cognitive performance, and blood biomarkers for AD, including Aβ1-40, Aβ1-42, Aβ1-42/Aβ1-40 ratio, pTau-181, NfL, and GFAP. Additionally, we examined differences in salivary mtDNA as a function of sex, APOE4 genotype, and family history of AD. Age and sex were included as covariates in all models.
To explore variations in cortical Aβ load with respect to salivary mtDNA and plasma pTau-181, we conducted vertex-wise multiple linear regression analyses on cortical surface data. These statistical models were adjusted for age and sex, and dependent variables underwent Box-Cox transformation. We also investigated the interaction between plasma pTau-181 and salivary mtDNA on cortical Aβ load. This statistical approach was similarly adjusted for age and sex.
Cortical surface-based analyses were performed using the SurfStat tool (https://www.math.mcgill.ca/keith/surfstat/). Multiple comparisons were controlled for by applying a hierarchical statistical model [27], which first adjusted for family-wise error rate within significant clusters over smoothed statistical maps using random field theory, followed by false discovery rate control within vertices of significant clusters on unsmoothed maps. The anatomical locations of corrected clusters were identified using the peak vertex positions on the Desikan-Killiany brain atlas [28]. After confirming significant effects, standardized local effect sizes (f2local or rholocal) were calculated for the regression models [29]. To estimate the precision of effect sizes, 95% confidence intervals (CI95%) were determined using the bootci function in Matlab with 10,000 bootstrap samples, applying a normal approximation to the interval based on bootstrapped bias and standard error.
In order to mitigate issues related to multiple comparisons, Bayesian linear regression was also performed on the peak vertex for each significant cortical cluster using JASP v0.12.2 (https://jasp-stats.org/). Bayesian models were specified using the Jeffreys-Zellner-Siow prior with an r scale of 0.354 [30]. The Bayes factor (BF10) was calculated to compare models with and without the predictor of interest (experimental versus null models). To interpret results, we follow the classification scheme by Lee and Wagenmakers [31]. Results were only reported when they met the following criteria: (i) the p-value from the frequentist analysis was <0.05 after correction for multiple comparisons, (ii) the local effect size was at least moderate, with the CI95% excluding zero, and (iii) there was at least moderate evidence in favor of the alternative hypothesis (BF10 ≥ 10).
Finally, we conducted simple mediation analyses with bootstrapping, using the R mediation package, to determine whether the relationship between salivary mtDNA and plasma pTau-181 was mediated by cortical Aβ load. We specifically tested whether Aβ burden in the region where pTau-181 was associated with salivary mtDNA acted as the mediator. The indirect effects were considered significant if the bootstrapped CI95% did not include zero. To ensure that spurious effects did not influence the hypothesized pathway, we also examined alternative models where cortical Aβ was the predictor, pTau-181 the mediator, and salivary mtDNA the outcome variable. Lastly, we assessed whether the relationship between salivary mtDNA and cortical Aβ burden was mediated by global Aβ or by other regions where pTau-181 might influence the Aβ-mtDNA association.
Results
Sample characteristics
Table 1 shows demographics, blood-based AD biomarkers, and global Aβ load. Table 2 shows results of cognitive testing. Salivary mtDNA was not associated with age, sex, APOE4 genotype, first-degree family history of AD, plasma Aβ1-40, Aβ1-42, Aβ1-42/Aβ1-40 ratio, serum NfL or GFAP (Figure S1). However, salivary mtDNA was significantly correlated with general cognitive ability (F3,87 = 4.1, p = 0.04; R2 = 0.06, β = −26.3, p = 0.04) and plasma pTau-181 (F3,87 = 4.6, p = 0.03; R2 = 0.06, β = 6.5, p = 0.03) adjusted for age and sex. The correlation with general cognitive ability was negative (r = −0.21, p = 0.04; Fig. 1A), while the correlation with plasma pTau-181 was positive (r = 0.22, p = 0.03; Fig. 1B).

A Correlations between salivary mtDNA and general cognitive ability (i.e., the g factor). B Correlations between salivary mtDNA and plasma pTau-181 levels. These analyses were adjusted for age and sex.
Associations between salivary mtDNA and cortical Aβ load
Salivary mtDNA was not significantly correlated with global Aβ load, but it was associated with FBB binding to Aβ deposits over posterior cortical regions (F5,85 = 15.7; p = 0.00002, f2local = 0.22 (moderate), CI95% [0.08 – 0.48]). Results are illustrated in Fig. 2. Specifically, salivary mtDNA levels were positively correlated with Aβ load in lateral occipital regions (r = 0.43, p = 0.0001) of the left hemisphere. The Bayesian analysis showed extreme evidence in favor of the alternative hypothesis (BF10 = 882).

(A) Significant results projected onto the cortical surface. (B) The scatter plot shows the relationship between salivary mtDNA levels and the peak vertex of cortical FBB binding, adjusted for age and sex. L: left.
Associations between plasma pTau-181 and cortical Aβ load
Plasma pTau-181 levels were significantly correlated with global Aβ load (F3,87 = 3.2, p = 0.027; R2 = 0.1, β = 0.01, p = 0.01). Bayesian analysis also provided very strong evidence in favor of the alternative hypothesis (BF10 = 60.6). This association was particularly noticeable over parieto-occipital regions in both hemispheres (left: F5,85 = 16.6; p = 0.001, f2local = 0.23 (moderate), CI95% [0.06 – 0.46]; right: F5,85 = 15.7; p = 0.003, f2local = 0.23 (moderate), CI95% [0.07 – 0.62]). In both cases, evidence favoring the alternative hypothesis was high (left BF10 > 2800; right BF10 > 500). These results are displayed in Fig. 3. We also analyzed associations between plasma Aβ1-42, Aβ1-40 levels and FBB binding. Plasma Aβ1-42, but not Aβ1-40, correlated negatively with Aβ load in both cortical hemispheres, affecting canonical AD regions such as posterior cingulate, inferior parietal, and insular cortex (Figure S2).

(A) Significant results projected onto the cortical surface. (B) Scatter plots showing the associations between plasma pTau-181 levels and the peak vertex of cortical FBB binding, adjusted for age and sex. L: left, R: Right.
Moderating effect of plasma pTau-181 on the relationship between salivary mtDNA and cortical Aβ load
Multiple regression analyses showed that the association between salivary mtDNA and cortical Aβ load was significantly moderated by plasma pTau-181 over regions of the temporal lobe in the left (F9,81 = 16.53; p = 0.0006, rholocal = 0.73 (large), CI95% [0.44 – 1.10]) and right hemisphere (F9,81 = 12.16; p = 0.00002, rholocal = 0.51 (large), CI95% [0.19 – 0.84]) (Fig. 4A). Evidence in favor of the alternative hypothesis was very strong for the left (BF10 = 87.4) and moderate for the right hemisphere (BF10 = 6.2). Post hoc analyses revealed that only those participants showing high plasma pTau-181 levels exhibited a positive association of salivary mtDNA with Aβ load in the left parahippocampal (r = 0.61, p = 0.0003) and right fusiform gyri (r = 0.39, p = 0.032) (Fig. 4B).

(A) Significant results projected onto the cortical surface. (B) Scatter plots depicting partial correlations between salivary mtDNA and peak vertices of cortical FBB binding in participants showing high (pink) and low (green) plasma pTau-181 levels, adjusted for age and sex. L: left, R: Right.
Mediation analyses
We conducted several mediation models to assess the hypothesis that cortical Aβ load mediates the association between plasma pTau-181 and salivary mtDNA. Results showed that this association was specifically mediated by FBB binding to Aβ deposits in the left lateral occipital cortex (Fig. 5), but not by global Aβ load (Figure S3A) or by Aβ burden in the temporal lobe (Figure S3,B). As illustrated in Fig. 5, mediation analysis revealed that plasma pTau-181 was indirectly related to salivary mtDNA through its relationship with Aβ accumulation in the left lateral occipital cortex, adjusted for age and sex. In this model, increased plasma pTau-181 was associated with increased Aβ (standardized β = 0.38, p = 0.0005), and increased Aβ was subsequently related to increased levels of salivary mtDNA (standardized β = 0.40, p = 0.0002). A 95% CI based on 10,000 bootstrapped samples of the indirect effect was entirely above zero (1.4 – 8.05), strengthening the significance of the hypothesized mediation pathway. There was no direct effect of plasma pTau-181 on lateral occipital Aβ load (standardized β = 0.08, p = 0.47). Similar results were found when salivary mtDNA was included as independent variable and pTau-181 as dependent variable in the model. In this case, the mediation effect was also significant (standardized β = 0.005, p = 0.004), contrary to the direct effect (standardized β = 0.003, p = 0.49). However, the reverse mediation, with plasma pTau-181 as mediator and temporal Aβ load as independent variable, was not significant (indirect effect = 0.03 [−84.2 – 180.5]) (Figure S3,C), confirming the Aβ-mediated effect.

Schematic representation of a simple mediation model exploring the relationship between FBB binding to Aβ deposits over the left lateral occipital cortex (i.e., the significant cluster displayed in Fig. 2A) as mediator variable, plasma pTau-181 levels as independent variable, and salivary mtDNA levels as dependent variable. Numbers along paths are standardized regression coefficients with the corresponding p value.
Discussion
Previous studies have reported the presence of mtDNA in human saliva [12,13,14,15,16,17,18], but its relationship with AD biomarkers has remained unexplored so far. Here, we showed that higher levels of salivary mtDNA were associated with greater Aβ burden in occipital regions, higher plasma pTau-181 levels, and lower cognitive ability in cognitively normal older adults. Remarkably, plasma pTau-181 levels moderated the association between salivary mtDNA and Aβ accumulation in regions of the inferior temporal lobe. In contrast, the relationship between salivary mtDNA and plasma pTau-181 was mediated by Aβ accumulation in occipital regions. Together, these results provide new evidence linking salivary mtDNA to well-established AD biomarkers, highlighting associations between salivary mtDNA and AD pathology before the onset of clinical symptoms.
Extensive research has supported mitochondrial dysfunction as an early feature of AD, either contributing to the development of Aβ pathology or mediating pathologic molecular cascades [32, 33]. Our results suggest that cortical Aβ load is accompanied by increased levels of salivary mtDNA. High concentrations of circulating mtDNA have been reported in a variety of pathological conditions, such as diabetes, cancer, myocardial infarction and major depressive disorder [3], suggesting that high mtDNA levels may be a potential biomarker and/or predictor of outcome in a variety of diseases. However, the positive correlation between salivary mtDNA and cerebral Aβ load found in our study differs from previous work showing decreased cf-mtDNA levels in asymptomatic patients with pathological levels of Aβ1-42 in CSF samples [8]. The source of cf-mtDNA in the CSF is likely circumscribed to brain parenchyma, while the content of mtDNA in saliva may originate from diverse cell types. We speculate that low levels of cf-mtDNA in CSF are the result of a blunted physiological release of byproducts of mitochondrial quality control due to Aβ aggregation, whereas high levels of salivary mtDNA may be due to increased systemic inflammation. Our mediation analyses supported the two hypotheses as the Aβ-mediated effect remained significant regardless of whether mtDNA was included as a dependent or independent variable in the model. Consequently, salivary mtDNA may hold promise as a non-invasive biomarker of Aβ accumulation in the brain. However, its diagnostic utility remains to be determined through future studies with larger cohorts, along the AD continuum.
In the present study, pTau-181 was the only blood-based AD biomarker that exhibited significant associations with salivary mtDNA. Although plasma Aβ species are commonly used as AD biomarkers, their concentrations are strongly influenced by peripheral production and binding interactions with blood components, which can obscure brain-derived signals and reduce their specificity for central Aβ pathology [34]. Our results are consistent with evidence that plasma pTau-181 is among the most AD-specific plasma biomarkers [35, 36], showing progressive increases across the AD continuum and accurately predicting tau and amyloid pathology in the brain [37, 38], while other plasma AD biomarkers do not perform as well as pTau181 in identifying Aβ pathology [39]. Interestingly, participants with the highest levels of plasma pTau-181 showed the strongest association between salivary mtDNA and cortical Aβ accumulation, consistent with evidence of a synergistic effect of Aβ and tau pathology on disease progression [40]. Indeed, Aβ can promote tau phosphorylation and aggregation [41, 42], and Aβ1-42-induced neurotoxicity depends on tau levels through the activation of glycogen synthase kinase-3 (GSK-3) [43, 44], while both pathologies disrupt mitochondrial integrity and function in a feed-forward loop [45, 46]. Conversely, increased mtDNA mutations have been shown to accelerate Aβ accumulation and propagation [47], worsening mitochondrial dynamics [48]. Despite these plausible mechanisms, we acknowledge that our data do not allow us to conclusively determine why associations with salivary mtDNA were selective for pTau-181. Further studies are needed to confirm this association and to clarify whether the preference for pTau-181 reflects biological mechanisms (e.g., tau-mediated mitochondrial stress and release of cell-free mtDNA) or methodological factors (e.g., differential assay sensitivity).
The biological origin of salivary mtDNA remains unclear [16]. While saliva contains buccal epithelial and white blood cells that could release mtDNA during isolation, previous studies have rule out cellular content as a major contributor to salivary mtDNA [16]. Alternatively, a fraction of salivary mtDNA may originate from blood, given that blood components can passively diffuse into the gingival crevicular fluid, which is in direct contact with saliva [49]. Indeed, many blood-derived compounds are detectable in saliva, as shown by comparative proteomic analysis [50]. Moreover, unprotected extracellular mtDNA passively released into the bloodstream [51] due to blood-brain barrier (BBB) leakage may act as an endogenous damage-associated molecular pattern (DAMP), triggering innate immune responses and potentially contributing to neuroinflammatory cascades [52, 53]. Future studies should clarify whether systemic inflammatory processes underlie the observed associations between salivary mtDNA and plasma pTau-181 levels.
This study has several limitations that should be considered. First, the sample size was relatively small, which may limit the generalizability of our findings. To address this limitation, we only reported results that satisfied both frequentist and Bayesian statistical thresholds and exhibited at least moderate local effect sizes. Future studies with larger and more diverse cohorts are needed to validate and extend these findings to broader contexts. Second, the cross-sectional design of this study precludes any assessment of the prognostic value of salivary mtDNA for AD progression. Follow-up studies are required to determine whether baseline salivary mtDNA levels are able to predict AD progression in cognitively normal older adults, and to examine whether salivary mtDNA levels change along the AD continuum. Finally, we only assessed correlations between salivary mtDNA and plasma pTau-181, limiting our ability to compare with other phosphorylated forms of tau (e.g., pTau-217) that have demonstrated superior accuracy in detecting Aβ pathology [54, 55].
To conclude, the results of the present study provide the first evidence of an association between salivary mtDNA and well-established AD biomarkers in cognitively normal older adults. These results open new avenues for using salivary mtDNA as a potential biomarker to identify individuals at risk for developing AD before the onset of clinical symptoms.
Data availability
Data used for this study are available upon reasonable request to the corresponding author.
References
Son JM, Lee C. Aging: all roads lead to mitochondria. Semin Cell Dev Biol. 2021;116:160–8. https://doi.org/10.1016/j.semcdb.2021.02.006
Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–12. https://doi.org/10.1126/science.120194
Miliotis S, Nicolalde B, Ortega M, Yepez J, Caicedo A. Forms of extracellular mitochondria and their impact in health. Mitochondrion. 2019;48:16–30. https://doi.org/10.1016/j.mito.2019.02.002
Picard M. Blood mitochondrial DNA copy number: What are we counting? Mitochondrion. 2021;60:1–11. https://doi.org/10.1016/j.mito.2021.06.010
Caicedo A, Benavides-Almeida A, Haro-Vinueza A, Peña-Cisneros J, Pérez-Meza ÁA, Michelson J, et al. Decoding the nature and complexity of extracellular mtDNA: types and implications for health and disease. Mitochondrion. 2024;75:101848. https://doi.org/10.1016/j.mito.2024.101848
Michelson J, Rausser S, Peng A, Yu T, Sturm G, Trumpff C, et al. MitoQuicLy: A high-throughput method for quantifying cell-free DNA from human plasma, serum, and saliva. Mitochondrion. 2023;71:26–39. https://doi.org/10.1016/j.mito.2023.05.001
Puigròs M, Calderon A, Martín-Ruiz D, Serradell M, Fernández M, Muñoz-Lopetegi A, et al. Mitochondrial DNA deletions in the cerebrospinal fluid of patients with idiopathic REM sleep behaviour disorder. EBioMedicine. 2024;102:105065. https://doi.org/10.1016/j.ebiom.2024.105065
Podlesniy P, Figueiro-Silva J, Llado A, Antonell A, Sanchez-Valle R, Alcolea D, et al. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol. 2013;74:655–68. https://doi.org/10.1002/ana.23955
Podlesniy P, Llorens F, Puigròs M, Serra N, Sepúlveda-Falla D, Schmidt C, et al. Cerebrospinal fluid mitochondrial DNA in rapid and slow progressive forms of Alzheimer’s disease. Int J Mol Sci. 2020;21:6298. https://doi.org/10.3390/ijms21176298
Gorham IK, Reid DM, Sun J, Zhou Z, Barber RC, Phillips NR. Blood-based mtDNA quantification indicates population-specific differences associated with Alzheimer’s disease-related risk. J Alzheimers Dis. 2024;97:1407–19. https://doi.org/10.3233/JAD-230880
Kaufman E, Lamster IB. The diagnostic applications of saliva–a review. Crit Rev Oral Biol Med. 2002;13:197–212. https://doi.org/10.1177/154411130201300209
Cai N, Chang S, Li Y, Li Q, Hu J, Liang J, et al. Molecular signatures of major depression. Curr Biol. 2015;25:1146–56. https://doi.org/10.1016/j.cub.2015.03.008
Chen Y, Hill HZ, Lange G, Falvo MJ. Salivary mitochondrial DNA copy number is associated with exercise ventilatory efficiency. J Strength Cond Res. 2017;31:2000–4. https://doi.org/10.1519/JSC.0000000000001932
Jiang WW, Rosenbaum E, Mambo E, Zahurak M, Masayesva B, Carvalho AL, et al. Decreased mitochondrial DNA content in posttreatment salivary rinses from head and neck cancer patients. Clin Cancer Res. 2006;12:1564–9. https://doi.org/10.1158/1078-0432.CCR-05-1471
Shin J, Kim KC, Lee DC, Lee HR, Shim JY. Association between salivary mitochondrial DNA copy number and chronic fatigue according to combined symptoms in Korean adults. Korean J Fam Med. 2017;38:206–12. https://doi.org/10.4082/kjfm.2017.38.4.206
Trumpff C, Rausser S, Haahr R, Karan KR, Gouspillou G, Puterman E, et al. Dynamic behavior of cell-free mitochondrial DNA in human saliva. Psychoneuroendocrinology. 2022;143:105852. https://doi.org/10.1016/j.psyneuen.2022.105852
Tymofiyeva O, Henje Blom E, Ho TC, Connolly CG, Lindqvist D, Wolkowitz OM, et al. High levels of mitochondrial DNA are associated with adolescent brain structural hypoconnectivity and increased anxiety but not depression. J Affect Disord. 2018;232:283–90. https://doi.org/10.1016/j.jad.2018.02.024
Sayal L, Hamadah O, Almasri A, Idrees M, Thomson P, Kujan O. Saliva-based cell-free DNA and cell-free mitochondrial DNA in head and neck cancers have promising screening and early detection role. J Oral Pathol Med. 2023;52:29–36. https://doi.org/10.1111/jop.13392
Gramunt N, Sánchez-Benavides G, Buschke H, Diéguez-Vide F, Peña-Casanova J, Masramon X, et al. The memory binding test: development of two alternate forms into Spanish and Catalan. J Alzheimers Dis. 2016;52:283–93. https://doi.org/10.3233/JAD-151175
Alegret M, Valero S, Ortega G, Espinosa A, Sanabria A, Hernández I, et al. Validation of the Spanish version of the Face Name Associative Memory Exam (S-FNAME) in cognitively normal older individuals. Arch Clin Neuropsychol. 2015;30:712–20. https://doi.org/10.1093/arclin/acv050
Puigròs M, Calderon A, Pérez-Soriano A, de Dios C, Fernández M, Colell A, et al. Cell-free mitochondrial DNA deletions in idiopathic, but not LRRK2, Parkinson’s disease. Neurobiol Dis. 2022;174:105885. https://doi.org/10.1016/j.nbd.2022.105885
Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA. 2010;97:11050–5. https://doi.org/10.1073/pnas.200033797
Greve DN, Salat DH, Bowen SL, Izquierdo-Garcia D, Schultz AP, Catana C, et al. Different partial volume correction methods lead to different conclusions: An (18)F-FDG-PET study of aging. Neuroimage. 2016;132:334–43. https://doi.org/10.1016/j.neuroimage.2016.02.042
Bernal-Rusiel JL, Atienza M, Cantero JL. Detection of focal changes in human cortical thickness: spherical wavelets versus Gaussian smoothing. Neuroimage. 2008;41:1278–92. https://doi.org/10.1016/j.neuroimage.2008.03.022
Mormino EC, Betensky RA, Hedden T, Schultz AP, Ward A, Huijbers W, et al. Amyloid and APOE epsilon4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology. 2014;82:1760–7. https://doi.org/10.1212/WNL.0000000000000431
Box GEP, Cox DR. An analysis of transformations. J R Stat Soc Ser B. 1964;26:211–43. https://doi.org/10.1111/j.2517-6161.1964.tb00553.x
Bernal-Rusiel JL, Atienza M, Cantero JL. Determining the optimal level of smoothing in cortical thickness analysis: a hierarchical approach based on sequential statistical thresholding. Neuroimage. 2010;52:158–71. https://doi.org/10.1016/j.neuroimage.2010.03.074
Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–80. https://doi.org/10.1016/j.neuroimage.2006.01.02
Cohen JE. Statistical Power Analysis for the Behavioral Sciences. Hillsdale, New Jersey: Lawrence Erlbaum Associates; 1988.
Liang F, Paulo R, Molina G, Clyde MA, Berger JO. Mixtures of g priors for Bayesian variable selection. J Am Stat Assoc. 2008;103:410–23. https://doi.org/10.1198/016214507000001337
Lee MD, Wagenmakers EJ. Bayesian Cognitive Modeling: A Practical Course. Cambridge University Press; 2013.
Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis. 2018;62:1403–16. https://doi.org/10.3233/JAD-170585
Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15:30. https://doi.org/10.1186/s13024-020-00376-6
Marcello A, Wirths O, Schneider-Axmann T, Degerman-Gunnarsson M, Lannfelt L, Bayer TA. Circulating immune complexes of Abeta and IgM in plasma of patients with Alzheimer’s disease. J Neural Transm. 2009;116:913–20. https://doi.org/10.1007/s00702-009-0224-y
Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26:379–86. https://doi.org/10.1038/s41591-020-0755-1
Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19:422–33. https://doi.org/10.1016/S1474-4422(20)30071-5
Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement. 2018;14:989–97. https://doi.org/10.1016/j.jalz.2018.02.013
Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387–97. https://doi.org/10.1038/s41591-020-0762-2
Kwon HS, Lee EH, Kim HJ, Park SH, Park HH, Jeong JH, et al. Predicting amyloid PET positivity using plasma p-tau181 and other blood-based biomarkers. Alzheimers Dement (Amst). 2023;15:e12502. https://doi.org/10.1002/dad2.12502
Pascoal TA, Mathotaarachchi S, Shin M, Benedet AL, Mohades S, Wang S, et al. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement. 2017;13:644–53. https://doi.org/10.1016/j.jalz.2016.11.005
Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–24. https://doi.org/10.1073/pnas.1017033108
De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol Aging. 2008;29:1334–47. https://doi.org/10.1016/j.neurobiolaging.2007.02.029
Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, et al. Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci Lett. 1996;203:33–6. https://doi.org/10.1016/0304-3940(95)12257-5
Koh SH, Noh MY, Kim SH. Amyloid-beta-induced neurotoxicity is reduced by inhibition of glycogen synthase kinase-3. Brain Res. 2008;1188:254–62. https://doi.org/10.1016/j.brainres.2007.10.064
Ittner LM, Götz J. Amyloid-β and tau–a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12:65–72. https://doi.org/10.1038/nrn2967
Torres AK, Jara C, Park-Kang HS, Polanco CM, Tapia D, Alarcón F, et al. Synaptic mitochondria: an early target of amyloid-β and tau in Alzheimer’s disease. J Alzheimers Dis. 2021;84:1391–414. https://doi.org/10.3233/JAD-215139
Kukreja L, Kujoth GC, Prolla TA, Van Leuven F, Vassar R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol Neurodegener. 2014;9:16. https://doi.org/10.1186/1750-1326-9-16
Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid β with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–509. https://doi.org/10.1093/hmg/ddr139
Subbarao KC, Nattuthurai GS, Sundararajan SK, Sujith I, Joseph J, Syedshah YP. Gingival crevicular fluid: An overview. J Pharm Bioallied Sci. 2019;11:S135–S139. https://doi.org/10.4103/JPBS.JPBS_56_19
Loo JA, Yan W, Ramachandran P, Wong DT. Comparative human salivary and plasma proteomes. J Dent Res. 2010;89:1016–23. https://doi.org/10.1177/0022034510380414
Gambardella S, Limanaqi F, Ferese R, Biagioni F, Campopiano R, Centonze D, et al. ccf-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders. Front Immunol. 2019;10:1064. https://doi.org/10.3389/fimmu.2019.01064
Frank MO. Circulating cell-free DNA differentiates severity of inflammation. Biol Res Nurs. 2016;18:477–88. https://doi.org/10.1177/1099800416642571
Newman LE, Shadel GS. Mitochondrial DNA release in innate immune signaling. Annu Rev Biochem. 2023;92:299–332. https://doi.org/10.1146/annurev-biochem-032620-104401
Jack CR Jr, Wiste HJ, Algeciras-Schimnich A, Weigand SD, Figdore DJ, Lowe VJ, et al. Comparison of plasma biomarkers and amyloid PET for predicting memory decline in cognitively unimpaired individuals. Alzheimers Dement. 2024;20:2143–54. https://doi.org/10.1002/alz.13651
Mendes AJ, Ribaldi F, Lathuiliere A, Ashton NJ, Janelidze S, Zetterberg H, et al. Head-to-head study of diagnostic accuracy of plasma and cerebrospinal fluid p-tau217 versus p-tau181 and p-tau231 in a memory clinic cohort. J Neurol. 2024;271:2053–66. https://doi.org/10.1007/s00415-023-12148-5
Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness, grants PID2023-149270OB-I00 (JLC), PID2023-151095NB-I00 (MA), and PID2023-153168OB-I00 (RT, PP); Instituto de Salud Carlos III “European Union NextGenerationEU/PRTR”, grant PMP22/00100 (RT), and CIBERNED (JLC, RT). We also thank the participants and their family for their collaboration in this study.
Author information
Authors and Affiliations
Contributions
JLC conceived the study and wrote the first draft. MA and JLC collected data, performed analysis, and prepared figures and tables. PP, MP and RT measured mtDNA content in saliva. All authors contributed to data interpretation and discussion of results, revised the article for important intellectual content, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cantero, J.L., Atienza, M., Podlesniy, P. et al. Salivary mitochondrial DNA is associated with biomarkers of Alzheimer’s disease in cognitively normal older adults. Transl Psychiatry 15, 355 (2025). https://doi.org/10.1038/s41398-025-03589-9
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03589-9
