
Abstract
Transposable elements (TEs) constitute over half of the human genome and have played a profound role in genome evolution. While most TEs have lost the ability to transpose, many retain functional elements that serve as drivers of genome innovation, including the emergence of novel genes and regulatory elements. Recent advances in experimental and bioinformatic methods have provided new insights into their roles in human biology, both in health and disease. In this review, we discuss the multifaceted roles of TEs in haematopoiesis, highlighting their contributions to both normal and pathological contexts. TEs influence gene regulation by reshaping gene-regulatory networks, modulating transcriptional activity, and creating novel regulatory elements. These activities play key roles in maintaining normal haematopoietic processes and supporting cellular regeneration. However, in haematological malignancies, TE reactivation can disrupt genomic integrity, induce structural variations, and dysregulate transcriptional programmes, thereby driving oncogenesis. By examining the impact of TE activity on genome regulation and variation, we highlight their pivotal roles in both normal haematopoietic processes and haematological cancers.
Similar content being viewed by others


Introduction
Transposable elements (TEs) are dispersed repetitive DNA sequences that have the ability to move within the host genome. First discovered in the 1940s by Barbara McClintock in the maize genome [1], TEs are now recognised across all organisms with varying composition and abundance. In humans, over 46% of the genome consists of TEs according to the telomere-to-telomere (T2T) CHM13 reference genome annotation (Fig. 1) [2]. Based on their mode of transposition, TEs are classified into two major classes [3]. Class I TEs (retrotransposons) transpose into the genome via reverse-transcribed RNA intermediates using a “copy-and-paste” mechanism. Retrotransposons are further subdivided into long terminal repeat (LTR) and non-LTR retrotransposons. LTR retrotransposons, including endogenous retroviruses (ERVs), are remnants of ancient integration events [4] and comprise around 9% of the human genome. Full-length ERVs harbour internal coding regions for viral proteins (gag, pol and env), flanked by two LTRs. However, 90% of ERVs exist as solo LTRs in the human genome [5]. Another major subclass of retrotransposons is long interspersed nuclear elements (LINEs). LINEs constitute ~21% of total DNA with ~500,000 copies [6]. Despite their abundance, only about 100 of these copies per genome remain fully functional and capable of retrotransposition [7, 8]. LINE-1 is the only autonomous non-LTR retrotransposon in the human genome, facilitating the mobilization of non-autonomous short interspersed nuclear elements (SINEs). SINEs, including the primate specific Alu elements, comprise around 13% of our DNA. While LINE-1s rely on RNA Polymerase II (Pol II) for their transcription [9], Alu elements encode a weak internal RNA Pol III promoter [10]. Class II TEs are DNA transposons that use a “cut-and-paste” mechanism for transposition and comprise ~3% of the human genome. There are no endogenous DNA transposons that are capable of transposition in the human genome [11].

TEs occupy 46% of the human genome based on the the telomere-to-telomere (T2T) CHM13 reference genome annotation (left). TEs are divided into two major classes based on their transposition mechanism- Class I TEs (retrotransposons) use copy-and-paste mechanism and are further subdivided into long terminal repeat (LTR), long interspersed nuclear element (LINE) and short interspersed nuclear element (SINE) superfamilies. LINE-1s are the only autonomous transposons currently active in the human genome. Class II TEs (DNA transposons) use cut-and-paste mechanism to integrate into the genome, however they have lost their ability for transposition in the human genome (right). Created in BioRender.
Although the vast majority of TEs in humans have lost their ability to retrotranspose due to accumulated mutations or host-mediated repression, many still retain functional sequences that have significantly impacted genome evolution [12]. This includes the domestication of TEs, a process by which originally mobile genetic elements are co-opted for new functions within the host genome. Notable examples of domesticated TEs include DNA transposon-derived piggyBac transposable element-derived protein 5 (PGBD5) [13], which induces site-specific DNA rearrangements in human cancers [14]; the recombination-activating genes RAG1 and RAG2, derived from Transib DNA transposons, which play a central role in the V(D)J recombination process [15]; and ERV-W-derived Syncytin gene, which contributes to placental development in mammals [16].
Beyond domesticated elements, other functional TEs continue to influence genome regulation, contributing to both genetic and epigenetic diversity with far-reaching consequences for both normal cellular processes and disease states. Derepression of TEs in cancer, for example, can lead to aberrant oncogene expression, inactivation of tumour suppressor genes or trigger genomic instability. Conversely, TE activation can contribute to cellular plasticity and shape lineage-specific transcriptome during normal haematopoiesis. In this review, we first provide an overview of the regulatory mechanisms that modulate TE activity (Box 1; Fig. 2). We then examine the multifaceted roles of TEs in shaping normal and pathological haematopoietic processes, focusing on their gene regulatory activities and contribution to structural genomic variations.

Various transcriptional and post-transcriptional mechanisms control the activity of TEs. Epigenetic repression of TEs is primarily achieved through the sequence-specific recognition of TE elements by KRAB-Zinc Finger Proteins (KRAB-ZFPs). These proteins recruit the KAP1 cofactor, which, in turn, facilitates the formation of a repressive chromatin structure by recruiting histone modifying enzymes, such as SETDB1 and histone deacetylase (HDAC), leading to histone methylation and deacetylation. DNA methyltransferases (DNMTs) deposit repressive DNA cytosine methylation marks on TE sequences, further silencing their activity. The maintenance of this silencing is supported by DNA methylation maintenance mechanisms during replication, primarily involving DNMT1 and UHRF1. Additionally, the human silencing hub (HUSH) complex targets young TE families and recruits SETDB1 to silence target TEs. Moreover, RNA modifications, including m6A and m5C, influence TE transcript stability, splicing, and translation, adding an additional layer of regulation. Created in BioRender.
Gene regulatory activity of TEs
The concept of TEs as influential components of gene regulators dates back to Barbara McClintock’s discovery of their “controlling elements” in maize, where she proposed their role in modulating gene activity [17]. This early insight was further advanced by Britten and Davidson, who suggested that TEs could introduce new regulatory sequences that drive the evolution of gene networks by spreading throughout the genome [18]. Today, we understand that TEs contribute significantly to the regulatory landscape of multicellular organisms, with recent studies from the ENCODE and Roadmap projects indicating that ~25% of candidate cis-regulatory elements in the human genome are derived from TEs [19, 20]. TEs contain their own cis-regulatory elements and transcription factor (TF) binding sites, which are essential for their transcription and transposition within the genome [20,21,22,–23]. Over evolutionary time, TEs can accumulate mutations, which eventually immobilizes them [24]. However, during this evolutionary process, stochastic mutations (such as CpG deamination) can randomly retain or introduce TF binding sites in TEs. Some of these changes may be preserved if they confer regulatory advantages and are favoured by natural selection, thereby allowing TEs to contribute to tissue- and lineage-specific regulatory networks [25, 26], underscoring their adaptive potential in shaping species- and cell-type-specific landscapes. In this section, we cover examples of TE-derived cis-regulatory elements and transcripts in the context of both normal and pathological haematopoiesis (Fig. 3).

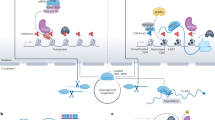
TEs provide a rich source of cis-regulatory elements that are spread across the genome. When active or accessible, the gene regulatory potential of TEs is exploited by the host genome in many ways in normal and malignant haematopoiesis. (i) TEs can act as enhancers through the recruitment of TFs and interacting with nearby promoters, modulating expression. Where this is a proto-oncogene, TEs can contribute to the malignant phenotypes. (ii) TEs can also produce lncRNAs, where 83% of lncRNAs are reported to contain a TE. lncRNAs can have diverse functions from stabilising promoter-enhancer interactions to the recruitment of epigenetic modifiers to chromatin. (iii) TEs can also function as alternative promoters, leading to generation of chimeric transcripts, which may promote a pathogenic phenotype. (iv) Alternative splicing events can occur from the exonisation of TEs, where they may provide alternate splice donor and acceptor sites. These aberrantly spliced transcripts may impair normal protein function, result in mRNA degradation or be translated into neoantigens. (v) TEs are a reservoir for CTCF binding sites and other anchor proteins, thereby retaining the potential to form neo-TADs which can bring cis-regulatory sequences to non-target genes and modulate their expression. This can be in the form of enhancer-promoter interactions to increase gene expression, or the inverse insulator-promoter interactions that decrease gene expression. Created in BioRender.
TE-derived alternative promoters
TEs can be integrated into various genomic regions. By introducing their own promoter elements, TEs can create alternative transcription start sites (TSSs) [27], thereby expanding the regulatory potential to neighbouring genes. In fact, it is estimated that between 18–31% of human TSSs overlap with TEs, emphasising their role as promoters in the human genome [28, 29]. Transcription initiated from TE promoters may extend into adjacent genes, generating chimeric transcripts that combine TE sequences with gene exons, enhancing transcriptomic diversity. For example, in murine erythroid cells, loss of transcriptional repressor KLF3 activates an ORR1A0 LTR that drives a chimeric PU.1 transcript with dominant negative activity, promoting erythroid differentiation [30].
In cancer, epigenetic dysregulation often reactivates TE-derived promoters, leading to widespread TE expression. Recent studies have highlighted specific TE expression patterns that correlate with disease subtypes and patient outcomes in haematological cancers, including AML and lymphoid malignancies, although the mechanisms underlying these associations remain unclear [31,32,33,–34].
In some cases, derepressed TEs drive the formation of cancer-specific chimeric transcripts leading to oncogene activation. An example of such a case was reported in Hodgkin’s Lymphoma, where the THE1B retrotransposon acts as an alternative promoter for the colony-stimulating factor 1 receptor (CSF1R) proto-oncogene. Transcription in these cells initiates from a THE1B LTR located ~6.2 kb upstream of the normal TSS. This ectopic expression produces a noncanonical transcript with an extended 5′ UTR, which is essential for tumour cell proliferation and survival [35]. Similarly, the LOR1a retrotransposon activates interferon regulatory factor 5 (IRF5) transcription in the same lymphoma type, promoting overexpression of this factor and supporting cancer cell proliferation [36]. In diffuse large B-cell lymphoma (DLBCL), a chimeric transcript initiated by the activation of an LTR2 element leads to the expression of the modified form of Fatty Acid-Binding Protein 7 (FABP7) [37]. This LTR-driven FABP7 isoform produces a chimeric protein with an altered N-terminus, which is essential for cell proliferation and growth in DLBCL cell lines.
Cancer-specific TE-derived chimeric transcripts have also been reported in AML, such as LTR2C-SAGE1 and LTR2B-RHEX transcripts, which are not expressed in healthy myeloid cells [38]. Further underscoring the impact of TE-derived chimeric transcripts, a recent study by Shah and colleagues [39] identified 1088 of such transcripts across nine different blood cancer types. Multiple myeloma exhibited the highest number with 662 events, followed by AML with 159 events, illustrating considerable variation among cancers. Interestingly, genes such as TERT, BUB1, and GABRA3 were frequently involved in chimeric transcript formation. LINEs accounted for 43.57% of these chimeric transcripts, followed by SINEs (29.14%) and LTRs (25.46%).
These findings highlight the diversity and significance of TE-driven chimeric transcripts across blood cancers, with links to disease phenotypes and clinical outcomes. However, ex vivo and in vivo models that directly demonstrate the oncogenic potential of such TE-mediated transcripts remain limited. Functional validation approaches, such as CRISPR/Cas9-mediated deletion or RNA silencing, are necessary to assess the contribution of TE promoters to cellular proliferation and survival. Recent advances in long-read sequencing technologies and computational tools (Box 2) offer greater precision in TE mapping and the identification of these transcripts, potentially revealing more TE-derived transcripts in blood cancers.
Beyond their role in transcriptomic diversity, in some cases TE-derived transcripts are translated into proteins, which may lead to the formation of novel TE-chimeric antigens or cryptic ORFs [39,40,41,–42], that are recognised by immune cells and influence immune responses in haematopoietic malignancies. Aberrant TE expression may also give rise to TE-derived double-stranded RNAs which can activate innate immune sensors such as RIG-I and MDA5, triggering inflammatory pathways. These TE-derived proteins and immunogenic RNAs may contribute to both tumour immunity and immune evasion in haematological cancers, which has been extensively reviewed elsewhere [43] and are not included in this review.
TE-derived lncRNAs
TEs also provide a major promoter source for long noncoding RNAs (lncRNAs). LncRNAs derived from TEs play a crucial role in cancer by modulating gene expression, influencing chromatin structure, and interacting with RNA-binding proteins, as reviewed in [44]. In contrast to protein coding genes, 83% of lncRNAs contain TE sequences with TEs comprising 30–42% of lncRNA sequence lengths [45, 46].
Although much is known about the involvement of lncRNAs in cancer and haematopoiesis, particularly in regulating key processes such as cell proliferation, differentiation, and drug resistance [47], specific regulatory mechanisms linking TEs and lncRNAs in these contexts are less understood and represent an emerging area of research. For instance, lncRNAs such as NALT [48] and UCA1 [49] have been well-documented for their roles in promoting cell proliferation in leukaemia via pathways such as NOTCH and p27kip1 suppression, respectively. Similarly, lncRNAs such as HOTAIRM1 [50] and LINC00173 [51] are crucial in regulating haematopoiesis by influencing lineage specification.
However, only a few studies have examined the specific roles of TE-derived lncRNAs in haematopoiesis and blood malignancies. For example, novel lncRNAs identified in B-cell lymphoma are enriched at super-enhancer regions and may utilise TE-derived polyadenylation signals, suggesting a complex regulatory interplay [52]. Additionally, some findings suggest that ERV9 retrotransposon-derived lncRNAs may act in cis to stabilize enhancer complexes in erythroid cells, indicating their potential in lineage-specific gene regulation [53]. Yet, the broader roles and mechanisms of TE-derived lncRNAs in blood malignancies remain largely unexplored. The role of TE-derived lncRNAs may remain a ‘hidden’ area: many transcripts annotated as lncRNAs might actually be TE-derived RNAs misannotated as lncRNAs. This represents a significant gap in our understanding, highlighting the need for further research to explore this connection.
TEs and alternative splicing
Another way that TEs contribute to the diversity of the transcriptome is through alternative splicing, enabling multiple mRNA species to be generated from a single gene. The role of TEs in alternative splicing in the human genome was first demonstrated in Alu elements, where intronic Alu elements generate new exons, thereby affecting expression patterns of numerous genes [54]. Pan-cancer studies have shown that TE-derived alternative splicing events are widespread in cancer [55, 56]. Alu and LINE-1 elements are particularly influential in this process, introducing novel splice acceptor and donor sites, contributing to cancer-specific and recurrent alternative splicing events [55, 56]. Interestingly, the splice donor and acceptor sites within TEs are not randomly distributed but instead cluster at specific hotspots across different TE subfamilies. For example, almost all splicing sites within L1Hs elements are near their 5′ end, whereas all splicing sites within Alus are on their antisense strands [57]. While pan-cancer studies have demonstrated the widespread nature of TE-derived alternative splice sites, these studies predominantly focus on solid tumours and largely exclude haematological cancers, mainly due to the lack of appropriate healthy control tissue. Although the role of TEs in haematological cancers has been less explored, notable examples exist, such as the exonization of an Alu element in the TIF-IA gene, which occurs predominantly in leukaemia cell lines and not in healthy cells [58]. Additionally, truncated forms of the ERBB4 oncogene in anaplastic lymphoma kinase-negative anaplastic large-cell lymphoma (ALK(-)ALCL) have been shown to arise from an endogenous LTR-driven alternative splicing event [59]. The authors reported that two ERBB4 transcript isoforms were generated through the activation of intronic TSSs derived from MLT1H2 and MLT1C elements. Furthermore, a recent comprehensive pan-cancer analysis across 34 cancer types demonstrated that AML samples exhibit among the highest TE-derived exonization events, with an average of 39 events per sample [57]. Moreover, such noncanonical exonization events mediated by TEs can encode tumour-specific immunogenic peptides as demonstrated in lung cancer [60]. Although not yet studied in haematological malignancies, similar immunogenic TE-mediated splicing events may contribute to immune surveillance and hold promise for targeted immunotherapies.
Given the numerous somatic mutations identified in components of the RNA splicing machinery, such as SF3B1, SRSF2, and U2AF1, in haematological malignancies and clonal heamatopoiesis [61], it is crucial to investigate how these mutations may exacerbate TE-derived alternative splicing events, potentially contributing to disease pathology. This highlights a critical need for further research to determine the extent to which TE activity contributes to splicing dysregulation in normal and malignant haematopoiesis, and whether these TE-derived splice variants hold potential as novel therapeutic targets or biomarkers.
TEs as enhancers
Beyond their roles in generating diverse transcripts, some TEs have been exapted to act as enhancers, influencing gene expression programmes. The first documented example of a TE-derived enhancer in haematopoiesis is the ERV9-LTR element which drives the expression of beta-globin in erythroid cells [62]. This element is conserved during primate evolution and gained enhancer function in embryonic and erythroid cells [63]. In humans, the ERV9 enhancer directly binds to NFY and GATA2 TFs, which subsequently interacts with haematopoietic TFs, facilitating long-range chromatin interactions that enhance beta-globin expression [64, 65]. Interestingly, this enhancer complex is stabilized by ERV9-derived lncRNAs, creating a positive feedback loop that reinforces enhancer-promoter interactions and sustains high levels of beta-globin expression during erythropoiesis [53]. Another TE-derived enhancer influencing normal haematopoiesis is a MIR element, integrated into the genome ~130–160 million years ago during the mammalian/marsupial branchpoint. This ancient MIR element transposed into the enhancer region of the HSC-associated TAL1 gene, contributing to its regulation in HSCs in humans and mice [66]. Beyond these individual TE examples, genome-wide analyses of the Roadmap Epigenomics Project have shown that TEs contribute to regions with epigenetic enhancer profiles more frequently in haematopoietic lineages than in other human tissues [19]. For instance, 84 TE subfamilies –including ERVs, SINEs, and LINE-2s– are overrepresented in enhancer domains of mouse CD8 + T cells, which are commonly shared with other immune lineages [67]. These immune cell-specific enhancers contain a higher density of TEs compared to active enhancers in non-immune tissues, and TEs are more abundant near immune-related genes. This suggests that TEs have played a critical role in the evolution of immune regulatory networks by integrating functional motifs that fine-tune the expression of immune genes, thereby providing adaptive advantages [67]. Yet, much of the evidence remains correlative. In addition, during the endothelial-to-haematopoietic transition (EHT)—a critical stage in HSC emergence — specific TE subtypes are differentially accessible and overlap with distal enhancer regions, indicating a potential role for TE-derived enhancers in driving HSC formation [68]. However, this study mostly relies on epigenomic signatures that may not directly correlate with functional activity in vivo and the causality of the enhancer roles of TEs in HSCs requires direct functional validation.
Given their contribution to haematopoietic enhancers under physiological conditions, it is plausible that dysregulated TE activity could contribute to the aberrant gene expression seen in haematological malignancies. In fact, genome-wide analyses in various blood cancers have attempted to link epigenetic dysregulations within TEs with their potential enhancer function. For instance, in a study of paediatric acute lymphocytic leukaemia (ALL), TEs, particularly SINEs and LTRs, were frequently found in ALL-specific differentially methylated regions compared to healthy controls. Although this does not provide direct evidence of TE contributions to ALL enhancers, an association was observed between focal hypomethylation in enhancer-marked loci and the abundance of specific TEs [69]. Similarly, another study of 119 chronic lymphocytic leukaemia (CLL) patients from the International Cancer Genome Consortium identified significant differential methylation in retrotransposons, namely LINE-1 and Alu elements [70]. Most of these regions showed hypomethylation with enrichment at enhancers and locus-specific hypomethylation correlated with the differential expression of proximal genes. Additionally, a large cohort study from AML demonstrated that MIR retrotransposons within enhancers of AML-associated genes were highly susceptible to DNA methylation changes in a subtype-specific manner, further linking TE methylation status to their gene regulatory role [71].
While these studies have established correlative links between TE hypomethylation and enhancer activity, direct evidence of TE-derived enhancers shaping gene regulatory networks in haematological cancers is still emerging. For example, Zeng et al. [72] validated the enhancer function of MIR elements, enriched within chromatin accessibility sites in AML patient samples, using a reporter assay in vitro. Furthermore, we previously provided direct evidence for several LTR elements functioning as active enhancers and contributing to transcriptional networks in AML [38]. Accordingly, we identified six LTR subfamilies, including LTR12C and LTR2/2B, that are differentially accessible and bear enhancer-like chromatin signatures in AML patient samples compared to healthy myeloid cells. CRISPR-Cas9 mediated locus-specific genetic deletion and family-level epigenetic silencing of these LTR-derived enhancers led to the downregulation of nearby AML-expressed genes in AML cell lines. Furthermore, we demonstrated that deletion of an LTR2 element within the enhancer region of the APOC1 gene resulted in significant downregulation of APOC1 expression, reduced cell proliferation, and increased apoptosis, revealing a direct role for TE-derived enhancers in AML cell survival and gene regulatory networks [38]. One limitation of these findings is that they were primarily based on cell line studies, and it remains unclear whether loss of a single enhancer would produce similar effects in primary AML cells, which exhibit greater heterogeneity and epigenetic plasticity. Notably, Grillo et al. further demonstrated that chromatin accessibility at one of these LTR subfamilies, LTR12C, is highly enriched in leukaemia stem cells (LSCs) and is essential for maintaining stemness properties [73]. By applying a CRISPR-dCas9 mediated targeting approach in an AML cell line, they showed that epigenetic silencing of the LTR12C subfamily significantly reduced LSC fractions, confirming that LTR12C accessibility plays a critical role in sustaining LSC identity and hierarchical cellular organisation [73]. These findings highlight how, beyond correlative studies, direct functional assays reveal the enhancer roles of TEs in AML, with implications that may extend to other blood cancers.
TEs and 3D genome organisation
The human genome is organized into topologically associated domains (TADs), which are regions of DNA anchored at distal sequences to facilitate cis interactions within the same domain. Within TADs, cis-regulatory elements interact with gene promoters to modulate their expression [74,75,–76]. Central to the formation of TADs and chromatin interactions are main architectural proteins and TFs such as CTCF, YY1 and ZNF143 [77, 78].
During haematopoiesis, the 3D structure of the genome undergoes dynamic changes to modulate long-range enhancer-promoter interactions that drive cell-specific transcriptional programmes. CTCF plays a crucial role in regulating these boundaries of higher-order chromatin structures during haematopoiesis [79, 80]. Importantly, CTCF’s role is not limited to normal haematopoiesis; alterations in CTCF binding sites are associated with changes in 3D chromatin conformation in AML [81]. A growing body of evidence suggests that many CTCF binding sites specific to humans and other species are derived from TEs [74, 82, 83]. For instance, Choudhary et al. identified a LINE-1 (L1MC) locus that forms a human-specific chromatin loop mediated by CTCF. Disruption of this LINE-1 element via CRISPR knockout alters gene expression at its interacting locus, directly demonstrating the functional importance of this TE-derived binding site [82]. Notably, TE-derived CTCF binding sites are cell-type specific, with over 80% of these sites specific to leukaemia cell lines compared to lymphoblast cells [84].
YY1 is another looping factor also associated with CTCF binding sites that are better conserved among mammals [85]. YY1 frequently binds to TE-derived sequences, particularly regulatory sequences of LINE-1 elements, with potential implications for transcriptional regulation [86,87,–88]. Interestingly, high YY1 expression has been linked to impaired differentiation in AML cells [89]. Similarly, ZNF143 also interacts with CTCF to regulate chromatin organisation [90]. In a mouse model, ZNF143 has been shown to be essential for embryonic HSC development and the formation of a subset of CTCF binding sites [78].
More directly, a recent study in human embryonic stem cells (ESCs) showed that TEs with active histone acetylation marks are significantly enriched at TAD borders and form chromatin loops with genes, suggesting their involvement in chromatin looping and 3D genome organisation [91]. YY1 is enriched at such acetylated LINE-1s, while such LTRs and Alu elements are enriched in the looping factor ZNF143. Moreover, the binding of other key factors such as CTCF and RAD21 were shown to be more pronounced at TEs marked with H4K16ac and H3K27ac, suggesting that histone modifications play a crucial role in TEs’ contribution to 3D chromatin structure via binding to these anchoring and looping factors. Notably, beyond these looping factors, TEs also contribute to chromatin organisation through the formation of insulator elements, independently of CTCF. For example, MIR-derived insulators have been shown to act as chromatin barriers and regulators of gene expression in T cells [92].
Together, these findings highlight the potential of TEs helping to partition the genome into regulatory domains, expanding the functional repertoire of TE-derived elements in 3D genome organisation. These elements may be integral to the dynamic regulation of chromatin organisation in both normal and malignant haematopoiesis. However, direct evidence and functional assays confirming these roles are still lacking. Targeted genetic and epigenetic editing studies are required to definitively establish how these TE-derived sequences modulate chromatin conformation in haematopoietic contexts.
TE-associated structural variations
Besides their gene regulatory roles, TEs also play a pivotal role in shaping genome structure through their contributions to structural variations. These elements drive genomic changes through various mechanisms including insertional mutagenesis via retrotransposition, generating polymorphisms, and inducing chromosomal rearrangements through recombination of homologous repeats. The following sections explore how each of these processes contributes to genomic instability and disease development, particularly in haematological malignancies.
Insertional mutagenesis
Although the vast majority of TEs in the human genome are inactive, some young LINE and SINE elements retain their capacity to mobilise, occasionally causing insertional mutagenesis through sporadic retrotransposition events [93]. One of the first described human disorders linked to LINE retrotransposition is haemophilia A, an X-linked blood clotting disorder resulting from a deficiency in Factor VIII [94]. In 1988, Kazazian and colleagues discovered de novo insertions of LINE-1 sequences in exon 14 of the Factor VIII gene in two unrelated patients, providing the first evidence that LINE-1 is an active mobile element in humans. This potential for insertional mutagenesis is particularly relevant in tumours, where epigenetic dysregulations can lead to increased TE activity, although such somatic integrations rarely contribute to oncogenesis. The first report and a unique instance of TE-driven oncogenesis involves a LINE-1 insertion that disrupts the tumour suppressor adenomatous polyposis coli (APC) gene, leading to the development of colon cancer [95]. Many studies have since systematically investigated retrotransposition events across various cancer types [96,97,98,99,–100]. Overall, these events are found in high proportions across many cancers, especially in epithelial tumours (such as lung, prostate, ovarian, and colon cancers). Most of these events occur in intergenic regions or are associated with passenger mutations, underscoring the rarity of TE somatic insertions that drive cancer development and progression. On the other hand, these pan-cancer studies demonstrated that blood cancers show very low to undetectable levels of retrotransposition events [96,97,98,–99]. In line with this, an unexpected tumour suppressive role for LINE-1 elements has recently been described in AML, where LINE-1 silencing is correlated with poor prognosis [101]. The authors showed that the loss of MPP8, a component of the HUSH complex, negatively impacts AML progression by promoting the retrotransposition of LINE-1s, which induces a DNA damage response and cell cycle exit. LINE-1 retrotransposition is initiated by the endonuclease activity of ORF2p, which cleaves a single strand of DNA to enable reverse transcription [102]. This process compromises DNA integrity and can lead to the formation of double-strand breaks, either directly or through cellular responses to accumulated DNA damage, potentially activating the DNA damage response and triggering genomic instability [103, 104]. In fact, Gu et al. demonstrated that LINE-1 reactivation in AML increases the formation of γH2A.X foci, a well-established marker for double-strand DNA breaks [101]. This link between high LINE-1 activation and genomic instability in AML may explain why haematological cancers exhibit low LINE-1 activity; clones with high LINE-1 retrotransposition face reduced fitness due to DNA damage, compromising their survival.
TE-derived polymorphism
While the above mentioned somatic retrotranspositions do not segregate among populations, germline retrotransposition of TEs can result in polymorphic TE loci, where individuals vary in the presence or absence of TE insertions. As such, around 20% of Alu and 15% of LINE-1–mediated deletions are polymorphic in the human population [105]. Recent studies have highlighted the significance of TE insertion polymorphisms, such as ERV-K in the 8q24.13 -8q24.21 region, which is associated with AML cases through disrupting key cancer driver genes such as MYC and PVT1 [106]. Similarly, polymorphic Alu insertions in MEF2C and TAX1BP1 genes have been linked to allele-specific reductions in transcriptional activity in ALL, potentially impacting nearby gene expression and contributing to leukaemogenesis [107].
The pangenomic approach is helpful for such polymorphic TE analyses, because it integrates the wide structural variations across different human populations [108]. Moreover, long-read sequencing technologies enable more precise detection and characterization of polymorphic TEs [109, 110]. Unfortunately, these advanced methodologies have yet to be applied in studies focusing on normal haematopoiesis and blood cancers.
The impact of TEs on genetic diversity is further complemented by single nucleotide polymorphisms (SNPs) within TEs, which can modify nearby gene expression. For instance, an SNP in an LTR5B-derived enhancer element disrupts a MAFK-binding motif, leading to reduced RPL7L1 expression, a gene that is upregulated in AML [38]. These examples underline the role of both insertion polymorphisms and SNPs within TEs as contributors to genetic and functional diversity in haematological cancers.
TE-mediated chromosomal rearrangements
TEs can contribute to chromosomal rearrangements through non-allelic homologous recombination and non-homologous repair mechanisms, leading to deletions, duplications, translocations, and inversions in the genome [105]. These TE-mediated chromosomal rearrangements and translocations have been implicated in various cancers, including blood cancers. Particularly, Alu elements act as hotspots for ectopic recombination events due to their high abundance and repetitive nature. These recombination events between non-allelic Alu elements can lead to gene duplications, as in the case of partial tandem duplication of the MLL gene frequently occurring in AML patients [111] and tandem duplication of the MYB oncogene in T-cell acute lymphoblastic leukemia (T-ALL) [112]. Alu elements have also been reported to be frequently located in close vicinity of breakpoints and implicated in more complex chromosomal rearrangements with various examples predominantly reported in haematological malignancies. Although these events are relatively rare compared to other mechanisms, the presence of Alu elements increases the likelihood of fusion formation, contributing to the pathogenesis of haematological malignancies. These Alu-associated fusions include MLL-ENL fusions in ALL [113], Philadelphia chromosome (BCR-ABL fusions) in chronic myeloid leukaemia [114] and MALT1-API2 fusions in mucosa-associated lymphoid tissue (MALT) lymphoma [115]. In addition, LINE-1 elements play a role in promoting large-scale chromosomal rearrangements. Notably, LINE-1 elements account for nearly 19% of chromosomal breakpoints across 17 whole human genomes, implicating them in chromosomal translocations, inversions, and large deletions [116]. Moreover, in Myc-induced mouse models of lymphoma, LINE-1 sequences were frequently found at break sites, with no significant sequence homology, further implicating these elements in driving translocations and other chromosomal rearrangements through erroneous repair mechanisms, similar to Alu-mediated recombination events noted in blood cancers [117]. Though less abundant in the human genome, DNA transposons, such as MER20, also contribute to chromosomal rearrangements. One notable example involves the MER20 transposon, which has been linked to the development of B-cell precursor ALL, with nearly half of the reported translocations in TCF3-PBX1 ALL involving this transposon [118].
In summary, the involvement of TEs such as Alu, LINE-1, and MER20 in driving chromosomal rearrangements in haematological malignancies suggests their potential role in cancer progression, warranting further investigation into their mechanistic contributions and therapeutic implications.
Conclusion
TEs have long been considered as genomic bystanders, yet their contributions to both normal and malignant haematopoiesis are increasingly recognised. By reshaping gene-regulatory networks and contributing to structural genome variation, TEs have played a dual role in biology: driving innovation in normal cellular processes and supporting haematopoietic regeneration, while also promoting genomic instability and oncogenesis in haematological malignancies. This review highlights the growing body of research on the importance of TEs in haematopoiesis in both health and disease. In normal haematopoiesis, TEs are instrumental in fine-tuning gene expression programmes, often acting as enhancers or regulatory elements that influence lineage commitment and the functional specialization of haematopoietic cells. In the malignant state, cancer cells can exploit the gene regulatory activities of TEs for their own fitness, promoting oncogene expression. Furthermore, TE-mediated structural variations, such as insertions, deletions, and translocations, are increasingly implicated in the pathogenesis of haematological malignancies, potentially linking their activity to clonal evolution and disease progression.
With advances in experimental and computational approaches, the field is poised to expand our understanding of the multifaceted activities of TEs and their implications in normal and malignant haematopoiesis. Future efforts will focus on establishing causative links of TE activity in these contexts. CRISPR-Cas9 genetic and epigenetic editing offer powerful tools to explore these links by enabling precise manipulation of their activity and allowing researchers to dissect specific TE functions in vitro and in vivo. Along with genome editing tools, future research using long-read sequencing (Boxes 2 and 3) and T2T-CHM13 will allow researchers to map TEs precisely and help to delineate the mechanisms through which TEs influence both normal and pathological processes. The growing recognition of TE-driven processes in haematological malignancies presents an opportunity to develop novel prognostic and therapeutic strategies. Given that TE expression is often cell- and tissue-specific, the activity of oncogenic TEs is typically uniquely activated in tumours, not only making them attractive targets but also potential biomarkers for diagnosis, prognosis and therapy response. From a therapeutic perspective, small-molecule inhibitors or epigenetic drugs that alter TE chromatin states represents a promising approach to modulate TE-driven oncogenic transcriptional programmes. More precisely, the use of lipid nanoparticle-based delivery systems for in vivo epigenetic editing [119] could enable targeted silencing of specific TEs in malignant cells, while RNA-based therapies, including antisense oligonucleotides, may allow selective degradation of TE-derived transcripts. As our understanding of TE-mediated oncogenic processes deepens, integrating these insights into therapeutic strategies has the potential to refine treatment paradigms and improve clinical outcomes in haematological malignancies.
References
McClintock B. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci. 1950;36:344–55.
Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science. 2022;376:44–53.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–82.
Blikstad V, Benachenhou F, Sperber GO, Blomberg J. Endogenous retroviruses: evolution of human endogenous retroviral sequences: a conceptual account. Cell Mol Life Sci. 2008;65:3348–65.
Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol. 2014;192:1343–9.
International Human Genome Sequencing Consortium, Whitehead Institute for Biomedical Research, Center for Genome Research, Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, et al. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–70.
Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci. 2003;100:5280–5.
Lavie L, Maldener E, Brouha B, Meese EU, Mayer J. The human L1 promoter: Variable transcription initiation sites and a major impact of upstream flanking sequence on promoter activity. Genome Res. 2004;14:2253–60.
Roy AM, West NC, Rao A, Adhikari P, Alemán C, Barnes AP, et al. Upstream flanking sequences and transcription of SINEs 1 1Edited by M. Gottesman. J Mol Biol. 2000;302:17–25.
Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–68.
Senft AD, Macfarlan TS. Transposable elements shape the evolution of mammalian development. Nat Rev Genet. 2021;22:691–711.
Henssen AG, Henaff E, Jiang E, Eisenberg AR, Carson JR, Villasante CM, et al. Genomic DNA transposition induced by human PGBD5. eLife. 2015;4:e10565.
Henssen AG, Koche R, Zhuang J, Jiang E, Reed C, Eisenberg A, et al. PGBD5 promotes site-specific oncogenic mutations in human tumors. Nat Genet. 2017;49:1005–14.
Kapitonov VV, Koonin EV. Evolution of the RAG1-RAG2 locus: both proteins came from the same transposon. Biol Direct. 2015;10:20.
Lavialle C, Cornelis G, Dupressoir A, Esnault C, Heidmann O, Vernochet C, et al. Paleovirology of ‘ syncytins ’, retroviral env genes exapted for a role in placentation. Philos Trans R Soc B Biol Sci. 2013;368:20120507.
McClintock B. Controlling elements and the gene. Cold Spring Harb Symp Quant Biol. 1956;21:197–216.
Britten RJ, Davidson EH. Repetitive and non-repetitive dna sequences and a speculation on the origins of evolutionary novelty. Q Rev Biol. 1971;46:111–38.
Pehrsson EC, Choudhary MNK, Sundaram V, Wang T. The epigenomic landscape of transposable elements across normal human development and anatomy. Nat Commun. 2019;10:5640.
Du AY, Chobirko JD, Zhuo X, Feschotte C, Wang T. Regulatory transposable elements in the encyclopedia of DNA elements. Nat Commun. 2024;15:7594.
Bourque G, Leong B, Vega VB, Chen X, Lee YL, Srinivasan KG, et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008;18:1752–62.
Trizzino M, Park Y, Holsbach-Beltrame M, Aracena K, Mika K, Caliskan M, et al. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res. 2017;27:1623–33.
Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 2010;42:631–4.
Hollister JD, Gaut BS. Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009;19:1419–28.
Zemojtel T, Kielbasa SM, Arndt PF, Chung HR, Vingron M. Methylation and deamination of CpGs generate p53-binding sites on a genomic scale. Trends Genet. 2009;25:63–6.
Żemojtel T, Kiełbasa SM, Arndt PF, Behrens S, Bourque G, Vingron M. CpG deamination creates transcription factor–binding sites with high efficiency. Genome Biol Evol. 2011;3:1304–11.
Gu X, Wang M, Zhang XO. TE-TSS: an integrated data resource of human and mouse transposable element (TE)-derived transcription start site (TSS). Nucleic Acids Res. 2024;52:D322–33.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8.
Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41:563–71.
Mak KS, Burdach J, Norton LJ, Pearson RC, Crossley M, Funnell AP. Repression of chimeric transcripts emanating from endogenous retrotransposons by a sequence-specific transcription factor. Genome Biol. 2014;15:3252.
Colombo AR, Triche T, Ramsingh G. Transposable element expression in acute myeloid leukemia transcriptome and prognosis. Sci Rep. 2018;8:16449.
Ferlita AL, Nigita G, Tsyba L, Palamarchuk A, Alaimo S, Pulvirenti A, et al. Expression signature of human endogenous retroviruses in chronic lymphocytic leukemia. Proc Natl Acad Sci. 2023;120:e2307593120.
Singh B, Dopkins N, Fei T, Marston JL, Michael S, Reyes-Gopar H, et al. Locus specific human endogenous retroviruses reveal new lymphoma subtypes. bioRxiv. 2023 Jun 8; 2023.06.08.544208.
Alcazer V, Bonaventura P, Tonon L, Michel E, Mutez V, Fabres C, et al. HERVs characterize normal and leukemia stem cells and represent a source of shared epitopes for cancer immunotherapy. Am J Hematol. 2022;97:1200–14.
Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D, et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med. 2010;16:571–9.
Babaian A, Romanish MT, Gagnier L, Kuo LY, Karimi MM, Steidl C, et al. Onco-exaptation of an endogenous retroviral LTR drives IRF5 expression in Hodgkin lymphoma. Oncogene. 2016;35:2542–6.
Lock FE, Rebollo R, Miceli-Royer K, Gagnier L, Kuah S, Babaian A, et al. Distinct isoform of FABP7 revealed by screening for retroelement-activated genes in diffuse large B-cell lymphoma. Proc Natl Acad Sci. 2014;111:E3534–43.
Deniz Ö, Ahmed M, Todd CD, Rio-Machin A, Dawson MA, Branco MR. Endogenous retroviruses are a source of enhancers with oncogenic potential in acute myeloid leukaemia. Nat Commun. 2020;11:3506.
Shah NM, Jang HJ, Liang Y, Maeng JH, Tzeng SC, Wu A, et al. Pan-cancer analysis identifies tumor-specific antigens derived from transposable elements. Nat Genet. 2023;55:631–9.
Attig J, Young GR, Hosie L, Perkins D, Encheva-Yokoya V, Stoye JP, et al. LTR retroelement expansion of the human cancer transcriptome and immunopeptidome revealed by de novo transcript assembly. Genome Res. 2019;29:1578–90.
Kong Y, Rose CM, Cass AA, Williams AG, Darwish M, Lianoglou S, et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat Commun. 2019;10:5228.
Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10:eaau5516.
Chour M, Porteu F, Depil S, Alcazer V. Endogenous retroelements in hematological malignancies: From epigenetic dysregulation to therapeutic targeting. Am J Hematol. 2025;100:116–30.
Nguyen TM, Alchalabi S, Oluwatoyosi A, Ropri AS, Herschkowitz JI, Rosen JM. New twists on long noncoding RNAs: from mobile elements to motile cancer cells. RNA Biol. 2020;17:1535–49.
Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. Hoekstra HE, editor. PLoS Genet. 2013;9:e1003470.
Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012;13:R107.
Wong NK, Huang CL, Islam R, Yip SP. Long non-coding RNAs in hematological malignancies: translating basic techniques into diagnostic and therapeutic strategies. J Hematol OncolJ Hematol Oncol. 2018;11:131.
Wang Y, Wu P, Lin R, Rong L, Xue Y, Fang Y. LncRNA NALT interaction with NOTCH1 promoted cell proliferation in pediatric T cell acute lymphoblastic leukemia. Sci Rep. 2015;5:13749.
Hughes JM, Legnini I, Salvatori B, Masciarelli S, Marchioni M, Fazi F, et al. C/EBPα-p30 protein induces expression of the oncogenic long non-coding RNA UCA1 in acute myeloid leukemia. Oncotarget. 2015;6:18534–44.
Zhang X, Lian Z, Padden C, Gerstein MB, Rozowsky J, Snyder M, et al. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood. 2009;113:2526–34.
Schwarzer A, Emmrich S, Schmidt F, Beck D, Ng M, Reimer C, et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat Commun. 2017;8:218.
Verma A, Jiang Y, Du W, Fairchild L, Melnick A, Elemento O. Transcriptome sequencing reveals thousands of novel long non-coding RNAs in B cell lymphoma. Genome Med. 2015;7:110.
Hu T, Pi W, Zhu X, Yu M, Ha H, Shi H, et al. Long non-coding RNAs transcribed by ERV-9 LTR retrotransposon act in cis to modulate long-range LTR enhancer function. Nucleic Acids Res. 2017;45:4479–4492.
Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3’ splice-site selection in Alu Exons. Science. 2003;300:1288–91.
Clayton EA, Rishishwar L, Huang TC, Gulati S, Ban D, McDonald JF, et al. An atlas of transposable element-derived alternative splicing in cancer. Philos Trans R Soc B Biol Sci. 2020;375:20190342.
Kim WR, Park EG, Lee YJ, Bae WH, Lee DH, Kim HS. Integration of TE induces cancer specific alternative splicing events. Int J Mol Sci. 2022;23:10918.
Lee B, Park J, Voshall A, Maury E, Kang Y, Kim YJ, et al. Pan-cancer analysis reveals multifaceted roles of retrotransposon-fusion RNAs. Cancer Biol. 2023;19:2023.10.16.562422.
Amit M, Sela N, Keren H, Melamed Z, Muler I, Shomron N, et al. Biased exonization of transposed elements in duplicated genes: A lesson from the TIF-IA gene. BMC Mol Biol. 2007;8:109.
Scarfò I, Pellegrino E, Mereu E, Kwee I, Agnelli L, Bergaggio E, et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood. 2016;127:221–32.
Burbage M, Rocañín-Arjó A, Baudon B, Arribas YA, Merlotti A, Rookhuizen DC, et al. Epigenetically controlled tumor antigens derived from splice junctions between exons and transposable elements. Sci Immunol. 2023;8:eabm6360.
Szelest M, Giannopoulos K. Biological relevance of alternative splicing in hematologic malignancies. Mol Med. 2024;30:62.
Long Q, Bengra C, Li C, Kutlar F, Tuan D. A long terminal repeat of the human endogenous retrovirus ERV-9 is located in the 5' boundary area of the human beta-globin locus control region. Genomics. 1998;54:542–55.
Ling J, Pi W, Bollag R, Zeng S, Keskintepe M, Saliman H, et al. The solitary long terminal repeats of ERV-9 endogenous retrovirus are conserved during primate evolution and possess enhancer activities in embryonic and hematopoietic cells. J Virol. 2002;76:2410–23.
Pi W, Zhu X, Wu M, Wang Y, Fulzele S, Eroglu A, et al. Long-range function of an intergenic retrotransposon. Proc Natl Acad Sci. 2010;107:12992–7.
Yu X, Zhu X, Pi W, Ling J, Ko L, Takeda Y, et al. The long terminal repeat (LTR) of ERV-9 human endogenous retrovirus binds to NF-Y in the assembly of an active LTR enhancer complex NF-Y/MZF1/GATA-2. J Biol Chem. 2005;280:35184–94.
Smith AM, Sanchez MJ, Follows GA, Kinston S, Donaldson IJ, Green AR, et al. A novel mode of enhancer evolution: the Tal1 stem cell enhancer recruited a MIR element to specifically boost its activity. Genome Res. 2008;18:1422–32.
Ye M, Goudot C, Hoyler T, Lemoine B, Amigorena S, Zueva E. Specific subfamilies of transposable elements contribute to different domains of T lymphocyte enhancers. Proc Natl Acad Sci. 2020;117:7905–16.
Feng C, Tie R, Xin S, Chen Y, Li S, Chen Y, et al. Systematic single-cell analysis reveals dynamic control of transposable element activity orchestrating the endothelial-to-hematopoietic transition. BMC Biol. 2024;22:143.
Almamun M, Levinson BT, Van Swaay AC, Johnson NT, McKay SD, Arthur GL, et al. Integrated methylome and transcriptome analysis reveals novel regulatory elements in pediatric acute lymphoblastic leukemia. Epigenetics. 2015;10:882–90.
Barrow TM, Nakjang S, Lafta F, Bilotkach K, Woodhouse L, Junge G, et al. Epigenome-wide analysis reveals functional modulators of drug sensitivity and post-treatment survival in chronic lymphocytic leukaemia. Br J Cancer. 2021;124:474–83.
Telonis AG, Yang Q, Huang HT, Figueroa ME. MIR retrotransposons link the epigenome and the transcriptome of coding genes in acute myeloid leukemia. Nat Commun. 2022;13:6524.
Zeng Y, Cao Y, Halevy RS, Nguyen P, Liu D, Zhang X, et al. Characterization of functional transposable element enhancers in acute myeloid leukemia. Sci China Life Sci. 2020;63:675–87.
Grillo G, Nadorp B, Qamra A, Mitchell A, Arlidge C, Nand A, et al. Transposable elements shape stemness in normal and leukemic hematopoiesis. bioRxiv. 2024 Jan 1;2021.02.16.431334.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80.
Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D Map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80.
Symmons O, Pan L, Remeseiro S, Aktas T, Klein F, Huber W, et al. The shh Topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Dev Cell. 2016;39:529–43.
Beagan JA, Duong MT, Titus KR, Zhou L, Cao Z, Ma J, et al. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017;27:1139–52.
Zhou Q, Yu M, Tirado-Magallanes R, Li B, Kong L, Guo M, et al. ZNF143 mediates CTCF-bound promoter–enhancer loops required for murine hematopoietic stem and progenitor cell function. Nat Commun. 2021;12:43.
Qi Q, Cheng L, Tang X, He Y, Li Y, Yee T, et al. Dynamic CTCF binding directly mediates interactions among cis -regulatory elements essential for hematopoiesis. Blood. 2021;137:1327–39.
Takayama N, Murison A, Takayanagi Sichiro, Arlidge C, Zhou S, Garcia-Prat L, et al. The transition from quiescent to activated states in human hematopoietic stem cells is governed by dynamic 3D genome reorganization. Cell Stem Cell. 2021;28:488–501.e10.
Xu J, Song F, Lyu H, Kobayashi M, Zhang B, Zhao Z, et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature. 2022;611:387–98.
Choudhary MNK, Quaid K, Xing X, Schmidt H, Wang T. Widespread contribution of transposable elements to the rewiring of mammalian 3D genomes. Nat Commun. 2023;14:634.
Diehl AG, Ouyang N, Boyle AP. Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes. Nat Commun. 2020;11:1796.
Sundaram V, Cheng Y, Ma Z, Li D, Xing X, Edge P, et al. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014;24:1963–76.
Schwalie PC, Ward MC, Cain CE, Faure AJ, Gilad Y, Odom DT, et al. Co-binding by YY1 identifies the transcriptionally active, highly conserved set of CTCF-bound regions in primate genomes. Genome Biol. 2013;14:R148.
Becker KG, Swergold G, Ozato K, Thayer RE. Binding of the ubiquitous nuclear transcription factor YY1 to a cis regulatory sequence in the human LINE-1 transposable element. Hum Mol Genet. 1993;2:1697–702.
Athanikar JN. A YY1-binding site is required for accurate human LINE-1 transcription initiation. Nucleic Acids Res. 2004;32:3846–55.
Sanchez-Luque FJ, Kempen MJHC, Gerdes P, Vargas-Landin DB, Richardson SR, Troskie RL, et al. LINE-1 Evasion of epigenetic repression in humans. Mol Cell. 2019;75:590–604.e12.
Noguera NI, Travaglini S, Scalea S, Catalanotto C, Reale A, Zampieri M, et al. YY1 knockdown relieves the differentiation block and restores apoptosis in AML cells. Cancers. 2023;15:4010.
Zhang M, Huang H, Li J, Wu Q. ZNF143 deletion alters enhancer/promoter looping and CTCF/cohesin geometry. Cell Rep. 2024;43:113663.
Pal D, Patel M, Boulet F, Sundarraj J, Grant OA, Branco MR, et al. H4K16ac activates the transcription of transposable elements and contributes to their cis-regulatory function. Nat Struct Mol Biol. 2023;30:935–47.
Wang J, Vicente-García C, Seruggia D, Moltó E, Fernandez-Miñán A, Neto A, et al. MIR retrotransposon sequences provide insulators to the human genome. Proc Natl Acad Sci. 2015;112:E4428-37.
Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215.
Jr HK, Wong C, Youssoufian H, Scottt AF, Phillips DG. Haemophilia A resulting from de novo insertion of Ll sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164-6.
Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, et al. Disruption of the APC gene by a retrotransposal insertion of li sequence in a colon cancer. Cancer Res. 1992;52:643-5.
Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337:967–71.
Tubio JMC, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, et al. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345:1251343.
Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, Zamora J, Supek F, Demeulemeester J, et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat Genet. 2020;52:306–19.
Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014;24:1053–63.
Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–61.
Gu Z, Liu Y, Zhang Y, Cao H, Lyu J, Wang X, et al. Silencing of LINE-1 retrotransposons is a selective dependency of myeloid leukemia. Nat Genet. 2021;53:672–82.
Thawani A, Ariza AJF, Nogales E, Collins K. Template and target-site recognition by human LINE-1 in retrotransposition. Nature. 2024;626:186–93.
Mendez-Dorantes C, Burns KH. LINE-1 retrotransposition and its deregulation in cancers: implications for therapeutic opportunities. Genes Dev. 2023;37:948–67.
Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol. 2006;357:1383–93.
Balachandran P, Walawalkar IA, Flores JI, Dayton JN, Audano PA, Beck CR. Transposable element-mediated rearrangements are prevalent in human genomes. Nat Commun. 2022;13:7115.
Camargo-Forero N, Orozco-Arias S, Perez Agudelo JM, Guyot R. HERV-K (HML-2) insertion polymorphisms in the 8q24.13 region and their potential etiological associations with acute myeloid leukemia. Arch Virol. 2023;168:125.
Komkov AY, Maschan MA, Shvets VI, Lebedev YuB. Functional analysis of polymorphic insertions of Alu retroelements in acute lymphoblastic leukemia patients. Russ J Bioorg Chem. 2012;38:306–18.
Liao WW, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, et al. A draft human pangenome reference. Nature. 2023;617:312–24.
Zhou W, Emery SB, Flasch DA, Wang Y, Kwan KY, Kidd JM, et al. Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Res. 2020;48:1146–63.
Ewing AD, Smits N, Sanchez-Luque FJ, Faivre J, Brennan PM, Richardson SR, et al. Nanopore sequencing enables comprehensive transposable element epigenomic profiling. Mol Cell. 2020;80:915–928.e5.
Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu- mediated homologous recombination in acute myeloid leukemia. Proc Natl Acad Sci. 1998;95:2390–5.
O’Neil J, Tchinda J, Gutierrez A, Moreau L, Maser RS, Wong KK, et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J Exp Med. 2007;204:3059–66.
Blanco JG, Dervieux T, Edick MJ, Mehta PK, Rubnitz JE, Shurtleff S, et al. Molecular emergence of acute myeloid leukemia during treatment for acute lymphoblastic leukemia. Proc Natl Acad Sci. 2001;98:10338–43.
Jeffs A. The BCR gene recombines preferentially with Alu elements in complex BCR- ABL translocations of chronic myeloid leukaemia. Hum Mol Genet. 1998;7:767–76.
Liu H, Hamoudi RA, Ye H, Ruskone‐Fourmestraux A, Dogan A, Isaacson PG, et al. t(11;18)(q21;q21) of mucosa‐associated lymphoid tissue lymphoma results from illegitimate non‐homologous end joining following double strand breaks. Br J Haematol. 2004;125:318–29.
Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, et al. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–47.
Rockwood LD, Felix K, Janz S. Elevated presence of retrotransposons at sites of DNA double strand break repair in mouse models of metabolic oxidative stress and MYC-induced lymphoma. Mutat Res Mol Mech Mutagen. 2004;548:117–25.
Rodić N, Zampella JG, Cornish TC, Wheelan SJ, Burns KH. Translocation junctions in TCF3-PBX1 acute lymphoblastic leukemia/lymphoma cluster near transposable elements. Mob DNA. 2013;4:22.
Cappelluti MA, Mollica Poeta V, Valsoni S, Quarato P, Merlin S, Merelli I, et al. Durable and efficient gene silencing in vivo by hit-and-run epigenome editing. Nature. 2024;627:416–23.
de Tribolet-Hardy J, Thorball CW, Forey R, Planet E, Khubieh B, Offner S, et al. Genetic features and genomic targets of human KRAB-Zinc finger proteins. Genome Res. 2023;33:1409–1423.
Rosspopoff O, Trono D. Take a walk on the KRAB side. Trends Genet. 2023;39:844–57.
Stoll GA, Pandiloski N, Douse CH, Modis Y. Structure and functional mapping of the KRAB‐KAP1 repressor complex. EMBO J. 2022;41:e111179.
Imbeault M, Helleboid PY, Trono D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature. 2017;543:550–4.
Ecco G, Imbeault M, Trono D. KRAB zinc finger proteins. Development. 2017;144:2719–29.
Jacobs FMJ, Greenberg D, Nguyen N, Haeussler M, Ewing AD, Katzman S, et al. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature. 2014;516:242–5.
Kosuge M, Ito J, Hamada M. Landscape of evolutionary arms races between transposable elements and krab-zfp family. Sci Rep. 2024;14:23358
Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, et al. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature. 2018;553:228–32.
Robbez-Masson L, Tie CHC, Conde L, Tunbak H, Husovsky C, Tchasovnikarova IA, et al. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 2018;28:836–45.
Tunbak H, Enriquez-Gasca R, Tie CHC, Gould PA, Mlcochova P, Gupta RK, et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat Commun. 2020;11:5387.
Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Göttgens B, et al. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science. 2015;348:1481–5.
Tchasovnikarova IA, Timms RT, Douse CH, Roberts RC, Dougan G, Kingston RE, et al. Hyperactivation of HUSH complex function by Charcot–Marie–Tooth disease mutation in MORC2. Nat Genet. 2017;49:1035–44.
Deniz Ö, Frost JM, Branco MR. Regulation of transposable elements by DNA modifications. Nat Rev Genet. 2019;20:417–31.
Yoder J, Colum W, Bestor T. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–40.
Zoch A, Auchynnikava T, Berrens RV, Kabayama Y, Schöpp T, Heep M, et al. SPOCD1 is an essential executor of piRNA-directed de novo DNA methylation. Nature. 2020;584:635–9.
De Fazio S, Bartonicek N, Di Giacomo M, Abreu-Goodger C, Sankar A, Funaya C, et al. The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature. 2011;480:259–63.
Ozata DM, Gainetdinov I, Zoch A, O’Carroll D, Zamore PD. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet. 2019;20:89–108.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640–50.
Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S, et al. m6A RNA methylation regulates the fate of endogenous retroviruses. Nature. 2021;591:312–6.
Liu J, Gao M, He J, Wu K, Lin S, Jin L, et al. The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature. 2021;591:322–6.
Xu W, Li J, He C, Wen J, Ma H, Rong B, et al. METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature. 2021;591:317–21.
Xiong F, Wang R, Lee JH, Li S, Chen SF, Liao Z, et al. RNA m6A modification orchestrates a LINE-1–host interaction that facilitates retrotransposition and contributes to long gene vulnerability. Cell Res. 2021;31:861–85.
Hwang SY, Jung H, Mun S, Lee S, Park K, Baek SC, et al. L1 retrotransposons exploit RNA m6A modification as an evolutionary driving force. Nat Commun. 2021;12:880.
Sun T, Xu Y, Xiang Y, Ou J, Soderblom EJ, Diao Y. Crosstalk between RNA m6A and DNA methylation regulates transposable element chromatin activation and cell fate in human pluripotent stem cells. Nat Genet. 2023;55:1324–35.
Zou Z, Dou X, Li Y, Zhang Z, Wang J, Gao B, et al. RNA m5C oxidation by TET2 regulates chromatin state and leukaemogenesis. Nature. 2024;634:986–94.
Sexton CE, Han MV. Paired-end mappability of transposable elements in the human genome. Mob DNA. 2019;10:29.
Panda K, Slotkin RK. Long-Read cDNA sequencing enables a “gene-like” transcript annotation of transposable elements. Plant Cell. 2020;32:2687–98.
Shahid S, Slotkin RK. The current revolution in transposable element biology enabled by long reads. Curr Opin Plant Biol. 2020;54:49–56.
Hoyt SJ, Storer JM, Hartley GA, Grady PGS, Gershman A, De Lima LG, et al. From telomere to telomere: the transcriptional and epigenetic state of human repeat elements. Science. 2022;376:eabk3112.
Wang T, Antonacci-Fulton L, Howe K, Lawson HA, Lucas JK, Phillippy AM, et al. The human pangenome project: a global resource to map genomic diversity. Nature. 2022;604:437–46.
Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–82.
Berrens RV, Yang A, Laumer CE, Lun ATL, Bieberich F, Law CT, et al. Locus-specific expression of transposable elements in single cells with CELLO-seq. Nat Biotechnol. 2022;40:546–54.
Adamopoulos PG, Tsiakanikas P, Stolidi I, Scorilas A. A versatile 5′ RACE-Seq methodology for the accurate identification of the 5′ termini of mRNAs. BMC Genomics. 2022;23:163.
Li X, Lu K, Chen X, Tu K, Xie D. capTEs enables locus-specific dissection of transcriptional outputs from reference and nonreference transposable elements. Commun Biol. 2023;6:974.
Maeng JH, Jang HJ, Du AY, Tzeng SC, Wang T. Using long-read CAGE sequencing to profile cryptic-promoter-derived transcripts and their contribution to the immunopeptidome. Genome Res. 2023;33:2143–55.
Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, et al. TT-seq maps the human transient transcriptome. Science. 2016;352:1225–8.
Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc. 2016;11:1455–76.
Bendall ML, De Mulder M, Iñiguez LP, Lecanda-Sánchez A, Pérez-Losada M, Ostrowski MA, et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLOS Comput Biol. 2019;15:e1006453.
Jin Y, Tam OH, Paniagua E, Hammell M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 2015;31:3593–9.
Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014;15:583.
Yang WR, Ardeljan D, Pacyna CN, Payer LM, Burns KH. SQuIRE reveals locus-specific regulation of interspersed repeat expression. Nucleic Acids Res. 2019;47:e27.
Rodríguez-Quiroz R, Valdebenito-Maturana B. SoloTE for improved analysis of transposable elements in single-cell RNA-Seq data using locus-specific expression. Commun Biol. 2022;5:1063.
Polimeni B, Marasca F, Ranzani V, Bodega B. IRescue: uncertainty-aware quantification of transposable elements expression at single cell level. Nucleic Acids Res. 2024;52:e93.
Morrissey A, Shi J, James DQ, Mahony S. Accurate allocation of multimapped reads enables regulatory element analysis at repeats. Genome Res. 2024;34:937–51.
Hu Y, Jiang Z, Chen K, Zhou Z, Zhou X, Wang Y, et al. scNanoATAC-seq: a long-read single-cell ATAC sequencing method to detect chromatin accessibility and genetic variants simultaneously within an individual cell. Cell Res. 2022;33:83–6.
Li Q, Guo Y, Wu Z, Xu X, Jiang Z, Qi S, et al. scNanoSeq-CUT&Tag: a single-cell long-read CUT&Tag sequencing method for efficient chromatin modification profiling within individual cells. Nat Methods. 2024;21:2044–57.
Yue X, Xie Z, Li M, Wang K, Li X, Zhang X, et al. Simultaneous profiling of histone modifications and DNA methylation via nanopore sequencing. Nat Commun. 2022;13:7939.
Shipony Z, Marinov GK, Swaffer MP, Sinnott-Armstrong NA, Skotheim JM, Kundaje A, et al. Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat Methods. 2020;17:319–27.
Leduque B, Edera A, Vitte C, Quadrana L. Simultaneous profiling of chromatin accessibility and DNA methylation in complete plant genomes using long-read sequencing. Nucleic Acids Res. 2024;52:6285–97.
Lanciano S, Philippe C, Sarkar A, Pratella D, Domrane C, Doucet AJ, et al. Locus-level L1 DNA methylation profiling reveals the epigenetic and transcriptional interplay between L1s and their integration sites. Cell Genomics. 2024;4:100498.
Deshpande AS, Ulahannan N, Pendleton M, Dai X, Ly L, Behr JM, et al. Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nat Biotechnol. 2022;40:1488–99.
Taylor D, Lowe R, Philippe C, Cheng KCL, Grant OA, Zabet NR, et al. Locus-specific chromatin profiling of evolutionarily young transposable elements. Nucleic Acids Res. 2022;50:e33.
Raviram R, Rocha PP, Luo VM, Swanzey E, Miraldi ER, Chuong EB, et al. Analysis of 3D genomic interactions identifies candidate host genes that transposable elements potentially regulate. Genome Biol. 2018;19:216.
Zhao S, Lu J, Pan B, Fan H, Byrum SD, Xu C, et al. TNRC18 engages H3K9me3 to mediate silencing of endogenous retrotransposons. Nature. 2023;623:633–42.
Altemose N, Maslan A, Smith OK, Sundararajan K, Brown RR, Mishra R, et al. DiMeLo-seq: a long-read, single-molecule method for mapping protein–DNA interactions genome wide. Nat Methods. 2022;19:711–23.
Acknowledgements
The authors could not cite the work from all the colleagues due to space limitations. The work in the Deniz laboratory is supported by Cancer Research UK Career Development Fellowship (RCCCDF-Nov21\100002), UKRI’s Horizon Europe Guarantee scheme (EP/Y030338/1), the European Hematology Association grants (RG-202111-01467). The figures were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
ÖD conceptualized the review. HT and EL prepared the figures. All the other authors DP, HT, EL, BD, FC, ÖD drafted, reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prokopov, D., Tunbak, H., Leddy, E. et al. Transposable elements as genome regulators in normal and malignant haematopoiesis. Blood Cancer J. 15, 87 (2025). https://doi.org/10.1038/s41408-025-01295-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-025-01295-9
