
Abstract
Background
KRASG12C alterations are present in ~13% of lung adenocarcinomas. AZD4625 is a covalent small molecule inhibitor that selectively binds and inhibits GDP-KRASG12C, leading to reduced cell viability and protein signaling responsible for tumor survival in models with this gain-of-function alteration.
Methods
We studied short-term changes in signaling and mechanisms of primary resistance to AZD4625 in twelve KRASG12C lung adenocarcinoma patient-derived xenografts (PDX) and six organoids derived from these twelve models.
Results
Sustained tumor regression in four (33%) PDXs was observed while the remaining eight models were intrinsically resistant to AZD4625. Organoid responses to AZD4625 were concordant with their derived PDXs. Acute AZD4625 exposure significantly decreased gene expression of the ERK1/2 negative regulator, DUSP6, in all models while protein MAPK and AKT/mTOR signals were downregulated more frequently in the AZD4625-sensitive than AZD4625-resistant cohorts. Analyzing PDX transcriptomes and proteomes identified mTOR signaling as a putative mechanism of primary resistance to AZD4625.
Conclusions
Our findings confirm AZD4625 as a highly active KRASG12C inhibitor. This data also supports the use of PDX models in understanding resistance mechanisms that may be leveraged to develop more active combination therapies.
Similar content being viewed by others


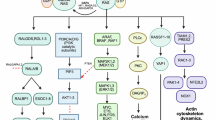
Background
RAS proteins are GTPases that cycle between their inactive GDP-bound and active GTP-bound forms, facilitating the activation of several signalling pathways involved in a variety of cellular processes [1]. Kirsten rat sarcoma viral oncogene homologue (KRAS), like other RAS proteins, possesses GTPase activating protein (GAP)-stimulated GTP hydrolysis in addition to intrinsic GTPase activity [2]. Gain-of-function genetic alterations in KRAS are present in ~25% of lung adenocarcinoma (LUAD) tumours [3,4,5], the most common form of non-small cell lung cancer (NSCLC). Around 90% of these alterations occur at the G12 residue of KRAS, which when mutated, impairs GAP-mediated and intrinsic hydrolysis, leading to dysregulated downstream effector pathway stimulation that contributes to the survival of cancer cells [6]. KRASG12C are present in ~13% of NSCLC [7], which represent ~40% of KRAS alterations in LUAD [8]. Over a decade ago, Ostrem et al. identified a pocket on the surface of GDP-KRASG12C suitable for lead-based drug design [9]. Since then, there has been extensive efforts to optimise these compounds for clinical use [1]. To date, two GDP-KRASG12C inhibitors (G12Ci) have been approved, sotorasib and adagrasib, as a second-line or greater therapy in local/advanced stage/metastatic KRASG12C NSCLC [10, 11]. These agents demonstrated real but modest radiographic response rates of 37% (sotorasib: 46/123 patients, NCT03600883) and 43% (adagrasib: 48/112 patients, NCT03785249), while patients achieved objective response rates of 53% and 63–78% with the newer divarasib [12] (NCT04449874) or olomorasib [13] (NCT04956640), respectively. Molecules designed to target the active, GTP-bound form of KRASG12C and wild-type KRAS are also under investigation as they may overcome clinical resistance to GDP-KRASG12C inhibitors [14, 15]. While these developments are promising, many patients still do not respond to these novel therapies.
Identifying mechanisms of resistance to G12Ci has become a major endeavour for preclinical and clinical studies. To date, several mechanisms have been identified along the MAPK signalling pathway axis, either through mRNA upregulation, protein upregulation, proteomic changes, or genetic alterations [1, 2]. Due to the complexity of these phenomena and how intertwined they are with one another, simplifying them as either primary, adaptive, or acquired means of resistance is warranted for the sake of conceptualisation [1, 16]. Primary resistance refers to pre-treatment resistance to a given therapy of choice whereas acquired resistance refers to the development of treatment resistance after initial response [17]. Unfortunately, clinical investigations of primary resistance are challenging due to the lack of assays that may pinpoint these mechanisms. As such, many studies of primary resistance occur preclinically. However, only a limited number of KRASG12C LUAD models (n = 9 cell lines) [18] are available for preclinical study of G12Ci. To this end, we have investigated primary resistance to a recently developed G12Ci, AZD4625 [19, 20], in a cohort of twelve KRASG12C PDX with multi-omics characterisation [21, 22].
Methods
Drugs
AZD4625 (CAS 2387927-95-7) was supplied by AstraZeneca (Cambridge, UK) [19, 20] and sotorasib (CAS 2296729-00-3) was purchased from UHN Shanghai (Shanghai, China) and Selleckchem (Houston, TX, USA).
Cell line studies
NCI-H358 cells were obtained from the American Type Culture Collection and authenticated using DNA fingerprinting using AmpFLSTR™ Identifiler™ PCR amplification Kit (Thermo Fisher Scientific, Waltham, MA, USA) and a 9 Clear mouse pathogens panel evaluation (Charles River Laboratories, Wilmington, MA, USA) every five passages. For cell viability assays, 5000 cells were seeded per well in a 96-well plate (Sarstedt, Nümbrecht, Germany) or a Corning® 96-well round bottom ultra-low attachment spheroid microplate (Corning Incorporated, Corning, NY, USA). Drug was added to wells at ten different concentrations after 24 h. After 72 h 20 µL of CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) were added to each well and incubated at 37 °C. Plates were read according to manufacturer’s protocol. Absorbances were log-transformed, normalised to DMSO controls and plotted using GraphPad Prism Version 8.4.3 (San Diego, CA, USA; RRID:SCR_002798).
Xenograft-derived organoid studies
Xenograft-derived organoids (XDO) were generated as previously described [23]. For drug screening, DMSO was limited to a maximum of 0.1% in culture. On day 1, AZD4625 or sotorasib were added to the plates. Twenty-one drug dilutions were dispensed using an HP T8+ Dispensehead Cassette (Palo Alto, CA, USA) on a Tecan D300e Digital Dispenser (Tecan, Männedorf, Switzerland). On Day 7, 30 µL of CellTiter-Glo 3D (Promega, Madison, WI, USA) was added to each well and plates read as previously described [23]. Absorbances were log-transformed, normalised to DMSO controls and plotted using GraphPad Prism Version 8.4.3 (RRID:SCR_002798).
Animal studies
PDX models were established from resected LUAD tumours as previously described [22, 24] and using protocols approved by the Research Ethics Board (REB 09-0510-T) and Animal Care Committee (AUP#743) in accordance to ARRIVE guidelines at the University Health Network. Authentication was by DNA fingerprinting using AmpFLSTR™ Identifiler™ PCR amplification Kit (Thermo Fisher Scientific), and histology evaluation. All tumours were murine pathogen-free by 9 Clear panel evaluation and used between passages 3–10 and included tumours from male and female patients of various ages. All models were derived from resected tumours of treatment-naïve, primary patient lung tumours.
Cryopreserved PDX tissue fragments were implanted at the subcutaneous flank of non-obese diabetic severe combined immunodeficient (NOD/SCID; RRID:IMSR_JAX:001303) mice, grown to 1500 mm3 [3], collected and divided to be used as fresh donor fragments into study mice replicates and maintained as previously described [25]. Tumours were randomised into study arms based on tumour volumes using a stratified method for equal distribution. AZD4625 was prepared in 0.5% hydroxypropyl methylcellulose/0.1% Tween-80 [19] and was administered at a dose of 100 mg/kg by oral gavage once per day, five times per week. Body weight was measured daily and tumour size with skin fold were measured twice weekly. Log-linear mixed effects regression was used to quantify the difference in tumour growth rates between treatment versus vehicle within each tumour model, as previously described [25].
A pharmacodynamic screen was performed on three AZD4625-sensitive and three resistant models, each implanted into twenty-one mouse replicates. Replicates of three tumours were collected at acute post-drug timepoints 0 hour (h) pre-dose, 4 h, 6 h, 24 h, 48 h and 120 h after a single AZD4625 dose. A chronic cohort of three mice were given one dose every 24 h for 5 days designated as T120C (120-h chronic), and tumours were collected 24 h after final drug dose.
Histologic images were taken using a Leica DM2500 LED optical microscope (Leica Microsystems, Wetzlar, Germany) using the Leica Microsystems LasX image acquisition software.
Somatic mutation calling
Whole-exome sequencing (WES) profiling on PDX models for somatic mutations was done as previously described [25]. Sequencing was performed on a Novaseq S4 flowcell (Illumina, San Diego, CA, USA) with an average of 200X coverage and referenced against either matched patient normal or population normal. KRAS mutations were verified using Sanger sequencing as previously described [25].
Bulk RNA sequencing
RNA was extracted and isolated from snap frozen PDX tumour tissues via homogenisation in Ambion™ TRIzol. Overnight RNA precipitation was done using isopropanol at −80 °C. Nucleic acid pellets were resuspended in 70% ethanol and added to a QIAGEN RNeasy Mini spin column (Hilden, Germany) on ice, followed by manufacturer instructions to elute RNA. 2 µg of RNA from each sample was run on a NovaSeq S4 flowcell at a sequencing depth of 200 M reads/sample (paired-end 150 bp reads). NEB Ultra I Directional PolyA mRNA library prep was utilised for bulk RNA-sequencing. Xenome (version 1.0.1) [26] was used to filter mouse reads from human reads. STAR (version 2.7.9) was the aligner used to align the human reads to genome reference hg19 and Cufflinks (version 2.2.1) [27] was used for transcript quantification as fragments per kilobase of transcripts per million mapped reads (FPKM). The FPKMs then were converted to transcripts per million (TPM) values. The log2(TPM + 1) were used for all statistical analysis. Principal component analysis plots were done using the top 2000 most variable genes. Differentially expressed genes were detected using limma (3.38.3) package [28].
Gene set enrichment analysis of bulk RNA sequencing
Hallmark gene sets were obtained from the Molecular Signature Database v2023.2.Hs [29]. Differentially expressed genes between baseline AZD4625-sensitive and AZD4625-resistant tumours were filtered using a nominal p value cutoff of p < 0.05 and a log2 fold change cutoff of <−0.5 or >0.5. These gene lists were input into RStudio/2023.06.1 build 524 and evaluated against Hallmark gene sets using the fgsea R package [30].
Quantitative RT-PCR
RNA was reverse transcribed using Superscript III (Invitrogen, Waltham, MA, USA). Primers for qRT-PCR included the DUSP6 forward primer: 5′-CGACTGGAACGAGAATACGG-3′; the DUSP6 reverse primer: 5′-TTGGAACTTACTGAAGCCACCT-3′. the TBP (housekeeping) forward primer: 5′- TGTGCACAGGAGCCAAGAGT -3′; the TBP reverse primer: 5′- ATTTTCTTGCTGCCAGTC TGG -3′. The tubes were cycled 95 °C for 10 min, 40 cycles of 95 °C for 30 s and 60 °C for 1 min, 95 °C for 1 min, 60 °C for 30 s, 95 °C for 30 s. Fold-change values were normalised to TBP housekeeping and plotted using GraphPad Prism Version 8.4.3 (RRID: SCR_002798).
Immunoblot analysis
Immunoblotting of snap frozen PDX tumour fragments were analysed as previously described [25]. Membranes were incubated overnight at 4 °C with 1:2000 dilutions of β-actin (CST #4967; RRID: AB_330288), vinculin (CST #4650; RRID:AB_10559207), phospho-S6 ribosomal protein (S235/236) (CST #2211; RRID:AB_331679), S6 ribosomal protein (S235/236) (CST #2217; RRID:AB_331355), phospho-p44/42 MAPK (T202/Y204) (CST #4370; RRID:AB_2315112), p44/42 MAPK (CST #4695; RRID:AB_390779), or 1:1000 dilution of KRAS (sc-30; RRID:AB_627865). Membranes were later incubated with secondary 1:2000 anti-rabbit IgG HRP-linked antibody (CST #7074; RRID:AB_2099233) or 1:2000 anti-mouse IgG HRP-linked antibody (CST #7076; RRID:AB_330924) for 1 h and bands were visualised by Clarity ECL (Bio-Rad, Hercules, CA) on a Konica SRX-101A film processor (Tokyo, Japan).
LC-MS/MS sample preparation
PDX snap frozen tumour fragments were cut into ~15 mg pieces on dry ice and manually pulverised on an autoclaved Biopulverizer (BioSpec, Bartlesville, OK, USA). Frozen lung tumour tissue samples were mixed with lysis buffer, sonicated, incubated at 95 °C for 20 min with mixing at 1000 rpm, centrifuged, and proteins pelleted by chloroform/methanol precipitation (Supplemental methods). Peptides were labelled with 16-plex tandem mass tag (TMTPro, ThermoFisher Scientific A44520) reagents according to manufacturer’s instructions into nine sets of fourteen samples, plus two samples of pooled samples (126, 127N, 127C, 128N, 128C, 129N, 129C, 130N, 130C, 131N, 131C, 132N, 132C, 133N, 133C, and 134N). High-pH reverse phase high-pressure liquid chromatography (HPLC) separated the proteome aliquots into 60 separate fractions [31].
LC-MS/MS analysis
HPLC fractions were dried by vacuum centrifugation, resuspended, and loaded onto Evotips (Evosep Biosystems, Denmark) as per manufacturer’s instructions. Peptides were analysed using an Evosep One LC system with 30 SPD method and 15 cm analytical column (Dr Maisch C18 AQ, 1.9 μm beads, 150 μm ID, PepSep) and Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer (Thermo Fisher Scientific) in the following order: set 1, 2, 3, 7, 9, 4, 8, 5, 6. Data for total proteome TMT sets was acquired using MultiNotch synchronous precursor selection MS3 scanning for TMT tags [32] (Supplemental methods).
Mass spectrometry search analysis
Acquired total proteome raw data was analysed using scripts and methods from Phillip Wilmarth’s GitHub PAW-pipeline repository (github.com/pwilmart/PAW_pipeline). Data was searched against a database containing the canonical mouse and human proteomes (human 20404 entries; mouse 17137 entries) using Comet search engine (v2016.01) with parent and fragment mass tolerances set to 1.25 Da and 1.0005 Da, respectively, two missed cleavages allowed, oxidation (M, +15.9949) as variable modification, and carbamidomethylation (C, +57.021461) and TMT labelling (Nterm peptide, K, +304.2071) as fixed modifications. Search results were filtered using a strict false discovery rate (FDR) of 0.005 and parsimonious protein inference and grouping employed. TMT sets were normalised using internal reference scaling [33] and by percentage of human reads per sample.
Mass spectrometry differential expression and statistical analyses
Intensity/abundance values were log2-transformed and were per-sample median normalised. All correlation estimates and p values were calculated using the ‘cor.test’ function. Differential expression was calculated using the Estimated Marginal Means package (emmeans_1.8.4-1) to calculate group means, estimates and p values from a linear mixed model fit (lme4_1.1–31) with drug sensitivity and time point as factors with an interaction term allowing for a different intercept per PDX. FDRs were estimated by calculating adjusted p values using Benjamini-Hochberg method per contrast. Gene set enrichment analysis (GSEA) was applied using a preranked list of Gene Symbols sorted based on estimated fold changes against the human ‘Cancer Hallmarks’ and ‘KEGG Medicus’ gene sets with minimum and maximum sizes of 25 and 200, respectively. Only Cancer Hallmarks with FDR < 25% were selected for display. Heatmap was generated using ComplexHeatmap_2.16.0 against the Cancer Hallmarks epithelial-to-mesenchymal transition (EMT) gene set.
Results
AZD4625 is comparable to sotorasib as KRAS G12C inhibitor
AZD4625 was first tested in vitro on KRASG12C LUAD NCI-H358 cells and had an IC50 of 356 nM (Supplemental Fig. S1A). Three-dimensional NCI-H358 spheroids had a nearly 10-fold increase in sensitivity to AZD4625 versus in 2D-monoculture [34], and disintegration of spheroids became noticeable around the IC50 drug concentration of 38 nM (Supplemental Fig. S1B, C). To evaluate AZD4625 further, we attempted to generate XDO.
Six of our twelve KRASG12C PDX models [24] were established as stable XDO [23] (Fig. 1a, Table 1) and WES confirmed KRASG12C retention from their originating PDX at similar mutant allele frequencies (Supplemental Fig. S2A). To ensure in vitro G12Ci selectivity, the six KRASG12C XDO and twelve KRASWT XDO were treated with sotorasib, and all mutant models were sensitive except for PHLC344 (Fig. 1b). Afterwards, the six KRASG12C XDO were treated with AZD4625 or sotorasib monotherapy to assess their comparability. For AZD4625, the dose response curves of XDO194 and XDO207 had IC50 values < 1 µM, and the other four models with an IC50 > 1 µM (Fig. 1c, d). Both compounds led to sub-micromolar IC50 values for XDO194 and XDO207, while XDO239 and XDO362 had higher IC50 values with sotorasib (IC50 = 6.57 µM and 6.07 µM, respectively) than AZD4625 (IC50 = 1.13 µM and 3.35 µM, respectively). These data warranted the in vivo assessment of AZD4625.

a Oncoprint of the six long-term KRASG12C PDX. b Comparing KRASG12C inhibitor activity in KRASG12C and KRASWT long-term XDO. c KRASG12C long-term XDO responses to AZD4625 and sotorasib. d Violin plot of IC50 values for the two drugs used in (c). Dashed line represents median value; dotted lines represent the 25% and 75% percentiles. e PHLC207 was treated with AZD4625 or sotorasib. Each arm had n = 3 replicates, except for vehicle (sotorasib) which had n = 2.
To ensure reproducibility of G12Ci sensitivity in vivo, NCI-H358 cell-derived xenografts were treated with AZD4625, which led to stable disease (Supplemental Fig. S1D). Also, the PDX PHLC207 was treated with AZD4625 or sotorasib for 4 weeks (Fig. 1e). Both monotherapies led to similar tumour regression and both compounds were well tolerated by mice, suggesting similar antitumor efficacy.
AZD4625 monotherapy is sensitising in subset of KRAS G12C PDX models
In our PDX cohort, nine of twelve models were KRAS-heterozygous, while three were homozygous and KRAS-zygosity did not correlate with KRAS protein expression nor with activated ERK1/2 expression (Supplemental Fig. S2B). KRASG12C somatic mutations were identified and confirmed using WES and direct sequencing (Supplemental Fig. S2C).
We evaluated AZD4625 antitumor activity in PDXs over 4 weeks, with no significant changes in mouse body weights between vehicle and treatment arms. PHLC82, PHLC151, PHLC194 and PHLC207 regressed on AZD4625 treatment (had a rate of change in tumour volume below 1, meaning volume was decreasing) and were categorised as AZD4625-sensitive, while the remaining eight models that continued to grow on AZD4625 were classified as AZD4625-resistant (Fig. 2a). While these eight models were resistant, each grew at a different rate. To better understand tumour growth kinetics, we utilised a log-linear mixed effects statistical model [25] to predict growth rates of control and treatment tumours (Table 2). Of the eight resistant models, four grew significantly slower on AZD4625 relative to vehicle while there was no difference between growth rates in the other four resistant models. KRAS amplification status and co-mutations with TP53, KEAP1 or STK11 were not determinants of AZD4625 response, nor were co-mutations with BRAF, FGFR1-4 or CDKN2A/B (Fig. 1a).

a Tumour log-linear mixed effects growth rates of KRASG12C PDX treated with AZD4625 (green) versus vehicle (black). For each model, log-tumour volume was regressed on treatment, time and the interaction between treatment and time, allowing differences in growth rates over time to be assessed. Models were adjusted for multiple correlated observations per mouse using a random intercept and slope to model tumour starting volume and growth rate per mouse, respectively. Average growth rates of each arm are represented in plots as thick solid-coloured lines, while the thin lines connected by dots are the raw tumour measurements per mouse. Treatment began on day 0 and continued until 4-weeks of dosing passed or humane endpoints were reached. The IDs of sensitive models are highlighted in green, resistant in red. b Tumour masses of nine PDX models after at least 4 weeks of treatment are shown for each treatment group. Masses for PHLC82, PHLC151 and PHLC334 were used for further downstream applications and data is not available.
Numerically slower average growth rates in tumour masses were observed in AZD4625- versus vehicle-treated tumours for all evaluable models (Fig. 2b, Supplemental Fig. S3A). However, certain tumour models presented as complex pathologies, complicating tumour measurements. For instance, PHLC362 presented with cysts, concealing the true sizes of these tumours (Supplemental Fig. S3B). Although tumour regression was noted in 33% (4/12) of the tumours evaluated, the cystic and mucinous growth of PHLC362 and PHLC256 may have led to the underreporting of tumour regression.
Baseline transcriptomics distinguishes AZD4625-sensitive and resistant tumour features
To understand the differential sensitivity of PDX models to AZD4625, we compared the baseline transcriptomic profiles of the AZD4625-sensitive and resistant tumours (Fig. 3a). Altogether 970 significantly differentially expressed genes were identified between the sensitive versus resistant models, with 359 and 611 genes being up- and down-regulated, respectively. GSEA analysis revealed that sensitive tumours were enriched for gene sets that are downregulated upon KRAS activation (KRAS_Signaling_DN), suggesting that KRAS signalling plays an important regulatory and potentially survival function in these tumours (Fig. 3b). In contrast, the resistant tumours were enriched for gene sets involved in cell cycle regulation and proliferation, including E2F_Targets, MYC_Targets_V1, G2M_Checkpoint, as well as cell survival pathways and energy production, highlighting potential primary resistance mechanisms to AZD4625 (Fig. 3c).

a Principal component 1 and principal component 2 plots of baseline bulk RNA sequencing. Gene set enrichment analysis plot of the top 30 hallmark gene sets within the AZD4625-sensitive (b) and AZD4625-resistant (c) group of tumours at baseline. Sets enriched with a nominal p value of <0.1 are highlighted.
Short-term AZD4625 pharmacodynamics reveals model-specific inhibition of MAPK and mTOR signalling
To uncover adaptive changes to AZD4625 in vivo, we conducted an acute pharmacodynamic screen of three AZD4625-sensitive and three resistant PDX models (Fig. 4a). In the sensitive models, decreases in pERK1/2 and pS6 (except PHLC194) began at 4-h through 6-h and rebounded to baseline levels at 24-h (Fig. 4b, Supplemental Fig. S4A, Supplemental Fig. S5A). Of the three resistant models, decreases in pERK1/2 only occurred in PHLC191 and PHLC344, pS6 decreased in PHLC191 and no changes occurred in PHLC277. To assess further the MAPK pathway activity, we analysed gene expression of DUSP6, a negative regulator of ERK1/2 activation [35]. DUSP6 gene expression was significantly reduced in all sensitive models at 4- and 6-h, with rebound of signal beginning at 24-h and signal increasing twofold relative to baseline at 120-h for some models (Fig. 4c, Supplemental Fig. S5B). The reduction at 4- and 6-h in the AZD4625-resistant tumours was dampened relative to the AZD4625-sensitive tumours (Fig. 4c). Chronically treated tumours of all models had sustained and significant inhibition of DUSP6 transcription despite inconsistent pERK1/2 protein activation, suggesting that resistance to AZD4625 may not be facilitated solely through the MAPK signalling axis.

a Acute and chronic AZD4625 dosing in six PDX models. b Immunoblot analysis of the six models. c qPCR of DUSP6 expression. DUSP6 expression was normalised to control and baseline levels (0-hour (hr) timepoint) and statistical comparisons using two-sided, unpaired t-tests were between the average sensitive (green bars) or resistant (red bars) tumours at baseline (0-h timepoint) and timepoints of interest. Data is from two independent experiments. *: p < 0.05; ***: p < 0.005; ****: p < 0.001. PO bol: single oral bolus of AZD4625; QDx5: once-per-day dosing of AZD4625 for a period of 5 days. Hr hour.
Global proteomics identifies putative AZD4625-resistance mechanisms
The global proteome of acutely and chronically treated tumours were analysed using liquid chromatography with tandem mass spectrometry (LC-MS/MS), where 4703 human proteins were mapped across all 126 samples. Samples from the same biological tumour clustered together, rather than by treatment timepoint (Supplemental Fig. S6A). However, the proteomic profiles of the untreated (baseline) tumours did not cluster according to AZD4625 response (Supplemental Fig. S6B) and is thus not predictive of AZD4625 sensitivity. However, GSEA highlighted an enrichment of the hallmark EMT gene set (64 proteins mapped) in the AZD4625-sensitive models, while AZD4625-resistant models were enriched for gene sets related to aerobic oxidation including Hallmark_Oxidative_Phosphorylation (Supplemental Fig. S6C).
As EMT is a putative resistance mechanism to KRASG12C inhibitors [36], we investigated the role of EMT in AZD4625-sensitivity by staining our entire KRASG12C PDX cohort with E-cadherin (epithelial marker), and the mesenchymal markers vimentin and twist. Strong E-cadherin staining was apparent in all models, while only PHLC194 (AZD4625-sensitive) and PHLC362 (AZD4625-resistant) highly expressed vimentin. Both sensitive and resistant models expressed variable levels of nuclear and/or cytoplasmic twist, with no apparent differences that are associated with drug sensitivity. (Supplemental Fig. S7).
We next aimed to understand the acute and chronic effects of AZD4625 on the tumour proteome. The largest effects relative to baseline were in the AZD4625-sensitive tumours at the T120C timepoint, where over 200 differentially expressed proteins (FDR < 0.05) were identified (Supplemental Table S1, Supplemental Fig. S8A). After chronic dosing, the proteins changing upon AZD4625 treatment were similar in the sensitive and resistant tumours as evidenced by a positive and significant correlation of changes in protein abundance between baseline and the T120C timepoint (R = 0.28; p < 2.2e-16) (Fig. 5a). However, the expression of the proteins that changed upon AZD4625 exposure were higher in the resistant versus sensitive tumours at baseline (R = 0.21; p < 2.2e-16) (Fig. 5b, c). This suggested that while AZD4625 similarly changes the proteomes of all exposed tumours, the higher baseline expression of certain proteins in the resistant rather than the sensitive tumours acts as a buffer to ensure tumour survival.

a Heatmap of protein expression changes in AZD4625-sensitive (S) tumours at baseline versus T120C (y-axis) compared to those changes in AZD4625-resistant (R) tumours (x-axis). b Heatmap of protein expression changes in AZD4625-sensitive (S) tumours at baseline versus T120C (y-axis) compared to the differences in baseline protein expression between resistant and sensitive tumours (x-axis). c Explanation of (a, b). R: Resistant tumours; S: Sensitive tumours; T0: Baseline tumours; T120C: Chronic-treated AZD4625 tumours. d Positive enrichment of Hallmark gene sets in AZD4625-sensitive tumours at baseline compared to sensitive T120C tumours
Finally, hallmark GSEA uncovered downregulated enriched sets including E2F_Targets, MTORC1 Signalling and MYC_Targets_V1/2 in AZD4625-sensitive tumours at T120C relative to baseline (Fig. 5d). Many of these gene sets overlapped with those being significantly positively enriched in the transcriptomes of AZD4625-resistant tumours at baseline (Fig. 3c). Supporting these data, we noted a moderate and significant positive correlation between the transcriptome and proteome platforms at baseline (R = 0.48, p < 2.2e-16) (Supplemental Fig. S8B). Based on the robust detection of these pathways, they are likely the major biological programmes altered by AZD4625, and high baseline protein or mRNA expression may predict primary resistance to AZD4625.
Discussion
Efforts to develop small-molecule inhibitors to target the inactive, GDP-bound form of KRASG12C have been fruitful [1]. However, only a handful of KRASG12C LUAD cell lines are commercially available for preclinical analyses [18]. As such, models of colorectal cancer, pancreatic ductal adenocarcinoma, lung squamous carcinoma and other lung subtypes are often used to demonstrate preclinical activity (Supplemental Table S2), although this limits the translatability of data to the LUAD patient population. Indeed, clinical LUAD resistance to these compounds necessitates the combination of therapies to achieve responses, which are rapidly evolving [1]. Here, we aimed to better understand primary resistance to KRASG12C inhibitors by evaluating the novel KRASG12C inhibitor, AZD4625, in our cohort of new KRASG12C LUAD PDX and their XDO models. Through a 1 month-long drug monotherapy treatment regimen, tumour regression occurred in 4/12 (33.3%) of our tumour models. In an acute pharmacodynamic screen, MAPK and PI3K/AKT signals non-uniformly fluctuated between AZD4625-sensitive and resistant tumours. Finally, transcriptomic and proteomic analyses of PDX tumours highlighted cellular programmes that may elicit resistance to AZD4625.
Intrinsic/primary resistance to KRASG12C inhibitors is not well understood [36,37,38,39]. shRNA KRAS knockdown studies have proposed the idea of KRAS- ‘dependence’ for growth and survival, whereas some cell lines thrive even after shRNA knockdown of KRAS (KRAS- ‘independent’) [40]. KRAS-dependence has been attributed to direct proportionality to KRAS protein expression [36, 40], although we did not find such an association within our PDX cohort. Indeed, four of our twelve PDX models regressed on AZD4625, despite all models exhibiting high KRAS expression. KRAS-independence has also been linked to EMT [40], which was previously suggested as a mechanism of G12Ci primary resistance [36]. However, global proteomics identified EMT as the most significantly enriched gene set in our AZD4625-sensitive tumours at baseline (Supplemental Fig. S6C). While the loss of E-cadherin and gain of vimentin expression is characteristic of EMT in lung cancer [41] and these markers have been used in the context of KRASG12C resistance [36, 39], our immunohistochemical analysis of these markers and twist failed to demonstrate a clear relationship between EMT and G12Ci (Supplemental Fig. S7). We believe more study is warranted to fully understand the nuances of EMT and its contribution to G12Ci primary resistance.
To understand short-term responses to AZD4625, we conducted an acute and chronic pharmacodynamic drug screen and assessed DUSP6 gene expression as well as MAPK and AKT/mTOR protein pathway outputs. Reduction and rebound in MAPK and AKT signal was uniform across the sensitive models (Fig. 4b, Supplemental Fig. S5A), which mirrored studies that used other G12Ci [36, 42]. Three-dimensional models including xenografts are known to have an enhanced sensitivity to G12Ci [34], stemming from an increased dependence on KRAS signalling [43]. However, significant reductions in gene expression of the ERK1/2 negative regulator, DUSP6, did not translate to a reduction in MAPK signalling in the resistant PDX models. In response to G12Ci inactivation of MAPK signalling, compensatory activation of the AKT/mTOR pathway has been reported [44, 45]. However, as this only occurred in one of three AZD4625-resistant models, other mechanisms must be activated to maintain high ERK1/2 protein activation.
Several hallmark gene sets of interest were enriched in the transcriptomes of AZD4625-resistant tumours, including E2F_Targets, MYC_Targets_V1, G2M_Checkpoint (Fig. 3c), which have been implicated in G12Ci resistance [36, 46, 47]. These gene sets were also identified as being negatively enriched in the transcriptomes of NCI-H358 cells (sensitive to G12Ci) treated with a KRASG12C inhibitor for 24-h [34], and in human PDX models treated for at least 1 month with a different G12Ci [48]. With chronic AZD4625 treatment, we identified downregulation of these same gene sets in AZD4625-sensitive tumours including E2F_Targets, MTORC1_Signaling, and MYC_Targets_V1/V2 (Fig. 5d). High gene expression of the E2F_Targets may predict the response of certain cancers to neoadjuvant chemotherapy and cyclin-dependent kinase inhibition [49], and our data suggests that it may also predict for AZD4625 response. mTOR has been implicated in G12Ci secondary resistance, and consistent with our finding, G12Ci treatment was found to upregulate mTOR gene expression in KRASG12C mutant lung cancer cell lines (H23, HOP-62, Calu-1) [50], possibly through YAP/TAZ pathway activation [51, 52].
In this study we sought to determine the antitumor activity of the KRASG12C inhibitor, AZD4625, in novel PDX and XDO LUAD models. We observed AZD4625 sensitivity with tumour regression in 33.3% of PDXs overall, and in the matching XDO models. Despite mostly being early-stage, successful engraftment of primary NSCLC tumours has been correlated with poorer clinical outcomes of the patients and tumour aggressiveness, thus somewhat reflecting the biology of advanced stage tumours [21, 24]. Our transcriptomic and proteomic analyses suggest that tumours with higher baseline gene and protein expression of mTOR and MYC signalling may be intrinsically resistant to AZD4625. However, MAPK signalling at baseline was not a determinant of sensitivity, although acute changes could differentiate sensitive and resistant tumours. Overall, we provide support for the continued investigation of AZD4625 for the indication of KRASG12C NSCLC and highlight the utility of PDXs for studying resistance to small molecule inhibitors. We underscore the need for further in vivo proteomic studies into overcoming primary resistance to KRASG12C inhibitors. As phosphorylation changes occur rapidly, we suggest that phospho-proteomic analyses may help uncover the intricacies of G12Ci intrinsic resistance [2]. Finally, as we demonstrated that transcriptomics and proteomics can provide complementary insights into G12Ci resistance, we suggest that future studies should combine the two approaches.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Rosen JC, Sacher A, Tsao MS. Direct GDP-KRASG12C inhibitors and mechanisms of resistance: the tip of the iceberg. Ther Adv Med Oncol. 2023;15:17588359231160141.
Solanki HS, Welsh EA, Fang B, Izumi V, Darville L, Stone B, et al. Cell type-specific adaptive signaling responses to KRASG12C inhibition. Clin Cancer Res. 2021;27:2533–48.
Chevallier M, Borgeaud M, Addeo A, Friedlaender A. Oncogenic driver mutations in non-small cell lung cancer: past, present and future. World J Clin Oncol. 2021;12:217–37.
Yu HA, Sima CS, Shen R, Kass S, Gainor J, Shaw A, et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J Thorac Oncol. 2015;10:431–7.
Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5:288–300.
Ricciuti B, Mira A, Andrini E, Scaparone P, Michelina SV, Pecci F, et al. How to manage KRAS G12C-mutated advanced non-small-cell lung cancer. Drugs Context. 2022;11:2022–7–4.
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371–81.
Garassino MC, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22:235–7.
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51.
FDA. FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC. FDA, 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sotorasib-kras-g12c-mutated-nsclc Accessed 3 Aug 2024.
ESMO. FDA Grants Accelerated Approval to Adagrasib for KRAS G12C-Mutated NSCLC, https://www.esmo.org/oncology-news/fda-grants-accelerated-approval-to-adagrasib-for-kras-g12c-mutated-nsclc Aaccessed 5 Aug 2024.
Sacher A, LoRusso P, Patel MR, Miller WH Jr, Garralda E, et al. Single-Agent divarasib (GDC-6036) in solid tumors with a KRAS G12C mutation. N Engl J Med. 2023;389:710–21.
Fujiwara Y, Burns T, Dragnev K, Murciano-Goroff Y, Lee D, Hollebecque A, et al. Efficacy and safety of olomorasib (LY3537982), a second-generation KRAS G12C inhibitor (G12Ci), in combination with pembrolizumab in patients with KRAS G12C-mutant advanced NSCLC. J Clin Oncol. 2024;42:195–195.
Jiang J, Jiang L, Maldonato BJ, Wang Y, Holderfield M, Aronchik I, et al. Translational and therapeutic evaluation of RAS-GTP inhibition by RMC-6236 in RAS-driven cancers. Cancer Discov. 2024;14:994–1017.
Nokin MJ, Mira A, Patrucco E, Ricciuti B, Cousin S, Soubeyran I, et al. RAS-ON inhibition overcomes clinical resistance to KRAS G12C-OFF covalent blockade. Nat Commun. 2024;15:7554.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23.
Mikubo M, Inoue Y, Liu G, Tsao MS. Mechanism of drug tolerant persister cancer cells: the landscape and clinical implication for therapy. J Thorac Oncol. 2021;16:1798–809.
Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a cancer dependency map. Cell. 2017;170:564–76.e16.
Chakraborty A, Hanson L, Robinson D, Lewis H, Bickerton S, Davies M, et al. AZD4625 is a potent and selective inhibitor of KRASG12C. Mol Cancer Ther. 2022;21:1535–46.
Kettle JG, Bagal SK, Bickerton S, Bodnarchuk MS, Boyd S, Breed J, et al. Discovery of AZD4625, a covalent allosteric inhibitor of the mutant GTPase KRASG12C. J Med Chem. 2022;65:6940–52.
Mirhadi S, Tam S, Li Q, Moghal N, Pham N-A, Tong J, et al. Integrative analysis of non-small cell lung cancer patient-derived xenografts identifies distinct proteotypes associated with patient outcomes. Nat Commun. 2022;13:1811.
Wang D, Pham NA, Tong J, Sakashita S, Allo G, Kim L, et al. Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int J Cancer. 2017;140:662–73.
Shi R, Radulovich N, Ng C, Liu N, Notsuda H, Cabanero M, et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin Cancer Res. 2020;26:1162–74.
John T, Kohler D, Pintilie M, Yanagawa N, Pham NA, Li M, et al. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin Cancer Res. 2011;17:134–41.
Rosen JC, Weiss J, Pham NA, Li Q, Martins-Filho SN, Wang Y, et al. Antitumor efficacy of XPO1 inhibitor Selinexor in KRAS-mutant lung adenocarcinoma patient-derived xenografts. Transl Oncol. 2021;14:101179.
Conway T, Wazny J, Bromage A, Tymms M, Sooraj D, Williams ED, et al. Xenome—a tool for classifying reads from xenograft samples. Bioinformatics. 2012;28:i172–178.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. cels. 2015;1:417–25.
Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. bioRxiv 2021. https://doi.org/10.1101/060012.
Krieger JR, Wybenga-Groot LE, Tong J, Bache N, Tsao MS, Moran MF. Evosep one enables robust deep proteome coverage using tandem mass tags while significantly reducing instrument time. J Proteome Res. 2019;18:2346–53.
McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84:7469–78.
Plubell DL, Wilmarth PA, Zhao Y, Fenton AM, Minnier J, Reddy AP, et al. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol Cell Proteom. 2017;16:873–90.
Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172:578–89.e17.
Zhang Z, Kobayashi S, Borczuk AC, Leidner RS, Laframboise T, Levine AD, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31:577–86.
Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R, et al. Epithelial-to-mesenchymal transition is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitor in KRAS G12C-mutant non-small cell lung cancer. Clin Cancer Res. 2020;26:5962–73.
Morimoto K, Yamada T, Hirai S, Katayama Y, Fukui S, Sawada R, et al. AXL signal mediates adaptive resistance to KRAS G12C inhibitors in KRAS G12C-mutant tumor cells. Cancer Lett. 2024;587:216692.
Baranyi M, Molnár E, Hegedűs L, Gábriel Z, Petényi FG, Bordás F, et al. Farnesyl-transferase inhibitors show synergistic anticancer effects in combination with novel KRAS-G12C inhibitors. Br J Cancer. 2024;130:1059–72.
Thatikonda V, Lyu H, Jurado S, Kostyrko K, Bristow CA, Albrecht C, et al. Co-targeting SOS1 enhances the antitumor effects of KRASG12C inhibitors by addressing intrinsic and acquired resistance. Nat Cancer. 2024;5:1–19.
Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500.
Nam MW, Kim CW, Choi KC. Epithelial-mesenchymal transition-inducing factors involved in the progression of lung cancers. Biomol Ther. 2022;30:213–20.
Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577:421–5.
Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316–29.
Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. 2019;25:796–807.
Ryan MB, Coker O, Sorokin A, Fella K, Barnes H, Wong E, et al. KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy. Cell Rep. 2022;39:110993.
Tong X, Patel AS, Kim E, Li H, Chen Y, Li S, et al. Adeno-to-squamous transition drives resistance to KRAS inhibition in LKB1 mutant lung cancer. Cancer Cell. 2024;42:413–28.e7.
Wang Y, Yao M, Li C, Yang K, Qin X, Xu L, et al. Targeting ST8SIA6-AS1 counteracts KRASG12C inhibitor resistance through abolishing the reciprocal activation of PLK1/c-Myc signaling. Exp Hematol Oncol. 2023;12:105.
Sudhakar N, Yan L, Qiryaqos F, Engstrom LD, Laguer J, Calinisan A, et al. The SOS1 inhibitor MRTX0902 blocks KRAS activation and demonstrates antitumor activity in cancers dependent on KRAS nucleotide loading. Mol Cancer Ther. 2024;23:1418–30.
Oshi M, Takahashi H, Tokumaru Y, Yan L, Rashid OM, Nagahashi M, et al. The E2F pathway score as a predictive biomarker of response to neoadjuvant therapy in ER+/HER2− breast cancer. Cells. 2020;9:1643.
Ito M, Miyata Y, Hirano S, Morihara N, Takemoto M, Irisuna F, et al. Dual inhibition of GTP-bound KRAS and mTOR in lung adenocarcinoma and squamous cell carcinoma harboring KRAS G12C. Cell Commun Signal. 2025;23:220.
Edwards AC, Stalnecker CA, Jean Morales A, Taylor KE, Klomp JE, Klomp JA, et al. TEAD inhibition overcomes YAP1/TAZ-driven primary and acquired resistance to KRASG12C inhibitors. Cancer Res. 2023;83:4112–29.
Mukhopadhyay S, Huang HY, Lin Z, Ranieri M, Li S, Sahu S, et al. Genome-wide CRISPR screens identify multiple synthetic lethal targets that enhance KRASG12C inhibitor efficacy. Cancer Res. 2023;83:4095–111.
Acknowledgements
This work is supported by a research grant from AstraZeneca UK as part of its Partner of Choice Program. The MS analyses at SPARC BioCentre were supported by CFI (#36294) and the Pan-Canadian Proteomics Centre Genomic Technology Platform Program (264PRO) of Genome Canada and Genome British Columbia. Dr. M.-S. Tsao was the M. Qasim Choksi Chair in Lung Cancer Translational Research at the Princess Margaret Cancer Centre. Xenograft and organoid methodologies were supported by Princess Margaret Living biobank. Portions of this work have been published as conference abstracts: JCR, N-AP, QL, PC, KH, TK, NR, AK, MN, SR, AS, M-ST. KRASG12C inhibitor resistance in patient-derived NSCLC models. Cancer Res 84 (6_Supplement) (abstract 6909) (2024). JCR, N-AP, QL, PC, KH, TK, NR, AK, MN, SR, AS, M-ST. The Use of Lung Adenocarcinoma Patient-Derived Xenografts and Organoids to Study GDP-KRAS G12C Inhibitor Resistance. J Thorac Oncol 17 (9_Supplement) (abstract EP08.02-079) (2022).
Funding
This project was supported by grants from AstraZeneca and the Canadian Institutes of Health Sciences (FDN-148395 to MST). MTW is supported by The Canada Graduate Research Scholarship—Master’s (CGRS-M). MS analyses at SPARC BioCentre were supported by CFI (#36294) and the Pan-Canadian Proteomics Centre Genomic Technology Platform Program (264PRO) of Genome Canada and Genome British Columbia.
Author information
Authors and Affiliations
Contributions
Project administration: Rosen JC, Pham N-A; Conceptualisation: Rosen JC, Pham N-A, Sacher A, Tsao M-S; Writing—original draft: Rosen JC, Pham N-A, Sacher A, Tsao M-S; Writing—review & editing: Rosen JC, Pham N-A, Li Q, Sacher A, Tsao M-S; Formal analysis: Rosen JC, Li Q, Waas M, Hueniken K; Wang M, Navab R; Methodology: Rosen JC, Cao P, Pham N-A Wybenga-Groot L, Moran MF; Investigation: Rosen JC, Cao P, Li Q, Waas M; Resources: Rosen JC, Li Q, Radulovich N, Wybenga-Groot L, Moran MF; Tsao M-S; Validation: Rosen JC, Pham N-A, Li Q, Hueniken K, Waas M; Supervision: Koers A, Niedbala M, Ross S, Moran MF, Sacher A, Tsao M-S; Funding acquisition: Koers A, Niedbala M, Ross S, Sacher A, Tsao M-S.
Corresponding authors
Ethics declarations
Competing interests
MN, AK and SR are AstraZeneca employees and shareholders. Dr. Sacher declares institutional Research & Clinical Trial PI from AstraZeneca, Amgen, Genentech, Merck, Lilly, Pfizer, BMS, Spectrum, GSK, Iovance, CRISPR Therapeutics, BridgeBio, HotSpot Therapeutics, AdaptImmune; Advisory committee (no personal fees): Genentech, Amgen, Merck; Travel expenses for clinical trial investigator meetings: Amgen, Merck, Genentech-Roche. MST received research grant (to institution) from AstraZeneca and Sanofi, and consultancy honoraria from AstraZeneca, Daiichi-Sankyo, Abbvie, Boehringer-Ingelheim and Sanofi. All other authors have nothing to declare.
Ethics approval
Study protocols were approved by the Research Ethics Board (REB 09-0510-T) and Animal Care Committee (AUP#743) in accordance to ARRIVE guidelines at the University Health Network. This study was performed in accordance with the ethical principles outlines in the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rosen, J.C., Cao, P., Pham, NA. et al. Modeling response to the KRAS-G12C inhibitor AZD4625 in KRASG12C NSCLC patient-derived xenografts reveals insights into primary resistance mechanisms. Br J Cancer 134, 165–174 (2026). https://doi.org/10.1038/s41416-025-03216-w
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41416-025-03216-w
