
Abstract
Glioblastoma (GBM) is a highly lethal disease with limited treatment options due to its infiltrative nature and the lack of efficient therapy able to cross the protective blood-brain barrier (BBB). GBMs are metabolically characterized by increased glycolysis and glutamine dependence. This study explores a novel metabolism-based therapeutic approach using a polyurea generation 4 dendrimer (PUREG4) surface functionalized with lactate (LA) (PUREG4-LA24), to take advantage of glucose-dependent monocarboxylate transporters (MCTs) overexpression, loaded with selenium-chrysin (SeChry) and temozolomide (TMZ) or complexed with anti-glutaminase (GLS1) siRNAs to abrogate glutamine dependence. The nanoparticles (PUREG4-LA24) were efficient vehicles for cytotoxic compounds delivery, since SeChry@PUREG4-LA24 and TMZ@PUREG4-LA24 induced significant cell death in GBM cell lines, particularly in U251, which exhibits higher MCT1 expression. The anti-GLS1 siRNA-dendriplex with PUREG4-LA12 (PUREG4-LA12-anti-GLS1-siRNA) knocked down GLS1 in the GBM cell lines. In two in vitro BBB models, these dendriplexes successfully crossed the BBB, decreased GLS1 expression and altered the exometabolome of GBM cell lines, concomitantly with autophagy activation. Our findings highlight the potential of targeting glucose and glutamine pathways in GBM using dendrimer-based nanocarriers, overcoming the BBB and disrupting key metabolic processes in GBM cells.

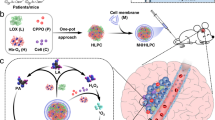
PUREG4-LA12-anti-GLS1-siRNA dendriplexes cross the blood–brain barrier (BBB) and impair glioblastoma (GBM) metabolism. The BBB is formed by a thin monolayer of specialized brain microvascular endothelial cells joined together by tight junctions that selectively control the passage of substances from the blood to the brain. It is a major obstacle in the treatment of GBM, since many chemotherapeutic drugs are unable to penetrate the brain. Therefore, we developed a strategy to overcome this obstacle: a lactate-coated polyurea dendrimer generation 4 (PUREG4) able to cross the BBB in vitro, that act as a nanocarrier of drugs and siRNA to the GBM cells. PUREG4-LA12 are nanoparticles functionalized with lactate (LA) to target MCT1, a lactate transporter highly expressed by GBM cells. Moreover, a complex of this nanoparticle with anti-GLS1 (glutaminase) siRNA (PUREG4-LA12-anti-GLS1-siRNA) was made, to target glutamine metabolism. It efficiently knocked down GLS1. Moreover, PUREG4-LA24 loaded with SeChry led to BBB disruption.
Similar content being viewed by others


Introduction
Glioblastomas (GBM) are grade 4 gliomas, that account for 14.2% of all central nervous system (CNS) tumors and 50.9% of all malignant CNS neoplasms [1]. These tumors are extremely aggressive, characterized by high infiltrative capacity of the surrounding brain tissue, making complete surgical removal very difficult [2]. The median overall survival (OS) is only 8 months after diagnosis [1, 3], mainly due to the difficulty to find new effective therapeutic strategies. The standard treatment remains the same since 2005, the Stupp protocol, which consists in maximal surgical resection, followed by radiotherapy with concomitant and adjuvant chemotherapy (Temozolomide – TMZ) [4]. Therefore, there is urgency in finding new targets and new drug formulations to tackle GBM.
One of the main obstacles of GBM treatment is the blood-brain barrier (BBB). This physiological barrier is composed of specialized endothelial cells that line the blood vessels in the brain, tightly regulating the passage of substances between the bloodstream and the brain tissue [5]. Pericytes, endfeet astrocytes and neurons are also involved in the BBB structure [6]. While the BBB protects the brain from harmful substances and pathogens, it also presents a challenge for delivering therapeutic agents to brain tumors [5, 6]. Therefore, many chemotherapeutic drugs and targeted therapies are unable to effectively penetrate the BBB, limiting their efficacy in treating GBM. Strategies to bypass or disrupt the BBB are being explored, such as nanotechnology [7, 8], to improve drug delivery to the brain tumor. Dendrimers are a class of nanocarriers able to cross the BBB and enhance the drugs internalization in the targeted brain tissues [9]. In particular, poly-amidoamine (PAMAM) dendrimers were found to cross the BBB in animal models [10] and very recently it was demonstrated their ability to cross the BBB when complexed with long non-coding RNAs [11].
Cancer metabolism is a rediscovered research field that generated a huge amount of knowledge in the last decade. However, very few have been applied for the improvement of cancer therapy. As described by our team, GBM cells suffer a metabolic remodeling, being dependent on glucose and glutamine [12]. Regarding glucose metabolism, it is observed an increased expression of glucose transporters (GLUT1 and GLUT3) [13] and an increased expression of monocarboxylate transporters (MCT1 and MCT4), responsible for lactate transport a main consequence of increased glycolysis [14, 15]. In the context of glutamine metabolism, the downregulation of glutamine synthetase (GS) in GBM cells, prompts their dependence on glutamine uptake [16,17,18] with the increased expression of glutamine transporters (ASCT2 and SNAT3) [19, 20]. Moreover, these tumors express significant amounts of glutaminase (GLS) [21], being able to fully metabolize glutamine that will serve as a substrate to supply bioenergetic and biosynthetic circuitries crucial for GBM cell survival [22,23,24,25].
Therefore, we hypothesized that a BBB-crossing dendrimer nanoformulation double-targeting glucose and glutamine metabolism would be able to impair GBM metabolism and survival. The polyurea generation 4 (PUREG4) dendrimer was already explored as nanocarrier of several drugs by our group in the context of ovarian [26, 27], lung [28], and breast cancer [29] and even in the context of endothelial cells activation and cancer angiogenesis [29]. These cationic dendrimers (displaying primary amines in the surface) are biocompatible and biodegradable nanocarriers that can encapsulate drugs or allow conjugation at the surface [30,31,32]. Here, we used PUREG4 partially functionalized with lactate molecules (LA) at the surface, a conjugation strategy to direct these nanoparticles to MCT1 expression related to increased glycolysis and complexed with anti-GLS1 small interference RNAs (anti-GLS1-siRNAs) to target glutamine metabolism in GBM cells. The complexation of siRNAs with polyurea dendrimers and efficient cytosolic delivery was already reported by us [33]. Therefore, we initially evaluated the efficacy of these dendrimers as drug nanocarriers for GBM in vitro. We encapsulated selenium-chrysin (SeChry) in PUREG4-LA24 (SeChry@PUREG4-LA24), a nanoformulation which inhibits cysteine metabolism and induces oxidative stress [26, 28, 34]. We also tested encapsulated TMZ (TMZ@PUREG4-LA24), the most used drug in the treatment of GBM [4]. Then we evaluated the dendriplex PUREG4-LA12-anti-GLS1-siRNA effect directly on GBM cells and after crossing two in vitro models of the BBB.
Materials and Methods
Cell culture
Two commercial GBM cell lines, U-87MG (HTB-14, American Type Culture Collection – ATCC, Manassas, Virginia, USA) and U251 (09063001, European Collection of Authenticated Cell Cultures – ECACC, Salisbury, United Kingdom) were used. The cell lines were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 Ham (DMEM/F-12; 11330-032, Gibco, Life Technologies, Grand Island, New York, USA), supplemented with 10% fetal bovine serum (FBS; P40-37500, PAN Biotech, Aidenbach, Germany), 1% Antibiotic-Antimycotic (AA; P06-07300, PAN Biotech) and 50 µg/mL gentamicin (15750-060, Gibco, Life Technologies).
For the BBB in vitro model, mouse BALB/c brain endothelioma cell line b.End3 was used (CRL.2299, ATCC). This cell line was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose (11965092, Gibco) supplemented with 10% FBS, 1% AA and 50 μg/mL Gentamicin. A normal human astrocyte cell line was also used, iNHA (NHA variant immortalized with SV40) (CC-2565, ATCC). This cell line was cultured in DMEM/F-12 supplemented with 10% FBS, 1% AA and 50 μg/mL Gentamicin.
Cells were maintained in a humidified environment of 5% CO2 at 37 °C, cultured until an optical confluence of 75-100%, detached with 0.05% trypsin-EDTA 1× (25300-054, Invitrogen, Thermo Fisher Scientific, Waltham, Massachusetts, USA), at 37 °C for approximately 5 min, and split to new plates according to the experimental procedures. During exposure to experimental conditions, cells were maintained in DMEM/F-12 with 1% FBS (GBM and iNHA cells) or DMEM 1%FBS (b.End3).
Synthesis of lactate-coated PUREG4 dendrimers
PUREG4 was synthesized following the reported supercritical-assisted polymerization methodology [35]. To prepare PUREG4 surface-functionalized with lactate (LA), PUREG4 was reacted with the activated lactic acid succinic ester, synthesized according to our protocol [36].
Two lactate-coated PUREG4 dendrimers were synthetized, one with 50% of LA in the surface (PUREG4-LA24, Fig. S1A) and another with 25% LA in the surface (PUREG4-LA12, Figure S1B and C). PUREG4-LA24 was used for the delivery of selenium-chrysin (SeChry) and TMZ (Temodal, temozolomide, Accord, Barcelona, Spain) (Fig. S2), and PUREG4-LA12 was used for the complex (dendriplex) formation with anti-GLS1 siRNA. Briefly, for the synthesis of PUREG4-LA24, in a round-bottom flask, 41 mg (0.456 mmol) of sodium lactate (LA; Sigma-Aldrich, Burlington, Massachusetts, USA) was dissolved in anhydrous N,N-dimethylformamide (DMF; Sigma-Aldrich, Burlington, Massachusetts, USA) (5 mL). After the addition of 105 mg (0.908 mmol) of N-hydroxysuccinimide (Sigma-Aldrich, Burlington, Massachusetts, USA), 103.6 mg (0.502 mmol) of N,N’-dicyclocarbodiimide (Sigma-Aldrich, Burlington, Massachusetts, USA), and 0.140 mL (1.04 mmol) of triethylamine (TEA; Sigma-Aldrich, Burlington, Massachusetts, USA), the reaction was stirred at room temperature overnight in the dark. After this period, TEA was evaporated and 150 mg of PUREG4 (0.019 mmol) in DMF anhydrous (0.75 mL) and 0.070 mL of TEA (0.502 mmol) were added and allowed to react overnight. The mixture was then filtered to remove precipitated solids and TEA was evaporated. The solution was then dialyzed (MWCO 100–500 Da), and the recovered solution was washed several times with diethyl ether (Sigma-Aldrich, Burlington, Massachusetts, USA) to remove residual DMF. After water evaporation, 194 mg (quantitative yield) of the product was obtained as a light-yellow sticky oil. By 1H-NMR analysis, a total of 24 lactate molecules incorporated in the dendrimer surface was confirmed. The PUREG4-LA12 dendrimer was synthesized following the same protocol using the same amount of PUREG4 and half of the lactic acid succinic ester reagent.
Preparation of SeChry@PUREG4-LA24 and TMZ@PUREG4-LA24 nanoformulations
SeChry was encapsulated in PUREG4-LA24 (Fig. S2A) nanoparticles following a modified protocol [31]. Briefly, a CHCl3 solution (0.5 mL) of SeChry (6.5 mg) was added to an aqueous solution (2 mL) of PUREG4-LA24 (125 mg). Next, CHCl3 was removed in a rotary evaporator and the mixture was allowed to stir at room temperature overnight. Then, the aqueous solution was extracted with CHCl3 to remove non-encapsulated SeChry. No SeChry was found in the CHCl3 extracts (control by thin-layer chromatography), thus confirming a full encapsulation. The release profile followed the usual pattern reported for this nanodelivery system.
Using a similar protocol [31], TMZ (Temozolomide Accord, 250 mg capsules containing 250 mg of TMZ and 154,3 mg of lactose as excipient) was encapsulated in PUREG4-LA24 (Fig. S2B). To an aqueous solution (5 mL) of PUREG4-LA24 (50 mg), were added 50 mg of the capsule content (25 mg of TMZ) and the mixture was allowed to stir at room temperature overnight. Next, the solution was dialyzed, and the nanoformulation obtained after water evaporation and drying under a vacuum.
Cells were exposed to different concentrations of SeChry@PUREG4-LA24 (25–400 μM of SeChry) and TMZ@PUREG4-LA24 (50–1000 μM of TMZ). The empty nanoparticle, PUREG4-LA24, was used as control in equimolar concentrations to SeChry@ PUREG4-LA24 and TMZ@ PUREG4-LA24.
Transfection with anti-GLS1 siRNA
Cells were seeded in 6-well plates (2 × 105 cells/well). Two anti-GLS1 siRNAs were mixed in the proportion 1:1 (AM16708, Assay ID 145342 and 145343, ThermoFisher Scientific): AM16708 siRNA anti-GLS1 (Assay ID: 145342): Sense strand: 5’ – GCACUCAAAUCUAC AGGAUtt – 3’; Antisense strand: 5’ – AUCCUGUAGAUUUGAGUGCtg – 3’. AM16708 siRNA anti-GLS1 (Assay ID: 145343): Sense strand: 5’ – GGCACAGACAUGGUUGGUAtt – 3’; Antisense strand: 5’ – UACCAACCAUGUCUGUGCCtt – 3’.
Cells were transfected with siRNA in lipofectamine 2000 (11668019, Invitrogen). Lipofectamine 2000 and siRNA were incubated separately with DMEM/F-12 without FBS for 5 min, 37 °C and then the two suspensions were mixed and incubated for 20 min, 37 °C. After incubation, cells were exposed to the mix, in a final concentration of 30 nM siRNA, according to the manufacturer’s protocol. GLS1 mRNA expression was then evaluated by RT-qPCR.
Preparation of the PUREG4-LA12-anti-GLS1-siRNA dendriplex
In order to improve the efficacy of the dendriplex PUREG4-LA12-anti-GLS1-siRNA formation, since 24 negatively charged lactate molecules (LA groups) can reduce the availability of amine groups on the surface of the dendrimer and interfere with siRNA binding and stability, a new nanoparticle with 12 LA groups on the surface was used. Two anti-GLS1 siRNAs (145342 and 145343, ThermoFisher Scientific) were mixed in the proportion 1:1. The complexation of PUREG4-LA12 with siRNA was performed as previously described [33]. PUREG4-LA12 and siRNA were mixed and incubated in nuclease-free water for 2 h at 37 °C. The formation of the dendriplex PUREG4-LA12-anti-GLS1-siRNA was performed based on Aydin and colleagues work [37], taking into account the ratio of positively charged amine groups (N) of the carrier and negatively charged phosphorus groups (P) of RNA. Two N/P ratios (positively charged amine groups of the carrier (N) of the dendrimer / negatively charged phosphorus (P) of siRNA) were tested: 1:1.5 (20.8 nM PUREG4-LA12, 29.6 nM siRNA) and 1:2 (20.8 nM PUREG4-LA12, 39.4 nM siRNA), and the dendriplexes formation was confirmed on a 2% (w/v) agarose gel electrophoresis.
Reverse transcription and quantitative PCR (RT-qPCR)
Total RNA was extracted using the RNeasy Mini Extraction kit (74104, Qiagen, Venlo, Netherlands), according to the manufacturer’s protocol. cDNA was synthesized from 0.5 µg RNA, using SuperScript II Reverse Transcriptase (18080e44, Invitrogen), according to the manufacturer’s protocol. Relative quantification using quantitative PCR (qPCR) was performed using Power SYBR™ Green PCR Master Mix (4368708, Applied Biosystems, Foster City, California, USA), according to the manufacturer’s protocol. Real-time PCR was carried out in a Lightcycler® 480 System instrument (05015243001, Roche, Basel, Switzerland). Primers used: for GLS1 gene Forward: 5’ – CTTCTACTTCCAGCTGTGCTC – 3’, Reverse: 5’ – CACCA GTAATTGGGCAGAAACC – 3’; for MCT1 coding gene (SLC16A1) Forward: 5’ – GCTGGGC AGTGGTAATTGGA – 3’, Reverse: 5’ – CAGTAATTGATTTGGGAAATGCA – 3’. The hypoxanthine-guanine phosphoribosyltransferase 1 gene (HPRT1) was used has a housekeeping gene for normalization purposes (Forward: 5’ – TGACAC TGGCAAAACAATGCA – 3’, Reverse: 5’ – GGTCCTTTTCACCAGCAAGCT – 3’). Experiments were performed in biological triplicates.
Cell death analysis by flow cytometry
To analyze the effects of the experimental conditions on cell viability, GBM cell lines were seeded in 24-well plates (1 × 105 cells/well) and b.End3 cell line in 48-well plates (5 × 104 cells/well). After 24, 48 or 72 h exposure to experimental conditions, the conditioned culture medium (supernatant) was collected, and adherent cells were detached with 0.05% trypsin-EDTA. Trypsinized cells and supernatant were collected in the same tube and centrifuged at 155 × g for 3 min. Then, cell pellets were incubated with 0.5 µL annexin V-fluorescein isothiocyanate labeled (annexin V-FITC; 640906, BioLegend, San Diego, California, USA), in annexin V binding buffer 1× (10 mM HEPES (pH 7.4; 391333, Millipore, Darmstadt, Germany), 140 mM sodium chloride (NaCl; 106404, Merck, Darmstadt, Germany), 2.5 mM calcium chloride (CaCl2; 449709, Sigma-Aldrich, Burlington, Massachusetts, USA) for 15 min in the dark, at room temperature. Cells were resuspended in 200 µL 0.1% (v/w) bovine serum albumin (BSA; A9647, Sigma-Aldrich) in PBS 1× and centrifuged at 155 × g for 2 min. The remaining pellets were resuspended in annexin V binding buffer 1× and 2.5 µL of 50 µg/mL propidium iodide (PI; P4170, Sigma-Aldrich). Flow cytometry was performed in a BD Accuri C6 Plus (Becton Dickinson, Franklin Lakes, New Jersey, USA) and data was analyzed in the respective software. Half maximal effective concentration (EC50) was calculated for SeChry@PUREG4-LA24, TMZ@PUREG4-LA24, and PUREG4-LA24 in U251 and U-87MG cell lines. Experiments were performed in biological triplicates.
Nuclear magnetic resonance (NMR) spectroscopy
After exposure to experimental conditions for 72 h, supernatants were collected and stored at -80 °C, to be analyzed by 1H-NMR spectroscopy. A 60 µL solution of 1.5 M potassium phosphate buffer (KPi, pH 7.4), NaN3 2 mM and 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (TSP) 0.16 mM in D2O was added to 540 µL of supernatant sample. 1H-NMR spectra were obtained at 25 °C in a magnetic field of 800 MHz in UltrashieldTM 800 Plus Spectrometer (Bruker, Billerica, Massachusetts) with a TCI cryo-probe. Spectra were acquired with a noesygppr1d pulse program, free induction decay size = 131k points, 40 scans, 20 ppm of spectral window, with 4 s of acquisition time and 10 ms of mixing time. TopSpin 3.6 software (Bruker) was used for spectra acquisition and processing. Compound identification on supernatants was made by resorting to the Human Metabolome Database (http://www.hmdb.ca/, accessed on 24 May 2024) and Chenomx NMR Suite software version 8.1 (Chenomx Inc.), which was also used for metabolite quantification. Data analysis was performed through MetaboAnalyst 6.0 platform (https://www.metaboanalyst.ca, accessed on 26 May 2024).
BBB in vitro model
A BBB in vitro model was established based on the literature [38,39,40,41]. In this model, a transwell insert (9321002, cellQART 24-Well Insert 1.0 µm pore PET translucent, Northeim, Germany) with a microporous semipermeable membrane was inserted in a 24-well plate and separated the upper (luminal, blood side) and the lower (abluminal, brain side) chambers.
In the BBB model with iNHA, the bottom of the inserts was precoated with 0.001% Poly-L-Lysine (L 7240, Biochrom AG, Darmstadt, Germany) for 45 min at room temperature. iNHA (4.455 × 104 cells/insert) were plated on the lower side of the insert, which was upside down until cells’ adherence. Then, b.End3 cells (1 × 104 cells/insert) were seeded on the inserts after 1 h coating with 2% matrigel DMEM without FBS and antibiotics, at 37 °C. After 3 days in culture, 550 nM of hydrocortisone (H4001, Sigma-Aldrich), was added as described [38]. To confirm the barrier tightness of the BBB model, Transendothelial Electrical Resistance (TEER) measure readings were performed using a chopstick electrode STX2 coupled to EVOM Volt/Ohm meter (World Precision Instruments, Sarasota, Florida, USA). Readings were collected until 14 days after b.End3 cells seeding, and TEER was calculated. Subtraction of the empty insert values was done, and the value was multiplied by the area of the insert (0.3 cm2). The results are expressed as Ω × cm2.
On the 6th day after seeding, 5 × 104 cells/well GBM cells were seeded in the 24-well plate, on the 7thy day the inserts with b.End3 cells were placed on top of the 24-well plate with the GBM cells, and experimental conditions were added.
Experiments were performed in biological triplicates.
Immunofluorescence
b.End3 cells (1 × 104 cells/insert) were cultured in transwells for 7 days. After fixation with 2% paraformaldehyde for 15 min, 4 °C, blocking was performed with 3% BSA - 0.1% triton X-100 in PBS 1 × (w/v/v) (AF938, R&D systems, Minneapolis, Minnesota, USA) for 30 min, room temperature. Cells incubated with anti-VE-Cad (sc-6458, Santa Cruz Biotechnology, Heidelberg. Germany: 1:50 in 3% BSA-0.1% triton X-100 PBS), overnight at 4 °C. After that, cells were incubated with the secondary antibody (Alexa Fluor 488 donkey anti-goat, 1:500 in 3% BSA-0.1% triton X-100 PBS; A11055, Thermo Fisher Scientific), for 2 h at room temperature. DAPI was not used to stain the nuclei since its adsorption to the transwell filter background impairs the visualization of VE-Cad- Alexa Fluor 488 signal.
GBM cells (1 × 105 cells/well) were seeded on glass slides with a 0.2% gelatin coating (G-1890, Sigma Aldrich), until 80% of confluence and then fixed in 4% paraformaldehyde for 15 min at 4 °C. After fixation, cells were incubated with 50 mM ammonium chloride (NH4Cl) for 10 min. Blocking was performed with PBS 1× − 0.5% BSA − 0.1% saponin (w/v/v), between incubations, cells were rinsed three times with PBS 1×, for 5 min. After blocking for 30 min, slides were incubated with rabbit anti-human MCT1 (ab315382, Abcam, Cambridge, United Kingdom; 1:100), rabbit anti-human GLS1 (56750 T, Cell Signaling, Danvers, Massachusetts, USA; 1:200) or goat anti-human ATG12 (AB0083, SICGEN; 1:100) in PBS 1× − 0.5% BSA − 0.1% saponin (w/v/v), overnight at 4 °C. After rinsing, slides were incubated with the secondary antibody Alexa Fluor 488 goat anti-rabbit (A-11008 Invitrogen - Thermo Fisher Scientific) or Alexa Fluor 488 donkey anti-goat (A-11055 Invitrogen - Thermo Fisher Scientific) 1:500 in PBS 1× − 0.5% BSA − 0.1% saponin (w/v/v) for 2 h at room temperature. The slides were mounted in VECTASHIELD media with DAPI (4′-6-diamidino-2-phenylindole) (H-1200, Vector Labs, Newark, California, USA).
All slides were examined by standard fluorescence microscopy under a Zeiss Imajer.Z1 AX10 microscope. Images were acquired and processed with CytoVision software (https://www.leicabiosystems.com/pt-pt/ihc-ish/fish-molecular-solutions/cytogenetics/) and quantified with ImageJ software (imagej.nih.gov/ij/). Experiments were performed in biological triplicates.
Western blotting
Total protein extracts were obtained after cell lysis in 20 mM MOPS (pH 6.5), 1% Triton X-100 (v/w) previously supplemented with 1 mM Na3VO4, 1 mM NaF, and 1 × protease inhibitors (11836170001, Roche). Bradford method was used to determine the protein concentration, using Bio-Rad protein assay reagent (500–0006, Bio-Rad, Hercules, California, USA) through spectrophotometric quantification (595 nm). After protein quantification, loading buffer containing 10% SDS, 0.5% bromophenol blue in Tris-HCL (pH 6.8), and 10% β-mercaptoethanol (M3148, Sigma) was added to each cell lysate and boiled at 95-100 °C for 10 min. Total protein (100 μg) was separated in a 12% (SDS-polyacrylamide gel electrophoresis (SDS-PAGE) in a MINIPROTEAN Tetra Electrophoresis System (Bio-Rad). Afterward, proteins were transferred onto nitrocellulose membranes (BioRad) in a Trans-Blot® Turbo TM Blotting system. Then, membranes were incubated with the primary antibody anti-MCT1 (1:500; ab179832, Abcam) at 4 °C, overnight. Blots were further incubated with anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000, 31460, Thermo Scientific) for 2 h at room temperature, and immunoreactive bands were detected using the enhanced chemiluminescence (ECL) method in a ChemiDoc XRS System (BioRad) with Image Lab software. β-actin (mouse anti-human β-actin, 1:5000, A5441, Sigma) was used as endogenous control. Experiments were performed in biological triplicates.
Statistical analysis
All in vitro data were analyzed using student’s t-test, one-way ANOVA, or two-way ANOVA in GraphPad Prism v8 software (www.graphpad.com/). The assays were performed with at least 3 biological replicates per condition, and the differences were determined statistically significant at p-value < 0.05.
MetaboAnalyst 6.0 (http://www.metaboanalyst.ca/) online platform was used for NMR bioinformatic analyses. This software was used for the multivariate statistical analysis, and samples were normalized by sample weight and data scaling. Multivariate analysis involved principal component analysis (PCA), an unsupervised method to assess variance between samples and their clustering, and partial least squares-discriminant analysis (PLS-DA), a supervised method.
Results
PUREG4-LA24 can efficiently deliver cytotoxic drugs SeChry and TMZ to GBM cells, inducing cell death
The nanoformulations were functionalized with lactate since MCT1 is commonly increased in GBM cells as reported [12]. Then, we assessed the MCT1 expression at the mRNA and protein levels in U251 and U-87MG cell lines and confirmed the MCT1 expression in both cell lines, with the U251 cell line expressing higher levels than U-87MG (Fig. 1A and B).

MCT1 expression is higher in U251 compared to U-87MG, at the mRNA (A) (relative to HPRT) and protein level (B) (relative to β-actin). Cells were exposed to these nanoformulations for 24 h. Selenium-chrysin encapsulated (SeChry@PUREG4-LA24) induced cell death in U251 (EC50: 113 μM, (C)) and in U-87MG (EC50: 121.4 μM, (D)). Temozolomide encapsulated (TMZ @PUREG4-LA24), also induced cell death in U251 (EC50: 406.8 μM, (E)) and in U-87MG (EC50: 501.3 μM, (F)). Moreover, empty PUREG4-LA24 induced low levels of cell death in U251 (G) and in U-87MG (H). I b.End3 cells were exposed to the EC50 of U251 and U-87MG: TMZ 1 – EC50 U251: 406.8 μM; TMZ 2 – EC50 U-87MG: 501.3 μM; SeChry 1 – EC50 U251: 113 μM; SeChry 2 – EC50 U-87MG: 121.4 μM; empty PUREG4-LA24 concentrations corresponded to the EC50 of each encapsulated compound in U251 (1) and U-87MG (2). SeChry@PUREG4-LA24 induced b.End3 cell death. All data are represented as mean ± SD. **p < 0.01, ****p < 0.0001.
To determine if PUREG4-LA24 were suitable drug nanocarriers, GBM cell lines were exposed for 24 h to SeChry@PUREG4-LA24 and TMZ@PUREG4-LA24. Both nanoformulations induced cell death in both GBM cell lines (Fig. 1C–F). Interestingly, U251 cell line was more sensitive to these nanoformulations (lower EC50 comparing to U-87MG). Moreover, the empty nanoparticle (PUREG4-LA24) did not induce cell death, in both GBM cell lines (Fig. 1G and H).
The EC50 determined in GBM cells for the nanoformulations SeChry@PUREG4-LA24 and TMZ@PUREG4-LA24 were tested in b.End3 cell line. SeChry@PUREG4-LA24, but not TMZ@PUREG4-LA24, significantly induced b.End3 cell death (Fig. 1I), and the empty PUREG4-LA24 did not induce b.End3 cell death (Fig. 1I).
Upon transfection, anti-GLS1 siRNA led to the knockdown of GLS1, increased cell death and induced alterations in the metabolic profile
The knockdown of GLS1 gene was confirmed until 96 h after transfection with anti-GLS1 siRNAs (Fig. 2A and B). The decrease in protein expression was also observed 72 h after transfection in both cell lines (Fig. 2C – F). Cell death was also assessed by flow cytometry, showing that GLS1 knockdown tended to induce cell death in U251 cell line comparing with control cells (Fig. 2G), while in U-87MG a similar trend was observed (Fig. 2H). A transfection with a scramble siRNA was also performed (Fig. S3A and B).

GBM cells were transfected with lipofectamine and anti-GLS1 siRNA and the expression of GLS1 was assessed by qPCR until 96 h post transfection (all data is relative to HPRT and normalized to the control- cells exposed to lipofectamine) (A and B) and the protein expression was assessed by immunofluorescence (C – F), magnification 630×. Anti-GLS1 siRNA induced cell death in U251 (G) and tends to induce cell death in U-87MG (H) after 48 h exposure. 33 metabolites were identified in U251, and 32 metabolites were identified in U-87MG exometabolome. I PCA of U251 cell line showing the separation between control and lipofectamine groups from anti-GLS1 siRNA treated group. Metabolite levels found on the supernatant of U251 cell line: (J) amino acids, (K) metabolites involved in glycolysis and TCA cycle, (L) other relevant metabolites. M PCA of U-87MG cell line showing the separation between control and lipofectamine groups from anti-GLS1 siRNA treated group. Metabolite levels found on the supernatant of U-87MG cell line: (N) amino acids, (O) metabolites involved in glycolysis and TCA cycle, (P) other relevant metabolites. Immunofluorescence revealed increased ATG12, an autophagy marker, after transfection with anti-GLS1 siRNA in U251 cell line (Q and R), magnification 630×. All data are represented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The effect of GLS1 knockdown in GBM cells exometabolome was assessed by 1H-NMR analysis of the conditioned media (extracellular metabolites). In U251 cells, a clear separation of the metabolic profile of the exometabolome was observed in a PCA analysis, when comparing U251 cells transfected with the anti-GLS1 siRNA and the controls (control and lipofectamine) (Fig. 2I). The statistical model was considered valid when the PLS-DA model (Figure S4A) adopted the criteria for success of R2 ≥ 0.7 and Q2 ≥ 0.4 [42], which was observed (Figure S4B).
In detail for U251, glucose, glutamine, glycine, serine and arginine were the metabolites that most contributed for the separation (Fig. S4C) according to the VIP score (VIP score ≥ 1). Regarding glutamine, arginine (Fig. 2J) and glucose (Fig. 2K), there are significantly higher levels in the extracellular levels due to the GLS1 knockdown. Lactate was also pointed out as a major contributor for the separation, but its levels were lower upon transfection (Fig. 2K). A trend for increased levels of glycine and serine (Fig. 2J) due to siRNA was observed. Besides these metabolites with the greatest contribution for the separation, more metabolites presented altered levels after the GLS1 knockdown comparing to control and lipofectamine, namely higher levels of leucine, threonine (Fig. 2J), acetate (Fig. 2K), 3-methyl-2-oxovalerate, formate, glutathione (GSH) and histamine (Fig. 2L).
Regarding U-87MG, a clear separation of the exometabolome was observed in the PCA analysis comparing U-87MG cells transfected with the anti-GLS1 siRNA and the controls (control and lipofectamine) (Fig. 2M). Statistical model was considered valid when the PLS-DA model (Fig. S4D) adopted the criteria for success of R2 ≥ 0.7 and Q2 ≥ 0.4 [42], which was observed (Fig. S4E). In U-87MG, eight metabolites presented VIP scores ≥ 1: lactate, glucose, pyruvate, alanine, glutamine, citrate, acetate and arginine (Fig. S4F). GLS1 knockdown led to lower levels of alanine, glutamate (Fig. 2N), pyruvate, lactate, citrate (Fig. 2O), and formate (Fig. 2P), while the levels of glutamine, lysine (Fig. 2N), and glucose were higher (Fig. 2O).
These results indicate that the knockdown of GLS1 affected the metabolic profile of both GBM cell lines, and in U251 cells autophagy was activated, as seen by the increased levels of ATG12 protein after GLS1 knockdown (Fig. 2Q and R).
PUREG4-LA12-anti-GLS1-siRNA led to the knockdown of GLS1
PUREG4-LA12 dendrimers functionalized with 12 lactate molecules were used to improve stability of dendriplexes. The formation of the dendriplex PUREG4-LA12-anti-GLS1-siRNA (Fig. 3A) was confirmed in an agarose gel to assess the electrophoretic retardation, and we used four N/P ratios: 1:0.5, 1:1, 1:1.5 and 1:2 (Figs. 3B and S5). The dendriplexes with higher N/P ratio showed more retention (dragging of the band) corresponding to the efficient binding of siRNA to PUREG4-LA12 (Fig. 3B and S5). A dendriplex with scramble siRNA was also synthesized, and retardation was also observed (Fig, S6A).

A Representation of PUREG4-LA12-anti-GLS1-siRNA dendriplex structure. B Agarose gel showing the retardation of PUREG4-LA12-anti-GLS1-siRNA dendriplexes with higher N/P ratios. GBM cells were exposed for 72 h to the dendriplexes and in both cell lines there was knockdown of GLS1 (relative to HPRT and normalized to the control) (C and D). PCA of U251 and U-87MG cell lines showing the exometabolic profile obtained by 1H-NMR: analysis between the three conditions (E and F) and only between samples exposed to PUREG4-LA12 and PUREG4-LA12-anti-GLS1-siRNA (G, H). All data are represented as mean ± SD. *p < 0.05, ***p < 0.001.
GBM cells were exposed to the dendriplex for 72 h, and PUREG4-LA12-anti-GLS1-siRNA efficiently delivered the siRNA to the cell, leading to the knockdown of GLS1 in both cell lines (Fig. 3C and D). Dendriplex with scramble siRNA did not induce GLS1 knockdown (Fig. S6B and C) and did not affect GLS1 protein expression (Fig. S6D–G).
GBM cells were directly exposed to the PUREG4-LA12-anti-GLS1-siRNA and the effect of the dendriplex in GBM cells exometabolome was assessed by 1H-NMR. For both cell lines no separation was observed between the metabolic profiles of cells upon exposure to PUREG4-LA12 and to PUREG4-LA12-anti-GLS1-siRNA (Fig. 3E and F). Comparing with the control the differences increase (Fig. 3G and H, Figure S7A–C and G–I), with valid statistical models indicated by the criteria for success of R2 ≥ 0.7 and Q2 ≥ 0.4 [42] (Fig. S7B and H).
Regarding U251 cell line exometabolome, the most important metabolites (Fig. S7C) for the separation that presented increased concentration after dendriplex exposure were isocitrate (Fig. S7E) and formate (Fig. S7F), while pyroglutamate presented decreased concentration (Fig. S7F). Other metabolites considered important for the discrimination that also presented a tendency to increase were methionine, proline, glutamine (Fig. S7D), citrate, lactate (Figure S7E), creatine phosphate, and isobutyrate (Fig. S7F), while alanine levels decreased (Fig. S7D).
In U-87MG exometabolome the most important metabolites (Fig. S7I) that contributed to the separation and that showed increased concentration after dendriplex exposure were acetate, succinate, lactate (Fig. S7K), and formate (Fig. S7L), while pyroglutamate (Fig. S7L) decreased.
PUREG4-LA12-anti-GLS1-siRNA promoted autophagy
The effect on cell death was also assessed, which was not affected by the PUREG4-LA12-anti-GLS1-siRNA at 72 h (Fig. 4A and B). However, autophagy was activated in GBM cells exposed to PUREG4-LA12-anti-GLS1-siRNA, suggested by the increased levels of autophagy related 12 (ATG12) protein, in both cell lines at 72 h (Fig. 4C–F). Moreover, ATG12 levels were also accessed after exposure to PUREG4-LA12-scramble, and there was no alteration in the protein levels (Fig. S8A–D).

U251 (A) and U-87MG (B) were exposed to two N/P ratios of PUREG4-LA12-anti-GLS1-siRNA dendriplexes, and cell viability was not affected. Immunofluorescence revealed increased ATG12, an autophagy marker, after exposure to PUREG4-LA12-anti-GLS1-siRNA 1:2 dendriplex in both cell lines (C–F), magnification 630×. All data are represented as mean ± SD. *p < 0.05.
PUREG4-LA12-anti-GLS1-siRNA was able to cross the in vitro BBB, altering the metabolism of GBM cells
A BBB in vitro model was established with b.End3 cell line. These cells were plated in a transwell and exposed to 550 nM of hydrocortisone on day 3, showing higher TEER values at day 7 (Fig. 5A).

Initially, we established an in vitro model of BBB with b.End3 cells seeded in a transwell. The tightness of the monolayer was assessed by TEER measurements (A), in which hydrocortisone increased TEER values, with higher TEER values at day 7. Changes in endothelial cell morphology were observed with VE-Cad staining (B) (magnification 200× and 400×). Dendriplexes did not induce b.End3 cell death (C). Once the model was established (D), we exposed it to the dendriplexes. E–H Dendriplexes decreased GLS1 protein expression (magnification 630×). 1H-NMR analysis (I, J) of the conditioned media of the basolateral side showed that the dendriplexes crossed the BBB. All data are represented as mean ± SD. *p < 0.05, ***p < 0.001.
VE-cadherin (VE-Cad) immunofluorescence in b.End3 transwell monolayers at day 7 showed a higher extension of cell elongation upon exposure to hydrocortisone (Fig. 5B), indicating a tight endothelial cell barrier [43, 44]. The effect of PUREG4-LA12-anti-GLS1-siRNA in b.End3 cells viability was assessed, and no alteration was observed (Fig. 5C). Since b.End3 cells are from murine origin the effect on GLS coding genes was not evaluated. Therefore, in the following assays we exposed b.End3 cells to 550 nM of hydrocortisone on day 3 and exposed the ‘blood side’ to the dendriplex at day 7, with GBM cells in the lower well (Fig. 5D). The dendriplex crossing through the b.End3 tight monolayer (BBB) to the ‘brain side’ was confirmed by the decreased expression of GLS1 protein (Fig. 5E–H) at 72 h.
1H-NMR analysis of the conditioned media of the basolateral side (Fig. S9A and B) showed that the nanoparticle and the dendriplex efficiently crossed the BBB, since new peaks appeared in the samples exposed to the nanoformulations that did not appear in the control (Fig. 5I and J). These peaks did not correspond to any metabolite, appearing due to the presence of PUREG4-LA12. Moreover, some peaks that appeared increased in the samples exposed to the nanoformulations (Fig. S9C and D) corresponded to N,N-dimethylformamide, a solvent used in the synthesis of PUREG4-LA12.
The effect of the PUREG4-LA12-anti-GLS1-siRNA on the exometabolome of GBM cells was also analyzed by 1H-NMR spectroscopy of the conditioned media of the “brain side”. Even though a complete separation of the metabolic profiles was not obtained in U251 (Fig. 6A, Fig. S10A), the statistical model was considered valid, since the PLS-DA model (Fig. S10A) adopted the criteria for success of R2 ≥ 0.7 and Q2 ≥ 0.4 [42] (Fig. S10B).

In both cell lines 40 metabolites were identified in the exometabolome (supernatants). A PCA of U251 cell line showing the metabolic profiles induced by the experimental conditions. Metabolite levels found on the supernatant of U251 cell line: (B) amino acids, (C) metabolites involved in glycolysis and TCA cycle, (D) other relevant metabolites. (E) PCA of U-87MG cell line showing the metabolic profiles induced by the experimental conditions. Metabolite levels found on the supernatant of U-87MG cell line: (F) amino acids, (G) metabolites involved in glycolysis and TCA cycle, (H) other relevant metabolites. All data are represented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Several metabolites showed different concentrations after U251 exposure to PUREG4-LA12-anti-GLS1-siRNA, decreased levels of alanine, arginine, cysteine, lysine, threonine (Fig. 6B), acetate, pyruvate and lactate (Fig. 6C) and including increased levels of formate (Fig. 6D) and succinate (Fig. 6C), a tendency for increased GSH levels (Fig. 6D). From these metabolites, formate, succinate and lysine were considered the most important metabolites that contribute to the separation (Fig. S10C). There was also a tendency for increased levels of glutamine (Fig. 6B), creatine phosphate and creatine (Fig. 6D) and decreased lysine (Fig. 6B) in U251 exposed to PUREG4-LA12-anti-GLS1-siRNA comparing to samples exposed to PUREG4-LA12.
In U-87MG, as it happened with U251, even without a complete separation of the metabolic profiles (Figs. 6E, S10D), the statistical model was considered valid, since the PLS-DA model (Fig. S10D) adopted the criteria for success of R2 ≥ 0.7 and Q2 ≥ 0.4 [42] (Fig.e S10E).
The levels of GSH, proline, leucine (Fig. 6F), glucose (Fig. 6G) and formate (Fig. 6H) were increased in the exometabolome of U-87MG, whereas lactate and acetate (Fig. 6G) were decreased. Formate, GSH, and leucine tended to be even more increased in samples exposed to the PUREG4-LA12-anti-GLS1-siRNA. Formate was significantly increased and was also one of the most important metabolites for the separation (Fig. S10F).
These results indicate that the exposure to PUREG4-LA12 and PUREG4-LA12-anti-GLS1-siRNA altered the metabolic profile of both GBM cell lines.
PUREG4-LA12-anti-GLS1-siRNA was able to cross an in vitro BBB model with astrocytes
Since endfeet astrocytes are also a component of the BBB, a more complex BBB in vitro model, involving the contribution of astrocytes (iNHA) in the “brain side” of the transwell, was tested (Fig. 7A). It was confirmed that astrocytes viability was not affected by PUREG4-LA12-anti-GLS1-siRNA (Fig. 7B). Therefore, the ‘blood side’ was exposed to PUREG4-LA12-anti-GLS1-siRNA at day 7, and GLS1 knockdown was confirmed by the significantly decreased GLS1 protein expression in U-87MG and a trend to decrease in U251 cell line (Fig. 7C–F) at 72 h.

We established a new in vitro model of BBB with b.End3 cells and astrocytes (iNHA) seeded in each side of a transwell (A). Dendriplexes did not induce iNHA cell death (B). Once the model was established, we exposed it to the dendriplexes, which decreased GLS1 protein expression in both GBM cell lines (C–F), magnification 630×. 1H-NMR analysis of the conditioned media of the basolateral side showed that the dendriplexes crossed the BBB, and induced changes in the exometabolome after 72 h (G, H). All data are represented as mean ± SD. **p < 0.01.
1H-NMR analysis of the conditioned media of the “brain side” showed the different metabolic profiles induced by the PUREG4-LA12-anti-GLS1-siRNA (Fig. 7G and H). In U251, the statistical model was considered valid since the PLS-DA model (Fig. S11A) adopted the criteria for success of R2 ≥ 0.7 and Q2 ≥ 0.4 [42], which was observed for U251 (Fig. S11B).
In U251, the metabolites that most contributed for the separation (VIP score ≥ 1) (Fig. S11C) after PUREG4-LA12-anti-GLS1-siRNA exposure were glutamine (Fig. S11D) and formate (Figure S11F), which increased, while arginine (Fig. S11D), isobutyrate and isopropanol decreased (Fig. S11F). Citrate (Fig. S11E) tended to increase while cysteine and serine (Fig. S11D) tended to decrease. Pyroglutamate also presented decreased levels in U251 after PUREG4-LA12-anti-GLS1-siRNA exposure.
The U-87MG statistical model was considered valid since the PLS-DA model (Figure S11G) adopted the criteria for success of R2 ≥ 0.7 and Q2 ≥ 0.4 [42], which was observed for U-87MG (Figure S11H).
In U-87MG the metabolites that most contributed for the separation (VIP score ≥ 1) (Fig. S11I) were histidine (Fig. S11J), pyruvate (Fig. S11K), and formate (Fig. S11L), which increased after PUREG4-LA12-anti-GLS1-siRNA exposure. Other metabolites with VIP score ≥ 1 were GSH (Fig. S11L) and citrate (Fig. S11K), which tended to increase, and isobutyrate (Fig. S11L), which tended to decrease after PUREG4-LA12-anti-GLS1-siRNA exposure.
Discussion
The altered glucose metabolism and dependence on glutamine is a therapeutic opportunity in GBM. The high MCT1 expression in GBM cells, particularly on U251 cell line (Fig. 1A and B), promotes the efficacy of lactate functionalized nanoparticles in drug delivery. In this study, the induction of cell death by SeChry@PUREG4-LA24 and TMZ@PUREG4-LA24, in GBM cells was evident (Fig. 1C–F), with a higher impact in U251, which corresponds to the higher levels of MCT1 expression in this cell line comparing with U-87MG. Importantly, it was also observed that the SeChry@PUREG4-LA24 nanoformulation induced cell death in b.End3 cell line (Fig. 1I). The disruption of the BBB has been faced as an opportunity to improve the delivery of drugs to the brain and in particular to malignant tumors [45,46,47]. Accordingly, the efficacy of some chemotherapeutical regimens is improved by the combination with mannitol, which disrupts the BBB [48,49,50]. Therefore, SeChry@PUREG4-LA24 may be a step forward in GBM management, thereby inducing GBM cell death while disrupting the BBB.
The transfection with anti-GLS1 siRNAs and the exposure to dendriplex PUREG4-LA12-anti-GLS1-siRNA were able to decrease GLS1 protein expression (Fig. 2C and D and Fig. 5E–H) and alter the exometabolic profile of the GBM cell lines (Fig. 2I-P and Fig. 6, Fig, S9A and B). The transfection assay clearly demonstrated a decrease in glutamine consumption due to reduced GLS1 levels, whereas this was not as apparent in the dendriplex PUREG4-LA12-anti-GLS1-siRNA assay. This difference may be attributed to the varying cell culture durations, with exometabolome analysis conducted after 96 h in the transfection assay and 72 h in the dendriplex assay. However, in U251 cells exposed to dendriplex PUREG4-LA12-anti-GLS1-siRNA, a trend toward increased glutamine levels was observed (Fig. 6B). The dendriplex assay was terminated at 72 h to prevent BBB disruption and to ensure that dendriplexes were effectively crossing the BBB. Accordingly, the overall exometabolome analysis showed metabolic adjustments in both GBM cell lines due to GLS1 decreased levels. The in vitro BBB models, with and without iNHA astrocytes (Fig. 7A), which also contribute to the structural integrity of the BBB [51], demonstrated that the dendriplexes induced the decrease of GLS1 protein levels after the exposure to PUREG4-LA12-anti-GLS1-siRNA (Fig. 7C–F), which indicates that the dendriplex was able to cross the BBB model and reach GBM cells.
The relevance of GLS1 in GBM cells survival was demonstrated in GBM cells transfected with anti-GLS1 siRNAs, by the significant reduction in GLS1 expression confirmed at mRNA and protein levels (Fig. 2A–F), and the concomitant induction of cell death in transfected cells (Fig. 2G and H). The fact that the increased cell death is more visible in U251 than in U-87MG cells points out that metabolic heterogeneity and plasticity among GBM cell subsets may influence treatment efficacy due to the adaptive capacity of cancer cells to metabolically stressful conditions. Our group has already shown that these cell lines rely differently in the glutamine-glutamate metabolism, presenting different metabolic profiles in the absence of glutamine and glucose [12]. Therefore, these cell lines possibly follow different adaptive strategies, highlighted by the metabolic profiles defined in GBM cells with GLS1 knockdown in all strategies used. In both cell lines, a constant observation was the increased or maintained high levels of glutamine and glucose (Figs. 2J and N; 6B and F; S4C and F; S7D, E, J and K, and S11D and J), indicating that cells are less competent in degrading glutamine [52]. This is reinforced by the decreased levels of glutamate and pyroglutamate, products of glutamine metabolism [53], in most GLS1 knockdown conditions and models. Moreover, glucose is being consumed less, which indicates that these cell lines rely more on glutamine, even substituting glucose as a carbon substrate. Moreover, some studies suggest that glutaminolysis is required for the uptake of glucose [54, 55]. Therefore, GLS1 has been pointed out as a ‘metabolic checkpoint’, controlling cell survival via a coordination between glutamine and glucose [56]. Decreased levels of alanine, lactate and pyruvate were often observed (Figs. 2J, N, K and O; 6B, C, F and G; S4C and F; Fig. S7D, E, J and K, and Fig. S11D and E), thereby emphasizing the decreased consumption of glucose and an attempt of cells to adapt to the lower ability to consume glucose upon GLS1 knockdown. This adjustment may resemble the alanine-glucose cycle, in which metabolic pathways, including gluconeogenesis, respond to limited glucose and glutamine availability [57], mimicked by the reduced capacity to degrade glutamine. Therefore, alanine can give rise to glutamate to compensate the lack of glutamine-derived glutamate to supply the TCA cycle [58, 59]. Additionally, alanine can be a source of pyruvate and lactate, which are suitable sources of organic acids in TCA cycle [58, 59]. Furthermore, to sustain the conversion of pyruvate to acetyl-CoA and its entry in the TCA cycle, due to the lack of glutamate induced by GLS1 knockdown, it implies the activation of other metabolic pathways. The increased levels of acetate in some conditions (Figs. 2N and O; 6C and G; S4C and F; S7E and K, and S11E) may indicate increased conversion of acetyl-CoA as a mechanism to maintain the viability considering that glycolysis and glutaminolysis are impaired. This is deeply connected to citrate availability, since citrate is a cataplerotic compound in TCA cycle, which derives from oxaloacetate and acetyl-CoA, but it can also be deviated to feed other metabolic pathways [60, 61]. A main source of acetate and citrate is the fatty acids β-oxidation, which may support TCA cycle functioning [61, 62]. On the other hand, increased levels of isocitrate and a tendency for high levels of citrate after dendriplex direct exposure may indicate a decreased flux into TCA cycle, with increased accumulation of these metabolites in the extracellular milieu. In addition, decreased citrate levels may indicate mitochondrial dysfunction [63].
In fact, glutamine deprivation triggers mitochondrial fusion, an adaptive mechanism of the cell to maximize efficiency, maintain the respiratory chain complexes and the integrity of mitochondrial DNA [64]. As TCA cycle and β−oxidation are mitochondrial pathways, citrate may have a pivotal role in metabolic adaptation arising from mitochondria assembly. Increased cysteine uptake may also stimulate mitochondria fusion to potentiate the OXPHOS capacity [65], and upon GLS1 knockdown cysteine was not detected in extracellular media or it was presented in decreased levels (Figs. 2J and N; 6B and F; S4C and F; S7D and J, and S11D and J). In the BBB model with astrocytes, increased glutamine levels (Fig. S11D) in the supernatant of U251 were observed, which may suggest an astrocytes involvement. Once the astrocytes are responsible for the recycling of the glutamate released by pre-synaptic neurons to the synaptic cleft, converting it into glutamine, which is imported by the neurons [66]. Therefore, due to the decreased GLS1 protein expression, GBM cell lines may be exporting more glutamate, which is recycled by astrocytes into glutamine. Hence, this could be an adaptive mechanism to overcome the effect of PUREG4-LA12-anti-GLS1-siRNA.
Dynamic patterns of amino acids were also identified upon GLS1 knockdown (Figs. 2J and N; 6B and F; S4C and F; S7D and J, and S11D and J), indicating adjustments in the intracellular metabolism. This highlights proteolysis as a source to compensate amino acids balance, as glutamine-derived glutamate is the main source for amino acids synthesis mainly through transamination processes [67, 68]. Proteolysis can supply various metabolic needs, such as to increase the glutamate pool to feed the TCA cycle. Moreover, this could also indicate increased autophagy, a process that cancer cells go through to survive under nutrient-deprived conditions, in which the export of amino acids occurs [69]. In fact, autophagy activation was confirmed in U251 and U-87MG cells by the increased levels of ATG12 protein (Fig. 2Q and R and 4C and D). ATG12 is a crucial protein in autophagy, involved in the formation of autophagosomes, a double-membrane vesicle that captures and delivers cellular components to lysosomes for degradation [70, 71]. Therefore, this suggests that the metabolic stress induced by GLS1 knockdown can trigger autophagy as a survival mechanism. This observation is aligned with previous studies indicating that in advanced stages, autophagy promotes the survival of tumor cells by alleviating stress in the tumor microenvironment - TME [72]. This phenomenon could justify the lack of cell death induction by the dendriplexes, since autophagy is a pro-survival process [73]. However, long autophagy duration can trigger cell death [74].
The increased levels of formate (Figs. 2N and O; 6C and G; S4C and F; S7E and K, and S11E and K) may indicate a decreased flux into one-carbon metabolism, resulting in formate export. Besides being a byproduct of one-carbon metabolism, formate is a precursor of glycine [75], which is one of the three components of GSH [76]. On another hand, the release of formate reinforces the activation of autophagy [77] observed upon GLS1 knockdown.
The increased levels of GSH (Figs. 2L; 6D and H; and S11F and L) observed in some conditions regarding GLS1 knockdown may contribute to the decreased levels of glutamate, glycine and cysteine [76], since these are the components of the GSH molecule. Therefore, it seems that GLS1 downregulation interferes with the GBM cell redox control. When analyzing samples exposed to PUREG4-LA12-anti-GLS1-siRNA from both cell lines, there was a tendency for increased extracellular levels of formate and GSH comparing to samples exposed to PUREG4-LA12 (Fig. 6D and H). We had previously observed increased extracellular GSH levels after anti-GLS1 siRNA transfection in U251 cell line (Fig. 2H). Increased export of GSH can be a part of GBM cells strategy to protect themselves from oxidative damage induced by this therapeutic approach, mitigating the harmful effects of ROS in the surrounding TME. Moreover, to perform the detoxification of chemotherapeutic compounds, GSH conjugates with drugs via the GSH-S-transferase and lead to its efflux by to multidrug resistance-associated protein (Mrp) transporters [78]. Therefore, GBM cells can be trying to eliminate the dendrimers.
Additionally, we also observed a tendency to increased levels of creatine phosphate, succinate and creatine comparing to samples exposed to PUREG4-LA12 (Fig. 6B–D and S7F and L). GLS1 knockdown can disrupt the balance between ATP production and consumption, leading to increased synthesis and breakdown of creatine phosphate, a reserve of high-energy phosphates in the brain [79]. This imbalance may cause excess creatine and creatine phosphate to accumulate and be exported out of the cell.
Another achievement of this study was the fact that we successfully established a BBB in vitro model, a tight endothelial barrier using hydrocortisone-treated b.End3 cells, to evaluate nanoformulations (Fig. 5A – D). Previous studies have shown that hydrocortisone induces brain endothelial cell tightness, increasing the TEER values of the confluent layer [38, 80]. The expression of VE-cadherin (VE-Cad), an adherens junction protein and a key regulator of endothelial barrier function [77], was assessed in this model, showing cell elongation, which indicates the presence of a tight endothelial-like monolayer [43, 44]. The developed strategy showed that the dendriplex PUREG4-LA12-anti-GLS1-siRNA was effective in crossing the in vitro BBB, as observed by NMR analysis (Figs. 5I – J, S9), without affecting b.End3 cells viability (Fig. 5C). This capability is pivotal, as it demonstrates the potential of dendrimer-based nanocarriers to overcome the BBB, the most significant barrier in CNS drug delivery. Therefore, depending on the therapeutic objective PUREG4-LA24/ PUREG4-LA12 can carry compounds that disrupt or preserve the BBB integrity, while also affecting GBM cells metabolism, allowing a flexible use of these carriers in nanoformulations.
Conclusion
Our study stresses the pivotal role of a new metabolism-based therapy using GLS1 as a target of a dendriplex PUREG4-LA12-anti-GLS1-siRNA proposed as a new tool for gene modulation therapy. This system, sustained by dendrimers as nanocarriers of drugs and siRNAs, can be adapted to cross or disrupt the BBB. By interfering with GLS1 expression, this system offers a dual approach that disrupts both glucose and glutamine pathways without affecting the BBB viability. In another hand, the encapsulated TMZ can be more efficiently delivered to GBM cells after crossing the BBB, while encapsulated SeChry efficiently triggers cell death in GBM cells and simultaneously disrupts the BBB, which can be beneficial to improve the therapy effectiveness. The setting of results obtained supports further pre-clinical research focused on refining dendrimer formulations, optimizing dosing regimens, and evaluating the therapeutic efficacy in vivo, within the complex brain microenvironment. In the future, exploring the combinatory potential of dendrimer-based delivery systems with existing treatments, such as the standard of care (Stupp protocol), could enhance therapeutic outcomes. Investigating the interaction of these nanocarriers with the TME and their long-term safety profiles will be crucial for translating these findings into clinical applications.
Data availability
Data will be available in a public repository at https://doi.org/10.21228/M8KV62.
Change history
10 July 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41417-025-00933-5
References
Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016—2020. Neuro Oncol. 2023;25:iv1–99.
Obara-Michlewska M, Szeliga M. Targeting glutamine addiction in gliomas. Cancers. 2020;12:310.
Stoyanov GS, Lyutfi E, Georgieva R, Georgiev R, Dzhenkov DL, Petkova L, et al. Reclassification of Glioblastoma Multiforme According to the 2021 World Health Organization Classification of Central Nervous System Tumors: A Single Institution Report and Practical Significance. Cureus. 2022;14:e21822.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005;352:987–96.
Ballabh P, Braun A, Nedergaard M. The blood–brain barrier: an overview. Neurobiol Dis. 2004;16:1–13.
Benz F, Liebner S Structure and Function of the Blood–Brain Barrier (BBB). In 2020. p. 3–31. Available from: https://link.springer.com/10.1007/164_2020_404.
Ceña V, Játiva P. Nanoparticle crossing of blood–brain barrier: a road to new therapeutic approaches to central nervous system diseases. Nanomedicine. 2018;13:1513–6.
Teleanu DM, Chircov C, Grumezescu AM, Volceanov A, Teleanu RI. Blood-brain delivery methods using nanotechnology. Pharmaceutics. 2018;10:1–16.
Palan F, Chatterjee B. Dendrimers in the context of targeting central nervous system disorders. J Drug Deliv Sci Technol. 2022;73:103474.
Srinageshwar B, Peruzzaro S, Andrews M, Johnson K, Hietpas A, Clark B, et al. PAMAM Dendrimers Cross the Blood–Brain Barrier When Administered through the Carotid Artery in C57BL/6J Mice. Int J Mol Sci. 2017;18:628.
Jin Z, Piao L, Sun G, Lv C, Jing Y, Jin R. Dual functional nanoparticles efficiently across the blood–brain barrier to combat glioblastoma via simultaneously inhibit the PI3K pathway and NKG2A axis. J Drug Target. 2021;29:323–35.
Martins F, van der Kellen D, Gonçalves LG, Serpa J. Metabolic Profiles Point Out Metabolic Pathways Pivotal in Two Glioblastoma (GBM) Cell Lines, U251 and U-87MG. Biomedicines. 2023;11:2041.
Libby CJ, Zhang S, Benavides GA, Scott SE, Li Y, Redmann M, et al. Identification of Compounds That Decrease Glioblastoma Growth and Glucose Uptake in Vitro. ACS Chem Biol. 2018;13:2048–57.
Park SJ, Smith CP, Wilbur RR, Cain CP, Kallu SR, Valasapalli S, et al. An overview of MCT1 and MCT4 in GBM: small molecule transporters with large implications. Am J Cancer Res. 2018;8:1967–76.
Kubelt C, Peters S, Ahmeti H, Huhndorf M, Huber L, Cohrs G, et al. Intratumoral Distribution of Lactate and the Monocarboxylate Transporters 1 and 4 in Human Glioblastoma Multiforme and Their Relationships to Tumor Progression-Associated Markers. Int J Mol Sci. 2020;21:6254.
Castegna A, Menga A. Glutamine Synthetase: Localization Dictates Outcome. Genes. 2018;9:108.
Márquez J, Alonso FJ, Matés JM, Segura JA, Martín-Rufián M, Campos-Sandoval JA. Glutamine Addiction In Gliomas. Neurochem Res. 2017;42:1735–46.
Natarajan SK, Venneti S Glutamine metabolism in brain tumors. Cancers. 2019;11.
Kobayashi M, Mizutani A, Nishi K, Nakajima S, Shikano N, Nishii R, et al. Differences in accumulation and the transport mechanism of l- and d-methionine in high- and low-grade human glioma cells. Nucl Med Biol. 2017;44:78–82.
Sidoryk M, Matyja E, Dybel A, Zielinska M, Bogucki J, Jaskólski DJ, et al. Increased expression of a glutamine transporter SNAT3 is a marker of malignant gliomas. Neuroreport. 2004;15:575–8.
Szeliga M, Matyja E, Obara M, Grajkowska W, Czernicki T, Albrecht J. Relative Expression of mRNAS Coding for Glutaminase Isoforms in CNS Tissues and CNS Tumors. Neurochem Res. 2008;33:808–13.
de los Santos-Jiménez J, Campos-Sandoval JA, Márquez-Torres C, Urbano-Polo N, Brøndegaard D, Martín-Rufián M, et al. Glutaminase isoforms expression switches microRNA levels and oxidative status in glioblastoma cells. J Biomed Sci. 2021;28:14.
Restall IJ, Cseh O, Richards LM, Pugh TJ, Luchman HA, Weiss S. Brain Tumor Stem Cell Dependence on Glutaminase Reveals a Metabolic Vulnerability through the Amino Acid Deprivation Response Pathway. Cancer Res. 2020;80:5478–90.
Miki K, Yagi M, Hatae R, Otsuji R, Miyazaki T, Goto K, et al. Glutaminolysis is associated with mitochondrial pathway activation and can be therapeutically targeted in glioblastoma. Cancer Metab. 2024;12:35.
Carbone D, Vestuto V, Ferraro MR, Ciaglia T, Pecoraro C, Sommella E, et al. Metabolomics-assisted discovery of a new anticancer GLS-1 inhibitor chemotype from a nortopsentin-inspired library: From phenotype screening to target identification. Eur J Med Chem. 2022;234:114233.
Santos I, Ramos C, Mendes C, Sequeira CO, Tomé CS, Fernandes DGH, et al. Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium–Chrysin Polyurea Dendrimer Nanoformulation. Nutrients. 2019;11:2523.
Cruz A, Mota P, Ramos C, Pires RF, Mendes C, Silva JP, et al. Polyurea Dendrimer Folate-Targeted Nanodelivery of l-Buthionine Sulfoximine as a Tool to Tackle Ovarian Cancer Chemoresistance. Antioxidants. 2020;9:133.
Hipólito A, Mendes C, Martins F, Lemos I, Francisco I, Cunha F, et al. H2S-Synthesizing Enzymes Are Putative Determinants in Lung Cancer Management toward Personalized Medicine. Antioxidants. 2023;13:51.
Lopes-Coelho F, Martins F, Hipólito A, Mendes C, Sequeira CO, Pires RF, et al. The Activation of Endothelial Cells Relies on a Ferroptosis-Like Mechanism: Novel Perspectives in Management of Angiogenesis and Cancer Therapy. Front Oncol [Internet]. 2021 May 10;11. Available from: https://www.frontiersin.org/articles/10.3389/fonc.2021.656229/full.
Cruz A, Barbosa J, Antunes P, Bonifácio VDB, Pinto SN A Glimpse into Dendrimers Integration in Cancer Imaging and Theranostics. Int J Mol Sci. 2023;24.
Restani RB, Conde J, Pires RF, Martins P, Fernandes AR, Baptista PV, et al. POxylated Polyurea Dendrimers: Smart Core-Shell Vectors with IC 50 Lowering Capacity. Macromol Biosci. 2015;15:1045–51.
Restani RB, Morgado PI, Ribeiro MP, Correia IJ, Aguiar-Ricardo A, Bonifácio VDB, et al. Biocompatible Polyurea Dendrimers with pH-Dependent Fluorescence. Angew Chem Int Ed. 2012;51:5162–5.
Restani RB, Conde J, Baptista PV, Cidade MT, Bragança AM, Morgado J, et al. Polyurea dendrimer for efficient cytosolic siRNA delivery. RSC Adv. 2014;4:54872–8.
Mendes C, Lemos I, Hipólito A, Abreu B, Freitas-Dias C, Martins F, et al. Metabolic profiling and combined therapeutic strategies unveil the cytotoxic potential of selenium-chrysin (SeChry) in NSCLC cells. Biosci Rep. 2024 Jul 31;44. Available from: https://portlandpress.com/bioscirep/article/44/7/BSR20240752/234693/Metabolic-profiling-and-combined-therapeutic.
Restani RB, Morgado PI, Ribeiro MP, Correia IJ, Aguiar-Ricardo A, Bonifácio VDB. Biocompatible Polyurea Dendrimers with pH-Dependent Fluorescence. Angew Chem Int Ed. 2012;51:5162–5.
Mota P, Pires RF, Serpa J, Bonifácio VDB. l-Buthionine Sulfoximine Detection and Quantification in Polyurea Dendrimer Nanoformulations. Molecules. 2019;24:3111.
Aydin O, Kanarya D, Yilmaz U, Tunç CÜ Determination of Optimum Ratio of Cationic Polymers and Small Interfering RNA with Agarose Gel Retardation Assay. In 2022. p. 117–28. Available from: https://link.springer.com/10.1007/978-1-0716-2010-6_7.
Garcia AR, Godinho-Pereira J, Figueira I, Malhó R, Brito MA Replicating the blood-brain barrier properties in an in vitro model: effects of hydrocortisone and/or shear stress. Off J Port Anat Soc. 2019;
Palmela I, Sasaki H, Cardoso FL, Moutinho M, Kim KS, Brites D, et al. Time-dependent dual effects of high levels of unconjugated bilirubin on the human blood-brain barrier lining. Front Cell Neurosci. 2012;6. Available from: http://journal.frontiersin.org/article/10.3389/fncel.2012.00022/abstract.
Aday S, Li W, Karp JM, Joshi N. An in vitro Blood-brain Barrier Model to Study the Penetration of Nanoparticles. BIO-PROTOCOL. 2022;12:1–7.
Czupalla CJ, Liebner S, Devraj K In Vitro Models of the Blood–Brain Barrier. In 2014. p. 415–37. Available from: https://link.springer.com/10.1007/978-1-4939-0320-7_34.
Jiménez-Contreras E, Torres-Salinas D, Moreno RB, Baños RR, López-Cózar ED. Response Surface Methodology and its application in evaluating scientific activity. Scientometrics. 2009;79:201–18.
Patabendige A, Skinner RA, Morgan L, Joan Abbott N. A detailed method for preparation of a functional and flexible blood–brain barrier model using porcine brain endothelial cells. Brain Res. 2013;1521:16–30.
Burek M, Salvador E, Förster CY. Generation of an Immortalized Murine Brain Microvascular Endothelial Cell Line as an In Vitro Blood Brain Barrier Model. J Vis Exp [Internet]. 2012 Aug 29;. Available from: https://www.jove.com/t/4022/generation-of-an-immortalized-murine-brain-microvascular-endothelial-cell-line-as-an-in-vitro-blood-brain-barrier-model.
Teleanu RI, Preda MD, Niculescu AG, Vladâcenco O, Radu CI, Grumezescu AM, et al. Current Strategies to Enhance Delivery of Drugs across the Blood–Brain Barrier. Pharmaceutics. 2022;14:987.
Wu D, Chen Q, Chen X, Han F, Chen Z, Wang Y. The blood–brain barrier: structure, regulation, and drug delivery. Signal Transduct Target Ther. 2023;8:217.
Stamp MEM, Halwes M, Nisbet D, Collins DJ. Breaking barriers: exploring mechanisms behind opening the blood–brain barrier. Fluids Barriers CNS. 2023;20:87.
Chakraborty S, Filippi CG, Wong T, Ray A, Fralin S, Tsiouris AJ, et al. Superselective intraarterial cerebral infusion of cetuximab after osmotic blood/brain barrier disruption for recurrent malignant glioma: phase I study. J Neurooncol. 2016;128:405–15.
Burkhardt JK, Riina H, Shin BJ, Christos P, Kesavabhotla K, Hofstetter CP, et al. Intra-Arterial Delivery of Bevacizumab after Blood-Brain Barrier Disruption for the Treatment of Recurrent Glioblastoma: Progression-Free Survival and Overall Survival. World Neurosurg. 2012;77:130–4.
Karmur BS, Philteos J, Abbasian A, Zacharia BE, Lipsman N, Levin V, et al. Blood-Brain Barrier Disruption in Neuro-Oncology: Strategies, Failures, and Challenges to Overcome. Front Oncol. 2020;10. Available from: https://www.frontiersin.org/article/10.3389/fonc.2020.563840/full.
Cabezas R, Õvila M, Gonzalez J, El-Bachá RS, Báez E, GarcÃa-Segura LM, et al. Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci. 2014;8. Available from: http://journal.frontiersin.org/article/10.3389/fncel.2014.00211/abstract.
Xiang L, Mou J, Shao B, Wei Y, Liang H, Takano N, et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019;10:40.
Mena FV, Baab PJ, Zielke CL, Huang Y, Zielke HR. Formation of extracellular glutamate from glutamine: Exclusion of pyroglutamate as an intermediate. Brain Res. 2005;1052:88–96.
Pan T, Gao L, Wu G, Shen G, Xie S, Wen H, et al. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem Biophys Res Commun. 2015;456:452–8.
Kaadige MR, Looper RE, Kamalanaadhan S, Ayer DE. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proc Natl Acad Sci. 2009;106:14878–83.
Song Z, Wei B, Lu C, Li P, Chen L. Glutaminase sustains cell survival via the regulation of glycolysis and glutaminolysis in colorectal cancer. Oncol Lett. 2017;14:3117–23.
Otto AM Metabolic Constants and Plasticity of Cancer Cells in a Limiting Glucose and Glutamine Microenvironment—A Pyruvate Perspective. Front. Oncol. 2020 Dec 8;10. Available from: https://www.frontiersin.org/articles/10.3389/fonc.2020.596197/full.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–34. https://doi.org/10.1038/nrc.2016.71.
Holeček M. Origin and Roles of Alanine and Glutamine in Gluconeogenesis in the Liver, Kidneys, and Small Intestine under Physiological and Pathological Conditions. Int J Mol Sci. 2024;25:7037.
Deja S, Crawford PA, Burgess SC. Krebs takes a turn at cell differentiation. Cell Metab. 2022;34:658–60.
Icard P, Coquerel A, Wu Z, Gligorov J, Fuks D, Fournel L, et al. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int J Mol Sci. 2021;22:6587.
Williams NC, O’Neill LAJ A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front Immunol. 2018 Feb 5;9. Available from: http://journal.frontiersin.org/article/10.3389/fimmu.2018.00141/full.
Chhimpa N, Singh N, Puri N, Kayath HP The Novel Role of Mitochondrial Citrate Synthase and Citrate in the Pathophysiology of Alzheimer’s Disease. Jagannatha Rao K, Ooi L, Hegde ML, Zhang Y, Ip NY, Nami M, et al., editors. J Alzheimer’s Dis. 2023 Jul 25;94:S453–72. Available from: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi=10.3233/JAD-220514.
Cai WF, Zhang C, Wu YQ, Zhuang G, Ye Z, Zhang CS, et al. Glutaminase GLS1 senses glutamine availability in a non-enzymatic manner triggering mitochondrial fusion. Cell Res. 2018;28:865–7.
Yao CH, Wang R, Wang Y, Kung CP, Weber JD, Patti GJ Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife. 2019 Jan 29;8. Available from: https://elifesciences.org/articles/41351.
Martins F, Gonçalves LG, Pojo M, Serpa J Take Advantage of Glutamine Anaplerosis, the Kernel of the Metabolic Rewiring in Malignant Gliomas. Biomolecules. 2020 26;10:1370. Available from: https://www.mdpi.com/2218-273X/10/10/1370.
Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. 2020;52:1496–516.
Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J [Internet]. 2017;36:1302–15.
Anderson CM, Macleod KF Autophagy and cancer cell metabolism. In 2019. p. 145–90. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1937644819300632.
Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19:12.
Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008;9:859–64.
Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front Oncol. 2020;10. Available from: https://www.frontiersin.org/article/10.3389/fonc.2020.578418/full.
Desantis V, Saltarella I, Lamanuzzi A, Mariggiò MA, Racanelli V, Vacca A, et al. Autophagy: A New Mechanism of Prosurvival and Drug Resistance in Multiple Myeloma. Transl Oncol. 2018;11:1350–7.
Liu S, Yao S, Yang H, Liu S, Wang Y. Autophagy: Regulator of cell death. Cell Death Dis. 2023;14:648.
Xu Y, Ren J, Wang W, Zeng A. Improvement of glycine biosynthesis from one-carbon compounds and ammonia catalyzed by the glycine cleavage system in vitro. Eng Life Sci. 2022;22:40–53.
Nunes SC, Serpa J Glutathione in ovarian cancer: A double-edged sword. Int J Mol Sci. 2018 Jun 26;19:1882. Available from: http://www.mdpi.com/1422-0067/19/7/1882.
WANG Y, XU SL, XU WJ, YANG HY, HU P, LI YX. Sodium formate induces autophagy and apoptosis via the JNK signaling pathway of photoreceptor cells. Mol Med Rep. 2016;13:1111–8.
Backos DS, Franklin CC, Reigan P. The role of glutathione in brain tumor drug resistance. Biochem Pharm. 2012;83:1005–12.
Forbes SC, Cordingley DM, Cornish SM, Gualano B, Roschel H, Ostojic SM, et al. Effects of Creatine Supplementation on Brain Function and Health. Nutrients. 2022;14:921.
Hoheisel D, Nitz T, Franke H, Wegener J, Hakvoort A, Tilling T, et al. Hydrocortisone Reinforces the Blood–Brain Barrier Properties in a Serum Free Cell Culture System. Biochem Biophys Res Commun. 1998;244:312–6.
Acknowledgements
This work benefited from access to CERMAX, ITQB-NOVA, Oeiras, Portugal with equipment funded by FCT, project AAC 01/SAICT/2016.
Funding
The institutions are funded by Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior (FCT/MCTES, Portugal) through national funds to iNOVA4Health (UIDB/04462/2020 and UIDP/04462/2020), to MOSTMICRO-ITQB (UIDB/04612/2020 and UIDP/04612/2020), to BioISI (Center Grant doi 10.54499/UIDB/04046/2020), and the Associated Laboratory LS4FUTURE (LA/P/0087/2020). Filipa Martins was funded by a FCT individual Ph.D. fellowship (2020.04780.BD). Luis G. Gonçalves was financed by a FCT contract according to DL57/2016, [SFRH/BPD/111100/2015]. Data availability in a public repository is supported by NIH U2C-DK119886 and OT2-OD030544 grants.
Author information
Authors and Affiliations
Contributions
F.M. wrote the first draft of the paper, planned and performed most of the experiments, revised and discussed the paper; R.A. planned and performed the experiments, revised and discussed the paper; H.B. performed the nanoparticles synthesis, revised and discussed the paper; P.M. coordinated the BBB monolayer tightness and TEER optimizing experiments, revised and discussed the paper; J.R. Produced and characterized the antibody for autophagy marker ATG12, revised and discussed the paper; V.C. coordinated experiments for dendriplex stability, revised and discussed the paper; V.D.B.B. coordinated the nanoparticles synthesis, revised and discussed the paper; L.G.G. coordinated NMR spectroscopy analysis, acquired funding, revised and discussed the paper; J.S. coordinated and supervised the whole project, responsible for acquiring funding, wrote the first draft of the paper, revised and discussed the paper. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. The project was approved regarding the in vitro models by the NOVA Medical School Ethical Committee (Ref: 158/2021/CEFCM). Since the project does not involve in vivo models or patient samples, ethical approval and consent from participants were not required.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Martins, F., Arada, R., Barros, H. et al. Lactate-coated polyurea-siRNA dendriplex: a gene therapy-directed and metabolism-based strategy to impair glioblastoma (GBM). Cancer Gene Ther 32, 690–705 (2025). https://doi.org/10.1038/s41417-025-00906-8
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41417-025-00906-8
This article is cited by
-
Soft nanoparticles as delivery agents for RNA-based anticancer therapy
Polymer Journal (2026)
-
Ioning out glioblastoma: ferroptosis mechanisms and therapeutic frontiers
Cell Death Discovery (2025)
