
Abstract
Recent advancements in ovarian cancer treatment, particularly with PARP inhibitors, have markedly enhanced the recurrence-free interval, shifting the treatment paradigm and increasing treatment success in patients with BRCA mutations or HRD (homologous recombination deficiency). However, a significant proportion of cases experience relapse, resulting in poorer long-term survival rates when compared to other female cancers, such as breast cancer. This review explores the potential of adeno-associated virus (AAV) vectors for gene therapy in ovarian cancer and examines rational gene therapy strategies by categorizing them based on target cells and target genes to determine the most effective approach for ovarian cancer treatment. Specifically, it examines strategies such as anti-angiogenesis and immune modulation, highlighting the strategy of gene supplementation to hinder ovarian cancer progression. Innovations in AAV capsid design now allow for targeted delivery, focusing on ovarian cancer stem cells (CSCs) identified by specific markers. Additionally, leveraging DNA sequencing technologies enhances the identification and incorporation of therapeutic genes into AAV vectors, promising new avenues for ovarian cancer gene therapy.
Similar content being viewed by others
Introduction
Globally, ovarian cancer is the eighth most common cancer in women, accounting for an estimated 3.7% of cases and 4.7% of cancer deaths in 2020 [1]. It is the leading cause of gynecological cancer-related deaths and the second most common gynecologic cancer [2]. Over 90% of ovarian cancers are epithelial ovarian cancers (EOC), with high-grade serous ovarian cancer (HGSOC) being the predominant subtype [3] (Hereafter, these will be referred to as ovarian cancer). Most ovarian cancers (75%) are diagnosed at an advanced stage (III or IV) and initially respond well to standard treatment (platinum-based chemotherapy and cytoreductive surgery) with a response rate of over 80% [4]. However, the recurrence rate is nearly 80% of advanced ovarian cancers, with progressively shorter progression-free intervals and repeated chemotherapy cycles [5]. Consequently, the 5-year survival rate is 26% for stage III and 14% for stage IV [6] (Fig. 1a). Treatment following recurrence is primarily ineffective, with a median survival time of only 2 years, making this disease essentially lethal [7].

A Typical anatomical presentation of FIGO (International Federation of Gynecology and Obstetrics) stage IIIC ovarian cancer. The 5-year survival rate for patients initially diagnosed with stage III ovarian cancer is ~26%. B Schematic representation of CA-125 levels and ovarian tumor burden in stage III high-grade serous ovarian cancer (HGSOC). The standard treatment regimen for ovarian cancer typically involves optimal cytoreductive surgery followed by six cycles of chemotherapy using carboplatin and paclitaxel. Post-chemotherapy, PARP inhibitors may be used as maintenance therapy to prolong progression-free survival (PFS). If the patient experiences a relapse more than 6 months after completing the initial chemotherapy, it is classified as a platinum-sensitive relapse, allowing for the same platinum- and taxane-based chemotherapy regimen to be reused. Conversely, if the relapse occurs within six months, it is considered a platinum-resistant relapse. In these cases, targeted therapies such as bevacizumab, combined with agents like pegylated liposomal doxorubicin (PLD), gemcitabine, or topotecan, are often utilized. Typically, PFS decreases with each successive relapse of ovarian cancer, eventually leading to a stage where no effective treatments remain, resulting in patient mortality. Therefore, extending the PFS following the initial chemotherapy is crucial for improving outcomes in ovarian cancer treatment. Promising gene therapy has the potential to significantly increase this initial PFS by targeting specific pathways involved in tumor progression.
Gene therapy encompasses the delivery of genes (DNA or RNA) to patients, aiming to inhibit the expression of oncogenes (gene silencing), restore mutated-tumor suppressor genes with normal genes (gene restoration), provide therapeutic genes to target cells (gene supplementation) [8]. Initially, research primarily concentrated on treating hereditary diseases with well-defined causes. However, current investigations have expanded to include a variety of diseases, such as neurodegenerative diseases [9], rheumatoid arthritis [10], cardiovascular diseases [11], infectious diseases [12], and aging-related diseases [13]. Moreover, the expanded understanding of genes implicated in cancer formation, growth, and metastasis has stimulated research [14] and clinical investigations [15] into cancer gene therapy, leading to notable advancements in the field.
For cancer gene therapy, two distinct groups of delivery vehicles exist viral and non-viral vectors. While both vectors have their advantages and limitations, the central challenge of vectors for cancer gene therapy is achieving efficient and safe gene delivery. Non-viral vectors include carriers such as lipid nanoparticles (LNPs), cationic liposomes, peptides, and cationic polymers like polyethyleneimine (PEI) [16]. Although non-viral vectors often face limitations in tumor-targeting specificity due to their relatively simple structures [17], several studies have demonstrated tissue-specific delivery [18]. Furthermore, similar to viral vectors, non-viral vectors can be functionalized with biomolecules (e.g., peptides, antibodies, or aptamers) to enhance their targeting ability and therapeutic efficacy. These advantages, however, are often outweighed by viral vectors’ inherent strengths. Compared to non-viral systems, viral vectors typically offer significantly longer-lasting transgene expression [19, 20] and intrinsic mechanisms for efficient cellular entry and genome integration or persistence [21]. These features make viral vectors especially effective for in vivo gene delivery [22], explaining why they remain the primary focus in a substantial portion of cancer gene therapy research [23]. Notably, viral vectors, such as retroviruses and adenoviruses, can potentially achieve cancer cell-specific delivery by modifying the proteins present in the viral envelope or, in the case of AAV (Adeno-associated virus), altering the structure of the viral capsid [14, 15].
AAV has a protein structure known as the viral capsid, allowing for AAV capsid engineering through systematic gene modifications [24]. This characteristic facilitates the development of AAV variants with specific tissue tropism for research purposes. As a result, AAVs have been developed that can cross the human blood-brain barrier (BBB) for CNS transduction [25], target human hepatocytes [26, 27], and transduce human cancer cells more efficiently than wild-type AAV [28]. As a viral vector capable of cell-type-specific or tissue tropism, AAV is relatively safe [29] and provides long-term expression [30], enhancing its potential for FDA approval in gene therapy compared to previously developed viral vectors. Over 200 completed and ongoing clinical trials use AAV as the gene transfer vector. The outstanding virtues of AAV—its relative safety and the ability to provide prolonged transgene expression—suggest that it will play a critical role as a strategic tool in cancer gene therapy aimed at controlling and treating cancer as a manageable disease. This review examines studies on AAV-mediated cancer gene therapy for the treatment of ovarian cancer and proposes future directions for this therapeutic approach.
Understanding ovarian cancer: diagnosis, genetics, initial surgery, and chemotherapy
Diagnosis
Ovarian cancer often presents insidiously, making early diagnosis challenging. Most women are diagnosed at stage III or IV, exhibiting symptoms such as abdominal pain or discomfort, menstrual irregularities, dyspepsia, other gastrointestinal disturbances, and urinary frequency or retention. In advanced stages, respiratory symptoms may occur due to ascites or pleural effusion, and bowel obstruction can also be present [31]. The late diagnosis is attributed to subtle onset and vague symptoms of the disease, which patients may mistake for ordinary changes related to childbearing, menopause, or aging. Additionally, these symptoms are often nonspecific and can mimic conditions like irritable bowel syndrome. This difficulty in recognizing ovarian cancer symptoms results in a prolonged and convoluted diagnostic process [32].
Genetics
Hereditary factors account for ~20% of ovarian cancers [33]. Most are due to pathogenic mutations in the BRCA1 or BRCA2 genes, which repair DNA double-stranded breaks via homologous recombination. Inherited mutations in these genes are major risk factors, with germline BRCA1 mutations increasing ovarian cancer risk by 20%–50% and BRCA2 mutations by 10%–20% [34]. These cancers typically occur at a younger age, especially in BRCA1 mutation carriers, with a median diagnosis age in the mid-40s [34]. For this reason, women with suspected hereditary cancer syndromes, such as BRCA1 or BRCA2 mutations, or those with a family history, young age at diagnosis, or high-grade ovarian cancer, should receive genetic testing and counseling. If a mutation is identified, risk-reducing bilateral salpingo-oophorectomy is considered an effective preventive strategy to reduce the risk of ovarian cancer [34]. While BRCA1 and BRCA2 are well-documented as key genetic factors in hereditary ovarian cancer, including mutations in other DNA repair genes, only around 20% of ovarian cancer cases are attributed to genetic causes [33]. The remaining 80% of cases remain unexplained by known genetic mutations.
Initial surgery and chemotherapy
The prognosis for ovarian cancer is largely determined by the maximum diameter of residual disease after cytoreductive surgery [35, 36]. The standard treatment protocol for ovarian cancer includes optimal cytoreductive surgery followed by a chemotherapy regimen of six cycles of carboplatin and paclitaxel, or docetaxel if paclitaxel is not tolerated [37] (Fig. 1b). In advanced-stage cases, the volume of residual disease post-surgery is the most important prognostic indicator [35, 36], necessitating a comprehensive surgical approach that includes total abdominal hysterectomy, bilateral salpingo-oophorectomy, omentectomy, and maximal cytoreduction [38]. Additional procedures involve peritoneal washings, multiple peritoneal biopsies, appendectomy in mucinous histology, and resection of bulky para-aortic and pelvic lymph nodes [38]. This standard treatment may also be employed in cases of primary suboptimal cytoreduction. For advanced-stage patients (IIIC or IV) with unresectable tumors, 2–3 cycles of neoadjuvant chemotherapy followed by surgical cytoreduction and further chemotherapy are required [39].
The catalytic activity of PARP1 (poly ADP-ribose polymerase 1) is crucial for mediating various DNA damage repair pathways, including stabilizing DNA replication forks [40, 41]. Additionally, its role in chromatin remodeling is closely linked to its function in DNA repair [42]. Consequently, inhibiting PARP1 is an effective strategy for treating cancers with deficiencies in the homologous recombination repair of DNA double-strand breaks. Studies have shown that BRCA mutations in ovarian cancers disrupt the homologous recombination pathway, leading to increased sensitivity to PARP inhibitors [43, 44]. Indeed, several clinical studies demonstrated the promising efficacy of PARP inhibitors in ovarian cancer patients [45,46,47]. There is growing evidence supporting that the use of maintenance therapy with PARP inhibitors after a response to platinum-based chemotherapy, in both first-line and second-line settings, has significantly extended the interval between response and disease relapse [48, 49] (Fig. 1b).
Although PARP inhibitors have increased progression-free survival and overall survival compared to control ovarian cancer treatments, patients ultimately experience disease relapse and develop resistance to PARP inhibitors, leading to mortality (Fig. 1b). The most presumptive resistance mechanism of PARP inhibitors is the restoration of BRCA1 or BRCA2 protein functionality through secondary mutations [50]. This mechanism is also shared in resistance to platinum-based treatments in cancer cells [51]. In PARP inhibitor-resistant human pancreatic cancer cell lines, new BRCA2 isoforms were made by an intragenic deletion of the frameshift mutation. This deletion restored the open reading frame (ORF) of the BRCA2 gene, enabling the cells to repair drug-induced DNA double-strand breaks via homologous recombination [52].
Although current treatment approaches are optimum, most women with advanced-stage ovarian cancer will relapse eventually due to resistance to platinum-based drugs, PARP inhibitors, or because of refractory cancer (Fig. 1b). This challenge has driven significant interest in developing new, more targeted strategies for treatment, with gene therapy presenting a potential new option.
Adeno-associated virus as a delivery vector: from basics to therapeutic applications
Adeno-associated virus (AAV) belongs to human Parvovirus with a single-stranded DNA genome and is one of the smallest known viruses (~25 nm). Although it can transduce human cells, it has not been identified as a causative agent of any specific disease [53, 54]. AAV does not replicate well within host cells unless co-infected with adenovirus [55]. Unlike adenovirus, AAV exhibits low immunogenicity and does not strongly elicit a host immune response [56].
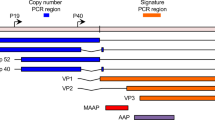
Wild-type AAV contains a single-stranded DNA viral genome flanked by inverted terminal repeat (ITR) sequences that form hairpin structures [57] (Fig. 2a). Between these ITR sequences, the AAV genome encodes the Rep gene, which is involved in the rescue and replication of the viral genome, and the Cap gene, which encodes the icosahedral capsid proteins responsible for packaging the ssDNA viral genome [58]. In contrast to wild-type AAV, recombinant AAV (rAAV) replaces the viral genes between the ITR sequences with a therapeutic transgene (Fig. 2b). To produce rAAV, host cells such as 293T cells are co-transfected with plasmids encoding the Rep and Cap genes, as well as adenovirus helper genes E4, E2a, and VA, to facilitate the assembly of AAV particles containing the therapeutic gene [59].

A Schematic representation of wild-type AAV, featuring its single-stranded viral genome. The wild-type AAV genome comprises single-stranded DNA with two inverted terminal repeats (ITRs) at either end and includes the essential genes Rep and Cap for viral replication. This genome produces at least three different transcript variants, each originating from distinct starting points. The p5 and p19 promoters are responsible for transcribing mRNA for the Rep78, Rep68, Rep52, and Rep40 proteins, while the p40 promoter transcribes the genes for the viral capsid proteins VP1, VP2, and VP3, as well as the non-structural proteins AAP and MAAP, which are crucial for virus production. B Production of recombinant AAV and its representative structure. To produce AAV, a eukaryotic cell, such as a HEK293T cell, is transfected with an AAV transgene plasmid containing the therapeutic gene intended for gene therapy, a Rep/Cap plasmid derived from wild-type AAV, and a helper plasmid derived from adenovirus that contains genes promoting efficient AAV production. After allowing sufficient time for the cells to produce AAV particles, AAV is harvested and purified from the cell lysate and supernatant. This process results in the generation of functional recombinant AAV capable of therapeutic gene expression in the host. Unlike wild-type AAV, recombinant AAV cannot replicate or propagate after transducing the host cell due to the absence of Rep/Cap genes.
A major limitation of using adeno-associated virus (AAV) for cancer gene therapy is its relatively small viral genome size compared to other viral vectors such as lentivirus and adenovirus [60]. The ideal size for a therapeutic gene delivered by AAV is generally <5 kb [61]. Although the wild-type AAV genome itself is ~4.7 kb (4675 bp) [57] (Fig. 1a), this size includes essential regulatory elements like promoters and poly-A regions, which leaves limited space for the therapeutic gene. Consequently, for genetic disorders such as cystic fibrosis (CFTR, 4443 bp) or Duchenne muscular dystrophy (DMD, 11,034 bp) where the size of the therapeutic gene exceeds 4 kb, AAV-based gene therapy may become either unfeasible or exceptionally challenging.
In addition to the limitation imposed by the AAV packaging capacity, a significant challenge in systemic AAV-mediated gene delivery—particularly in the context of repeated dosing—is the host immune system’s recognition of the viral capsid [62]. Pre-existing neutralizing antibodies (NAbs), which develop from natural exposure to wild-type AAV, can bind to the recombinant AAV vectors and prevent it from reaching target cells [56]. These antibodies block viral entry by interfering with receptor interactions and promote rapid clearance through the reticuloendothelial system [63]. Notably, seroprevalence studies indicate that around half of the population develops AAV-specific NAbs by the age of two, and once formed, these antibodies typically persist throughout life [64,65,66]. Given this high prevalence, efforts to develop effective AAV-based gene therapies for widespread applications, such as anti-tumor treatments, must also address the challenge posed by pre-existing NAbs. In the context of cancer, where broad patient applicability is essential, strategies to evade or overcome AAV neutralization—through capsid engineering, immune modulation, or alternative serotype selection—are likely to be critical for clinical success.
A notable advancement in AAV technology is capsid engineering, which allows modification of the virus’s infection efficiency and tissue tropism [67]. One of the most significant achievements in this area is the development of AAV capsids capable of crossing the blood-brain barrier (BBB) to achieve central nervous system (CNS) transduction in several animal models, including mouse and NHP (non-human primate) [25, 68, 69]. Subsequently, a breakthrough study translated animal model research into a human context by engineering an AAV capsid that specifically targets the human transferrin receptor on the blood-brain barrier [70]. When tested in transgenic mice expressing this receptor, the engineered AAV was shown to effectively traverse the barrier and deliver a therapeutically relevant gene to the CNS [70]. These efforts hold great potential for treating CNS disorders.
The US FDA approved the most successful AAV-based gene therapy in 2019 for treating spinal muscular atrophy, marketed as Zolgensma (onasemnogene abeparvovec) [71, 72]. Subsequently, multiple other AAV-based gene therapies have also gained approval, solidifying AAV’s position as a promising viral vector in the gene therapy market [58].
Challenges of gene therapy in ovarian cancer
Gene therapy represents a promising strategy for treating ovarian cancer, as it enables the introduction of various genes that regulate molecular processes. This approach can inhibit tumor growth, angiogenesis, invasion, and metastasis, and modulate immune response [73]. Nonetheless, at least two significant challenges must be addressed to ensure the success of these therapeutic strategies.
Firstly, current knowledge of the molecular mechanisms driving tumorigenesis and cancer progression remains incomplete. For example, High-grade serous ovarian cancer (HGSOC), which constitutes over two-thirds of ovarian cancers, is characterized by high intra-tumoral heterogeneity [74]. This genomic instability leads to the emergence of tumor subclones with competitive advantages, such as faster growth rates or resistance to chemotherapy. These advantageous subclones ultimately dominate the tumor through a process known as “clonal expansion”, and an elevated extent of clonal expansion has a detrimental effect on a patient’s overall survival [75]. This observation of drug resistance driven by clonal expansion through intra-tumoral heterogeneity underscores the current limitations in our understanding of the specific stages of ovarian cancer progression and the identification of optimal cellular (e.g., cancer stem cells) or genetic targets for effective gene therapy.
Second, the erratic blood supply in ovarian cancer presents substantial challenges for the direct delivery of therapeutic genes to ovarian cancer cells in vivo. Uncontrolled tumor growth contributes to the development of abnormal vasculature through rapid and uncoordinated vascular expansion [76]. As tumors grow uncontrollably, the demand for oxygen and nutrients in hypoxic status increases, leading to the formation of haphazardly arranged blood vessels that lack proper structure and function. The aggressive expansion of tumor mass often outpaces the capacity of the newly formed blood vessels to provide sufficient blood supply, resulting in a chaotic and ineffective vascular network [77]. In addition, dysregulated angiogenesis signaling pathways in ovarian cancer lead to the secretion of pro-angiogenic factors, including vascular endothelial growth factor (VEGF), angiopoietin (ANGPT), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF) [78]. These factors facilitate the formation of new yet structurally disorganized blood vessels, characterized by increased permeability and suboptimal vessel maturation, ultimately undermining the effectiveness of therapeutic gene delivery to ovarian cancer cells via systemic administration.
Thus, for the successful implementation of gene therapy in ovarian cancer, selecting the appropriate target cells and therapeutic genes tailored to the stages of ovarian cancer progression is necessary. Furthermore, taking advantage of the unique anatomical location of ovarian cancer within the abdominal cavity (Fig. 1a), it is essential to assess the efficacy of intraperitoneal gene therapy when combined with traditional systemic chemotherapy.
Strategies for targeting ovarian cancer cells in gene therapy
The theoretical strategies for targeting ovarian cancer cells in gene therapy can be broadly categorized based on two main criteria: the target cell types for therapeutic gene expression and the therapeutic gene types used to suppress cancer cell growth (Fig. 3).

Schematic representation of gene therapy strategies targeting ovarian cancer cells using AAV. The strategies are categorized based on the target cells and the specific gene therapy approaches employed. The diagram highlights the engineering of AAV capsids to selectively target ovarian cancer stem cells or immune cells. Additionally, functional screening of therapeutic genes could be conducted using an in vivo ovarian cancer model with AAV to identify potential therapeutic genes, including antibodies, anti-angiogenic factors, cytokines, and secretory tumor suppressors.
The first criterion focuses on the types of cells within the tumor microenvironment and other organs or systemic compartments that are targeted to receive the therapeutic gene expression. Three main strategies fall under this criterion.
The first strategy focuses on selectively targeting cancer cells within the tumor mass, with the goal of directing gene therapy predominantly to malignant cells while limiting effects on surrounding non-cancerous components. While not exclusively specific, this approach enhances targeting toward cancer cells, potentially limiting unintended effects on surrounding normal cells or non-malignant cell types within the tumor microenvironment. For example, intratumoral injection of an AAV vector encoding SaCas9-KKH and sgRNA has been reported to reduce tumor growth and moderately improve overall survival in an orthotopic glioblastoma mouse model [79]. In addition, AAV capsid engineering has been shown to enhance tumor cell-specific transduction. One study developed AAV6 vectors incorporating RGD peptides and capsid mutations (Y705-731F, T492V, K531E), which significantly increased transduction efficiency in integrin-overexpressing cancer cells and improved tumor specificity in vivo [28]. Another study engineered AAV2 capsids to display a plectin-1 targeting peptide (PTP), resulting in a 37-fold preference for pancreatic ductal adenocarcinoma (PDAC) tumors over liver tissue, thus demonstrating the potential of engineered capsids for tumor-targeted gene delivery [80].
The second strategy broadens the scope to include the cancer cells, the tumor microenvironment, and other organs or systemic compartments. This approach leverages the supportive roles that these cells play in tumor growth, aiming to disrupt the tumor’s supportive microenvironment. By simultaneously targeting cancer cells and their surrounding support systems, this strategy potentially enhances therapeutic efficacy and contributes to a more hostile environment for tumor survival. For example, systemic administration of AAV8 vectors encoding soluble VEGF receptors (sVEGFR2 and sVEGFR3) led to transgene expression in both ovarian tumor cells and their surrounding microenvironment. This approach effectively reduced intratumoral angiogenesis and, when combined with chemotherapy, suppressed tumor growth and ascites formation, ultimately improving overall survival in an ovarian cancer model [81, 82]. Additionally, AAV capsid engineering has produced RGD-modified vectors with enhanced muscle-specific transduction after systemic delivery [83]. These capsids outperformed natural serotypes in mouse models of genetic muscle disease and showed conserved efficacy in non-human primates, highlighting their therapeutic potential. This approach enables a strategy to maximize anti-tumor effects by increasing specificity toward cancer cells or their surrounding microenvironment.
The third strategy targets the surrounding tissue cells within the tumor microenvironment, as well as cells in other organs or systemic compartments, deliberately excluding the cancer cells. Here, the therapeutic genes are introduced into the surrounding supportive cells in the tumor microenvironment. By modifying these supportive cells, this strategy can indirectly impair cancer growth by weakening the resources and support that the tumor relies on. A representative example of this approach is an AAV-based vaccine that, despite being delivered intramuscularly to normal tissue, induced strong and durable antigen-specific T and B cell responses. This immune activation led to effective tumor suppression in mouse models, including the poorly immunogenic B16/F10-Ova melanoma, highlighting the potential of targeting the tumor microenvironment rather than cancer cells directly [84, 85].
The second criterion for categorizing gene therapy strategies in ovarian cancer pertains to the type of therapeutic genes used. These genes fall into three main types: gene silencing, gene restoration, and gene supplementation. Gene silencing aims to deactivate oncogenes or genes that play a role in cancer progression, such as mutant KRAS [86], CLDN3 [87], and EGFR [88]. By silencing these cancer-promoting genes, this approach effectively reduces the proliferative and invasive potential of cancer cells.
Gene restoration is another therapeutic approach that involves reintroducing or restoring the function of tumor suppressor genes, such as TP53 [89, 90] and PTEN [91, 92], which are often mutated or inactivated in cancer. Restoring these genes can re-enable cellular mechanisms that prevent uncontrolled cell division and tumor growth, contributing to cancer suppression.
Finally, gene supplementation involves the addition of therapeutic genes that aid in cancer treatment, including genes encoding cytokines [93,94,95], anti-angiogenic proteins [96, 97], and immune checkpoint inhibitors such as bevacizumab [98] and pembrolizumab [99]. This supplementation strategy introduces additional therapeutic agents to the tumor environment, aiming to improve the immune response against cancer cells, inhibit blood vessel formation needed for tumor growth, and enhance overall therapeutic outcomes.
Limitations of gene silencing and gene restoration strategies
The necessity of categorizing effective anticancer gene therapy strategies based on target cells and therapeutic genes arises from the inherent limitation that gene delivery vectors cannot realistically transduce 100% of ovarian cancer cells. As previously mentioned, it is practically challenging to achieve complete transduction of the ovarian cancer mass, which can grow to several centimeters in size [100], with poorly formed blood vessels leading to hypoxic necrotic regions [101]. In particular, when employing gene silencing strategies, the specificity and efficiency of vector-mediated transduction of cancer cells become critically important. In this context, the cancer cell transduction rate of the delivery vector becomes a crucial determinant for the success of gene therapy.
Even if not all ovarian cancer cells are transduced, effectively inhibiting and killing the transduced cells to extend progression-free survival by several months can still be considered beneficial. However, given the high production and distribution costs of gene therapies, these treatments must demonstrate substantial efficacy, akin to the significant improvements in overall survival seen with PARP1 inhibitors in ovarian cancer treatment [46, 47].
From this perspective, although gene supplementation strategies are employed, approaches such as expressing suicide genes specifically in cancer cells [102] (Fig. 3), using the CRISPR-Cas9 system to knock out overexpressed oncogenes [103], or inducing the expression of microRNAs to inhibit oncogene translation [104] within cancer cells may fail due to the realistic limitation of not being able to transduce all cancer cells.
The gene restoration strategy, which involves delivering intact tumor suppressor genes, similarly faces the intrinsic limitation that the transduction efficiency of the delivery vector cannot reach 100%. Even if the theoretical inhibition efficiency of genes delivered for gene silencing and gene restoration is 100%, if not all cancer cells are eradicated, the clonal expansion of un-transduced cancer cells may prevent patients from experiencing the full therapeutic benefits [105, 106]. Therefore, the issue of delivery efficiency significantly undermines the gene silencing and gene restoration strategies, which rely on specifically transducing cancer cells (Fig. 3).
TP53 gene therapy as a representative example of gene restoration therapy
TP53 mutations are nearly ubiquitous in ovarian cancer, with a prevalence of 96% [107]. Initial gene therapy efforts focused on delivering normal TP53 to cancer cells via adenovirus, aiming to restore its function and induce growth arrest and apoptosis [108]. While these approaches were effective in vitro and in vivo, they did not translate successfully to clinical trials [109]. Two main reasons for this failure are the limited understanding of TP53’s biological functions and the inadequacy of delivery vectors and routes. The assumption that delivering normal TP53 would restore its function overlooked the dominant-negative effect of mutant TP53. In cancer cells, endogenous mutant p53 exerts a dominant-negative effect on wild-type (WT) p53, thereby inhibiting the tumor-suppressing activities of WT p53 [110]. TP53 functions as a tetramer [111], and in the presence of mutant TP53 or without a significantly higher amount of normal TP53, functional restoration is insufficient. Moreover, adenovirus vectors, used in clinical trials for TP53 delivery, induce only transient expression of the therapeutic gene [112], which is inadequate for sustained anticancer effects in ovarian cancer. Additionally, adenoviruses elicit strong immune responses, limiting their delivery efficiency to cancer cells when administered via peritoneal injection [89].
Two independent studies have investigated the restoration of wild-type TP53 using AAV vectors. One study demonstrated that AAV-mediated expression of WT TP53 inhibited cancer cell proliferation in vitro [113]. Another study showed tumor growth suppression after cancer cells, transduced with AAV expressing WT TP53, were injected subcutaneously into immunodeficient mice [114]. However, these experimental designs do not provide sufficient evidence to evaluate the in vivo transduction efficiency of AAV in tumor cells. Therefore, based on these findings, the strategy of TP53 gene restoration may need to be reconsidered for its applicability in in vivo gene therapy.
Proposed ovarian cancer gene therapy: gene supplementation
Anti-angiogenic gene supplementation using AAV in ovarian cancer
Considering the failures of adenovirus-mediated TP53 gene therapy in ovarian cancer, gene supplementation emerges as a promising alternative (Fig. 3). AAV vectors have been employed in ovarian cancer gene therapy to induce the expression of anti-angiogenesis factors or VEGFR-inhibiting antibodies [54]. Gene supplementation aligns with the mechanisms of modern immunotherapies [115] and CAR-T therapy [116], which strengthen host immune responses against cancer cells rather than targeting cancer cells directly. These therapies fall under the category of targeting cells within the tumor microenvironment, excluding cancer cells (Fig. 3). From this perspective, studies that have demonstrated the efficacy of ovarian cancer gene therapy using AAV by expressing secretory proteins as gene supplements are as follows.
AAV encoding the human soluble FMS-like tyrosine kinase receptor 1 (sFlt-1), which functions by both sequestering vascular endothelial growth factor (VEGF) and forming inactive heterodimers with other membrane-spanning VEGF receptors, leads to stable expression and significantly inhibits the growth of angiogenesis-dependent human ovarian cancer cells in a mouse xenograft model [96, 97] Compared to the control group, treatment with AAV-sFlt-1 resulted in an 80% reduction in tumor size and protected 83% of mice from death [96]. Kringle 5 (K5), a fragment of plasminogen, serves as an effective endogenous inhibitor of angiogenesis by both inhibiting endothelial cell proliferation and migration and inducing endothelial cell apoptosis [117, 118]. Administration of AAV-K5 led to a significant reduction in both subcutaneous and intraperitoneal growth of human ovarian cancer cells [119]. Compared to the control group, treatment with AAV encoding K5 resulted in a 60% reduction in tumor size and a 30% decrease in tumor weight [119]. A single intramuscular administration of AAV encoding angiostatin and endostatin inhibits intraperitoneal ovarian cancer growth in a preclinical mouse model [120]. Compared to the control mice, those treated with AAV encoding sFlt-1 exhibited significant tumor-free survival, reduced ascites volume, and lower levels of vascular endothelial growth factor (VEGF) in the ascites. Specifically, AAV-sFlt-1 treatment led to a 20% reduction in tumor-induced ascites volume, a 50% decrease in tumor weight, and protected 30% of the mice from tumor-related death [120]. Similarly, when AAV-encoding endostatin and angiostatin were administered intraperitoneally, it also showed sustained antitumor effects on the growth and dissemination of epithelial ovarian cancer in a mouse model [121]. Surprisingly, while AAV-mediated expression of endostatin and angiostatin alone extended mouse survival by ~25 days, the combination of AAV-mediated endostatin and angiostatin expression with paclitaxel treatment provided 90% protection from ovarian cancer–related death over a 240-day observation period [121]. Likewise, using AAV to express a point-mutated endostatin (P125A-endostatin) with enhanced endothelial cell binding and antiangiogenic activity resulted in sustained expression for 9 weeks from a single intramuscular administration and significantly inhibited the growth of human ovarian cancer cells in athymic nude mice, leading to an 80% reduction in tumor volume [122]. Treatment with AAV-P125A-endostatin in combination with carboplatin resulted in 60% of the animals remaining tumor-free for over 200 days, which was significantly better than treatment with AAV-vehicle and/or carboplatin alone [123].
Thrombospondin-1 (TSP-1) exhibits potent antiangiogenic activity due to its three type 1 repeat (3TSR) domain binding to CD36 on endothelial cells [124, 125]. Additionally, the KRFK motif in the second of these repeats is involved in activating the TGF-β pathway and demonstrates antitumor activity [126]. Pretreatment with 3TSR combined with chemotherapy significantly induced ovarian tumor regression, normalized tumor vasculature, and improved drug uptake, resulting in a reduction in ovarian tumor size compared to PBS controls and enhanced survival in advanced-stage ovarian cancer [127]. This combination also reduced tumor hypoxia by 50% and decreased tumor weight by 70%. In addition, the AAV-mediated delivery of 3TSR effectively decreases the formation of primary and secondary lesions and significantly prolongs survival in a murine model of orthotopic epithelial ovarian cancer at 90 days post-tumor implantation [128]. Furthermore, AAV-mediated delivery of 3TSR and the Fc domain-modified Fc3TSR, which is designed to prolong transgene expression, significantly enhances survival in a mouse model of epithelial ovarian cancer, although it did not outperform AAV-Bevacizumab in terms of survival benefit [129].
Bevacizumab, a humanized monoclonal antibody that targets and neutralizes VEGF, effectively inhibits tumor angiogenesis and exhibits significant anticancer activity [130]. The use of AAV vectors for delivering bevacizumab has shown significant results in cancer therapy. AAV-mediated delivery to the pleura effectively inhibits metastatic lung tumors [131], and persistent AAV-mediated bevacizumab therapy demonstrates notable tumor growth suppression in ovarian cancer models, reducing tumor weight by more than 80% and enabling the treated mice to survive twice as long as the control mice [132]. Furthermore, the use of AAV-mediated sVEGFR (soluble VEGFR) decoy gene therapy effectively inhibits intra-tumoral angiogenesis and tumor growth in an ovarian cancer model by targeting the VEGF/VEGFR signaling pathway, resulting in a twofold increase in survival rate. This approach demonstrates the potential of antiangiogenic gene therapy as a viable treatment for ovarian cancer [81].
AAV gene supplementation for enhancing immune responses against ovarian cancer
Utilizing AAV for the expression of cytokines represents a promising approach for the development of immune-based cancer immunotherapies [93,94,95]. IL-12 is a cytokine known for its potent immune-stimulating activity and plays a critical role in initiating and augmenting cell-mediated immunity [133]. IL-12 is primarily synthesized by immune cells such as dendritic cells and macrophages, and it plays a crucial role in driving Th1 cell differentiation, stimulating activation of T, B, and NK cells, and inducing the reprogramming of immunosuppressive cells [134]. Indeed, AAV-mediated delivery of IL-12 and tumor-associated cell-based vaccines represents an innovative approach in ovarian cancer immunotherapy [135].
While preclinical studies have demonstrated strong anti-tumor activity of AAV vectors carrying IL-12 [136,137,138,139], systemic administration of recombinant IL-12 in mice and humans has been associated with severe adverse effects [140, 141]. To reduce the toxicity associated with IL-12 and achieve controlled transgene expression, the Tet-On system has been employed for regulatable AAV-mediated IL-12 expression [142].
AAV-Tet-On-IL-12 has demonstrated high efficacy in preventing the establishment of metastasis and inducing a robust T-cell memory response against tumor cells. Additionally, a tetracycline-dependent riboswitch has been developed, allowing potent regulation of AAV-based transgene expression via a tetracycline aptamer [143]. The Tet-On system enables inducible gene expression in the presence of doxycycline or tetracycline, allowing temporal control over transgene activation. This system is designed to enable protein production from mRNA only in the presence of tetracycline, offering reversibility and repeatable induction capabilities for treating hepatocellular cancer in mice [144]. Considering the long-term expression characteristic of AAV in cancer gene therapy, a system that enables AAV transgene expression only when treatment is needed could be a valuable application for future AAV-mediated therapeutics.
AAV-based antigen loading of dendritic cells generates efficient cytotoxic T-cell responses in vitro [145, 146]. A quantitative transcriptomic-based investigation demonstrated that AAV particles are efficiently internalized without inducing detectable transcriptomic changes in monocyte-derived dendritic cells, in contrast to adenoviral infection, which upregulates anti-viral pathways. This report indicates that AAV has an immunologically favorable profile for the activation of dendritic cells [147]. AAV-mediated Her-2/neu expression in dendritic cells robustly stimulated cytotoxic T cells targeting ovarian cancer cells [148]. Dendritic cells transduced with an AAV-expressing Sperm protein 17 (Sp17) have been shown to generate a robust antigen-specific cytotoxic T-cell response against ovarian cancer cells. Additionally, these transduced dendritic cells significantly enhance the differentiation of cytotoxic T-cells, demonstrating their potential efficacy in immunotherapeutic strategies against ovarian cancer [149].
Additional considerations for successful ovarian cancer gene therapy using AAV
Significant advancements in AAV biotechnology, particularly in AAV capsid engineering and transgene expression, can be applied to ovarian cancer gene therapy (Fig. 3). This integration can enhance ovarian cancer gene therapy from the following two perspectives:
Capsid engineering for ovarian cancer stem cell-specific transduction
Recent studies have demonstrated that AAV capsid engineering can enhance cancer cell-specific transduction efficiency [28, 80, 150,151,152], thereby increasing antitumor effects while reducing cytotoxicity to normal cells [153]. Although these studies have shown promising antitumor effects in both in vitro and in vivo models, the practical challenges of achieving comprehensive transduction of all cancer cells, particularly due to the clonal expansion of cancer cells and drug resistance associated with cancer stem cells and intra-tumoral heterogeneity, highlight the limitations of strategies targeting the entire tumor mass in ovarian cancer gene therapy.
One of the major challenges in treating ovarian cancer is overcoming chemoresistance, which is often caused by intra-tumor heterogeneity [154, 155]. A significant concern is cancer stem cells, which are considered to be a source of tumor heterogeneity [156, 157]. These cells can survive initial chemotherapy and lead to relapse through clonal expansion [156, 157]. Cancer organoids, derived from cells with stemness, are valuable for their ability to accurately replicate the complexities and heterogeneity of primary tumors. They retain the genetic and molecular characteristics of the original tumor, including mutations, gene expression profiles, and drug resistance [158]. Thus, ovarian cancer stem cell-derived organoids serve as valuable models for screening therapeutic transgenes to overcome chemoresistance [159], as it is known that organoids form from ovarian cancer stem cells [160,161,162]. These ovarian cancer organoids are already used for drug screening in personalized cancer therapy [163,164,165]; however, the limited availability of existing anticancer drugs poses a significant challenge to organoid-assisted precision personalized therapy. In this situation, gene therapy could offer a broader range of therapeutic genes, potentially fulfilling the concept of personalized cancer treatment more effectively.
AAV vectors can be engineered to transduce specific cell types, due to their protein-based capsid structure. By modifying the AAV capsid genes, vectors can be designed to target ovarian cancer stem cells specifically, addressing chemoresistance and preventing ovarian cancer relapse. Thus, research focused on engineering AAV capsids for specific transduction of ovarian cancer stem cells represents a promising direction (Fig. 3). This approach holds promise as it may enable targeted therapy against ovarian cancer stem cells, which are critical in tumor relapse and drug resistance. Considering that organoid-based drug screening using patient-derived ovarian cancer stem cells is currently being pursued [163,164,165], developing AAV capsids with the capability to specifically transduce ovarian cancer stem cells represents a feasible and strategic research direction. Similarly, a study reported that combining capsid engineering with microRNA-dependent gene editing enhances tumor specificity. Specifically, a mosaic-capsid AAV vector—composed of AAV2 and AAV.CAP-B22, the latter known for its ability to cross the mouse blood-brain barrier—was used to target glioblastoma-initiating cells (GICs). This approach selectively eliminated GICs, reduced tumor growth, and extended survival in mice. Compared to the control group, which had a median survival of 32 days, treated mice survived for 55 days—an increase of ~72%. In addition, tumor volume was reduced by about 60%, highlighting the potential of this strategy for precision cancer therapy. These findings suggest that capsid engineering can be effectively utilized to target cancer stem cell populations, providing a viable strategy for overcoming therapeutic resistance. This supports the feasibility of applying similar approaches to ovarian cancer stem cells, reinforcing the promise of AAV capsid engineering as a precision tool for targeting the root of tumor relapse and progression [166].
Furthermore, evaluating therapeutic transgenes utilizing such AAVs to target ovarian cancer stem cells is crucial, as overcoming chemotherapy resistance remains a significant challenge in the effective treatment of ovarian cancer.
Screening for the most effective therapeutic genes deliverable by AAV
Screening for the most effective anticancer genes to be delivered via AAV is challenging. Traditionally, research approaches in cancer gene therapy have focused on delivering genes with well-defined mechanisms of action, such as Inhibition of angiogenic pathways, or immune activation [153]. The advantage of these studies lies in the ease of hypothesis formulation and the straightforward interpretation of results. However, the major drawback is the difficulty in adopting an unbiased approach, making it impossible to discover entirely new therapeutic methods unpredictably.
Recently, research utilizing the CRISPR-Cas9 system for genetic screening has been actively conducted to elucidate the mechanisms underlying drug resistance in cancer and to identify related genes [167, 168]. These studies employed lentivirus as a delivery vector, which allows for easy control of the MOI (multiplicity of infection), and utilized barcodes and UMIs (unique molecular identifier) to track delivered sgRNAs (single guide RNA), with data analysis facilitated by NGS (next-generation sequencing) techniques. Similarly, constructing AAVs loaded with various potential anticancer genes as transgenes and conducting screenings in vivo or in vitro could establish a critical strategic screening platform for discovering novel anticancer genes previously unknown [169, 170].
Conclusion and future direction
Ovarian cancer, similar to pancreatic cancer, often lacks clear symptoms in its early stages, making early detection very difficult [32]. As a result, it is usually diagnosed at an advanced stage. Due to the ovary’s anatomical structure, which is directly exposed to the peritoneal cavity, and the nature of cancers originating from ovarian epithelium, ovarian cancer is often found with peritoneal metastasis, frequently accompanied by significant ascites. Despite this dismal situation, the initial response rate to treatment for ovarian cancer is relatively high. This achievement is due to a well-established standard protocol that includes maximal cytoreductive surgery combined with platinum-based chemotherapy, as well as PARP inhibitor maintenance therapy. While complete remission is sometimes observed, over 80% of patients with advanced ovarian cancer experience recurrence and eventually develop resistance to all administered chemotherapies, leading to a high mortality rate [5].
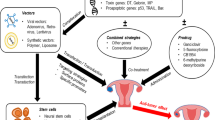
To address the therapeutic limitations associated with ovarian cancer, a range of innovative strategies has been investigated. This review focuses on the potential of AAV as a gene delivery vector for ovarian cancer therapy, with particular emphasis on approaches involving anti-angiogenic and immune-modulating genes. Specifically, AAV-mediated gene therapy for ovarian cancer should be considered for administration either in combination with, or sequentially following, standard chemotherapy regimens involving paclitaxel and carboplatin after maximal cytoreductive surgery (Fig. 4a). The primary objective of this therapeutic strategy would ideally be to significantly prolong progression-free survival beyond two years post-surgery and, ultimately, to achieve complete prevention of disease recurrence.

A This schematic illustrates a conceptual framework for incorporating AAV-based gene therapy into the current standard treatment regimen for ovarian cancer. Following maximal cytoreductive surgery and adjuvant chemotherapy with paclitaxel and carboplatin, AAV gene therapy—targeting angiogenesis or immune modulation pathways—is administered either in parallel with or sequentially after chemotherapy. The addition of gene therapy aims to enhance the durability of treatment response and significantly extend the duration of PFS beyond what is typically achieved with chemotherapy alone. This strategy addresses the critical need for therapeutic interventions that can delay relapse and improve long-term outcomes in ovarian cancer patients. B This illustration depicts the proposed molecular mechanism of AAV-mediated gene therapy targeting ovarian cancer. AAV vectors can be administered either intravenously (IV) or intraperitoneally (IP). Although the evaluation of administration routes lies beyond the scope of this review, it remains a critical consideration due to the anatomical characteristics of ovarian cancer, which predominantly resides within the peritoneal cavity. Determining the more effective route for AAV delivery—IV or IP—may significantly influence therapeutic efficacy. Engineered AAV capsids capable of efficiently infecting residual, microscopic ovarian cancer cells following maximal cytoreductive surgery can facilitate the delivery and expression of therapeutic genes within both ovarian tumor cells and cancer stem cells. Ideally, the anti-cancer proteins encoded by these genes should exert not only cell-autonomous anti-tumor effects in transduced cells, but also paracrine effects on neighboring malignant cells, thereby enhancing the overall therapeutic response.
This approach underscores the practical applicability of gene supplementation strategies in the context of ovarian cancer suppression. For optimal efficacy, the therapeutic gene encoded within the AAV vector should demonstrate preferential transduction of ovarian tumor cells and cancer stem cells over normal tissue. Moreover, genes capable of exerting anti-tumor effects not only within directly transduced cells but also on neighboring malignant cells would represent ideal candidates for inclusion in such gene therapy platforms (Fig. 4b).
Recent advances in capsid engineering have shown that AAV can target specific tissues or cells. By taking advantage of this property, developing AAV that targets cancer stem cells using markers known to identify ovarian cancer stem cells (CD24, CD44, CD133, CD117, ALDH1, SOX2, and OCT4) [171] presents an effective and promising gene therapy strategy, given the role of cancer stem cells in drug resistance and tumor heterogeneity. Additionally, with the rapid advancements in DNA sequencing technologies, research aimed at identifying therapeutic genes for ovarian cancer gene therapy through functional screening and incorporating them into AAV vectors could lead to new directions for ovarian cancer gene therapy.
References
Webb PM, Jordan SJ. Global epidemiology of epithelial ovarian cancer. Nat Rev Clin Oncol. 2024;21:389–400.
Lee B, Chang SJ, Kwon BS, Son JH, Lim MC, Kim YH, et al. Clinical guidelines for ovarian cancer: the Korean Society of Gynecologic Oncology guidelines. J Gynecol Oncol. 2024;35:e43.
Wang Y, Duval AJ, Adli M, Matei D. Biology-driven therapy advances in high-grade serous ovarian cancer. J Clin Investig. 2024;134:e174013.
Konstantinopoulos PA, Matulonis UA. Clinical and translational advances in ovarian cancer therapy. Nat Cancer. 2023;4:1239–57.
Giornelli GH. Management of relapsed ovarian cancer: a review. Springerplus. 2016;5:1197.
Gaitskell K, Hermon C, Barnes I, Pirie K, Floud S, Green J, et al. Ovarian cancer survival by stage, histotype, and pre-diagnostic lifestyle factors, in the prospective UK Million Women Study. Cancer Epidemiol. 2022;76:102074.
Pignata S, Cecere SC, Du Bois A, Harter P, Heitz F. Treatment of recurrent ovarian cancer. Ann Oncol. 2017;28:viii51–6.
Cesur-Ergun B, Demir-Dora D. Gene therapy in cancer. J Gene Med. 2023;25:e3550.
Ling Q, Herstine JA, Bradbury A, Gray SJ. AAV-based in vivo gene therapy for neurological disorders. Nat Rev Drug Discov. 2023;22:789–806.
Li W, Sun J, Feng SL, Wang F, Miao MZ, Wu EY, et al. Intra-articular delivery of AAV vectors encoding PD-L1 attenuates joint inflammation and tissue damage in a mouse model of rheumatoid arthritis. Front Immunol. 2023;14:1116084.
Bar C, Bernardes de Jesus B, Serrano R, Tejera A, Ayuso E, Jimenez V, et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat Commun. 2014;5:5863.
Fischer A. Gene therapy for inborn errors of immunity: past, present and future. Nat Rev Immunol. 2023;23:397–408.
Hosseinkhani H, Domb AJ, Sharifzadeh G, Nahum V. Gene therapy for regenerative medicine. Pharmaceutics. 2023;15:856.
Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021;6:53.
Zhao Z, Anselmo AC, Mitragotri S. Viral vector-based gene therapies in the clinic. Bioeng Transl Med. 2022;7:e10258.
Wang C, Pan C, Yong H, Wang F, Bo T, Zhao Y, et al. Emerging non-viral vectors for gene delivery. J Nanobiotechnol. 2023;21:272.
Bhatia SN, Chen X, Dobrovolskaia MA, Lammers T. Cancer nanomedicine. Nat Rev Cancer. 2022;22:550–6.
Kim J, Eygeris Y, Ryals RC, Jozic A, Sahay G. Strategies for non-viral vectors targeting organs beyond the liver. Nat Nanotechnol. 2024;19:428–47.
Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. 2021;39:47–55.
Jadlowsky JK, Hexner EO, Marshall A, Grupp SA, Frey NV, Riley JL, et al. Long-term safety of lentiviral or gammaretroviral gene-modified T cell therapies. Nat Med. 2025;31:1134–44.
Greig JA, Martins KM, Breton C, Lamontagne RJ, Zhu Y, He Z, et al. Integrated vector genomes may contribute to long-term expression in primate liver after AAV administration. Nat Biotechnol. 2024;42:1232–42.
Taghdiri M, Mussolino C. Viral and non-viral systems to deliver gene therapeutics to clinical targets. Int J Mol Sci. 2024;25:7333.
Hadi M, Qutaiba BAO, Jabari M, Jasoor AM, Naderloo O, Yasamineh S, et al. Recent advances in various adeno-associated viruses (AAVs) as gene therapy agents in hepatocellular carcinoma. Virol J. 2024;21:17.
Zolotukhin S, Vandenberghe LH. AAV capsid design: a Goldilocks challenge. Trends Mol Med. 2022;28:183–93.
Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34:204–9.
Paulk NK, Pekrun K, Zhu E, Nygaard S, Li B, Xu J, et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Molecular Ther J Am Soc Gene Ther. 2018;26:289–303.
Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–6.
Sayroo R, Nolasco D, Yin Z, Colon-Cortes Y, Pandya M, Ling C, et al. Development of novel AAV serotype 6 based vectors with selective tropism for human cancer cells. Gene Ther. 2016;23:18–25.
Srivastava A. AAV vectors: are they safe?. Hum Gene Ther. 2020;31:697–9.
Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O’Malley KL, et al. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet. 1994;8:148–54.
Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol. 2023;41:4065–76.
Bankhead CR, Kehoe ST, Austoker J. Symptoms associated with diagnosis of ovarian cancer: a systematic review. BJOG. 2005;112:857–65.
Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2:482–90.
Berek JS, Renz M, Kehoe S, Kumar L, Friedlander M. Cancer of the ovary, fallopian tube, and peritoneum: 2021 update. Int J Gynaecol Obstet. 2021;155:61–85.
Griffiths CT, Parker LM, Fuller AF Jr. Role of cytoreductive surgical treatment in the management of advanced ovarian cancer. Cancer Treat Rep. 1979;63:235–40.
Hoskins WJ, Bundy BN, Thigpen JT, Omura GA. The influence of cytoreductive surgery on recurrence-free interval and survival in small-volume stage III epithelial ovarian cancer: a Gynecologic Oncology Group study. Gynecol Oncol. 1992;47:159–66.
Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21:3194–200.
Armstrong DK, Alvarez RD, Bakkum-Gamez JN, Barroilhet L, Behbakht K, Berchuck A, et al. Ovarian cancer, version 2.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2021;19:191–226.
Vergote I, Trope CG, Amant F, Kristensen GB, Ehlen T, Johnson N, et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. New Engl J Med. 2010;363:943–53.
Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. Proteome-wide identification of poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol Cell. 2013;52:272–85.
Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336:728–32.
Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18:610–21.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–61.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. New Engl J Med. 2016;375:2154–64.
Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1274–84.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87.
Li N, Zhu J, Yin R, Wang J, Pan L, Kong B, et al. Treatment with niraparib maintenance therapy in patients with newly diagnosed advanced ovarian cancer: a phase 3 randomized clinical trial. JAMA Oncol. 2023;9:1230–7.
Jiang X, Li X, Li W, Bai H, Zhang Z. PARP inhibitors in ovarian cancer: sensitivity prediction and resistance mechanisms. Journal Cell Mol Med. 2019;23:2303–13.
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20.
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5.
Ponnazhagan S, Curiel DT, Shaw DR, Alvarez RD, Siegal GP. Adeno-associated virus for cancer gene therapy. Cancer Res. 2001;61:6313–21.
Santiago-Ortiz JL, Schaffer DV. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Control Release. 2016;240:287–301.
Hoggan MD, Blacklow NR, Rowe WP. Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc Natl Acad Sci USA. 1966;55:1467–74.
Verdera HC, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther J Am Soc Gene Ther. 2020;28:723–46.
Srivastava A, Lusby EW, Berns KI. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J Virol. 1983;45:555–64.
Wang JH, Gessler DJ, Zhan W, Gallagher TL, Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct Target Ther. 2024;9:78.
Dobrowsky T, Gianni D, Pieracci J, Suh J. AAV manufacturing for clinical use: Insights on current challenges from the upstream process perspective. Current Opin Biomed Eng. 2021;20:100353.
Li X, Le Y, Zhang Z, Nian X, Liu B, Yang X. Viral vector-based gene therapy. Int J Mol Sci. 2023;24:7333.
Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther J Am Soc Gene Ther. 2010;18:80–6.
Doshi BS, Markmann CA, Novak N, Juarez Rojas S, Davidson R, Chau JQ, et al. Use of CD19-targeted immune modulation to eradicate AAV-neutralizing antibodies. Mol Ther J Am Soc Gene Ther. 2025;S1525-0016:00177–7.
Dhungel BP, Winburn I, Pereira CDF, Huang K, Chhabra A, Rasko JEJ. Understanding AAV vector immunogenicity: from particle to patient. Theranostics. 2024;14:1260–88.
Li C, Narkbunnam N, Samulski RJ, Asokan A, Hu G, Jacobson LJ, et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2012;19:288–94.
Calcedo R, Wilson JM. AAV natural infection induces broad cross-neutralizing antibody responses to multiple AAV serotypes in chimpanzees. Hum Gene Ther Clin Dev. 2016;27:79–82.
Wei C, Li D, Zhang M, Zhao Y, Liu Y, Fan Y, et al. Prevalence of adeno-associated virus-9-neutralizing antibody in chinese patients with Duchenne muscular dystrophy. Hum Gene Ther. 2024;35:26–35.
Lopez-Gordo E, Chamberlain K, Riyad JM, Kohlbrenner E, Weber T. Natural adeno-associated virus serotypes and engineered adeno-associated virus capsid variants: tropism differences and mechanistic insights. Viruses. 2024;16:442.
Chen X, Ravindra Kumar S, Adams CD, Yang D, Wang T, Wolfe DA, et al. Engineered AAVs for non-invasive gene delivery to rodent and non-human primate nervous systems. Neuron. 2022;110:2242–57.e6.
Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20:1172–9.
Huang Q, Chan KY, Wu J, Botticello-Romero NR, Zheng Q, Lou S, et al. An AAV capsid reprogrammed to bind human transferrin receptor mediates brain-wide gene delivery. Science. 2024;384:1220–7.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. New Engl J Med. 2017;377:1713–22.
Alecu I, Mumneh N. The Zolgensma journey: a groundbreaking therapy for SMA. Dev Gene Ther Chapman Hall/CRC. 391–406.
Ayen A, Jimenez Martinez Y, Marchal JA, Boulaiz H. Recent progress in gene therapy for ovarian cancer. Int J Mol Sci. 2018;19:1930.
Pu T, Zhang C, Su B, Li L, Fu J. Research progress in intratumoral heterogeneity and clinical significance of ovarian cancer. Medicine. 2024;103:e36074.
Schwarz RF, Ng CK, Cooke SL, Newman S, Temple J, Piskorz AM, et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: a phylogenetic analysis. PLoS Med. 2015;12:e1001789.
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31.
Dudley AC, Griffioen AW. Pathological angiogenesis: mechanisms and therapeutic strategies. Angiogenesis. 2023;26:313–47.
Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. 2023;8:198.
Nieland L, Vrijmoet AB, Jetten IW, Rufino-Ramos D, de Reus A, Breyne K, et al. CRISPR targeting of mmu-miR-21a through a single adeno-associated virus vector prolongs survival of glioblastoma-bearing mice. Mol Ther J Am Soc Gene Ther. 2025;33:133–51.
Konkalmatt PR, Deng D, Thomas S, Wu MT, Logsdon CD, French BA, et al. Plectin-1 Targeted AAV Vector for the Molecular Imaging of Pancreatic Cancer. Front Oncol. 2013;3:84.
Kujala A, Valkonen E, Sallinen H, Tuppurainen L, Laakso H, Yla-Herttuala E, et al. AAV8-mediated sVEGFR2 and sVEGFR3 gene therapy combined with chemotherapy reduces the growth and microvasculature of human ovarian cancer and prolongs the survival in mice. Front Med. 2022;9:1018208.
Sanchez-Martinez C, Grueso E, Calvo-Lopez T, Martinez-Ortega J, Ruiz A, Almendral JM. VEGF-Virus interactions: pathogenic mechanisms and therapeutic applications. Cells. 2024;13:1642.
Tabebordbar M, Lagerborg KA, Stanton A, King EM, Ye S, Tellez L, et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell. 2021;184:4919–38.e22.
Franke AC, Hardet R, Prager L, Bentler M, Demeules M, John-Neek P, et al. Capsid-modified adeno-associated virus vectors as novel vaccine platform for cancer immunotherapy. Mol Ther Methods Clin Dev. 2023;29:238–53.
Krotova K, Kuoch Yoshitomi H, Caine C, Aslanidi G. Tumor antigen-loaded AAV vaccine drives protective immunity in a melanoma animal model. Mol Ther Methods Clin Dev. 2023;28:301–11.
Kim MJ, Lee SJ, Ryu JH, Kim SH, Kwon IC, Roberts TM. Combination of KRAS gene silencing and PI3K inhibition for ovarian cancer treatment. J Control Release. 2020;318:98–108.
Huang YH, Bao Y, Peng W, Goldberg M, Love K, Bumcrot DA, et al. Claudin-3 gene silencing with siRNA suppresses ovarian tumor growth and metastasis. Proc Natl Acad Sci USA. 2009;106:3426–30.
Dickerson EB, Blackburn WH, Smith MH, Kapa LB, Lyon LA, McDonald JF. Chemosensitization of cancer cells by siRNA using targeted nanogel delivery. BMC Cancer. 2010;10:10.
Wallis B, Bowman KR, Lu P, Lim CS. The challenges and prospects of p53-based therapies in ovarian cancer. Biomolecules. 2023;13:159.
Hassin O, Oren M. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discov. 2023;22:127–44.
Takei Y, Saga Y, Mizukami H, Takayama T, Ohwada M, Ozawa K, et al. Overexpression of PTEN in ovarian cancer cells suppresses i.p. dissemination and extends survival in mice. Mol Cancer Ther. 2008;7:704–11.
Minaguchi T, Mori T, Kanamori Y, Matsushima M, Yoshikawa H, Taketani Y, et al. Growth suppression of human ovarian cancer cells by adenovirus-mediated transfer of the PTEN gene. Cancer Res. 1999;59:6063–7.
Reul J, Frisch J, Engeland CE, Thalheimer FB, Hartmann J, Ungerechts G, et al. Tumor-specific delivery of immune checkpoint inhibitors by engineered AAV vectors. Front Oncol. 2019;9:52.
Liu JQ, Zhu J, Hu A, Zhang A, Yang C, Yu J, et al. Is AAV-delivered IL-27 a potential immunotherapeutic for cancer?. Am J Cancer Res. 2020;10:3565–74.
Yu YL, Wei CW, Chen YL, Chen MH, Yiang GT. Immunotherapy of breast cancer by single delivery with rAAV2-mediated interleukin-15 expression. Int J Oncol. 2010;36:365–70.
Mahendra G, Kumar S, Isayeva T, Mahasreshti PJ, Curiel DT, Stockardt CR, et al. Antiangiogenic cancer gene therapy by adeno-associated virus 2-mediated stable expression of the soluble FMS-like tyrosine kinase-1 receptor. Cancer Gene Ther. 2005;12:26–34.
Takei Y, Mizukami H, Saga Y, Yoshimura I, Hasumi Y, Takayama T, et al. Suppression of ovarian cancer by muscle-mediated expression of soluble VEGFR-1/Flt-1 using adeno-associated virus serotype 1-derived vector. Int J Cancer. 2007;120:278–84.
Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. New Engl J Med. 2011;365:2473–83.
Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol. 2019;30:1080–7.
Xiao F, Zhang L, Yang S, Peng K, Hua T, Tang G. Quantitative analysis of the MRI features in the differentiation of benign, borderline, and malignant epithelial ovarian tumors. J Ovarian Res. 2022;15:13.
Jiang H, Feng Y. Hypoxia-inducible factor 1alpha (HIF-1alpha) correlated with tumor growth and apoptosis in ovarian cancer. Int J Gynecol Cancer. 2006;16:405–12.
Nguyen QM, Dupre PF, Haute T, Montier T, d’Arbonneau F. Suicide gene strategies applied in ovarian cancer studies. Cancer Gene Ther. 2023;30:812–21.
Sherazi SAM, Rafique F, Haris M, Arshad A, Qaiser H, Uzair M, et al. Applications of CRISPR Cas-9 in ovarian cancer research. Protein Pept Lett. 2023;30:653–67.
Ankasha SJ, Shafiee MN, Abdul Wahab N, Raja Ali RA, Mokhtar NM. Oncogenic role of miR-200c-3p in high-grade serous ovarian cancer progression via targeting the 3’-untranslated region of DLC1. Int J Environ Res Public Health. 2021;18:5741.
McPherson A, Roth A, Laks E, Masud T, Bashashati A, Zhang AW, et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet. 2016;48:758–67.
Zhang AW, McPherson A, Milne K, Kroeger DR, Hamilton PT, Miranda A, et al. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell. 2018;173:1755–69.e22.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Zeimet AG, Riha K, Berger J, Widschwendter M, Hermann M, Daxenbichler G, et al. New insights into p53 regulation and gene therapy for cancer. Biochem Pharmacol. 2000;60:1153–63.
Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer?. Lancet Oncol. 2003;4:415–22.
Vinyals A, Peinado MA, Gonzalez-Garrigues M, Monzo M, Bonfil RD, Fabra A. Failure of wild-type p53 gene therapy in human cancer cells expressing a mutant p53 protein. Gene Ther. 1999;6:22–33.
Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22:741–53.
Trivedi PD, Byrne BJ, Corti M. Evolving horizons: adenovirus vectors’ timeless influence on cancer, gene therapy and vaccines. Viruses. 2023;15:2378.
Ruifa H, Liwei L, Binxin L, Ximing L. Additional gene therapy with rAAV-wt-p53 enhanced the efficacy of cisplatin in human bladder cancer cells. Urologia Int. 2006;77:355–61.
Qazilbash MH, Xiao X, Seth P, Cowan KH, Walsh CE. Cancer gene therapy using a novel adeno-associated virus vector expressing human wild-type p53. Gene Ther. 1997;4:675–82.
Oliveira G, Wu CJ. Dynamics and specificities of T cells in cancer immunotherapy. Nat Rev Cancer. 2023;23:295–316.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl J Med. 2017;377:2531–44.
Li L, Yao YC, Gu XQ, Che D, Ma CQ, Dai ZY, et al. Plasminogen kringle 5 induces endothelial cell apoptosis by triggering a voltage-dependent anion channel 1 (VDAC1) positive feedback loop. J Biol Chem. 2014;289:32628–38.
Cao Y, Chen A, An SS, Ji RW, Davidson D, Llinas M. Kringle 5 of plasminogen is a novel inhibitor of endothelial cell growth. J Biol Chem. 1997;272:22924–8.
Bui Nguyen TM, Subramanian IV, Xiao X, Nguyen P, Ramakrishnan S. Adeno-associated virus-mediated delivery of kringle 5 of human plasminogen inhibits orthotopic growth of ovarian cancer. Gene Ther. 2010;17:606–15.
Isayeva T, Ren C, Ponnazhagan S. Recombinant adeno-associated virus 2-mediated antiangiogenic prevention in a mouse model of intraperitoneal ovarian cancer. Clin Cancer Res. 2005;11:1342–7.
Isayeva T, Ren C, Ponnazhagan S. Intraperitoneal gene therapy by rAAV provides long-term survival against epithelial ovarian cancer independently of survivin pathway. Gene Ther. 2007;14:138–46.
Subramanian IV, Ghebre R, Ramakrishnan S. Adeno-associated virus-mediated delivery of a mutant endostatin suppresses ovarian carcinoma growth in mice. Gene Ther. 2005;12:30–8.
Subramanian IV, Bui Nguyen TM, Truskinovsky AM, Tolar J, Blazar BR, Ramakrishnan S. Adeno-associated virus-mediated delivery of a mutant endostatin in combination with carboplatin treatment inhibits orthotopic growth of ovarian cancer and improves long-term survival. Cancer Res. 2006;66:4319–28.
Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, et al. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA. 1990;87:6624–8.
Henkin J, Volpert OV. Therapies using anti-angiogenic peptide mimetics of thrombospondin-1. Expert Opin Ther Targets. 2011;15:1369–86.
Miao W-M, Lin Seng W, Duquette M, Lawler P, Laus C, Lawler J. Thrombospondin-1 type 1 repeat recombinant proteins inhibit tumor growth through transforming growth factor-β-dependent and -independent mechanisms1. Cancer Res. 2001;61:7830–9.
Russell S, Duquette M, Liu J, Drapkin R, Lawler J, Petrik J. Combined therapy with thrombospondin-1 type I repeats (3TSR) and chemotherapy induces regression and significantly improves survival in a preclinical model of advanced stage epithelial ovarian cancer. FASEB J. 2015;29:576–88.
Yu DL, Stegelmeier AA, Chow N, Rghei AD, Matuszewska K, Lawler J, et al. AAV-mediated expression of 3TSR inhibits tumor and metastatic lesion development and extends survival in a murine model of epithelial ovarian carcinoma. Cancer Gene Ther. 2020;27:356–67.
Stegelmeier AA, Santry LA, Guilleman MM, Matuszewska K, Minott JA, Yates JGE, et al. AAV-vectored expression of the vascular normalizing agents 3TSR and Fc3TSR, and the anti-angiogenic bevacizumab extends survival in a murine model of end-stage epithelial ovarian carcinoma. Biomedicines. 2022;10:362.
Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–9.
Watanabe M, Boyer JL, Crystal RG. AAVrh.10-mediated genetic delivery of bevacizumab to the pleura to provide local anti-VEGF to suppress growth of metastatic lung tumors. Gene Ther. 2010;17:1042–51.
Xie Y, Hicks MJ, Kaminsky SM, Moore MA, Crystal RG, Rafii A. AAV-mediated persistent bevacizumab therapy suppresses tumor growth of ovarian cancer. Gynecol Oncol. 2014;135:325–32.
Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA, et al. Localized Interleukin-12 for Cancer Immunotherapy. Front Immunol. 2020;11:575597.
Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, et al. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007;13:4677–85.
Chang MC, Chen YL, Chiang YC, Chen TC, Tang YC, Chen CA, et al. Mesothelin-specific cell-based vaccine generates antigen-specific immunity and potent antitumor effects by combining with IL-12 immunomodulator. Gene Ther. 2016;23:38–49.
Chiu TL, Wang MJ, Su CC. The treatment of glioblastoma multiforme through activation of microglia and TRAIL induced by rAAV2-mediated IL-12 in a syngeneic rat model. J Biomed Sci. 2012;19:45.
Chiu TL, Peng CW, Wang MJ. Enhanced anti-glioblastoma activity of microglia by AAV2-mediated IL-12 through TRAIL and phagocytosis in vitro. Oncol Rep. 2011;25:1373–80.
Lo CH, Chang CM, Tang SW, Pan WY, Fang CC, Chen Y, et al. Differential antitumor effect of interleukin-12 family cytokines on orthotopic hepatocellular carcinoma. J Gene Med. 2010;12:423–34.
Chiu TL, Lin SZ, Hsieh WH, Peng CW. AAV2-mediated interleukin-12 in the treatment of malignant brain tumors through activation of NK cells. Int J Oncol. 2009;35:1361–7.
Gil-Farina I, Di Scala M, Salido E, Lopez-Franco E, Rodriguez-Garcia E, Blasi M, et al. Transient expression of transgenic IL-12 in mouse liver triggers unremitting inflammation mimicking human autoimmune hepatitis. J Immunol. 2016;197:2145–56.
Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–8.
Vanrell L, Di Scala M, Blanco L, Otano I, Gil-Farina I, Baldim V, et al. Development of a liver-specific Tet-on inducible system for AAV vectors and its application in the treatment of liver cancer. Mol Ther J Am Soc Gene Ther. 2011;19:1245–53.
Strobel B, Duchs MJ, Blazevic D, Rechtsteiner P, Braun C, Baum-Kroker KS, et al. A small-molecule-responsive riboswitch enables conditional induction of viral vector-mediated gene expression in mice. ACS Synth Biol. 2020;9:1292–305.
Duchs MJ, Kratzer RF, Vieyra-Garcia P, Strobel B, Schonberger T, Gross P, et al. Riboswitch-controlled IL-12 gene therapy reduces hepatocellular cancer in mice. Front Immunol. 2024;15:1360063.
Liu Y, Chiriva-Internati M, Grizzi F, Salati E, Roman J, Lim S. Rapid induction of cytotoxic T-cell antigen gene delivery into human dendritic cells by an adeno-associated virus vector. Cancer Gene Ther. 2001;8:948–57.
Ponnazhagan S, Mahendra G, Curiel DT, Shaw DR. Adeno-associated virus type 2-mediated transduction of human monocyte-derived dendritic cells: implications for ex vivo immunotherapy. J Virol. 2001;75:9493–501.
Masri S, Carre L, Jaulin N, Vandamme C, Couzinie C, Guy-Duche A, et al. Transcriptomic analysis reveals the inability of recombinant AAV8 to activate human monocyte-derived dendritic cells. Int J Mol Sci. 2023;24:10447.
Yu Y, Pilgrim P, Zhou W, Gagliano N, Frezza EE, Jenkins M, et al. rAAV/Her-2/neu loading of dendritic cells for a potent cellular-mediated MHC class I restricted immune response against ovarian cancer. Viral Immunol. 2008;21:435–42.
Mirandola L, Chiriva-Internati M, Bresalier R, Piccotti L, Grizzi F, Marincola FM. A novel method for efficient generation of antigen-specific effector T-cells using dendritic cells transduced with recombinant adeno-associated virus and p38 kinase blockade. J Transl Med. 2019;17:424.
Stiefelhagen M, Sellner L, Kleinschmidt JA, Jauch A, Laufs S, Wenz F, et al. Application of a haematopoetic progenitor cell-targeted adeno-associated viral (AAV) vector established by selection of an AAV random peptide library on a leukaemia cell line. Genet Vaccines Ther. 2008;6:12.
Michelfelder S, Lee MK, deLima-Hahn E, Wilmes T, Kaul F, Muller O, et al. Vectors selected from adeno-associated viral display peptide libraries for leukemia cell-targeted cytotoxic gene therapy. Exp Hematol. 2007;35:1766–76.
Grifman M, Trepel M, Speece P, Gilbert LB, Arap W, Pasqualini R, et al. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol Ther J Am Soc Gene Ther. 2001;3:964–75.
Hacker UT, Bentler M, Kaniowska D, Morgan M, Buning H. Towards clinical implementation of adeno-associated virus (AAV) vectors for cancer gene therapy: current status and future perspectives. Cancers. 2020;12:1889.
Yang L, Xie HJ, Li YY, Wang X, Liu XX, Mai J. Molecular mechanisms of platinum‑based chemotherapy resistance in ovarian cancer (Review). Oncol Rep. 2022;47:82.
Ortiz M, Wabel E, Mitchell K, Horibata S. Mechanisms of chemotherapy resistance in ovarian cancer. Cancer Drug Resist. 2022;5:304–16.
Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–20.
Hu L, McArthur C, Jaffe RB. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br J Cancer. 2010;102:1276–83.
Fan H, Demirci U, Chen P. Emerging organoid models: leaping forward in cancer research. J Hematol Oncol. 2019;12:142.
Kopper O, de Witte CJ, Lohmussaar K, Valle-Inclan JE, Hami N, Kester L, et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat Med. 2019;25:838–49.
Maenhoudt N, Defraye C, Boretto M, Jan Z, Heremans R, Boeckx B, et al. Developing organoids from ovarian cancer as experimental and preclinical models. Stem cell Rep. 2020;14:717–29.
Garson K, Vanderhyden BC. Epithelial ovarian cancer stem cells: underlying complexity of a simple paradigm. Reproduction. 2015;149:R59–70.
Chen YA, Lu CY, Cheng WF, Kuo KT, Yu CW, Ho HN, et al. An experimental model for ovarian cancer: propagation of ovarian cancer initiating cells and generation of ovarian cancer organoids. BMC Cancer. 2022;22:967.
de Witte CJ, Espejo Valle-Inclan J, Hami N, Lohmussaar K, Kopper O, Vreuls CPH, et al. Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter- and intrapatient drug responses. Cell Rep. 2020;31:107762.
Hu H, Sun C, Chen J, Li Z. Organoids in ovarian cancer: a platform for disease modeling, precision medicine, and drug assessment. J Cancer Res Clin Oncol. 2024;150:146.
Driehuis E, Kretzschmar K, Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat Protoc. 2020;15:3380–409.
Wang Z, Zou P, Chen Z, Hou J, Son YL, Yamashita D, et al. Novel strategy to target glioblastoma-initiating cells using a braintropic adeno-associated virus carrying a miR-dependent genome-editing system. Bri J Cancer. 2025;132:1100–9.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7.
Dong MB, Tang K, Zhou X, Zhou JJ, Chen S. Tumor immunology CRISPR screening: present, past, and future. Trends Cancer. 2022;8:210–25.
Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy or glioblastoma. Nat Biotechnol. 2019;37:1302–13.
Cappelletto A, Alfi E, Volf N, Vu TVA, Bortolotti F, Ciucci G, et al. EMID2 is a novel biotherapeutic for aggressive cancers identified by in vivo screening. J Exp Clin Cancer Res CR. 2024;43:15.
Ottevanger PB. Ovarian cancer stem cells more questFions than answers. Semin Cancer Biol. 2017;44:67–71.
Funding
This work was supported by the Chung-Ang University Research Grants (20230339) and supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2024-00412879). Open Access funding enabled and organized by Chung-Ang University.
Author information
Authors and Affiliations
Contributions
HCY Conceived, drafted, and wrote the manuscript; SL and JYP reviewed the manuscript for essential intellectual content; EL supported the draft design and provided revisions and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yoo, H.C., Lee, S., Park, J.Y. et al. AAV for ovarian cancer gene therapy. Cancer Gene Ther 32, 817–830 (2025). https://doi.org/10.1038/s41417-025-00926-4
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41417-025-00926-4
