
Abstract
Ferroptosis is an oxidative, non-apoptotic cell death frequently inactivated in cancer, but the underlying mechanisms in oncogene-specific tumors remain poorly understood. Here, we discover that lactate dehydrogenase (LDH) B, but not the closely related LDHA, subunits of active LDH with a known function in glycolysis, noncanonically promotes ferroptosis defense in KRAS-driven lung cancer. Using murine models and human-derived tumor cell lines, we show that LDHB silencing impairs glutathione (GSH) levels and sensitizes cancer cells to blockade of either GSH biosynthesis or utilization by unleashing KRAS-specific, ferroptosis-catalyzed metabolic synthetic lethality, culminating in increased glutamine metabolism, oxidative phosphorylation (OXPHOS) and mitochondrial reactive oxygen species (mitoROS). We further show that LDHB suppression upregulates STAT1, a negative regulator of SLC7A11, thereby reducing SLC7A11-dependent GSH metabolism. Our study uncovers a previously undefined mechanism of ferroptosis resistance involving LDH isoenzymes and provides a novel rationale for exploiting oncogene-specific ferroptosis susceptibility to treat KRAS-driven lung cancer.
Similar content being viewed by others


Introduction
Oncogenic KRAS mutations are common in non-small cell lung cancer (NSCLC) and other human cancers [1]. Despite advances in targeting KRAS directly or indirectly and the advent of immunotherapy, effective therapies for KRAS-mutant NSCLC remain elusive [2]. Mutant KRAS reprograms cancer metabolism [3,4,5,6] to meet the increased energetic, biosynthetic and redox demands of tumor cells and promote KRAS-induced tumorigenicity [7]. In particular, KRAS-mutant cancer has been shown to produce high levels of reactive oxygen species (ROS) and has evolved sophisticated antioxidant programs to overcome the oxidative stress barrier during tumorigenesis [3, 8], on which tumor cells exquisitely depend for survival. Consequently, disruption of ROS defense would be selectively toxic for cancer cells [9, 10].
LDHB (LDH1) and LDHA (LDH2) are subunits of the active tetrameric LDH, which catalyzes the interconversion of lactate/pyruvate and NAD+/NADH in glycolysis and plays an important role in ATP generation and energy homeostasis in both anaerobic glycolysis and aerobic glycolysis known as the Warburg effect [11,12,13,14]. Despite considerable sequence and structural homology, LDHA and LDHB differ in their subcellular localization and substrate affinities, resulting in distinct functional roles [13, 15,16,17]. While LDHA predominantly converts pyruvate to lactate, supporting glycolysis under both anaerobic and aerobic conditions, thereby supporting the Warburg effect in cancer cells, LDHB has a higher affinity for lactate, catalyzing its conversion back to pyruvate, which fuels OXPHOS by linking it to the tricarboxylic acid (TCA) cycle [13, 18, 19]. Intriguingly, both LDHA and LDHB have been shown to play critical roles in KRAS-mutant cancers, required for tumor progression by regulating mitochondrial activities and stem cell properties, and are viable therapeutic targets for KRAS-dependent NSCLC [20,21,22]. LDH may also have functions independent of lactate/pyruvate metabolism [13, 17, 23, 24], although the precise mechanisms underlying the role of LDH in cancer remain to be elucidated.
Ferroptosis is an oxidative form of non-apoptotic cell death activated by ROS- and iron-dependent lipid peroxidation of polyunsaturated fatty acids (PUFAs) [25,26,27], which is often inactivated in cancer [28, 29]. The susceptibility of cancer cells to ferroptosis is finely balanced by the cellular metabolism that triggers lipid oxidation and the antioxidant systems that counteract it [29]. The cystine/glutamate antiporter subunit SLC7A11 (also known as xCT) and the selenium-dependent hydroperoxidase glutathione peroxidase 4 (GPX4) are the most potent antioxidant hubs defending against ferroptosis [30, 31]. Whereas SLC7A11 imports cysteine for GSH biosynthesis, GPX4 utilizes GSH to detoxify lipid peroxides and suppress ferroptosis [32]. Consequently, blockade of the SLC7A11/GPX4 axis with inhibitors (e.g. erastin and RSL3) leads to uncontrolled accumulation of lipid peroxides at the plasma membrane and endomembranes, ultimately inducing ferroptosis [29, 32]. Escape from ferroptosis has been shown to contribute to Kras-driven tumor development and progression [33, 34]. However, the antioxidant adaptations specific to oncogenic KRAS, such as the cellular processes that impinge on key antioxidant proteins and thereby modulate ferroptosis sensitivity of KRAS-dependent NSCLC, remain poorly understood [35].
In this study, we report for the first time that LDHB, but not LDHA, plays a role in protecting KRAS-mutant NSCLC from ferroptosis. LDHB modulates GSH metabolism through a noncanonical role in the regulation of SLC7A11, and as a result, LDHB suppression sensitizes cancer cells to SLC7A11/GPX4 inhibition by unleashing ferroptosis-mediated synthetic lethality in vitro and in vivo, which is mechanistically driven by increased glutamine metabolism, OXPHOS and mitoROS. Our results reveal a novel mechanism of ferroptosis defense involving LDH isoenzymes and provide a viable rationale for exploiting oncogene-specific ferroptosis susceptibility to treat KRAS-mutant lung cancer.
Results
LDHB silencing impairs GSH metabolism in KRAS-dependent lung cancer cells
We have recently shown that LDHB plays an important role in tumor-initiating cells and targeting LDHB affects mitochondrial metabolism in NSCLC [20]. To better understand the metabolic pathways underlying LDHB function in KRAS-driven NSCLC [20, 22], we performed unbiased metabolomics of A549 cells using LC-MS. LDHB knockdown (KD) by siRNAs significantly downregulated a number of cellular metabolites enriched in multiple metabolic processes (Fig. 1a, b; Fig. S1a; Original data file 1). LDHB KD A549 cells also showed reduced levels of intracellular lactate (Fig. S1b), consistent with its role in the Warburg effect [12, 14]. In particular, several intermediate metabolites of the de novo synthesis of GSH (Fig. 1c), such as cysteine (Cys), γ-glutamylcystine (γ-GC), glutamine (Gln), glutamate (Glu), and GSH itself, as well as the ratio of GSH to glutathione disulfide (GSSG), the oxidized form of GSH, were significantly decreased upon LDHB KD, although GSSG per se was not significantly altered in LDHB KD A549 cells (siLDHB) compared to control A549 cells (siNT) (Fig. 1d, e). In support of the metabolomics results, re-analysis of our transcriptomic data from A549 cells [20] showed that LDHB silencing significantly reduced the GSH gene signature and the mRNA levels of several key enzymes involved in GSH synthesis (Fig. 1c; Fig. 1f; Fig. S1c). Importantly, we confirmed that LDHB KD in KRAS-dependent A549 and H838 cells (Table S1) significantly reduced the GSH/GSSG ratio, a standard measure of cellular oxidative stress (Fig. 1g), but did not significantly alter total ROS in LDHB KD compared to control A549 cells (Fig. S1d), consistent with the metabolomics results (Fig. 1e). These results indicate that LDHB silencing impairs GSH metabolism, suggesting a novel role for LDHB in regulating antioxidant programs in KRAS-dependent NSCLC.

a Metabolomic analysis (LC-MS) of LDHB KD (siLDHB) and control (siNT) A549 cells (48 h post-transfection). Heat map showing the top 30 metabolites significantly different between LDHB KD and control A549 cells (n = 12). Relative abundance is scaled between 2 to -2. b Pathway enrichment analysis shows significantly downregulated metabolic processes in LDHB KD compared to control A549 cells. c Schematic of de novo GSH synthesis and the effect of LDHB KD on the pathway. Highlighted in blue are the genes and metabolites significantly altered by LDHB KD in A549 cells. Cys, cysteine; Cysta, cystathionine; γ-GC, γ-glutamylcysteine; Gly, glycine; Gln, glutamine; Glu, glutamate; GSH, glutathione; GSSG, glutathione disulfide; GCLC/GCLM, glutamate-cysteine ligase (GCL) catalytic and modifier subunits; GSS, glutamine synthetase; GSR, glutathione reductase; xCT, SLC7A11. d Heat map illustrating the abundance of key GSH metabolites in LDHB KD and control A549 cells. e The abundance of GSH, GSSG, and GSH/GSSG ratio in LDHB KD and control A549 cells. The analysis was based on the LC-MS data of A549 cells, p values by Student’s t-test. f LDHB KD downregulates GSH metabolism gene signature. GSEA was based on the transcriptome of LDHB KD (siLDHB) and control (siNT) A549 cells. g Ratios of GSH/GSSG in LDHB KD and control cells transfected with siRNAs for 48 h. Data are shown as mean ± s.d. (n = 3), with p values by Student’s t-test.
LDHB suppression sensitizes KRAS-dependent NSCLC cells to blockade of GSH-dependent ferroptosis defense
GSH is produced by the two-step synthesis of a tripeptide L-glutamic acid, cysteine, and glycine, with the cysteine required for GSH synthesis being obtained by SLC7A11-mediated uptake. We therefore hypothesized that LDHB KD would induce dependence on the SLC7A11/GSH antioxidant program. Indeed, a synthetic lethal chemical screen using small molecule inhibitors (n = 22) targeting multiple metabolic and oncogenic pathways, including the SLC7A11 inhibitor erastin (Table S2), showed that erastin and to a lesser extent sorafenib and dasatinib, preferentially suppressed the viability of LDHB KD lung cancer cells (A549, H838, H460 and H2122) harboring KRAS alterations, as measured by their AUC (area under curve) decrease in LDHB KD cells compared to control cells (Original data file 2). Interestingly, the increased susceptibility to erastin upon LDHB depletion was only observed in KRAS-dependent NSCLC cells, but not in EGFR-mutant PC9 (NSCLC), KRAS-mutant AsPC1 (pancreatic cancer), HT1080 (fibrosarcoma) or BEAS-2B, a normal epithelial cell line (Fig. 2a–c), suggesting that the antioxidant role of LDHB is oncogene- and lineage-specific. Confirming this finding, LDHB suppression sensitized KRAS-mutant A549, H838, H460 and H2122 cells to erastin in clonogenic assay (Fig. 2d), which was accompanied by a significant increase in oxidized lipids (Fig. 2e). Moreover, LDHB KD significantly increased the sensitivity of A549 cells to genetic inhibition of not only SLC7A11 but also GPX4 (Fig. 2f), which utilizes GSH to detoxify lipid peroxidation, further supporting a GSH-dependent role for LDHB. In sharp contrast, LDHB silencing (siRNA) failed to sensitize several KRAS wild-type NSCLC cell lines (H1299, H522 and Calu-3) to erastin (Fig. S2a).

a Immunoblots of the indicated cells transfected with siNT or siLDHB for 48 h. b Heat map showing relative viability of LDHB KD cells treated for 72 h with the indicated compounds dosed at IC80/IC90 in control cells. Data are expressed as percentages of viable LDHB KD cells normalized to the corresponding control cells. c Sensitivity of LDHB KD cells and control cells to erastin dosed at IC80/90 in control cells. Drug sensitivity is determined by the area under curve (AUC) calculated by Graphpad 9.1. Data are presented as mean ± s.d. (n = 3), with p < 0.05 by Student’s t-test. d Clonogenic assay of the indicated human KRAS-mutant NSCLC cells transfected with siLDHB and siNT and treated for 72 h with erastin (A549, 1 μM; H838, 0.25 μM; H460, 10 μM; H2122, 5 μM) or DMSO. e A549 cells transfected with siNT or siLDHB for 36 h were treated with DMSO or erastin (5 μM) for another 14 h before stained with C11 BODIPY 581/591. Scale bars, 100 μm. f Viability assay of A549 and H838 cells transfected with siNT or siLDHB for 24 h and subsequently transfected with siNT, siSLC7A11 or siGPX4 for additional 48 h. Data are shown as mean ± s.d. (n = 3), with the statistical analyses by one-way ANOVA. ns, no significant difference. g Cell death and lipid peroxidation assay of the indicated cells transfected with siNT and siLDHB for 48 h followed by further treatment for 16 h with sulfasalazine (SSZ; A549, 1 mM; H838, 0.5 mM; H460, 2.5 mM; H2122, 2 mM), alone or with Liproxstatin-1 (LIP1; 3 µM). Data are shown as mean ± s.d. (n = 3). ** p < 0.01; ***p < 0.001; ****p < 0.0001 by one-way ANOVA.
Similar results were obtained with sulfasalazine (SSZ), an FDA-approved drug with ferroptosis-inducing activity via inhibition of cystine/glutamate antiporter (xCT), which induced a significantly greater cell death and lipid peroxidation in LDHB KD A549, H838, H460 and H2122 cells than in control cells (Fig. 2g). Importantly, SSZ-induced cell death and lipid peroxidation in LDHB KD cells could be robustly reversed by liproxstatin-1 (LIP1) (Fig. 2g), a ferroptosis inhibitor that eliminates lipid hydroperoxides and has an anti-ferroptotic effect similar to GPX4 [30, 32], suggesting that SSZ-induced synthetic lethality in LDHB KD cells is mediated by ferroptosis.
This finding was validated by several independent assays in which shRNA-mediated KD of LDHB (Fig. 3a) sensitized A549, H838 and murine KP (KrasG12D/+; p53-/-) cells to erastin and the GPX4 inhibitor RSL3, accompanied by a significant increase in PTGS2 (prostaglandin endoperoxide synthase 2) expression, a ferroptosis biomarker [36], and lipid peroxidation (Fig. 3b–g). Importantly, the erastin- and RSL3-induced upregulation of PTGS2, loss of cell viability and increase in lipid peroxidation in LDHB KD cells were almost completely rescued by FER1, but not by inhibitors of necrosis (NEC), autophagy (HCQ) or apoptosis (ZVF) (Fig. 3c–g; Fig. S2b–d). Notably, the antioxidant N-acetylcysteine (NAC), a ROS scavenger, could also largely attenuate the erastin- or SSZ-induced suppression of cell viability in LDHB KD cells (Fig. 3h). These results accommodate evidence for a novel role of LDHB in GSH-associated ferroptosis denfense, and its suppression sensitizes KRAS-dependent NSCLC cells to blockade of the SLC7A11/GSH/GPX4 antioxidant program.

a Immunoblot of the indicated cells stably transduced with shNT or shRNAs against LDHB. b Clonogenic assay of murine KP cells transduced with shLdhb#b and shNT and further treated for 72 h with erastin (15 μM) or DMSO. c Quantitative analysis (qPCR) of PTGS2 mRNA in the indicated cells treated for 14 h with DMSO or erastin (A549, 5 µM; H838, 2.5 µM) with or without FER1 (3 µM). d–f Viability assay of shNT- or shLDHB-transduced cells after treated with RSL3 or erastin, alone or in combination with 2 μM Ferrostatin-1(FER1). g Flow cytometry of C11 BODIPY mean fluorescence intensity ratio of oxidative channel (FITC 488 nm) versus non-oxidative channel (PE-TEXAS RED 610 nm) in shNT- or shLDHB-transduced cells. A549 cells were treated with 0.5 μM RSL3 or 5 μM erastin, alone or in combination with 2 μM FER1 for 6 h and 14 h, respectively. H838 cells were treated with 0.25 μM RSL3 or 2.5 μM erastin, alone or in combination with 2 μM FER1 for 6 h and 14 h, respectively. Data are shown as mean ± s.d (n = 3), with statistical analyses by two-way ANOVA. h Colony assay of A549 and H838 shNT- or shLDHB-transduced cells treated with erastin (A549, 5 µM; H838, 2.5 µM) or sulfasalazine (SSZ; A549, 0.25 mM; H838, 1 mM) for 24 h in the presence or absence of NAC (10 mM).
Distinct roles of LDHA and LDHB in KRAS-driven NSCLC
Like LDHB, LDHA is required for the Warburg effect [35] and promotes tumor cell survival by protecting against ROS [18, 37]. Importantly, LDHA has been shown to play an essential role in KRAS-driven NSCLC, as a lack of LDHA results in reduced tumorigenesis, disease regression, reprogramming of pyruvate metabolism and a reduction in lactate accumulation in a mouse model of Kras-mutant NSCLC [21]. Furthermore, LDHA activity is important for the development of RAS-induced fibrosarcoma [38]. We therefore tested whether LDHA has a similar function to LDHB in the regulation of ferroptosis. However, in sharp contrast to the LDHB scenario, siRNA-based LDHA KD (Fig. S3a) and inhibitors (GSK2837808A, R-GNE-140) of LDHA showed no apparent effect on the sensitivity of A549 cells to RSL3 or erastin (Fig. S3b-d). To rule out a cell line-specific effect, we further tested KRAS-driven H838 and H460 cells and found similar results to A549 cells (Fig. S3e). Thus, LDHA, although closely related to LDHB, is not involved in the control of ferroptosis, suggesting that the two LDH isoenzymes have distinct roles in KRAS-mutant NSCLC.
LDHB knockdown suppresses SLC7A11 expression through the upregulation of STAT1
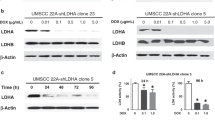
Next, we investigated the mechanism by which LDHB promotes GSH-associated ferroptosis defense. Re-analysis of our transcriptomic data from A549 cells [20] revealed that siRNA-mediated KD of LDHB most significantly downregulated SLC7A11 among other ferroptsosis-related genes (Fig. 4a), which we independently confirmed at the protein level: LDHB KD reduced the SLC7A11 protein in A549, H838, H460, H2122 and murine KP (KrasG12D/+; p53-/-) cells (Fig. 4b, c), as well as in A549 xenografts carrying LDHB-targeting shRNAs (Fig. S4a). Importantly, forced overexpression of SLC7A11 almost completely overcame the erastin- and SSZ-induced suppression of cell viability in LDHB KD A549 and H838 cells, accompanied by a significant decrease in lipid peroxidation (Fig. 4d–f). These results suggest that LDHB promotes GSH-associated ferroptosis defense by regulating SLC7A11, supporting a role for LDHB beyond glycolysis in KRAS-mutant NSCLC.

a Heat maps showing the mRNA fold change of ferroptosis-related genes (n = 25) in siLDHB cells (KD) compared to siNT cells (WT) (based on RNA-seq data). b, c Immunoblots of the indicated cells transfected for 48 h with siNT (-) or siLDHB (+) or stably expressing shRNAs. d Immunoblot of A549 stably expressing shNT or shLDHB were further transduced with either an empty vector or a SLC7A11-expressing plasmid (pCMV-SLC7A11). e, f Viability and lipid ROS assay of the indicated cells treated with erastin (A549, 5 µM; H838, 2.5 µM) or sulfasalazine (SSZ; A549, 0.25 mM; H838, 1 mM) for 24 h. g Immunoblots of A549 transfected (48 h) with siLDHB or siNT and further treated (16 h) with erastin (5 uM) or Sulfasalazine (1 mM). Murine KP cells expressing Ldhb shRNA or control shRNA were treated with erastin (10 uM) or Sulfasalazine (1 mM) for 24 h. h, i Immunoblots h and viability assay i of A549 cells transfected with siLDHB, siSTAT1 or siNTs for 24 h, followed by further treatment with erastin (10 µM) for another 24 h. j SLC7A11 mRNA fold change in A549 cells transfected (72 h) with siSTAT1 compared to A549 cells transfected with siNT. **p < 0.01 by Student’s t-test. k ChIP of STAT1 in A549 cells transfected with siLDHB or siNT, or treated with IFNγ. STAT1 binding to the SLC7A11 promoter (GAS2 domain) was quantified by qPCR. Results are expressed as fold change in site occupancy over IgG control and shown as mean ± SD from three independent experiments (n = 3). **p < 0.01 by two-way ANOVA.
Recent studies have shown that some metabolic enzymes have non-canonical functions to promote cancer progression, in addition to their known roles in metabolism [39]. Notably, our RNAseq data revealed that LDHB KD significantly upregulated the interferon α/γ (IFNα/γ) response genes in A549 cells [20]. Similar results were observed in erastin-treated A549 cells, where the IFNα/γ pathway was among the top candidates most significantly upregulated in erastin-treated LDHB KD vs. erastin-treated control A549 cells (Fig. S4b, c; Original data file 3). IFNα/γ and the downstream STAT1 promote ferroptosis through transcriptional repression of SLC7A11 [40], and we confirmed that LDHB KD alone and in combination with erastin or SSZ markedly increased STAT1 protein levels and concomitantly decreased SLC7A11 in A549, H358, and murine KP cells, whereas erastin or SSZ alone had no effect on STAT1 expression (Fig. 4g, h). STAT3, SCD1, C-MYC and ACSL4, previously shown to contextually regulate ferroptosis, were also not affected under the same conditions (Fig. S4d). Interestingly, we observed that LDHB KD, but not its combination with erastin, slightly increased the protein gasdermin D (GSDMD) in A549 cells (Fig. S4e), which is a key mediator of inflammasome-dependent pyroptotic cell death [41, 42].
Importantly, siRNA KD of STAT1 reversed and largely overcame the inhibition of erastin on the viability of LDHB KD A549 cells (Fig. 4i), suggesting that STAT1 activity is functionally required and sufficient for erastin-induced ferroptosis in LDHB KD cells. STAT1 KD significantly upregulated SLC7A11 mRNA levels (Fig. 4j), consistent with previous findings that STAT1 transcriptionally represses SLC7A11 [43]. Moreover, STAT1 chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR) demonstrated the enrichment of GAS – an element within the SLC7A11 promoter that physically interacts with STAT1 and is associated with IFNγ-mediated SLC7A11 transcriptional repression [43] - when STAT1 antibodies were used, compared to IgG control (Fig. 4k). Notably, LDHB KD further enhanced STAT1 occupancy at the GAS2 site (Fig. 4k), mirroring the effect seen with IFNγ treatment (Fig. 4k). These findings confirm that STAT1 directly binds the SLC7A11 promoter and indicate that LDHB modulates the STAT1/GAS interaction. Collectively, these results suggest that LDHB KD downregulates SLC7A11 expression via the upregulation of STAT1.
LDHB/SLC7A11 inhibition induces ferroptosis by activating glutamine metabolism
To elucidate the metabolic process underlying ferroptosis upon LDHB/SLC7A11 inhibition, we performed metabolomic analysis and found that erastin inhibits glutathione metabolism but activates glutamine metabolism in LDHB KD cells, evidenced by a significant decrease in cysteine (Cys), cysteinylglycine (Cys-Gly) and GSSG but an increase in intracellular glutamine and glutamate in LDHB KD A549 cells compared to control A549 cells (Fig. 5a–c; Original data file 4), in agreement with our results from LDHB KD cells (Fig. 1). Notably, erastin-treated LDHB KD A549 cells also showed a significant accumulation of γ-glutamyl-peptides such as glutamylalanine and L-glutamyltaurine (Original data file 4), in line with the recent finding that cysteine deprivation promotes the synthesis of γ-glutamyl peptides due to a non-canonical activity of the glutamate-cysteine ligase catalytic subunit (GCLC) [44].

a Volcano plot showing the metabolomics profile of A549 cells transfected for 48 h with siLDHB or siNT and further treated with erastin (5 µM) or vehicle for 20 h. b The metabolic pathways most significantly upregulated in erastin-treated LDHB KD cells compared to those in vehicle-treated control cells. c Schematic of glutamine metabolism and glutamine-fueled glutaminolysis. Inhibitors that mitigate LDHB/SLC7A11 inhibition-induced ferroptosis are highlighted in green. d, e Oxygen consumption rate (OCR) measurement d and quantification e of A549 cells transfected with siLDHB or siNT for 48 h and further treated for 20 h with DMSO or erastin (5 μM). Cell numbers normalized to 50 ng/DNA. Data represent the average of basal respiration, maximal respiration, and ATP production taken at multiple time points during the respective phases of the Seahorse assay, and are shown as mean ± s.e.m (n = 6). f, g, OCR measure f and quantification g of A549 KD cells transfected with siLDHB or siNT for 48 h and further treated for 20 h with DMSO, erastin (5 μM), CB839 (0.5 μM), BPTES (2 μM), and GPNA (50 μM), alone or in combination. Normalization was based on vehicle-treated siNT and siLDHB groups (set as 100%), respectively. Data represent the average of basal respiration, maximal respiration, and ATP production taken at multiple time points during the respective phases of the Seahorse assay, and are shown as mean ± s.e.m (n = 6). h MitoSox (mitochondrial ROS marker) quantification (left) and flow cytometry (right) analysis of A549 cells transfected and treated as above d–g; MitoROS is shown as mean fluorescence intensity (MFI) ± s.d. (n = 3), with the siNT group used for normalization. i Viability assay of A549 cells transfected with siLDHB or siNT for 48 h and further treated for 20 h with DMSO, erastin (5 μM), CB839 (0.5 μM), BPTES (2 μM), and GPNA (50 μM), alone or in combination. Normalization was based on vehicle-treated siNT and siLDHB groups (set as 100%), respectively.
Glutamine and glutamine-fueled glutaminolysis play a versatile role in cellular metabolism to provide glutamate for the tricarboxylic acid (TCA) cycle and GSH biosynthesis, thereby orchestrating mitochondrial oxidative phosphorylation (OXPHOS), a major source of mitochondrial ROS [45]. Both glutaminolysis and mitoROS have been shown to be crucial for the execution of ferroptosis [46]. Accordingly, we analyzed the real-time oxygen consumption rate (OCR) to assess mitochondrial OXPHOS activity. As expected, LDHB KD alone significantly decreased OCR, consistent with LDHB’s well-known role in mitochondrial function [13, 15, 20]. However, in erastin-treated LDHB KD cells, we observed a significant increase in OCR compared to the siLDHB group (Fig. 5d, e). Notably, pharmacological inhibition of GLS (CB839; BPTES) or SLC1A5 (GPNA), which suppresses glutamine uptake and subsequent glutaminolysis (Fig. 5c), significantly abrogated the erastin-induced increase in OCR (Fig. 5f, g) and mitoROS (Fig. 5h) in LDHB KD cells, but not in control A549 cells (Fig. S5a, b). Importantly, this decrease in OCR and mitoROS was accompanied by an attenuated toxicity of erastin, as measured by a significant rescue of cell viability suppression in erastin-treated LDHB KD cells but not in erastin-treated control A549 cells (Fig. 5i), although the inhibitors alone had no or only a mild effect on the viability of LDHB KD or control A549 cells (Fig. 5i). Thus, the erastin-induced increase in OCR and mitoROS in LDHB KD cells is driven by enhanced glutaminolysis, and these metabolic alterations are directly linked to ferroptotic cell death.
In summary, our results suggest that the inhibition of LDHB and SLC7A11 induces ferroptosis-dependent metabolic synthetic lethality by activating glutamine metabolism. This metabolic shift fuels mitochondrial OXPHOS and increases mitoROS production, creating a heightened dependency on antioxidant defenses. However, due to limited GSH availability, this defense mechanism fails, ultimately resulting in ferroptotic cell death.
In vivo effects of LDHB/SLC7A11 inhibition in KRAS-driven NSCLC
We validated our in vitro results in KRAS-mutant NSCLC xenografts and in a genetically engineered mouse (GEM) model of KrasG12D-induced NSCLC model, which closely resembles the human disease [10]. In A549 and H460 xenografts, erastin (30 mg/kg) and SSZ (150 mg/kg) significantly and consistently suppressed the growth of LDHB KD tumors but not control tumors, despite no apparent toxicities (Fig. 6a, b; Fig. S6a). Notably, erastin upregulated the lipid peroxidation marker 4-HNE, but not the apoptotic marker caspase-3, in residual LDHB KD tumors, supporting that the in vivo effect of LDHB/SLC7A11 inhibition is driven by ferroptosis (Fig. S6b–d).

a, b Tumor development of A549 a and H460 b xenografts treated with erastin (30 mg/kg/day) and SSZ (150 mg/kg/day). c Micro-CT images of LSL-KrasG12D/WT; p53fl/fl mice and LSL-KrasG12D/WT; p53fl/fl; LDHBfl/fl mice at the indicated time points. H&E staining of lung tissue sections after 9-week treatment. Scale bar, 20 μm. d–h Lung volume d, tumor burden e, tumor number f, average tumor size g, and survival fraction h after SSZ treatment. Data are shown as the mean ± s.d. *p < 0.05; **p < 0.01; ***p < 0.001 by one-way ANOVA. NS not significant.
Similar results were observed in the GEM model, where SSZ had only mild effects on KP (KrasG12D/WT; p53fl/fl) tumors, but significantly suppressed KPL (KrasG12D/WT; p53fl/fl; Ldhb−/−) tumor growth (Fig. 6c). This was demonstrated by a substantial reduction in tumor burden, as evidenced by preserved lung volume and reduced tumor size and numbers (Fig. 6d–g). Additionally, SSZ-treated KPL mice exhibited a significantly improved survival rate compared to SSZ-treated KP mice (Fig. 6h). These in vivo results corroborate our in vitro data and suggest that LDHB/SLC7A11 inhibition is a viable strategy for targeting KRAS-driven NSCLC.
In conclusion, our in vitro and in vivo results support a model that LDHB noncanonically promotes GSH-dependent ferroptosis defense through SLC7A11. Targeting LDHB and the GSH axis induces synthetic lethality in KRAS-driven lung cancer by activating glutaminolysis, elevating mitoROS, and ultimately triggering ferroptosis (Fig. 7).

LDHB noncanonically protects KRAS-driven lung cancer from ferroptosis by promoting the SLC7A11/GSH axis. LDHB blockade is synthetic lethal with SLC7A11 inhibitors due to hyperactivation of glutamine metabolism and mitoROS-dependent ferroptosis.
Discussion
Here, we provide evidence that the glycolytic enzyme LDHB, but not LDHA, non-canonically promotes the GSH-dependent ferroptosis defense in KRAS-driven NSCLC and that inhibition of LDHB and the SLC7A11/GPX4 axis confers ferroptosis-mediated metabolic synthetic lethality.
It is widely accepted that oncogenic KRAS dysregulates metabolism to promote tumorigenesis [7, 47]. A major metabolic manifestation of KRAS-driven cancer is the abnormal ROS production [5], although the ROS surveillance mechanisms specifically co-opted by KRAS remain incompletely understood. Here, we report for the first time that LDHB is part of the antioxidant program utilized by KRAS-mutant NSCLC to protect against ferroptosis, a ROS-dependent mode of cell death driven by uncontrolled lipid peroxidation [25]. We show that LDHB silencing impairs GSH biosynthesis, which is mediated by the upregulation of STAT1, the transcription factor previously shown to negatively regulate SLC7A11 expression [40], shedding mechanistic light on the critical role of LDHB in this disease [20, 22]. Our findings are consistent with the increasingly appreciated consensus that bypass or silencing of ferroptosis is a hallmark of cancer and that the SLC7A11/GSH axis is a key anti-ferroptosis program co-opted by mutant KRAS to overcome the oxidative barrier during tumor development and progression [33, 34, 48,49,50,51].
Our finding that STAT1 plays an important role in LDHB-mediated regulation of SLC7A11 is reminiscent of the well-established mechanisms by which immune cell-derived IFNs activate the IFNα/γ-STAT pathway, leading to the suppression of SLC7A11 and promoting ferroptosis in cancer cells [40]. Notably, our previous research has shown that LDHB silencing activates INF response pathways in KRAS-mutant NSCLC cells [20], aligning with the protumor activities of IFN within cancer cells [52]. In this study, we show that STAT1 directly regulates SLC7A11 expression by binding to the GAS2 domain of its promoter, a binding event significantly enhanced by LDHB KD. These findings suggest that LDHB KD may activate IFN signaling, resulting in increased STAT1 expression or enhanced protein stability. Given LDHB’s metabolic role, particularly in lactate metabolism, another plausible mechanism involves changes in specific metabolites following LDHB KD that could modulate STAT1 transcription, translation, or post-translational modifications (PTMs). For instance, PTMs such as lactylation, known to impact gene expression, might contribute to this regulatory network [53, 54]. The non-canonical functions of metabolic enzymes, as described for LDHA, underscores the broader regulatory capacities of these enzymes beyond their conventional metabolic functions [17, 23, 24, 39]. Although further experimental validation is needed, our findings highlight a novel role for LDHB in ferroptosis defense that extends beyond its traditional enzymatic activities.
Given that KRAS mutations reprogram cancer metabolism [3, 6], contextually co-opted metabolic dependencies in KRAS-mutant cancers have been widely pursued for their therapeutic potential [10, 33, 34, 55,56,57]. Here, we identify and validate metabolic synthetic lethality through inhibition of the LDHB and GSH-dependent antioxidant program. Mechanistically, inhibition of LDHB and SLC7A11 converges on increased glutamine metabolism, glutaminolysis and OXPHOS, which abnormally upregulates mitochondrial ROS and in turn triggers ferroptosis. Our results are consistent with previous findings that mitochondria play a critical role in cysteine-deprivation-induced ferroptosis [45, 46], that mitochondrial metabolism regulates ferroptosis [58], and that LDHB regulates mitochondrial activity [15, 20], and extend the prior work by demonstrating that the SLC7A11/GSH antioxidant axis is an effector of LDHB and that LDHB/SLC7A11 inhibition induces metabolic synthetic lethality, providing a novel, ferroptosis-based strategy for the treatment of KRAS-driven NSCLC.
LDHA/B are subunits of the active LDH enzyme, traditionally recognized for their roles in ATP production and energy homeostasis in both anaerobic and aerobic glycolysis [13]. LDH is crucial for cancer progression, with both LDHA and LDHB being essential for the development of KRAS-mutant lung cancer by regulating various aspects of tumor biology [20, 21]. Surprisingly, our findings show that only LDHB, and not the closely related LDHA, promotes ferroptosis defense in KRAS-driven NSCLC, highlighting distinct roles for these isoenzymes in the disease. The functional differences between LDHA and LDHB are influenced by several biological factors, including sequence and structural variations, substrate preferences, and distinct metabolic roles. While LDHA and LDHB share sequence and structural similarities, they differ in specific amino acid residues that impact their catalytic properties. LDHA has a higher affinity for pyruvate and is optimized for reducing pyruvate to lactate, supporting anaerobic glycolysis. In contrast, LDHB preferentially catalyzes the conversion of lactate back to pyruvate, feeding into mitochondrial OXPHOS [13]. The role of LDHB in mitochondrial metabolism is particularly significant in KRAS-driven cancers, which rely heavily on oxidative metabolism and robust antioxidant systems to maintain redox balance [5,6,7,8,9]. This reliance may explain LDHB’s unique contribution to ferroptosis defense in these cancer cells [20, 45]. In addition, LDHA and LDHB differ in their subcellular localization, which could impact their non-canonical functions [13, 15, 20]. In our study, we demonstrate that LDHB supports redox homeostasis by regulating GSH metabolism, offering new mechanistic insights into its essential role in KRAS-driven NSCLC [22]. Future research should explore whether the distinct roles of LDHA and LDHB in redox regulation are conserved across other cancer types. Such investigations may reveal deeper insights into the unique functions of LDH isoenzymes beyond their traditional metabolic activities.
In conclusion, we uncover a KRAS-specific mechanism of resistance to ferroptosis. In addition, our results provide strong mechanistic support for combining inhibition of LDHB and the SLC7A11/GSH/GPX4 nexus for the treatment of KRAS-driven lung cancer.
Materials and methods
Cell culture and reagents
Human NSCLC cell lines (Table S1) were obtained from American Type Culture Collection (Manassas, VA, USA). BEAS-2B and murine KP cells (KrasG12D;Trp53−/−) were described previously [59]. Cells were cultured in RPMI-1640 medium or Medium 199 (#8758; #4540; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS (#10270-106; Life Technologies, Grand Island, NY, USA) and 1% penicillin/streptomycin solution (#P0781, Sigma-Aldrich). All cells were authenticated by DNA fingerprinting and confirmed free from mycoplasma (Microsynth, Bern, Switzerland). Inhibitors were listed in Table S2.
Cell viability, cell death and clonogenic survival assay
Cells seeded in 96-well plates (2500 cells/well) were dosed 24 h later and cell viability were determined by APH assay as previously described [10, 55]. The efficacy of drugs on cell growth was normalized to untreated control. Each data point was generated in triplicate and each experiment was done three times (n = 3). Best-fit curve was generated in GraphPad Prism [(log (inhibitor)) vs. response (-variable slope four parameters)]. Error bars are mean ± SD. Cell death was determined using SYTOX dead cell stain sampler Kit (ThermoFisher Scientific, S34862) according to the manufacturer’s instructions. Clonogenic assay was performed as we described previously [20, 59].
Gene silencing by small interfering RNA (siRNA), short hairpin RNAs (shRNA) and single-guide RNAs (sgRNA)
Cells at 50-70% confluency were transfected with control or specific pooled siRNA Oligo Duplex (Origene Technologies, Rockville, MD, USA) using Lipofectamine 2000 transfection reagent (Invitrogen, Eugene, OR, USA). Stable knockdown was achieved by lentiviral shRNAs, with lentiviruses produced in HEK293T cells co-transfected with pCMV-VSV-G, pCMV-dR8.2 and shRNA constructs. All shRNAs and siRNAs used in this study are listed in Tables S3 and S4.
Drug screening
Cells transfected with siLDHB or siNT were re-seeded 24 h later into 96-well plates, treated with the indicated drugs (Table S2) for 72 h before viability assay to determine IC50 of each drug in siLDHB and siNT cells. To identify the drugs that differentially affect LDHB KD and siNT cell viability, the IC10–IC20 dose of each drug in siNT groups (80–90% viability) were used to treat LDHB KD cells. Data analysis, including the calculation of the area under the curve (AUC) was performed using GraphPad Prism 9.1.
Quantitive PCR, RNA sequencing and analysis
Total RNA was isolated and purified using RNeasy Mini Kit (Qiagen, Hilden, Germany). Complementary DNA was synthesized by the High capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) per manufacturer’s instructions. Real-time PCR (RT-PCR) was performed on a 7500 Fast RealTime PCR System (Applied Biosystems) using TaqMan primer/probes (Table S5). Normalization was based on the ΔΔCT method.
RNA sequencing was performed as previously described [20]. Briefly, sequencing libraries were made using an Illumina TruSeq Stranded mRNA Library Prep kit (#20020595; Illumina) combined with TruSeq RNA UD Indexes (#20022371; Illumina). Pooled cDNA libraries were sequenced paired-end using an Illumina NovaSeq 6000 SP Reagent Kit (#20028401, 100 cycles; Illumina) on an Illumina NovaSeq 6000 instrument. The quality of the sequencing run was assessed using Illumina Sequencing Analysis Viewer (Illumina version 2.4.7) and all base call files were demultiplexed and converted into FASTQ files using Illumina bcl2fastq conversion software v2.20. Pathway enrichment analysis was performed using Metascape (RRID: SCR_016620), and the Gene Set Enrichment Analysis (GSEA) using GSEA software (SeqGSEA, RRID: SCR_005724).
Immunoblotting, immunohistochemistry and immunofluorescence
Western blot analysis were performed as described [10, 55]. In brief, proteins resolved by SDS-PAGE (#4561033; Bio-Rad Laboratories, Hercules, CA, USA) were transferred onto nitrocellulose membranes (#170-4158; Bio-Rad), which was blocked by blocking buffer (#927-4000; Li-COR Biosciences, Bad Homburg, Germany), incubated with primary antibodies (Table S6) and IRDye 680LT-conjugated goat anti-mouse IgG (#926-68020) and IRDye 800CW-conjugated goat anti-rabbit IgG (#926-32211). Membrane-bound secondary antibodies were imaged using the Odyssey infrared Imaging System (Li-COR Biosciences).
For immunofluorescence, cells grown on poly-lysine-treated coverslides were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100/PBS before incubated with primary antibodies and appropriate secondary antibodies: Alexa Fluor 647 goat anti-mouse IgG (#A21236) or Alexa Fluor 488 goat anti-Rabbit IgG (#A11034) from Invitrogen. Nuclei were counterstained with DAPI. Images were acquired using a ZEISS Axioplan 2 imaging microscope (Carl Zeiss MicroImaging, Göttingen, Germany) and processed by Adobe illustrator CC 2017 (Adobe Systems, San Jose, CA, USA).
Immunohistochemistry (IHC) was performed as described [10, 55]. In brief, formalin-fixed and paraffin-embedded (FFPE) tumors were sectioned, deparaffinized, rehydrated and stained with hematoxylin and eosin (H&E) and appropriate antibodies (Table S6) using the automated system BOND RX (Leica Biosystems, Newcastle, UK) and visualized by the Bond Polymer Refine Detection kit (Leica Biosystems) as per the manufacturer. Images were acquired by PANNORAMIC® whole slide scanners, processed by Case Viewer (3DHISTECH Ltd.) and quantified by QuPath software.
Chromatin immunoprecipitation (ChIP) Assay
Chromatin immunoprecipitation was performed using a ChIP assay kit (EMD Millipore 17-295) according to the manufacturer’s protocol. Briefly, one million cells were harvested, and histone DNA was cross-linked with 1% formaldehyde in cell culture medium at 37 °C for 8 min. The formaldehyde was quenched with 125 mM glycine. The DNA of the cells was then sheared to lengths of 200–1000 base pairs using the Bioruptor® Plus sonication device. The sonicated cell supernatants were immunoprecipitated overnight with the STAT1 antibody (Cell Signaling Technology 14994S) or the IgG control antibody after being cleared with protein A agarose/salmon sperm DNA. After three washes of the protein A-agarose/antibody/histone complex, the histone-DNA crosslinks were eluted and recovered by heating at 65 °C for 4 h. DNA was then recovered by phenol/chloroform extraction and ethanol precipitation. Finally, the DNA was analyzed by RT-PCR using specific primers (Table S5).
Liquid chromatography–mass spectrometry (LC-MS) and metabolomics data analysis
Cells seeded in six-well plates were treated with indicated drugs. Cells were then washed twice with PBS and a solvent (75 mM ammonium carbonate, pH 7.4) and pre-cooled (−20 °C) extraction solvent (40% acetonitrile, 40% methanol, 20% nanopure water) was immediately added to the plates. Cells were then scraped from the dish on the ice, vortexed for 30 s and immediately stocked at –20 °C for 1 h and then at –80 °C. LC–MS measurement and analysis were described previously [60], with metabolomics data analysis with MetaboAnalyst 5.0 software.
OCR measurements
Cells seeded in Seahorse XF96 V3PS cell culture microplates (Agilent Technologies, 101085-004) were treated, washed twice with PBS and resuspended in Seahorse XF DMEM or RPMI medium (Agilent Technologies, 103680-100, 103681-100), containing 10 mM glucose, 0.5 mM pyruvate, and 2 mM glutamine, with the pH adjusted to 7.4, incubated in a CO2-free incubator for 1 h before successively adding 1 μM oligomycin, 1.0 μM and 1.5 μM FCCP, a mixture of 1 μM rotenone and 1 μM antimycin. The data were analyzed using Seahorse Wave (Agilent Technologies). Cell numbers quantification were normalized to 50 ng DNA, which is quantified by CyQUANT™ Cell Proliferation Assay kit (Thermo Fisher Scientific, C7026) according to the manufacturer’s protocol.
Lipid peroxidation and MitoSOX measurements
Cells in 6-well plates (0.1–0.3 ×106) or 12-well plates (0.05–0.2 ×106) were mixed with 2.5 μM C11_BODIPY (#D3861, Invitrogen) or 5 μM MitoSOX™ Red Mitochondrial Superoxide Indicator (#M36008, ThermoFisher Scientific) followed by flow cytometry (FC) analysis using FACS LSRII instrument (BD Biosciences) to determine lipid peroxidation and mitoROS, respectively. FlowJo V10 was used for data analysis.
In vivo mouse study
Mouse studies were conducted in accordance with Institutional Animal Care and Ethical Committee-approved animal guidelines and protocols. Xenograft tumors of A549, H838 and H460 cells expressing shLDHB or shNT in NSG (NOD-scid IL2Rγnull) mice were generated and treated as described (31,32), with tumor size calculated as follows: (length × width2)/2. KrasLSL-G12D/WT;p53flox/flox; Ldhb+/+ (KP) and KrasLSL-G12D/WT;p53flox/flox; Ldhb−/− (KPL) mice were described previously [10, 20]. MicroCT images were processed and analyzed using Fiji and 3D Slicer (version 4.13) as we previously described [20]. The sample size was chosen based on standard practice in similar studies to ensure sufficient animals or samples per group for reliable results. Animals were randomly assigned to experimental groups to minimize bias. Tumor induction, drug administration, and outcome assessments (mouse scores, survival monitoring, CT imaging, and tumor measurements) were performed by different investigators to ensure blinding.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.01 (GraphPad Software Inc., San Diego, CA, USA) unless otherwise indicated. In all studies, data represent biological replicates (n) and are depicted as mean values ± SD or mean values ± SEM as indicated. In all analyses, P values less than 0.05 were considered statistically significant.
Data availability
The RNA sequencing data have been deposited in GEO under the accession number GSE224098. The metabolomics data are available within the Source Data File. All other data are available from the corresponding authors upon request.
References
Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res. 2020;80:2969–74.
Punekar SR, Velcheti V, Neel BG, Wong KK. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. 2022;19:637–55.
Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–3.
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–9.
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–93.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9.
Mukhopadhyay S, Vander Heiden MG, McCormick F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat Cancer. 2021;2:271–83.
Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra215.
Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci USA. 2011;108:8773–8.
Yang Z, Liang SQ, Saliakoura M, Yang H, Vassella E, Konstantinidou G, et al. Synergistic effects of FGFR1 and PLK1 inhibitors target a metabolic liability in KRAS-mutant cancer. EMBO Mol Med. 2021;13:e13193.
Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
Cheng A, Zhang P, Wang B, Yang D, Duan X, Jiang Y, et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat Commun. 2019;10:5566.
Claps G, Faouzi S, Quidville V, Chehade F, Shen S, Vagner S, et al. The multiple roles of LDH in cancer. Nat Rev Clin Oncol 2022;19:749-762
Zdralevic M, Brand A, Di Ianni L, Dettmer K, Reinders J, Singer K, et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J Biol Chem. 2018;293:15947–61.
Chen YJ, Mahieu NG, Huang X, Singh M, Crawford PA, Johnson SL, et al. Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol. 2016;12:937–43.
Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685–92.
Urbanska K, Orzechowski A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int J Mol Sci 2019;20:2085.
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010;107:2037–42.
Zha X, Wang F, Wang Y, He S, Jing Y, Wu X, et al. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. 2011;71:13–18.
Deng H, Gao Y, Trappetti V, Hertig D, Karatkevich D, Losmanova T, et al. Targeting lactate dehydrogenase B-dependent mitochondrial metabolism affects tumor initiating cells and inhibits tumorigenesis of non-small cell lung cancer by inducing mtDNA damage. Cell Mol Life Sci. 2022;79:445.
Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase-a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014;19:795–809.
McCleland ML, Adler AS, Deming L, Cosino E, Lee L, Blackwood EM, et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res. 2013;19:773–84.
Liu J, Zhang C, Zhang T, Chang CY, Wang J, Bazile L, et al. Metabolic enzyme LDHA activates Rac1 GTPase as a noncanonical mechanism to promote cancer. Nat Metab. 2022;4:1830–46.
Brisson L, Banski P, Sboarina M, Dethier C, Danhier P, Fontenille MJ, et al. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell. 2016;30:418–31.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137–47.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620.
Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920–37.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Hu K, Li K, Lv J, Feng J, Chen J, Wu H, et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest. 2020;130:1752–66.
Zdralevic M, Vucetic M, Daher B, Marchiq I, Parks SK, Pouyssegur J. Disrupting the ‘Warburg effect’ re-routes cancer cells to OXPHOS offering a vulnerability point via ‘ferroptosis’-induced cell death. Adv Biol Regul. 2018;68:55–63.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Chen Y, Wu G, Li M, Hesse M, Ma Y, Chen W, et al. LDHA-mediated metabolic reprogramming promoted cardiomyocyte proliferation by alleviating ROS and inducing M2 macrophage polarization. Redox Biol. 2022;56:102446.
Nilsson LM, Forshell TZ, Rimpi S, Kreutzer C, Pretsch W, Bornkamm GW, et al. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012;8:e1002573.
Pan C, Li B, Simon MC. Moonlighting functions of metabolic enzymes and metabolites in cancer. Mol Cell. 2021;81:3760–74.
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Wang W, Kryczek I, Dostál L, Lin H, Tan L, Zhao L, et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. 2016;165:1092–105.
Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 2021;33:174–189.e177.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354–363.e353.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
Kimmelman AC. Metabolic dependencies in RAS-driven cancers. Clin Cancer Res. 2015;21:1828–34.
Bartolacci C, Andreani C, Vale G, Berto S, Melegari M, Crouch AC, et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat Commun. 2022;13:4327.
Müller F, Lim JKM, Bebber CM, Seidel E, Tishina S, Dahlhaus A, et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2022;30:442-456
Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci USA. 2019;116:9433–42.
Yang H, Zhao L, Gao Y, Yao F, Marti TM, Schmid RA, et al. Pharmacotranscriptomic analysis reveals novel drugs and gene networks regulating ferroptosis in cancer. Cancers. 2020;12:3273
Cheon H, Wang Y, Wightman SM, Jackson MW, Stark GR. How cancer cells make and respond to interferon-I. Trends Cancer. 2023;9:83–92.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–80.
Chen Y, Wu J, Zhai L, Zhang T, Yin H, Gao H, et al. Metabolic regulation of homologous recombination repair by MRE11 lactylation. Cell 2023;187:294–311
Yang Z, Liang SQ, Zhao L, Yang H, Marti TM, Hegedus B, et al. Metabolic synthetic lethality by targeting NOP56 and mTOR in KRAS-mutant lung cancer. J Exp Clin Cancer Res. 2022;41:25.
Koundinya M, Sudhalter J, Courjaud A, Lionne B, Touyer G, Bonnet L, et al. Dependence on the pyrimidine biosynthetic enzyme DHODH Is a synthetic lethal vulnerability in mutant KRAS-driven cancers. Cell Chem Biol. 2018;25:705–717.e711.
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–89.
Yao X, Li W, Fang D, Xiao C, Wu X, Li M, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci. 2021;8:e2100997.
Liang SQ, Buhrer ED, Berezowska S, Marti TM, Xu D, Froment L, et al. mTOR mediates a mechanism of resistance to chemotherapy and defines a rational combination strategy to treat KRAS-mutant lung cancer. Oncogene. 2019;38:622–36.
Alseekh S, Aharoni A, Brotman Y, Contrepois K, D’Auria J, Ewald J, et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat Methods. 2021;18:747–56.
Acknowledgements
We thank Christelle Dubey for animal studies and technical support, the Translational Research Unit at the Institute of Tissue Medicine and Pathology, University of Bern for IHC and the Next Generation Sequencing (NGS) Platform, the Flow Cytometry and Cell Sorting Facility, the Live Cell Imaging Core Facility, the Microscopy Imaging Center, the Experimental Animal Center at University of Bern.
Funding
This work was supported by grants from the Swiss National Science Foundation (SNSF; #310030_192648 to RWP; 310030_212766 to TMM), the Swiss Cancer Research Foundation (#KFS-4851-08-2019 to RWP; KFS-5405-08-2021 to TMM), and the Stiftung zur Krebsbekämpfung (to PD), PhD fellowships from the China Scholarship Council supported JZ and HZ. Open access funding provided by University of Bern.
Author information
Authors and Affiliations
Contributions
LZ conceived the project, designed and conducted the experiments, analyzed the data and drafted the manuscript. HD generated the stable LDHB knockdown cell lines and the Ldhb-deficient mouse model and performed the revision experiments. JZ assisted with FACS experiments and supported in vivo studies. NZ performed metabolomics analyses and contributed to data interpretation. HY, GY, ZY, DX and HZ participated in in vitro and in vivo studies. GAG and RB carried out RNA-seq data analysis and provided statistical expertise. QZ and RAS supplied essential resources for the study. TMM contributed to Ldhb-deficient mouse model development and secured grant funding. PD provided critical resources and financial support. RWP conceptualized the project, supervised the study, secured funding and co-wrote the manuscript. All authors reviewed, revised, and approved the final version of the manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Mouse studies were approved by the Veterinary Office of the Canton Bern, Switzerland, and conducted in accordance with Institutional Animal Care.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, L., Deng, H., Zhang, J. et al. Lactate dehydrogenase B noncanonically promotes ferroptosis defense in KRAS-driven lung cancer. Cell Death Differ 32, 632–645 (2025). https://doi.org/10.1038/s41418-024-01427-x
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41418-024-01427-x
This article is cited by
-
ppAT drives glutamine-dependent purine biosynthesis and malignant progression in hepatocellular carcinoma
BMC Cancer (2026)
-
Oxysophoridine promotes osteoarthritis repair via GSH system activation and ROS suppression
Scientific Reports (2026)
-
Targeting metabolic-epigenetic-immune axis in cancer: molecular mechanisms and therapeutic implications
Signal Transduction and Targeted Therapy (2026)
-
The moonlighting functions of glycolytic enzymes in tumorigenesis
Cellular Oncology (2026)
-
The integration of transcriptomics and metabolomics elucidates the antitumor mechanisms of mycophenolic acid in bladder cancer cells
BMC Cancer (2025)
