
Abstract
The ductular reaction (DR) is a dynamic adaptive cellular response within the liver, triggered by various hepatic insults and characterized by an expansion of dysmorphic biliary epithelial cells and liver progenitors. This complex response presents a dual role, playing a pivotal function in liver regeneration but, paradoxically, contributing to the progression of liver diseases, depending upon specific contextual factors and signaling pathways involved. This comprehensive review aims to offer a holistic perspective on the DR, focusing into its intricate cellular and molecular mechanisms, highlighting its pathological significance, and exploring its potential therapeutic implications. An up-to-date understanding of the DR in the context of different liver injuries is provided, analyzing its contributions to liver regeneration, inflammation, fibrosis, and ultimately carcinogenesis. Moreover, the review highlights the role of multiple microenvironmental factors, including the influence of extracellular matrix, tissue mechanics and the interplay with the intricate hepatic cell ecosystem in shaping the DR’s regulation. Finally, in vitro and in vivo experimental models of the DR will be discussed, providing insights into how researchers can study and manipulate this critical cellular response. By comprehensively addressing the multifaceted nature of the DR, this review contributes to a more profound understanding of its pathophysiological role in liver diseases, thus offering potential therapeutic avenues for hepatic disorders and improving patient outcomes.
Similar content being viewed by others

Facts
-
In response to liver injury, hepatic progenitor cells become activated through a process known as the ductular reaction (DR).
-
DR plays a dual role in liver pathophysiology, supporting regeneration while also contributing to disease progression.
-
DR is regulated by various signaling pathways and microenvironmental factors, along with the composition and mechanical properties of the extracellular matrix.
-
DR is a promising therapeutic target for preventing liver disease progression.
Open questions
-
What specific factors determine whether DR plays a regenerative or pathological role in liver disease?
-
What are the precise contributions of different signaling pathways on DR, and can selective modulation of these pathways provide therapeutic benefit?
-
How do DR cells interact with different immune cell types to contribute to chronic inflammation and fibrosis in liver diseases?
-
Can we identify reliable biomarkers from DR activity for non-invasive diagnosis and monitoring of liver disease progression?
Introduction
The term Ductular reaction (DR) was coined by Hans Popper in 1957 and refers to a complex cellular response characterized by bile duct hyperplasia observed in the liver during various hepatic insults and pathological conditions [1]. The DR is a dynamic process that occurs in response to bile ducts injury or parenchymal damage, mainly when regenerative capacity of hepatocytes is impaired or overwhelmed, such as in chronic liver diseases. It serves as an adaptive mechanism aimed at restoring tissue integrity and functions through a process of progenitor-dependent epithelial regeneration [2]. This process mainly involves hepatic progenitor cells (HPC) mobilization from their quiescent state or activation of biliary epithelial cells (BEC, aka cholangiocytes) undergoing de-differentiation toward HPC and proliferation (Fig. 1). HPC were first described in rat liver in 1937 by Kinosita as cholangiocyte-like cells belonging to what was termed by Farber later in 1956 the ‘oval cell compartment’, due to the morphological feature of clusters of small ovoid cells with high nuclear:cytoplasmic ratio [3]. HPC reside in the portal areas of the liver, in close proximity to the bile ducts and canals of Hering (Fig. 1). HPC are a heterogeneous and highly plastic population of cells with stem/progenitor properties displaying the ability to proliferate, self-renew and terminally differentiate into both BEC and hepatocytes, thus acting as facultative bi-potent epithelial stem cells [4]. Furthermore, in response to injury, certain BEC and HPC may undergo senescence caused by chronic inflammation, where these metabolically active cells no longer respond to mitogenic stimuli but instead activate a pro-inflammatory response known as “senescence-associated secretory phenotype” (SASP), releasing soluble factors similar to those produced by inflammatory cells [5]. The origin of DR cells is still debated and likely depends on the nature of the liver injury. Indeed, in different pathologic settings, they have been shown to originate either from HPC; de-differentiation of adult BEC; or from hepatocyte ductular metaplasia [6] (Fig. 1). Histologically, DR is characterized by the activation, proliferation, and eventually differentiation of HPC, leading to the formation of ductular-like structures within the portal area or the liver parenchyma [7]. The DR can present in various forms, including the emergence of multiple small ducts within the portal area with variable luminal definition; the formation of multilayered ductular structures; or the presence of “cords” of BEC lacking a lumen and infiltrating the hepatic parenchyma facing the portal mesenchyme (Fig. 1). These proliferating ductules exhibit characteristics of both cholangiocytes and hepatocytes, but displaying mainly markers of BEC, such as cytokeratins (e.g., CK7, CK19) and epithelial cell adhesion molecule (EpCAM).

In response to liver injury, HPC in the canal of Hering or quiescent cholangiocytes in the portal space undergo activation, leading to the formation of reactive ductules that proliferate and eventually infiltrate the parenchyma. HPC can also derive from the liver parenchyma as a consequence of hepatocyte metaplasia characterized by acquisition of a biliary phenotype, while mature cholangiocyte can derive from peri-portal hepatocyte trans-differentiation. DR structures can take various forms, including multiple small ducts, multilayered ductular structures, or HPC cords infiltrating the hepatic parenchyma. HPC can differentiate back to cholangiocytes and hepatocytes in order to regenerate the tissue. HA hepatic artery, PV portal vein, BD bile duct.
The DR has both beneficial and detrimental effects on liver pathology, depending on the type of injury and environmental factors. Although the effective contribution of DR in liver regeneration remains largely unproved in humans, it is believed to serve as an attempt to restore liver function and promote tissue repair (Fig. 2). On the contrary, in certain chronic liver diseases such as cholangiopathies, alcoholic hepatitis, Metabolic dysfunction-Associated SteatoHepatitis (MASH), and cirrhosis, persistent and dysregulated DR has been demonstrated to contribute to inflammation, fibrogenesis, and liver dysfunction [7] (Fig. 2). For this reason, DR has recently emerged as a promising pharmacological target for chronic liver diseases, offering opportunities for therapeutic interventions aimed at preventing the progression of these conditions [8].

An overview of the opposing functions exhibited by DR cells in regeneration (left) or disease progression (right). The figure underscores the complexity of these cellular responses and emphasizes key molecular pathways governing these divergent functions. On the left side of the figure, DR cells act as agents of tissue repair and regeneration. They engage in various processes that collectively contribute to the restoration and renewal of damaged tissues. Conversely, the right side of the figure illustrates the disease-related functions of chronically-activated DR cells. In this context, DR cells are implicated in processes that exacerbate diseases. The molecular pathways associated with this function involve release of cholangiokynes from proliferating HPC cells or SASP from senescent HPC that can enhance inflammation, facilitate fibrosis, and promote liver disease progression, including cancer.
Ductular reaction in liver regeneration
In normal conditions, hepatic injury leads to the death of some hepatocytes, allowing healthy hepatocytes to proliferate and replace the lost cells. In this context, DR contribution to parenchymal regeneration is minimal. However, in cases of chronic injury, when hepatocyte proliferation is compromised due to mechanisms such as senescence, epigenetic/signaling abnormalities, or other mechanisms, BEC or HPC plastically re-enter the cell cycle, expand, and ultimately acquire biliary or hepatocyte identity, thus supporting parenchymal repair and re-establishment of liver functions (Fig. 2). In support to this hypothesis, careful lineage-tracing studies have shown that, HPC or BEC terminally differentiate into functional hepatocytes in mice with liver injury [9,10,11]. Loss of β1-integrin in hepatocytes of mice with liver damage triggers a DR, resulting in the significant emergence of BEC-derived hepatocytes that coordinate a substantial parenchymal regeneration [12]. Loss of β-catenin in hepatocytes of mice placed on the CDE diet develops severe liver injury that stimulates BEC to differentiate into hepatocytes and drive regeneration [13]. Of note, DR-derived hepatocytes are characterized by higher proliferative capability, less apoptosis, a lower proportion of highly polyploid nuclei and ultimately are able to eliminate DNA damage, making DR an interesting target for regenerative medicine [9,10,11,12,13,14,15]. In addition, HPC transplanted in mouse and human livers demonstrated regenerative capacity in vivo, thus highlighting their therapeutic potential for cell therapies [14, 16]. Moreover, BEC-derived hepatocytes upon chronic injury gain a Hnf4α+CK19+ bi-phenotypic state in periportal regions and fibrotic septa, which can also be detected in cirrhotic human livers [9].
DR can be reversed when the causative trigger is eliminated through mechanisms involving apoptosis of ductular structures leading to improvement of the disease progression. On the contrary, in the presence of chronic liver diseases, the DR transitions into a chronic state, significantly limiting its regenerative capacity for the liver parenchyma and activating a chronic wound-healing response characterized by pro-inflammatory and fibrogenic signals that promote disease progression (Fig. 2). Thus, the impact of DR on liver diseases is a double-edged sword and understanding the intricate cellular and molecular mechanisms underlying the DR during liver regeneration is essential for developing effective strategies to enhance liver regeneration in various liver diseases and conditions.
Ductular reaction in chronic liver diseases
Although DR can support the activation and differentiation of HPC and aid in the repair of liver injury in some experimental setting, its effect in chronic liver disease may not consistently yield positive outcomes, and, on the contrary, contribute to the occurrence and progression of inflammation and liver fibrosis (Fig. 2). In line with this notion, DR occurs in association with a large spectrum of human liver diseases and in numerous experimental animal models of hepatic injury and, importantly, selective ablation of DR cells prevents disease progression in mouse models of cholestasis [17, 18]. Chronic viral hepatitis, particularly hepatitis B (HBV) and hepatitis C (HCV) infections, are major causes of DR, which plays a significant role in the progression of these diseases [19, 20]. Here, the DR is activated by viral proteins from HBV and HCV and observed in areas of liver tissue where hepatocyte necrosis and inflammation occur [21]. For example, HBV X protein (HBx) and HCV core protein can activate signaling pathways involved in HPC activation and proliferation [22].
Alcohol-Associated Liver Disease (ALD) is a consequence of chronic and excessive alcohol consumption which leads to hepatocyte damage, inflammation, and oxidative stress, resulting in chronic liver injury. In response to alcohol-induced liver injury, the DR is initiated as a reparative process but progresses as a maladaptive wound-healing response which correlates with disease severity, prognosis and mortality [23,24,25].
Metabolic dysfunction-Associated Steatotic Liver Disease (MASLD) encompasses a spectrum of conditions characterized by the accumulation of fat in the liver in the absence of excessive alcohol consumption. MASH represents a more advanced stage of MASLD characterized by liver inflammation and ballooning degeneration of hepatocytes. The DR is frequently observed in MASLD and is implicated in the progression of MASLD to MASH. In this pathological context, the DR is particularly evident in parenchymal areas of liver inflammation and fibrosis and may derive from hepatocyte ductular metaplasia. Of note, correlative studies have demonstrated that DR associates with fibrosis stage in MASH patients [26,27,28,29].
Chronic cholestatic liver diseases such as Primary biliary cholangitis (PBC), Primary sclerosing cholangitis (PSC) and Biliary atresia (BA) are characterized by inflammation and damage of the bile ducts. In such cholangiopathies, the DR is an important component of the pathophysiology and has a prognostic role by predicting response to therapy and disease progression [30,31,32,33].
The DR has been recognized as a precursor lesion for hepatocellular carcinoma (HCC), cholangiocarcinoma (CCA) and mixed HCC-CCA, the most common forms of primary liver cancer [34, 35]. Lineage-tracing studies of EpCAM-expressing biliary proliferating cells, demonstrated that these cells can develop into HCC [36]. Furthermore, lineage-tracing experiments using Mdr2-KO mice revealed that HPC serve as the source of mixed HCC-CCA. In this model, chronic liver inflammation and fibrosis triggered HPC activation, and the ablation of HPC significantly reduced mixed HCC-CCA formation [37].
The involvement of HPC in intrahepatic CCA has also been shown and mechanistically linked to specific genetic mutations. Mutant IDH (Isocitrate dehydrogenase 1) proteins disrupt hepatocyte differentiation by suppressing HNF-4α, a master regulator of hepatocyte identity, thereby driving HPC proliferation. The combined action of IDH and Kras mutations promotes the expansion of HPC, the development of premalignant biliary lesions, and progression to metastatic CCA [38].
The clinical relevance of DR cells as a cell of origin for CCA is further supported by studies in PSC where the chronic activation of DR cells provides an ideal setting for the acquisition of genetic alterations or epigenetic changes and the development of neoplastic features. The progressive accumulation of genetic and epigenetic changes in DR cells can lead to the formation of dysplastic nodules, which can further progress to liver cancer. Consequently, PSC patients exhibit a significantly elevated lifetime risk of developing CCA, estimated to exceed 10%. This clinical evidence strongly links PSC, DR, and CCA, highlighting the oncogenic potential of DR cells and the critical interplay between chronic liver injury and biliary tumorigenesis [39]. Experimental models have further demonstrated the tumorigenic potential of DR cells in vitro. Organoids derived from HPC, which recapitulate key features of the DR, have been genetically modified to harbor cancer-associated mutations such as BAP1 loss and FGFR2 fusions. These engineered organoids exhibit abnormal proliferation and differentiation defects, mimicking early tumorigenic events. When transplanted orthotopically into mouse models, these organoids reliably progress to CCA, directly demonstrating the oncogenic potential of genetic alterations in DR cells [40, 41].
In conclusion, while DR plays a crucial role in liver regeneration, its persistent activation in chronic liver diseases contributes to fibrosis, inflammation, and carcinogenesis, making it both a potential therapeutic target and a key factor in disease progression (Fig. 2).
Origin and fate of ductular reaction cells in different etiologies
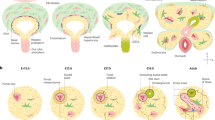
DR cellular origin and fate can diverge based on the underlying injurious stimuli and disease context. In cholestatic conditions, such as PBC, PSC, BA and bile duct obstruction, persistent damage to cholangiocytes triggers extensive ductular proliferation, which aims to restore the surface area of bile drainage. The origin of DR cells in these conditions primarily includes HPC located in the canals of Hering and cholangiocytes that re-enter the cell cycle. Histologically, these cells express biliary markers such as CK19 and CK7. The fate of DR cells in cholestatic injuries is predominantly biliary differentiation, maintaining a ductular phenotype to repair damaged biliary ducts, with no significant differentiation into hepatocytes [2] (Fig. 2).
In hepatocellular injuries, including chronic HBV, HCV, alcohol-related liver disease, or MASH, DR is less intense and DR cells originate from HPC in the periportal regions and aim to re-establish the connection between damaged hepatocytes, the destroyed canaliculi, and the biliary tree. These cells exhibit the capacity to differentiate into both cholangiocytes and hepatocytes, depending on the extent of injury and local microenvironmental cues. In experimental models of widespread hepatocyte necrosis or dysfunction, HPC-derived cells can shift toward a hepatocytic fate, replenishing damaged parenchyma. However, ongoing inflammation and fibrosis often restrict their hepatocytic potential [2] (Fig. 2).
In toxin-induced liver injuries, such as those caused by carbon tetrachloride (CCl4) or acetaminophen (APAP) in experimental models, DR cells originate from both HPC and proliferating cholangiocytes. The fate of DR cells in these models varies with the severity of the injury. While biliary differentiation is predominant, there can also be hepatocytic differentiation under conditions of severe parenchymal damage.
In ischemia/reperfusion injuries, DR cells originate mainly from HPC in the periportal regions, which become activated in response to localized necrosis. These cells generally remain confined to a biliary phenotype, as the injury is often restricted to periportal areas where biliary structures dominate. Of note, in ischemia and fatty liver disease, DR can be seen also in pericentral location, caused by hepatocyte trans-differentiation [2] (Fig. 2).
Across all these conditions, the local microenvironment plays a crucial role in shaping both the origin and fate of DR cells. These differences are evident at the molecular level, as demonstrated by a recent study analyzing the transcriptomic profiles of HPC derived from livers affected by hepatitis C virus infection and PSC, which serve as models for hepatocellular and biliary injury, respectively. This study revealed notable disparities in the expression of more than 300 genes among the DR cells in these two diseases, providing insights into how these variations affect the recruitment and localization of inflammatory cells [42].
Understanding the disease-specific properties of DR is not a trivial distinction but a critical step toward precision medicine in hepatology. However, the molecular, cellular, and inflammatory differences in DR across various liver diseases remain poorly defined, and their impact on disease progression is not fully understood. Further research in this area could help unlock new treatment paradigms, making it a high-priority open question in liver pathology.
Cellular and molecular mechanisms of ductular reaction
The DR involves intricate cellular and molecular mechanisms regulated by a network of signaling pathways and microenvironmental cues that drive the activation, proliferation, and differentiation of HPC. Notch signaling plays a crucial role in the fate determination of HPC, promoting their differentiation into cholangiocyte-like cells. DR in human BA and cholestasis induced by 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) in mice are associated with Notch signaling activation, which induces the expression of biliary markers and drives the formation of ductular structures. Moreover, inhibition of recombination signal binding protein immunoglobulin kappa J (RBP-jκ), a common DNA-binding partner of Notch receptors, reduces DR and biliary fibrosis in DDC mice [43]. Similarly, it has been found that the Notch-RBPJ signaling axis regulates biliary regeneration by coordinating the fate decision of HPC toward cholangiocytes [44] while Notch inhibition in steatohepatitis models reduced DR and cholestatic liver fibrosis [45, 46]. During biliary regeneration, expression of Jagged 1 (a Notch ligand) by myofibroblasts promoted Notch signaling in HPC and thus their biliary specification to cholangiocytes [47]. Finally, in cholangiocytes, CCN1 activated NF-κB through integrin αvβ5/αvβ3, leading to Jag1 expression, JAG1/NOTCH signaling, and proliferation [48].
The Wnt/β-catenin signaling is also implicated in promoting HPC proliferation and expansion [49]. Upon partial hepatectomy, in the absence of β-catenin a dramatic reduction in HPC number has been observed, thus demonstrating a crucial role of Wnt/β-catenin in HPC biology [50]. Wnts regulate HPC proliferation and hepatocyte-to-cholangiocyte metaplasia. Specifically, Wnt7b is induced in DR cells in DDC mice [51]. However, macrophage-derived Wnt proteins have been shown to prevent DR by opposing the Notch signaling [47, 52]. The Wnt target gene Leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5) is activated in DR cells that appear near bile ducts upon damage, characterized by robust activation of Wnt signaling. Of note, Lgr5+ cells are able to regenerate both hepatocytes and cholangiocytes in vivo [53]. However, dysregulation of Wnt/β-catenin signaling can lead to aberrant HPC expansion and contribute to liver pathologies [54], including fibrosis and HCC.
The Hedgehog (Hh) signaling is well-known to control proliferation of fetal liver progenitors and adult HPC activation, proliferation, and differentiation [55]. In MASH, ductular cells express OPN, downstream of the Hh pathway, which contributes to hepatic stellate cells (HSC) activation and fibrogenesis [56, 57]. In PBC, the Hh pathway is activated in HPC during the fibroproliferative response to chronic cholestatic liver injury [58]. Furthermore, markers of canonical Hh signaling in human cirrhosis were predominantly found to be confined to the DR cells and the Hh transducer Gli supports HPC survival [59] and regeneration during chronic injury [60].
Recent studies have shown that the Hippo pathway plays a pivotal role in the activation of HPC and is instrumental to activate a DR response. Upon liver injury, the Hippo nuclear transducers YAP/TAZ translocate into the nucleus in HPC [61,62,63,64,65] and initiate a transcriptional program associated with acquisition of progenitor features, through activating the Notch signaling [66,67,68]. Conversely, inhibition of YAP/TAZ can suppress cholangiocyte differentiation and bile duct morphogenesis and YAP/TAZ deletion in adult bile ducts causes severe defects and delay in liver regeneration.
TROP2, is expressed exclusively in HPC, establishing TROP2 as a reliable marker to distinguish DR cells from normal cholangiocytes [69]. Moreover, single-cell RNA sequencing (scRNAseq) from normal liver tissue identified a TROP2int progenitor population with strong potential to form bipotent liver organoids in vitro [70]. Recombinant hepatic growth factor (HGF) is known to promote regeneration in rodents after partial hepatectomy [71]. The HGF/c-MET axis promotes the migratory and invasive phenotype of HPC as well as DR and progenitor-mediated liver regeneration [72, 73]. Of note, c-Met stimulation promotes the trans-differentiation of hepatocytes toward biliary cells [74].
Cell metabolism plays a crucial role in DR cells, as an effect of the increased demand for energy and biosynthetic precursors to support the proliferation of HPC. Additionally, changes in cellular metabolism can influence the fate and plasticity of HPC, determining whether they differentiate into hepatocytes or cholangiocytes. In this context, a recent CRISPR screen identified the metabolic master regulator mammalian target of rapamycin (mTOR) as a crucial player in HPC growth in vitro [62]. Furthermore, lipid overload has been found to prime the reprogramming of a subset of BEC to a “progenitor” state in early stages of MASLD [75].
In addition to these well-known signaling pathways, other key proteins, including CD44 [76]; CD133(PROM1) [18]; SOX9 [77]; and OV6 [78], seem to have a prominent role in DR and are considered markers of HPC.
Overall, the cellular and molecular mechanisms of DR activation are complex and tightly regulated processes and the interplay between different signaling pathways governs the activation, proliferation, and differentiation of HPCs, ultimately shaping the DR and its implications in liver pathology (Fig. 2).
Inflammatory and fibrogenic functions of ductular reaction cells
Cholangiokines represent a group of secreted factors released by quiescent, senescent and proliferating cholangiocytes or HPC during DR and include cytokines, chemokines and growth factors (Fig. 2) [79]. When activated, HPC display a distinct secretory profile that significantly influences the surrounding microenvironment by regulating the recruitment of immune cells and the migration and activation of mesenchymal cells [80]. Cholangiokines secretion is influenced by different factors such as tissue inflammation, infection, and metabolic dysregulation and actively maintain the portal area’s microenvironment contributing to immune response and inflammation [81]. Cholangiocytes’ secretory activity functions in autocrine, paracrine, and endocrine manners to uphold biliary homeostasis and influence other cell types, including hepatocytes, HSCs, and macrophages [82], ultimately supporting different pathological states [83]. IL-6 and IL-17, secreted by reactive cholangiocytes, facilitate differentiation of T-helper (Th) cells. Histological and single cell RNA sequencing data from individuals with PSC indicate the accumulation of Th cells in the liver, specifically proximal to DR cells [84,85,86]. DR cells, in turn, release CX3CL1 and CXCL-1, which attract monocytes and T cells [87, 88]. Upon biliary injury, reactive cholangiocytes secrete TNFα and IL-6 [89]. The former activates naïve and effector T cells, while the latter regulates B cell terminal differentiation and the balance between Th17 and Treg cells. The anti-inflammatory drug dexamethasone efficiently prevents HPC activation via TNFα and IL-6 inhibition [90]. Human cholangiocytes also secrete CCL-20 and CCL-2, promoting infiltration of Th17 cells, monocytes, and dendritic cells into portal tracts [91, 92]. Moreover, substantial biliary damage triggers pronounced CX3CL1 release, attracting natural killer (NK) cells [93]. Immune factors released by DR cells, including IL-1b, IL-6, IL-23, CXCL-1/2/3/6/8, CCL-2/20, are overexpressed in PSC patients [85]. The inflammatory mediator CXCL-5 is released by DR cells and is overexpressed in patients with alcoholic hepatitis [94]. IFNγ prompts a shift in cholangiocyte cytokine secretion, transitioning from acute to chronic inflammation and decreases IL-8 secretion while elevating the release of diverse cytokines such as MCP-1, monokine, ITAC, and IP10. Additionally, both IFNγ and IL-6 stimulate nitric oxide (NO) production in cholangiocytes by upregulating nitric oxide synthase-2 (NOS-2) expression [95,96,97,98]. IFNα reduces the number of HPC upon CDE diet [99]. During Helicobacter bilis or flukes infection, cholangiocytes proliferate and substantially release IL-6 and IL-8, initiating the activation of innate mucosal immunity against microorganisms [100, 101].
Beyond the role in portal inflammation, DR cell’s secretome orchestrates dynamic interaction between reactive ductular cells and portal myofibroblast cells, known as epithelial–mesenchymal crosstalk, that directly stimulates hepatic stellate cells (HSC) activation, collagen deposition and fibrosis. Of note, previous analyses of chronically diseased human livers have demonstrated a correlation between the rate of DR and the severity of fibrosis [26, 102,103,104,105]. DR cells play a crucial role as a source of the fibrogenic connective tissue growth factor (CTGF), which is also triggered by YAP in response to tissue stiffening during fibrosis progression [106, 107]. Recent studies have unveiled a prominent role of cholangiocytes in biliary fibrosis, particularly through their communication with HSC, driven by TGFβ-mediated signaling and the epigenetic activation of a fibrogenic gene network involving H3K9 acetylation [108]. Moreover, recent data indicate that portal fibroblasts trans-differentiation into myofibroblasts, driven by the MCP-1/CCL2 axis, acts in a paracrine loop with BEC [109]. TGF-β2 expression in DR cells is evident in fibrotic liver and correlate with fibrosis activity and bile duct proliferation [110]. IL-8 levels are high in the bile of PSC patients, and stimulate cholangiocyte proliferation and expression of pro-fibrotic genes [111]. During chronic cholestasis, BEC have the capacity to produce platelet-derived growth factor-B (PDGF-B) which activates HSC leading to apoptosis susceptibility and fibrogenesis [112]. These data also demonstrated the efficacy of the BH3 mimetic A-1331852 in reducing both senescent BEC and activated stromal fibroblasts, thereby ameliorating biliary fibrosis in an experimental PSC model [113]. In a recent study Zhang et al. have described the pivotal role of NF-κB-inducing kinase (NIK) in orchestrating DR and fibrogenesis in chronic liver diseases [114]. The upregulation of biliary NIK, mediated by insults like DDC, ANIT, or BDL treatment, plays a crucial role in promoting DR, liver injury, inflammation, and fibrosis. NIK’s influence on immune cells and HSC via cholangiokines contributes to the proinflammatory and profibrotic milieu. In both BDL and Mdr2 knockout (Mdr2-/-) mouse models, exosomes enriched with lncRNA-H19 and secreted by cholangiocytes, were readily taken up by HSC, accelerating liver fibrosis. Additionally, these exosomes were found to enhance the trans-differentiation of primary mouse HSC, promoting their activation and inducing proliferation and extracellular matrix production [115]. In cholestatic liver injury, PROM1-expressing HPC promote biliary fibrosis associated with activation of myofibroblasts [18].
Integrin αvβ6, up-regulated in proliferating HPC during tissue injury, is essential for their function by activating TGFβ1 and promoting their adhesion to fibronectin [116, 117]. Inhibiting αvβ6 using the 3G9 antibody or genetic disruption not only hampers HPC activation in chronic biliary injury mouse models but also provides protection against liver fibrosis and tumorigenesis. The neuroendocrine secretin signaling promotes DR by stimulating cholangiocyte proliferation through PKA-ERK1/2 axis, thus contributing to increased cholangiocyte expansion observed in PSC patients [118]. Moreover, after liver injury, secretin produced by cholangiocytes reduces microRNA 125b and let7a levels, resulting in up-regulation of VEGF and NGF [119] and modulation of TGF-β1, which can be inhibited by secretin receptor antagonism, thus preventing fibrosis progression [120].
In conclusion, HPC orchestrates liver injury responses through diverse secretory phenotypes, influencing immune cell recruitment, inflammation, stromal activation, and fibrosis thus highlighting their crucial role in liver pathogenesis and offering potential therapeutic targets for preventing cholangiopathies and fibrosis.
Role of portal ECM in ductular reaction
The specialized niche where the HPC get activated and from which DR expands is the portal zone, which consists of a bile duct, a branch of the hepatic artery, and a branch of the portal vein, commonly referred to as “portal triad” (Figs. 1 and 3). The portal triad is structurally supported by a complex network of ECM proteins that build up a specialized matrix called the “space of Mall” that resembles that of fetal and neonatal liver, containing collagen fibers, fibronectin, proteoglycans, laminin and playing instructive roles in determining HPC fate. It is generally accepted that HPC are located close to the canals of Hering within peri-portal zone 1, a link between the hepatocyte canalicular system and the biliary tree, and this location is consistent with their bi-potential property. Spatially, proliferative cholangiocytes are mainly located in the peripheral ductules and a classification of DR based on its geometrical configuration has been proposed Desmet [121, 122]. Type 1 (typical) DR, remains confined within the portal area and originates from pre-existing cholangiocytes; type 2 (atypical) A, mostly periportal, caused by HPC or ductal metaplasia of peri-portal hepatocytes; type 2B, occurs in the parenchyma hypoxic areas; and type 3 DR, caused by activation and proliferation of HPC located in the canals of Hering in response to massive hepatocyte necrosis, which extends further into the parenchyma [123]. Clerbaux further extended this classification based on grade of invasiveness into the parenchyma: type 1 non invading DR; type 2 invades the parenchyma within approximately 80 µm beyond the portal mesenchyme; type 3 invasive DR [123]. It has been suggested that invasive DR might play a role in restoring hepatobiliary junctions and ensuring bile excretion during hepatocellular injury [122, 123].

The dynamic interplay between the ECM and DR during disease progression. On the left (healthy), the tissue under normal physiological conditions. The portal ECM is depicted with a sparse distribution of collagen and elastic fibers, adhesive peptides and proteoglycans, which mirrors its typical composition in health where BEC are in a quiescent state. In the middle (transient injury): the ECM undergoes modifications, resulting in a mild accumulation of collagen fibers and a subtle increase in stiffness. These changes in ECM properties support a vigorous DR promoting activation and marked proliferation of HPC. In the right (chronic damage): during portal fibrosis collagen accumulation increases significantly, leading to an aberrantly high level of tissue stiffness which reduces the HPC replicative potential and promotes an inflammatory phenotype.
Fibrillar collagens are abundant in the portal region and provide tensile strength to the portal space and help maintain its structural integrity (Fig. 3). The portal space provides a favorable environment for BEC to acquire an activated state in response to tissue damage, in terms of both biochemical and physical signals. In healthy livers, Type I and III collagens constitute 95% of the collagens whereas type IV, V and VI collagens constitute 1%, 2–5% and 0.1%, respectively [124]. The portal ECM is composed mainly of collagen I and mature elastic bundles with a sharp edge facing the limiting plate, the first line of hepatocytes facing the portal region. The peri-sinusoidal ECM, instead, is made mainly of collagen III with no elastic fibers [125]. Compelling data from Leclerq’s lab have elegantly demonstrated the requirement of an ECM-rich microenvironment for efficient HPC expansion and lobular invasion [126] and, in particular, collagen I and laminin seem to be required to support an efficient DR [126, 127]. Therefore, HPC are embedded in a defined fibrillar ECM matrix that functions as a scaffold to support HPC activation to drive their infiltration into the parenchyma during DR (Fig. 3) [126].
Non-collagenous glycoproteins are also abundant in the portal ECM and include laminin, entactin, fibronectin, nidogen and tenascin [128]. These ECM proteins play a crucial role in both DR onset and propagation. Signalling via integrin α6β1 is suggested to play a critical role in HPC survival and growth, possibly through interaction with laminin and fibronectin in the portal ECM and activation of the ERK1/2 and p38 MAPKs pathways [129, 130]. Moreover, the activation and spreading of DR cells into the hepatic parenchyma is closely associated with the accumulation of laminin, fibronectin and collagen during hepatic regeneration [126, 131,132,133]. However, ECM proteins can also control the fate of HPC with laminin and collagen IV promoting their self-renew, while fibronectin and collagen I supporting their quiescence/differentiation state [128, 133]. Of note, different ECM compositions across liver zones might influence HPC behavior. For example, the distinct composition of the discontinuous basement membrane at the canal of Hering and parenchyma interface, characterized by laminin-rich biliary ECM and loosely aggregated type IV collagen fibers in adjacent hepatocytes’ space of Disse, may influence the unique niche functionality in this region within the hepato-biliary linkage context [134]. CD44, a transmembrane glycoprotein family, is expressed on HPC’s surface and binds to ECM components such as the glycosaminoglycan hyaluronic acid (HA), collagen type I, fibronectin, and laminin. CD44 aids HPC proliferation and infiltration [76, 135]. Moreover, CD44v6, a Met co-receptor, enhances HPC responses to HGF, suggesting a role in HPC-HGF interactions.
Finally, proteoglycans and their attached glycosaminoglycan chains have a role in the regulation of liver ECM properties through their water retention capabilities [136] and serve as a reservoir in portal zone for various growth factors and cytokines potentially impacting the DR response.
In summary, the periportal zone and its peculiar biochemical composition are vital for HPC activation and expansion during the DR.
Regulation of ductular reaction by tissue mechanics
The liver is a highly complex organ with unique tissue mechanics, particularly in the portal space, that plays a critical role in regulating DR [137]. Tissue mechanics are constantly modulated by mechanical forces resulting from blood flow, ECM elasticity, and cell contractility. These biomechanical cues have been associated with the activation of specific signaling pathways (e.g., ERK, Hippo, Notch.) and are correlated with growth factor presentation [138,139,140]. Although healthy liver consists of relatively soft tissue with stiffness values lower than 1 kilopascal (kPa) in mice, local stiffness has been shown to be greater near the periportal region compared to the parenchymal and peri-central area [141], indicating that HPC are exposed to a peculiar mechanical niche able to potentially regulate their activation (Fig. 3). In addition, liver stiffness can substantially increase progressively during liver fibrosis, due to aberrant accumulation of collagen I fibers and such conditions are often associated with increased DR, suggesting that matrix remodeling can directly control HPC activation/mobilization [142, 143]. However, as the disease progresses, the liver stiffness can escalate to as high as 20 kPa, a pathological condition associated with reduced activation and proliferation of HPC. This evidence suggests that during chronic liver diseases, HPC are subjected to an increasingly rigid ECM that initially promotes their activation until a threshold elasticity is reached. Beyond this point, the abnormally stiff ECM inhibits HPC expansion, leading to detrimental effects on their regenerative capacity (Fig. 3). In line with this, Kallis et al. used the metalloprotease-resistant collagen Ia1(r/r) mouse, which has an exaggerated fibrotic response to liver injury, to investigate the link between ECM stiffness and HPC response and found that these mice displayed significantly reduced HPC response in chronic liver injuries compared to wild-type, associated with persistent collagen I and impaired laminin deposition [144]. Building on this ground, we have recently developed a standardized method to study the behavior of HPC in a defined mechanical environment, using mechanically-tunable synthetic polyethylene glycol (PEG) hydrogels. We were able to accurately model the aberrant mechanical properties of the fibrotic liver, providing evidence that, while physiological elasticity of the microenvironment is required for full HPC activation, an aberrantly stiff ECM negatively impacts liver progenitor proliferation [145]. Collectively, this evidence aligns with the notion that a physiologically elastic ECM is required for the complete induction of a DR. Studies on the molecular mechanisms by which ECM mechanics impact HPC biology suggest a key role of the nuclear mechano-transducers YAP/TAZ. YAP is essential for identity of BEC and acts as a mechanical sensor of bile canaliculi remodeling during liver regeneration [65, 146]. In response to liver injury, YAP/TAZ get activated mainly in the portal region and induce activation of quiescent biliary epithelial cells or metaplasia of hepatocytes into proliferating progenitors, required for DR-dependent liver regeneration [62, 65, 147]. Moreover, periportal hepatocytes which are close to the portal space, are specialized in the biosynthesis of lipids, known activators of YAP/TAZ, thus metabolically fueling their activation [148]. The CD44/RhoA as well as the integrin/FAK/Src axis act as upstream inputs to control YAP/TAZ activation in HPC during liver regeneration [145, 149]. Importantly, increased mechano-transduction in vivo by inhibition of Capzb (capping actin protein of muscle z-line subunit beta) leads YAP activation and HPC expansion around the portal area [150]. Moreover, increased stiffness induced by collagen accumulation during MASH progression, mechanically activates TAZ in vivo leading to MASH progression to HCC [151].
In summary, the specialized mechanics of the periportal zone influence DR activation and propagation, while changes in portal tissue mechanics primarily activate HPCs through regulation of the mechano-sensitive YAP/TAZ pathway.
Role of the portal cell ecosystem in ductular reaction activation
The portal space can be considered a complex cell ecosystem, where various cell types communicate, interact, and influence each other’s functions. The DR involves not only activation of HPC but also dynamic interactions with other cell types, including hepatocytes, hepatic stellate cells/myofibroblasts, portal fibroblasts, immune cells, and endothelial cells. In human, a 50% loss of hepatocytes is required for robust DR activation [152] and injured hepatocytes play a key role in supporting the DR, releasing paracrine factors that influence HPC activation and differentiation, such as Hh ligands and HGF [153,154,155], thus driving cholangiocyte activation and hepatic regeneration (Fig. 4) [72]. HSC, located in the space of Disse and responsible for ECM biosynthesis, maintain a quiescent phenotype in normal liver but they can get activated in response to multiple hepatic injuries. HSC are actively involved in DR regulation mainly controlling ECM deposition, thus creating a mechano-chemical niche for HPC activation and modulating their response to injury. HPC, in turn, can influence HSC activation through paracrine signaling, establishing bidirectional crosstalk between these cell populations. Of note, the number of HSC is associated with the DR stage [28]. In addition, HSC promote differentiation of HPC through inducing the Notch and Hh pathways, thus leading to the regeneration of bile ducts. In a recent study, Cordero-Espinoza et al. revealed that a specific group of SCA1+ periportal mesenchymal cells plays a dual influence on DR. The authors found that BEC proliferation can be either induced by soluble factors or completely abolished by contacts with mesenchymal cells, partially mediated by the Notch signaling, thus highlighting the importance of both soluble factors and physical cell-cell contacts as key regulatory cues that govern liver regeneration [156].

Upon liver injury, various cell types orchestrate the activation of biliary cells or HPC, contributing to the establishment of a ductular reaction. Neutrophils, lymphocytes, mast cells, macrophages, endothelial cells, hepatocytes, and myofibroblasts release specific signals—including cytokines, growth factors, and extracellular matrix components—that modulate HPC behavior. These interactions collectively drive HPC activation, proliferation, differentiation, and niche remodeling, ultimately shaping the DR response.
Immune cells, such as macrophages and lymphocytes, are integral components of the liver microenvironment and inflammation during liver injury can significantly impact the DR. Immune cells release cytokines and growth factors that can promote or inhibit HPC activation and differentiation, shaping the outcome of the DR and liver regeneration. Infiltrated portal macrophages induce HPC proliferation in mouse liver via secretion of the TNF family member called TWEAK (TNF-like weak inducer of apoptosis), acting as a selective HPC mitogen through its receptor Fn14 expressed in HPC. Moreover, Fn14 knockout mice exposed to CDE diet showed reduced DR in a NFkB-dependent manner [157,158,159,160]. Macrophages were found to promote HPC differentiation into hepatocytes in the DDC diet mouse model, and the Wnt/β-catenin pathway was the key mechanism in this process. After phagocytosis of the hepatocyte debris, macrophages increase the expression and secretion of Wnt3a, activating the β-catenin pathway in HPC and inducing differentiation into hepatocytes [47, 161]. In MASLD patients, macrophages increase significantly in the DR area, and macrophage infiltration correlates with fibrosis stage, indicating the potential role of the DR-associated macrophages in disease progression [162]. Kupffer cells, the hepatic resident macrophages, have been found to modulate the invasive behavior of DR cells, through modulation of the density of extracellular matrix as well as via Notch and Wnt signaling pathways [47, 163].
Recent studies suggest that the development of chronic liver diseases is influenced by the interaction of DR cells and mast cells (MC). MC can promote MASLD progression by activating Kupffer cells and HSC through histamine release or by direct secretion of TGF-β1. Inhibiting MC has been shown to effectively reduce DR and hepatobiliary damage [164,165,166,167]. FXR signaling in MC has been linked to liver injury and DR in cholestasis models [168]. However, the exact mechanism of how MC influence HPC activation remains unclear.
In alcoholic hepatitis, cells involved in DR exhibit an inflammatory profile actively attracting neutrophils mainly via the inflammatory mediator CXCL5 and very recent research unveiled a functional role of DR-associated neutrophils (DRANs) in chronic liver diseases, contributing to maladaptive tissue healing through elastase secretion [25, 94].
The role of lymphocytes in DR is poorly understood. Genetic experiments using immunodeficient mice and adoptive transfer of T cells point to a major role for natural killer (NK) and T cells in HPC expansion. In wild-type mice, HPC proliferation is accompanied by an intrahepatic inflammatory response, with lymphocytes producing TH1 proinflammatory cytokines that stimulate a DR through local secretion of IFN-γ and TNF-α [169]. During biliary injury, Hh pathway activation has been found to induce cholangiocyte production of chemokines that recruit NK cells to portal tracts [153, 170]. Finally, biliary proliferation is controlled by lymphocytes through IL-33 and IL-17 [171, 172].
DR often involves increased surrounding vascular structures, with DR cells expressing angiogenesis-related genes via Slit2-Robo1 and Endothelin pathways, in liver injury mouse models [173,174,175]. In this context, bile duct ligation leads to an expansion of the peribiliary plexus to support the increased biliary volume and nutrient supply. Notably, PBC patients show correlation between DR-induced angiogenesis and VEGF-expressing hepatic progenitor cells [176]. DR cells secrete angiogenic factors (VEGF-A, PDGF-B, TGF-b2, endothelin-1), often in synergy with portal myofibroblast-produced VEGF-A, driving biliary repair remodeling. A recent study has identified a therapeutic role for VEGFA in promoting progenitor-to-hepatocyte conversion in acute and chronic liver diseases [177]. These findings imply HPC-endothelial cell communication mediated by autocrine and paracrine VEGF effects [175].
Thus, understanding the intricate interaction between HPC and the liver cell ecosystem is crucial to deciphering DR mechanisms and their impact on regeneration and disease (Fig. 4).
In vivo and in vitro models to study DR
In vivo and in vitro models of DR provide valuable tools for studying the behavior of HPC and investigating the cellular and molecular processes involved in this pathological response and allow to assess the functional consequences of DR on liver fibrosis, carcinogenesis, and overall liver function.
The CDE model utilizes a choline-deficient diet and ethionine to induce liver damage and promote DR and HPC expansion. After three weeks of CDE feeding, the liver shows signs of steatosis, hepatocellular injury, and DR cells expansion in the portal area and gradually invading the parenchyma. These DR cells exhibit the capacity to differentiate into hepatocytes, although only a small proportion of hepatocytes are derived from DR in this model. In the CDE model, DR cells have only a vague or no lumen, and comprise small elongated cells with little cytoplasm extending in the periportal parenchyma and associated with dense collagen fibers recapitulating the HPC activation typical of MASLD and hepatitis [178]. The DDC model involves a diet enriched with 3,5-diethoxycarbonyl-1,4-dihydrocollidine, causing accumulation of protoporphyrin and biliary damage. After 3 weeks of DDC feeding, dysmorphic cholangiocytes proliferate in the portal area, leading to the expansion of DR within the portal mesenchyme. The DR pattern observed in DDC livers resembles that of PSC and PBC, where DR proliferation is abundant within the portal area and is accompanied by periportal fibrosis.
Another commonly used in vivo model of the DR is the bile duct ligation (BDL) model, where obstruction of the bile duct leads to cholestasis, inducing strong proliferation of cholangiocytes and HPC, resulting in extensive DR, portal inflammation and rapid establishment of biliary fibrosis [179]. This model allows for the study of HPC activation and differentiation in the context of portal injury and fibrosis. The genetically-modified Mdr2 knockout mouse model is widely utilized to study DR. In this model, the Mdr2 gene, which encodes for a phospholipid transporter essential for biliary phospholipid secretion, is knocked out. This disruption leads to impaired biliary excretion and results in the accumulation of toxic bile acids within the liver, triggering liver injury and the activation of DR. The Mdr2 knockout model provides valuable insights into the pathogenesis of liver diseases involving DR and biliary injury
The induction of acute liver damage using acetaminophen (APAP), also known as paracetamol, represents a frequently employed experimental approach for inducing acute liver injury in mice. Given that APAP prompts a DR, the similarity in mechanisms and histology of acetaminophen-induced injury between mouse and human livers renders it a valuable model for studying liver regeneration and the involvement of progenitor cells in this phenomenon [180].
In vitro models, on the other hand, involve the culture of isolated liver cells or cell lines under defined conditions. These models allow for a more controlled examination of the cellular and molecular mechanisms underlying the DR. Primary BEC or HPC can be isolated and cultured, providing a platform to investigate their behavior, differentiation potential, and responses to various stimuli [181,182,183]. Co-culture systems, where HPC are cultured with other cell types such as hepatocytes or hepatic stellate cells, can mimic the in vivo cellular environment and facilitate the study of paracrine signaling and bidirectional crosstalk. More recently, advancements in tissue engineering have led to the development of organ-on-a-chip, three-dimensional (3D) assembloids and organoid models, that better mimic the native liver tissue architecture and microenvironment. These models allow for the study of HPC behavior and differentiation in a more physiologically-relevant context.
Cholangiocyte organoids, derived from human and mouse adult liver tissue or induced pluripotent stem cells (iPSCs), offer a valuable in vitro model to study DR [53, 184,185,186]. These organoids, representing stable HPC cultures, express stem-cell genes and recapitulate the profiles of regenerative HPC in injured mouse liver. They can differentiate into functional hepatocytes in vitro and show engraftment potential in the damaged mouse and human liver upon transplantation, demonstrating their high plasticity [16, 184]. Cholangiocyte organoids from cirrhotic patients mimic liver DR and faithfully recapitulate DR-associated inflammatory phenotype [94]. Cholangiocyte organoids can repair different regions of the biliary tree when transplanted, making them a promising tool for regenerative medicine and studying DR in vitro [16]. The recent implementation of hydrogels to replicate the mechanical characteristics of the fibrotic liver microenvironment in 3D cultures, represents a significant advancement in this field [145]. This advanced model enables a more comprehensive investigation of DR in a highly relevant and realistic context, providing valuable insights into liver disease progression and potential therapeutic strategies.
Therapeutic implications
The DR is emerging as a promising therapeutic target for managing liver diseases. Manipulating this response can yield substantial benefits, including promoting liver regeneration, mitigating or preventing fibrosis, and potentially preventing liver cancer. Approaches to modulate the DR involve influencing specific signaling pathways, growth factors, and cytokines that govern HPC activation and differentiation. By targeting the underlying cellular and molecular processes, therapeutic strategies could be designed to control the activation, expansion, and differentiation of the DR. The recent studies suggesting that mature hepatocytes are the principal source of newly formed liver cells, with the DR cells contributing minimally to true parenchymal regeneration [187], together with the evidence that expanding DR cells sustain an inflammatory and profibrogenic milieu, has directed the focus of translational investigations toward mitigating or preventing the DR itself.
Several pharmacological interventions demonstrate that dampening the DR can yield tangible therapeutic benefits. In the Mdr2–/– mouse, treatment with 24-norursodeoxycholic acid (norUDCA) substantially reduces ductular proliferation and inflammation, leading to less portal fibrosis and improved liver pathology [188]. In this context, genetic and pharmacological inhibition of Farnesoid X receptor (FXR), have also shown efficacy in downregulating ductular proliferation and reducing hepatic fibrogenesis, reflecting broad antifibrotic and anti-DR effects in cholestatic contexts [189, 190]. Likewise, in the DDC-fed mouse, pharmacological Hedgehog pathway inhibition diminishes DR and collagen deposition, thereby curbing fibrosis [191]. Additionally, administration of S63845, a proapoptotic BH3-mimetic therapy, significantly reduced the DR-cell population and markers of inflammation and liver fibrosis in Mdr2–/– mice, while targeted removal of senescent cholangiocytes via AP20187 or fisetin leads to a reduction in the expression of inflammatory, fibrotic, and senescence markers associated with the disease [17, 192].
In conclusion, targeting the DR in liver diseases carries significant therapeutic potential. Modulating it, through pharmacological approaches, offers promise for regeneration, fibrosis alleviation, and cancer prevention. However, there are currently no FDA-approved therapies designed to target the DR in a highly specific manner. This highlights the need for deeper mechanistic insights into how reactive cholangiocytes expand, with the aim to identifying new vulnerabilities within the DR. Such strategies may not only improve liver architecture and function but also help preventing tumorigenesis in the chronically injured liver, such as in PSC patients.
Future perspectives and open questions
Despite significant advances in the field, there remain numerous open questions regarding the DR that call for deeper investigation. First, while it is known that various etiologies can trigger a DR, it is not fully understood whether these divergent insults give rise to fundamentally distinct DR cell populations or whether they represent different states of a common progenitor pool. Clarifying the lineage relationships and plasticity of these cells is essential, as such insights could reveal universal versus disease-specific therapeutic targets. Second, the dual nature of the DR remains a major conundrum. Determining the molecular and cellular cues that tip the DR from a beneficial, reparative role to a maladaptive, proinflammatory state is thus a central priority. In particular, dissecting the interactions between DR cells and other key players—such as hepatic stellate cells, macrophages, and diverse immune cell populations—will help illuminate the mechanisms by which the microenvironment shapes DR outcomes. Similarly, the role of the DR in carcinogenesis, particularly in creating a permissive environment for the development of HCC and CCA, raises important questions about the genetic and epigenetic changes that underlie this transition. Third, a suite of powerful new technologies now promises to bring unprecedented detail to DR research. Single-nucleus RNA sequencing (snRNA-seq) can identify rare or transitional cell states within a complex tissue, multiplex imaging can reveal the spatial organization and cell-cell interactions, and spatial transcriptomics can map gene expression to specific anatomical niches. Together, these tools may help researchers clarify spatially resolved mechanisms of distinct DR phenotypes and regulatory networks. Fourth, an important step forward to address these questions, will be the development of advanced multi-cellular in vitro models—such as organoids grown in defined hydrogels or precision-cut liver slices, that include a native ECM components and intact histological architecture. These organotypic cultures may capture critical paracrine and biomechanical signals that are lost in simpler monolayer systems, enabling more accurate dissection of DR mechanisms, as well as preclinical screening of therapeutic strategies aimed at controlling its proinflammatory and profibrogenic effects. In terms of clinical application, the potential to identify reliable biomarkers derived from DR cells, such as secreted cholangiokines, could revolutionize the non-invasive diagnosis and monitoring of liver disease progression. Finally, the biomechanical properties of the extracellular matrix and their influence on DR activation and propagation are emerging and present intriguing possibilities for therapeutic intervention, particularly in modulating mechano-transduction to support regeneration while limiting pathological effects.
A comprehensive understanding of these unresolved questions will not only enhance our knowledge of DR biology but also pave the way for precision medicine approaches that can selectively modulate DR depending on the disease context—either promoting its regenerative role in liver repair or, more realistically, inhibiting its pro-fibrotic and tumorigenic effects in chronic liver disease.
References
Popper H, Kent G, Stein R. Ductular cell reaction in the liver in hepatic injury. J Mt Sinai Hosp N. Y. 1957;24:551–6.
Jain R, Clark I. The Differential Diagnosis of Intrahepatic Ductular Reaction in Medical Liver Biopsy. Adv Anat Pathol. 2021;28:72–80.
Farber E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3’-methyl-4-dimethylaminoazobenzene. Cancer Res. 1956;16:142–8.
Köhn-Gaone J, Gogoi-Tiwari J, Ramm GA, Olynyk JK, Tirnitz-Parker JEE. The role of liver progenitor cells during liver regeneration, fibrogenesis, and carcinogenesis. American J Physiol-Gastrointest Liver Physiol. 2016;310:G143–54.
Meadows V, Baiocchi L, Kundu D, Sato K, Fuentes Y, Wu C, et al. Biliary Epithelial Senescence in Liver Disease: There Will Be SASP. Frontiers in Molecular Biosciences [Internet]. 2021 [cited 2023 Aug 18];8. https://www.frontiersin.org/articles/10.3389/fmolb.2021.803098.
Grompe M. Chapter 72 - Adult Liver Stem Cells. In: Lanza R, Atala A, editors. Handbook of Stem Cells (Second Edition) [Internet]. San Diego: Academic Press; 2013 [cited 2023 Jul 1]. p. 873–87. https://www.sciencedirect.com/science/article/pii/B978012385942600072X.
Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology. 2019;69:420–30.
Sun T, Annunziato S, Tchorz JS. Hepatic ductular reaction: a double-edged sword. Aging (Albany NY). 2019;11:9223–4.
Deng X, Zhang X, Li W, Feng RX, Li L, Yi GR, et al. Chronic Liver Injury Induces Conversion of Biliary Epithelial Cells into Hepatocytes. Cell Stem Cell. 2018;23:114–22.e3.
Manco R, Clerbaux LA, Verhulst S, Bou Nader M, Sempoux C, Ambroise J, et al. Reactive cholangiocytes differentiate into proliferative hepatocytes with efficient DNA repair in mice with chronic liver injury. Journal Hepatol. 2019;70:1180–91.
Español-Suñer R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology. 2012;143:1564–75.e7.
Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017;547:350–4.
Russell JO, Lu WY, Okabe H, Abrams M, Oertel M, Poddar M, et al. Hepatocyte-Specific β-Catenin Deletion During Severe Liver Injury Provokes Cholangiocytes to Differentiate Into Hepatocytes. Hepatology. 2019;69:742–59.
Lu WY, Bird TG, Boulter L, Tsuchiya A, Cole AM, Hay T, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol. 2015;17:971–83.
Stueck AE, Wanless IR. Hepatocyte buds derived from progenitor cells repopulate regions of parenchymal extinction in human cirrhosis. Hepatology. 2015;61:1696–707.
Sampaziotis F, Muraro D, Tysoe OC, Sawiak S, Beach TE, Godfrey EM, et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science. 2021;371:839–46.
Azad AI, Krishnan A, Troop L, Li Y, Katsumi T, Pavelko K, et al. Targeted Apoptosis of Ductular Reactive Cells Reduces Hepatic Fibrosis in a Mouse Model of Cholestasis. Hepatology. 2020;72:1013–28.
Fenlon M, Short C, Xu J, Malkoff N, Mahdi E, Hough M, et al. Prominin-1-expressing hepatic progenitor cells induce fibrogenesis in murine cholestatic liver injury. Physiological Rep. 2020;8:e14508.
Sun C, Jin XL, Xiao JC. Oval cells in hepatitis B virus-positive and hepatitis C virus-positive liver cirrhosis: histological and ultrastructural study. Histopathology. 2006;48:546–55.
Sclair SN, Fiel MI, Wu HS, Doucette J, Aloman C, Schiano TD. Increased hepatic progenitor cell response and ductular reaction in patients with severe recurrent HCV post-liver transplantation. Clin Transpl. 2016;30:722–30.
Prakoso E, Tirnitz-Parker JEE, Clouston AD, Kayali Z, Lee A, Gan EK, et al. Analysis of the intrahepatic ductular reaction and progenitor cell responses in hepatitis C virus recurrence after liver transplantation. Liver Transpl. 2014;20:1508–19.
Wang HY, Yang SL, Liang HF, Li CH. HBx Protein Promotes Oval Cell Proliferation by Up-Regulation of Cyclin D1 via Activation of the MEK/ERK and PI3K/Akt Pathways. International J Mol Sci. 2014;15:3507–18.
Sancho-Bru P, Altamirano J, Rodrigo-Torres D, Coll M, Millán C, José Lozano J, et al. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology. 2012;55:1931–41.
Dubuquoy L, Louvet A, Lassailly G, Truant S, Boleslawski E, Artru F, et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut. 2015;64:1949–60.
Ariño S, Aguilar-Bravo B, Coll M, Lee WY, Peiseler M, Cantallops-Vilà P, et al. Ductular reaction-associated neutrophils promote biliary epithelium proliferation in chronic liver disease. J Hepatol. 2023S01688278:00423-3.
Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander–Tetri BA, Bhathal PS, et al. Progressive Fibrosis in Nonalcoholic Steatohepatitis: Association With Altered Regeneration and a Ductular Reaction. Gastroenterology. 2007;133:80–90.
Skoien R, Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, et al. Heterogeneity of fibrosis patterns in non-alcoholic fatty liver disease supports the presence of multiple fibrogenic pathways. Liver Int. 2013;33:624–32.
Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014;59:1393–405.
Zhao L, Westerhoff M, Pai RK, Choi WT, Gao ZH, Hart J. Centrilobular ductular reaction correlates with fibrosis stage and fibrosis progression in non-alcoholic steatohepatitis. Mod Pathol. 2018;31:150–9.
Crosby HA, Hubscher SG, Joplin RE, Kelly DA, Strain AJ. Immunolocalization of OV-6, a putative progenitor cell marker in human fetal and diseased pediatric liver. Hepatology. 1998;28:980–5.
Kinugasa Y, Nakashima Y, Matsuo S, Shono K, Suita S, Sueishi K. Bile ductular proliferation as a prognostic factor in biliary atresia: an immunohistochemical assessment. J Pediatr Surg. 1999;34:1715–20.
Nyholm I, Sjöblom N, Pihlajoki M, Hukkinen M, Lohi J, Heikkilä P, et al. Deep learning quantification reveals a fundamental prognostic role for ductular reaction in biliary atresia. Hepatol Commun. 2023;7:e0333.
Jörs S, Jeliazkova P, Ringelhan M, Thalhammer J, Dürl S, Ferrer J, et al. Lineage fate of ductular reactions in liver injury and carcinogenesis. J Clin Invest. 2015;125:2445–57.
Cai X, Zhai J, Kaplan DE, Zhang Y, Zhou L, Chen X, et al. Background Progenitor Activation Is Associated With Recurrence After Hepatectomy of Combined Hepatocellular-Cholangiocarcinoma. Hepatology. 2012;56:1804–16.
Komuta M, Spee B, Vander Borght S, De Vos R, Verslype C, Aerts R, et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology. 2008;47:1544–56.
Matsumoto T, Takai A, Eso Y, Kinoshita K, Manabe T, Seno H, et al. Proliferating EpCAM-Positive Ductal Cells in the Inflamed Liver Give Rise to Hepatocellular Carcinoma. Cancer Res. 2017;77:6131–43.
Rosenberg N, Van Haele M, Lanton T, Brashi N, Bromberg Z, Adler H, et al. Combined hepatocellular-cholangiocarcinoma derives from liver progenitor cells and depends on senescence and IL-6 trans-signaling. J Hepatol. 2022;77:1631–41.
Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–4.
Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis - a comprehensive review. J Hepatol. 2017;67:1298–323.
Cristinziano G, Porru M, Lamberti D, Buglioni S, Rollo F, Amoreo CA, et al. FGFR2 fusion proteins drive oncogenic transformation of mouse liver organoids towards cholangiocarcinoma. J Hepatol. 2021;75:351–62.
Artegiani B, van Voorthuijsen L, Lindeboom RGH, Seinstra D, Heo I, Tapia P, et al. Probing the Tumor Suppressor Function of BAP1 in CRISPR-Engineered Human Liver Organoids. Cell Stem Cell. 2019;24:927–43.e6.
Govaere O, Cockell S, Van Haele M, Wouters J, Van Delm W, Van den Eynde K, et al. High-throughput sequencing identifies aetiology-dependent differences in ductular reaction in human chronic liver disease. J Pathol. 2019;248:66–76.
Lee SJ, Kim KH, Pak SC, Kang YN, Yoon GS, Park KK. Notch signaling affects biliary fibrosis via transcriptional regulation of RBP-jκ in an animal model of chronic liver disease. Int J Clin Exp Pathol. 2015;8:12688–97.
Lu J, Zhou Y, Hu T, Zhang H, Shen M, Cheng P, et al. Notch Signaling Coordinates Progenitor Cell-Mediated Biliary Regeneration Following Partial Hepatectomy. Sci Rep. 2016;6:22754.
Zhang X, Du G, Xu Y, Li X, Fan W, Chen J, et al. Inhibition of notch signaling pathway prevents cholestatic liver fibrosis by decreasing the differentiation of hepatic progenitor cells into cholangiocytes. Lab Investig. 2016;96:350–60.
Morell CM, Fiorotto R, Meroni M, Raizner A, Torsello B, Cadamuro M, et al. Notch signaling and progenitor/ductular reaction in steatohepatitis. PLOS ONE. 2017;12:e0187384.
Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18:572–9.
Kim KH, Chen CC, Alpini G, Lau LF. CCN1 induces hepatic ductular reaction through integrin αvβ5–mediated activation of NF-κB. J Clin Invest. 2015;125:1886–900.
Wang N, Kong R, Han W, Lu J. Wnt/β-catenin signalling controls bile duct regeneration by regulating differentiation of ductular reaction cells. J Cell Mol Med. 2020;24:14050–8.
Apte U, Thompson MD, Cui S, Liu B, Cieply B, Monga SPS. Wnt/beta-catenin signaling mediates oval cell response in rodents. Hepatology. 2008;47:288–95.
Okabe H, Yang J, Sylakowski K, Yovchev M, Miyagawa Y, Nagarajan S, et al. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology. 2016;64:1652–66.
Irvine KM, Clouston AD, Gadd VL, Miller GC, Wong WY, Melino M, et al. Deletion of Wntless in myeloid cells exacerbates liver fibrosis and the ductular reaction in chronic liver injury. Fibrogenesis Tissue Repair. 2015;8:19.
Huch M, Dorrell C, Boj SF, van Es JH, van de Wetering M, Li VSW, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–50.
Spee B, Carpino G, Schotanus BA, Katoonizadeh A, Vander Borght S, Gaudio E, et al. Characterisation of the liver progenitor cell niche in liver diseases: potential involvement of Wnt and Notch signalling. Gut. 2010;59:247–57.
Hirose Y, Itoh T, Miyajima A. Hedgehog signal activation coordinates proliferation and differentiation of fetal liver progenitor cells. Exp Cell Res. 2009;315:2648–57.
Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, et al. Osteopontin is Induced by Hedgehog Pathway Activation and Promotes Fibrosis Progression in Nonalcoholic Steatohepatitis. Hepatology. 2011;53:106–15.
X W, A L, X G, Y L, N K, R U, et al. Osteopontin induces ductular reaction contributing to liver fibrosis. Gut [Internet]. 2014 Nov [cited 2024 Oct 12];63(11). https://pubmed.ncbi.nlm.nih.gov/24496779/.
Jung Y, McCall SJ, Li YX, Diehl AM. Bile ductules and stromal cells express hedgehog ligands and/or hedgehog target genes in primary biliary cirrhosis. Hepatology. 2007;45:1091–6.
Grzelak CA, Sigglekow ND, Tirnitz-Parker JEE, Hamson EJ, Warren A, Maneck B, et al. Widespread GLI expression but limited canonical hedgehog signaling restricted to the ductular reaction in human chronic liver disease. PLOS ONE. 2017;12:e0171480.
Peng J, Li F, Wang J, Wang C, Jiang Y, Liu B, et al. Identification of a rare Gli1+ progenitor cell population contributing to liver regeneration during chronic injury. Cell Discov. 2022;8:1–18.
Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, et al. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J Hepatol. 2015;63:962–70.
Planas-Paz L, Sun T, Pikiolek M, Cochran NR, Bergling S, Orsini V, et al. YAP, but Not RSPO-LGR4/5, Signaling in Biliary Epithelial Cells Promotes a Ductular Reaction in Response to Liver Injury. Cell Stem Cell. 2019;25:39–53.e10.
Gurda GT, Zhu Q, Bai H, Pan D, Schwarz KB, Anders RA. The use of Yes-associated protein expression in the diagnosis of persistent neonatal cholestatic liver disease. Hum Pathol. 2014;45:1057–64.
Bai H, Zhang N, Xu Y, Chen Q, Khan M, Potter JJ, et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology. 2012;56:1097–107.
Pepe-Mooney BJ, Dill MT, Alemany A, Ordovas-Montanes J, Matsushita Y, Rao A, et al. Single-Cell Analysis of the Liver Epithelium Reveals Dynamic Heterogeneity and an Essential Role for YAP in Homeostasis and Regeneration. Cell Stem Cell. 2019;25:23–38.e8.
Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–38.
Verboven E, Moya IM, Sansores-Garcia L, Xie J, Hillen H, Kowalczyk W, et al. Regeneration Defects in Yap and Taz Mutant Mouse Livers Are Caused by Bile Duct Disruption and Cholestasis. Gastroenterology. 2021;160:847–62.
Molina LM, Zhu J, Li Q, Pradhan-Sundd T, Krutsenko Y, Sayed K, et al. Compensatory hepatic adaptation accompanies permanent absence of intrahepatic biliary network due to YAP1 loss in liver progenitors. Cell Rep. 2021;36:109310.
Okabe M, Tsukahara Y, Tanaka M, Suzuki K, Saito S, Kamiya Y, et al. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development. 2009;136:1951–60.
Aizarani N, Saviano A, Sagar S, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572:199–204.
Ishii T, Sato M, Sudo K, Suzuki M, Nakai H, Hishida T, et al. Hepatocyte growth factor stimulates liver regeneration and elevates blood protein level in normal and partially hepatectomized rats. J Biochem. 1995;117:1105–12.
Ishikawa T, Factor VM, Marquardt JU, Raggi C, Seo D, Kitade M, et al. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology. 2012;55:1215–26.
Suárez-Causado A, Caballero-Díaz D, Bertrán E, Roncero C, Addante A, García-Álvaro M, et al. HGF/c-Met signaling promotes liver progenitor cell migration and invasion by an epithelial-mesenchymal transition-independent, phosphatidyl inositol-3 kinase-dependent pathway in an in vitro model. Biochim Biophys Acta. 2015;1853:2453–63.
Limaye PB, Bowen WC, Orr AV, Luo J, Tseng GC, Michalopoulos GK. Mechanisms of hepatocyte growth factor-mediated and epidermal growth factor-mediated signaling in transdifferentiation of rat hepatocytes to biliary epithelium. Hepatology. 2008;47:1702–13.
Yildiz E, Alam GE, Perino A, Jalil A, Denechaud PD, Huber K, et al. Hepatic lipid overload potentiates biliary epithelial cell activation via E2Fs [Internet]. bioRxiv; 2022 [cited 2023 Feb 14]. p. 2022.07.26.501569. https://www.biorxiv.org/content/10.1101/2022.07.26.501569v1.
He Y, Wu GD, Sadahiro T, Noh SI, Wang H, Talavera D, et al. Interaction of CD44 and hyaluronic acid enhances biliary epithelial proliferation in cholestatic livers. American J Physiol-Gastrointest Liver Physiol. 2008;295:G305–12.
Kawaguchi Y. Sox9 and programming of liver and pancreatic progenitors. J Clin Investig. 2013;123:1881–6.
Roskams T, De Vos R, Van Eyken P, Myazaki H, Van Damme B, Desmet V. Hepatic OV-6 expression in human liver disease and rat experiments: evidence for hepatic progenitor cells in man. Journal Hepatol. 1998;29:455–63.
Boyer JL, Soroka CJ. Bile formation and secretion: An update. J Hepatol. 2021;75:190–201.
Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol. 2019;16:269–81.
Cai X, Tacke F, Guillot A, Liu H. Cholangiokines: undervalued modulators in the hepatic microenvironment. Front Immunol. 2023;14:1192840.
Fabris L, Spirli C, Cadamuro M, Fiorotto R, Strazzabosco M. Emerging concepts in biliary repair and fibrosis. Am J Physiol Gastrointest Liver Physiol. 2017;313:G102–16.
Guicciardi ME, Trussoni CE, LaRusso NF, Gores GJ. The Spectrum of Reactive Cholangiocytes in Primary Sclerosing Cholangitis. Hepatology. 2020;71:741–8.
Poch T, Krause J, Casar C, Liwinski T, Glau L, Kaufmann M, et al. Single-cell atlas of hepatic T cells reveals expansion of liver-resident naive-like CD4+ T cells in primary sclerosing cholangitis. J Hepatol. 2021;75:414–23.
Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol. 2009;157:261–70.
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40.
Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, et al. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41:506–16.
Mohamad Zaki NH, Shiota J, Calder AN, Keeley TM, Allen BL, Nakao K, et al. C-X-C motif chemokine ligand 1 induced by Hedgehog signaling promotes mouse extrahepatic bile duct repair after acute injury. Hepatology. 2022;76:936–50.
Yasoshima M, Kono N, Sugawara H, Katayanagi K, Harada K, Nakanuma Y. Increased expression of interleukin-6 and tumor necrosis factor-alpha in pathologic biliary epithelial cells: in situ and culture study. Lab Investig. 1998;78:89–100.
Nagy P, Kiss A, Schnur J, Thorgeirsson SS. Dexamethasone inhibits the proliferation of hepatocytes and oval cells but not bile duct cells in rat liver. Hepatology. 1998;28:423–9.
Kunzmann LK, Schoknecht T, Poch T, Henze L, Stein S, Kriz M, et al. Monocytes as Potential Mediators of Pathogen-Induced T-Helper 17 Differentiation in Patients With Primary Sclerosing Cholangitis (PSC). Hepatology. 2020;72:1310–26.
Harada K, Shimoda S, Ikeda H, Chiba M, Hsu M, Sato Y, et al. Significance of periductal Langerhans cells and biliary epithelial cell-derived macrophage inflammatory protein-3α in the pathogenesis of primary biliary cirrhosis. Liver Int. 2011;31:245–53.
Okamura A, Harada K, Nio M, Nakanuma Y. Participation of natural killer cells in the pathogenesis of bile duct lesions in biliary atresia. J Clin Pathol. 2013;66:99–108.
Aguilar-Bravo B, Rodrigo-Torres D, Ariño S, Coll M, Pose E, Blaya D, et al. Ductular Reaction Cells Display an Inflammatory Profile and Recruit Neutrophils in Alcoholic Hepatitis. Hepatology. 2019;69:2180–95.
Morland CM, Fear J, McNab G, Joplin R, Adams DH. Promotion of leukocyte transendothelial cell migration by chemokines derived from human biliary epithelial cells in vitro. Proc Assoc Am Physicians. 1997;109:372–82.
Morland CM, Fear J, Joplin R, Adams DH. Inflammatory cytokines stimulate human biliary epithelial cells to express interleukin-8 and monocyte chemotactic protein-1. Biochem Soc Trans. 1997;25:232S.
Spirlì C, Fabris L, Duner E, Fiorotto R, Ballardini G, Roskams T, et al. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology. 2003;124:737–53.
Mazzi V. Mig chemokine in primary biliary cirrhosis. Clin Ter. 2019;170:e211–5.
Lim R, Knight B, Patel K, McHutchison JG, Yeoh GC, Olynyk JK. Antiproliferative effects of interferon alpha on hepatic progenitor cells in vitro and in vivo. Hepatology. 2006;43:1074–83.
Yamashita M, Adachi T, Ono S, Yoshino K, Imamura H, Matsushima H, et al. Helicobacter bilis infection induces oxidative stress in and enhances the proliferation of human cholangiocytes. Helicobacter. 2022;27:e12908.
Ninlawan K, O’Hara SP, Splinter PL, Yongvanit P, Kaewkes S, Surapaitoon A, et al. Opisthorchis viverrini excretory/secretory products induce toll-like receptor 4 upregulation and production of interleukin 6 and 8 in cholangiocyte. Parasitol Int. 2010;59:616–21.
Xiao JC, Jin XL, Ruck P, Adam A, Kaiserling E. Hepatic progenitor cells in human liver cirrhosis: immunohistochemical, electron microscopic and immunofluorencence confocal microscopic findings. World J Gastroenterol. 2004;10:1208–11.
Lowes KN, Brennan BA, Yeoh GC, Olynyk JK. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am J Pathol. 1999;154:537–41.
Clouston AD, Powell EE, Walsh MJ, Richardson MM, Demetris AJ, Jonsson JR. Fibrosis correlates with a ductular reaction in hepatitis C: roles of impaired replication, progenitor cells and steatosis. Hepatology. 2005;41:809–18.
Roskams T, Yang SQ, Koteish A, Durnez A, DeVos R, Huang X, et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol. 2003;163:1301–11.
Pi L, Robinson PM, Jorgensen M, Oh SH, Brown AR, Weinreb PH, et al. Connective Tissue Growth Factor and Integrin αvβ6: a New Pair of Regulators Critical for Ductular Reaction and Biliary Fibrosis. Hepatology. 2015;61:678–91
Sedlaczek N, Jia JD, Bauer M, Herbst H, Ruehl M, Hahn EG, et al. Proliferating bile duct epithelial cells are a major source of connective tissue growth factor in rat biliary fibrosis. Am J Pathol. 2001;158:1239–44.
Aseem SO, Jalan-Sakrikar N, Chi C, Navarro-Corcuera A, De Assuncao TM, Hamdan FH, et al. Epigenomic Evaluation of Cholangiocyte Transforming Growth Factor-β Signaling Identifies a Selective Role for Histone 3 Lysine 9 Acetylation in Biliary Fibrosis. Gastroenterology. 2021;160:889–905.e10.
Kruglov EA, Nathanson RA, Nguyen T, Dranoff JA. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. American J Physiol-Gastrointest Liver Physiol. 2006;290:G765–71.
Milani S, Herbst H, Schuppan D, Stein H, Surrenti C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am J Pathol. 1991;139:1221–9.
Zweers SJ, Shiryaev A, Komuta M, Vesterhus M, Hov JR, Perugorria MJ, et al. Elevated interleukin-8 in bile of patients with primary sclerosing cholangitis. Liver Int. 2016;36:1370–7.
Grappone C, Pinzani M, Parola M, Pellegrini G, Caligiuri A, DeFranco R, et al. Expression of platelet-derived growth factor in newly formed cholangiocytes during experimental biliary fibrosis in rats. J Hepatol. 1999;31:100–9.
Moncsek A, Al-Suraih MS, Trussoni CE, O’Hara SP, Splinter PL, Zuber C, et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology. 2018;67:247.
Zhang Z, Zhong X, Shen H, Sheng L, Liangpunsakul S, Lok AS, et al. Biliary NIK promotes ductular reaction and liver injury and fibrosis in mice. Nat Commun. 2022;13:5111.
Liu R, Li X, Zhu W, Wang Y, Zhao D, Wang X, et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology. 2019;70:1317–35.
Peng ZW, Ikenaga N, Liu SB, Sverdlov DY, Vaid KA, Dixit R, et al. Integrin αvβ6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology. 2016;63:217–32.
Pi L, Robinson PM, Jorgensen M, Oh SH, Brown AR, Weinreb PH, et al. Connective tissue growth factor and integrin αvβ6: A new pair of regulators critical for ductular reaction and biliary fibrosis in mice. Hepatology. 2015;61:678–91.
Glaser S, Lam IP, Franchitto A, Gaudio E, Onori P, Chow BK, et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology. 2010;52:204–14.
Glaser S, Meng F, Han Y, Onori P, Chow BK, Francis H, et al. Secretin stimulates biliary cell proliferation by regulating expression of microRNA 125b and microRNA let7a in mice. Gastroenterology. 2014;146:1795–808.e12.
Wu N, Meng F, Invernizzi P, Bernuzzi F, Venter J, Standeford H, et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-β1 biliary secretion in mice. Hepatology. 2016;64:865–79.
Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications. II. Ontogenic liver growth in childhood. Virchows Arch. 2011;458:261–70.
Kamimoto K, Kaneko K, Kok CYY, Okada H, Miyajima A, Itoh T eLife. eLife Sciences Publications Limited; 2016 [cited 2023 Jul 10]. Heterogeneity and stochastic growth regulation of biliary epithelial cells dictate dynamic epithelial tissue remodeling. Available from: https://elifesciences.org/articles/15034/figures.
Clerbaux LA, Manco R, Van Hul N, Bouzin C, Sciarra A, Sempoux C, et al. Invasive Ductular Reaction Operates Hepatobiliary Junctions upon Hepatocellular Injury in Rodents and Humans. Am J Pathol. 2019;189:1569–81.
Nyström H. Extracellular matrix proteins in metastases to the liver – Composition, function and potential applications. Seminars Cancer Biol. 2021;71:134–42.
Hall A, Cotoi C, Luong TV, Watkins J, Bhathal P, Quaglia A. Collagen and elastic fibres in acute and chronic liver injury. Sci Rep. 2021;11:14569.
Van Hul NKM, Abarca-Quinones J, Sempoux C, Horsmans Y, Leclercq IA. Relation between liver progenitor cell expansion and extracellular matrix deposition in a CDE-induced murine model of chronic liver injury. Hepatology. 2009;49:1625–35.
Vestentoft PS, Jelnes P, Andersen JB, Tran TAT, Jørgensen T, Rasmussen M, et al. Molecular constituents of the extracellular matrix in rat liver mounting a hepatic progenitor cell response for tissue repair. Fibrogenesis Tissue Repair. 2013;6:21.
Zhu C, Coombe DR, Zheng MH, Yeoh GCT, Li L. Liver progenitor cell interactions with the extracellular matrix. J Tissue Eng Regen Med. 2013;7:757–66.
Suzuki A, Zheng Y, Kondo R, Kusakabe M, Takada Y, Fukao K, et al. Flow-cytometric separation and enrichment of hepatic progenitor cells in the developing mouse liver. Hepatology. 2000;32:1230–9.
Vellón L, Royo F, Matthiesen R, Torres-Fuenzalida J, Lorenti A, Parada LA. Functional blockade of α5β1 integrin induces scattering and genomic landscape remodeling of hepatic progenitor cells. BMC Cell Biol. 2010;11:81.
Zhang W, Chen XP, Zhang WG, Zhang F, Xiang S, Dong HH, et al. Hepatic non-parenchymal cells and extracellular matrix participate in oval cell-mediated liver regeneration. World J Gastroenterol. 2009;15:552–60.
Paku S, Schnur J, Nagy P, Thorgeirsson SS. Origin and structural evolution of the early proliferating oval cells in rat liver. Am J Pathol. 2001;158:1313–23.
Lorenzini S, Bird TG, Boulter L, Bellamy C, Samuel K, Aucott R, et al. Characterisation of a stereotypical cellular and extracellular adult liver progenitor cell niche in rodents and diseased human liver. Gut. 2010;59:645–54.
Theise ND, Dollé L, Kuwahara R. Low Hepatocyte Repopulation From Stem Cells: A Matter of Hepatobiliary Linkage Not Massive Production. Gastroenterology. 2013;145:253–4.
Chen Q, Kon J, Ooe H, Sasaki K, Mitaka T. Selective proliferation of rat hepatocyte progenitor cells in serum-free culture. Nat Protoc. 2007;2:1197–205.
Barkovskaya A, Buffone A, Žídek M, Weaver VM Proteoglycans as Mediators of Cancer Tissue Mechanics. Frontiers in Cell and Developmental Biology [Internet]. 2020 [cited 2023 Jul 8];8. https://www.frontiersin.org/articles/10.3389/fcell.2020.569377.
Mitten EK, Baffy G. Mechanotransduction in the pathogenesis of non-alcoholic fatty liver disease. Journal Hepatol. 2022;77:1642–56.
Rosenfeldt H, Grinnell F. Fibroblast Quiescence and the Disruption of ERK Signaling in Mechanically Unloaded Collagen Matrices*. Journal Biol Chem. 2000;275:3088–92.
Klein EA, Yin L, Kothapalli D, Castagnino P, Byfield FJ, Xu T, et al. Cell-Cycle Control by Physiological Matrix Elasticity and In Vivo Tissue Stiffening. Current Biol. 2009;19:1511–8.
Li Z, Dranoff JA, Chan EP, Uemura M, Sévigny J, Wells RG. Transforming growth factor-β and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–56.
Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, et al. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology. 2016;64:261–75.
Yeh WC, Li PC, Jeng YM, Hsu HC, Kuo PL, Li ML, et al. Elastic modulus measurements of human liver and correlation with pathology. Ultrasound Med Biol. 2002;28:467–74.
Yin M, Talwalkar JA, Glaser KJ, Manduca A, Grimm RC, Rossman PJ, et al. Assessment of Hepatic Fibrosis With Magnetic Resonance Elastography. Clinical Gastroenterol Hepatol. 2007;5:1207–1213.e2.
Kallis YN, Robson AJ, Fallowfield JA, Thomas HC, Alison MR, Wright NA, et al. Remodelling of extracellular matrix is a requirement for the hepatic progenitor cell response. Gut. 2011;60:525–33.
Sorrentino G, Rezakhani S, Yildiz E, Nuciforo S, Heim MH, Lutolf MP, et al. Mechano-modulatory synthetic niches for liver organoid derivation. Nat Commun. 2020;11:103416.
Meyer K, Morales-Navarrete H, Seifert S, Wilsch-Braeuninger M, Dahmen U, Tanaka EM, et al. Bile canaliculi remodeling activates YAP via the actin cytoskeleton during liver regeneration. Molecular Syst Biol. 2020;16:e8985.
Aloia L, McKie MA, Vernaz G, Cordero-Espinoza L, Aleksieva N, van den Ameele J, et al. Epigenetic remodelling licences adult cholangiocytes for organoid formation and liver regeneration. Nat Cell Biol. 2019;21:1321–33.
Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16:357–66.
Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45.
Pocaterra A, Santinon G, Romani P, Brian I, Dimitracopoulos A, Ghisleni A, et al. F-actin dynamics regulates mammalian organ growth and cell fate maintenance. Journal Hepatol. 2019;71:130–42.
Filliol A, Saito Y, Nair A, Dapito DH, Yu LX, Ravichandra A, et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature. 2022;610:356–65.
Katoonizadeh A, Nevens F, Verslype C, Pirenne J, Roskams T. Liver regeneration in acute severe liver impairment: a clinicopathological correlation study. Liver Int. 2006;26:1225–33.
Omenetti A, Diehl AM. Hedgehog Signaling In Cholangiocytes. Curr Opin Gastroenterol. 2011;27:268–75.
Sicklick JK, Li YX, Melhem A, Schmelzer E, Zdanowicz M, Huang J, et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am J Physiol Gastrointest Liver Physiol. 2006;290:G859–870.
Jung Y, Witek RP, Syn WK, Choi SS, Omenetti A, Premont R, et al. Signals from dying hepatocytes trigger growth of liver progenitors. Gut. 2010;59:655–65.
Cordero-Espinoza L, Dowbaj AM, Kohler TN, Strauss B, Sarlidou O, Belenguer G, et al. Dynamic cell contacts between periportal mesenchyme and ductal epithelium act as a rheostat for liver cell proliferation. Cell Stem Cell. 2021;28:1907–21.e8.
Bird TG, Lu WY, Boulter L, Gordon-Keylock S, Ridgway RA, Williams MJ, et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated TWEAK signaling. Proceedings Natl Acad Sci. 2013;110:6542–7.
Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest. 2005;115:2330–40.
Xiang S, Dong HH, Liang HF, He SQ, Zhang W, Li CH, et al. Oval Cell Response Is Attenuated by Depletion of Liver Resident Macrophages in the 2-AAF/Partial Hepatectomy Rat. PLOS ONE. 2012;7:e35180.
Tirnitz-Parker JEE, Viebahn CS, Jakubowski A S, Klopcic BR, Olynyk JK, Yeoh GCT, et al. Tumor necrosis factor–like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology. 2010;52:291–302.
Carpino G, Nobili V, Renzi A, Stefanis CD, Stronati L, Franchitto A, et al. Macrophage Activation in Pediatric Nonalcoholic Fatty Liver Disease (NAFLD) Correlates with Hepatic Progenitor Cell Response via Wnt3a Pathway. PLOS ONE. 2016;11:e0157246.
Guillot A, Winkler M, Silva Afonso M, Aggarwal A, Lopez D, Berger H, et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology. 2023;78:150.
Van Hul N, Lanthier N, Español Suñer R, Abarca Quinones J, van Rooijen N, Leclercq I. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am J Pathol. 2011;179:1839–50.
Lombardo J, Broadwater D, Collins R, Cebe K, Brady R, Harrison S. Hepatic mast cell concentration directly correlates to stage of fibrosis in NASH. Human Pathol. 2019;86:129–35.
Kennedy L, Meadows V, Sybenga A, Demieville J, Chen L, Hargrove L, et al. Mast Cells Promote Nonalcoholic Fatty Liver Disease Phenotypes and Microvesicular Steatosis in Mice Fed a Western Diet. Hepatology. 2021;74:164–82.
Kennedy L, Hargrove L, Demieville J, Bailey JM, Dar W, Polireddy K, et al. Knockout of l-Histidine Decarboxylase Prevents Cholangiocyte Damage and Hepatic Fibrosis in Mice Subjected to High-Fat Diet Feeding via Disrupted Histamine/Leptin Signaling. Am J Pathol. 2018;188:600–15.
Hargrove L, Kennedy L, Demieville J, Jones H, Meng F, DeMorrow S, et al. Bile duct ligation-induced biliary hyperplasia, hepatic injury, and fibrosis are reduced in mast cell-deficient KitW-sh mice. Hepatology. 2017;65:1991–2004.
Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125:825–38.
Strick-Marchand H, Masse GX, Weiss MC, Di Santo JP. Lymphocytes Support Oval Cell-Dependent Liver Regeneration1. Journal Immunol. 2008;181:2764–71.
Omenetti A, Syn WK, Jung Y, Francis H, Porrello A, Witek RP, et al. Repair-related activation of hedgehog signaling promotes cholangiocyte chemokine production. Hepatology. 2009;50:518–27.
Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. 2014;124:3241–51.
Guillot A, Gasmi I, Brouillet A, Ait-Ahmed Y, Calderaro J, Ruiz I, et al. Interleukins-17 and 27 promote liver regeneration by sequentially inducing progenitor cell expansion and differentiation. Hepatol Commun. 2018;2:329–43.
Coll M, Ariño S, Martínez-Sánchez C, Garcia-Pras E, Gallego J, Moles A, et al. Ductular reaction promotes intrahepatic angiogenesis through Slit2-Roundabout 1 signaling. Hepatology. 2022;75:353–68.
Owen T, Carpino G, Chen L, Kundu D, Wills P, Ekser B, et al. Endothelin Receptor-A Inhibition Decreases Ductular Reaction, Liver Fibrosis, and Angiogenesis in a Model of Cholangitis. Cellular and Molecular Gastroenterology and Hepatology [Internet]. 2023 Jun 17 [cited 2023 Jul 30]. https://www.sciencedirect.com/science/article/pii/S2352345X23001194.
Mariotti V, Fiorotto R, Cadamuro M, Fabris L, Strazzabosco M. New insights on the role of vascular endothelial growth factor in biliary pathophysiology. JHEP Rep. 2021;3:100251.
Franchitto A, Onori P, Renzi A, Carpino G, Mancinelli R, Alvaro D, et al. Expression of vascular endothelial growth factors and their receptors by hepatic progenitor cells in human liver diseases. Hepatobiliary Surg Nutr. 2013;2:68–77.
Rizvi F, Lee YR, Diaz-Aragon R, Bawa PS, So J, Florentino RM, et al. VEGFA mRNA-LNP promotes biliary epithelial cell-to-hepatocyte conversion in acute and chronic liver diseases and reverses steatosis and fibrosis. Cell Stem Cell. 2023;30:1640–1657.e8.
Clerbaux LA, Hul NV, Gouw ASH, Manco R, Español-Suñer R, Leclercq IA, et al. Relevance of the CDE and DDC Mouse Models to Study Ductular Reaction in Chronic Human Liver Diseases. In: Experimental Animal Models of Human Diseases - An Effective Therapeutic Strategy [Internet]. IntechOpen; 2017 [cited 2023 Aug 2]. https://www.intechopen.com/chapters/55853.
Mariotti V, Strazzabosco M, Fabris L, Calvisi DF. Animal models of biliary injury and altered bile acid metabolism. Biochim Biophys Acta. 2018;1864:1254–61.
Kofman AV, Morgan G, Kirschenbaum A, Osbeck J, Hussain M, Swenson S, et al. Dose- and time-dependent oval cell reaction in acetaminophen-induced murine liver injury. Hepatology. 2005;41:1252–61.
Gaudio E, Carpino G, Cardinale V, Franchitto A, Onori P, Alvaro D. New insights into liver stem cells. Dig Liver Dis. 2009;41:455–62.
Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc. 2016;11:1724–43.
Du Y, de Jong IEM, Gupta K, Waisbourd-Zinman O, Har-Zahav A, Soroka CJ, et al. Human vascularized bile duct-on-a chip: a multi-cellular micro-physiological system for studying cholestatic liver disease. Biofabrication. 2023;16:015004.
Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MMA, et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell. 2015;160:299–312.
Sampaziotis F, Justin AW, Tysoe OC, Sawiak S, Godfrey EM, Upponi SS, et al. Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids. Nat Med. 2017;23:954–63.
Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B, et al. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat Biotechnol. 2015;33:853–61.
Yanger K, Knigin D, Zong Y, Maggs L, Gu G, Akiyama H, et al. Adult Hepatocytes Are Generated by Self-Duplication Rather than Stem Cell Differentiation. Cell Stem Cell. 2014;15:340–9.
Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–81.
Dean AE, Jungwirth E, Panzitt K, Wagner M, Anakk S. Hepatic farnesoid X receptor is necessary to facilitate ductular reaction and expression of heme biosynthetic genes. Hepatol Commun. 2023;7:e0213.
Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med. 2016;375:631–43.
Omenetti A, Yang L, Li YX, McCall SJ, Jung Y, Sicklick JK, et al. Hedgehog-mediated mesenchymal–epithelial interactions modulate hepatic response to bile duct ligation. Lab Invest. 2007;87:499–514.
Alsuraih M, O’Hara SP, Woodrum JE, Pirius NE, LaRusso NF. Genetic or pharmacological reduction of cholangiocyte senescence improves inflammation and fibrosis in the Mdr2 -/- mouse. JHEP Rep. 2021;3:100250.
Acknowledgements
I would like to acknowledge B.Anfuso, D. Selvestrel and S. Velnati for fruitful discussions and suggestions. I apologize with authors whose excellent work could not be cited due to space limitations.
Funding
The author’s research is funded by grants from: AIRC (Start-Up, 24322); Telethon (GGP20031); Italian Ministry of University and Research (Progetti di rilevante interesse nazionale, PWKZXE; P2022A9J9L); Worldwide Cancer Research (24-0361); PNRR NextGenerationEU Project CN00000041-National Center for Gene Therapy and Drugs based on RNA Technology.
Author information
Authors and Affiliations
Contributions
GS wrote and revised the manuscript, and prepared figures. Drawings created with BioRender.com.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Mauro Piacentini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sorrentino, G. Microenvironmental control of the ductular reaction: balancing repair and disease progression. Cell Death Dis 16, 246 (2025). https://doi.org/10.1038/s41419-025-07590-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-025-07590-4
This article is cited by
-
Mechanisms and targeted prevention of abnormal ductular reaction caused by a low concentration of Benzo(a)pyrene
Cell Death & Disease (2025)
