
Abstract
Mitochondrial fission is a critical physiological process in eukaryotic cells, participating in various vital activities such as mitosis, mitochondria quality control, and mitophagy. Recent studies have revealed a tight connection between mitochondrial fission and the mitochondrial metabolism, as well as apoptosis, which involves multiple cellular events and interactions between organelles. As a pivotal molecule in the process of mitochondrial fission, the function of DRP1 is regulated at multiple levels, including transcription, post-translational modifications. This review follows the guidelines for Human Gene Nomenclature and will focus on DRP1, discussing its activity regulation, its role in mitochondrial fission, and the relationship between mitochondrial fission and apoptosis.
Similar content being viewed by others
Facts
-
The function of DRP1 in cells is precisely regulated at multiple levels including transcription and translation, etc.
-
Under certain stress conditions, mitochondrial fission and apoptosis often occur concomitantly.
-
DRP1 is the pivotal molecule at the crossroads between mitochondrial fission and apoptosis processes.
Open questions
-
In the process of apoptosis under varied conditions, which occurs earlier, mitochondrial fission or the change in the permeability of the mitochondrial outer membrane?
-
What are the differences in the pathophysiological processes between apoptosis induced by mitochondrial fission and that induced by changes in mitochondrial permeability?
-
Can the occurrence of apoptosis be regulated by adjusting mitochondrial fission and fusion (It may be a feasible therapy for the treatment of cancers and neurodegenerative diseases)?
Introduction
As the “power plant” of eukaryotic cells [1], mitochondria not only provide energy support for various cellular activities but also participate in multiple biosynthetic and catabolic reactions, playing a crucial role in apoptosis and other cellular events [2, 3]. Mitochondria are organelles that constantly undergo dynamic changes by the means of fission and fusion at all times [4, 5]. And the dynamic balance between fission and fusion shapes the morphological structure of mitochondria, which in turn affects mitochondrial function, adapting to various demands of cells [6]. Mitochondrial fusion is mediated by proteins Mfn1 and Mfn2 involved in outer membrane fusion, and Opa1 involved in inner membrane fusion [7, 8]. In contrast, mitochondrial fission is mediated by molecules such as DRP1, Mff, Mid49/51, and Fis1 [9, 10], among which DRP1 stands in dominant position [11, 12].
Under stress or various pathological conditions, mitochondrial fission serves as the driving force inducing mitochondria to evolve towards “fragmented” state [13, 14], which excludes damaged ones from the mitochondrial system known as mitochondria quality control (MQC), and it also affects mitochondrial metabolism and mitophagy [15, 16]. Additionally, previous studies revealed that apoptosis often occurs concurrently with mitochondrial fragmentation [17], suggesting a connection between mitochondrial fission and apoptosis. And the connection may contribute to the processes of neurodegenerative diseases and tumors [18, 19]. Research on DRP1 will help to elucidate the mechanism of multiple diseases and provide new insights for their diagnosis and therapy.
Characteristics of the DRP1 gene and molecular structure
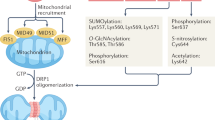
The gene encoding the DRP1 protein, DNM1L, is located at 12p11.21 and consists of 21 exons with a total length of 66,451 base pairs [20]. The encoded protein, Dynamin-related protein 1 (DRP1) has nine splice variants [21, 22]. And based on its functions, DRP1 can be divided into four domains structurally [12](Fig. 1): 1. GTPase domain: The domain is primarily responsible for binding and hydrolyzing GTP [23]; 2. Middle domain: It is mainly involved in the self-assembly and polymerization of DRP1 molecules [24, 25]; 3. Variable domain(VD): It participates in the regulation of DRP1 polymerization and influences the curvature of the DRP1 complex helix. Additionally, most of the post-translational modification sites of the DRP1 are located within the VD region [26, 27]; 4. GED domain: In the 3D-structure, this domain folds back and interacts with the GTPase domain to regulate and activate the GTPase activity of DRP1 [24, 28]. GTPase domain acts as the catalytic domain, while stalk is comprised of middle domain and GTPase effector domain (GED), which is consistent to other member of Dynamin superfamily proteins (DSPs) [12]. In addition, fission DSPs include an additional bundle signaling element (BSE) connecting the stalk and G domain [29]. What’s more, VD is the unique intervening sequence adjacent to the stalk [30].

The structure of DRP1.
Mitochondrial fission
Mitochondrial fission is a complex process involving multiple factors. Although some mechanisms remain elusive, existed studies could outline the process to us. The mechanism involves the following steps: 1. Endoplasmic reticulum (ER) constrains mitochondria. At the site of mitochondrial fission, the ER-mitochondria contact interface, the ER-associated actin regulatory factor INF2 and Spire1C work together to facilitate actin polymerization [31, 32]. Myosin II participates by binding to actin filaments, constricting mitochondria into a tubular shape at the fission site [33]. Actin-binding proteins such as Cofilin, Cortactin, and the Arp2/3 complex may also be involved in this process [34]. 2. DRP1 receptor recruits DRP1. Current DRP1 receptors on the mitochondrial surface consist of Mff, Mid49/51, and Fis1. Although it is found in in vitro that Fis1 can bind to DRP1, knockout of Fis1 has little effect on the mitochondrial fission process. However, some studies have found that Fis1 participates in the formation of a complex containing DRP1, Mff and MAMs-related proteins, and then plays role in the stress-induced mitochondrial fission process [35]. Both Mff and Mids can recruit DRP1 independently, but given the absence of Mff having the most significant impact on mitochondrial fission, which suggests that Mff is thought to play a dominant role in DRP1 recruitment to mitochondria [36]. Studies on the functions of Mff and Mids revealed that they play a synergistic effect in mitochondrial fission. Cells lacking either Mff or Mids exhibit impaired mitochondrial fission ability, while cells lacking all three are incompetent to undergo mitochondrial fission [37]. Previous research has shown that Mff can recruit activated, oligomerized DRP1, while Mids inactive, dimerized DRP1 [38]. In a liposome model, it was found that the binding of Mff to DRP1 can upregulate DRP1’s GTPase activity, while the binding of Mid51 to the opposite [39]. This depicts a model that dimerized DRP1, upon binding to Mids, whose GTPase activity is restricted, ensures that DRP1 conjuncts with unhydrolyzed GTP consistently. Once DRP1 assembly is completed, Mff, which shares the same spatial localization with Mids [40], binds to DRP1 and activates its GTPase activity, facilitating the scission action at the fission site. Other studies have also reported that the assembly and recruitment of DRP1 at the mitochondrial fission site involve phosphorylation regulation at multiple sites of the DRP1 [41] and the recruitment and activation of polymerized actin, Myosin2, and INF2 by DRP1 [42].
The regulation of DRP1 activity
The activity of the DRP1 is regulated at multiple levels and in complex manners, and the intricate regulatory network ensures the precise execution of DRP1 function temporally and spatially.
Regulation at transcriptional and translational levels
The existing body of evidence regarding Drp1 transcriptional regulation is hitherto scarce. Some studies have reported that P53 bonds to the promoter region of DNM1L to upregulate DRP1 transcription, and repressing P53 impeded mitochondrial fission and the activation of apoptosis [43, 44]. Additionally, c-Myc also upregulated DRP1 expression at the transcriptional level through miR-373-3p, a process that played a role in the pathophysiology of hepatic cancer development [45].
At the translational level, the RNA-binding protein Hu antigen R(HuR) can bind to the 3’ untranslated region of DRP1 RNA, ensuring DRP1 translation, and knocking down HuR definitely downregulate DRP1 expression and promoting mitochondrial fusion [46]. The heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) also regulates DRP1 expression by interacting with DRP1 mRNA at its 3’UTR region directly, enhancing translation process without affecting mRNA stability [47]. Inhibition of hnRNP A1 results in mitochondrial fusion, while overexpression of hnRNP A1 promotes mitochondrial fission. Further research is needed to elucidate the mechanisms of DRP1 expression at the transcriptional and translational levels.
Post-translational modifications
Recent studies have revealed that the regulation of DRP1 function is primarily achieved through post-translational modifications, among which phosphorylation is the most extensively studied one.
Phosphorylation modification
(1) The orderly temporal and spatial regulation of DRP1 function is essential for cell proliferation and development, and the phosphorylation modification of DRP1 plays crucial role in the mechanism.
It was found that during mitosis, the cyclin B1/Cdk1 complex phosphorylates DRP1 at the Ser-585 site to promote DRP1 aggregation on mitochondria membrane, maintaining a short rod-like shape of mitochondria, which facilitates their distribution to daughter cells. After mitosis, the mitochondria regain their reticular shape in daughter cells [48]. PINK1 phosphorylates DRP1 at the Ser616 site to contribute to the maturation of spinal dendrites and axons [49, 50], a critical process of neuron development. Under physiological conditions, CDK19/Cdk8 phosphorylates DRP1 at the Ser616 site to drive mitochondrial fission, maintaining the normal function of the mitochondrial system. What’s more, studies have found that CDK19/Cdk8 can rescue the DRP1 disfunction caused by PINK1 mutations, suggesting that mutations or deficiencies in CDK19/Cdk8 may be a cause of Parkinson’s disease [51]. These physiological roles are just the tip of the iceberg for DRP1 functions, and more details remain to be revealed.
(2) Under various stresses or pathological conditions, the phosphorylation status of DRP1 is a critical factor affecting mitochondrial function and is tightly related to apoptosis, which plays role in the onset and progression of multiple diseases.
Cyclin-dependent kinase (CDK) is an important enzyme in the cell cycle [52]. Current research has found that it plays a role in various diseases and pathophysiological processes by phosphorylating multiple sites on the DRP1. In the radiation-induced optic neuropathy, CDK5 phosphorylates DRP1 at the Ser616 site, promoting mitochondrial fission and affecting mitochondrial function, which is tightly associated with the progression of the disease [53,54,55]. Studies have also reported that in NMDA-induced neuron loss, CDK phosphorylates DRP1 at the Ser585 site, forcing mitochondrial fission [56], and interfering with this process can alleviate neuron death, reversing the pathophysiological process. In Alzheimer’s disease (AD) model, CDK5 also phosphorylate DRP1 at the Ser579 site, which promotes mitochondrial fission and enhances neuron sensitivity to Aβ, leading to neurodegenerative diseases [57, 58].
What’s more, varied kinases could phosphorylate specific sites on DRP1 (for instance the Ser616 site) participating in various pathophysiological processes. In the pathology of osteoarthritis (OA), the activation of TANK-binding kinase 1 (TBK1) and the following phosphorylation of DRP1 at the Ser616 site contribute to the progress of the disease [59]. In the pathological process of renal fibrosis, TGF-β-induced phosphorylation of DRP1 at the Ser616 site plays a significant role [60]. And existing research has found that in LPS-induced inflammatory responses, signal transducers and activators of transcription 2 (Stat2) phosphorylates DRP1 at the Ser616 site, promoting the accumulation of DRP1 on mitochondria, which plays an important role in macrophage differentiation [61]. Further, in various inflammatory responses involving macrophages, Protein Kinase C delta (δPKC)phosphorylates DRP1 at the Ser616 site, inducing mitochondrial fission [62]. It also found that mitogen-activated protein kinase 1 (MAPK1) phosphorylates DRP1 at the Ser616 site leaving fragmented mitochondria, which participates in the pathology of Huntington’s disease (HD) [63]. Under radiation stress, CaMKII phosphorylates DRP1 at the Ser-616 site, mediating the occurrence of mitochondrial fission, which plays a role in the apoptosis procedure of cells under radiation stress [64, 65].
Additionally, the phosphorylation of varied sites on DRP1 has been found in the progression of various diseases. IFN-β can phosphorylate and activate STAT5 that is an up-regulator of PGAM5 expression, and the following upregulation of PGAM5 promotes the dephosphorylation of DRP1 at the Ser643 site; and the dephosphorylation at the S643 site provides privilege for CaMKIIa to phosphorylate DRP1 at the Ser622 site, promoting mitochondrial fission [66], which participates in the pathological processes of multiple neurodegenerative diseases. Rho-associated, coiled-coil-containing protein kinases (ROCK1) phosphorylates DRP1 at the Ser600 site, promoting the mitochondrial fission [67], which promotes mtROS generation and apoptosis in glomerular cells, playing an important role in the pathology of microvascular lesions induced by hyperglycemia. In muscle cells, AMPK phosphorylates DRP1 at the Ser637 site, promoting mitochondrial fusion [68], which can counteract the mitochondrial fission and dysfunction under high-fat diet (HFD) treatment, and it takes part in the pathological process of fatty liver [69]. Glycogen synthase kinase 3β (GSK3β) can phosphorylate the Ser40/44 sites to promote mitochondrial fission, increasing neuron sensitivity to Aβ and promoting neuronal apoptosis [70]; however, under oxidative stress, it can phosphorylate the Ser693 site, promoting mitochondrial fusion and enhancing the stress resistant ability of cells to stress [71]. And c-Abl can phosphorylate threonine at the 266, 368, and 449 sites, driving mitochondrial fission and apoptosis [72], which contribute to the neuron loss under oxidative stress.
(3) In addition to kinases, phosphatases also play a significant role in the regulation of DRP1 activity.
ROCK1 upregulate the activity of PP1 and PP2A to dephosphorylate DRP1 at the Ser637 site, activating DRP1 activity and promoting its aggregation on mitochondria and resulting in mitochondrial fission [73,74,75]. In an MPTP-induced Parkinson’s disease (PD) model, ROCK1 also participates in the dephosphorylation of DRP1 at the Ser656 site, contributing to mitochondrial fission [76]. Phosphoglycerate mutase 5 (PGAM5) can dephosphorylate DRP1 at Ser-637 site, promoting the translocation of DRP1 to mitochondria, achieving mitochondrial fission [77], which plays an important role in the programmed necrosis. In RGCs cells, A-kinase anchoring protein 1 (AKAP1) conducts the dephosphorylation of DRP1 at the Ser637 site, promoting mitochondrial fission and participating in the pathophysiology of glaucoma [78].
In addition to the classic phosphorylation regulation of DRP1 by kinases or phosphatases, the state of DRP1 phosphorylation is also related to other types of modifications. Those modifications may alter the conformation of the DRP1, affecting the phosphorylation status of DRP1. For instance, PDI-induced S-nitrosylation of DRP1 can promote the phosphorylation of DRP1 at the Ser616 site, resulting in mitochondrial fission [79].
Under various conditions, multiple factors affect the phosphorylation status of amino acid residues at multiple sites of DRP1 through phosphorylation, dephosphorylation, or the altered conformation of DRP1, thereby regulating mitochondrial function and even affecting cell fate. These sites are distributed across different domains of the DRP1, and the Ser616 and Ser637 sites located in the GED region were extensively studied [26] (see the Table 1 below for details). However, the commonalities of these sites after phosphorylation have not been thoroughly elucidated.
SUMOylation modification
Small ubiquitin-like modifier (SUMO) is a conserved molecule that conjugates to the lysine residue of substrates in the post-translational modification (PTM) [80]. There exist five SUMO isoforms in mammalian cells, named SUMO1, 2, 3, 4, and 5, with SUMO2 and SUMO3 sharing 96% homology. Current research suggests that sumoylation can promote the stability of DRP1 attaching to the mitochondrial surface and contribute to mitochondrial fission [81, 82]. Studies revealed that SUMO ligase MAPL SUMOylates DRP1 at K532, K535, K558, K568, K594, K597, K606, and K608 sites in VD region, promoting its anchoring on mitochondria and mitochondrial fission [83, 84]. In contrast, Sentrin/SUMO-specific protease 5 (SENP5) inhibits mitochondrial fission by de-sumoylating DRP1 [85]. However, SENP3 can promote DRP1 binding to MFF and induce mitochondrial fission, cytochrome c release, and apoptosis by desumoylating DRP1 [86,87,88,89]. Additionally, different SUMO modifications on the same substrate lead to different pathophysiological effects. Inhibiting SENP3 enhances the covalent binding of DRP1 to SUMO2/3, impeding the cytochrome c release and protecting against ischemia-induced cell death [89]. Overexpressing SUMO1 can promote mitochondrial fission and stabilize DRP1 on mitochondria [90], and SENP5 can reverse the mitochondrial fission induced by SUMO1 through deSUMOylating DRP1 [85].
Furthermore, ubiquitination can also affect the activity of DRP1. For example, Parkin promotes the degradation of DRP1 through ubiquitination [91], while the anaphase-promoting complex/cyclosome and its coactivator Cdh1 (APC/C) can regulate the mitochondrial fission during mitosis by ubiquitinating and degrading DRP1 [92]. Reversely, OTUD6A cleaves off ubiquitin residues and increases DRP1 stability in the cell [93].
Relationship between mitochondrial fission and apoptosis
Mitochondrial fission is temporally and spatially correlated with apoptosis. Under physiological conditions, activation of about 3% DRP1 is sufficient to maintain the balance between fusion and fission of the mitochondrial system [94]. However, excessive activation of DRP1 and its aggregation on mitochondria can affect mitochondrial function and even initiate apoptosis [95, 96].
Existing studies indicate that inducing mitochondrial fission can promote apoptosis [97,98,99], while mitochondrial fusion may be a means for cancer cells to evade apoptosis induced by chemotherapy [100, 101]. Inhibiting DRP1 activity and mitochondrial fission, whether through chemical or genetic knockout methods, can exert anti-apoptotic effects [102,103,104]. In a study on lung adenocarcinoma, inducing mitochondrial fusion under chemotherapy can elevate chemo-resistant ability of cancer cells [105]; inhibiting DRP1 activity through inhibitors or genetic intervention can exert anti-apoptotic effects [103, 104](the most commonly used inhibitors of DRP1 listed in Table 2). In addition, in PINK1 double knockout and MPTP drug-induced PD models, inhibiting DRP1 activity significantly reduces the loss of substantia nigra neurons and promotes neuron survival [102].
It is currently believed that cells with fragmented mitochondria are more sensitive to apoptosis-related stimuli [106, 107], which may be attributed to the inefficient energy synthesis state of fission mitochondria [106, 108]. Inhibiting mitochondrial fission and promoting mitochondrial fusion can enhance the efficiency of the mitochondrial electron transport chain and increase oxidative phosphorylation capacity [109,110,111], increasing the stress-resistant ability, which is consistent with our previous study [112]. In addition, the mitochondrial fission can induce mitochondrial MOMP and genomic instability, which may also be vital factor involved [113].
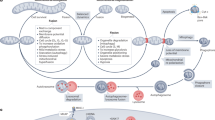
Further, research has found that DRP1 can promote the release of mitochondrial cytochrome c and the activation of Caspases [104]; DRP1 interacts with Bcl2 family, promoting the translocation of pro-apoptosis molecule BAX to mitochondria [114] or directly suppressing the activity of anti-apoptosis Bcl2 [115] to execute apoptosis. Some studies suggest that DRP1 can bind to the N-terminal domain of BAX, which simultaneously promotes the activity of BAX and DRP1, on the one hand, promoting the oligomerization of BAX to drive the apoptosis, and on the other hand, accelerating the translocation of DRP1 to mitochondria to promote mitochondrial fission [116], and this interaction between BAX and DRP1 may be achieved in a PGAM5 dependent manner [117]. In addition, during the apoptosis, BAX and BAK can stabilize DRP1 on the mitochondrial outer membrane by promoting the sumoylation of DRP1 [118], and the colonization of DRP1 on the mitochondrial membrane in turn conducts mitochondrial fission and the apoptosis [119, 120], thus forming a positive feedback process to induce apoptosis, which may be one of the reasons why mitochondrial fission and apoptosis are associated(Fig. 2). In other studies, with DRP1 knockout, a single BAX oligomer ring cannot launch the release of cytochrome c from inter mitochondrial membrane space (IMS) [121, 122]. Suppressing DRP1 activity can counteract mitochondrial fragmentation, impede the release of cytochrome c, and prevent apoptosis, and this process is independent of the translocation of BAX to mitochondria [123], suggesting that DRP1 not only participates in the activation of BAX but also in the MOMP. It should be noted that during the apoptosis, DRP1 can anchor to the mitochondrial outer membrane in a BAX/BAK-dependent manner, and which does not trigger mitochondrial fission [84], suggesting that although the apoptotic role of DRP1 is closely related to mitochondrial fission, there still exists independence between each other to some extent.

Mitochondrial-localized DRP1 not only functions in mitochondrial fission but also influences the apoptotic process by interacting with BCL2 family members and regulating the mitochondrial permeability transition pore (mPTP).
In addition, Duan et al. found that DRP1 conduct the opening of the mitochondrial permeability transition pore (mPTP) to induce apoptosis. Under hypoxic conditions, first, DRP1 recognizes BAX and PiC and contacts with mPTP; then, DRP1 recruits and inactivates LRRK2, leading to the inactivation and dissociation of HK2, which induces an alteration in the conformation of mPTP and result in excessive opening [124]. Moreover, other studies suggest that the formation of the complex of PGAM5-L, BAX, and DRP1 is a fundamental step in the occurrence of intrinsic apoptosis [117]. To the contrary, some studies found that mitochondrial fission exhibits anti-apoptotic properties [125, 126]. For example, in brain tumor research, knocking down DRP1 in tumor-initiating cells can suppress cell growth and induce apoptosis [125], which may be attributed to specific cell types or physiological conditions (Fig. 2).
It is currently believed that during the mitochondrial fission, activated DRP1 can promote BAX activity, alter mitochondrial membrane permeability, and induce pro-apoptotic substances release, promoting apoptosis.
Conclusion
As the key player in the process of mitochondrial fission, the activity of DRP1 is regulated at multiple levels, in various ways to manipulate mitochondrial function precisely and adjust it to the demands of cells under varied physiological conditions, external stress, and pathological processes. Firstly, DRP1 is an essential molecule for normal physiological processes such as mitosis and mitochondrial quality control. Secondly, the mitochondrial fission is closely related to the apoptosis; and under external/inner stress such as ischemia-hypoxia or chemotherapy, DRP1 act as the balancer, which determines the fate of cells and plays vital role in the progress of diseases. In summary, elucidating the regulatory mechanisms of DRP1 under different conditions is of great significance, as it will help clarify the coordinated interaction between various cellular events and provide new perspectives for the diagnosis and treatment of multiple diseases. However, to achieve this goal, more in-depth research is still needed.
References
Devulder J. Mitochondria in diseases: from cell power plants to DAMPs. Thorax. 2023;78:116–7. https://doi.org/10.1136/thorax-2022-219076
Roy S, Das A, Bairagi A, Das D, Jha A, Srivastava AK, et al. Mitochondria act as a key regulatory factor in cancer progression: current concepts on mutations, mitochondrial dynamics, and therapeutic approach. Mutat Res Rev Mutat Res. 2024;793:108490 https://doi.org/10.1016/j.mrrev.2024.108490
Lacombe A, Scorrano L. The interplay between mitochondrial dynamics and autophagy: From a key homeostatic mechanism to a driver of pathology. Semin Cell Dev Biol. 2024;161-162:1–19. https://doi.org/10.1016/j.semcdb.2024.02.001
Fogo GM, Anzell AR, Maheras KJ, Raghunayakula S, Wider JM, Emaus KJ, et al. Machine learning-based classification of mitochondrial morphology in primary neurons and brain. Sci Rep. 2021;11:5133 https://doi.org/10.1038/s41598-021-84528-8
Heine KB, Hood WR. Mitochondrial behaviour, morphology, and animal performance. Biol Rev Camb Philos Soc. 2020;95:730–7. https://doi.org/10.1111/brv.12584
Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. 2020;15:235–59. https://doi.org/10.1146/annurev-pathmechdis-012419-032711
Jenkins BC, Neikirk K, Katti P, Claypool SM, Kirabo A, McReynolds MR, et al. Mitochondria in disease: changes in shapes and dynamics. Trends Biochem Sci. 2024;49:346–60. https://doi.org/10.1016/j.tibs.2024.01.011
Reiss AB, Gulkarov S, Jacob B, Srivastava A, Pinkhasov A, Gomolin IH, et al. Mitochondria in Alzheimer’s disease pathogenesis. Life. 2024;14. https://doi.org/10.3390/life14020196.
Chen C, Merrill RA, Jong CJ, Strack S. Driving mitochondrial fission improves cognitive, but not motor deficits in a mouse model of ataxia of Charlevoix-Saguenay. Cerebellum. 2024. https://doi.org/10.1007/s12311-024-01701-1.
Beg MA, Huang M, Vick L, Rao KNS, Zhang J, Chen Y. Targeting mitochondrial dynamics and redox regulation in cardiovascular diseases. Trends Pharm Sci. 2024;45:290–303. https://doi.org/10.1016/j.tips.2024.02.001
Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharm Sin. 2021;42:655–64. https://doi.org/10.1038/s41401-020-00518-y
Rochon K, Bauer BL, Roethler NA, Buckley Y, Su CC, Huang W, et al. Structural basis for regulated assembly of the mitochondrial fission GTPase Drp1. Nat Commun. 2024;15:1328 https://doi.org/10.1038/s41467-024-45524-4
Dong WT, Long LH, Deng Q, Liu D, Wang JL, Wang F, et al. Mitochondrial fission drives neuronal metabolic burden to promote stress susceptibility in male mice. Nat Metab. 2023;5:2220–36. https://doi.org/10.1038/s42255-023-00924-6
Shimura D, Nuebel E, Baum R, Valdez SE, Xiao S, Warren JS, et al. Protective mitochondrial fission induced by stress-responsive protein GJA1-20k. Elife. 2021;10. https://doi.org/10.7554/eLife.69207.
Zhao T, Niu D, Chen Y, Fu P. The role of mitochondrial quality control mechanisms in chondrocyte senescence. Exp Gerontol. 2024;188:112379 https://doi.org/10.1016/j.exger.2024.112379
Liu YB, Hong JR, Jiang N, Jin L, Zhong WJ, Zhang CY, et al. The role of mitochondrial quality control in chronic obstructive pulmonary disease. Lab Investig. 2024;104:100307 https://doi.org/10.1016/j.labinv.2023.100307
Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion. 2010;10:640–8. https://doi.org/10.1016/j.mito.2010.08.005
Sun F, Fang M, Zhang H, Song Q, Li S, Li Y, et al. Drp1: focus on diseases triggered by the mitochondrial pathway. Cell Biochem Biophys. 2024. https://doi.org/10.1007/s12013-024-01245-5
Sbai O, Bazzani V, Tapaswi S, McHale J, Vascotto C, Perrone L. Is Drp1 a link between mitochondrial dysfunction and inflammation in Alzheimer’s disease? Front Mol Neurosci. 2023;16:1166879 https://doi.org/10.3389/fnmol.2023.1166879
DNM1L dynamin 1 like [Homo sapiens (human)] - Gene - NCBI (nih.gov) https://www.ncbi.nlm.nih.gov/gene/10059#genomic-regions-transcripts-products.
Banerjee R, Mukherjee A, Nagotu S. Mitochondrial dynamics and its impact on human health and diseases: inside the DRP1 blackbox. J Mol Med. 2022;100:1–21. https://doi.org/10.1007/s00109-021-02150-7
Rosdah AA, Smiles WJ, Oakhill JS, Scott JW, Langendorf CG, Delbridge LMD, et al. New perspectives on the role of Drp1 isoforms in regulating mitochondrial pathophysiology. Pharm Ther. 2020;213:107594 https://doi.org/10.1016/j.pharmthera.2020.107594
Francy CA, Alvarez FJ, Zhou L, Ramachandran R, Mears JA. The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. J Biol Chem. 2015;290:11692–703. https://doi.org/10.1074/jbc.M114.610881
Sesaki H, Adachi Y, Kageyama Y, Itoh K, Iijima M. In vivo functions of Drp1: lessons learned from yeast genetics and mouse knockouts. Biochim Biophys Acta. 2014;1842:1179–85. https://doi.org/10.1016/j.bbadis.2013.11.024
Frohlich C, Grabiger S, Schwefel D, Faelber K, Rosenbaum E, Mears J, et al. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013;32:1280–92. https://doi.org/10.1038/emboj.2013.74
Adhikary A, Mukherjee A, Banerjee R, Nagotu S. DRP1: at the crossroads of dysregulated mitochondrial dynamics and altered cell signaling in cancer cells. ACS Omega. 2023;8:45208–23. https://doi.org/10.1021/acsomega.3c06547
Macdonald PJ, Francy CA, Stepanyants N, Lehman L, Baglio A, Mears JA, et al. Distinct splice variants of dynamin-related protein 1 differentially utilize mitochondrial fission factor as an effector of cooperative GTPase activity. J Biol Chem. 2016;291:493–507. https://doi.org/10.1074/jbc.M115.680181
Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem. 2004;279:35967–74. https://doi.org/10.1074/jbc.M404105200
Chappie JS, Mears JA, Fang S, Leonard M, Schmid SL, Milligan RA, et al. A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell. 2011;147:209–22. https://doi.org/10.1016/j.cell.2011.09.003
Mahajan M, Bharambe N, Shang Y, Lu B, Mandal A, Madan Mohan P, et al. NMR identification of a conserved Drp1 cardiolipin-binding motif essential for stress-induced mitochondrial fission. Proc Natl Acad Sci USA. 2021;118. https://doi.org/10.1073/pnas.2023079118.
Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, et al. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife. 2015;4. https://doi.org/10.7554/eLife.08828.
Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–7. https://doi.org/10.1126/science.1228360
Korobova F, Gauvin TJ, Higgs HN. A role for myosin II in mammalian mitochondrial fission. Curr Biol. 2014;24:409–14. https://doi.org/10.1016/j.cub.2013.12.032
Li S, Xu S, Roelofs BA, Boyman L, Lederer WJ, Sesaki H, et al. Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J Cell Biol. 2015;208:109–23. https://doi.org/10.1083/jcb.201404050
Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JT, Wang C, et al. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell. 2014;25:145–59. https://doi.org/10.1091/mbc.E13-09-0525
Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–67. https://doi.org/10.1091/mbc.E12-10-0721
Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. 2016;212:531–44. https://doi.org/10.1083/jcb.201508099
Liu R, Chan DC. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol Biol Cell. 2015;26:4466–77. https://doi.org/10.1091/mbc.E15-08-0591
Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, et al. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci. 2016;129:2170–81. https://doi.org/10.1242/jcs.185165
Elgass KD, Smith EA, LeGros MA, Larabell CA, Ryan MT. Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J Cell Sci. 2015;128:2795–804. https://doi.org/10.1242/jcs.169136
Atkins K, Dasgupta A, Chen KH, Mewburn J, Archer SL. The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: implications for human disease. Clin Sci. 2016;130:1861–74. https://doi.org/10.1042/CS20160030
Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife. 2015;4:e11553 https://doi.org/10.7554/eLife.11553
Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6:e1000795 https://doi.org/10.1371/journal.pgen.1000795
Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2010;20:14–24. https://doi.org/10.1016/j.tcb.2009.10.002
Wang D, Tian J, Yan Z, Yuan Q, Wu D, Liu X, et al. Mitochondrial fragmentation is crucial for c-Myc-driven hepatoblastoma-like liver tumors. Mol Ther. 2022;30:1645–60. https://doi.org/10.1016/j.ymthe.2022.01.032
Bae JE, Park SJ, Hong Y, Jo DS, Lee H, Park NY, et al. Loss of RNA binding protein, human antigen R enhances mitochondrial elongation by regulating Drp1 expression in SH-SY5Y cells. Biochem Biophys Res Commun. 2019;516:713–8. https://doi.org/10.1016/j.bbrc.2019.06.091
Park SJ, Lee H, Jo DS, Jo YK, Shin JH, Kim HB, et al. Heterogeneous nuclear ribonucleoprotein A1 post-transcriptionally regulates Drp1 expression in neuroblastoma cells. Biochim Biophys Acta. 2015;1849:1423–31. https://doi.org/10.1016/j.bbagrm.2015.10.017
Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–9. https://doi.org/10.1074/jbc.M607279200
Shou J, Huo Y. PINK1 phosphorylates Drp1(S616) to improve mitochondrial fission and inhibit the progression of hypertension-induced HFpEF. Int J Mol Sci. 2022;23. https://doi.org/10.3390/ijms231911934.
Gao Q, Tian R, Han H, Slone J, Wang C, Ke X, et al. PINK1-mediated Drp1(S616) phosphorylation modulates synaptic development and plasticity via promoting mitochondrial fission. Signal Transduct Target Ther. 2022;7:103 https://doi.org/10.1038/s41392-022-00933-z
Liao JZ, Chung HL, Shih C, Wong KKL, Dutta D, Nil Z, et al. Cdk8/CDK19 promotes mitochondrial fission through Drp1 phosphorylation and can phenotypically suppress pink1 deficiency in Drosophila. Nat Commun. 2024;15:3326 https://doi.org/10.1038/s41467-024-47623-8
Wood DJ, Endicott JA. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018;8. https://doi.org/10.1098/rsob.180112.
Rong R, Xia X, Peng H, Li H, You M, Liang Z, et al. Cdk5-mediated Drp1 phosphorylation drives mitochondrial defects and neuronal apoptosis in radiation-induced optic neuropathy. Cell Death Dis. 2020;11:720 https://doi.org/10.1038/s41419-020-02922-y
He M, Wang X, Liu Z, Cui Q, Chen Y, Geng W, et al. CDK5 mediates proinflammatory effects of microglia through activated DRP1 phosphorylation in rat model of intracerebral hemorrhage. Dis Markers. 2022;2022:1919064 https://doi.org/10.1155/2022/1919064
Cho B, Cho HM, Kim HJ, Jeong J, Park SK, Hwang EM, et al. CDK5-dependent inhibitory phosphorylation of Drp1 during neuronal maturation. Exp Mol Med. 2014;46:e105 https://doi.org/10.1038/emm.2014.36
Jahani-Asl A, Huang E, Irrcher I, Rashidian J, Ishihara N, Lagace DC, et al. CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum Mol Genet. 2015;24:4573–83. https://doi.org/10.1093/hmg/ddv188
Xu D, Yang P, Yang ZJ, Li QG, Ouyang YT, Yu T, et al. Blockage of Drp1 phosphorylation at Ser579 protects neurons against Abeta(1‑42)‑induced degeneration. Mol Med Rep. 2021;24. https://doi.org/10.3892/mmr.2021.12296.
Guo MY, Shang L, Hu YY, Jiang LP, Wan YY, Zhou QQ, et al. The role of Cdk5-mediated Drp1 phosphorylation in Abeta(1-42) induced mitochondrial fission and neuronal apoptosis. J Cell Biochem. 2018;119:4815–25. https://doi.org/10.1002/jcb.26680
Hu SL, Mamun AA, Shaw J, Li SL, Shi YF, Jin XM, et al. TBK1-medicated DRP1 phosphorylation orchestrates mitochondrial dynamics and autophagy activation in osteoarthritis. Acta Pharm Sin. 2023;44:610–21. https://doi.org/10.1038/s41401-022-00967-7
Wang Y, Lu M, Xiong L, Fan J, Zhou Y, Li H, et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020;11:29 https://doi.org/10.1038/s41419-019-2218-5
Yu W, Wang X, Zhao J, Liu R, Liu J, Wang Z, et al. Stat2-Drp1 mediated mitochondrial mass increase is necessary for pro-inflammatory differentiation of macrophages. Redox Biol. 2020;37:101761 https://doi.org/10.1016/j.redox.2020.101761
Lin AJ, Joshi AU, Mukherjee R, Tompkins CA, Vijayan V, Mochly-Rosen D, et al. deltaPKC-mediated DRP1 phosphorylation impacts macrophage mitochondrial function and inflammatory response to endotoxin. Shock. 2022;57:435–43. https://doi.org/10.1097/SHK.0000000000001885
Roe AJ, Qi X. Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem Biophys Res Commun. 2018;496:706–11. https://doi.org/10.1016/j.bbrc.2018.01.114
Yang D, Rong R, Yang R, You M, Wang M, Li H, et al. CaMK II-induced Drp1 phosphorylation contributes to blue light-induced AIF-mediated necroptosis in retinal R28 cells. Biochem Biophys Res Commun. 2021;559:113–20. https://doi.org/10.1016/j.bbrc.2021.04.082
Bo T, Yamamori T, Suzuki M, Sakai Y, Yamamoto K, Inanami O. Calmodulin-dependent protein kinase II (CaMKII) mediates radiation-induced mitochondrial fission by regulating the phosphorylation of dynamin-related protein 1 (Drp1) at serine 616. Biochem Biophys Res Commun. 2018;495:1601–7. https://doi.org/10.1016/j.bbrc.2017.12.012
Tresse E, Riera-Ponsati L, Jaberi E, Sew WQG, Ruscher K, Issazadeh-Navikas S. IFN-beta rescues neurodegeneration by regulating mitochondrial fission via STAT5, PGAM5, and Drp1. EMBO J. 2021;40:e106868 https://doi.org/10.15252/embj.2020106868
Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200. https://doi.org/10.1016/j.cmet.2012.01.009
Merz KE, Hwang J, Zhou C, Veluthakal R, McCown EM, Hamilton A, et al. Enrichment of the exocytosis protein STX4 in skeletal muscle remediates peripheral insulin resistance and alters mitochondrial dynamics via Drp1. Nat Commun. 2022;13:424 https://doi.org/10.1038/s41467-022-28061-w
Du J, Wang T, Xiao C, Dong Y, Zhou S, Zhu Y. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates hepatic steatosis in vitro. Curr Mol Med. 2024. https://doi.org/10.2174/0115665240275594231229121030.
Yan J, Liu XH, Han MZ, Wang YM, Sun XL, Yu N, et al. Blockage of GSK3beta-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiol Aging. 2015;36:211–27. https://doi.org/10.1016/j.neurobiolaging.2014.08.005
Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC, et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One. 2012;7:e49112 https://doi.org/10.1371/journal.pone.0049112
Zhou L, Zhang Q, Zhang P, Sun L, Peng C, Yuan Z, et al. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 2017;8:e3117 https://doi.org/10.1038/cddis.2017.524
Dickey AS, Strack S. PKA/AKAP1 and PP2A/Bbeta2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci. 2011;31:15716–26. https://doi.org/10.1523/JNEUROSCI.3159-11.2011
Hu J, Zhang H, Li J, Jiang X, Zhang Y, Wu Q, et al. ROCK1 activation-mediated mitochondrial translocation of Drp1 and cofilin are required for arnidiol-induced mitochondrial fission and apoptosis. J Exp Clin Cancer Res. 2020;39:37 https://doi.org/10.1186/s13046-020-01545-7
Merrill RA, Slupe AM, Strack S. N-terminal phosphorylation of protein phosphatase 2A/Bbeta2 regulates translocation to mitochondria, dynamin-related protein 1 dephosphorylation, and neuronal survival. FEBS J. 2013;280:662–73. https://doi.org/10.1111/j.1742-4658.2012.08631.x
Zhang Q, Hu C, Huang J, Liu W, Lai W, Leng F, et al. ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s disease. Exp Mol Med. 2019;51:1–13. https://doi.org/10.1038/s12276-019-0318-z
Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–43. https://doi.org/10.1016/j.cell.2011.11.030
Edwards G, Perkins GA, Kim KY, Kong Y, Lee Y, Choi SH, et al. Loss of AKAP1 triggers Drp1 dephosphorylation-mediated mitochondrial fission and loss in retinal ganglion cells. Cell Death Dis. 2020;11:254 https://doi.org/10.1038/s41419-020-2456-6
Lee DS, Kim JE. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018;9:869 https://doi.org/10.1038/s41419-018-0910-5
Sheng Z, Zhu J, Deng YN, Gao S, Liang S. SUMOylation modification-mediated cell death. Open Biol. 2021;11:210050 https://doi.org/10.1098/rsob.210050
Huang J, Xie P, Dong Y, An W. Inhibition of Drp1 SUMOylation by ALR protects the liver from ischemia-reperfusion injury. Cell Death Differ. 2021;28:1174–92. https://doi.org/10.1038/s41418-020-00641-7
Adaniya SM, O-Uchi J, Cypress MW, Kusakari Y, Jhun BS. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am J Physiol Cell Physiol. 2019;316:C583–C604. https://doi.org/10.1152/ajpcell.00523.2018
Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell. 2015;59:941–55. https://doi.org/10.1016/j.molcel.2015.08.001
Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–50. https://doi.org/10.1083/jcb.200610042
Zunino R, Schauss A, Rippstein P, Andrade-Navarro M, McBride HM. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci. 2007;120:1178–88. https://doi.org/10.1242/jcs.03418
Yamada S, Sato A, Ishihara N, Akiyama H, Sakakibara SI. Drp1 SUMO/deSUMOylation by Senp5 isoforms influences ER tubulation and mitochondrial dynamics to regulate brain development. iScience. 2021;24:103484 https://doi.org/10.1016/j.isci.2021.103484
Guo C, Wilkinson KA, Evans AJ, Rubin PP, Henley JM. SENP3-mediated deSUMOylation of Drp1 facilitates interaction with Mff to promote cell death. Sci Rep. 2017;7:43811 https://doi.org/10.1038/srep43811
Anderson CA, Blackstone C. SUMO wrestling with Drp1 at mitochondria. EMBO J. 2013;32:1496–8. https://doi.org/10.1038/emboj.2013.103
Guo C, Hildick KL, Luo J, Dearden L, Wilkinson KA, Henley JM. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 2013;32:1514–28. https://doi.org/10.1038/emboj.2013.65
Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–5. https://doi.org/10.1016/j.cub.2004.02.004
Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–22. https://doi.org/10.1038/sj.embor.7400790
Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell. 2011;22:1207–16. https://doi.org/10.1091/mbc.E10-07-0567
Shi L, Liu J, Peng Y, Zhang J, Dai X, Zhang S, et al. Deubiquitinase OTUD6A promotes proliferation of cancer cells via regulating Drp1 stability and mitochondrial fission. Mol Oncol. 2020;14:3169–83. https://doi.org/10.1002/1878-0261.12825
Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–56. https://doi.org/10.1091/mbc.12.8.2245
Chang X, Niu S, Shang M, Li J, Guo M, Zhang W, et al. ROS-Drp1-mediated mitochondria fission contributes to hippocampal HT22 cell apoptosis induced by silver nanoparticles. Redox Biol. 2023;63:102739 https://doi.org/10.1016/j.redox.2023.102739
Yi L, Shang XJ, Lv L, Wang Y, Zhang J, Quan C, et al. Cadmium-induced apoptosis of Leydig cells is mediated by excessive mitochondrial fission and inhibition of mitophagy. Cell Death Dis. 2022;13:928 https://doi.org/10.1038/s41419-022-05364-w
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–8. https://doi.org/10.1073/pnas.0808249105
Cereghetti GM, Costa V, Scorrano L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010;17:1785–94. https://doi.org/10.1038/cdd.2010.61
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–66. https://doi.org/10.1038/ncb1907
Kong B, Wang Q, Fung E, Xue K, Tsang BK. p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J Biol Chem. 2014;289:27134–45. https://doi.org/10.1074/jbc.M114.594812
Farrand L, Kim JY, Im-Aram A, Suh JY, Lee HJ, Tsang BK. An improved quantitative approach for the assessment of mitochondrial fragmentation in chemoresistant ovarian cancer cells. PLoS One. 2013;8:e74008 https://doi.org/10.1371/journal.pone.0074008
Rappold PM, Cui M, Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L, et al. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat Commun. 2014;5:5244 https://doi.org/10.1038/ncomms6244
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. https://doi.org/10.1016/j.devcel.2007.11.019
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25. https://doi.org/10.1016/s1534-5807(01)00055-7
Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. 2017;1858:686–99. https://doi.org/10.1016/j.bbabio.2017.01.012
Ong SB, Hausenloy DJ. Mitochondrial dynamics as a therapeutic target for treating cardiac diseases. Handb Exp Pharm. 2017;240:251–79. https://doi.org/10.1007/164_2016_7
Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem. 2004;279:52726–34. https://doi.org/10.1074/jbc.M408910200
Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–98. https://doi.org/10.1038/ncb2220
Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. 2016;41:261–73. https://doi.org/10.1016/j.tibs.2016.01.001
Gao T, Zhang X, Zhao J, Zhou F, Wang Y, Zhao Z, et al. SIK2 promotes reprogramming of glucose metabolism through PI3K/AKT/HIF-1alpha pathway and Drp1-mediated mitochondrial fission in ovarian cancer. Cancer Lett. 2020;469:89–101. https://doi.org/10.1016/j.canlet.2019.10.029
Javed Z, Shin DH, Pan W, White SR, Kim YS, Elhaw AT, et al. Alternative splice variants of the mitochondrial fission protein DNM1L/Drp1 regulate mitochondrial dynamics and tumor progression in ovarian cancer. bioRxiv 2024. https://doi.org/10.1101/2023.09.20.558501
Wang N, Huang R, Yang K, He Y, Gao Y, Dong D. Interfering with mitochondrial dynamics sensitizes glioblastoma multiforme to temozolomide chemotherapy. J Cell Mol Med. 2022;26:893–912. https://doi.org/10.1111/jcmm.17147
Cao K, Riley JS, Heilig R, Montes-Gomez AE, Vringer E, Berthenet K, et al. Mitochondrial dynamics regulate genome stability via control of caspase-dependent DNA damage. Dev Cell. 2022;57:1211–1225 e6. https://doi.org/10.1016/j.devcel.2022.03.019
Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. https://doi.org/10.1016/j.cell.2010.08.017
Kowaltowski AJ, Cosso RG, Campos CB, Fiskum G. Effect of Bcl-2 overexpression on mitochondrial structure and function. J Biol Chem. 2002;277:42802–7. https://doi.org/10.1074/jbc.M207765200
Jenner A, Pena-Blanco A, Salvador-Gallego R, Ugarte-Uribe B, Zollo C, Ganief T, et al. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 2022;41:e108587 https://doi.org/10.15252/embj.2021108587
Xu W, Jing L, Wang Q, Lin CC, Chen X, Diao J, et al. Bax-PGAM5L-Drp1 complex is required for intrinsic apoptosis execution. Oncotarget. 2015;6:30017–34. https://doi.org/10.18632/oncotarget.5013
Wang P, Wang P, Liu B, Zhao J, Pang Q, Agrawal SG, et al. Dynamin-related protein Drp1 is required for Bax translocation to mitochondria in response to irradiation-induced apoptosis. Oncotarget. 2015;6:22598–612. https://doi.org/10.18632/oncotarget.4200
Balog J, Mehta SL, Vemuganti R. Mitochondrial fission and fusion in secondary brain damage after CNS insults. J Cereb Blood Flow Metab. 2016;36:2022–33. https://doi.org/10.1177/0271678X16671528
Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–68. https://doi.org/10.1016/j.bbamcr.2013.02.002
Dai CQ, Guo Y, Chu XY. Neuropathic pain: the dysfunction of Drp1, mitochondria, and ROS homeostasis. Neurotox Res. 2020;38:553–63. https://doi.org/10.1007/s12640-020-00257-2
Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016;35:402–13. https://doi.org/10.15252/embj.201592789
Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–11. https://doi.org/10.1091/mbc.e04-04-0294
Duan C, Kuang L, Hong C, Xiang X, Liu J, Li Q, et al. Mitochondrial Drp1 recognizes and induces excessive mPTP opening after hypoxia through BAX-PiC and LRRK2-HK2. Cell Death Dis. 2021;12:1050 https://doi.org/10.1038/s41419-021-04343-x
Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–10. https://doi.org/10.1038/nn.3960
Inoue-Yamauchi A, Oda H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem Biophys Res Commun. 2012;421:81–5. https://doi.org/10.1016/j.bbrc.2012.03.118
Lu YT, Li LZ, Yang YL, Yin X, Liu Q, Zhang L, et al. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis. 2018;9:672 https://doi.org/10.1038/s41419-018-0708-5
Zaja I, Bai X, Liu Y, Kikuchi C, Dosenovic S, Yan Y, et al. Cdk1, PKCdelta and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem Biophys Res Commun. 2014;453:710–21. https://doi.org/10.1016/j.bbrc.2014.09.144
Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase Cdelta under oxidative stress conditions in vivo. Mol Biol Cell. 2011;22:256–65. https://doi.org/10.1091/mbc.E10-06-0551
Perdiz D, Lorin S, Leroy-Gori I, Pous C. Stress-induced hyperacetylation of microtubule enhances mitochondrial fission and modulates the phosphorylation of Drp1 at (616)Ser. Cell Signal. 2017;39:32–43. https://doi.org/10.1016/j.cellsig.2017.07.020
Xiong X, Hasani S, Young LEA, Rivas DR, Skaggs AT, Martinez R, et al. Activation of Drp1 promotes fatty acids-induced metabolic reprograming to potentiate Wnt signaling in colon cancer. Cell Death Differ. 2022;29:1913–27. https://doi.org/10.1038/s41418-022-00974-5
Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. 2015;57:521–36. https://doi.org/10.1016/j.molcel.2015.01.003
Ke W, Wang B, Liao Z, Song Y, Li G, Ma L, et al. Matrix stiffness induces Drp1-mediated mitochondrial fission through Piezo1 mechanotransduction in human intervertebral disc degeneration. J Transl Med. 2023;21:711 https://doi.org/10.1186/s12967-023-04590-w
Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57:537–51. https://doi.org/10.1016/j.molcel.2015.01.002
Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–44. https://doi.org/10.1038/sj.embor.7401062
Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, et al. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44:532–44. https://doi.org/10.1016/j.molcel.2011.08.045
Ko HJ, Tsai CY, Chiou SJ, Lai YL, Wang CH, Cheng JT, et al. The phosphorylation status of Drp1-Ser637 by PKA in mitochondrial fission modulates mitophagy via PINK1/Parkin to exert multipolar spindles assembly during mitosis. biomolecules 2021;11. https://doi.org/10.3390/biom11030424.
Park JE, Kim YJ, Lee SG, Kim JY, Chung JY, Jeong SY, et al. Drp1 phosphorylation is indispensable for steroidogenesis in Leydig cells. Endocrinology. 2019;160:729–43. https://doi.org/10.1210/en.2019-00029
Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, et al. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–85. https://doi.org/10.1083/jcb.200802164
Jhun BS, O-Uchi J, Adaniya SM, Mancini TJ, Cao JL, King ME, et al. Protein kinase D activation induces mitochondrial fragmentation and dysfunction in cardiomyocytes. J Physiol. 2018;596:827–55. https://doi.org/10.1113/JP275418
Pennanen C, Parra V, Lopez-Crisosto C, Morales PE, Del Campo A, Gutierrez T, et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J Cell Sci. 2014;127:2659–71. https://doi.org/10.1242/jcs.139394
Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, et al. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–26. https://doi.org/10.1096/fj.12-226225
Rosdah AA, Abbott BM, Langendorf CG, Deng Y, Truong JQ, Waddell HMM, et al. A novel small molecule inhibitor of human Drp1. Sci Rep. 2022;12:21531 https://doi.org/10.1038/s41598-022-25464-z
Qi X, Qvit N, Su YC, Mochly-Rosen DA. novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802. https://doi.org/10.1242/jcs.114439
Lackner LL, Nunnari J. Small molecule inhibitors of mitochondrial division: tools that translate basic biological research into medicine. Chem Biol. 2010;17:578–83. https://doi.org/10.1016/j.chembiol.2010.05.016
Rios L, Pokhrel S, Li SJ, Heo G, Haileselassie B, Mochly-Rosen D. Targeting an allosteric site in dynamin-related protein 1 to inhibit Fis1-mediated mitochondrial dysfunction. Nat Commun. 2023;14:4356 https://doi.org/10.1038/s41467-023-40043-0
Wu D, Dasgupta A, Chen KH, Neuber-Hess M, Patel J, Hurst TE, et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020;34:1447–64. https://doi.org/10.1096/fj.201901467R
Mallat A, Uchiyama LF, Lewis SC, Fredenburg RA, Terada Y, Ji N, et al. Discovery and characterization of selective small molecule inhibitors of the mammalian mitochondrial division dynamin, DRP1. Biochem Biophys Res Commun. 2018;499:556–62. https://doi.org/10.1016/j.bbrc.2018.03.189
Bonner MY, Karlsson I, Rodolfo M, Arnold RS, Vergani E, Arbiser JL. Honokiol bis-dichloroacetate (Honokiol DCA) demonstrates activity in vemurafenib-resistant melanoma in vivo. Oncotarget. 2016;7:12857–68. https://doi.org/10.18632/oncotarget.7289
Acknowledgements
The authors would like to thank the Department of Neurosurgery of Jilin University for the generous support to the research that has been conducted.
Funding
This work was funded by: The Science and Technology Department Project Foundation of Jilin Province(YDZJ202301ZYTS035). National Natural Science Foundation of China (NSFC)(32300594). The Finance Department of Jilin Province(2023SCZ60).
Author information
Authors and Affiliations
Contributions
Nan Wang: Writing-Original draft. Xinwai Wang: Visualization. Beiwu Lan: Writing-Reviewing & Editing. Yuanyuan Cai: Supervision & Editing. Yufei Gao: Supervision & Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, N., Wang, X., Lan, B. et al. DRP1, fission and apoptosis. Cell Death Discov. 11, 150 (2025). https://doi.org/10.1038/s41420-025-02458-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-025-02458-0
This article is cited by
-
Mitochondrial transfer in cancer: mechanisms, immune evasion, and therapeutic opportunities
Genomics & Informatics (2026)
-
Chronic gut inflammation differentially modulates mitochondrial and antioxidant transcriptional programs in limbic brain structures
Scientific Reports (2025)
-
Research on the Application of Mesenchymal Stem Cells for Addressing Mitochondrial Damage in Neurodegenerative Diseases
Cellular and Molecular Neurobiology (2025)
