
Abstract
Recent evidence consolidates the deleterious impact of environmental exposure on testicular damage. Environmental exposures can instigate testicular toxicity, causing damage to the Sertoli-Sertoli cell-mediated blood-testis barrier (BTB) integrity, alterations in hormone levels orchestrated by aberrant Leydig cells, and disruption of spermatogenesis. Despite diverse study designs and methodologies, a consensus is emerging on how environmental factors induce oxidative stress by elevating ROS levels, affecting autophagy through pathways such as the ROS-mediated mTOR signaling pathway, ultimately culminating in testicular damage. This review synthesizes existing literature on how environmental exposures, including metals, air pollutants, industrial contaminants, and pesticides, disturb testicular homeostasis via autophagy-mediated oxidative stress, highlighting recent significant advancements. It also explores interventions like antioxidant support and autophagy regulation to alleviate testicular damage. These findings underscore the importance of elucidating the mechanisms of autophagy influenced by environmental exposures in disrupting the equilibrium of oxidative stress, identifying potential drug targets, and establishing a groundwork for enhancing future treatments and clinical management of testicular injuries.
Similar content being viewed by others

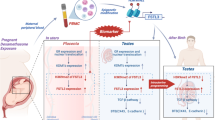
Facts
-
An increase in male reproductive disorders characterized by diminished sperm quality and quantity, attributing these patterns to environmental exposure.
-
Diverse environmental contaminants have the potential to interfere with spermatogenesis by adjusting cellular autophagy pathways, resulting in testicular harm.
-
Mitigation tactics for testicular damage primarily involve antioxidative mechanisms and anti-autophagic methodologies.
Introduction
The preservation of typical testicular function is meticulously overseen by genetic and environmental determinants [1,2,3], where persistent exposure to environmental stimuli can result in various testicular impairments [4]. Environmental exposure-triggered testicular harm primarily encompasses the compromise of Sertoli-Sertoli-cell-mediated blood-testis barrier (BTB) integrity, alterations in hormone levels orchestrated by aberrant Leydig cells, and disruption of spermatogenesis [5,6,7,8]. Environmental exposure impacts spermatogenesis, leading to the formation of vacuoles in seminiferous tubules, depletion of testicular germ cells, a significant decrease in sperm count, an increase in reactive oxygen species (ROS), culminating in apoptosis and irregularities in spermatogenesis [9,10,11,12].
Autophagy, an indispensable pathway for degradation and recycling, plays a vital role in maintaining cellular equilibrium, determining cell destiny, and fostering reproductive development [13]. It comprises three categories: macroautophagy, microautophagy, and chaperone-mediated autophagy, among which macroautophagy represents a major form [14] (Fig. 1). Accumulating evidence underscores the pivotal role of autophagy in diverse cellular processes within the male reproductive system. During spermatogenesis, autophagy is essential for both the formation of critical structures and the degradation of specific components, ensuring successful spermatid development [15]. In the autophagic process, the preautophagosome isolation membrane encapsulates protein aggregates or damaged organelles, leading to autophagosome formation, followed by fusion with the lysosome for cargo degradation by lysosomal enzymes [16]. The principal autophagic process revolves around autophagosome formation, regulated by an array of autophagy-related genes and pivotal signaling pathways, which include the mammalian target of rapamycin complex 1 (mTORC1), the serine/threonine-protein kinase ULK1/2, Beclin 1, autophagy proteins, class III phosphatidylinositol 3-kinases (PI3K), and microtubule-associated protein 1 A/1B-light chain 3-I/II (LC3-I/II) [17].

Macroautophagy entails the generation of a phagophore that envelops the cargo, culminating in the formation of an autophagosome. This entity subsequently merges with a lysosome to create an autolysosome where degradation transpires. Microautophagy is portrayed by the direct engulfment of the substrate via the invagination of lysosomal or late endosomal membranes. Chaperone-mediated autophagy involves the recognition by the lysosome-associated receptor LAMP2A of specific cytosolic proteins harboring a KFERQ-like motif. The chaperone heat shock cognate protein 70 (Hsc70) aids in the translocation of these proteins into the lysosome for degradation.
Various endogenous sources like mitochondria, peroxisomes, and phagocytic cells, along with exogenous factors such as pollution, UV exposure, xenobiotic compounds, and cigarette smoke, can generate reactive oxygen and nitrogen species [18]. The generation of ROS in male germ cells, exceeding antioxidant defenses, leads to oxidative stress, triggering apoptosis, autophagy, and DNA damage, all of which are critical factors contributing to male infertility [19]. During spermatogenesis, the mitochondrial count diminishes to a range of 20-80 within the mature spermatozoon, and this limited mitochondrial population plays a crucial role in male fertility, as any disturbance in structures located at the tail of spermatids can impair fertility [20]. Mitochondria, being the primary sites of ROS production, possess the ability to regulate and modulate autophagy [21]. Mitophagy, responsible for mitochondrial quality control, involves the engulfment of dysfunctional mitochondria by autophagosomes, followed by lysosomal digestion post-fusion [22]. Diverse mechanisms are utilized by cells for mitophagy, particularly through the PINK1/Parkin partnership for depolarized mitochondria turnover, stress-induced BCL2 interacting protein 3 (BNIP3), BCL2 interacting protein 3 like (BNIP3L), and FUN14 domain containing 1 (FUNDC1) molecular adaptors that directly interact with the LC3 protein to facilitate mitophagy [23]. It is hypothesized that impaired mitochondria release increased ROS level, which in turn influence autophagy through mTOR-dependent pathways in the cytoplasm, either activating autophagy by inhibiting the PI3K-AKT-mTOR axis or by stimulating AMPK to suppress the mTOR signaling pathway [16, 24,25,26]. Furthermore, p53 triggers AMPK activation, leading to subsequent mTOR inhibition [16] (Fig. 2).

Exposure to environmental stressors induces the production of reactive oxygen species (ROS), thereby impacting autophagy through mTOR-dependent routes. Autophagy activation is achieved by inhibiting the PI3K-AKT-mTOR axis or by stimulating AMPK to suppress the mTOR signaling pathway. Moreover, p53 instigates AMPK activation and can lead to mTOR inhibition. The antioxidant factor Nrf2 and the autophagy-associated protein p62 mutually enhance each other’s expression.
Oxidative stress denotes an increase in intracellular ROS levels, resulting in harm to lipids, proteins, and DNA [27, 28]. In response to oxidative stress, NFE2 like bZIP transcription factor 2 (Nrf2) regulates antioxidant defenses by recognizing and binding to antioxidant response elements (ARE) sequences [29]. Keap1 inhibits nuclear activation of ARE by Nrf2, sequestering Nrf2 in the cytoplasm through the Keap1-Cul3 complex for ubiquitin-proteasome degradation, with oxidants or electrophiles modifying Keap1 to prevent Nrf2 ubiquitination and promote its nuclear translocation [29, 30]. Nrf2 binds to the ARE motif in the p62 promoter, enhancing p62 mRNA expression to stimulate cytoplasmic autophagy as a post-transcriptional regulatory mechanism, while p62 reciprocally boosts NRF2 transcription [31] (Fig. 2). Studies have demonstrated that an excess of p62 or autophagy deficiency disrupts the Nrf2 and Keap1 interaction, resulting in Nrf2 stabilization and subsequent transcriptional activation of Nrf2 target genes [32]. Additionally, the FUNDC1 phosphatase PGAM family member 5, mitochondrial serine/threonine protein phosphatase (PGAM5) serves as a Keap substrate, suggesting a connection between PGAM5 ubiquitination and degradation, as well as FUNDC1 dephosphorylation, culminating in the initiation of FUNDC1-dependent mitophagy [29].
Recent epidemiological data demonstrate a rise in male reproductive disorders with decreased sperm quality and quantity, linking these trends to environmental exposure based on increasing evidence from human and animal studies [33, 34]. Human activities like mining, industrial discharges, and electronic waste recycling have escalated metal contamination, with harmful metals gathering in vital organs such as the testis, heart, liver, kidneys, and brain [35, 36]. Predominantly comprising carbonaceous particulate matter (PM) and gases like NO2, SO2, CO, and volatile organic compounds (VOCs), air pollution stems from varied sources like residential energy consumption, industrial emissions, and vehicular exhaust [37]. Synthetic chemicals pose risks to post-polymerization, with leachable monomers or additives like bisphenols potentially diffusing into the environment, leading to inadvertent exposure [38]. Pesticides, omnipresent in our environment, exert deleterious effects on human health, potentially contributing to male infertility by adversely affecting non-target organs and reproductive health upon prolonged exposure [39, 40].
Research suggests that various environmental pollutants can disrupt spermatogenesis by modulating cellular autophagy pathways, leading to testicular damage. To better understand the subject, this review used many keywords, including ‘testicular damage’, ‘ROS’, ‘oxidative stress’, ‘apoptosis’, ‘autophagy’, and ‘male infertility’, along with related keywords in major online databases. Therefore, this paper systematically reviews the research progress on how environmental exposures, such as metals, air pollutants, industrial contaminants, and pesticides disrupt testicular homeostasis via autophagy-mediated oxidative stress (Table 1).
Metal
Cadmium
A primary source of exposure for the general population is cadmium, which is widespread in numerous human food items due to its high soil-to-plant transfer rates [41]. The current global standards for tolerable intake and acceptable excretion of cadmium are set at 0.83 µg/kg body weight per day and 5.24 µg/g creatinine, respectively [42]. Various studies have highlighted the heightened susceptibility of mammalian testes to cadmium, leading to toxicity in male reproductive organs, specifically affecting the testicles and sperm parameters [43]. Within the Sertoli cells of the testis, cadmium has been observed to notably elevate the levels of autophagy markers such as LC3, p62, ATG7, Beclin-1, and ATG5, along with the lysosomal membrane protein LAMP2 [44]. Excessive autophagy activation can culminate in cell death under conditions of oxidative stress and metal toxicity [44]. The mTOR pathway, functioning as a central checkpoint that negatively modulates autophagy, assumes a critical role upstream of autophagy during oxidative stress, implying that the mTOR signaling pathway may represent a primary conduit through which cadmium amplifies autophagy and induces reproductive toxicity [45]. This underscores the significance of the ROS signal in governing the cadmium-disrupted autophagy process in Leydig cells [46]. Notably, emerging reports have suggested that exposure to cadmium in testicular tissue can instigate defective autophagy flux, despite the upregulation of several autophagy-related factors, including ATG3, ATG5, p62/sequestosome-1 (SQSTM1), and Beclin 1 [47].
Arsenic
Arsenic, a naturally occurring element in the earth’s crust, is found as a contaminant in a diverse range of metal ores [48]. This element poses a significant health hazard, with estimates indicating that over 100 million individuals worldwide are exposed to arsenic levels deemed carcinogenic, primarily through the consumption of drinking water taken from arsenic-contaminated aquifers [49, 50]. Arsenic compounds As (III) and As (V) are classified as non-threshold Class I carcinogens with acute toxicities ranging from 15 to 42 mg/kg body mass. In contrast, simple methylated arsenicals exhibit intermediate toxicity levels, while the tetraalkylated compound arsenobetaine (AB), a common dietary source of arsenic, is considered non-toxic with a lethal dose 50% (LD50) surpassing 10,000 mg/kg body weight, primarily excreted intact in urine by humans [51]. Studies have shown that arsenic, known to trigger ROS production, disrupts spermatogenesis by impeding spermatid elongation, notably impacting semen quality and prompting endocrine dysfunction [52, 53]. Moreover, arsenic exposure has been linked to a remarkable increase in protein expression levels of Beclin-1, LC3, ATG7 and p62, and knockdown of beclin-1 has been shown to attenuate the alterations induced by arsenic treatment in MLTC-1 cells [54]. Furthermore, arsenic accumulation in testes, leading to increased oxidative stress markers like malondialdehyde (MDA), superoxide dismutase (SOD), and methionine sulfoxide reductases (MsrA), may effectively trigger autophagy and apoptosis processes [55].
Copper
Essential for maintaining overall health and fertility, copper can however exhibit toxicity when present in excessive amounts, with detrimental effects on male fertility [56]. Research indicates correlations between copper levels in seminal plasma and sperm quality parameters like motility, viability, and morphology [57]. Studies in chicken testes show increased expression of genes related to mitochondrial fission alongside decreased levels of fusion-related genes [58]. Copper exposure in mouse testes induces oxidative stress, characterized by increased ROS, MDA, and lactate dehydrogenase (LDH), coupled with reduced catalase (CAT) activity and glutathione (GSH) levels [59]. This oxidative environment leads to mitochondrial dysfunction, as indicated by lowered transmembrane potential and ATP levels, with upregulated autophagy-related genes and proteins pointing towards copper-triggered cell death and autophagy mediated by oxidative stress-induced mitochondrial dysfunction [59]. Moreover, copper overload in Drosophila also impacts testicular aging, highlighting the interplay between copper overload, long non-coding RNAs (lncRNAs), and the induction of cuproptosis and ferroptosis pathways through the mitochondrial tricarboxylic acid (TCA) cycle [56].
Hexavalent chromium
Hexavalent chromium emerges as the most potent carcinogen among hazardous heavy metal(loid)s contaminants in agricultural soil, water, and air, commonly associated with activities like metallurgic industries, tanneries, paint manufacturing, and petroleum refineries [60]. As per the North Carolina Health Department in the United States, the concentration of 0.07 µg/L equates to a 1-in-1-million lifetime cancer risk [61]. Consequently, the numerous instances of surpassing this threshold suggest a potentially significant portion of the population at risk. Treatment with hexavalent chromium disrupts spermatogenesis, resulting in the accumulation of prematurely released spermatocytes, spermatids, and uni- and multinucleate giant cells within the seminiferous tubules [62]. Studies conducted on rat testes have revealed that hexavalent chromium suppresses the Sirtuin1 (SIRT1)/peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha) pathway, leading to mitochondrial dynamics disorder characterized by elevated mitochondrial division and inhibited mitochondrial fusion [63]. Furthermore, downregulation of the downstream effector Nrf2 from Sirt1 exacerbates oxidative stress, leading to mitochondrial dynamics disorder and Nrf2 inhibition, ultimately contributing to abnormal testicular mitochondrial dynamics and enhancing apoptosis and autophagy [63].
Metallic nanomaterials (NMs)
Metallic NMs are employed in industrial contexts for various applications that often result in environmental release. These applications encompass antimicrobial coatings, fuel cells, water electrolysis, air and water purification, as well as biomedical imaging contrast agents [64]. Numerous nanoparticles (NP) have been identified to adversely impact spermatogenesis [65]. Gold nanoparticles (AuNPs), specifically noted for their catalytic attributes and biomedical uses, have sparked a growing interest in comprehending the potential toxic repercussions of such particles [66,67,68]. Liu et al., demonstrated that AuNPs (5 nm) can penetrate into the endosomes/lysosomes in Leydig cells, induce autophagosome formation, elevate ROS production, and interfere with the cell cycle in the S phase, thereby inducing concentration-dependent cytotoxicity and DNA impairment [69]. Meanwhile, biogenic nanocopper (BNC) agents exhibit robust anticancer, antimicrobial, and antiparasitic properties, and due to their minimal impact on normal cells, they are favored for treating various ailments [70]. Exposure to nanocopper increases autophagy-related factors like LC3, ATG5, ATG7, ATG12, and Beclin-1, while reducing p62 levels, linking nanocopper-induced damage in testicular tissues and spermatogenesis to cell apoptosis and autophagy via Akt/mTOR signaling and oxidative stress [71]. Silver nanoparticles (AgNPs) find utility in a broad array of products, spanning electronics, biosensors, textiles, the food industry, coatings, sunscreens, cosmetics, and medical devices [72]. AgNPs as stressors elevate ROS production, potentially upregulating p53, Bax, and Caspase-3 in response to stress factors in testicular tissues, leading to programmed cell death [73].
Air pollutant
PM
As a prominent constituent of air pollution, the health implications of PM2.5 (aerodynamic diameter ≤2.5 μm) represent a critical public health concern [74]. Research reveals a notable 2.0% decline in fertility rates per 10 mg/m³ rise in PM2.5 levels, as illustrated by PM2.5 mapping data in China [75]. Subsequent evaluation of testicular impact post-PM2.5 exposure demonstrated a significant increase in abnormal sperm morphology rates in both low and high PM2.5 dosage groups compared to controls [76]. Oxidative stress emerges as a pivotal initiator of reproductive impairment induced by PM2.5, with its ability to escalate cellular oxidative stress through excessive ROS generation, culminating in cell demise [76, 77]. Following PM2.5 exposure, heightened expression levels of PI3K and pAKT were observed in Sertoli cells, suggesting a potential role of ROS as signaling molecules activating Nrf2-mediated defenses against PM2.5-induced oxidative stress via the PI3K/AKT pathway [78, 79]. Conversely, in GC-spg cells, PM2.5 exposure led to a significant reduction in PI3K phosphorylation at 5 μg/cm², with both PI3K and AKT phosphorylation diminishing at 20 μg/cm², accompanied by elevated autophagy activity [80]. Despite stable Beclin-1 and p-p62 levels, elevated ATG7 and LC3 expression suggested PM2.5 could promote autophagosome formation [80].
Tobacco
Nicotine, a toxic alkaloid from tobacco plants, has been detected in various water sources, including surface waters, ground waters, industrial waste waters, and bottled waters [81, 82]. Studies show that ingestion of nicotine gums up to 6 mg/kg can induce intoxication symptoms in humans without causing fatality [83]. In rats, nicotine exposure has dose-dependently reduced sperm count and motility, while inducing seminiferous tubule and spermatogenic disruptions in the testes, possibly via excessive mitochondrial fusion triggering oxidative stress [84, 85]. Chronic low-dose nicotine exposure is implicated in oxidative stress through free radical-mediated lipid peroxidation (LPO) and protein oxidation in the testes and prostate, potentially due to altered mitochondrial dynamics favoring fusion over mitophagy [85, 86]. Moreover, nicotine treatment marginally increased autophagosome formation but hindered their fusion with lysosomes, accompanied by elevated LC3II/LC3I ratio and p62 levels, indicating impaired autophagic turnover in Leydig cells [87].
Aldehydes
Formaldehyde (FA) is a prevalent environmental pollutant encountered in various settings such as outdoors, indoors, workplaces, and residences, primarily through inhalation exposure [88]. Evidence from human and animal studies suggests that FA exposure can induce reproductive toxicity, inhibiting spermatogenesis with seminiferous tubule degeneration, spermatogenic cell apoptosis, lowered testosterone levels, and disrupted testicular antioxidant defenses [89, 90]. FA exposure elevates ROS levels and reduces SOD and GSH activities, while increasing MDA in rat testes, indicating oxidative stress-induced testicular damage [91]. Moreover, FA exposure dose-dependently induces autophagy, evidenced by LC3-I/LC3-II conversion and elevated LC3-II expression in testes [90]. Recent research suggests that FA exposure suppresses mTOR expression in testicular tissue, correlating with increased testicular autophagy levels in an mTOR-dependent manner [89].
Acrolein exposure is prevalent, originating from sources such as cigarette smoke, industrial pollution, and other environmental exposures that are known to increase the production of ROS [92]. Acrolein, one of the most reactive lipid aldehydes generated during the LPO process in oxidatively stressed spermatozoa, inhibits sperm motility and escalates ROS production, LPO, oxidative DNA damage, and Caspase activation [93]. Exposure of mouse Sertoli cells to acrolein resulted in a concentration-dependent elevation in cell mortality, with the deleterious impact associated with oxidative stress via p38 activation [94]. In Leydig cells, acrolein induces toxic ROS production and reduces superoxide dismutase activity, leading to increased lipid oxidation reflected by elevated MDA level, thereby initiating oxidative stress [95]. This exposure also initiates AKT protein expression under oxidative stress, triggering early-stage autophagy via the PI3K/AKT/mTOR pathway, which progresses to activate apoptosis-related pathways, culminating in programmed cell death in Leydig cells [95].
Synthetic chemicals
Nano-plastics
Nano-plastics, originating as byproducts from the degradation and manufacturing processes of plastic items, possess colloidal properties and range in size from 1 to 1000 nanometers (nm) [96]. Individuals inadvertently consume or inhale nano-plastics face potential health risks, with an estimated annual exposure to 39,000–52,000 nano-plastics [97]. Studies have demonstrated that when mice are orally administered polystyrene nanoparticles (PS-NPs) measuring an average size of 38.92 nm, at doses of 1, 3, 6, and 10 mg/kg/day for 35 days, these NP accumulate in various organs including the testes, intestines, liver, kidney, and brain, with the highest accumulation observed in the testes [98]. Concurrently, another study observed a notable reduction in sperm concentration was observed in groups exposed to PS-NPs at equivalent concentrations and durations, with a notably higher percentage of sperm showing abnormal morphology compared to the control group [99]. Numerous studies have indicated that ROS overproduction serves as the primary event triggering male reproductive toxicity induced by nanoplastics in mammals, subsequently instigating oxidative stress [100]. The surge in ROS initiates multiple cascading events at various levels, encompassing cellular oxidative stress, mitochondrial dysfunction, sperm DNA damage, endoplasmic reticulum (ER) stress, apoptosis, and autophagy of testicular cells [100]. ROS can compromise the integrity of mitochondrial DNA (mtDNA), establishing a detrimental cycle where compromised mitochondria, due to oxidized mtDNA, become dysfunctional, leading to excessive ROS production, further exacerbating mitochondrial impairment and ultimately resulting in severe nuclear DNA damage and cell demise [21]. Notably, nano-plastics significantly augment the expression of the autophagy biomarkers LC3-II and p62 while concurrently suppressing mTORC expression, indicating that nano-plastics could induce excessive autophagy by modulating mTORC signaling in spermatocyte cells [101]. Following exposure to PS-NPs in spermatocyte cells, a significant decrease in Nrf2 and HO-1 expression was noted, leading to the activation of mitochondrial apoptosis and autophagy pathways [100]. When autophagy is initiated by ROS, p62 undergoes degradation, disrupting the feedforward loop linking Nrf2 and p62, leading to a direct decrease in antioxidant capacity and an increase in ROS levels [102].
Bisphenol A (BPA)
BPA, a commonly used plasticizer, is readily absorbed by both animals and humans, exerting toxic effects on a range of tissues such as the liver, intestine, heart, kidney, testes, and ovary [103, 104]. The estimated daily intake of BPA for adults via drinking tap water is 148 ng/day [105]. BPA induces a range of testicular impairments, affecting seminiferous tubules, sperm quality, germ cells, and the BTB, intricately linked to molecular processes involving ROS, apoptosis, and autophagy [106]. Exposure to BPA induces oxidative stress in the testicular niche cells, as evidenced by elevated levels of MDA and reduced SOD activity, consequently heightening ROS levels in vitro [106]. Exposure to BPA activates the AKT pathway and inhibits the mTOR pathway, leading to concurrent apoptosis and autophagy in adolescent testes [107]. High-dose exposure results in increased autophagosomes in seminiferous epithelial cells, displaying irregular shapes with cytoplasm, damaged ER, and abnormal mitochondria, surrounded by secondary lysosomes, suggesting active phagocytosis post-BPA exposure [107]. Moreover, p62, a Keap1-Nrf2 pathway component, is degraded through autophagy, disrupting the feedback loop, resulting in reduced antioxidant capacity and increased ROS levels [102].
Di-(2-ethylhexyl) phthalate (DEHP)
Phthalate esters (PAEs) are commonly utilized organic chemicals as plasticizers in various industrial applications [108]. Among these, DEHP is extensively employed in plastics, rubber, adhesives, and other materials, yet concerns arise due to its leakage into greenhouse vegetables, dust, and medical equipment, leading to significant acute exposure levels [109]. Notably, DEHP exposure results in decreased serum testosterone levels, potentially harming Leydig cell function [110]. Upon entry into the body, DEHP induces ROS overproduction, triggering oxidative stress marked by elevated MDA and reduced GSH levels, implicated in apoptosis and autophagy of Leydig cells [110]. DEHP exposure also disrupts Sertoli cell function and compromises the BTB integrity [111]. Furthermore, DEHP exposure elevates ROS levels and simultaneously increases the number of autophagosomes by impairing autophagy degradation [111]. On the other hand, DEHP can also induce testicular injury through the excessive generation of ROS in immature testes and DEHP-triggered autophagy may lead to the hyperactivation of the NLRP3 inflammasome, resulting in germ cell impairment via the ROS/mTOR/NLRP3 pathway [112].
Perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA)
PFOS and PFOA have found application in numerous fields, including waxes and polishes, fabric protection, stain repellents for textiles and leather and food packaging coatings that resist oil [113]. PFOS and PFOA exposures can occur through multiple pathways, including consumption from food packaging, food migration, or direct ingestion [114]. Studies on male reproductive toxicity have indicated that PFOA and PFOS can lead to reduced serum testosterone levels, testes weights, increased abnormal sperm counts, and disruption of the BTB in rodent models [115,116,117,118]. PFOA and PFOS exposures have been linked to the ROS generation via oxidative stress imbalance in mammalian cells [119]. PFOS exerts its reproductive toxicity by disrupting the BTB and also increases ROS production, leading to the decreased activity of the PI3K/AKT/mTOR pathway to induce autophagy in Sertoli cells [116]. Exposure to PFOA leads to elevated ROS production, heightened p53 levels inhibiting NRF2, resulting in testicular oxidative stress characterized by raised LPO, germ cell apoptosis, and diminished antioxidants in mouse testes [119]. Moreover, PFOA hinders autophagic breakdown by blocking autophagosome-lysosome fusion, linked to reduced α-SNAP expression, which reduces the levels of TJ-related proteins (Occludin and Claudin-11), GJ-related protein (connexin-43 or CX43), and ES-related proteins (N-cadherin and β-catenin) in Sertoli cells [120]. Within Leydig cells, PFOA diminishes LC3-II levels and elevates p62 levels in a dose-dependent manner, indicating a suppression of autophagosome formation, yet autophagy activation mitigates PFOA-triggered apoptosis, underscoring PFOA’s cell injury induction through autophagosome formation inhibition [115].
4-Nonylphenol (4-NP)
4-NP, a persistent environmental contaminant within the alkylphenol group, is widely employed in various products like detergents, lubricants, cosmetics, pesticides, plastics, paints, and wetting agents [121]. It is prevalent in food sources such as fish, animal tissues, milk, cereals, vegetables, and fruits due to its lipophilic nature and extended half-life, leading to bioaccumulation in aquatic organisms and humans [122]. Research on rodents indicates that 4-NP detrimentally affects male reproductive function, causing testicular apoptosis, seminiferous tubule degeneration, reduced testicular germ cell and Sertoli cell counts, sperm abnormalities, and diminished sperm quality, count, and viability [123]. Sertoli cells have been identified as targets of 4-NP, with studies showing its potential to induce apoptosis and autophagy in these cells upon early exposure [123]. 4-NP exposure triggers oxidative stress by increasing ROS and MDA levels while reducing SOD and CAT activities, culminating in ROS-induced AMPK activation that suppresses mTOR activity, boosts Beclin-1 expression, and enhances the LC3-II/LC3-I ratio, thereby initiating Sertoli cell autophagy potentially through the ROS-mediated AMPK-mTOR pathway [124, 125].
Tributyltin chloride (TBTCL)
Extensively employed as a biocide in antifouling paints and agricultural products, TBTCL has caused environmental and marine pollution, with human exposure which mainly stems from tainted seafood consumption, fungicide use on crops, and potential contact with organotin-stabilized polyvinyl chloride in various products like food packaging, plastics, and water pipes [126]. Exposure to TBTCL elicits a cellular response marked by increased calcium levels that stimulate the ROS generation from mitochondrial ATP production systems, disrupting the ROS-GSH balance, causing oxidative stress, and ultimately leading to cell death in Sertoli-germ cell cocultures [127]. TBTCL exposure is also linked to decreased testosterone production, triggering ER stress and inhibiting autophagy flux, leading to apoptosis and cell cycle arrest in Leydig cells [128].
Acrylamide
Acrylamide, a highly toxic compound utilized in plastics, paper production, dyes, and water treatment, is also a significant by-product formed during the high-temperature cooking of starchy foods, representing a primary source of human exposure estimated at around 1 μg/kg bw/day [129]. This environmental chemical exerts detrimental effects on biological systems, contributing to human infertility [130]. Farag et al., demonstrates that exposure to acrylamide can result in decreased sperm quality, testicular degeneration, epididymis weight loss, and disrupted steroidogenic signaling [131]. Studies further indicate that acrylamide exposure in rat testes leads to a notable increase in MDA level alongside reduced GSH level [132]. Subsequent to oxidative stress, acrylamide administration prompts a significant elevation in testicular AMPK gene expression and phosphorylated protein levels, which downregulate PI3K and mTOR, as well as pAKT content, ultimately instigating an autophagic apoptosis process in testes [133].
Pesticide
Pesticides encompass a range of classifications, such as insecticides, herbicides, fungicides, rodenticides, acaricides, and fumigants [134]. One method of categorization is based on the LD50 value, which indicates the level of toxicity of a pesticide. Pesticides are stratified as highly toxic, moderately toxic, slightly toxic, or relatively nontoxic based on their LD50 values [135]. Exposure to pesticides can transpire through four primary pathways: oral, dermal, respiratory, and ocular exposure [136]. Prior studies have revealed notable correlations between human exposure to pesticide and decreased sperm quality [137]. This review delves into the realm of insecticides, herbicides, and fungicides.
Insecticides
Globally recognized as one of the most potent pyrethroid insecticides, lambda cyhalothrin, also known as cyhalothrin, exhibits a wide range of applications and is characterized by its potency and fast action as both an insecticide and acaricide [138]. Concentrations of lambda-cyhalothrin in surface water have been documented to range from 0.35 to 0.80 μg/L, heightening the potential risk of human exposure to this substance [138]. Lambda-cyhalothrin exposure may result in the abnormal ROS accumulation, potentially leading to oxidative stress [139], which can directly damage DNA by oxidizing nucleoside groups, including the formation of 8-oxoguanine [138]. Elevated oxidative stress levels can trigger mitochondrial dysfunction during cyfluthrin-induced testicular injury, accompanied by decreased p62 levels at both protein and mRNA levels and a gradual increase in LC3 expression, indicating a potential elevation in autophagy levels [140].
The pyrethroid insecticide cypermethrin, which is extensively employed, holds the potential to provoke adverse endocrine-disrupting impacts on the male reproductive system [141]. Cypermethrin has been found to disrupt mitochondrial membrane integrity and slightly increase the levels of Sqstm1/p62 protein in the mitochondria of mouse Leydig and Sertoli cells, indicating its potential to impair mitochondrial function and inhibit mitophagy [142]. Owing to their lipophilic characteristics, pyrethroids like cypermethrin can accumulate in cellular membranes, fostering the ROS generation, which can induce oxidative harm in animals [143].
Avermectins represent broad-spectrum antiparasitic agents wildly used in agriculture and for the treatment of domestic animals [144]. Furthermore, multiple studies have illustrated the adverse impacts of avermectins on male fertility [145, 146]. Abamectin exposure at levels pertinent to both occupational and environmental settings has been associated with decreased sperm quality parameters, particularly a decline in sperm concentration [147]. Abamectin has been evidenced to instigate oxidative stress, eliciting ER stress, inflammation, apoptosis, and autophagy [148]. Oxidative stress is regarded as a pivotal element in the cytotoxicity induced by avermectins [149]. It has been postulated that exposure to avermectins prompts apoptosis and autophagy in Leydig cells by accumulating ROS, which orchestrates the suppression of the PI3K/AKT/mTOR signaling pathway [150].
Imidacloprid, known chemically as 1-(6-chloro-3-pyridylmethyl)-N-nitroimidazolidin-2-ylideneamine, is a systemic insecticide classified under the neonicotinoid group, designated by the World Health Organization (WHO) as a Class II hazardous pesticide due to its enduring presence and toxicity, it poses risks to ecosystems by potentially disrupting food chains and biogeochemical cycles [151]. Imidacloprid is known to elicit male reproductive toxicity in mammals [152]. The male reproductive organs are particularly vulnerable to the harmful impacts of ROS, with imidacloprid’s detrimental effects on the male rats’ reproductive system thought to stem from inducing oxidative stress in the testes [153]. Lysosomes and distinct autophagic vacuoles containing damaged mitochondria and other cellular organelles can be observed in Leydig cells exposed to 400 and 500 µM imidacloprid [152]. Additionally, a study elucidates that the oxidative stress induced by imidacloprid leads to mitochondrial dysfunction, subsequently activating the nuclear factor-kappa B (NF-κB)/c-Jun N-terminal kinase (JNK) pathway to modulate mitochondrial apoptosis and BNIP3-mediated mitophagy [154].
Herbicides
Glyphosate functions as a broad-spectrum herbicide and stands as one of the extensively researched pesticides [155]. The Food and Agriculture Organization (FAO) has emphasized glyphosate’s potential toxicological risks from residue accumulation in the food chain, but stated that the risk of dietary exposure is unlikely if the maximum daily intake stays below 1 mg/kg of body weight [155]. Glyphosate exposure has been linked to male reproductive impairment through the disruption of the BTB, deterioration of sperm quality and seminal parameters, and inhibition of testosterone production [156]. Previous research on sperm has indicated that impaired spermatogenesis following glyphosate exposure is associated with oxidative stress induced by excessive ROS [157]. Glyphosate-induced dysfunction in TM3 cells, featuring abnormal mitochondria, disrupted dynamics, increased mitochondrial ROS production, decreased steroidogenic enzyme levels, and suppressed testosterone synthesis, was linked to heightened autophagic flux and mitophagy, the latter dependent on Parkin activation [158].
During the cultivation of various crops like winter wheat, potatoes, sunflowers, sugar canes and cotton, a pyrrolidone herbicide known for its selective properties, namely flurochloridone, is utilized to regulate a diverse range of broadleaf weeds and grasses [159]. Studies suggest that fluorochloridone may function as a potential endocrine disruptor, exerting adverse effects on the reproductive functions and hormonal equilibrium in male rats [160]. Fluorochloridone exposure has been associated with ROS buildup, mitochondrial dysfunction, and the initiation of cell apoptosis in Sertoli cells [161]. In live organisms, fluorochloridone promotes the formation of autophagosomes and elevates the levels of LC3II/LC3I, Beclin-1, and p62 proteins, correlating with autophagic degradation [162].
Fungicides
Thiram, a member of the dimethyldithiocarbamate (DDC) fungicide group, is used in rubber, plastic, and agricultural industries to protect crops and seeds from fungal infections [163]. Improper management or storage of thiram in chemical facilities and warehouses can lead to environmental contamination, given its widespread use in agriculture [163]. Prolonged exposure to thiram can cause sensitization and reproductive issues, as evidenced by altered gene expression in testicular cells, indicating potential autophagy induction via the mTOR/Atg5/p62 pathway, particularly at higher concentrations [164]. The recent study emphasizes that elevated thiram levels can adversely affect the testes by disrupting the BTB, leading to testicular tissue damage marked by decreased ZO-1 and Occludin mRNA expression, fibrosis promotion, and increased intercellular space [164]. Treatment with Thiram has also been demonstrated to elevate ROS production, reduce GSH levels, and induce oxidative stress [165].
Intervention strategies for testicular injury mediated by environmental exposures
Currently, numerous antioxidants are utilized to ameliorate testicular injury by modulating oxidative stress or autophagy. In cases of testicular damage induced by environmental exposure through oxidative stress-mediated autophagy, therapeutic interventions typically employ various strategies to mitigate testicular harm: (1) through antioxidative mechanisms, involving the neutralization of free radicals, activation of antioxidant enzymes, and modulation of regulatory transcription factors, such as the Nrf2/HO-1 signaling pathway. (2) via anti-autophagic approaches, including the inhibition of autophagy-related proteins like Beclin-1 and LC3II, as well as modulation of signaling pathways influencing the autophagy process, such as the AMPK/mTOR signaling pathways.
Antioxidants form a comprehensive defense mechanism against ROS
Antioxidant defense systems, incorporating enzymatic and non-enzymatic elements, serve to protect cellular and organ integrity from the deleterious impacts of free radicals. These antioxidants can either be endogenously synthesized or obtained from external sources like dietary intake or nutritional supplements [166]. Dietary and endogenous enzymatic and non-enzymatic antioxidants employ various strategies, such as electron donation, catalytic removal, or radical binding, as well as gene expression regulation, to counteract the deleterious impacts of free radicals [20].
-
(1)
Free Radical Neutralization: Antioxidants play a crucial role in neutralizing free radicals by donating electrons, thereby shielding against damage. Vitamin E, in its various forms, exhibits potent antioxidant properties by neutralizing lipid peroxyl radicals through hydrogen provision from the phenolic group on the chromanol ring [167]. Vitamin E can potentially mitigate the surge in free radical production induced by acrylamide in testicular tissues [168]. Additionally, Vitamin E treatment significantly reduces the impact of formaldehyde exposure on testicular structure, sperm quantity, and quality, attributed to its direct free radical scavenging abilities and interaction with membrane phospholipid bilayers to halt ROS-initiated chain reactions [90, 91]. Additionally, the hydroxyl group within the structure of carvacrol also contributes to mitigating spermatid differentiation disorders by reducing sodium arsenite-induced oxidative stress, inflammation, apoptosis, and autophagy [169].
-
(2)
Activation of Antioxidant Enzymes: Enzymes like SOD, CAT, glutathione peroxidase (GPx), among others, are integral components of the antioxidant defense system [170]. Quercetin, an antioxidant, effectively counteracts the decrease in activities of GSH, SOD, and GPx induced by cadmium exposure [171]. Furthermore, Açai berry demonstrates a significant elevation in Nrf2 and HO-1 levels, thereby enhancing the physiological antioxidant response by boosting CAT and GSH activity in cyclophosphamide-induced genitourinary damage [172].
-
(3)
Regulatory Transcription Factor: Nrf2 orchestrates antioxidant responses by activating defensive genes like heme oxygenase-1 (HO-1), glutathione-S-transferases, and NADPH quinone oxidoreductase 1, crucial for scavenging ROS and safeguarding cells from oxidative stress damage [173]. Studies indicate that azoramide treatment can inhibit the upregulation of Nrf2 induced by cadmium, hinting at its potential to suppress ROS production and mitigate cadmium induced mitochondrial injury [174]. Açai berry has also been shown to elevate Nrf2 and HO-1 levels in testes [172]. Eugenol exhibits the ability to reduce acrylamide induced ROS overproduction by facilitating testicular Nrf2 nuclear translocation and AKT phosphorylation, contrasting the effects observed in the acrylamide challenged group [133]. Additionally, the administration of Naringin holds promise in improving testicular damages by reversing the expression levels of p53, MAPK14, Caspase-3, and Bax proteins [132].
Regulation of autophagy-related proteins and pathways
Proteins implicated in autophagy play distinct roles in governing this cellular process. Beclin-1 and LC3 act as pivotal markers of autophagic flux. Nano‑selenium has demonstrated efficacy in mitigating cadmium-induced disruption of autophagy by modulating signaling pathways associated with Beclin-1 and LC3 in Leydig cells [46]. Lactoferrin significantly alleviated spermatogenetic dysfunction by reducing the heightened ratios of BAX/BCL2 and LC3II/LC3I, and p62 protein expression [106]. In rats treated with carvacrol, the autophagy and inflammation triggered by sodium arsenite in testes were notably diminished, attributed to the downregulation of biomarkers such as LC3, MAPK-14, NF-κB, TNF-α, IL-1β, iNOS, and COX-2 [170]. Some pharmaceutical agents can alleviate testicular damage by boosting autophagy, as indicated by the decrease in p62 level and LC3II/LC3I ratio in the cadmium+quercetin group, implying quercetin’s ameliorative effect on cadmium-induced autophagy [171]. Resveratrol treatment enhanced cell viability, SOD activity, and anti-apoptotic effects in nicotine-exposed Leydig cells, potentially offering cytoprotective benefits against oxidative damage through autophagy activation via the AMPK/mTOR pathway [87]. Meanwhile, eugenol treatment significantly enhanced sperm quality parameters through the improvement of ROS-mediated autophagy, apoptosis, and BTB remodeling [133]. Furthermore, sitagliptin, acting as a selective DPP4 inhibitor, can attenuate the activity of the ERK and AKT pathways while suppressing the AMPK signaling cascade, all of which are involved in autophagy regulation [175].
Conclusion
In summary, this review consolidates existing literature on how environmental exposures contribute to testicular damage by disrupting cellular autophagy and oxidative stress balance, showcasing recent significant progress. Despite varying study designs and methodologies, a consensus is emerging on how environmental factors trigger oxidative stress by elevating ROS levels, impacting autophagy via pathways like ROS-mediated mTOR pathway, ultimately leading to testicular damage. Moreover, the interplay between autophagy and oxidative stress, particularly the relationship between p62 and Nrf2, underscores their crucial roles. Furthermore, certain drugs demonstrate the potential to mitigate testicular damage by modulating oxidative stress and autophagy. These discoveries emphasize the necessity of elucidating the mechanisms of autophagy influenced by environmental exposures in disrupting oxidative stress equilibrium, pinpointing drug targets, and laying a foundation for optimizing future treatments and clinical management of testicular injuries.
References
Yang W, Hua R, Cao Y, He X. A metabolomic perspective on the mechanisms by which environmental pollutants and lifestyle lead to male infertility. Andrology. 2024;12:719–39.
Chen X, Zhang X, Jiang T, Xu W. Klinefelter syndrome: etiology and clinical considerations in male inf ertility. Biol Reprod. 2024;111:516–28.
Bhattacharya I, Sharma SS, Majumdar SS. Etiology of Male Infertility: an Update. Reprod Sci. 2024;31:942–65.
Wong EW, Cheng CY. Impacts of environmental toxicants on male reproductive dysfunction. Trends Pharm Sci. 2011;32:290–9.
Li H, Wang XR, Hu YF, Xiong YW, Zhu HL, Huang YC, et al. Advances in immunology of male reproductive toxicity induced by common environmental pollutants. Environ Int. 2024;190:108898.
Sharpe RM. Environmental/lifestyle effects on spermatogenesis. Philos Trans R Soc Lond B Biol Sci. 2010;365:1697–712.
Zhang Y, Song JY, Sun ZG. Exploring the impact of environmental factors on male reproductive health through epigenetics. Reprod Toxicol. 2025;132:108832.
Eslami H, Askari FR, Mahdavi M, Taghavi M, Ghaseminasab-Parizi M. Environmental arsenic exposure and reproductive system toxicity in male and female and mitigatory strategies: a review. Environ Geochem Health. 2024;46:420.
Qin Z, Song J, Huang J, Jiang S, Zhang G, Huang M, et al. Mitigation of triptolide-induced testicular Sertoli cell damage by mel atonin via regulating the crosstalk between SIRT1 and NRF2. Phytomedicine. 2023;118:154945.
Huynh PN, Hikim AP, Wang C, Stefonovic K, Lue YH, Leung A, et al. Long-term effects of triptolide on spermatogenesis, epididymal sperm f unction, and fertility in male rats. J Androl. 2000;21:689–99.
Ma B, Zhang J, Zhu Z, Bao X, Zhang M, Ren C, et al. Aucubin, a natural iridoid glucoside, attenuates oxidative stress-induced testis injury by inhibiting JNK and CHOP activation via Nrf2 up-regulation. Phytomedicine. 2019;64:153057.
Yang X, He L, Li X, Wang L, Bu T, Yun D, et al. Triptolide exposure triggers testicular vacuolization injury by disrupting the Sertoli cell junction and cytoskeletal organization via the AKT/mTOR signaling pathway. Ecotoxicol Environ Saf. 2024;279:116502.
Gao H, Khawar MB, Li W. Autophagy in reproduction. Adv Exp Med Biol. 2019;1206:453–68.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41.
Zhu Y, Yin Q, Wei D, Yang Z, Du Y, Ma Y. Autophagy in male reproduction. Syst Biol Reprod Med. 2019;65:265–72.
Ornatowski W, Lu Q, Yegambaram M, Garcia AE, Zemskov EA, Maltepe E, et al. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020;36:101679.
Thévenod F, Lee WK. Live and let die: roles of autophagy in cadmium nephrotoxicity. Toxics. 2015;3:130–51.
Janciauskiene S. The beneficial effects of antioxidants in health and diseases. Chronic Obstr Pulm Dis. 2020;7:182–202.
Sharma P, Kaushal N, Saleth LR, Ghavami S, Dhingra S, Kaur P. Oxidative stress-induced apoptosis and autophagy: balancing the contrary forces in spermatogenesis. Biochim Biophys Acta Mol Basis Dis. 2023;1869:166742.
Almansa-Ordonez A, Bellido R, Vassena R, Barragan M, Zambelli F. Oxidative stress in reproduction: a mitochondrial perspective. Biology (Basel). 2020;9:269.
Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377–88.
Sachdev U, Lotze MT. Perpetual change: autophagy, the endothelium, and response to vascular injury. J Leukoc Biol. 2017;102:221–35.
Springer MZ, Macleod KF. In Brief: mitophagy: mechanisms and role in human disease. J Pathol. 2016;240:253–5.
Ling Y, Li Y, Zhu R, Qian J, Liu J, Gao W, et al. Hydroxamic acid derivatives of β-carboline/hydroxycinnamic acid hybrids inducing apoptosis and autophagy through the PI3K/Akt/mTOR pathways. J Nat Prod. 2019;82:1442–50.
Zhu P, Qian J, Xu Z, Meng C, Liu J, Shan W, et al. Piperlonguminine and piperine analogues as TrxR inhibitors that promote ROS and autophagy and regulate p38 and Akt/mTOR signaling. J Nat Prod. 2020;83:3041–9.
Xu X, Su YL, Shi JY, Lu Q, Chen C. MicroRNA-17-5p promotes cardiac hypertrophy by targeting Mfn2 to inhibit autophagy. Cardiovasc Toxicol. 2021;21:759–71.
Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–462.
Zhang L, Qin Z, Li R, Wang S, Wang W, Tang M, et al. The role of ANXA5 in DBP-induced oxidative stress through ERK/Nrf2 pathway. Environ Toxicol Pharm. 2019;72:103236.
Garza-Lombó C, Pappa A, Panayiotidis MI, Franco R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion. 2020;51:105–17.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86.
Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35:615–21.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23.
Han X, Huang Q. Environmental pollutants exposure and male reproductive toxicity: The role of epigenetic modifications. Toxicology. 2021;456:152780.
Bellingham M, Evans N. Impact of real-life environmental exposures on reproduction: biosolids and male reproduction. Reproduction. 2024;168:e240119.
Marini HR, Micali A, Squadrito G, Puzzolo D, Freni J, Antonuccio P, et al. Nutraceuticals: a new challenge against cadmium-induced testicular injury. Nutrients. 2022;14:663.
Xiao C, Lai D. Impact of oxidative stress induced by heavy metals on ovarian function. J Appl Toxicol. 2024;45:107–16.
Roychoudhury S, Chakraborty S, Choudhury AP, Das A, Jha NK, Slama P, et al. Environmental factors-induced oxidative stress: hormonal and molecular pathway disruptions in hypogonadism and erectile dysfunction. Antioxidants (Basel). 2021;10:837.
Estévez-Danta A, Montes R, Prieto A, Santos MM, Orive G, Lertxundi U, et al. Wastewater-based epidemiology methodology to investigate human exposure to bisphenol A, bisphenol F and bisphenol S. Water Res. 2024;261:122016.
Hassanin HM, Kamal AA, Ismail OI. Resveratrol ameliorates atrazine-induced caspase-dependent apoptosis and fibrosis in the testis of adult albino rats. Sci Rep. 2024;14:17743.
Saka WA, Adeogun AE, Adisa VI, Olayioye A, Igbayilola YD, Akhigbe RE. L-arginine attenuates dichlorvos-induced testicular toxicity in male Wistar rats by suppressing oxidative stress-dependent activation of caspase 3-mediated apoptosis. Biomed Pharmacother. 2024;178:117136.
Satarug S, Garrett SH, Sens MA, Sens DA. Cadmium, environmental exposure, and health outcomes. Environ Health Perspect. 2010;118:182–90.
Satarug S, Vesey DA, Gobe GC, Phelps KR. Estimation of health risks associated with dietary cadmium exposure. Arch Toxicol. 2023;97:329–58.
Ali W, Ma Y, Zhu J, Zou H, Liu Z. Mechanisms of cadmium-induced testicular injury: a risk to male fertility. Cells. 2022;11:3601.
Ali W, Deng K, Sun J, Ma Y, Liu Z, Zou H. A new insight of cadmium-induced cellular evidence of autophagic-associated spermiophagy during spermatogenesis. Environ Sci Pollut Res Int. 2023;30:101064–74.
Wang M, Wang XF, Li YM, Chen N, Fan Y, Huang WK, et al. Cross-talk between autophagy and apoptosis regulates testicular injury/recovery induced by cadmium via PI3K with mTOR-independent pathway. Cell Death Dis. 2020;11:46.
Hu X, Lin R, Zhang C, Pian Y, Luo H, Zhou L, et al. Nano-selenium alleviates cadmium-induced mouse leydig cell injury, via the inhibition of reactive oxygen species and the restoration of autophagic flux. Reprod Sci. 2023;30:1808–22.
Arab HH, Gad AM, Reda E, Yahia R, Eid AH. Activation of autophagy by sitagliptin attenuates cadmium-induced testicular impairment in rats: targeting AMPK/mTOR and Nrf2/HO-1 pathways. Life Sci. 2021;269:119031.
Chandra A, Shah KA. Chronic arsenic poisoning. N Engl J Med. 2022;387:1414.
Moore LE, Karami S, Steinmaus C, Cantor KP. Use of OMIC technologies to study arsenic exposure in human populations. Environ Mol Mutagen. 2013;54:589–95.
Rahman MM, Ng JC, Naidu R. Chronic exposure of arsenic via drinking water and its adverse health impacts on humans. Environ Geochem Health. 2009;31:189–200.
Buekers J, Baken K, Govarts E, Martin LR, Vogel N, Kolossa-Gehring M, et al. Human urinary arsenic species, associated exposure determinants and potential health risks assessed in the HBM4EU Aligned Studies. Int J Hyg Environ Health. 2023;248:114115.
Renu K, Madhyastha H, Madhyastha R, Maruyama M, Vinayagam S, Valsala Gopalakrishnan A. Review on molecular and biochemical insights of arsenic-mediated male reproductive toxicity. Life Sci. 2018;212:37–58.
Liu Y, Tang J, Yuan J, Yao C, Hosoi K, Han Y, et al. Arsenite-induced downregulation of occludin in mouse lungs and BEAS-2B cells via the ROS/ERK/ELK1/MLCK and ROS/p38 MAPK signaling pathways. Toxicol Lett. 2020;332:146–54.
Liang C, Feng Z, Manthari RK, Wang C, Han Y, Fu W, et al. Arsenic induces dysfunctional autophagy via dual regulation of mTOR pathway and Beclin1-Vps34/PI3K complex in MLTC-1 cells. J Hazard Mater. 2020;391:122227.
Wu S, Zhong G, Wan F, Jiang X, Tang Z, Hu T, et al. Evaluation of toxic effects induced by arsenic trioxide or/and antimony on autophagy and apoptosis in testis of adult mice. Environ Sci Pollut Res Int. 2021;28:54647–60.
Huang Q, Li J, Qi Y, He X, Shen C, Wang C, et al. Copper overload exacerbates testicular aging mediated by lncRNA:CR43306 deficiency through ferroptosis in Drosophila. Redox Biol. 2024;76:103315.
Tvrda E, Peer R, Sikka SC, Agarwal A. Iron and copper in male reproduction: a double-edged sword. J Assist Reprod Genet. 2015;32:3–16.
Shao Y, Zhao H, Wang Y, Liu J, Zong H, Xing M. Copper-mediated mitochondrial fission/fusion is associated with intrinsic apoptosis and autophagy in the testis tissues of chicken. Biol Trace Elem Res. 2019;188:468–77.
Kang Z, Qiao N, Liu G, Chen H, Tang Z, Li Y. Copper-induced apoptosis and autophagy through oxidative stress-mediated mitochondrial dysfunction in male germ cells. Toxicol Vitr. 2019;61:104639.
Azeez NA, Dash SS, Gummadi SN, Deepa VS. Nano-remediation of toxic heavy metal contamination: Hexavalent chromium [Cr(VI)]. Chemosphere. 2021;266:129204.
Coyte RM, McKinley KL, Jiang S, Karr J, Dwyer GS, Keyworth AJ, et al. Occurrence and distribution of hexavalent chromium in groundwater from North Carolina, USA. Sci Total Environ. 2020;711:135135.
Aruldhas MM, Subramanian S, Sekar P, Vengatesh G, Chandrahasan G, Govindarajulu P, et al. Chronic chromium exposure-induced changes in testicular histoarchitecture are associated with oxidative stress: study in a non-human primate (Macaca radiata Geoffroy). Hum Reprod. 2005;20:2801–13.
Wang R, Huang Y, Yu L, Li S, Li J, Han B, et al. The role of mitochondrial dynamics imbalance in hexavalent chromium-induced apoptosis and autophagy in rat testis. Chem Biol Interact. 2023;374:110424.
Griffitt RJ, Luo J, Gao J, Bonzongo JC, Barber DS. Effects of particle composition and species on toxicity of metallic nanomaterials in aquatic organisms. Environ Toxicol Chem. 2008;27:1972–8.
Yang Q, Li F, Miao Y, Luo X, Dai S, Liu J, et al. CdSe/ZnS quantum dots induced spermatogenesis dysfunction via autophagy activation. J Hazard Mater. 2020;398:122327.
Alanazi FK, Radwan AA, Alsarra IA. Biopharmaceutical applications of nanogold. Saudi Pharm J. 2010;18:179–93.
Taylor U, Barchanski A, Garrels W, Klein S, Kues W, Barcikowski S, et al. Toxicity of gold nanoparticles on somatic and reproductive cells. Adv Exp Med Biol. 2012;733:125–33.
Wilner OI, Willner B, Willner I. DNA nanotechnology. Adv Exp Med Biol. 2012;733:97–114.
Liu Y, Li X, Xiao S, Liu X, Chen X, Xia Q, et al. The effects of gold nanoparticles on leydig cells and male reproductive function in mice. Int J Nanomed. 2020;15:9499–514.
Duran B, Perçin E, Aydın A, Destegül A, Aydin D, Özpınar N, et al. Biogenic nanocopper: eco-friendly synthesis, characterization, and their anti-trichomonas vaginalis, anticancer, and antimicrobial effects. J Med Food. 2022;25:787–92.
Chen H, Wang Y, Luo J, Kang M, Hou J, Tang R, et al. Autophagy and apoptosis mediated nano-copper-induced testicular damage. Ecotoxicol Environ Saf. 2022;229:113039.
Ahamed M, Alsalhi MS, Siddiqui MK. Silver nanoparticle applications and human health. Clin Chim Acta. 2010;411:1841–8.
Nazari M, Shabani R, Ajdary M, Ashjari M, Shirazi R, Govahi A, et al. Effects of Au@Ag core-shell nanostructure with alginate coating on male reproductive system in mice. Toxicol Rep. 2023;10:104–16.
Jiang L, Lin X, Jiang J, Qiu C, Zheng S, Zhao N, et al. METTL3-m6A-SIRT1 axis affects autophagic flux contributing to PM(2.5)-induced inhibition of testosterone production in Leydig cells. Sci Total Environ. 2024;918:170701.
Zhou L, Su X, Li B, Chu C, Sun H, Zhang N, et al. PM2.5 exposure impairs sperm quality through testicular damage dependent on NALP3 inflammasome and miR-183/96/182 cluster targeting FOXO1 in mouse. Ecotoxicol Environ Saf. 2019;169:551–63.
Wei Y, Cao XN, Tang XL, Shen LJ, Lin T, He DW, et al. Urban fine particulate matter (PM2.5) exposure destroys blood-testis barrier (BTB) integrity through excessive ROS-mediated autophagy. Toxicol Mech Methods. 2018;28:302–19.
Liu X, Ai Y, Xiao M, Wang C, Shu Z, Yin J, et al. PM 2.5 juvenile exposure-induced spermatogenesis dysfunction by triggering testes ferroptosis and antioxidative vitamins intervention in adult male rats. Environ Sci Pollut Res Int. 2023;30:111051–61.
Cao XN, Yan C, Liu DY, Peng JP, Chen JJ, Zhou Y, et al. Fine particulate matter leads to reproductive impairment in male rats by overexpressing phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway. Toxicol Lett. 2015;237:181–90.
Deng X, Rui W, Zhang F, Ding W. PM2.5 induces Nrf2-mediated defense mechanisms against oxidative stress by activating PIK3/AKT signaling pathway in human lung alveolar epithelial A549 cells. Cell Biol Toxicol. 2013;29:143–57.
Gu HJ, Ahn JS, Ahn GJ, Shin SH, Ryu BY. Restoration of PM2.5-induced spermatogonia GC-1 cellular damage by parthenolide via suppression of autophagy and inflammation: An in vitro study. Toxicology. 2023;499:153651.
Zarrelli A, DellaGreca M, Parolisi A, Iesce MR, Cermola F, Temussi F, et al. Chemical fate and genotoxic risk associated with hypochlorite treatment of nicotine. Sci Total Environ. 2012;426:132–8.
de Franco MA, da Silva WL, Bagnara M, Lansarin MA, Dos Santos JH. Photocatalytic degradation of nicotine in an aqueous solution using unconventional supported catalysts and commercial ZnO/TiO2 under ultraviolet radiation. Sci Total Environ. 2014;494-495:97–103.
Mayer B. How much nicotine kills a human? Tracing back the generally accepted lethal dose to dubious self-experiments in the nineteenth century. Arch Toxicol. 2014;88:5–7.
Price LR, Martinez J. Cardiovascular, carcinogenic and reproductive effects of nicotine exposure: A narrative review of the scientific literature. F1000Res. 2019;8:1586.
Zhang Z, Jiang Z, Cheng J, Price CA, Yang L, Li Q. Nicotine induces senescence in spermatogonia stem cells by disrupting homeostasis between circadian oscillation and rhythmic mitochondrial dynamics via the SIRT6/Bmal1 pathway. Life Sci. 2024;352:122860.
Budin SB, Kho JH, Lee JH, Ramalingam A, Jubaidi FF, Latif ES, et al. Low-dose nicotine exposure induced the oxidative damage of reproductive organs and altered the sperm characteristics of adolescent male rats. Malays J Med Sci. 2017;24:50–57.
Liu S, Sun Y, Li Z. Resveratrol protects Leydig cells from nicotine-induced oxidative damage through enhanced autophagy. Clin Exp Pharm Physiol. 2018;45:573–80.
Benedict B, Kristensen SM, Duxin JP. What are the DNA lesions underlying formaldehyde toxicity?. DNA Repair (Amst). 2024;138:103667.
Fang J, Li DH, Yu XQ, Lv MQ, Bai LZ, Du LZ, et al. Formaldehyde exposure inhibits the expression of mammalian target of rapamycin in rat testis. Toxicol Ind Health. 2016;32:1882–90.
Han SP, Zhou DX, Lin P, Qin Z, An L, Zheng LR, et al. Formaldehyde exposure induces autophagy in testicular tissues of adult male rats. Environ Toxicol. 2015;30:323–31.
Zhou DX, Qiu SD, Zhang J, Tian H, Wang HX. The protective effect of vitamin E against oxidative damage caused by formaldehyde in the testes of adult rats. Asian J Androl. 2006;8:584–8.
Moghe A, Ghare S, Lamoreau B, Mohammad M, Barve S, McClain C, et al. Molecular mechanisms of acrolein toxicity: relevance to human disease. Toxicol Sci. 2015;143:242–55.
Moazamian R, Polhemus A, Connaughton H, Fraser B, Whiting S, Gharagozloo P, et al. Oxidative stress and human spermatozoa: diagnostic and functional significance of aldehydes generated as a result of lipid peroxidation. Mol Hum Reprod. 2015;21:502–15.
Mao Z, Li H, Zhao XL, Zeng XH. Hydrogen sulfide protects Sertoli cells against toxicant Acrolein-induced cell injury. Food Chem Toxicol. 2023;176:113784.
Gu YP, Yang XM, Luo P, Li YQ, Tao YX, Duan ZH, et al. Inhibition of acrolein-induced autophagy and apoptosis by a glycosaminoglycan from Sepia esculenta ink in mouse Leydig cells. Carbohydr Polym. 2017;163:270–9.
Gigault J, Halle AT, Baudrimont M, Pascal PY, Gauffre F, Phi TL, et al. Current opinion: What is a nanoplastic?. Environ Pollut. 2018;235:1030–4.
Kumar R, Manna C, Padha S, Verma A, Sharma P, Dhar A, et al. Micro(nano)plastics pollution and human health: How plastics can induce carcinogenesis to humans?. Chemosphere. 2022;298:134267.
Ma T, Liu X, Xiong T, Li H, Zhou Y, Liang J. Polystyrene nanoplastics aggravated dibutyl phthalate-induced blood-testis barrier dysfunction via suppressing autophagy in male mice. Ecotoxicol Environ Saf. 2023;264:115403.
Amereh F, Babaei M, Eslami A, Fazelipour S, Rafiee M. The emerging risk of exposure to nano(micro)plastics on endocrine disturbance and reproductive toxicity: From a hypothetical scenario to a global public health challenge. Environ Pollut. 2020;261:114158.
Hu Y, Shen M, Wang C, Huang Q, Li R, Dorj G, et al. A meta-analysis-based adverse outcome pathway for the male reproductive toxicity induced by microplastics and nanoplastics in mammals. J Hazard Mater. 2024;465:133375.
Li S, Ma Y, Ye S, Su Y, Hu D, Xiao F. Endogenous hydrogen sulfide counteracts polystyrene nanoplastics-induced mitochondrial apoptosis and excessive autophagy via regulating Nrf2 and PGC-1α signaling pathway in mouse spermatocyte-derived GC-2spd(ts) cells. Food Chem Toxicol. 2022;164:113071.
Tian Y, Song W, Xu D, Chen X, Li X, Zhao Y. Autophagy induced by ROS aggravates testis oxidative damage in diabetes via breaking the feedforward loop linking p62 and Nrf2. Oxid Med Cell Longev. 2020;2020:7156579.
Jiao JH, Gao L, Yong WL, Kou ZY, Ren ZQ, Cai R, et al. Resveratrol improves estrus disorder induced by bisphenol A through attenuating oxidative stress, autophagy, and apoptosis. J Biochem Mol Toxicol. 2022;36:e23120.
Wang K, Qiu L, Zhu J, Sun Q, Qu W, Yu Y, et al. Environmental contaminant BPA causes intestinal damage by disrupting cellular repair and injury homeostasis in vivo and in vitro. Biomed Pharmacother. 2021;137:111270.
Li X, Ying GG, Su HC, Yang XB, Wang L. Simultaneous determination and assessment of 4-nonylphenol, bisphenol A and triclosan in tap water, bottled water and baby bottles. Environ Int. 2010;36:557–62.
He H, Chen X, Li X, Yang K, Li J, Shi H. Lactoferrin alleviates spermatogenesis dysfunction caused by bisphenol A and cadmium via ameliorating disordered autophagy, apoptosis and oxidative stress. Int J Biol Macromol. 2022;222:1048–62.
Quan C, Wang C, Duan P, Huang W, Chen W, Tang S, et al. Bisphenol a induces autophagy and apoptosis concurrently involving the Akt/mTOR pathway in testes of pubertal SD rats. Environ Toxicol. 2017;32:1977–89.
Fu G, Dai J, Li Z, Chen F, Liu L, Yi L, et al. The role of STAT3/p53 and PI3K-Akt-mTOR signaling pathway on DEHP-induced reproductive toxicity in pubertal male rat. Toxicol Appl Pharm. 2020;404:115151.
Yang L, Jiang L, Sun X, Li J, Wang N, Liu X, et al. DEHP induces ferroptosis in testes via p38α-lipid ROS circulation and destroys the BTB integrity. Food Chem Toxicol. 2022;164:113046.
Sun Y, Shen J, Zeng L, Yang D, Shao S, Wang J, et al. Role of autophagy in di-2-ethylhexyl phthalate (DEHP)-induced apoptosis in mouse Leydig cells. Environ Pollut. 2018;243:563–72.
Yi WEI, Xiang-Liang T, Yu Z, Bin L, Lian-Ju S, Chun-Lan L, et al. DEHP exposure destroys blood-testis barrier (BTB) integrity of immature testes through excessive ROS-mediated autophagy. Genes Dis. 2018;5:263–74.
Hong Y, Zhou Y, Shen L, Wei Y, Long C, Fu Y, et al. Exposure to DEHP induces testis toxicity and injury through the ROS/mTOR/NLRP3 signaling pathway in immature rats. Ecotoxicol Environ Saf. 2021;227:112889.
Antonopoulou M, Spyrou A, Tzamaria A, Efthimiou I, Triantafyllidis V. Current state of knowledge of environmental occurrence, toxic effects, and advanced treatment of PFOS and PFOA. Sci Total Environ. 2024;913:169332.
Wan HT, Lai KP, Wong CKC. Comparative Analysis of PFOS and PFOA Toxicity on Sertoli Cells. Environ Sci Technol. 2020;54:3465–75.
Ou J, Song Y, Zhong X, Dai L, Chen J, Zhang W, et al. Perfluorooctanoic acid induces Leydig cell injury via inhibition of autophagosomes formation and activation of endoplasmic reticulum stress. Sci Total Environ. 2024;917:169861.
Chen Z, Chen Z, Gao S, Shi J, Li X, Sun F. PFOS exposure destroys the integrity of the blood-testis barrier (BTB) through PI3K/AKT/mTOR-mediated autophagy. Reprod Biol. 2024;24:100846.
Qiu L, Wang H, Dong T, Huang J, Li T, Ren H, et al. Perfluorooctane sulfonate (PFOS) disrupts testosterone biosynthesis via CREB/CRTC2/StAR signaling pathway in Leydig cells. Toxicology. 2021;449:152663.
Qu J, Han Y, Zhao Z, Wu Y, Lu Y, Chen G, et al. Perfluorooctane sulfonate interferes with non-genomic estrogen receptor signaling pathway, inhibits ERK1/2 activation and induces apoptosis in mouse spermatocyte-derived cells. Toxicology. 2021;460:152871.
Liu W, Yang B, Wu L, Zou W, Pan X, Zou T, et al. Involvement of NRF2 in perfluorooctanoic acid-induced testicular damage in male mice. Biol Reprod. 2015;93:41.
Yang Y, Fu G, Zhao X, Wu X, Zhu K, Liu S, et al. Perfluorooctanoic acid induces tight junction injury of Sertoli cells by blocking autophagic flux. Food Chem Toxicol. 2023;173:113649.
Ying GG, Williams B, Kookana R. Environmental fate of alkylphenols and alkylphenol ethoxylates-a review. Environ Int. 2002;28:215–26.
Duan P, Hu C, Butler HJ, Quan C, Chen W, Huang W, et al. 4-Nonylphenol induces disruption of spermatogenesis associated with oxidative stress-related apoptosis by targeting p53-Bcl-2/Bax-Fas/FasL signaling. Environ Toxicol. 2017;32:739–53.
Duan P, Hu C, Quan C, Yu T, Zhou W, Yuan M, et al. 4-Nonylphenol induces apoptosis, autophagy and necrosis in Sertoli cells: Involvement of ROS-mediated AMPK/AKT-mTOR and JNK pathways. Toxicology. 2016;341-343:28–40.
Tao S, Yao Z, Li H, Wang Y, Qiao X, Yu Y, et al. Exposure to 4-nonylphenol compromises Leydig cell development in pubertal male mice. Ecotoxicol Environ Saf. 2023;266:115612.
Duan P, Hu C, Quan C, Yu T, Huang W, Chen W, et al. 4-Nonylphenol induces autophagy and attenuates mTOR-p70S6K/4EBP1 signaling by modulating AMPK activation in Sertoli cells. Toxicol Lett. 2017;267:21–31.
Beyer J, Song Y, Tollefsen KE, Berge JA, Tveiten L, Helland A, et al. The ecotoxicology of marine tributyltin (TBT) hotspots: a review. Mar Environ Res. 2022;179:105689.
Mitra S, Srivastava A, Khandelwal S. Tributyltin chloride induced testicular toxicity by JNK and p38 activation, redox imbalance and cell death in sertoli-germ cell co-culture. Toxicology. 2013;314:39–50.
Chen P, Song Y, Tang L, Zhong W, Zhang J, Cao M, et al. Tributyltin chloride (TBTCL) induces cell injury via dysregulation of endoplasmic reticulum stress and autophagy in Leydig cells. J Hazard Mater. 2023;448:130785.
Matoso V, Bargi-Souza P, Ivanski F, Romano MA, Romano RM. Acrylamide: a review about its toxic effects in the light of developmental origin of health and disease (DOHaD) concept. Food Chem. 2019;283:422–30.
Alturki HA, Elsawy HA, Famurewa AC. Silymarin abrogates acrylamide-induced oxidative stress-mediated testicular toxicity via modulation of antioxidant mechanism, DNA damage, endocrine deficit and sperm quality in rats. Andrologia. 2022;54:e14491.
Farag OM, Abd-Elsalam RM, El Badawy SA, Ogaly HA, Alsherbiny MA, Ahmed KA. Portulaca oleracea seeds’ extract alleviates acrylamide-induced testicular dysfunction by promoting oxidative status and steroidogenic pathway in rats. BMC Complement Med Ther. 2021;21:122.
Sengul E, Gelen V, Yildirim S, Cinar İ, Aksu EH. Effects of naringin on oxidative stress, inflammation, some reproductive parameters, and apoptosis in acrylamide-induced testis toxicity in rat. Environ Toxicol. 2023;38:798–808.
Saleh DO, Baraka SM, Jaleel GAA, Hassan A, Ahmed-Farid OA. Eugenol alleviates acrylamide-induced rat testicular toxicity by modulating AMPK/p-AKT/mTOR signaling pathway and blood-testis barrier remodeling. Sci Rep. 2024;14:1910.
Tudi M, Daniel Ruan H, Wang L, Lyu J, Sadler R, Connell D, et al. Agriculture development, pesticide application and its impact on the environment. Int J Environ Res Public Health. 2021;18:1112.
El-Nahhal I, El-Nahhal Y. Data on estimation of health hazards associated with pesticide residues in drinking water. Data Brief. 2022;41:107830.
Tănăsescu EC, Lite MC. Harmful health effects of pesticides used on museum textile artifacts - overview. Ecotoxicol Environ Saf. 2022;247:114240.
Knapke ET, Magalhaes DP, Dalvie MA, Mandrioli D, Perry MJ. Environmental and occupational pesticide exposure and human sperm parameters: A Navigation Guide review. Toxicology. 2022;465:153017.
Xu X, Yu Y, Ling M, Ares I, Martínez M, Lopez-Torres B, et al. Oxidative stress and mitochondrial damage in lambda-cyhalothrin toxicity: a comprehensive review of antioxidant mechanisms. Environ Pollut. 2023;338:122694.
Dong B, Liu XY, Li B, Li MY, Li SG, Liu S. A heat shock protein protects against oxidative stress induced by lambda-cyhalothrin in the green peach aphid Myzus persicae. Pestic Biochem Physiol. 2022;181:104995.
Zhao J, Ma LY, Xie YX, Zhu LQ, Ni WS, Wang R, et al. The role of stimulator of interferon genes-mediated AMPK/mTOR/P70S6K autophagy pathway in cyfluthrin-induced testicular injury. Environ Toxicol. 2023;38:727–42.
Li S, Wang Y, Zou C, Zhu Q, Wang Y, Chen H, et al. Cypermethrin inhibits Leydig cell development and function in pubertal rats. Environ Toxicol. 2022;37:1160–72.
Ham J, Jang H, Song G, Lim W. Cypermethrin induces endoplasmic reticulum stress and autophagy, leads to testicular dysfunction. Sci Total Environ. 2023;902:166167.
Ileriturk M, Kandemir O, Kandemir FM. Evaluation of protective effects of quercetin against cypermethrin-induced lung toxicity in rats via oxidative stress, inflammation, apoptosis, autophagy, and endoplasmic reticulum stress pathway. Environ Toxicol. 2022;37:2639–50.
Cordeiro F, Gonçalves V Jr, Moreira N, Slobodticov JI, de Andrade Galvão N, de Souza Spinosa H, et al. Ivermectin acute administration impaired the spermatogenesis and spermiogenesis of adult rats. Res Vet Sci. 2018;117:178–86.
Ahmed AE, Alshehri A, Al-Kahtani MA, Elbehairi SEI, Alshehri MA, Shati AA, et al. Vitamin E and selenium administration synergistically mitigates ivermectin and doramectin-induced testicular dysfunction in male Wistar albino rats. Biomed Pharmacother. 2020;124:109841.
El-Nahas AF, El-Ashmawy IM. Effect of ivermectin on male fertility and its interaction with P-glycoprotein inhibitor (verapamil) in rats. Environ Toxicol Pharm. 2008;26:206–11.
Martenies SE, Perry MJ. Environmental and occupational pesticide exposure and human sperm parameters: a systematic review. Toxicology. 2013;307:66–73.
Gur C, Kandemir O, Kandemir FM. Investigation of the effects of hesperidin administration on abamectin-induced testicular toxicity in rats through oxidative stress, endoplasmic reticulum stress, inflammation, apoptosis, autophagy, and JAK2/STAT3 pathways. Environ Toxicol. 2022;37:401–12.
Radi AM, Mohammed ET, Abushouk AI, Aleya L, Abdel-Daim MM. The effects of abamectin on oxidative stress and gene expression in rat liver and brain tissues: Modulation by sesame oil and ascorbic acid. Sci Total Environ. 2020;701:134882.
Zhu S, Zhou J, Sun X, Zhou Z, Zhu Q. ROS accumulation contributes to abamectin-induced apoptosis and autophagy via the inactivation of PI3K/AKT/mTOR pathway in TM3 Leydig cells. J Biochem Mol Toxicol. 2020;34:e22505.
Gautam P, Kumar Dubey S. Biodegradation of imidacloprid: Molecular and kinetic analysis. Bioresour Technol. 2022;350:126915.
Ibrahim M, Ferreira G, Venter EA, Botha CJ. Cytotoxicity, morphological and ultrastructural effects induced by the neonicotinoid pesticide, imidacloprid, using a rat Leydig cell line (LC-540). Environ Toxicol Pharm. 2023;104:104310.
Bal R, Türk G R, Tuzcu M, Yilmaz O, Kuloglu T, Gundogdu R, et al. Assessment of imidacloprid toxicity on reproductive organ system of adult male rats. J Environ Sci Health B. 2012;47:434–44.
Miao Z, Miao Z, Wang S, Wu H, Xu S. Exposure to imidacloprid induce oxidative stress, mitochondrial dysfunction, inflammation, apoptosis and mitophagy via NF-kappaB/JNK pathway in grass carp hepatocytes. Fish Shellfish Immunol. 2022;120:674–85.
González-Moscoso M, Meza-Figueroa D, Martínez-Villegas NV, Pedroza-Montero MR. GLYPHOSATE IMPACT on human health and the environment: Sustainable alternatives to replace it in Mexico. Chemosphere. 2023;340:139810.
Lu L, Lian CY, Lv YT, Zhang SH, Wang L, Wang L. Glyphosate drives autophagy-dependent ferroptosis to inhibit testosterone synthesis in mouse Leydig cells. Sci Total Environ. 2024;914:169927.
Xing C, Chen S, Wang Y, Pan Z, Zou Y, Sun S, et al. Glyphosate exposure deteriorates oocyte meiotic maturation via induction of organelle dysfunctions in pigs. J Anim Sci Biotechnol. 2022;13:80.
Lu L, Liu JB, Wang JQ, Lian CY, Wang ZY, Wang L. Glyphosate-induced mitochondrial reactive oxygen species overproduction activates parkin-dependent mitophagy to inhibit testosterone synthesis in mouse leydig cells. Environ Pollut. 2022;314:120314.
Liu L, Zhang Y, Chang X, Li R, Wu C, Tang L, et al. Fluorochloridone perturbs blood-testis barrier/Sertoli cell barrier function through Arp3-mediated F-actin disruption. Toxicol Lett. 2018;295:277–87.
Shi J, Xie C, Liu H, Krausz KW, Bewley CA, Zhang S, et al. Metabolism and Bioactivation of Fluorochloridone, a Novel Selective Herbicide, in Vivo and in Vitro. Environ Sci Technol. 2016;50:9652–60.
Sun W, Ni Z, Li R, Chang X, Li W, Yang M, et al. Flurochloridone induces Sertoli cell apoptosis through ROS-dependent mitochondrial pathway. Ecotoxicol Environ Saf. 2021;216:112183.
Ni Z, Sun W, Li R, Yang M, Zhang F, Chang X, et al. Fluorochloridone induces autophagy in TM4 Sertoli cells: involvement of ROS-mediated AKT-mTOR signaling pathway. Reprod Biol Endocrinol. 2021;19:64.
Liu K, Li Y, Iqbal M, Tang Z, Zhang H. Thiram exposure in environment: a critical review on cytotoxicity. Chemosphere. 2022;295:133928.
Chen Y, Tian P, Li Y, Tang Z, Zhang H. Thiram exposure: disruption of the blood-testis barrier and altered apoptosis-autophagy dynamics in testicular cells via the Bcl-2/Bax and mTOR/Atg5/p62 pathways in mice. Pestic Biochem Physiol. 2024;203:106010.
Salam S, Arif A, Mahmood R. Thiram-induced cytotoxicity and oxidative stress in human erythrocytes: an in vitro study. Pestic Biochem Physiol. 2020;164:14–25.
Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. 2016;15:71.
Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic Biol Med. 2014;72:76–90.
Üremiş MM, Gültekin S, Üremiş N, Şafak T, Çiğremiş Y, Gül M, et al. Protective role of vitamin E against acrylamide-induced testicular toxicity from pregnancy to adulthood: insights into oxidative stress and aromatase regulation. Naunyn Schmiedebergs Arch Pharm. 2024;397:829–41.
Gur C, Akarsu SA, Akaras N, Tuncer SC, Kandemir FM. Carvacrol reduces abnormal and dead sperm counts by attenuating sodium arsenite-induced oxidative stress, inflammation, apoptosis, and autophagy in the testicular tissues of rats. Environ Toxicol. 2023;38:1265–76.
Demirci-Çekiç S, Özkan G, Avan AN, Uzunboy S, Çapanoğlu E, Apak R. Biomarkers of oxidative stress and antioxidant defense. J Pharm Biomed Anal. 2022;209:114477.
Wang J, Zhu H, Wang K, Yang Z, Liu Z. Protective effect of quercetin on rat testes against cadmium toxicity by alleviating oxidative stress and autophagy. Environ Sci Pollut Res Int. 2020;27:25278–86.
Siracusa R, D’Amico R, Fusco R, Impellizzeri D, Peritore AF, Gugliandolo E, et al. Açai berry attenuates cyclophosphamide-induced damage in genitourinary axis-modulating Nrf-2/HO-1 pathways. Antioxidants (Basel). 2022;11.
He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. 2020;21:4777.
Zhang L, Zhang J, Zhou Y, Xia Q, Xie J, Zhu B, et al. Azoramide ameliorates cadmium-induced cytotoxicity by inhibiting endoplasmic reticulum stress and suppressing oxidative stress. PeerJ. 2024;12:e16844.
You F, Li C, Zhang S, Zhang Q, Hu Z, Wang Y, et al. Sitagliptin inhibits the survival, stemness and autophagy of glioma cells, and enhances temozolomide cytotoxicity. Biomed Pharmacother. 2023;162:114555.
Acknowledgements
We wish to thank all study participants, research staff, and students who assisted with this work. This work was supported by Natural Science Foundation of Jiangsu Province (BK20221376), Fund of Xuzhou Science and Technology (KC20092), Xuzhou Pengcheng Talents-Medical Youth Reserve Talent Project (XWRCSL20220162), Open Fund of Jiangsu Key Laboratory for New Drug Research and Clinical Pharmacy, Nantong Project of Science and Technology (MS2024066), Qinglan Project of Jiangsu Province of China.
Author information
Authors and Affiliations
Contributions
XK, XW, QX, QH, WY: Writing—original draft. QH, JL, CW, ZL, YL: Literature investigation. XT, BZ, YQ, JY: Writing—review & editing, Funding acquisition, Data curation, Conceptualization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kong, X., Wang, X., Xia, Q. et al. Unveiling the nexus between environmental exposures and testicular damages: revelations from autophagy and oxidative stress imbalance. Cell Death Discov. 11, 258 (2025). https://doi.org/10.1038/s41420-025-02543-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-025-02543-4
This article is cited by
-
Investigating the time-dependent withdrawal effects of sofosbuvir and/or ribavirin on male mice: a histological and histophotometrical approach
Naunyn-Schmiedeberg's Archives of Pharmacology (2026)
-
PRDX1 promotes testosterone synthesis and attenuates aging via redox regulation of ATG4B to modulate lipophagy
Nature Communications (2025)
