
Abstract
Breast cancer, the most prevalent malignancy among females, threatens public health worldwide. Patients with breast cancer need personalised treatment strategies on the basis of their distinct molecular characteristics due to the unique epidemiological patterns and high heterogeneity of breast cancer, which limits therapeutic efficacy and poses significant challenges to current treatments. The underlying reasons may involve complex interactions and alterations in various cell death pathways. Currently, most studies and therapeutic agents focus on a single type of cell death, whereas opportunities related to other cell death pathways are typically overlooked. Therefore, identifying the predominant type of cell death, understanding the transitions between different cell death modalities during treatment, and developing novel therapies are crucial. In this review, we summarise the dynamic balance between reactive oxygen species (ROS) production and clearance, as well as the characteristics of various forms of cell death induced by ROS, including pyroptosis, apoptosis, necroptosis, autophagy, ferroptosis, cuproptosis, disulfidoptosis, oxeiptosis, and epigenetic regulation of these types of cell death. Additionally, we explored a novel cell death pathway called PANoptosis. This review sheds new light on the treatment of breast cancer from the perspective of nanotechnology and the development of combination therapies.
Similar content being viewed by others

Facts
-
Introducing PANoptosis and constructing a comprehensive network of cell death mechanisms
-
The key role of ROS metabolic imbalance in the occurrence, development and treatment resistance of breast cancer
-
Taking ROS as the core node connecting cell death pathways and therapeutic strategies
-
Exploring innovative therapeutic strategies targeting the ROS-cell death network, providing a new direction for the precise treatment of breast cancer
Open questions
-
Can we design ‘epigenetic triggers’ to selectively prime resistant cells for programmed cell death execution?
-
How to precise molecular thresholds determining the shift from pro-survival signalling to lethal programmed cell death execution?
-
How can we engineer ‘ programmed cell death -cascade nanoparticles’ that leverage tumour microenvironment signals?
Introduction
Despite the overall decline in cancer mortality rates, the annual morbidity of breast cancer is increasing, and breast cancer is currently the most prevalent cancer globally and the second leading cause of cancer-related deaths among females [1]. A variety of treatment modalities are available for breast cancer, including surgical intervention, antiestrogen therapy for hormone receptor (HR)-positive cases, chemotherapy, targeted therapy, and the integration of immunotherapy [2]. Given its high degree of heterogeneity, treatment strategies must be tailored to specific molecular characteristics [3]. Recent research has identified oxidative hotspots of hydrogen, laying a foundation for understanding cancer cell migration [4]. Reactive oxygen species have been determined to be critical inducers of various forms of cell death, which are collectively termed oxidative cell death [5]. Given its intimate association with ROS, cell death can be triggered by genetically programmed mechanisms (such as apoptosis, necrosis, and pyroptosis), metabolic dysfunction, including ferroptosis, cuproptosis, oxytosis, disulfidptosis [6] and the recently identified sodium-dependent cell death [7]. Strategies that target ROS and utilise nanoparticles to increase cell death have emerged as innovative strategies for cancer treatment [8].
The initiation, progression, and drug resistance mechanisms of tumours are dynamically regulated by multiple programmed cell death (PCD) pathways [9]. Epigenetic regulatory mechanisms, as pivotal molecular switches, mediate numerous biological processes, including cell death [9]. This study delves into the mechanisms underlying breast cancer and ROS-related cell death, along with the associated epigenetic regulatory network, and systematically analyses the impact of different PCD modalities on tumour evolution and treatment, shedding new light on the optimisation of clinical treatment strategies and the development of emerging nanoparticle-targeted cell death therapies.
ROS
ROS are signalling molecules composed of oxygen-containing reactive entities such as the superoxide anion (O2−), singlet oxygen (1O2), the hydroxyl radical (·OH) and hydrogen peroxide (H2O2), and primarily originate from electron leakage in the mitochondrial electron transport chain [10].
Mechanisms of ROS production
In addition to mitochondrial sources, ROS generation involves various enzymatic pathways: transmembrane electron transfer mediated by NADPH oxidase (NOX), substrate oxidation catalysed by xanthine oxidoreductase (XOR), and byproducts from lipoxygenase (LOXs) and cyclooxygenase (COX) during arachidonic acid metabolism [11].
NOX4, a key source of ROS, serves as the predominant NADPH oxidase enzyme in breast cancer and facilitates oxidative stress regulation [12]. A distinctive feature of NOX4 lies in its ability to generate ROS within the inner membrane via the p22phox protein without activating cytoplasmic oxidase proteins or GTPase Rac [13], thereby promoting the metastasis of breast cancer through lymphangiogenesis [12]. Peroxisomes contain key enzymes such as catalase and flavoprotein oxidase, which are involved in fatty acid α-oxidation, fatty acid β-oxidation, and purine metabolism, as well as the biosynthesis of glycerides and bile acids. In addition to its role in purine catabolism, XOR exhibits nitrite reductase activity to produce nitric oxide and NADH oxidase activity to generate ROS under hypoxic, acidic, and inflammatory conditions [14]. The metabolites produced by arachidonic acid (AA) via the LOX and COX pathways can induce the production of ROS by activating NOX [14, 15]. Specifically, bioactive molecules such as leukotrienes and prostaglandins produced during AA metabolism via LOX and COX can directly or indirectly increase NOX complex activity, leading to the catalysis of intracellular ROS synthesis, including O₂−, thereby participating in the regulation of oxidative stress and cell signalling pathways [15]. Notably, ionising radiation significantly elevates intracellular endogenous ROS levels through DNA damage in a dose-dependent manner [16].
Elimination of ROS
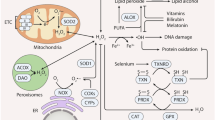
Excessive ROS production can induce oxidative stress despite its crucial physiological roles in the immune system and in cell signalling. Organisms have evolved a sophisticated antioxidant defence mechanism to counteract the detrimental effects of ROS [17]. This multitiered defence system comprises an endogenous antioxidant enzyme system (e.g., SOD/CAT/GPx) and small-molecule free radical scavengers such as vitamin C/E and β-carotene, which are supported by trace elements such as selenium and zinc, collectively maintaining redox homeostasis [18]. Therefore, in Fig. 1, we summarise the production and regulation of ROS, including both intracellular and extracellular ROS generation, as well as the associated regulatory systems.

NOX1, NOX2, NOX4, NOX5 and other enzymes are located on the cell membrane and catalyse the production of superoxide anions, with FAD and NADPH acting as cofactors. P22phox is a component of the NOX complex. Peroxidase 4 (GPX4) is involved in this process. The endoplasmic reticulum (ER), selenium (Se), Zn2+ and vitamins are involved in regulating the process of antioxidation. In mitochondria, SOD2 converts superoxide anions to hydrogen peroxide. If the balance between oxidation and antioxidation is disrupted, oxidative stress can be triggered. O₂−: Superoxide anions; O₂: Oxygen; H₂O₂: Hydrogen peroxide; ONOO−: Peroxynitrite; NO₂−: Nitrite; SOD: Superoxide Dismutase; LOX: Lipoxygenase; NOX: NADPH oxidase; cytC: Cytochrome C; IR: Ionising radiation. The figure was created with BioRender.com.
Superoxide dismutase (SOD)
SOD is an integral part of the endogenous antioxidant defence system in organisms and figures prominently in regulating ROS homeostasis by catalysing the disproportionation of O₂− into O₂ and H₂O₂ [19]. SOD reduces intracellular superoxide anion levels, thereby mitigating ROS-induced damage [20]. Specifically, studies have shown that SOD1 is abnormally overexpressed in breast cancer and is significantly correlated with ErbB2 oncogene activation and elevated ROS levels [21]. Notably, despite the upregulation of SOD1 in the high-ROS subgroup of ErbB2-positive breast cancer cells, normal cellular proliferation still relies on SOD1 activity, indicating its dual role in promoting cancer progression and maintaining basal metabolism [22]. Furthermore, the function of SOD2 is concentration dependent: when the intracellular O₂− level exceeds a specific threshold, the activity of SOD2 may exacerbate oxidative damage [22]. Therefore, regulating tumour progression hinges on maintaining ROS below the critical threshold that supports oncogene dependence [23], on the basis of which targeted regulation of SOD activity is regarded as a potential strategy in cancer treatment [24]. SOD-3 is a downstream effector of VEGFC, and VEGFC mediates SOD-3 through Nrp2 [24, 25]. SOD-3 and VEGFC are involved in the metastasis of breast cancer, and their expression is positively correlated with cancer [25].
Catalase (CAT)
CAT, an enzyme that decomposes H2O2 into O2 and H2O, is involved in the antioxidant defence mechanism [25]. In MCF-7 cancer cells, the overexpression of CAT affects their proliferation and migration and reduces their sensitivity to anticancer treatment [25]. Research has shown that CAT is associated with the luminal B subtype of breast cancer [26]. Reduced CAT activity has been observed in breast cancer; however, its activity is positively correlated with the advanced invasion and metastasis phenotypes of breast cancer in vivo [18].
Glutathione peroxidase (GPX)
The GPX family comprises eight members (GPX1–GPX8), which serve as classic antioxidant enzymes to mitigate oxidative stress and maintain redox homeostasis [27]. GPX specifically catalyses the reduction of lipid hydroperoxides (LOOHs) and organic hydroperoxides (ROOHs) through glutathione (GSH)-dependent reactions, thereby minimising oxidative damage and regulating prostaglandin biosynthesis [28]. Selenoproteins can catalyse the decomposition of peroxynitrite (ONOO−) into nitrite (NO₂−) and significantly reduce ONOO−-mediated protein nitration and DNA damage, thereby increasing cellular resistance to nitrogen stress [29].
Glutathione reductase (GR)
GR plays a crucial role in maintaining optimal glutathione levels by catalysing the reduction of oxidised glutathione (GSSG) to its reduced form, GSH, which is the primary antioxidant within the cellular antioxidant system [29], thereby preserving cellular redox homeostasis [30]. Studies have demonstrated that GR is highly expressed in MCF-7 breast cancer cells and that elevated GR activity is associated with increased resistance to radiotherapy; thus, inhibiting GR sensitises cells to oxidative stress [31].
Small-molecule free radical scavengers and trace elements
Vitamin D exerts its antioxidant effects by counteracting the activity of NADPH oxidase, which is responsible for ROS production [32]. Vitamin D also enhances the overall antioxidant capacity by upregulating the activity of peroxidases such as superoxide dismutase [32, 33]. Zn2+ regulates oxidative stress by inhibiting NOX [34]. Studies have revealed that the accumulation of zinc can increase the production of mitochondrial ROS, which in turn activates NF-κB and consequently modulates NOX1 expression [35].
Breast cancer
The molecular classification of breast cancer is determined by the status of hormone receptors. The prognostic outcomes for each subtype exhibit substantial variability [36], making it imperative to establish differentiated follow-up monitoring strategies on the basis of molecular typing, particularly for HER2-positive and triple-negative subtypes, which require enhanced posttreatment imaging surveillance and circulating tumour DNA detection. Future research should concentrate on analysing intrasubtype molecular heterogeneity and developing novel targeted therapies [37], especially therapeutic breakthroughs in triple-negative breast cancer [38]. Table 1 summarises the regulatory mechanisms of noncoding RNAs in the PCD of breast cancer.
Molecular mechanisms of different cell death pathways
PANoptosis
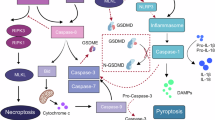
PANoptosis represents an integrated form of cell death encompassing apoptosis, pyroptosis, and necrosis [39], which is orchestrated by the PANoptosome complex. To date, four primary types of PANoptosomes have been identified: the ZBP1-PANoptosome (comprising ZBP1, NLRP3, ASC, caspase-1, caspase-6, caspase-8, RIPK1 and RIPK3) [39], AIM2-PANoptosome (comprising AIM2, Pyrin, ZBP1, ASC, caspase-1, caspase-8, FADD, RIPK1 and RIPK3) [40], RIPK1-PANoptosome (comprising RIPK1, RIPK3, NLRP3, ASC, caspase-1 and caspase-8) [41] and NLRP12-PANoptosome (comprising NLRP12, ASC, caspase-8 and RIPK3) [42]. These complexes facilitate the activation of caspase-3/7, cleavage of GSDMD and GSDME, and phosphorylation of MLKL, leading to membrane pore formation and the progression of PANoptosis [39]. Given the high heterogeneity of breast cancer, continuous refinement of existing molecular typing systems and prognostic assessment tools is needed. A robust prognostic prediction model, validated across multiple dimensions, has been developed [43]. This model can effectively guide the prediction of chemotherapy sensitivity and optimisation of targeted-immune combination therapies, thereby providing molecular tools to advance precise medicine paradigms in breast cancer management [44]. PANoptosis cannot be suppressed by pyroptosis, apoptosis or necroptosis. The formation and activation of the PANoptosome within a single cell also support PANoptosis. The molecular and regulatory mechanisms of this process are shown in Fig. 2.

The PANoptosome can concurrently participate in three key programmed cell death modes: apoptosis, pyroptosis and necroptosis. When triggered by certain factors, sensors, including ZBP1, AIM2, RIPK1 and NLRP12, can interact and recruit several other molecules to form PANoptosomes, which then induce the progression of PANoptosis. AIM2: absent in melanoma 2; ASC: Apoptosis-associated speck-like protein containing a caspase recruitment domain; CASP: Caspase; FADD: Fas-associated death domain protein; GSDMD: Gasdermin D; GSDME: Gasdermin E; GSDMC: Gasdermin C; MLKL: Mixed lineage kinase domain-like pseudokinase; RIPK: Receptor-interacting serine/threonine-protein kinase; ZBP1: Z-DNA binding protein 1; MOMP: Membrane permeabilisation. The figure was created with BioRender.com.
Apoptosis
As the central regulatory hub of apoptosis, mitochondria mediate signal transduction through both intrinsic and extrinsic pathways. The extrinsic pathway is initiated by death receptor-mediated or granzyme-dependent mechanisms [45]. Specifically, the extrinsic pathway begins with the binding of death receptors, members of the TNFR superfamily such as Fas and DR4/5, to their respective ligands, such as FasL and TRAIL. This binding subsequently recruits and activates initiator caspases-8/10 via adaptor proteins such as FADD/TRADD, leading to downstream activation of the effector caspases-3/7 and culminating in apoptosis [43]. The intrinsic pathway involves mitochondria-related and endoplasmic reticulum stress pathways, where the Bcl-2 protein family plays a pivotal role by modulating mitochondrial outer membrane permeabilisation (MOMP) [46]. This family includes antiapoptotic members (e.g., Bcl-2 and Bcl-xL) and proapoptotic members (e.g., Bax and Bak). Upon activation, proapoptotic proteins form pore complexes on the mitochondrial membrane, resulting in the loss of the mitochondrial membrane potential and the release of apoptotic factors such as cytochrome C, thereby activating the caspase-9/3 cascade [46]. Dysregulated apoptosis signalling pathways in breast cancer cells can be reprogrammed to re-enter the apoptotic cycle, representing a critical therapeutic strategy. In breast cancer, overexpression of the antiapoptotic protein MCL1 impedes apoptosis by inhibiting mitochondrial NOX4 function. However, BH3 mimetics targeting MCL1 have demonstrated promising therapeutic potential [47]. Moreover, p53, a key tumour suppressor, exerts dual regulatory effects on ROS-induced apoptosis. Wild-type p53 promotes apoptosis through the transcriptional activation of proapoptotic genes such as Bax and PUMA [48]. In triple-negative breast cancer (TNBC), mutant p53 induces nonclassical apoptosis via aberrant interaction with MDM2, independent of wild-type p53 [49]. Compound G613 promotes the apoptosis of MCF-7 cells by inhibiting the formation of the p53-MDM2 complex and upregulating p53 expression [50]. Additionally, increasing the bioavailability of bile acid promotes the apoptosis of breast cancer cells by activating the caspase-8/Bid/ROS pathway, suggesting a novel therapeutic target for hormone receptor-negative breast cancer [51].
Apoptotic pathways interactively regulate other cell death modalities. In ferroptosis–apoptosis cross-regulation, long noncoding RNA LINC00618 regulates cell death through a dual mechanism: downregulating SLC7A11 to inhibit ferroptosis and upregulating BAX expression to promote apoptosis [52]. In autophagy-dependent apoptosis, high ROS levels in tumour cells enhance the sensitivity to apoptosis by inducing mitochondrial hyperpolarisation, curcumin or DNA damage [53].
Necroptosis
Necroptosis is a caspase-independent form of cell death that significantly regulates the initiation and progression of breast cancer [54]. Necroptosis is a precisely regulated form of programmed necrosis, and its core mechanism hinges on the assembly of the RIPK1/RIPK3 kinase complex and subsequent phosphorylation–oligomerisation of MLKL. In triple-negative breast cancer, RIPK1 facilitates vascular mimicry by activating the AKT/eIF4E signalling pathway [55]. Moreover, RIPK3 expression exhibits spatiotemporal heterogeneity [56]. Owing to hypermethylation of the promoter, RIPK3 is silenced in primary tumours but significantly upregulated in recurrent lesions, with an enhanced dependence on cysteine metabolism, suggesting that targeting RIPK3 may inhibit tumour recurrence [56].
Notably, the DNA damage response protein MRE11 activates the cGAS‒STING pathway, thereby driving necroptosis mediated by the ZBP1‒RIPK3‒MLKL axis [57]. Low ZBP1 expression in TNBC is significantly associated with genomic instability, an immunosuppressive microenvironment, and a dismal prognosis, indicating its potential as a biomarker for functional defects in this pathway [58]. Necroptosis plays a dual role in tumour immune regulation, as it can activate antigen-specific immune responses by releasing damage-associated molecular patterns, while RIPK3/MLKL-mediated IL-1α release can suppress T-cell function and promote tumour immune escape [59]. In light of this dichotomy, novel targeted nanocomplexes have been developed to induce immunogenic cell death by specifically increasing RIPK3 phosphorylation and MLKL tetramerisation, thereby enhancing the cytotoxic effect of T cells on triple-negative breast cancer [60]. This paradoxical immune regulation underscores the need for precise modulation of necroptosis according to the treatment stage: leveraging its immune-stimulatory properties in the early stages while inhibiting its metastasis-promoting effects in the later stages [61]. For example, lung metastasis can be significantly reduced by inhibiting MLKL [62]. Epigenetic regulation also plays a crucial role in necroptosis. Z-DNA binding protein 1 has emerged as a potential therapeutic target for modulating the necrosis process in advanced tumours [63]. Moreover, necroptosis-related lncRNAs, such as LINC00472, which regulates RIPK1, have demonstrated their predictive value for the prognosis of patients with breast cancer and their guiding value for personalised treatment strategies [63].
Pyroptosis
Pyroptosis is a form of PCD, and its classical activation pathway is mediated by caspase-1 and executed through the formation of membrane pores by proteins from the gasdermin (GSDM) family [64]. The canonical pyroptosis pathway is initiated by the activation of the NLRP3 inflammasome complex, which includes NLRP3, the adaptor protein ASC containing the CARD domain, and pro-caspase-1 [65]. Notably, inflammatory responses play a dual role in tumorigenesis: they can both promote malignant transformation and metastasis and exert an antitumour effect via pyroptosis induction [66]. The nonclassical pyroptosis pathway is mediated by caspase-4/5/11. Although these caspases cannot directly cleave the precursors of IL-1β/IL-18, they can still trigger pyroptosis by cleaving GSDMD-NT, a process specifically activated by bacterial lipopolysaccharides [67, 68]. Additionally, caspase-8 regulates pyroptosis under specific conditions: nucleus-localised PD-L1 induces pyroptosis via caspase-8-mediated cleavage of GSDMC in the hypoxic tumour microenvironment [69]. Docetaxel cleaves GSDME via the ROS/JNK/caspase-3 pathway, and its pyroptotic effect can be enhanced by demethylation of the DFNA5 gene, highlighting the role of epigenetic regulation in chemosensitisation [70]. In the context of triple-negative immunotherapy for breast cancer, the combination of pyroptosis induction with immune checkpoint inhibitors has significant synergistic potential [62]. For example, TAT3 inhibitor nanoparticles (MPNPs) in conjunction with oncolytic viruses can activate GSDME-dependent pyroptosis and markedly enhance the efficacy of anti-PD-1 therapy [62]. The dopamine receptor DRD2 has been identified as a novel therapeutic target to remodel the immunosuppressive tumour microenvironment by promoting M1 macrophage polarisation, inhibiting the NF-κB signalling pathway, and inducing pyroptosis in breast cancer cells [62], which provides a promising new treatment direction for breast cancer. These findings establish a robust theoretical foundation and translational strategy for precision medicine approaches targeting pyroptosis regulation in breast cancer. Epigenetic modifications also play crucial roles in mediating tumour growth and metastasis by regulating pyroptosis-related pathways [71].
Ferroptosis
Ferroptosis is a form of iron-dependent PCD characterised by the aberrant accumulation of lipid peroxides and specific adjustments, as shown in Fig. 3 [72]. This process is specifically triggered by the peroxidation of polyunsaturated fatty acids (PUFAs), rather than a generalised increase in ROS [72]. Its regulatory network includes three core axes: the Xc−/GSH/GPX4 antioxidant system, ACSL4/LPCAT3/15-LOX pathway, and FSP1/CoQ10/NAD(P)H axis, which counteract ferroptosis by inhibiting lipid peroxidation [73]. Moreover, pathways mediating ferroptosis in breast cancer include T-cell inhibition via GCH1-BH4 [74] and ferroptosis triggered by DHODH-CoQH2 following GPX4 inactivation [75]. To date, 8 glutathione peroxidases have been identified, among which GPX4 directly reduces lipid peroxides [27], making it a potential therapeutic target for breast, ovarian, liver, and prostate cancers [76]. In breast cancer, the Xc−/GSH/GPX4 system is essential for maintaining redox homeostasis [77]. Cystine enters cells through Xc− and is reduced to cysteine via the cystine reduction pathway dependent on GSH or thioredoxin reductase 1 (TXNRD1), thereby promoting GSH synthesis [78]. GSH, as a cofactor of GPX4, facilitates the reduction of phospholipid hydroperoxides (PLOOHs) to their corresponding alcohols (PLOHs), thereby maintaining redox balance [33].

The synthesis of PUFA-PL, iron metabolism and mitochondrial metabolism can cause lipid peroxidation and induce ferroptosis. Conversely, the GPX4-GSH, FSP1-CoQH and DHODH-CoQH2 systems can inhibit lipid formation and thereby suppress ferroptosis. Imbalance of copper homeostasis can lead to an increase in intracellular Cu+ concentration and alter a series of cellular signalling pathways. Additionally, noncoding RNA is involved in copper metabolism. FSP1: Ferroptosis suppressor; GSSG: Glutathione; GSR: Glutathione-disulfide reductase; PUFA: Polyunsaturated fatty acid; PL-OOH: Phospholipid hydroperoxide; ROS: Reactive oxygen species; TCA: Mitochondrial TCA cycle. The figure was created with BioRender.com.
Targeted inhibition of the Xc−/GSH/GPX4 system axis effectively induces ferroptosis [79]. For example, DMOCPTL regulates EGR1 in TNBC cells and induces GPX4 ubiquitination to reduce EGR4 protein levels, thereby regulating mitochondria-mediated apoptosis [80]. In the FSP1/CoQ10/NAD(P)H axis, FSP1 reduces CoQ10 to generate the antioxidant CoQH2 independently of GPX4, inhibits lipid peroxidation [81], and cooperates with vitamin K metabolism to protect cells [82]. Ferroptosis in breast cancer involves subtype-specific regulation. In triple-negative breast cancer, the expression level of GPX4 is significantly associated with prognosis. Studies have demonstrated that GPX4 is markedly upregulated in the luminal androgen receptor subtype of TNBC, thereby conferring resistance to ferroptosis [83]. Conversely, the basal-like subtype is more susceptible to ferroptosis induction due to elevated expression levels of ACSL4 and FADS1/2 [84]. Notably, the sensitivity of BRCA1-deficient breast cancer cells to PARP inhibitors can be increased by inhibiting GPX4, thereby providing a novel strategy to overcome drug resistance [75].
In luminal breast cancer, ER/PR positivity is positively correlated with GPX4 expression, while m6A demethylation activates SLC7A11 by inhibiting the FGFR4/GSK-3β pathway, thereby increasing sensitivity to ferroptosis [85]. In HER2-positive breast cancer, overactivation of HER2 upregulates SLC7A11 via the PI3K/AKT/mTOR pathway to increase the antioxidant capacity [85]. The combination of chemotherapeutic drugs and ferroptosis inducers can reverse drug resistance in HER2+ breast cancer [86]. For clinical strategies targeting ferroptosis, existing interventions include GPX4 inhibitors, such as red ginseng polysaccharides, which induce ferroptosis by downregulating GPX4. Their combination with immunotherapy can enhance therapeutic efficacy [87]. Epigenetic regulation is prominent in breast cancer ferroptosis. DNA methylation, microRNAs (miRNAs), and m6A modifications differentially regulate the expression of ferroptosis-related genes [88]. High methylation of the SLC7A11 promoter suppresses its expression and increases cellular resistance to ferroptosis. m6A demethylation activates the SLC7A11/FPN1 axis by inhibiting the FGFR4/GSK-3β/β-catenin pathway, suggesting a novel strategy to overcome resistance to HER2 treatment [85]. The METTL3 inhibitor STM2457 promotes GPX4 degradation by reducing m6A modification, inducing ferroptosis and inhibiting metastasis in TNBC [89]. Noncoding RNAs and ferroptosis have complex relationships in cancer progression. Different ncRNAs can either promote or inhibit ferroptosis in cancer cells. ncRNA-mediated regulation of ferroptosis may represent a new therapeutic direction for treating breast cancer. Long noncoding RNAs (lncRNAs) contribute to improving prognosis and identifying potential therapeutic targets in breast cancer [90]. LncFASA promotes lipid droplet formation and inhibits lipid peroxidation by activating the SLC7A11‒GPX4 axis [91]. The mechanism of tumour suppression involves multiple pathways. LINC00152 confers tamoxifen resistance by inhibiting ferroptosis through BACH1, and sensitivity to endocrine therapy can be restored by silencing LINC00152 [92]. Additionally, the lncRNA H19 regulates chemotherapy resistance in breast cancer via the DNA damage response pathway [93].
Cuproptosis
Cuproptosis is a recently identified form of copper-dependent PCD that differs from apoptosis, necroptosis, and ferroptosis. Specifically, the accumulated copper ions within cells directly bind to lipoylated proteins in the TCA cycle, leading to their inactivation and subsequent cell death (Fig. 3) [94]. Notably, breast cancer cells exhibit elevated aerobic respiration due to their highly active mitochondrial metabolism, which may potentiate tumour angiogenesis via the cuproptosis pathway, thereby fostering a tumour-promoting microenvironment [95]. Copper homeostasis has a dual nature in that it enhances the capacity of copper ions as cofactors for SOD to scavenge reactive oxygen species [96]. Conversely, excessive Cu⁺ can generate ·OH via the Fenton reaction, inducing oxidative stress and cellular damage [97]. In this context, GSH can remarkably maintain copper homeostasis by neutralising ROS toxicity through reduction reactions [98]. During the cuproptosis process, the key target FDX1 interacts with components of the tumour microenvironment. The mitochondrial protein FDX1 has been validated as a target of the anticancer drug Elesclomol, which induces specific copper-mediated cell death by facilitating copper ion delivery [99]. Notably, the hypoxic microenvironment characteristic of TNBC markedly suppresses FDX1 expression. Under these conditions, lipoic acid synthase sustains TCA cycle function through Fe‒S cluster-mediated acylations [100].
In epigenetics, lncRNAs associated with cuproptosis significantly regulate the proliferation and metastasis of breast cancer cells and potentially predict prognosis and sensitivity to various therapies [101]. The copper transporter SLC31A1 is aberrantly overexpressed in breast cancer cells and is significantly positively correlated with tumour immune infiltration and the expression of immune checkpoint molecules such as PD-L1 [102]. SLC31A1 is significantly correlated with diverse immune cell infiltrations, immune cell biomarkers, and immune checkpoints in breast cancer and is regulated by the LINC01640/miR-204-5p/SLC31A1 axis, which may be central to copper-induced cell death in breast cancer, suggesting that SLC31A1 may serve as a potential target for adjuvant immunotherapy [103].
Oxeiptosis and disulfidptosis
Oxeiptosis
Oxeiptosis is a noninflammatory form of cell death initiated by oxidative stress and is sensitive to ROS [104]; this form of cell death is carried out primarily through the KEAP1/PGAM5/AIFM1 signalling pathway, thereby protecting the organism from ROS-induced damage and inflammation [105]. As shown in Fig. 4a, excessive accumulation of ROS triggers KEAP1 activation and enables KEAP1 to interact with PGAM5. This interaction facilitates the dephosphorylation of the intermembrane space protein AIFM1 by PGAM5, which in turn triggers the release and nuclear translocation of AIFM1, ultimately leading to large-scale DNA fragmentation [106]. Alloimperatorin enhances the expression of KEAP1 without altering the expression levels of PGAM5 or AIFM1, thereby inhibiting the proliferation and invasion of breast cancer cells [106].

a Schematic diagram of the oxeiptosis pathway. After being phosphorylated, AIFM1 in mitochondria is translocated to the nucleus under the action of PGAM5, where it causes DNA damage and ultimately leads to cell death. This process is regulated by KEAP1. b Schematic diagram of the disulfidptosis pathway. Under glucose-deprivation conditions, the supply of NADPH becomes limited, leading to excessive accumulation of cysteine and other disulfide-containing molecules in SLC7A11-high cells. This ultimately induces a disulfide stress state, which triggers rapid disulfide-dependent cell death. c Schematic diagram of the autophagy pathway. Nutrients and insulin signals can activate mammalian targets of mTOR. DDIT4-AS1 can inhibit mTOR, thereby activating ULK1 and promoting the formation of autophagosomes. In addition, H19 can affect the autophagy process by regulating SAHH and DNMT3B or through the HER2/HuR/LINC00969/trastuzumab axis. d Schematic diagram of the paraptosis pathway. Endoplasmic reticulum stress leads to Ca²⁺ depletion and the accumulation of misfolded proteins. Mitochondrial dysfunction causes Ca²⁺ overload and ROS generation, resulting in mitochondrial vacuolisation and ultimately cell death mediated by AIP-1/Alix. MANF: Neurotrophic factor; MAPK: Mitogen-activated protein kinase. AIFM1: Apoptosis-inducing factor mitochondrial 1; P: Phosphorylated; PGAM5: Phosphoglycerate mutase 5; PPP: Pentose phosphate pathway; NADPH: Nicotinamide adenine dinucleotide phosphate; NADP⁺: Nicotinamide adenine dinucleotide phosphate; Ca²⁺: Calcium ion; ROS: Reactive oxygen species. The figure was created with BioRender.com.
Disulfidptosis
When glucose is depleted, the cellular redox state becomes insufficient. Mesencephalic astrocyte-derived neurotrophic factor (MANF) mitigates protein oxidation and restores phagocytosis mediated by E3 ligase activity, thereby promoting the survival of BC cells under glucose starvation [106]. Cystine transported by SLC7A11 induces the response of actin cytoskeleton proteins to disulfide stress, leading to disulfide-induced apoptosis [107]. Central genes associated with disulfide-related immune checkpoints can predict the prognosis of breast cancer [108]. Breast cancer subtypes can be predicted by lncRNAs involved in disulfide metabolism. Specifically, in the basal subtype, LINC02188 is highly expressed, whereas LINC01488 and GATA3-AS1 exhibit the lowest expression levels [109]. LINC00511 is expressed at the highest level in the Her2 subtype, and GATA3-AS1 is expressed at the highest level in the LumA, LumB, and normal-like subtypes [108]. These findings provide a novel direction for identifying clinical therapeutic targets for breast cancer (Fig. 4b) [109].
Autophagy
Autophagy eliminates damaged organelles and abnormal proteins via the lysosomal degradation pathway to maintain intracellular homeostasis and suppress genomic instability [110]. Under physiological conditions, moderate levels of autophagy effectively mitigate both endogenous and exogenous inflammatory responses by inhibiting NLRP3 inflammasome activation [111]. Notably, breast cancer cells achieve metabolic adaptation by hijacking the autophagy mechanism: under glucose-deprivation conditions, MANF-mediated mitophagy eliminates dysfunctional mitochondria through the PRKN/Parkin-dependent pathway, maintains the ATP supply and promotes tumour survival [112], suggesting that targeting the MANF‒PRKN interaction can break the drug resistance barrier of breast cancer under metabolic stress.
Autophagy and ferroptosis involve complex metabolic interactions: autophagy-dependent degradation of ferritin leads to the accumulation of free iron and promotes lipid peroxidation through the Fenton reaction. Meanwhile, mitochondrial damage mediated by PCD protein 2 can increase the generation of mitochondrial reactive oxygen species, synergistically inducing autophagy and ferroptosis [113]. Notably, the combination of autophagy inhibitors (such as chloroquine) and ferroptosis inducers (such as erastin) can significantly inhibit breast cancer proliferation, suggesting the potential of synergistic therapy for dual death pathways [114]. Noncoding RNAs exert regulatory functions within the autophagy network [115]. LncRNAs significantly modulate autophagy plasticity via epigenetic and posttranscriptional mechanisms. For example, DDIT4-AS1 promotes protective autophagy by inhibiting mTORC1 complex activity, thereby facilitating the progression of triple-negative breast cancer. Its silencing enhances sensitivity to cisplatin-induced DNA damage [116]. H19 activates autophagy through the H19/SAHH/DNMT3B axis, potentially contributing to tamoxifen resistance in breast cancer (Fig. 4c) [117].
Paraptosis
Paraptosis represents an atypical form of PCD [118]. The key features that distinguish paraptosis from classical apoptosis include its independence from caspase-9 activation, the absence of typical apoptotic markers such as chromatin condensation or DNA laddering, and cytoplasmic vacuolisation as the predominant morphological feature positively modulated by the mitogen-activated protein kinase (MAPK) signalling pathway [119]. Additionally, paraptosis can be specifically inhibited by the apoptosis inhibitory protein AIP-1/Alix, indicating a distinct regulatory network separate from the apoptotic pathway [120]. The occurrence of paraptosis is closely associated with dysfunction in the ER‒mitochondrial axis [121]. The accumulation of misfolded proteins within the ER lumen leads to osmotic imbalance, resulting in water efflux and expansion of the ER compartment. Persistent ER stress promotes the extensive release of Ca2+ into the cytoplasm via the IP3R channel. Ca2+ subsequently enters the mitochondrial matrix through the mitochondrial calcium uniporter to cause mitochondrial swelling and membrane potential collapse, ultimately leading to cell death (Fig. 4d) [122, 123]. Research has demonstrated that celastrol induces paraptosis in breast cancer cells via the IP3R-dependent Ca2+ signalling pathway, highlighting the critical role of Ca2+ homeostasis disruption in this death pattern [124]. Increasing attention has been given to emerging therapeutic strategies based on the paraptosis mechanism [125].
Difficulties and innovative treatments in breast cancer therapy
The central role of reactive oxygen species in drug resistance mechanisms
ROS serve as a central regulatory hub in the development of drug resistance phenotypes in breast cancer. The mechanisms promoting drug resistance include the following: first, at the pharmacokinetic level, the overactivation of drug efflux pumps is mediated by ATP-binding cassette transporters (ABC transporters); second, at the molecular biology level, this involves epigenetic modifications of drug targets, activation of intracellular drug-metabolising enzymes, and abnormal activation of epithelial‒mesenchymal transition processes and DNA damage repair pathways regulated by key signalling pathways such as the PI3K/AKT and NF-κB pathways [126]. Notably, clinical observations indicate that endocrine therapy resistance is closely associated with oestrogen-dependent redox signal dysregulation mediated by ROS, suggesting that targeting ROS homeostasis may represent a novel breakthrough for reversing drug resistance [127]. Myeloperoxidase (MPO) generates ROS [128]. In a rigorous clinical trial, the Southwest Oncology Group SWOG-8897 found that the MPO genotype of breast cancer patients undergoing chemotherapy was associated with a lower risk of recurrence, and a high-activity MPO genotype was related to a higher survival rate among women treated with cyclophosphamide [129]. Memo is a copper-dependent redox enzyme that facilitates the production of ROS [130]. A greater than 40% increase in Memo expression has been linked to adverse clinical parameters in primary breast cancer and serves as an independent prognostic marker for early distant metastasis [121].
Nanodelivery systems enhancing photodynamic therapy
Photodynamic therapy (PDT) relies on ROS generation through photosensitizers under specific wavelength excitation. PDT can kill tumour cells but is limited by the low bioavailability of phototherapeutic agents. Nanoparticles have emerged as ideal carriers for optimising PDT due to their precise targeting and high drug loading efficiency [131]. Nanoparticles have become powerful and multifunctional tools for inducing ROS to achieve effective cancer therapies [132]. Preclinical studies have demonstrated that the sequential application of photodynamic therapy and radiotherapy significantly improves therapeutic outcomes for triple-negative breast cancer, highlighting the potential of combined therapies [133]. In a clinical trial, PDT was applied to 7 patients with breast cancer recurrence on the chest wall to relieve chest wall pain [134]. The results showed that the overall response rate was 91%, with a complete response rate of 73% and a partial response rate of 18%, indicating that this treatment plan has significant clinical efficacy [134].
Multiple catalytic effects of nanozymes
Nanozymes, characterised by their biomimetic catalytic activity, high stability, and programmable nature, offer a novel strategy to address the limitations of traditional enzyme preparations [135]. Specifically, nanozymes can induce immunogenic cell death through various mechanisms, such as alloy nanoparticles like Cu-Ag NPs [136] or FeCu-DA [137], as well as nanozyme platforms [138]. Notably, the PtMnIr nanozyme exhibits multiple enzymatic activities, integrating OXD, CAT, SOD, POD, and GPX functionalities, thereby promoting ferroptosis and apoptosis in tumour cells while inhibiting tumour metastasis by establishing an “endogenous GSH consumption–ROS cyclic generation” mechanism [139]. In addition, several studies have demonstrated the vital role of nanozymes in the research and treatment of breast cancer [140]. Currently, the catalytic activity of nanozymes is utilised to locally amplify ROS stress in breast cancer, and targeted therapy is achieved through carrier design [141]. The specific nanozymes and their corresponding mechanisms are illustrated in Table 2.
Tumour microenvironment (TME)-targeted nanotherapy
Reprogramming the TME is a critical direction in nanotherapy, with strategies including immune cell regulation and metabolic intervention. Stimulatory nanoparticles can reprogramme antigen-presenting cells and enhance T-cell activation, as demonstrated in the 4T1 model [142]. Paclitaxel nanoformulations induce macrophage polarisation to the M1 phenotype via the TLR4/cGAS-STING pathway, thereby reversing the immunosuppressive microenvironment [143,144,145]. Compared with standard paclitaxel, biologically interactive albumin-bound paclitaxel in solvent-free nanoparticles demonstrated greater efficacy and good safety [146]. The PFTT@CM nanosystem developed by Pan et al. enhances PDT efficacy by depleting GSH and inducing ferroptosis [147]. Additionally, ILA@Lip creates a highly hypoxic tumour microenvironment, reduces angiogenesis and inhibits tumour metastasis [148]. These advancements underscore that nanoparticles can precisely regulate TME, which is of great significance for personalised treatment [149].
Sonodynamic-nano synergistic therapy
The fundamental principle of sonodynamic therapy (SDT) involves utilising ultrasound to stimulate sonosensitizers to activate cavitation activity and promote the generation of ROS, followed by inducing the apoptosis of harmful cells such as tumour cells and bacteria [150]. SDT employs ultrasound to activate sonosensitizers for ROS production, with tissue penetration superior to that of photodynamic therapy. However, its efficacy is constrained by the heterogeneity of the tumour microenvironment [151]. Consequently, developing new sonosensitizers with enhanced sonosensitizing activity remains a significant challenge for SDT technology. The therapeutic efficacy against TNBC can be improved using tumour microenvironment-responsive nanoparticles and epigenetic reprogramming [152]. For instance, constructed HER3-modulated nanobioparticles can cross the blood‒brain barrier and target HER3 [153]. Given the hypoxic nature of the tumour microenvironment, clinical application of SDT remains challenging. Wu et al. proposed a synergistic strategy that combines oxygen-enhanced sonodynamic therapy with ferroptosis via engineered exosomes, demonstrating higher efficacy in anticancer treatment [154]. Moreover, the combination of sonodynamic therapy with localised chemotherapy can mitigate the adverse effects of chemotherapeutic agents on healthy tissue [155]. Currently, clinical trials investigating sonodynamic therapy are limited to brain tumours [156], and research on its application in breast cancer remains to be further explored.
Discussion
This review systematically elucidates the pivotal role of ROS in regulating multiple PCD pathways and highlights the potential therapeutic value of targeting the ROS–PCD network. ROS serves as both a pro-cancer signalling molecule and an executor of cell death, making it a critical node linking tumour metabolism, epigenetic regulation, and microenvironmental modulation. A comprehensive understanding of the balance between oxidants and antioxidants in breast cancer is essential for developing effective treatment strategies. The various PCD pathways in breast cancer are interconnected through shared molecular nodes, forming a complex regulatory network. Beyond apoptosis, other forms of cell death crosstalk also exist in breast cancer. The same stimulus can induce distinct forms of cell death depending on the specific situation. For example, lapatinib induces ferroptosis via iron-dependent ROS generation in the early stages but promotes protective autophagy to facilitate drug resistance in the later stages [157]. High concentrations of copper ions can activate autophagy through lysosome-dependent mechanisms [158]. Additionally, the long noncoding RNA LINC00618 orchestrates cell fate through dual mechanisms; LINC00618 inhibits cystine uptake by downregulating SLC7A11 to promote ferroptosis and simultaneously induces apoptosis by upregulating BCL-2-BAX expression and activating caspase-3 [52]. This multitarget regulatory pattern underscores the central role of epigenetic modifications in maintaining the dynamic equilibrium of PCD pathways. Therefore, investigating epigenetic regulation across different cell death states to unravel the progression of breast cancer is crucial. Based on current advancements in oxidative stress research in breast cancer, we propose several innovative therapeutic approaches targeting the ROS–PCD network for future development: (1) Oxidative stress-directed small-molecule drugs: Natural compounds such as curcumin can induce apoptosis by triggering mitochondrial ROS bursts and DNA damage while enhancing ferroptosis sensitivity through GPX4 inhibition. However, their low bioavailability and off-target effects limit their clinical application. Nanodelivery systems are required to optimise the targeting efficacy. Compared with free curcumin, curcumin encapsulated in NPs demonstrates enhanced anti-proliferative effects [159], offering a promising strategy for breast cancer treatment. (2) Epigenetic intervention: Targeting epigenetic modifications can reshape the expression profiles of PCD-related genes. DNA methyltransferase activates long noncoding RNA PHACTR2-AS1 to suppress PH20 expression, thereby controlling the growth and metastasis regulatory axis of epigenetic regulation across different cell death states [160]. The DNMT1-PAS1-PH20 axis represents a potential therapeutic target for breast cancer [160]. (3) Nanoimmune combination therapy: Multifunctional nanoplatforms can overcome TME barriers by integrating catalytic activity and immune modulatory functions. For instance, cascade multienzyme nanoparticles (e.g., PtMnIr) can deplete GSH, continuously generate ROS, and release damage-associated molecular patterns (DAMPs) to promote dendritic cell maturation and cytotoxic T lymphocyte infiltration [161]. The combination of these platforms with immune checkpoint inhibitors (e.g., α-PD-L1) significantly suppresses primary tumours and distant metastases, shedding new light on immunotherapy in “cold tumours” [161]. A search for “breast cancer” and “nanoparticles” in the ClinicalTrials.gov database reveals the number of clinical trials focusing on nanotechnology-based breast cancer treatments. As of 2025, a total of 81 clinical trials have been initiated in this area, with four of them having been withdrawn. In recent years, the number of clinical studies investigating nanoparticle-based therapies for breast cancer has steadily increased (ClinicalTrials.gov, 2025). However, complex interactions and safety concerns exist between nanoparticles and sonodynamic therapy. Consequently, no clinical trials have been initiated in this area to date, and significant challenges remain in the clinical translation of nanomedicines. In summary, despite the challenges posed by tumour heterogeneity, diverse microenvironmental adaptability, and safety concerns, targeted therapies aimed at ROS and related PCD pathways offer new perspectives for precision medicine in breast cancer. Future efforts should focus on full-chain innovation from mechanistic exploration to therapeutic breakthroughs through advanced drug design and closed-loop preclinical-clinical translation.
Conclusion
Breast cancer, the leading cause of cancer-related deaths among females worldwide, faces significant challenges due to its high heterogeneity and drug resistance. This article systematically examines the dual role of ROS in the initiation, progression, and treatment resistance of breast cancer and explores innovative therapeutic strategies targeting ROS-related PCD. In conclusion, targeting ROS and associated PCD pathways offers a promising breakthrough for the treatment of breast cancer. However, clinical success hinges on the deep integration of interdisciplinary technological innovation within the framework of precision medicine. Future research should focus on establishing a closed loop from mechanism to technology and clinical translation, ultimately achieving substantial improvements in the survival of patients. Although there remain several issues to be carefully considered, the enormous potential of these nanoparticles deserves further investigation. We hope this review can provide comprehensive insights to facilitate the therapeutic progress of breast cancer.
References
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63.
Schneider BJ, Naidoo J, Santomasso BD, Lacchetti C, Adkins S, Anadkat M, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO guideline update. J Clin Oncol. 2021;39:4073–126.
Xiang L, Yang J, Rao J, Ma A, Liu C, Zhang Y, et al. Integrating machine learning and bulk and single-cell RNA sequencing to decipher diverse cell death patterns for predicting the prognosis of neoadjuvant chemotherapy in breast cancer. Int J Mol Sci. 2025;26:3682.
Ueda Y, Kiyonaka S, Selfors LM, Inoue K, Harada H, Doura T, et al. Intratumour oxidative hotspots provide a niche for cancer cell dissemination. Nat Cell Biol. 2025;27:530–43.
Apel K, Hirt H. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol. 2004;55:373–99.
Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187:235–56.
Fu W, Wang J, Li T, Qiao Y, Zhang Z, Zhang X, et al. Persistent activation of TRPM4 triggers necrotic cell death characterized by sodium overload. Nat Chem Biol. 2025;21:1238–49.
Abdel-Rashid RS, El-Leithy ES, Ibrahim IT, Attallah KM. Radiolabelling and bioequivalence of modified Tamoxifen solid lipid nanoparticles as a targeted chemotherapeutic drug. Drug Deliv Transl Res. 2025; Epub ahead of print.
Mao C, Wang M, Zhuang L, Gan B. Metabolic cell death in cancer: ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 2024;15:642–60.
Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab. 2009;20:332–40.
Cho KJ, Seo JM, Kim JH. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol Cells. 2011;32:1–5.
Wang X, Liu Z, Sun J, Song X, Bian M, Wang F, et al. Inhibition of NADPH oxidase 4 attenuates lymphangiogenesis and tumor metastasis in breast cancer. FASEB J. 2021;35:e21531.
Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82.
Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–51.
Kawamura K, Qi F, Kobayashi J. Potential relationship between the biological effects of low-dose irradiation and mitochondrial ROS production. J Radiat Res. 2018;59:ii91–ii7.
Cheung EC, Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer. 2022;22:280–97.
He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44:532–53.
Radenkovic S, Milosevic Z, Konjevic G, Karadzic K, Rovcanin B, Buta M, et al. Lactate dehydrogenase, catalase, and superoxide dismutase in tumor tissue of breast cancer patients in respect to mammographic findings. Cell Biochem Biophys. 2013;66:287–95.
Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–49.
Luo M, Shen N, Shang L, Fang Z, Xin Y, Ma Y, et al. Simultaneous targeting of NQO1 and SOD1 eradicates breast cancer stem cells via mitochondrial futile redox cycling. Cancer Res. 2024;84:4264–82.
Gomez ML, Shah N, Kenny TC, Jenkins EC Jr, Germain D. SOD1 is essential for oncogene-driven mammary tumor formation but dispensable for normal development and proliferation. Oncogene. 2019;38:5751–65.
Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, et al. A novel HIF-2α targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. 2022;29:1769–89.
Palma FR, He C, Danes JM, Paviani V, Coelho DR, Gantner BN, et al. Mitochondrial superoxide dismutase: what the established, the intriguing, and the novel reveal about a key cellular redox switch. Antioxid Redox Signal. 2020;32:701–14.
Wang CA, Harrell JC, Iwanaga R, Jedlicka P, Ford HL. Vascular endothelial growth factor C promotes breast cancer progression via a novel antioxidant mechanism that involves regulation of superoxide dismutase 3. Breast Cancer Res. 2014;16:462.
Nandi A, Yan LJ, Jana CK, Das N. Role of catalase in oxidative stress- and age-associated degenerative diseases. Oxid Med Cell Longev. 2019;2019:9613090.
Timofeeva А, Minina VI, Torgunakova AV, Soboleva О, Тitov R, Zakharova Y, et al. Polymorphic variants of the hOGG1, APEX1, XPD, SOD2, and CAT genes involved in DNA repair processes and antioxidant defense and their association with breast cancer risk. Vavilovskii Zh Genet Selektsii. 2024;28:424–32.
Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–303.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Alehagen U, Opstad TB, Alexander J, Larsson A, Aaseth J. Impact of selenium on biomarkers and clinical aspects related to ageing. A review. Biomolecules. 2021;11:1478.
Ding H, Zhou C, Li T. Nanomedicines with versatile GSH-responsive linkers for cancer theranostics. ACS Biomater Sci Eng. 2024;10:5977–94.
Lin JH, Liu CC, Liu CY, Hsu TW, Yeh YC, How CK, et al. Selenite selectively kills lung fibroblasts to treat bleomycin-induced pulmonary fibrosis. Redox Biol. 2024;72:103148.
Kim DH, Meza CA, Clarke H, Kim JS, Hickner RC. Vitamin D and endothelial function. Nutrients. 2020;12:575.
Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan X, et al. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials. 2021;277:121110.
Li MS, Adesina SE, Ellis CL, Gooch JL, Hoover RS, Williams CR. NADPH oxidase-2 mediates zinc deficiency-induced oxidative stress and kidney damage. Am J Physiol Cell Physiol. 2017;312:C47–c55.
Salazar G, Huang J, Feresin RG, Zhao Y, Griendling KK. Zinc regulates Nox1 expression through a NF-κB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic Biol Med. 2017;108:225–35.
Zubair M, Wang S, Ali N. Advanced approaches to breast cancer classification and diagnosis. Front Pharmacol. 2020;11:632079.
Turner NC, Swift C, Jenkins B, Kilburn L, Coakley M, Beaney M, et al. Results of the c-TRAK TN trial: a clinical trial utilising ctDNA mutation tracking to detect molecular residual disease and trigger intervention in patients with moderate- and high-risk early-stage triple-negative breast cancer. Ann Oncol. 2023;34:200–11.
Tsang JYS, Tse GM. Molecular classification of breast cancer. Adv Anat Pathol. 2020;27:27–35.
Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol. 2019;9:406.
Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti TD. RIPK1 distinctly regulates Yersinia-induced inflammatory cell death, PANoptosis. Immunohorizons. 2020;4:789–96.
Chen W, Gullett JM, Tweedell RE, Kanneganti TD. Innate immune inflammatory cell death: PANoptosis and PANoptosomes in host defense and disease. Eur J Immunol. 2023;53:e2250235.
Sundaram B, Pandian N, Mall R, Wang Y, Sarkar R, Kim HJ, et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell. 2023;186:2783–801.e20.
Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23:1625–37.
Chen JW, Gong RH, Teng C, Lin YS, Shen LS, Lin Z, et al. Identification of a PANoptosis-related prognostic model in triple-negative breast cancer, from risk assessment, immunotherapy, to personalized treatment. Heliyon. 2024;10:e38732.
Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2024;25:379–95.
Czabotar PE, Garcia-Saez AJ. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat Rev Mol Cell Biol. 2023;24:732–48.
Demelash A, Pfannenstiel LW, Liu L, Gastman BR. Mcl-1 regulates reactive oxygen species via NOX4 during chemotherapy-induced senescence. Oncotarget. 2017;8:28154–68.
Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–35.
On JL, Ghaderi S, Rittmann C, Hoffmann G, Gier F, Woloschin V, et al. Pharmacological inhibition of MDM2 induces apoptosis in p53-mutated triple-negative breast cancer. Int J Mol Sci. 2025;26:1078.
Saxena R, Gupta G, Manohar M, Debnath U, Popli P, Prabhakar YS, et al. Spiro-oxindole derivative 5-chloro-4’,5’-diphenyl-3’-(4-(2-(piperidin-1-yl) ethoxy) benzoyl) spiro[indoline-3,2’-pyrrolidin]-2-one triggers apoptosis in breast cancer cells via restoration of p53 function. Int J Biochem Cell Biol. 2016;70:105–17.
Lan M, Kong Z, Liu F, Zou T, Li L, Cai T, et al. Activating caspase-8/Bid/ROS signaling to promote apoptosis of breast cancer cells by folate-modified albumin baicalin-loaded nanoparticles. Nanotechnology. 2022;33.
Wang Z, Chen X, Liu N, Shi Y, Liu Y, Ouyang L, et al. A nuclear long non-coding RNA LINC00618 accelerates ferroptosis in a manner dependent upon apoptosis. Mol Ther. 2021;29:263–74.
Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8.
Sarkar E, Kotiya A, Bhuyan R, Raza ST, Misra A, Ahmad R, et al. Curcumin chemo-sensitizes intrinsic apoptosis through ROS-mediated mitochondrial hyperpolarization and DNA damage in breast cancer cells. Cell Signal. 2025;128:111637.
Lin CC, Mabe NW, Lin YT, Yang WH, Tang X, Hong L, et al. RIPK3 upregulation confers robust proliferation and collateral cystine-dependence on breast cancer recurrence. Cell Death Differ. 2020;27:2234–47.
Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–25.
Amusan OT, Wang S, Yin C, Koehler HS, Li Y, Tenev T, et al. RIPK1 is required for ZBP1-driven necroptosis in human cells. PLoS Biol. 2025;23:e3002845.
Cho MG, Kumar RJ, Lin CC, Boyer JA, Shahir JA, Fagan-Solis K, et al. MRE11 liberates cGAS from nucleosome sequestration during tumorigenesis. Nature. 2024;625:585–92.
Hänggi K, Li J, Gangadharan A, Liu X, Celias DP, Osunmakinde O, et al. Interleukin-1α release during necrotic-like cell death generates myeloid-driven immunosuppression that restricts anti-tumor immunity. Cancer Cell. 2024;42:2015–31.e11.
Liang J, Tian X, Zhou M, Yan F, Fan J, Qin Y, et al. Shikonin and chitosan-silver nanoparticles synergize against triple-negative breast cancer through RIPK3-triggered necroptotic immunogenic cell death. Biomaterials. 2024;309:122608.
Jiao D, Cai Z, Choksi S, Ma D, Choe M, Kwon HJ, et al. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018;28:868–70.
Baik JY, Liu Z, Jiao D, Kwon HJ, Yan J, Kadigamuwa C, et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat Commun. 2021;12:2666.
Zhang Y, Yue Q, Cao F, Li Y, Wei Y. Necroptosis-related lncRNA signatures determine prognosis in breast cancer patients. Sci Rep. 2022;12:11268.
De Schutter E, Roelandt R, Riquet FB, Van Camp G, Wullaert A, Vandenabeele P. Punching holes in cellular membranes: biology and evolution of gasdermins. Trends Cell Biol. 2021;31:500–13.
Liu X, Xia S, Zhang Z, Wu H, Lieberman J. Channelling inflammation: gasdermins in physiology and disease. Nat Rev Drug Discov. 2021;20:384–405.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99.
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92.
Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol. 2015;45:2911–7.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75.
Zhang Z, Zhang H, Li D, Zhou X, Qin Q, Zhang Q. Caspase-3-mediated GSDME induced Pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J Cell Mol Med. 2021;25:8159–68.
Yang X, Weng X, Yang Y, Jiang Z. Pyroptosis-related lncRNAs predict the prognosis and immune response in patients with breast cancer. Front Genet. 2021;12:792106.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–8.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Cronin SJF, Seehus C, Weidinger A, Talbot S, Reissig S, Seifert M, et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature. 2018;563:564–8.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Lee J, Roh JL. Targeting GPX4 in human cancer: implications of ferroptosis induction for tackling cancer resilience. Cancer Lett. 2023;559:216119.
Li FJ, Long HZ, Zhou ZW, Luo HY, Xu SG, Gao LC. System X(c) (-)/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. 2022;13:910292.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14:19.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608:778–83.
Yang F, Xiao Y, Ding JH, Jin X, Ma D, Li DQ, et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35:84–100.e8.
Lorito N, Subbiani A, Smiriglia A, Bacci M, Bonechi F, Tronci L, et al. FADS1/2 control lipid metabolism and ferroptosis susceptibility in triple-negative breast cancer. EMBO Mol Med. 2024;16:1533–59.
Zou Y, Zheng S, Xie X, Ye F, Hu X, Tian Z, et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat Commun. 2022;13:2672.
Nath P, Alfarsi LH, El-Ansari R, Masisi BK, Erkan B, Fakroun A, et al. The amino acid transporter SLC7A11 expression in breast cancer. Cancer Biol Ther. 2024;25:2291855.
Zhai FG, Liang QC, Wu YY, Liu JQ, Liu JW. Red ginseng polysaccharide exhibits anticancer activity through GPX4 downregulation-induced ferroptosis. Pharm Biol. 2022;60:909–14.
Liu Z, Zhao Q, Zuo ZX, Yuan SQ, Yu K, Zhang Q, et al. Systematic analysis of the aberrances and functional implications of ferroptosis in cancer. iScience. 2020;23:101302.
Wu K, Li S, Hong G, Dong H, Tang T, Liu H, et al. Targeting METTL3 as a checkpoint to enhance T cells for tumour immunotherapy. Clin Transl Med. 2024;14:e70089.
Zhang K, Ping L, Du T, Liang G, Huang Y, Li Z, et al. A ferroptosis-related lncRNAs signature predicts prognosis and immune microenvironment for breast cancer. Front Mol Biosci. 2021;8:678877.
Fan X, Liu F, Wang X, Wang Y, Chen Y, Shi C, et al. LncFASA promotes cancer ferroptosis via modulating PRDX1 phase separation. Sci China Life Sci. 2024;67:488–503.
Saatci O, Alam R, Huynh-Dam KT, Isik A, Uner M, Belder N, et al. Targeting LINC00152 activates cAMP/Ca(2+)/ferroptosis axis and overcomes tamoxifen resistance in ER+ breast cancer. Cell Death Dis. 2024;15:418.
Zhao J, Xu J, Wu M, Wang W, Wang M, Yang L, et al. LncRNA H19 regulates breast cancer DNA damage response and sensitivity to PARP inhibitors via binding to ILF2. Int J Mol Sci. 2023;24:9157.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Michalczyk K, Cymbaluk-Płoska A. The role of zinc and copper in gynecological malignancies. Nutrients. 2020;12:3732.
Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Asp Med. 2005;26:268–98.
Balamurugan K, Schaffner W. Copper homeostasis in eukaryotes: teetering on a tightrope. Biochim Biophys Acta. 2006;1763:737–46.
Saporito-Magriñá CM, Musacco-Sebio RN, Andrieux G, Kook L, Orrego MT, Tuttolomondo MV, et al. Copper-induced cell death and the protective role of glutathione: the implication of impaired protein folding rather than oxidative stress. Metallomics. 2018;10:1743–54.
Tsvetkov P, Detappe A, Cai K, Keys HR, Brune Z, Ying W, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol. 2019;15:681–9.
Dreishpoon MB, Bick NR, Petrova B, Warui DM, Cameron A, Booker SJ, et al. FDX1 regulates cellular protein lipoylation through direct binding to LIAS. J Biol Chem. 2023;299:105046.
Wu X, Zhang Y, Liang G, Ye H. Cuproptosis-related lncRNAs potentially predict prognosis and therapy sensitivity of breast cancer. Front Pharmacol. 2023;14:1199883.
Li L, Li L, Sun Q. High expression of cuproptosis-related SLC31A1 gene in relation to unfavorable outcome and deregulated immune cell infiltration in breast cancer: an analysis based on public databases. BMC Bioinform. 2022;23:350.
Wu JH, Cheng TC, Zhu B, Gao HY, Zheng L, Chen WX. Identification of cuproptosis-related gene SLC31A1 and upstream LncRNA-miRNA regulatory axis in breast cancer. Sci Rep. 2023;13:18390.
Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. 2018;19:130–40.
Zhang J, Gao RF, Li J, Yu KD, Bi KX. Alloimperatorin activates apoptosis, ferroptosis, and oxeiptosis to inhibit the growth and invasion of breast cancer cells in vitro. Biochem Cell Biol. 2022;100:213–22.
Xiong Z, Yang L, Zhang C, Huang W, Zhong W, Yi J, et al. MANF facilitates breast cancer cell survival under glucose-starvation conditions via PRKN-mediated mitophagy regulation. Autophagy. 2025;21:80–101.
Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25:404–14.
Wang Y, Deng Y, Xie H, Cao S. Hub gene of disulfidptosis-related immune checkpoints in breast cancer. Med Oncol. 2023;40:222.
Xia Q, Yan Q, Wang Z, Huang Q, Zheng X, Shen J, et al. Disulfidptosis-associated lncRNAs predict breast cancer subtypes. Sci Rep. 2023;13:16268.
Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31.
Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. 2021;54:437–53.
Zhou TJ, Zhang MM, Liu DM, Huang LL, Yu HQ, Wang Y, et al. Glutathione depletion and dihydroorotate dehydrogenase inhibition actuated ferroptosis-augment to surmount triple-negative breast cancer. Biomaterials. 2024;305:122447.
Li Y, Lin H, Sun Y, Zhao R, Liu Y, Han J, et al. Platycodin D2 mediates incomplete autophagy and ferroptosis in breast cancer cells by regulating mitochondrial ROS. Phytother Res. 2025;39:581–92.
Valashedi MR, Roushandeh AM, Tomita K, Kuwahara Y, Pourmohammadi-Bejarpasi Z, Kozani PS, et al. CRISPR/Cas9-mediated knockout of Lcn2 in human breast cancer cell line MDA-MB-231 ameliorates erastin-mediated ferroptosis and increases cisplatin vulnerability. Life Sci. 2022;304:120704.
Li P, Lin Q, Sun S, Yang N, Xia Y, Cao S, et al. Inhibition of cannabinoid receptor type 1 sensitizes triple-negative breast cancer cells to ferroptosis via regulating fatty acid metabolism. Cell Death Dis. 2022;13:808.
Jiang T, Zhu J, Jiang S, Chen Z, Xu P, Gong R, et al. Targeting lncRNA DDIT4-AS1 Sensitizes Triple Negative Breast Cancer to Chemotherapy via Suppressing of Autophagy. Adv Sci. 2023;10:e2207257.
Wang J, Xie S, Yang J, Xiong H, Jia Y, Zhou Y, et al. The long noncoding RNA H19 promotes tamoxifen resistance in breast cancer via autophagy. J Hematol Oncol. 2019;12:81.
Kunst C, Tümen D, Ernst M, Tews HC, Müller M, Gülow K. Paraptosis-a distinct pathway to cell death. Int J Mol Sci. 2024;25:11478.
Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–81.
Sperandio S, Poksay K, de Belle I, Lafuente MJ, Liu B, Nasir J, et al. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004;11:1066–75.
MacDonald G, Nalvarte I, Smirnova T, Vecchi M, Aceto N, Dolemeyer A, et al. Memo is a copper-dependent redox protein with an essential role in migration and metastasis. Sci Signal. 2014;7:ra56.
Shubin AV, Demidyuk IV, Komissarov AA, Rafieva LM, Kostrov SV. Cytoplasmic vacuolization in cell death and survival. Oncotarget. 2016;7:55863–89.
Limonta P, Moretti RM, Marzagalli M, Fontana F, Raimondi M, Montagnani Marelli M. Role of endoplasmic reticulum stress in the anticancer activity of natural compounds. Int J Mol Sci. 2019;20:961.
Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, et al. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget. 2014;5:6816–31.
Lee SY, Kim TH, Choi WG, Chung YH, Ko SG, Cheon C, et al. SH003 causes ER stress-mediated apoptosis of breast cancer cells via intracellular ROS production. Cancer Genom Proteom. 2023;20:88–116.
Li Y, Zhang X, Wang Z, Li B, Zhu H. Modulation of redox homeostasis: a strategy to overcome cancer drug resistance. Front Pharmacol. 2023;14:1156538.
Penney RB, Roy D. Thioredoxin-mediated redox regulation of resistance to endocrine therapy in breast cancer. Biochim Biophys Acta. 2013;1836:60–79.
Mihalic ZN, Kloimböck T, Cosic-Mujkanovic N, Valadez-Cosmes P, Maitz K, Kindler O, et al. Myeloperoxidase enhances the migration and invasion of human choriocarcinoma JEG-3 cells. Redox Biol. 2023;67:102885.
Ambrosone CB, Barlow WE, Reynolds W, Livingston RB, Yeh IT, Choi JY, et al. Myeloperoxidase genotypes and enhanced efficacy of chemotherapy for early-stage breast cancer in SWOG-8897. J Clin Oncol. 2009;27:4973–9.
Marone R, Hess D, Dankort D, Muller WJ, Hynes NE, Badache A. Memo mediates ErbB2-driven cell motility. Nat Cell Biol. 2004;6:515–22.
Zhong YT, Cen Y, Xu L, Li SY, Cheng H. Recent progress in carrier-free nanomedicine for tumor phototherapy. Adv Health Mater. 2023;12:e2202307.
Foglietta F, Serpe L, Canaparo R. ROS-generating nanoplatforms as selective and tunable therapeutic weapons against cancer. Discov Nano. 2023;18:151.
Silva CR, Vieira DP, de Freitas AZ, Ribeiro MS. Photodynamic therapy as a strategic ally in radiotherapy for triple-negative breast cancer: the importance of treatment order. Breast Cancer Res Treat. 2025;210:687–97.
Cuenca RE, Allison RR, Sibata C, Downie GH. Breast cancer with chest wall progression: treatment with photodynamic therapy. Ann Surg Oncol. 2004;11:322–7.
Jiang D, Ni D, Rosenkrans ZT, Huang P, Yan X, Cai W. Nanozyme: new horizons for responsive biomedical applications. Chem Soc Rev. 2019;48:3683–704.
Cao C, Yang N, Su Y, Zhang Z, Wang C, Song X, et al. Starvation, ferroptosis, and prodrug therapy synergistically enabled by a cytochrome c oxidase like nanozyme. Adv Mater. 2022;34:e2203236.
Ning S, Zhang Z, Ren Y, Hou Y, Li D, Chen J, et al. A synergistic dual-atom sites nanozyme augments immunogenic cell death for efficient immunotherapy. Adv Sci. 2025;12:e2414734.
Liu Y, Niu R, Deng R, Song S, Wang Y, Zhang H. Multi-enzyme co-expressed dual-atom nanozymes induce cascade immunogenic ferroptosis via activating interferon-γ and targeting arachidonic acid metabolism. J Am Chem Soc. 2023;145:8965–78.
Li D, Ha E, Zhou Z, Zhang J, Zhu Y, Ai F, et al. “Spark” PtMnIr nanozymes for electrodynamic-boosted multienzymatic tumor immunotherapy. Adv Mater. 2024;36:e2308747.
Shahid S, Khan A, Shahid W, Rehan M, Asif R, Nisar H, et al. Nanoenzymes: a radiant hope for the early diagnosis and effective treatment of breast and ovarian cancers. Int J Nanomed. 2024;19:5813–35.
Huang G, Chen H, Dong Y, Luo X, Yu H, Moore Z, et al. Superparamagnetic iron oxide nanoparticles: amplifying ROS stress to improve anticancer drug efficacy. Theranostics. 2013;3:116–26.
Covarrubias G, Moon TJ, Loutrianakis G, Sims HM, Umapathy MP, Lorkowski ME, et al. Comparison of the uptake of untargeted and targeted immunostimulatory nanoparticles by immune cells in the microenvironment of metastatic breast cancer. J Mater Chem B. 2022;10:224–35.
Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018;78:5891–900.
Hu Y, Manasrah BK, McGregor SM, Lera RF, Norman RX, Tucker JB, et al. Paclitaxel induces micronucleation and activates pro-inflammatory cGAS-STING signaling in triple-negative breast cancer. Mol Cancer Ther. 2021;20:2553–67.
Esser AK, Ross MH, Fontana F, Su X, Gabay A, Fox GC, et al. Nanotherapy delivery of c-myc inhibitor targets protumor macrophages and preserves antitumor macrophages in breast cancer. Theranostics. 2020;10:7510–26.
Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–803.
Pan WL, Tan Y, Meng W, Huang NH, Zhao YB, Yu ZQ, et al. Microenvironment-driven sequential ferroptosis, photodynamic therapy, and chemotherapy for targeted breast cancer therapy by a cancer-cell-membrane-coated nanoscale metal-organic framework. Biomaterials. 2022;283:121449.
Pan Y, Liu L, He Y, Ye L, Zhao X, Hu Z, et al. NIR diagnostic imaging of triple-negative breast cancer and its lymph node metastasis for high-efficiency hypoxia-activated multimodal therapy. J Nanobiotechnol. 2023;21:312.
Rahimkhoei V, Alzaidy AH, Abed MJ, Rashki S, Salavati-Niasari M. Advances in inorganic nanoparticles-based drug delivery in targeted breast cancer theranostics. Adv Colloid Interface Sci. 2024;329:103204.
Neshastehriz A, Hormozi-Moghaddam Z, Kichi ZA, Taheri SM, Amini SM, Aghaei A. Overcoming breast cancer cell treatment resistance by optimizing sonodynamic therapy and radiation sensitizers on lncRNA PVT1 and miR-1204 expression. Photodiagnosis Photodyn Ther. 2025;51:104433.
Son S, Kim JH, Wang X, Zhang C, Yoon SA, Shin J, et al. Multifunctional sonosensitizers in sonodynamic cancer therapy. Chem Soc Rev. 2020;49:3244–61.
Yu L, Gao L, Liang B, Zhang L, Wu M, Liu J. Polymer-based nanodrugs enhance sonodynamic therapy through epigenetic reprogramming of the immunosuppressive tumor microenvironment. J Control Release. 2025;380:125–37.
Alonso-Valenteen F, Mikhael S, Wang H, Sims J, Taguiam M, Teh J, et al. Systemic HER3 ligand-mimicking nanobioparticles enter the brain and reduce intracranial tumour growth. Nat Nanotechnol. 2025;20:683–96.
Wu M, Zhang Z, Li D, Ruan X, Yang J, Chen S, et al. Integrating oxygen-boosted sonodynamic therapy and ferroptosis via engineered exosomes for effective cancer treatment. Theranostics. 2025;15:68–85.
Maghsoudian S, Yektakasmaei MP, Shaabani A, Perseh S, Fatahi Y, Nouri Z, et al. Synergistic effects of doxorubicin loaded silk fibroin nanoparticles and Cu-TiO(2) nanoparticles for local chemo-sonodynamic therapy against breast cancer. Int J Biol Macromol. 2025;289:138910.
Huangfu L, Zha B, Li P, Wang L, Liu X, Cui H, et al. A phase I clinical trial of sonodynamic therapy combined with radiotherapy for brainstem gliomas. Int J Cancer. 2025;156:1005–14.
Ma S, Dielschneider RF, Henson ES, Xiao W, Choquette TR, Blankstein AR, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE. 2017;12:e0182921.
Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, Chen X. Copper metabolism in cell death and autophagy. Autophagy. 2023;19:2175–95.
Iranpoor N, Jafari-Gharabaghlou D, Abdulzehra S, Dashti MR, Gorbanzadeh F, Zarghami N. Development and characterization of curcumin loaded pegylated niosomal nanoparticles: potential anti-cancer effect on breast cancer cells through RFC gene expression. Asian Pac J Cancer Prev. 2025;26:1017–26.
Fu Y, Zhang X, Liu X, Wang P, Chu W, Zhao W, et al. The DNMT1-PAS1-PH20 axis drives breast cancer growth and metastasis. Signal Transduct Target Ther. 2022;7:81.
Wang R, Qiu M, Zhang L, Sui M, Xiao L, Yu Q, et al. Augmenting immunotherapy via bioinspired MOF-based ROS homeostasis disruptor with nanozyme-cascade reaction. Adv Mater. 2023;35:e2306748.
Yin D, Yang L, Chen Y. Circ_0022587 regulates tumor properties of human breast cancer cells by targeting miR-335-5p/phosphoglycerate kinase 1 pathway. J Biochem Mol Toxicol. 2025;39:e70205.
Vishnubalaji R, Awata D, Alajez NM. LURAP1L-AS1 long noncoding RNA promotes breast cancer progression and associates with poor prognosis. Noncoding RNA Res. 2025;12:1–9.
Zhi Y, Zhang W, Wu Z, Chen Y, Feng L, He J, et al. miR-223-3p targets KIF4A and promotes the oxidative stress-mediated apoptosis of breast cancer cells. Cancer Biother Radiopharm. 2025;40:323–38.
Ma P, Kang S, Li H, Li M, Zhao Y, Yuan H, et al. A novel lncRNA AC112721.1 promotes the progression of triple-negative breast cancer by directly binding to THBS1 and regulating miR-491-5p/C2CD2L axis. Sci Rep. 2024;14:32056.
Lu Y, Wang K, Peng Y, Chen M, Zhong L, Huang L, et al. Hsa-miR-214-3p inhibits breast cancer cell growth and improves the tumor immune microenvironment by downregulating B7H3. Oncol Res. 2025;33:103–21.
Tahtasakal R, Hamurcu Z, Oz AB, Balli M, Dana H, Gok M, et al. miR-484 as an “OncomiR” in breast cancer promotes tumorigenesis by suppressing apoptosis genes. Ann Surg Oncol. 2025;32:2994–3008.
Gao Y, Huang Y, Zhao Y, Hu P. Cancer-associated fibroblast-secreted exosomal miR-454-3p inhibits lipid metabolism and ferroptosis in breast cancer by targeting ACSL4. Naunyn Schmiedebergs Arch Pharm. 2025;398:3925–37.
Nalla LV, Khairnar A. Empagliflozin drives ferroptosis in anoikis-resistant cells by activating miR-128-3p dependent pathway and inhibiting CD98hc in breast cancer. Free Radic Biol Med. 2024;220:288–300.
Li J, Li PT, Wu W, Ding BN, Wen YG, Cai HL, et al. POU2F2-mediated upregulation of lncRNA PTPRG-AS1 inhibits ferroptosis in breast cancer via miR-376c-3p/SLC7A11 axis. Epigenomics. 2024;16:215–31.
Huang X, Wu J, Wang Y, Xian Z, Li J, Qiu N, et al. FOXQ1 inhibits breast cancer ferroptosis and progression via the circ_0000643/miR-153/SLC7A11 axis. Exp Cell Res. 2023;431:113737.
Zhang C, Xu L, Li X, Chen Y, Shi T, Wang Q. LINC00460 facilitates cell proliferation and inhibits ferroptosis in breast cancer through the miR-320a/MAL2 axis. Technol Cancer Res Treat. 2023;22:15330338231164359.
Wang Y, Gong Y, Li X, Long W, Zhang J, Wu J, et al. Targeting the ZNF-148/miR-335/SOD2 signaling cascade triggers oxidative stress-mediated pyroptosis and suppresses breast cancer progression. Cancer Med. 2023;12:21308–20.
Liu W, Zhang J, Zhang J, Ye Y, Zhu J, Yu Q, et al. EIF4A3-induced circ_0022382 promotes breast cancer cell progression through the let-7a-5p/PI3K/AKT/mTOR signaling pathway and SLC7A11 axis. Front Oncol. 2024;14:1476731.
Tao S, Ji Y, Li R, Xiao Y, Wu H, Ye R, et al. Layered double hydroxide LDH-loaded miR-141-3p targets RAB10 suppressing cellular autophagy to reverse paclitaxel resistance in breast cancer. ACS Omega. 2025;10:5886–99.
Funding
This work was supported by the start-up funds for research from China Medical University (110/1210616002).
Author information
Authors and Affiliations
Contributions
Yiqiao Wen: Writing-original draft. Zhixuan Lin: Writing-original draft. Zhongwei Jiang: Supervision, Formal analysis. Yang Li: Writing-review & editing. Tianyi Wu: Writing-review & editing, Conceptualisation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wen, Y., Lin, Z., Jiang, Z. et al. Targeting the redox-programmed cell death axis in breast cancer: from molecular mechanisms to therapeutic resistance. Cell Death Discov. 11, 441 (2025). https://doi.org/10.1038/s41420-025-02743-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-025-02743-y
