
Abstract
Emerging research underscores the pivotal role of the gut–immune–brain axis, a dynamic bidirectional communication system involving intricate interactions between the gut microbiota, immune responses, and the central nervous system. Gut microbes and their metabolites have profound effects on immune and neurological homeostasis, influencing the development and function of multiple physiological systems. Disruption of the composition of the gut microbiota and barrier integrity has been implicated in various neurological and psychiatric disorders, including autism spectrum disorder, Alzheimer’s disease, Parkinson’s disease, depression, and anxiety. Most insights into these host–microbiota interactions come from preclinical models, revealing both the complexity and potential therapeutic opportunities of the gut–brain communication pathways. This review synthesizes the current understanding of these intricate interactions, exploring how microbiota-driven modulation of the gut and brain barriers, immune signaling, and neuronal pathways, such as those through the vagus nerve, contributes to health and disease. We further explore therapeutic implications, including personalized precision microbiota interventions, microbiome-derived biomarkers, and barrier-strengthening strategies. Advancing this field offers transformative potential for developing innovative, personalized therapies tailored to individual microbiomes and immune profiles, ultimately redefining clinical approaches to neurological and immune-mediated diseases.
Similar content being viewed by others
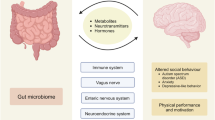
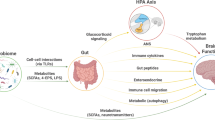
Introduction
The human body harbors a vast and diverse community of commensal microbes, with the gut microbiota playing a crucial role in regulating host physiology. Recent research has demonstrated that gut microbes not only influence metabolism and host immunity, but also modulate neurodevelopment and brain function [1, 2]. The concept of a gut‒brain axis has emerged as a major paradigm shift in neuroscience, revealing bidirectional communication pathways between the gastrointestinal tract and the central nervous system (CNS) [2]. While this axis was initially thought to rely on neural and endocrine signaling, accumulating evidence now suggests that the immune system is a critical intermediary in gut‒brain communication, forming what is increasingly recognized as the gut‒immune‒brain axis. The first evidence linking the gut microbiota to brain function came from studies in the early 21st century using germ-free (GF) mice. These studies revealed that the absence of the gut microbiota leads to alterations in stress responses, neurotransmitter levels, and neurodevelopment [3, 4]. The colonization of GF mice with specific microbiota restores normal levels of stress hormones and brain-derived neurotrophic factor (BDNF), highlighting the influence of microbes on neuronal function [5]. However, these early studies largely focused on direct neuronal interactions, leaving the potential role of the immune system largely unexplored.
Historically, the brain has been considered an immune-privileged organ that is shielded from peripheral immune interactions by the blood‒brain barrier (BBB) [6]. However, recent findings have challenged this notion, showing that immune cells actively infiltrate the brain and play essential roles in neurodevelopment, homeostasis, and disease [7, 8]. The discovery of tissue-resident T cells and border-associated macrophages in the CNS has further underscored the presence of a functional neuroimmune network [9, 10]. These findings, combined with evidence that systemic inflammation contributes to neurodegenerative and neuropsychiatric disorders, suggest that immune mechanisms are central to neural health and disease. Concurrently, research into gut microbiota–immune interactions has revealed intricate pathways by which microbial metabolites shape immune function [11]. The gut microbiota is essential for the development and regulation of both innate and adaptive immunity, producing bioactive metabolites such as short-chain fatty acids (SCFAs), tryptophan derivatives, and secondary bile acids, which modulate systemic inflammation and immune responses [12]. Importantly, these microbial signals have been shown to impact brain functions through both direct metabolic pathways and by shaping immune cell activity in the periphery, which in turn influences neuroinflammation [13].
Despite the increasing recognition of the gut‒immune‒brain axis, many mechanistic questions remain unresolved. The precise pathways through which gut-derived immune signals influence the CNS, the relative contributions of different immune populations, and the extent to which microbiota-targeted interventions can modulate brain function are still unclear. Recent studies suggest that gut microbiota dysbiosis may drive neuroinflammation in multiple neurological disorders, highlighting the translational relevance of this field [2].
In this review, we integrate findings from the gut microbiota, immunology, and neurobiology fields to elucidate the gut‒immune‒brain axis (Fig. 1). We first outline the fundamental principles of mucosal immunology and microbiota‒immune interactions, and then examine the roles of CNS‒resident and CNS‒infiltrating immune populations in brain function. Finally, we synthesize emerging evidence to propose a mechanistic framework for gut-derived immune modulation of the CNS and discuss its implications for neurological disorders. As this field continues to expand, a deeper understanding of these interactions will provide novel therapeutic avenues for neuroinflammatory and neuropsychiatric diseases.

The gut harbors a complex microbiota capable of producing metabolites that regulate host physiology. These metabolites, such as SCFAs, can influence host immune maturation and the balance between pro- and anti-inflammatory responses. On the other hand, immune signals, such as IgA and the Treg/Th17 balance, can regulate microbial tolerance. Microbial metabolites can modulate neuroinflammation and homeostasis by directly affecting brain-resident immune cells and neurons and by eliciting peripheral immune responses that cascade into the brain. Owing to bidirectional crosstalk, the brain itself can modulate the gut microbiota through the vagus nerve and the HPA axis
Gut–immune interactions
Early-life development of the gut microbiota and host immunity
Early-life development presents a crucial window during which the commensal microbiota shapes the maturation and functional programming of the host immune system [14]. The first immune cells of the fetus arise from the yolk sac and include premacrophages from erythro-myeloid progenitors, mast cells, natural killer cells, and innate lymphoid cells [15,16,17]. Neuroimmune cells, such as microglia, can be found in fetal neural tissue as early as 4.5 gestational weeks [18]. Moreover, T-cell development occurs later in the fetal thymus after 6 gestational weeks, with memory phenotypes arising in the second trimester [19, 20]. The maternal microbiome undergoes significant changes throughout pregnancy, with notable enrichment of Proteobacteria and Actinobacteria, as well as overall lower richness toward the third trimester [21, 22]. The progressive shifts in the maternal microbiome correspond to changes in the metabolite pool and the maternal immune system [23, 24]. Notably, maternal microbiota-derived metabolites, including secondary bile acids, have been identified in fetal intestines and may shape the infant immune system [25]. Moreover, a study comparing the fetal metabolome of specific pathogen-free (SPF) and GF mice demonstrated drastic differences among metabolites known to harbor immunological potential [26].
The impact of the maternal microbiota on offspring is further compounded at birth, when neonatal mucosal surfaces are rapidly colonized by microbial genera such as Lactobacillus, Staphylococcus, and Streptococcus [27, 28]. Throughout infancy, the microbiota composition is dynamically shaped by dietary factors, antibiotic exposure, infections, and environmental stimuli [28]. For example, a large body of literature demonstrates the impact of breastfeeding on infant gut microbial compositions [29]. Moreover, breastfed infants have been reported to harbor greater frequencies of blood leukocytes [30]. The essential role of the gut microbiota in immune development is highlighted in GF mice, which exhibit significant reductions in immune cell populations, including macrophages, dendritic cells, neutrophils, T cells, and B cells, along with lower cytokine production [31, 32]. Mice treated with antibiotics early in life also retain microbial, immunological, and neurophysiological disruptions well into adolescence [33]. Notably, microbiota reintroduction into GF mice partially restores immune cell populations and function, depending on bacterial strain composition and the timing of colonization [34, 35]. With the transition into adolescence, the gut microbiota composition develops further, which may be in part due to changes in sex hormone levels [36]. Adolescents undergo immunological changes, including functional changes in glial cells and the BBB, although it is not clear whether these changes are tied to those in the gut microbiota [37]. Importantly, this evidence highlights the critical windows during which microbiota‒immune interactions take place and their potential implications for later life.
Effects of the commensal microbiota on mucosal immunity
The mucosa-associated lymphoid tissue (MALT), which spans the oral, nasal, pulmonary, cutaneous, and gastrointestinal tracts, serves as a key interface for microbial‒immune interactions. Among these, the gastrointestinal mucosa harbors the most extensive immune network, continuously engaging with commensal microbes to regulate immune homeostasis [38]. In GF mice, the absence of commensal bacteria specifically leads to underdeveloped mucosal lymphoid structures, including Peyer’s patches and germinal centers, as well as reduced secretory immune factors [35, 39]. Microbiota-derived signals are essential for the differentiation and function of various immune cell populations. Microbial-derived peptides and polysaccharides promote the expansion of intestinal regulatory T cells (Tregs) and their production of anti-inflammatory IL-10, whereas segmented filamentous bacteria (SFB) drive Th (T helper) 17 differentiation and their production of proinflammatory IL-17 [40, 41]. Similarly, the composition of innate lymphoid cells (ILCs) is influenced by the microbiota, with Clostridium rodentiae enhancing ILC1 populations, Helicobacter pylori driving ILC2 expansion, and Salmonella typhimurium increasing ILC3 populations [42]. A critical mechanism through which the microbiota shapes mucosal immunity is IgA production, the most abundant antibody at mucosal surfaces [43, 44]. IgA not only neutralizes pathogens but also plays a key role in shaping the microbiota composition. Its production is regulated by T follicular helper (Tfh) cells, whose activity is modulated by specific commensal microbes, such as Anaeroplasma species [45]. T follicular regulatory cells further ensure IgA homeostasis, preventing excessive or nonspecific antibody responses while maintaining microbial equilibrium [46]. Thus, the gut microbiota and mucosal immune cells engage in a dynamic and reciprocal relationship in which microbial signals drive immune maturation, and in turn, immune responses shape the microbiota composition and function.
Mechanisms of microbiota‒immune cell crosstalk
SCFAs, which are produced primarily by microbial fermentation of dietary fiber, are essential regulators of innate and adaptive immunity [47]. SCFAs interact with G protein-coupled receptors (GPRs), such as GPR41, GPR43, and GPR109A, suppressing NF-κB activation and thereby modulating inflammatory cytokine production [48,49,50,51]. Additionally, SCFAs act as histone deacetylase (HDAC) inhibitors to regulate T-cell differentiation, promoting Treg differentiation and influencing inflammatory responses [52]. In addition to metabolic signaling, microbial-associated molecular patterns (MAMPs) engage the immune system through recognition by toll-like receptors (TLRs) on antigen-presenting cells, lymphocytes, fibroblasts, and epithelial cells [53]. TLRs serve as key sensors in immune surveillance, detecting microbial components and initiating context-dependent immune responses. For example, TLR4 recognizes bacterial lipopolysaccharides (LPS) and activates the NF-κB and interferon pathways, driving proinflammatory cytokine production [54]. Moreover, TLR2, which responds to surface polysaccharides from probiotic bacteria, promotes anti-inflammatory pathways by inducing Tregs [40]. TLR2 signaling is also crucial for Tfh cell function, thereby influencing IgA-mediated microbiota regulation [55]. In this manner, the direct activation of different TLRs by their respective microbial components can elicit targeted immune responses to maintain homeostasis, clear pathogens, and serve as avenues for therapeutics.
Effects of commensal microbes on systemic immunity
The gut microbiota influences not only mucosal immunity but also the development and regulation of systemic immune responses during both homeostasis and disease [56]. As in mucosal tissues, commensal microbes are essential for the proper maturation and function of systemic immune cells. Compared with their SPF counterparts, young GF mice exhibit reduced thymus sizes and decreased numbers of lymphocytes, indicating a critical role for the microbiota in conventional T-cell development [57, 58]. Gut microbiota-derived antigens are transported to the thymus via migratory dendritic cells, where they facilitate the expansion of antigen-specific T cells [59]. GF mice also exhibit impaired proliferation and TCR signaling in unconventional T cells, including mucosal-associated invariant T (MAIT) cells, highlighting the necessity of microbiota-derived metabolites for MAIT cell development [60]. Similarly, the expression of an autoimmune regulator (Aire) in thymic epithelial cells is necessary for the training and regulation of self-reactive T cells [61]. In GF mice, thymic epithelial cells exhibit reduced Aire expression, which can be restored by colonization with specific gram-positive bacteria, suggesting that microbial cues are required for proper T-cell clonal selection [61, 62]. These findings provide compelling evidence that the gut microbiota can drive systemic immune modulation, offering novel insights into host–microbiota crosstalk and potential therapeutic interventions.
Key players in neuroimmune interactions during health
Although the CNS has long been considered an immune-privileged organ, recent studies have begun to highlight the pivotal role of the immune system in brain development, homeostasis, and disease. Immune cells of the CNS are primarily compartmentalized within the meninges, choroid plexus, and parenchyma, each with distinct structures and barriers that regulate molecular exchange and immune accessibility. The meninges, consisting of the dura, arachnoid, and pia mater, serve as immunological barriers encasing the brain and the spinal cord. The meningeal lymphatic system facilitates CNS immune surveillance by draining cerebrospinal fluid (CSF) and macromolecules, monitoring CNS-derived antigens to sustain homeostasis [63]. The dura mater, a highly vascularized layer with fenestrated capillaries, harbors border-associated macrophages (BAMs), T cells, B cells, and dendritic cells that detect and respond to peripheral immune signals. The leptomeninges (arachnoid and pia mater) house fewer immune cells but contain CSF and cytokines. Unlike the dura, leptomeningeal blood vessels lack fenestrations and are sealed by endothelial tight junctions [64]. The arachnoid mater is also reinforced by tight junctions and demarcates the dura from the subarachnoid space. Dural molecules can only enter the subarachnoid space through arachnoid cuff exit points, which are localized permeable regions [65]. The choroid plexus, located in the brain ventricles, serves as both a CSF producer and a critical neuroimmune interface. Like the dura, it is highly vascularized with fenestrated capillaries, permitting selective molecular exchange. It harbors macrophages, dendritic cells, and T cells, which contribute to CNS immune surveillance. The blood–CSF barrier in the choroid plexus controls molecular and immune cell entry into the CSF, ensuring CNS homeostasis [66]. These structures collectively establish a dynamic CNS immunological landscape in which immune cells interact with other brain cells to regulate CNS functions. This section examines key neuroimmune players, including classical glial cells (microglia and astrocytes) and recently identified CNS-resident immune cells, with a focus on their roles in development, health, and disease (Table 1).
Microglia in health and homeostasis
Microglia are the primary resident immune cells of the CNS and play critical roles in its development and maintenance by continuously surveying the environment and responding to danger signals [67,68,69,70,71,72,73,74]. Microglia feature both common myeloid markers, such as CD11b, F4/80, CX3CR1, and IBA1 [75, 76],, and microglia-specific markers, including TREM119, P2RY12, and Siglec-H [77,78,79]. Recent single-cell RNA-seq studies have revealed substantial transcriptional heterogeneity among microglia in homeostasis and disease [80,81,82]. For example, disease-associated microglia (DAMs) play a dual role in neurodegeneration, clearing toxic aggregates such as amyloid-beta (Aβ) in Alzheimer’s disease (AD) and sustaining chronic inflammation that contributes to neuronal damage [82]. During development, microglia expand rapidly to regulate neuronal fate through the phagocytosis of neural precursor cells [83]. They also secrete neurotrophic factors to support neurodevelopment and eliminate dysfunctional synapses through “find-me” and “eat-me” signals [84,85,86]. In the context of neuroimmune interactions, microglia can respond to peripheral immune changes by detecting cytokines such as IL-4 and IFN-γ, shaping their functional state accordingly [87]. Specifically, IL-4 promotes an anti-inflammatory phenotype by upregulating Arg1 [88], whereas IFN-γ induces a proinflammatory state, leading to the release of TNF-α and IL-6 and the upregulation of MHC-II, which contributes to neuroinflammation [89].
Astrocytes in health and homeostasis
Astrocytes are the most abundant glial cell type within the CNS; they exhibit context-dependent heterogeneity and respond dynamically to both physiological and pathological stimuli [90,91,92,93,94]. Astrocytes mediate neuroinflammation by acting as key intermediaries between the CNS and immune system, expressing cytokine receptors for IL-1α, IL-6, TNF-α, and IFN-γ, enabling them to sense immune signals [95].
During neurodevelopment, astrocytes share similar phagocytic capabilities as microglia do, clearing apoptotic neurons and myelin debris [96, 97]. In the absence of microglia, astrocytes enhance their phagocytic activity and release proinflammatory signals as a compensatory response [98]. The interaction between astrocytes and microglia is fundamental for CNS development. Specifically, astrocyte-derived IL-33 recruits and directs microglial synaptic pruning, a critical step in neural circuit maturation [99]. During development, astrocytes extend branched processes that directly ensheathe multiple synapses. This structural support is essential for proper synaptic excitability and the maintenance of conduction velocity [100,101,102,103]. Furthermore, the perivascular endfeet of astrocytes directly support BBB integrity [104, 105]. Even after the neurodevelopmental period, astrocytes continue to maintain synaptic integrity, neurotransmitter signaling, and water intake, all of which are necessary for healthy neural plasticity [106, 107]. Collectively, astrocytes contribute to CNS homeostasis through their roles in releasing neurotrophic factors, phagocytosing injured neurons, and maintaining a supportive environment necessary for optimal neural function.
Brain-associated adaptive immunity in health
The CNS harbors resident adaptive immune cells, including specialized populations of T cells, which play vital roles in immune surveillance, neuroprotection, and inflammation regulation. In the dural meninges, γδ T cells accumulate from birth and persist lifelong, whereas the number of αβ T cells increases significantly after weaning [108, 109]. Meningeal T cells are composed mainly of CD44+CD62L− memory T cells, with tissue-resident markers such as CD69 [108,109,110]. Early studies demonstrated cognitive impairments in T cell-deficient mouse models, which were reversed by repopulation with functional T cells [111]. These findings led to further investigations into the role of meningeal T cells in regulating brain function. For example, meningeal γδ T cells, which secrete high levels of IL-17A, play a crucial role in modulating anxiety-like behaviors by signaling to neurons with IL-17RA within the medial prefrontal cortex (mPFC) [112]. Tregs also exist in the meninges. A recent study revealed meningeal Tregs with Th1 and Tfh-like transcriptional signatures, which regulate adult hippocampal neurogenesis by controlling IFN-γ signaling [113].
In addition to the meninges, CD4+ conventional T cells (Tconvs) and Tregs are also present in mouse and human leptomeninges and the brain parenchyma [10, 114]. Approximately 2000 CD4+ T cells, including approximately 150 Tregs, have been identified in the brains of healthy mice, with brain-resident Tconvs featuring activation and residency markers such as CD44, CD69, and CD103 [10, 114]. Similarly, brain-resident Tregs are distinct from their circulating counterparts, exhibiting increased levels of the activation markers CTLA-4 and ICOS and the residency markers ST2 and CD69 [114]. Notably, the absence of brain-resident CD4+ T cells disrupts microglial maturation and synaptic pruning functions, resulting in abnormal behavioral phenotypes [114].
Recent research has demonstrated that distinct T cell subtypes differentially regulate neuronal function through cytokine-mediated mechanisms. Neurons and glial cells express receptors for T-cell-derived cytokines. For example, IL-4 receptor alpha (IL-4Rα), which binds to both IL-4 and IL-13, has been found on excitatory and inhibitory neurons of the hippocampus, and signaling along this axis is necessary for proper excitatory and inhibitory potentials in both humans and mice [115]. IL-4-deficient mice also exhibit a proinflammatory meningeal myeloid cell phenotype and cognitive deficits, which can be reversed by the transfer of T cells from wild-type mice, restoring cognitive function [116]. Additionally, GABAergic neurons carry IFN-γ receptor 1, linking IFN-γ to inhibitory signaling and cognitive behaviors [111, 117]. IL-17 has gained particular interest because of its capacity to serve as a neuronal signaling molecule and act beyond its traditional role as a proinflammatory cytokine. In Caenorhabditis elegans, IL-17 has been shown to directly bind to its receptor on neurons to increase synaptic activity and modulate behavior [118]. Accordingly, in mice, IL-17A and IL-17C can activate basolateral amygdala neurons with IL-17 receptor A, promoting anxiety-like behaviors, whereas IL-10 opposes this pathway. [119] Furthermore, IL-17A produced by meningeal γδ T cells modulates glutamatergic synaptic plasticity through glial BDNF production, influencing memory processes by balancing short- and long-term memory retention [108]. Collectively, these studies highlight the critical importance of brain-resident adaptive immune cells, particularly T cells, and their cytokine signals in CNS homeostasis, neuronal function, and cognitive and behavioral regulation.
B cells represent another critical adaptive immune cell type within the CNS. Constituting approximately 15–30% of CD45high cells in the meninges, these B cells span diverse developmental stages, from pro-B cells to mature B cells [110, 120, 121]. While dura mater B cells, including immature B2 subsets, originate primarily from skull bone marrow, antigen-experienced B cells in the peripheral blood can infiltrate the meninges during aging [120]. Interestingly, autoreactive immature B cells that target CNS-specific antigens, such as myelin oligodendrocyte glycoprotein (MOG), undergo negative selection within the meninges, preventing their accumulation and potential autoimmune consequences [122].
B cells are also detected within neonatal brains, where their abundance peaks at P1 and remains elevated throughout early development [123]. Unlike predominantly immature dural B cells, neonatal brain B cells primarily consist of mature B-1a cells, whereas adult brain B cells are predominantly B-2 cells. The infiltration of neonatal B-1a cells is driven by the CXCL13–CXCR5 pathway, and these cells have been shown to promote oligodendrocyte precursor cell proliferation via IgM–Fcα/μR signaling [123]. This noncanonical role of B cells in the brain highlights their potential contribution to CNS regulation, suggesting a broader and previously underappreciated importance of B cells in brain function.
Brain-associated innate immune cells in health and homeostasis
The CNS harbors distinct innate immune populations, notably BAMs and innate lymphocytes. BAMs constitute a diverse group of myeloid cells distributed across the meninges, choroid plexus, and perivascular spaces. Unlike parenchymal microglia, BAMs act as immune sentinels at CNS borders, interacting with both peripheral and CNS-resident cells. The majority of BAMs originate during developmental stages, with the exception of the choroid plexus and dural macrophages, which are replenished by monocytes in adulthood [124, 125]. These cells are characterized by the expression of macrophage-associated markers, such as CD206, CD169, CD163, CD36, and LYVE1, and exhibit a distinct transcriptional signature from parenchymal microglia, characterized by the expression of genes, including Ms4a7, Ms4a6c, Tgfbi, and Lyz2 [126,127,128]. At key immune surveillance sites, such as the dural sinus and choroid plexus, MHC-IIhigh BAMs capture brain-derived antigens and engage in interactions with T cells [129, 130]. In the meninges, dural BAMs also communicate with other cells, including mural cells, to regulate antigen-dependent T cell responses [131]. By modulating vascular integrity, immune signaling, and BBB function, perivascular macrophages contribute to CNS homeostasis [132, 133].
Despite being one of the least studied lymphocyte populations in neuroimmune interactions, a few studies have reported the presence of natural killer (NK) cells and ILCs in CNS tissue under homeostatic conditions [110, 134, 135]. CNS-resident NK cells display distinct phenotypes from those of peripheral NK cells and are characterized by increased IL-2R and CD27 levels and a greater CD62L+ population, which is associated with NK maturity and enhanced cytotoxic function [110, 136]. The majority of these cells in the choroid plexus are NK/ILC1s and ILC2s, with only a few ILC3s being detected within the CNS [137]. A recent landmark study highlighted the critical role of meningeal ILC2-derived IL-13 in inhibitory synapse maturation and social behavior during early neurodevelopment through direct interactions with neuronal IL-13 receptors [135]. Furthermore, ILC2s accumulate in the choroid plexus of aged mouse brains, and intracerebroventricular transfer of activated ILC2s was found to rejuvenate the aged brain and enhance cognitive function in aged mice, highlighting the lifespan-regulating role of ILC2s in CNS health and aging [137].
Neuroimmune interactions during disease
Microglia and astrocytes in neuroinflammation and disease
Glial cells harbor a dual role as regulators of neurodevelopment and homeostasis and drive neuroinflammation and neurological disorders. Both microglia and astrocytes are capable of producing proinflammatory cytokines, including IL-6, IL-1β, IL-23, IL-18, and TNF-α, and IL-1β, TNF-α, and IL-17A, respectively [138,139,140]. These interactions often form proinflammatory feedback loops. Specifically, microglia-derived IL-1α, TNF, and complement component 1q activate astrocytes via the NF-κB signaling pathway, resulting in reactive astrogliosis characterized by impaired neuronal support, reduced synaptic functions, and increased neuronal vulnerability [93]. This microglia‒astrocyte crosstalk is implicated in neurodegenerative diseases such as AD, Parkinson’s disease (PD), and Huntington’s disease (HD) [93]. Conversely, reactive astrocytes produce TNF-α and IL-6, further enhancing microglial activation and inflammation [141,142,143,144]. This glial-driven inflammation can drive neuronal dysfunction and degeneration. For example, TNF-α directly promotes neuronal death through the activation of caspase cascades, while IL-1β erodes synaptic sites by inducing pre- and post- synaptic dysfunction [145, 146]. Additionally, the quantity of proinflammatory cytokines is directly correlated with the accumulation of Aβ plaques in AD, partly due to reduced Aβ clearance as well as increased astrocytic Aβ production [147, 148]. In addition to cytokines, glial cells are potent generators of reactive oxygen species (ROS) and NO, which further contribute to neurotoxicity and tissue damage [149]. Finally, chemokines produced by activated glial cells, such as CCL5, recruit peripheral immune populations, including T and B cells, to the brain [150].
In severe CNS injuries, such as ischemic stroke, microglia display fascinating heterogeneity in their function. Immediately following stroke, microglia take on an anti-inflammatory and tissue repair-associated phenotype, expressing CD206, Arg1, Ccl22, Il10, and Tgfb1, with enhanced phagocytic activity for clearing damaged tissue and supporting neuronal recovery and reinnervation [151]. However, approximately one week postinjury, a shift in the microglial phenotype occurs at the damage site, resulting in reduced phagocytosis and increased production of proinflammatory cytokines and chemokines, as well as the recruitment of other immune populations [151, 152]. Concurrently, ischemic stroke in Sprague–Dawley rats also induces pronounced reactive astrogliosis, characterized by elevated GFAP levels [153]. These reactive astrocytes are essential for reducing reactive oxygen species buildup following stroke and preventing further tissue damage [154]. Furthermore, astrocytes at injury sites are reprogrammed permanently to upregulate genes involved in wound healing, immune signaling, cell adhesion, border maturation, and microbial defense [155]. Astrocyte deficiency significantly disrupts vascular repair and remodeling, impairing motor function recovery after stroke and underscoring their crucial reparative role [156]. Together, microglia and astrocytes play dual roles: they are indispensable in maintaining CNS health, but are also pivotal contributors to neuroinflammation and the progression of neurological diseases.
Brain-resident adaptive immunity in tissue repair and neuroinflammation
During neurotraumatic disorders, T cells are essential mediators of tissue homeostasis, where they significantly influence neuroprotection and tissue repair [157, 158]. In ischemic stroke and traumatic brain injury, diverse immune populations, including T cells, infiltrate the CNS. Following brain injury, CD4⁺ T cells differentiate into various effector subsets, each of which secretes distinct cytokines that exert either proinflammatory or anti-inflammatory effects [157, 158]. In contrast, CD8⁺ T cells contribute to neuronal damage by releasing cytotoxic molecules, such as perforin and granzyme, which induce cell death and exacerbate neuroinflammation [157, 158]. In particular, the roles of Tregs in brain tissue repair are well established. Ito et al. demonstrated that ischemic stroke in mice leads to substantial accumulation of Tregs at the damage site, which is mediated by CCL1 and CCL20 [159]. These Tregs express unique signatures, such as the serotonin receptor 5-HT7, and are essential for producing amphiregulin to suppress neurotoxic astrogliosis [159]. Similarly, in a mouse model of neuromyelitis optica spectrum disorder, Tregs accumulate around lesion sites and, more specifically, colocalize with inflammatory microglia and macrophages [160]. These Tregs reduce the proinflammatory chemokines CCL1, CCL2, and CCL5, as well as the proinflammatory cytokines TNF-α and IFN-γ, through the production of IL-10 and TGF-β [160]. Earlier studies also support the notion of Treg–glial cell interactions, with one showing that Tregs are capable of restraining the LPS-induced inflammatory phenotypes of microglia and macrophages [161].
On the other hand, T cells can drive neuroinflammation. During multiple sclerosis (MS), autoreactive CD4⁺ T cells recognize myelin antigens, leading to CNS inflammation and demyelination [162]. B cells contribute by acting as antigen-presenting cells, sustaining T cell activation, and producing autoantibodies [163]. The interplay among these adaptive immune cells exacerbates neuroinflammation, leading to progressive neurodegeneration and functional impairment in MS.
Brain-resident T cells during neurological disorders
Adaptive immune cells play an active role in neurodegenerative diseases, such as AD. A recent study in a mouse model of tauopathy demonstrated that T cells infiltrate the brain, driven by an expanded population of CD8⁺ effector T cells [164]. Notably, depletion of either microglia or T cells significantly blocks tau-induced neurodegeneration [164]. Clinical studies have further noted lymphocyte infiltration in the brains of patients with AD and PD, highlighting adaptive immune infiltration as a conserved feature of neurodegenerative disorders and a potential target for therapeutic intervention [165, 166].
Adaptive immune cells may also drive neurodevelopmental disorders. T cell cytokines have been identified in the postmortem brain and serum of autism spectrum disorder (ASD) patients [167, 168]. Animal models, such as the maternal immune activation (MIA) model, have begun to reveal the neuroimmune mechanisms underlying neurodevelopmental disorders. MIA in pregnant mice is induced by LPS or poly(I:C), which are recognized by TLR3 and TLR4, respectively, and trigger pathogen-associated molecular patterns and damage-associated molecular patterns [169,170,171]. While the MIA model was initially linked to only IL-6, nearly a decade of research has revealed that a more robust immune response occurs within the MIA model [172]. In particular, MIA induced by poly(I:C) increases the serum levels of IL-6, TNF-α, and IL-1β in pregnant mice, but more importantly, IL-17A from Th17 cells is the key cause of MIA-associated behaviors [173, 174]. This mechanism is directly dependent on the presence of SFB in the maternal gut microbiota [174]. Gut microbial signals activate meningeal γδ T cells to produce IL-17A, resulting in IL-17A receptor signaling on neurons and subsequent abnormal anxiety-like behaviors [112]. Additionally, IL-17A secreted by meningeal T cells can activate border-associated macrophages, inducing ROS production and resulting in impaired cognitive function [175]. Interestingly, while IL-17A has been alluded to as a pathogenic cytokine in ASD, studies have also demonstrated a paradoxical capacity for IL-17A to restore neurotypical behaviors in MIA mice [173, 176]. To date, studies have demonstrated only insults from peripheral or meningeal T cells during neurodevelopmental disorders. It is yet to be determined whether parenchymal T cells also contribute to neuroinflammation. Taken together, these studies underscore the critical involvement of adaptive immune responses in diverse neurological conditions, revealing new avenues for therapeutic development targeting neuroimmune interactions.
Innate immune cells in neuroinflammation and disease
During neuroinflammation, BAMs act as early responders, rapidly initiating immune responses. Upon inflammatory activation, BAMs upregulate critical antigen-presenting molecules, such as MHC-II and CD44, to increase T-cell activation and contribute to the modulation of inflammatory processes [129, 177]. BAMs have also garnered significant attention in the context of neurodegenerative diseases, as they play essential roles in waste clearance, antigen recognition and presentation, and BBB regulation. For example, studies using the TgCRND8 AD mouse model have demonstrated that increased perivascular BAM turnover significantly promotes the clearance of Aβ peptides from cerebral vessels, implicating these macrophages in mitigating AD pathology [178]. Additionally, in a mouse model of PD characterized by alpha-synuclein (α-syn) accumulation, BAMs, rather than microglia, were identified as the key antigen-presenting cells responsible for mediating CD4⁺ T cell-driven neuroinflammation [179]. BAMs also regulate BBB permeability through the modulation of vascular oxidative stress, specifically by NADPH oxidase 2 (NOX2), highlighting their critical influence on vascular integrity and neuroimmune communication [180]. Collectively, these findings underscore BAMs as central regulators of CNS inflammation and homeostasis, coordinating peripheral immune signals and leukocyte trafficking during pathological states.
Innate lymphocytes similarly exert significant regulatory effects during neuroinflammation. In experimental autoimmune encephalomyelitis (EAE), NKp46⁺ ILCs are essential for initiating CNS infiltration via pathogenic CD4⁺ Th17 cells [181]. Specifically, genetic deletion of the transcription factor T-bet in NKp46⁺ ILCs significantly reduces Th17-mediated neuroinflammation, demonstrating their critical role in orchestrating adaptive immune responses [181]. Conversely, meningeal ILC2s exhibit neuroprotective properties, particularly following CNS injury, by upregulating calcitonin gene-related peptide (CGRP) and other neurotrophic molecules, thereby contributing to tissue repair and inflammation resolution [182]. These studies highlight innate lymphocytes as dynamic regulators of inflammatory and protective immune responses within the CNS, suggesting promising targets for therapeutic intervention in neuroinflammatory diseases.
Signaling mechanisms from the gut to the brain
With the rise of correlative studies linking the gut to the brain, the major question in the field is related to the mechanisms by which the gut can influence the brain. Numerous studies in recent decades have elucidated several distinct pathways by which the brain is linked with the brain (Fig. 2). Direct mechanisms include microbiota-derived metabolites that are taken up by neurons, whereas indirect mechanisms involve the activation of host immunity, which can influence neuroinflammation.

Bidirectional crosstalk between the gut and brain occurs through numerous pathways. Gut microbiota-derived metabolites can directly affect neurons within the brain. While large metabolites cannot cross the BBB, precursors such as tryptophan and levodopa are capable of entering the brain. Additionally, BBB transporters, including MCT-1, can mediate the entry of SCFAs into the brain. The BBB itself can be affected by metabolites, as TMAO and SCFAs can enhance BBB integrity, whereas various bile acids can induce barrier disruption by modulating the expression of tight junction proteins. Once within the brain, SCFAs and bile acids can interact with microglia via TLR and TGR signaling, respectively, promoting an anti-inflammatory microglial phenotype, particularly under injury or inflammatory conditions. Finally, the vagus nerve acts as a major highway for gut‒brain crosstalk, facilitating bidirectional regulation. Gut-derived signals are transmitted via vagal afferents to the brainstem, where they are integrated and processed within interconnected nuclei, primarily the nucleus tractus solitarius (NTS) and the dorsal motor nucleus of the vagus (DMV). The NTS handles sensory input, whereas the DMV sends efferent signals that influence gut immunity and microbiota composition
Neurotransmitters from the gut
The gut microbiota significantly influences the production and metabolism of major neurotransmitters, including dopamine, serotonin, glutamate, and gamma-aminobutyric acid (GABA). Specific microbes such as Enterococcus faecalis and Enterococcus faecium synthesize dopamine via levodopa decarboxylation, whereas serotonin synthesis in the gut is modulated primarily by species such as Clostridium, which influences enterochromaffin cells [183,184,185]. Additionally, Lactobacillus and Bifidobacterium species produce GABA through enzymatic glutamate decarboxylation [186]. However, interpreting the direct impact of gut-derived neurotransmitters on CNS function requires caution, as most peripheral neurotransmitters cannot freely cross the BBB under homeostatic conditions [187]. Nonetheless, several microbiota-derived precursor molecules, such as tryptophan and its metabolite 5-hydroxytryptophan (5-HTP), which are precursors to serotonin, as well as tyrosine and levodopa (L-DOPA), which are precursors to dopamine, can cross the BBB and subsequently modulate CNS neurotransmitter availability [187].
The role of short-chain fatty acids on neurons
SCFAs produced by commensal microbes are absorbed by colonocytes, enter portal circulation, and become an important source of energy for peripheral tissues, including the brain [188, 189]. SCFAs are capable of crossing the BBB through monocarboxylate transporter 1 (MCT-1), and their concentrations have been detected in human CSF and brain tissues [189,190,191]. Importantly, SCFAs regulate neuronal development and activity. For example, in human neural progenitor cell cultures, treatment with a mixture of acetate, propionate, and butyrate increased the expression of genes related to neurogenesis and proliferation while reducing the activity of apoptotic pathways [192]. In vivo, carbon-labeled acetate from fermentation in the gut was shown to transverse into the brain by crossing the BBB, upon which it is utilized by hypothalamic neurons for the generation of GABA and lactate [193]. Acetate can directly change the production of neuropeptides as well as hypothalamic AMP-activated protein kinase phosphorylation, ultimately regulating appetite behavior [193]. Similarly, butyrate can also suppress orexigenic neurons of the hypothalamus and neuron activity within the brainstem, reducing appetite and food intake in mice [194]. Appetite modulation in mice has also been demonstrated by SCFA receptors on vagal sensory neurons of the gut, particularly those for propionate [195].
In addition to feeding behaviors, SCFAs can control sympathetic responses to the gut microenvironment. The binding of propionate and acetate to GPR41 and GPR43 on the sympathetic superior cervical ganglion neurons of the spinal cord regulates calcium channel voltage and norepinephrine release, suggesting a mechanism of gastrointestinal sensing [196, 197]. Finally, GABA signaling in neurons may also be dependent on propionate. For example, in mice, propionate can enter GABAergic neurons and induce the accumulation of extracellular GABA and neuronal histone acetylation, a mechanism that may involve the inhibition of GABA transaminase [198].
Butyrate is among the best studied SCFAs because of its impact on neurons. Sodium butyrate, a histone deacetylase inhibitor, is capable of inducing long-term potentiation in the hippocampus, enhancing long-term memory [199]. In line with this finding, sodium butyrate is well documented to improve cognition and memory in several neurodegenerative disorder models by enhancing histone acetylation in the brain, including AD, HD, and PD [200,201,202]. In a mouse model of amyotrophic lateral sclerosis, phenylbutyrate inhibited programmed motor neuron apoptosis by acetylating NF-κB p50, resulting in elevated Bcl-2 expression and consequently blocking caspase activation [203]. Butyrate administration also provides neuroprotection following brain injuries, such as ischemic stroke. Sodium butyrate and moderate doses of acetate can reduce the area of tissue damage following middle cerebral artery occlusion (MCAO) by increasing GPR41, PI3K, and phosphorylated Akt levels and inhibiting neuronal apoptosis [204]. Moreover, butyrate supplementation was shown to improve angiogenesis following stroke in aged mice and improve BBB integrity by promoting IL-22 production by brain-associated ILCs and CD4+ T cells [205]. Specifically, IL-22 receptor activation on brain endothelial cells increases tight junction protein expression and endothelial cell proliferation, thus reducing inflammation-driven BBB permeability [205].
Short-chain fatty acids on brain-resident immune cells
Microglia exhibit significant phenotypic differences between GF and SPF mice. New studies have demonstrated that these differences may be due in part to commensal SCFAs. Mice lacking Ffar2 (which encodes GPR43) for SCFAs exhibit microglial phenotypes similar to those of GF mice [206]. Feeding GF mice a mixture of SCFAs was sufficient to restore healthy microglial morphology and maturation signatures [206]. Specific SCFAs have the ability to modulate microglial function. Acetate restrains LPS-induced IL-1β, IL-6, and TNF-α production among microglia in vivo and in vitro through reversing NF-κB p65 phosphorylation while simultaneously increasing TGF-β and IL-4 production [207]. Acetate was also shown to have similar effects on primary astrocyte cultures by reversing MAPK p38 and NF-κB p65 phosphorylation [208].
Butyrate profoundly modulates microglial responses during neuroinflammation. During MCAO-induced ischemic stroke, sodium butyrate can induce histone modification in activated microglia to reduce the activity of proinflammatory genes while increasing the expression of genes involved in anti-inflammatory pathways [209]. Specifically, sodium butyrate can attenuate the LPS-induced expression of inflammatory Tnf, Il6, Nos2, and Irf1 in microglia [209]. This may be due in part to TLR2 and TLR4 signaling, as they recognize microbial cell wall components and LPS. Notably, TLR2 and TLR4 have been identified on microglia and are implicated in microglial activation, phagocytic activity, and proinflammatory cytokine production [210,211,212]. TLR-mediated neuroinflammation is particularly common in neurodegenerative disorders such as AD, with microglial TLR2 and TLR4 recognition of HMGB1 or Tau exacerbating age-associated disorders [213, 214]. While the direct activation of microglial TLRs by microbial metabolites remains to be tested, neuraminidase from bacteria and viruses was shown to induce inflammatory phenotypes in microglia through TLR2 and TLR4 [215].
The role of bile acids in neurons
Bile acids represent another significant group of commensal-associated metabolites that impact CNS function. Gut microbes convert primary bile acids to secondary bile acids, which influence various neurological outcomes [2]. Tauroursodeoxycholic acid (TUDCA), a secondary bile acid, has demonstrated neuroprotective effects in 3-nitropropionic acid and R6/2 mouse models of HD [216, 217]. TUDCA has also been shown to ameliorate injury and improve motor function after stroke by reducing NF-κB and BCL-2 levels while enhancing the activity of the Akt- and Bad-mediated antiapoptotic pathways in rat neurons [218]. Moreover, TUDCA treatment ameliorates neurotransmitter imbalance and promotes hippocampal neuronal survival in a corticosterone-induced mouse model of depression [219].
However, bile acids also have neurotoxic potential. For example, during the early stages of AD, deoxycholic acid accumulates in the brain and can directly impair cognition by binding with the bile acid receptor Takeda G protein-coupled receptor 5 (TGR5) on excitatory neurons, leading to STAT3 phosphorylation, Aph1 transcription, and increased Aβ production [220]. Interestingly, bile acids have also been linked with behaviors such as itching and scratching in mice [221]. During cholestasis, the stalling of bile acid transport and the accumulation of bile acids can activate TGR5 on dorsal root ganglia neurons, causing hyperexcitability and the production of gastrin-releasing peptide and leucine-enkephalin, which induce itch and analgesia [222]. Other behaviors, such as circadian rhythm sleep disorders during liver diseases, have been attributed to elevated bile acid levels within the suprachiasmatic nucleus and TGR5 binding to neurons [223]. These findings highlight bile acids as dynamic neuromodulators originating from the gut microbiota that are capable of both protective and detrimental effects on neuronal function.
Bile acids on brain-resident immune cells
Among neuroimmune cells, bile acids have beneficial and anti-inflammatory properties. For example, activated microglia express TGR5, which restricts neuroinflammation by stunting their activation, proliferation, and production of IL-1β, IL-6, and TNF-α [224]. This mechanism could be due to blockade of the NF-κB pathway, activation of nuclear factor-erythroid 2-related factor-2 (NRF2) signaling, and enhanced mitochondrial repair within activated microglia, which results in the suppression of the TNF-α positive feedback loop and CCL3 and CCL6 signaling [225, 226]. In this manner, ursodeoxycholic acid was shown to restrain inflammatory microglia during middle cerebral artery occlusion (MCAO) and improve neuron recovery and cognitive behavior in mice [227]. NF-κB inhibition by the binding of taurochenodeoxycholic acid to TGR5 has also been shown to drastically reduce inflammatory phenotypes among astrocytes during experimental autoimmune encephalomyelitis [228]. In the APP/PS1 mouse model of AD, TUDCA was able to restrain microglial and astrocyte activation and proinflammatory polarization and reduce Aβ generation, thus improving the rescue of cognitive deficits [229].
The dynamics of how bile acids impact the brain were recently characterized in depth. Profiling bile acids from various brain regions of young and aged mice has revealed a specific age-associated bile acid, tauro-β-muricholic acid (TβMCA), which can directly induce inflammatory microglia and subsequent neuroinflammation and cognitive decline [230]. Surprisingly, TβMCA accumulation and aging were correlated with a decline in gut microbes with bile salt hydrogenase, and these outcomes could be reversed by the introduction of young microbiota [230]. These dynamics indicate that bile acids should not be viewed as simply beneficial or harmful. However, each bile acid can trigger distinct responses in the brain and its various cell types. Neurons, endothelial cells, and glial cells may react differently to each bile acid, and altering the bile acid pool through the modulation of the gut microbiota could offer therapeutic potential.
Effects of commensal metabolites on BBB integrity
The gut microbiota profoundly influences BBB development and maintenance. GF mice display disrupted BBB integrity, characterized by reduced tight junction protein expression and increased permeability, underscoring the essential role of the microbiota in barrier homeostasis [231]. Various commensal-derived metabolites facilitate this crosstalk. Trimethylamine-N-oxide (TMAO) is the byproduct of dietary methylamine metabolism by commensal microbes from the Anaerococcus, Clostridium, Escherichia, Proteus, Providencia, and Edwardsiella genera [232]. Circulating TMAO enhances BBB integrity in both homeostasis and LPS-induced inflammation by increasing annexin A1 signaling in endothelial cells and protecting against inflammation-associated memory impairment in wild-type mice [233]. On the other hand, commensal metabolites can also be antagonistic to BBB integrity. For example, during cholestasis, elevated bile acid concentrations result in increased BBB permeability [234]. More specifically, chenodeoxycholic acid and deoxycholic acid directly reduce ZO-1 and OCLN tight junction proteins and disrupt the BBB [234]. Similar findings were reported in azoxymethane-induced mouse models of acute liver failure, in which serum bile acid levels increase, resulting in increased BBB permeability, increased bile acid concentrations in the brain, and neurological decline [235, 236].
An emerging hypothesis proposes a correlation between intestinal and BBB permeability. In one mouse model of sepsis-associated encephalopathy, disease onset is initiated by intestinal barrier disruption, which results in sepsis and neurological dysfunction [237]. Surprisingly, prophylactic treatment with a mixture of acetate, propionate, and butyrate in drinking water not only improved the intestinal barrier structure and integrity but also prevented BBB damage, neuroinflammation, and behavioral deficits [237]. Interestingly, sepsis reduces ZO-1 and OCLN levels in both the intestines and the brain, which SCFA treatment could rescue, suggesting a potential link between these two tissue barriers [237]. Similarly, colitis disrupts the intestinal barrier and results in gastrointestinal inflammation, which increases BBB permeability and neuroinflammation [238, 239]. A recent study using dextran sulfate sodium salt-induced colitis mice demonstrated that an enrichment in beneficial microbes, including Lactobacillus, Akkermansia, and Faecalibaculum, as well as elevated production of butyrate from the gut, and upregulated ZO-1 and OCLN levels in the BBB, enhancing its barrier function [240]. Finally, propionate has been shown to bind to GPR41 on BBB endothelial cells to maintain their integrity against nonspecific infections and oxidative stress [241]. Taken together, these findings emphasize that gut-derived microbial metabolites play critical roles in modulating BBB integrity, suggesting promising avenues for therapeutic intervention in neurological conditions associated with compromised barrier function.
Gut-activated peripheral immunity on the brain
The gut microbiota and its metabolites may indirectly impact the brain through the modulation of peripheral immunity (Fig. 3). Gut-derived neurotransmitters impact not only neurons but also peripheral immune populations. Peripheral immune cells express various neurotransmitter receptors. For example, T cells have dopamine and serotonin receptors, which influence their proliferation, differentiation, cytokine secretion, and migration [242, 243]. Specifically, dopamine receptor activation can either enhance or suppress T cell activity, depending on the receptor subtype engaged [244]. GABA inhibits activation-induced calcium signaling and suppresses NF-κB activity in immune cells [245]. Similarly, glutamate receptor signaling enhances T cell migration, influencing T cell localization and function during immune challenges [246, 247]. Serotonin receptor signaling in T cells modulates their inflammatory responses, promoting regulatory T cell differentiation and immunosuppressive functions [248]. Notably, a recent study demonstrated that gut microbiota-derived serotonin directly signals to intestinal T cells, promoting Treg differentiation during neonatal development [249].

Gut-mediated immune signaling regulates brain function. In the intestine, Th17 cells are induced by SFB and IL-17A during early brain development and are linked to ASD-associated behaviors. During inflammatory conditions such as EAE, IFN-γ-producing NK cells are recruited to the brain in a gut microbiome-dependent manner, where they promote anti-inflammatory astrocyte responses. Dysbiosis induces the production of proinflammatory cytokines that disrupt BBB integrity. Conversely, butyrate promotes IL-22 production by ILCs and CD4⁺ T cells, enhancing BBB stability via direct effects on endothelial cells. CNS-resident T cells secrete cytokines to modulate brain function: IFN-γ regulates neuronal activity, IL-4 regulates microglial synaptic pruning, and IL-4 and IL-13 aid in inhibitory synapse formation. Peripheral cytokines, including IL-2, which drives Treg expansion, and IL-33, which suppresses proinflammatory Th17/Th1 responses, also influence CNS-resident immune cells. The role of the gut microbiota in these processes remains to be elucidated
Gut-activated immune responses can influence the brain by transmitting inflammatory signaling molecules, such as cytokines, through the bloodstream. While the BBB typically restricts the direct entry of cytokine molecules into the brain, multiple studies have demonstrated the significant impact that peripheral cytokines can still have on brain function. IL-1β can signal through endothelial cells lining brain blood vessels, inducing the expression of Cox2, which contributes to sickness behavior and facilitates neutrophil infiltration into the brain [250]. Additionally, circulating proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 can compromise BBB integrity by disrupting epithelial tight junctions, potentially allowing greater permeability to inflammatory molecules [251]. These circulating factors can elicit responses in CNS-resident immune cells. For example, in a house dust mite-induced allergic asthma mouse model, IL-4 can alter microglial phenotypes, resulting in reduced synaptic pruning capability and subsequently causing abnormal neural circuitry within the cerebellum [252]. Additionally, peripheral injection of IL-33 has been shown to mitigate EAE pathology in mice by shifting the Th17/Th1 balance toward a Th2 phenotype [253]. Similarly, the IL-2-mediated expansion of peripheral Tregs in humans and mice has demonstrated efficacy in various neurological disorder models, including amyotrophic lateral sclerosis [254]. Whether circulating cytokines, particularly those influenced by the gut microbiota, can modulate brain-resident immune cells under homeostatic conditions remains an open question and warrants further investigation.
A recent study demonstrated interorgan immune communication, showing that gut-derived immune cells migrate to distant tissues [255]. Gut-educated immune cells can directly migrate into the CNS, enabling them to interact with brain-resident cells. For example, a distinct subset of astrocytes characterized by LAMP1 and TRAIL1 (LAMP1+ TRAIL+ astrocytes) plays a critical role in limiting CNS inflammation in EAE mice [256]. During homeostasis, the development of these astrocytes relies on IFN-γ produced by meningeal NK cells [256]. Using photoconvertible Kaede mice, researchers revealed that these IFN-γ-producing meningeal NK cells originate from the gut, with the microbiota directly modulating IFN-γ production [256].
Significant alterations in CNS-resident immune cells often follow changes in the gut microbiota. For example, meningeal IgA-secreting plasma cells are notably reduced in GF mice but can be restored upon gut microbiota recolonization [257]. Similarly, in GF mice, brain-resident conventional CD4⁺ T cells undergo significant expansion following colonization with the wild-type microbiota, indicating that peripheral T cell activation by the microbiota promotes CNS infiltration [114]. Once in the CNS, these peripherally activated CD4⁺ T cells support the maturation and functional regulation of brain-resident microglia [114]. The gut also clearly contributes immune cells to pathological conditions that modulate neuroinflammation, as demonstrated by the migration of gut-derived IgA+ plasma cells into the CNS during EAE and the infiltration of intestinal T cells and monocytes into the CNS in a mouse model of stroke [258, 259]. Collectively, these findings underscore the gut as a critical reservoir and regulator of peripheral immune cells that populate and influence the CNS.
Signaling mechanisms from the brain to the gut
English phrases such as ‘butterflies in your stomach’ during moments of anxiety and nervousness or ‘gut-wrenching feelings’ during upsetting moments allude to rudimentary ideas that the brain may influence our gut. Since ancient times, stress has often been implicated in gastrointestinal distress, including heartburn, constipation, and diarrhea. Today, emerging studies are beginning to identify several mechanisms by which signals from the brain act on the gastrointestinal tract to regulate digestion, the gut microbiota composition, the microbially derived metabolite pool, and gastrointestinal immunity.
Bidirectional crosstalk through the vagus nerve: Gut-to-brain
The vagus nerve is the tenth cranial nerve that runs from the brainstem down into the abdomen, innervating muscular and mucosal tissues of the gastrointestinal tract [260]. The vagus nerve regulates the parasympathetic control of the digestive tract, as well as the delivery of sensory information from the gut to the brain. The vagus nerve is suspected to be one mechanism by which the gut microbiota regulates the brain. For example, vagus nerve activity and its ability to stimulate brainstem neurons are dependent on the presence of the gut microbiota and its metabolites [261]. Additionally, in mice treated with antibiotics, vagotomy is capable of reducing antibiotic-induced anxiety and depression [262, 263]. Similarly, vagotomy suppresses LPS-induced inflammation-mediated depression in C57BL/6 mice by reducing synaptic activity in the brain and suppressing proinflammatory cytokine levels [264]. A similar protective effect of vagotomy was also shown in Lactobacillus and IL-6-induced mouse depression models, suggesting that the vagus nerve plays a crucial role in gut-immune-brain crosstalk [265]. Interestingly, bacteria from the same genus have different neurological impacts through the vagus nerve. For example, while L. instinalis and L. reuteri strains induce depression via the vagus nerve, L. rhamnosus JB-1 is capable of reducing stress-associated anxiety and depression in mice by regulating synaptic activity within the gut and the brain [265,266,267]. These differences might be attributed to metabolic regulation by the vagus nerve.
Serotonin receptors (specifically the 5-HT3 receptor) are present within the vagus nerve, and serotonin itself can activate these neurons [268]. A recent study demonstrated that the gut microbiota of stress-induced mice activates the vagus nerve to elicit serotonin and dopamine dysregulation in the brainstem and hippocampus and depressive behaviors [269]. Additionally, it was recently proposed that even microbial extracellular vesicles can reduce serotonin and BDNF signaling by translocating into the brain through the vagus nerve, causing depression and neuroinflammation [270]. Vagotomy can ameliorate these effects by promoting neurogenesis and suppressing TNF-α, IL-6, and IL-1β in the hippocampus [269, 270]. Furthermore, vagotomy can abrogate other metabolic and neurotransmitter signals from the gut to the brain, including SCFAs, BDNF, and GABA, demonstrating that the vagus nerve is required for gut-to-brain metabolic signaling [261, 266, 271, 272].
Bidirectional crosstalk through the vagus nerve: Brain-to-gut
While the vagus nerve is largely known for its role in mediating gut-to-brain signals, the brain also has the potential to regulate the gut although the vagus nerve. Early reports demonstrated that vagus nerve stimulation can activate STAT3 signaling in intestinal macrophages and suppress intestinal inflammation [273]. More recently, an all-encompassing murine study elegantly illustrated a broader understanding of how the CNS senses peripheral immune activation through the vagus nerve and, in turn, can modulate immune homeostasis [274]. Briefly, neurons in the brainstem are activated by peripheral immune insult through the vagus nerve, as vagotomy is capable of eliminating brainstem activation [274]. Surprisingly, two separate vagal neurons were identified, one with the capacity to respond to proinflammatory cytokines to induce a negative feedback response and the other that can respond to anti-inflammatory cytokines to boost a positive feedback loop [274]. All of these responses are dependent on GABAergic neuron activation within the brainstem to suppress LPS-mediated inflammation [274].
In addition to immune modulation, the vagus nerve can directly control the gastrointestinal tract and the gut microbiota. For example, the activation of the dorsal motor nucleus in vagus neurons increases fat absorption and weight gain [275]. This mechanism is attributed to GABA receptor activity among vagal neurons, which regulates jejunal microvillus length and reduces the surface area by which fat can be absorbed [275]. This capacity to alter the intestinal structure also implies changes in the gut microbiota. Indeed, in the loperamide and restraint-stress model for murine irritable bowel syndrome (IBS), vagotomy improved IBS symptoms by increasing defecation, fecal water content, and constipation levels [276]. These changes were accompanied by shifts in the microbiota composition and microbial activity [276]. Similar observations were made in an MCAO-induced ischemic stroke model, in which vagotomy ameliorated both intestinal and BBB damage by suppressing mast cell activation in these regions [277]. Vagotomy in mice subjected to MCAO altered the gut microbiota composition by increasing the Firmicutes/Bacteroidetes ratio; altering the composition of the flora; reducing LPS endotoxin levels in the serum, brain, and colon; and suppressing the serum levels of TNF-α, IL-6, and IL-1β [277]. Taken together, the vagus nerve is a bidirectional pathway in which gut–immune–brain crosstalk is transmitted and regulated.
The effects of the hypothalamic‒pituitary‒adrenal axis on the regulation of the gut
Stress is colloquially thought to weaken the immune system, which is scientifically evidenced by cancer studies demonstrating stress-induced T cell suppression and tumor growth [278,279,280]. The hypothalamic‒pituitary‒adrenal (HPA) axis is a neuroendocrine system that regulates stress responses via hormone feedback loops between the hypothalamus, pituitary gland, and adrenal gland [281]. Briefly, stress triggers the release of corticotrophin-releasing hormone (CRH) from the hypothalamus, which causes the pituitary glands to secrete adrenocorticotrophic hormone (ACTH). ACTH travels to the adrenal glands, where it stimulates cortisol production. Cortisol acts as a negative feedback loop to suppress hypothalamus and pituitary gland activity. Various cell types throughout the gastrointestinal tract express cortisol receptors, suggesting that cortisol and the stress response may act directly on the regulation of the gut and its microbiota [282].
Among patients with infantile epileptic spasms syndrome (IESS), a form of drug-resistant epilepsy syndrome, the serum levels of IL-2, IL-4, IL-6, and IL-17a are elevated, along with CRH and the abundance of Sutterellaceae and Sutterella in the gut, indicating immunological, neuroendocrine, and gut microbiota disturbances in this neurological disorder [283]. Surprisingly, treatment with ACTH not only reduces CRH, ACTH, and cortisol but also lowers cytokine levels in the serum [283]. The stress response, particularly the levels of ACTH and cortisol, are well documented for their role in systemic immune regulation. ACTH therapy can lower lymphocyte counts in the serum, resulting in greater suppression of Th cells than of cytotoxic T cells [284]. In fact, in vitro, ACTH has the opposite effect on cytotoxic T cells by enhancing memory T cell cytotoxic responses and IFN-γ production [285].
Cortisol also has immunosuppressive effects, particularly among innate immune populations, and reduces TNF-α and IL-6 levels [286]. In particular, clinical studies on cortisol levels following exercise or circadian rhythm also suggest that elevated serum cortisol levels are correlated with suppressed lymphocyte presence and proinflammatory cytokine levels [287, 288]. Traumatic brain injuries can elicit a similar response in wild-type mice, causing elevated plasma cortisol levels and decreased circulating T cell populations, suggesting that the brain can regulate immune responses through the HPA axis [289]. These systemic immune modulations may directly impact gastrointestinal immune homeostasis, although more research is necessary in this regard.
The changes to the immune system caused by the HPA axis may involve crosstalk between host immunity and the gut microbiota. In IESS patients, ACTH treatment decreases Lachnospiraceae incertae sedis abundances while increasing Alistipes and Rikenellaceae abundances [283]. Following ACTH therapy, the changes in Alistipes and Rikenellaceae are directly correlated with IL-17a, IFN-γ, IL-6 and IFN-α levels, whereas Sutterellaceae is positively correlated with CRH levels and negatively correlated with IL-2 and TNF-α [283].
The HPA axis may also mediate maternal stress in infants, altering their microbiome. In a clinical study of maternal stress and depression, both prenatal and postnatal maternal stress directly regulated cortisol levels in mothers and correlated with infant microbiota composition [290]. In particular, high maternal stress and depression during pregnancy result in lower infant microbial diversity, lower Lachnospiraceae, and higher Enterobacteriaceae abundances [290]. On the other hand, maternal postpartum stress and depression reduce the abundance of Bifidobacterium and Lachnospiraceae and increase the abundance of Streptococcaceae in infants, presumably due to the effects of breastfeeding [290]. Importantly, some of these infant microbiota changes are correlated with maternal and infant cortisol levels during these periods, with elevated maternal cortisol during pregnancy correlating with lower Bacteroides in infants and increasing the abundance of the genus postpartum [290]. This study reveals an interesting dynamic in how maternal stress, both during pregnancy and after birth, affects infant microbiota composition through the HPA axis.
Neurotransmitters that regulate gut permeability and microbes
The enteric nervous system (ENS) regulates digestion and gastrointestinal activity through the secretion of various neurotransmitters. Epithelial cells of the intestinal lining and smooth muscles throughout the gut express receptors for neurotransmitters, allowing the modulation of barrier integrity, peristalsis, and lumen maintenance [291]. For example, acetylcholine induces smooth muscle contractions necessary for intestinal motility and the progression of digestion [292]. The gut microbiota is correlated with intestinal motility, with the enrichment of certain microbes, such as Rumioncoccaceae, Eubacterium, Butyricimonas, and Bacteroidetes, during constipation and the depletion of Prevotella, Faecalibacterium, and Dialister during diarrhea [293, 294]. This is also evidenced by observations that changes in intestinal motility and the gut microbiota are key factors in IBS, along with inflammatory immune activation, all three factors that converge in a feedback loop [295].
ENS neurotransmitters are also tied directly to the regulation of intestinal barrier functions, which control both the gut microbiota composition and immune responses. Acetylcholine has been well studied for its ability to regulate mucus levels in the intestinal lumen by inducing mucus secretion in goblet cells, regulating microbial populations [296,297,298]. One mechanism may involve the regulation of stress responses against intestinal epithelial cells, as acetylcholine has been shown to block mitochondrial unfolded protein responses in C. elegans [299]. In a mouse model of DSS-induced colitis, acetylcholine receptors on epithelial cells enhanced protection against colitis progression by increasing the expression of tight junction proteins and inhibiting NF-κB [300]. This mechanism is in part due to the ability of acetylcholine to negate TNF-α signaling in epithelial cells, thus demonstrating its immunomodulatory potential [300]. Overall, acetylcholine is a key mediator of gastrointestinal disorders.
Serotonin is capable of reducing ileal inflammation in mice by inducing acetylcholine release from myenteric plexus neurons, which bind to acetylcholine receptors on macrophages to reduce infiltration [301]. Furthermore, in C. elegans, microbial infections can stimulate acetylcholine production in neurons, which activates Wnt signaling and the production of antimicrobial peptides in gastrointestinal epithelial cells [302]. The gut microbiota is recognized to directly produce neurotransmitters that can affect intestinal function. For example, in Drosophila melanogaster, L. plantarum was found to produce acetylcholine, which directly altered gut motility [303]. These findings suggest bidirectional communication through neurotransmitter signaling, in which both the brain and commensal microbes can regulate the gastrointestinal tract as well as responses to gastrointestinal inflammation.
Evidence for a gut‒immune‒brain axis in neurological disorders
To date, this review has addressed evidence for gut microbiota-mediated regulation of host immunity, the roles of brain-associated immune cells in health and disease, and various pathways by which commensals influence and are influenced by the brain. While very few studies have comprehensively combined all three aspects in a single model, we synthesized available studies to hypothesize that the gut microbiota can mediate neurological disorders through the activation of host immunity. In this section, we discuss the current evidence highlighting how the gut microbiota influences neurodevelopmental and neurodegenerative disorders by modulating host immune pathways (Table 2).
The gut–immune brain axis in neurodevelopmental disorders
Numerous microbiota profiling studies from clinical and preclinical settings have identified alterations in microbial compositions among ASD patients, suggesting gut microbial signatures as potential biomarkers. A meta-analysis of 18 studies encompassing 493 ASD patients and 404 neurotypical individuals revealed distinct gut microbiota patterns in ASD, characterized by elevated levels of Bacteroides, Parabacteroides, Clostridium, Faecalibacterium, and Phascolarctobacterium and reduced Coprococcus and Bifidobacterium abundances [304]. In a cohort of ASD patients in China, Clostridium, Streptococcus, Acinetobacter, and Alcaligenaceae were enriched in ASD patients, along with arginase metabolism, which has been proposed as a biomarker for ASD [305]. A separate ASD cohort from China also revealed fewer butyrate-producing bacteria, including Faecalibacterium and Coproccus, among ASD patients and disrupted SCFA metabolism and neurotoxin degradation pathways, which were suggested to be ASD biomarkers [306].
Interestingly, geographic variations significantly influence microbiota profiles. A study conducted among Lebanese ASD patients and their neurotypical siblings revealed lower Bacteroidetes and higher Catenibacterium and Tenericutes abundances in ASD patients, whereas a separate study in Saudi Arabia reported higher Bacteroides and lower Bifidobacterium and Prevotella abundances in ASD patients than in their siblings [307, 308]. Such drastic differences in microbiota compositions across geography highlight the challenges in using gut microbiota compositions as a sole diagnostic biomarker. While cross-cohort studies do exist, such as the one comparing the Shenzhen and Moscow ASD cohorts, the vast differences in diets, culture, and other lifestyle factors result in underpowered analyses, with the example study identifying only two species, Eubacterium_sp_CAG_248 and P. copri, as shared ASD-associated biomarkers [309]. Thus, a more overarching approach, one that takes into consideration the underlying mechanistic pathways, is needed.
Metabolomic studies provide deeper insight into the functional impact of the gut microbiota during ASD pathogenesis. Metabolite analyses have revealed increased blood levels of glutamate, citrulline, acetylcarnitine, lactate, choline, ornithine, glycine, histidine, and free fatty acids among ASD patients [310,311,312,313]. Additionally, metagenomic analysis of ASD patients from the United States of America revealed that dysfunctional dopamine, GABA, and serotonin pathways are correlated with specific enzymes and oral microbes that may be responsible for these effects [314]. For example, GABA degradation into butyrate was enriched in ASD, which is mediated by 4-aminobutyrate transaminase, glutamate dehydrogenase, and ButCoA acetyl transferase, and these enzymes were correlated with the abundances of Actinomyces, Cardiobacterium, and Streptococcus species among ASD patients [314].
A recent longitudinal metabolomic study comparing newborn and 5-year-old ASD patients revealed that the majority of the metabolic differences in ASD patients were accounted for by 14 key biochemical pathways [315]. In this study, metabolic signatures were nearly identical between pre-ASD newborns and neurotypical newborns, and differential signatures were observed only in 5-year-olds, suggesting that ASD-associated metabolic signatures develop with age and the maturation of their respective gut microbiota [315]. In pre-ASD newborns, several sphingolipid metabolic pathways and chenodeoxyglycocholic and taurodeoxycholic bile acids are increased, whereas serotonin, dopamine, niacin, vitamin B3, and flavin adenine dinucleotide are decreased [315]. Among 5-year-olds, ASD was correlated with lower sphingomyelin, bile salt, tryptophan, serotonin, vitamin, and L-carnitine metabolic pathways [315]. Taken together, ASD metabolic signatures are correlated with decreased anti-inflammatory and antioxidant responses, with decreased glutathione, carnosine, 5’-methyltetrahydrofolic acid, and CoQ10 levels, whereas the levels of metabolites involved in stress response pathways, such as lactate, glycerol, cholesterol, and ceramides, are increased [315].
Immunological phenotypes, particularly with respect to patients’ gut microbiota and metabolic composition, may also prove to be robust biomarkers. Inflammation is a hallmark of ASD, with elevated levels of IL-1β, IL-6, IL-8, IL-12, IL-17, IFN-γ, and TNF-α in plasma and serum samples [316,317,318]. IL-1β, IL-6, IL-17, and TNF-α are also elevated in the postmortem brains of ASD patients [319, 320]. Interestingly, a correlative analysis between metabolites altered in ASD patients revealed that glutamine, serine, proline, histidine, lysine, taurine, 5-aminolevulinic acid, and 2-hydroxyglutaric acid, among others, are involved in inflammatory pathways [321]. For example, glutamine, glycine, and taurine all have anti-inflammatory and antioxidant effects [322,323,324]. An imbalance in such metabolites during ASD may be causative for enhanced neuroinflammation. With this concept, meta-analyses are necessary to link microbiota compositions with metabolic pools and identify pathways involved in the regulation of inflammation. Studies applying this approach are slowly emerging, with one metagenomic study linking the abundance of Ruminococcus in individuals with ASD with increased middle chain fatty acid production, which is known to induce proinflammatory IL-12 [325]. Additionally, the present study revealed lower Prevotella abundance among constipated ASD patients, a genus that has often decreased across ASD studies [308, 325]. Prevotella are important producers of succinic acid, which has anti-inflammatory effects by suppressing IL-6, TNF-α, IL-1β, and nitric oxide production in macrophages, further supporting a microbe‒metabolite‒immune link in ASD [325, 326].
The gut–immune–brain axis in neurodegenerative disorders
Compared with healthy individuals, AD patients have a reduced richness in the gut microbiota composition, with lower Firmicutes and higher Bacteroidetes [327]. According to previous studies, Clostridiaceae, Dialister, Clostridium, Erysipelotrichaceae, Lachnoclostridium, and Subdoligranulum are decreased in AD patients, whereas Blautia, Phascolarctobacterium, Gemella, Bacteroides, Alistipes, Odoribacter, and Barnesiella are increased [327,328,329]. In particular, elevated genera such as Bacteroides and Blautia are positively correlated with higher CSF concentrations of tau and Aβ, whereas less abundant genera, including Dialister, are negatively correlated with these genera [327]. SCFA-producing bacteria, including B. massiliensis, B. pseudocatenulatum, and Fusicatenibacter saccharivorans, are also indicative of better cognitive function among elderly individuals with mild cognitive impairment [330]. It was hypothesized that B. pseudocatenulatum, a producer of acetate, could participate in anti-inflammatory signaling and the maintenance of intestinal immune homeostasis [330, 331]. Notably, the microbiota of AD patients can impair neurogenesis in the hippocampus as well as cognitive and memory capacity in adult male Sprague‒Dawley rats [332]. Furthermore, the human AD gut microbiota promotes an intestinal inflammatory state by lowering P-glycoprotein expression and slightly elevating MRP2 protein levels in cultured intestinal epithelial cells, thus modulating inflammatory responses against commensals [329].
Human primary brain cells cultured with B. fragilis-derived LPS drive NF-κB, which may help promote neuroinflammation and pathogenesis during AD [333]. Interestingly, in a GF APPPS1 mouse model of AD, a reduced Aβ load was directly associated with diminished levels of SCFAs in the serum, and supplementation with an acetate, butyrate, and propionate mixture was capable of doubling Aβ plaque accumulation within the brain [334]. SCFAs were shown to enhance microglial localization to Aβ plaques; however, SCFAs also reduced microglial uptake of Aβ and upregulated APOE, suggesting an immunosuppressive function of SCFAs in the context of AD [334]. While neuroinflammation is considered a pathogenic hallmark of AD, few studies have directly connected the microbial and metabolic signatures of AD with their immunomodulatory capacities. Future research in AD will need to address whether the gut microbiota and its metabolites are capable of modulating neuroinflammation and thereby affecting the outcome of AD pathology.
The gut–immune brain axis in psychiatric disorders
In addition to neurodevelopmental and neurodegenerative disorders, the interplay of the gut-immune-brain axis is also observable in neuropsychiatric disorders. A genome-wide association study among depression patients revealed significant correlations between gut microbes and their predicted metabolic pathways, immune dysfunction, and incidence of depression [335]. Specifically, Paramecium excretum and Parasutterella were identified as having causal associations with depression, along with the lysine biosynthesis and methionine biosynthesis pathways [335]. CD8+ T cells, CD11b+ monocytic myeloid-derived suppressor cells, and CD27+ B cells are also correlated with depression, with CD27+ B cells specifically mediating the effects of the microbial methionine biosynthesis III pathway during disorders [335].
Depression patients also present increased TNF, macrophage colony-stimulating factor (MCSF), and IL-12 in the intestinal mucosa and reduced abundances of Clostridium, Roseburia, Haemophilus, SMB53, and Turicibacter, which are positively correlated with propionate and butyrate levels [336]. Interestingly, the gut microbiota of patients with treatment-resistant inflammatory depression differed from that of patients with other types of depression in that inflammation-associated Bacteroides were enriched, whereas SCFA-producing Clostridium were decreased [336]. This was consistent with reduced butanoate metabolism; increased TLR-4, NF-κB, NLRP3, Caspase-1, TNF, and MCSF; and decreased ZO-1 and OCLN levels in the intestinal mucosa [336]. Notably, these phenotypes could be recapitulated in wild-type mice that received fecal microbiota transplantation (FMT) of inflammatory depression patients’ feces, and C. butyricum treatment could ameliorate depression phenotypes by reducing inflammation, demonstrating a causal effect [336]. Furthermore, in a chronic social defeat stress mouse model of depression, inhibiting intestinal γδ T cells with TCR γ and δ antibodies could directly prevent stress-induced γδ T-cell accumulation in the meninges and subsequent behavioral deficits [337]. This evidence highlights both the immunomodulatory function of the gut microbiota and the capacity for immune modulation to directly affect depressive disorders, supporting the theory of a gut–immune–brain mechanism in neuropsychiatric disorders.
Targeting the gut–immune–brain axis for therapeutics
The gut–immune–brain axis has emerged as a crucial target for developing novel therapies for neurological disorders, with increasing evidence underscoring its role in neuroinflammation, immune regulation, and brain function. Therapeutic strategies aimed at this axis focus primarily on two approaches: immune-based interventions that modulate inflammatory pathways and microbiota-based therapeutics (MBTs) that leverage the gut microbiome to influence brain health. Understanding how immune dysfunction and microbial imbalances contribute to neurological conditions provides valuable insights for precision medicine. In this section, we explore current strategies targeting immune and MBTs and their potential to restore homeostasis within the gut–immune–brain axis, ultimately improving neurological outcomes (Table 3).
Immune therapies for neurological disorders
Currently, there are no FDA-approved immunomodulatory therapies for stroke. Despite promising results from medical and physical interventions in experimental stroke models, successful translation into clinical applications remains a challenge [338, 339]. Among potential candidates, fingolimod, a sphingosine-1-phosphate receptor (S1P) modulator that prevents lymphocyte egress from the lymph nodes, has shown encouraging outcomes in preclinical and early clinical studies [340]. Additionally, anakinra, an IL-1 receptor antagonist, has demonstrated potential therapeutic efficacy in several clinical trials [341]. Recent preclinical studies have identified Tregs as promising therapeutic targets for stroke. The expansion of Tregs via IL-2/IL-2 antibody complexes and brain-targeted IL-2 overexpression in astrocytes via AAV vectors have shown beneficial effects in MCAO and cortical impact mouse models of stroke [342, 343]. The modulatory effects of Treg expansion may be mediated through the regulation of glial cell activation, facilitated by Treg-derived molecules such as SPP1 and AREG [159, 344].
Immune-based therapies for AD primarily target Aβ [345]. In particular, immunization with Aβ peptides and the administration of anti-Aβ antibodies have emerged as key therapeutic strategies for AD. Notably, two monoclonal antibodies, aducanumab and lecanemab, have received traditional FDA approval for the treatment of AD [346,347,348]. Furthermore, masitinib, a tyrosine kinase inhibitor, has demonstrated neuroprotective effects through the inhibition of mast cells and microglia/macrophage activity, and clinical trials have shown significant cognitive improvement [349]. Additionally, PD-1/PD-L1 immune checkpoint inhibitors are currently under clinical investigation for AD. Preclinical studies have shown that PD-1/PD-L1 blockade can mobilize monocyte-derived macrophages, enabling their infiltration into the brain to reduce pathology and combat cognitive decline in 5XFAD AD mice [350, 351]. An antibody targeting PD-L1 has been developed to stimulate the immune system by inhibiting immune checkpoint proteins, and this approach is currently in early-phase clinical trials for AD.
The aggregation of α-syn into Lewy bodies is a pathological hallmark of PD. Both passive and active immunization against α-syn aggregates are being explored in ongoing trials [352]. Additional immune-targeted strategies include the use of Sargramostim, a human recombinant granulocyte‒macrophage colony-stimulating factor that increases the number of Tregs, which can modestly improve motor functions [353]. Furthermore, the immunosuppressant azathioprine has been reported to improve cognition in PD patients by inhibiting DNA synthesis [354]. Moreover, AKST4290, a chemokine inhibitor that targets C-C chemokine receptor 3 to reduce immune cell infiltration, is currently being evaluated as a potential PD treatment [355].
In addition to neurotraumatic and neurodegenerative diseases, neurodevelopmental disorders, such as pediatric epilepsies, are increasingly recognized as targets for immunotherapy, particularly in cases where immune dysfunction contributes to seizure pathology. In IESS and Lennox–Gastaut syndrome (LGS), ACTH and steroids have demonstrated therapeutic efficacy through immune suppression [356, 357]. Additionally, cytokine-targeting therapies, including tocilizumab (an anti-IL-6 receptor inhibitor) and anakinra (an anti-IL-1 receptor antagonist), have been shown to be effective in treating immune-mediated epileptic syndromes, such as new-onset refractory status epilepticus (NORSE) and febrile infection-related epilepsy syndrome (FIRES) [358, 359]. Natalizumab, an anti-α4-integrin antibody that blocks leukocyte migration to the brain, is also currently under clinical investigation for drug-resistant epilepsy, with some successful cases reported in Rasmussen’s encephalitis [360, 361]. These advancements underscore the potential of immunotherapies as promising treatment strategies for pediatric and drug-resistant epilepsies.
Finally, ASD represents another promising area for immune-based intervention. Clinical observations have revealed reduced frequencies of Tregs in individuals with ASD [362, 363]. Accordingly, therapeutic strategies aimed at selectively expanding Tregs through IL-2/anti-IL-2 complex therapies are currently being explored as potential approaches to alleviate ASD-associated inflammation and symptoms [364].
Microbiota-based therapeutics for neurological disorders
MBTs come in multiple forms, including probiotic live bacteria, prebiotics, synbiotics, postbiotics, and microbial metabolites [1]. In recent years, numerous reviews and meta-analyses have characterized clinical and animal studies testing the efficacy of MBTs in various neurodevelopmental, neurodegenerative, and neuropsychiatric disorders [2]. However, studies that address all three aspects of the gut–immune–brain axis simultaneously remain limited. In the following section, we highlight the latest studies in which MBTs are utilized specifically for their immunomodulatory potential in the context of neurological disorders.
MBTs have been extensively studied in ASD, where inflammation and microbial dysbiosis are key pathological features. For example, the BTBR and valproic acid mouse models for ASD are characterized by systemic inflammation and weakened barrier functions [365]. In these mice, treatment with SCFA-producing C. butyricum improved intestinal barrier integrity by increasing the levels of TREK1, CLDN1, CNDN3, and OCLN tight junction proteins and reducing the levels of IL-6, TNF-α, and IFN-γ in the colon, resulting in reduced ASD-associated behaviors [365]. Similar findings have also been reported in a rat model of propionic acid-induced ASD, in which treatment with a mixture of L. rhamnosus and luteolin (an antioxidant ingredient of artichokes) reduces proinflammatory TNF-α and IL-6 levels and elevates GABA, glutathione, and glutathione peroxidase levels in the brain [366]. Additionally, B. longum BB536 mirrors these effects in propionic acid-induced ASD rats by reducing IFN-γ levels within the brain [367]. In the MIA mouse model of ASD, P. goldsteinii MTS01 ameliorated antisocial and anxiety-like behaviors by reducing colonic TNF-α, IL-6, and IL-1β in the intestines while increasing the activity of antioxidant pathways and decreasing glutamate receptor signaling in the hippocampus [368]. Finally, prebiotics, such as 3% galacto-oligosaccharide/fructo-oligosaccharide, have been shown to prevent valproic acid-induced ASD-associated social and cognitive deficits by suppressing microglial activation, lowering Th1 and Th17 and increasing Treg populations, improving intestinal barrier function, and normalizing the gut microbiota composition and SCFA pool [369].
MBTs are also being explored for the treatment of neurodegenerative diseases, such as AD and PD. In an APP/PS1 mouse model for AD, a mixture of L. paracasei D3.5, L. rhamnosus D4.4, L. plantarum D6.2, L. rhamnosus D7.5, L. plantarum D13.4, E. raffinosus D24.1, E. INBio D24.2, E. avium D25.1, E. avium D25.2 and E. avium D26.1 improved cognitive function by enhancing BBB and intestinal barrier integrity, reducing activated microglia, and reducing IL-6 and TNF-α within the brain, plasma, and intestines [370]. This study reveals a clear link between barrier function and immune modulation across the intestines, periphery, and brain. Furthermore, in female 3xTg-AD mice, a mixture of Lactobacillus plantarum KY1032 and Lactobacillus curvatus HY7601 is capable of improving memory by reducing microglia and astrocyte densities while increasing neuron populations [371]. MBTs have also demonstrated therapeutic capacity in PD. A synbiotic combination of L. paracasei DG and inulin fiber improved cognitive function among PD patients [372]. This mechanism could be immune-modulated, as other Lactobacillus strains, such as L. rhamnosus E9, are capable of reducing PD-associated inflammation by improving intestinal barrier integrity [373]. Furthermore, a consortium of L. rhamnosus, E. faecium, L. acidophilus, and L. plantarum can significantly reduce plasma IL-6 and TNF-α levels in PD patients [374]. Similar results were found in in vitro cultures of peripheral blood mononuclear cells from PD patients, which showed suppressed inflammatory cytokine and ROS production when these cells were treated with six individual probiotic strains, supporting a probiotic–immune link in PD [375].
Neuropsychiatric disorders may also be targeted through MBTs. In a chronic unpredictable stress model of murine depression, a synbiotic combination of L. plantarum ATCC 793 (Lp793), B. longum ATCC 15707 (Bl15707), and a botanical-derived polyphenolic preparation (BDPP) promoted CTLA4+ Treg expansion, lowered Th17 cells in the gut, lowered IL-1β and IL-17a levels in the serum, and lowered prefrontal cortex IL-1β levels, which attenuated depression and anxiety-like behaviors [376]. Another probiotic mixture containing L. lactis, L. cremoris, L. diacetylactis, L. acidophilus, and lactic yeasts also confers protection against LPS-induced depression and anxiety in CD1 mice by reducing the serum levels of IL-12, TNF-α, IL-1β, and TNF-α in the brain [377]. In a clinical trial for ADHD, Synbiotic 2000, a mixture of Pediococcus pentosaceus 5-33:3/16:1, L. casei subsp. paracasei F19, L. plantarum 2362 and beta-glucan, inulin, pectin and resistant starch fibers, was capable of lowering neuroinflammatory IL-12/IL-23p40 and soluble intercellular adhesion molecule 1 while increasing propionic acid levels in the blood [378]. Notably, while Synbiotic 2000 did not improve ADHD symptoms compared with placebo, Synbiotic 2000 showed efficacy in improving autism symptoms in children and emotion regulation among adults [379]. Collectively, these studies suggest the potential of MBTs as immunomodulatory therapeutics for neurological conditions. However, many studies are still merely correlative. Future investigations should explicitly test causality by systematically inhibiting individual components of the gut–immune–brain axis to establish direct therapeutic mechanisms.
Conclusion and future directions
The intricate interplay among the gut microbiota, immune system, and CNS has emerged as a compelling area of research, offering new perspectives on pathogenesis and potential treatment strategies for neurological disorders. The evidence discussed throughout this review highlights not only how the gut microbiota and its metabolites shape immune and neurological homeostasis but also how immune and neural signals reciprocally modulate microbial communities.
In neurological disorders, increasing amounts of data highlight correlations between disease progression, gut microbial dysbiosis, altered metabolite pools, and inflammation. This leads to the hypothesis that neurological disorders may be induced by a gut-immune mechanism. Aberrations in the gut microbial composition can lead to dysregulated immune signaling, increased neuroinflammation, and disruptions in blood‒brain barrier integrity, all of which can exacerbate neurological dysfunction. This growing body of evidence underscores the therapeutic potential of targeting the gut microbiota and immune pathways to develop novel interventions for neurological diseases.
To achieve this, several critical directions must be pursued. One promising approach is the identification of new biomarkers. Microbial metabolites and immune signatures have potential for predicting disease onset, disease progression, and therapeutic response in several neurological disorders. Validating and clinically applying these biomarkers could significantly improve diagnostic precision and enable personalized treatment strategies. With respect to therapeutic targets, current findings suggest that the compromised BBB, a hallmark of various neurological disorders, may be effectively addressed through novel interventions. These could include barrier-enhancing microbiota-derived metabolites or probiotics to restore BBB integrity. Additionally, targeting microbial metabolites and immune factors may also be effective in treating neurotraumatic diseases by modulating local immune responses and promoting neuronal recovery. Another crucial consideration is the interindividual variability in microbiome composition and immune profiles. Patient-specific interventions, such as personalized dietary strategies, CRISPR–Cas9–based microbiome editing, and tailored probiotic formulations, are essential for effective treatment. Alongside these clinical efforts, mechanistic studies must continue to unravel the complex interactions within the axis.
Promoting interdisciplinary collaboration among immunologists, neuroscientists, microbiologists, bioengineers, and clinicians will be crucial for driving innovative, integrative discoveries. As research in this field continues to evolve, the gut–immune–brain axis is likely to redefine our understanding of neurological disorders, paving the way for innovative microbiome-based and immune-modulating therapies.
References
O’Riordan, KJ, Moloney, GM, Keane, L, Clarke, G & Cryan, JF The gut microbiota-immune-brain axis: Therapeutic implications. Cell Rep Med 6 (2025). https://doi.org/10.1016/j.xcrm.2025.101982.
Park JC, Im SH. The gut-immune-brain axis in neurodevelopment and neurological disorders. Microbiome Res Rep. 2022;1:23. https://doi.org/10.20517/mrr.2022.11.
Gaykema RPA, Goehler LE, Lyte M. Brain response to cecal infection with Campylobacter jejuni: analysis with Fos immunohistochemistry. Brain Behav Immun. 2004;18:238–45. https://doi.org/10.1016/j.bbi.2003.08.002.
Sudo N. Stress and gut microbiota: Does postnatal microbial colonization programs the hypothalamic‒pituitary‒adrenal system for stress response?. Int Congr Ser. 2006;1287:350–4. https://doi.org/10.1016/j.ics.2005.12.019.
Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, et al. Postnatal microbial colonization programs the hypothalamic–pituitary–adrenal system for stress response in mice. J Physiol. 2004;558:263–75. https://doi.org/10.1113/jphysiol.2004.063388.
Nutma E, Willison H, Martino G, Amor S. Neuroimmunology – the past, present and future. Clin Exp Immunol. 2019;197:278–93. https://doi.org/10.1111/cei.13279.
Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 2004;28:254–60. https://doi.org/10.1002/jnr.490280213.
Ludowyk PA, Willenborg DO, Parish CR. Selective localization of neuro-specific T lymphocytes in the central nervous system. J Neuroimmunol. 1992;37:237–50. https://doi.org/10.1016/0165-5728(92)90008-9.
Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci. 2010;107:17872–9. https://doi.org/10.1073/pnas.1010201107.
Smolders J, Heutinck KM, Fransen NL, Remmerswaal E, Hombrink P, Ten Berge I, et al. Tissue-resident memory T cells populate the human brain. Nat Commun. 2018;9:4593. https://doi.org/10.1038/s41467-018-07053-9.
Byun S, Lee J, Choi YH, Ko H, Lee C, Park JC, et al. Gut microbiota defines functional direction of colonic regulatory T cells with unique TCR repertoires. J Immunol. 2024;213:886–97. https://doi.org/10.4049/jimmunol.2300395.
Yang W, Cong Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol Immunol. 2021;18:866–77. https://doi.org/10.1038/s41423-021-00661-4.
Sun X, Shukla M, Wang W, Li S. Unlocking gut-liver-brain axis communication metabolites: energy metabolism, immunity and barriers. npj Biofilms Microbiomes. 2024;10:136. https://doi.org/10.1038/s41522-024-00610-9.
Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–44. https://doi.org/10.1126/science.aad9378.
Mass, E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science 353 (2016). https://doi.org/10.1126/science.aaf4238
Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity. 2018;48:1160–.e1165. https://doi.org/10.1016/j.immuni.2018.04.025.
Popescu D-M, Botting RA, Stephenson E, Green K, Webb S, Jardine L, et al. Decoding human fetal liver hematopoiesis. Nature. 2019;574:365–71. https://doi.org/10.1038/s41586-019-1652-y.
Andjelkovic AV, Nikolic B, Pachter JS, Zecevic N. Macrophages/microglia in human central nervous system during development: an immunohistochemical study. Brain Res. 1998;814:13–25. https://doi.org/10.1016/s0006-8993(98)00830-0.
Feyaerts D, Urbschat C, Gaudilliere B, Stelzer IA. Establishment of tissue-resident immune populations in the fetus. Semin Immunopathol. 2022;44:747–66. https://doi.org/10.1007/s00281-022-00931-x.
Stras SF, Werner L, Toothaker JM, Olaloye OO, Oldham AL, McCourt CC, et al. Maturation of the human intestinal immune system occurs early in fetal development. Dev Cell. 2019;51:357–.e355. https://doi.org/10.1016/j.devcel.2019.09.008.
Nuriel-Ohayon M, Neuman H, Koren O. Microbial changes during pregnancy, birth, and infancy. Front Microbiol. 2016;7:1031. https://doi.org/10.3389/fmicb.2016.01031.
Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–80. https://doi.org/10.1016/j.cell.2012.07.008.
Gao Y, Zhang J, Chen H, Jin X, Lin Z, Fan C, et al. Dynamic changes in the gut microbiota during three consecutive trimesters of pregnancy and their correlation with abnormal glucose and lipid metabolism. Eur J Med Res. 2024;29:117. https://doi.org/10.1186/s40001-024-01702-0.
Aghaeepour, N, Ganio EA, Mcilwain D, Tsai AS, Tingle M, Van Gassen S, et al. An immune clock of human pregnancy. Sci Immunol 2 (2017). https://doi.org/10.1126/sciimmunol.aan2946
Li, Y, Toothaker JM, Ben-Simon S, Ozeri L, Schweitzer R, McCourt BT, et al. In utero human intestine harbors unique metabolome, including bacterial metabolites. JCI Insight 5 (2020). https://doi.org/10.1172/jci.insight.138751
Pessa-Morikawa T, Husso A, Kärkkäinen O, Koistinen V, Hanhineva K, Iivanainen A, Niku M. Maternal microbiota-derived metabolic profile in fetal murine intestine, brain and placenta. BMC Microbiol. 2022;22:46. https://doi.org/10.1186/s12866-022-02457-6.
Yao, Y, Cai X, Ye Y, Wang F, Chen F, Zheng C. The Role of Microbiota in Infant Health: From Early Life to Adulthood. Front Immunol 12 (2021). https://doi.org/10.3389/fimmu.2021.708472
Tamburini S, Shen N, Wu HC, Clemente JC. The microbiome in early life: implications for health outcomes. Nat Med. 2016;22:713–22. https://doi.org/10.1038/nm.4142.
Ho NT, Li F, Lee-Sarwar KA, Tun HM, Brown BP, Pannaraj PS, et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat Commun. 2018;9:4169. https://doi.org/10.1038/s41467-018-06473-x.
Hamdan TA, Alkhateeb S, Oriquat G, Alzoubi A, Ahmed KA-A. Impact of breastfeeding and formula feeding on immune cell populations and blood cell parameters: an observational study. J Int Med Res. 2024;52:3000605241307217 https://doi.org/10.1177/03000605241307217.
Bauer H, Horowitz RE, Levenson SM, Popper H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am J Pathol. 1963;42:471–83.
Kennedy EA, King KY, Baldridge MT. Mouse microbiota models: comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front Physiol. 2018;9:1534 https://doi.org/10.3389/fphys.2018.01534.
Lynch C, Cowan C, Bastiaanssen T, Moloney GM, Theune N, van de Wouw M, et al. Critical windows of early-life microbiota disruption on behavior, neuroimmune function, and neurodevelopment. Brain, Behav Immun. 2023;108:309–27. https://doi.org/10.1016/j.bbi.2022.12.008.
Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–93. https://doi.org/10.1016/j.cell.2012.04.037.
McDermott AJ, Huffnagle GB. The microbiome and regulation of mucosal immunity. Immunology. 2014;142:24–31. https://doi.org/10.1111/imm.12231.
Korpela K, Kallio S, Salonen A, Hero M, Kukkonen AK, Miettinen PJ, et al. Gut microbiota develop toward an adult profile in a sex-specific manner during puberty. Sci Rep. 2021;11:23297. https://doi.org/10.1038/s41598-021-02375-z.
Brenhouse HC, Schwarz JM. Immunoadolescence: Neuroimmune development and adolescent behavior. Neurosci Biobehav Rev. 2016;70:288–99. https://doi.org/10.1016/j.neubiorev.2016.05.035.
Shi N, Li N, Duan X, Niu H. Interaction between the gut microbiome and mucosal immune system. Mil Med Res. 2017;4:14. https://doi.org/10.1186/s40779-017-0122-9.
Pietrzak, B, Tomela, K, Olejnik-Schmidt, A, Mackiewicz, A & Schmidt, M. Secretory IgA in intestinal mucosal secretions as an adaptive barrier against microbial cells. Int J Mol Sci 21 (2020). https://doi.org/10.3390/ijms21239254
Verma, R, Lee C, Jeun EJ, Yi J, Kim KS, Ghosh A, et al. Cell surface polysaccharides of Bifidobacterium bifidum induce the generation of Foxp3(+) regulatory T cells. Sci Immunol 3 (2018). https://doi.org/10.1126/sciimmunol.aat6975
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell. 2009;139:485–98. https://doi.org/10.1016/j.cell.2009.09.033.
Guo Y, Liu Y, Rui B, Lei Z, Ning X, Liu Y et al. Crosstalk between the gut microbiota and innate lymphoid cells in intestinal mucosal immunity. Front Immunol 14 (2023). https://doi.org/10.3389/fimmu.2023.1171680
Lee, C, Lee, H, Park, JC & Im, S-H. Microbial components and effector molecules in T helper cell differentiation and function. Immune Netw 23 (2023). https://doi.org/10.4110/in.2023.23.e7
Zhou X, Wu Y, Zhu Z, Lu C, Zhang C, Zeng L, et al. Mucosal immune response in biology, disease prevention and treatment. Signal Transduct Target Ther. 2025;10:7 https://doi.org/10.1038/s41392-024-02043-4.
Beller A, Kruglov A, Durek P, von Goetze V, Werner K, Heinz GA, et al. Specific microbiota enhances intestinal IgA levels by inducing TGF-β in T follicular helper cells of Peyer’s patches in mice. Eur J Immunol. 2020;50:783–94. https://doi.org/10.1002/eji.201948474.
Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, et al. Foxp3+ T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity. 2014;41:152–65. https://doi.org/10.1016/j.immuni.2014.05.016.
Yoon J-H, Do J-S, Velankanni P, Lee C-G, Kwon H-K. Gut Microbial metabolites on host immune responses in health and disease. Immune Netw. 2023;23:6 https://doi.org/10.4110/in.2023.23.e6.
Tazoe H, Otomo Y, Kaji I, Tanaka R, Karaki SI, Kuwahara A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharm. 2008;59:251–62.
Liu XF, Shao JH, Liao YT, Wang LN, Jia Y, Dong PJ, et al. Regulation of short-chain fatty acids in the immune system. Front Immunol 14 (2023). https://doi.org/10.3389/fimmu.2023.1186892
Zhang D, Jian YP, Zhang YN, Li Y, Gu LT, Sun HH, et al. Short-chain fatty acids in diseases. Cell Commun Signal. 2023;21:212. https://doi.org/10.1186/s12964-023-01219-9.
Hung TV, Suzuki T. Short-chain fatty acids suppress inflammatory reactions in Caco-2 cells and mouse colons. J Agric Food Chem. 2017;66:108–17. https://doi.org/10.1021/acs.jafc.7b04233.
Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, Kim CH. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol. 2015;8:80–93. https://doi.org/10.1038/mi.2014.44.
Rolfo C, Giovannetti E, Martinez P, McCue S, Naing A. Applications and clinical trial landscape using Toll-like receptor agonists to reduce the toll of cancer. npj Precis Oncol. 2023;7:26. https://doi.org/10.1038/s41698-023-00364-1.
Park BS, Lee J-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66–e66. https://doi.org/10.1038/emm.2013.97.
Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O'Connell RM, Round JL. MyD88 Signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe. 2015;17:153–63. https://doi.org/10.1016/j.chom.2014.12.009.
Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30:492–506. https://doi.org/10.1038/s41422-020-0332-7.
Ennamorati M, Vasudevan C, Clerkin K, Halvorsen S, Verma S, Ibrahim S, et al. Intestinal microbes influence development of thymic lymphocytes in early life. Proc Natl Acad Sci. 2020;117:2570–8. https://doi.org/10.1073/pnas.1915047117.
Hebbandi Nanjundappa R, Sokke Umeshappa C, Geuking MB. The impact of the gut microbiota on T-cell ontogeny in the thymus. Cell Mol Life Sci. 2022;79:221 https://doi.org/10.1007/s00018-022-04252-y.
Zegarra-Ruiz DF, Kim DV, Norwood K, Kim M, Wu WH, Saldana-Morales FB, et al. Thymic development of gut-microbiota-specific T cells. Nature. 2021;594:413–7. https://doi.org/10.1038/s41586-021-03531-1.
Legoux F, Bellet D, Daviaud C, El Morr Y, Darbois A, Niort K, et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science. 2019;366:494–9. https://doi.org/10.1126/science.aaw2719.
Nakajima A, Negishi N, Tsurui H, Kadowaki-Ohtsuji N, Maeda K, Nanno M et al. Commensal bacteria regulate thymic aire expression. PLoS ONE 9 (2014). https://doi.org/10.1371/journal.pone.0105904
Kim, KS Regulation of T-cell repertoires by commensal microbiota. Front Cell Infect Microbiol 12 (2022). https://doi.org/10.3389/fcimb.2022.1004339
Papadopoulos Z, Herz J, Kipnis J. Meningeal lymphatics: from anatomy to central nervous system immune surveillance. J Immunol. 2020;204:286–93. https://doi.org/10.4049/jimmunol.1900838.
Mastorakos, P & McGavern, D The anatomy and immunology of vasculature in the central nervous system. Sci Immunol 4 (2019). https://doi.org/10.1126/sciimmunol.aav0492
Smyth L, Xu D, Okar SV, Dykstra T, Rustenhoven J, Papadopoulos Z, et al. Identification of direct connections between the dura and the brain. Nature. 2024;627:165–73. https://doi.org/10.1038/s41586-023-06993-7.
Solár P, Zamani A, Kubícková L, Dubovy P, Joukal M. Choroid plexus and the blood-cerebrospinal fluid barrier in disease. Fluids Barriers CNS. 2020;17:35 https://doi.org/10.1186/s12987-020-00196-2.
Rossi F, Lewis C. Microglia’s heretical self-renewal. Nat Neurosci. 2018;21:455–6. https://doi.org/10.1038/s41593-018-0123-3.
del Rio-Hortega, P. Microglia. In Cytology and cellular pathology of the nervous system. 483–534 (PB Hoeber, 1932).
Rezaie P, Male D. MESOGLIA & MICROGLIA – A historical review of the concept of mononuclear phagocytes within the central nervous system. J Hist Neurosci. 2010;11:325–74. https://doi.org/10.1076/jhin.11.4.325.8531.
Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2006;55:233–8. https://doi.org/10.1002/glia.20459.
Mordelt A, de Witte LD. Microglia-mediated synaptic pruning as a key deficit in neurodevelopmental disorders: Hype or hope?. Curr Opin Neurobiol. 2023;79:102674 https://doi.org/10.1016/j.conb.2022.102674.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–8. https://doi.org/10.1126/science.1202529.
Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, Yamashita T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16:543–51. https://doi.org/10.1038/nn.3358.
Li QY, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18:225–42. https://doi.org/10.1038/nri.2017.125.
Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience. 1985;15:313–26. https://doi.org/10.1016/0306-4522(85)90215-5.
Prinz M, Mildner A. Microglia in the CNS: Immigrants from another world. Glia. 2010;59:177–87. https://doi.org/10.1002/glia.21104.
Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. 2016;113:E1738–E1746. https://doi.org/10.1073/pnas.1525528113.
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat Neurosci. 2013;17:131–43. https://doi.org/10.1038/nn.3599.
Konishi H, Kobayashi M, Kunisawa T, Imai K, Sayo A, Malissen B, et al. Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia. 2017;65:1927–43. https://doi.org/10.1002/glia.23204.
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50:253–.e256. https://doi.org/10.1016/j.immuni.2018.11.004.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–.e1217. https://doi.org/10.1016/j.cell.2017.05.018.
Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173:1073–81. https://doi.org/10.1016/j.cell.2018.05.003.
Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–47. https://doi.org/10.1016/s0896-6273(04)00069-8.
Rusin, D, Vahl Becirovic L, Lyszczarz G, Krueger M, Benmamar-Badel A, Vad Mathiesen C, et al. Microglia-derived insulin-like growth Factor 1 is critical for neurodevelopment. Cells 13 (2024). https://doi.org/10.3390/cells13020184
Komori T, Okamura K, Ikehara M, Yamamuro K, Endo N, Okumura K, et al. Brain-derived neurotrophic factor from microglia regulates neuronal development in the medial prefrontal cortex and its associated social behavior. Mol Psychiatry. 2024;29:1338–49. https://doi.org/10.1038/s41380-024-02413-y.
Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: New Roles for the Synaptic Stripper. Neuron. 2013;77:10–18. https://doi.org/10.1016/j.neuron.2012.12.023.
Butovsky O, Bukshpan S, Kunis G, Jung S, Schwartz M. Microglia can be induced by IFN-γ or IL-4 to express neural or dendritic-like markers. Mol Cell Neurosci. 2007;35:490–500. https://doi.org/10.1016/j.mcn.2007.04.009.
Rossi C, Cusimano M, Zambito M, Finardi A, Capotondo A, Garcia-Manteiga JM, et al. Interleukin 4 modulates microglia homeostasis and attenuates the early slowly progressive phase of amyotrophic lateral sclerosis. Cell Death Dis. 2018;9:250 https://doi.org/10.1038/s41419-018-0288-4.
Kann O, Almouhanna F, Chausse B. Interferon γ: a master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci. 2022;45:913–27. https://doi.org/10.1016/j.tins.2022.10.007.
Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311–21. https://doi.org/10.1038/nrn3484.
Rossi D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. 2015;130:86–120. https://doi.org/10.1016/j.pneurobio.2015.04.003.
Wei, DC & Morrison, EH in StatPearls (2025).
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–7. https://doi.org/10.1038/nature21029.
Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–25. https://doi.org/10.1038/s41593-020-00783-4.
Lee, H-G, Lee, J-H, Flausino, LE & Quintana, FJ Neuroinflammation: An astrocyte perspective. Sci Transl Med 15 (2023). https://doi.org/10.1126/scitranslmed.adi7828
Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, et al. Megf10 Is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J Neurosci. 2016;36:5185–92. https://doi.org/10.1523/jneurosci.3850-15.2016.
Ponath G, Ramanan S, Mubarak M, Housley W, Lee S, Sahinkaya FR, et al. Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain. 2017;140:399–413. https://doi.org/10.1093/brain/aww298.
Konishi H, Okamoto T, Hara Y, Komine O, Tamada H, Maeda M, et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 2020;39:104464 https://doi.org/10.15252/embj.2020104464.
Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269–73. https://doi.org/10.1126/science.aal3589.
Séjourné G, Eroglu C. Astrocyte-neuron crosstalk in neurodevelopmental disorders. Curr Opin Neurobiol. 2024;89:102925. https://doi.org/10.1016/j.conb.2024.102925.
Chung WS, Allen NJ, Eroglu C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol. 2015;7:a020370 https://doi.org/10.1101/cshperspect.a020370.
Dutta DJ, Woo DH, Lee PR, Pajevic S, Bukalo O, Huffman WC, et al. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc Natl Acad Sci. 2018;115:11832–7. https://doi.org/10.1073/pnas.1811013115.
Lezmy J, Arancibia-Cárcamo IL, Quintela-López T, Sherman DL, Brophy PJ, Attwell D. Astrocyte Ca 2+ -evoked ATP release regulates myelinated axon excitability and conduction speed. Science. 2021;374:2858 https://doi.org/10.1126/science.abh2858.
Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia. 2010;58:1094–103. https://doi.org/10.1002/glia.20990.
Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. https://doi.org/10.1038/nrn1824.
Adermark L, Lagström O, Loftén A, Licheri V, Havenäng A, Loi EA, et al. Astrocytes modulate extracellular neurotransmitter levels and excitatory neurotransmission in dorsolateral striatum via dopamine D2 receptor signaling. Neuropsychopharmacology. 2021;47:1493–502. https://doi.org/10.1038/s41386-021-01232-x.
Woo J, Kim JE, Im JJ, Lee J, Jeong HS, Park S, et al. Astrocytic water channel aquaporin-4 modulates brain plasticity in both mice and humans: a potential gliogenetic mechanism underlying language-associated learning. Mol Psychiatry. 2017;23:1021–30. https://doi.org/10.1038/mp.2017.113.
Ribeiro, M, Brigas HC, Temido-Ferreira M, Pousinha PA, Regen T, Santa C, et al. Meningeal gammadelta T-cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci Immunol 4 (2019). https://doi.org/10.1126/sciimmunol.aay5199
Kim YC, Ahn JH, Jin H, Yang MJ, Hong SP, Yoon JH, et al. Immaturity of immune cells around the dural venous sinuses contributes to viral meningoencephalitis in neonates. Sci Immunol. 2023;8:eadg6155. https://doi.org/10.1126/sciimmunol.adg6155.
Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci. 2017;20:1300–9. https://doi.org/10.1038/nn.4610.
Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-gamma in regulating neuronal connectivity and social behavior. Nature. 2016;535:425–9. https://doi.org/10.1038/nature18626.
Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal gammadelta T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol. 2020;21:1421–9. https://doi.org/10.1038/s41590-020-0776-4.
Marin-Rodero M, Cintado E, Walker AJ, Jayewickreme T, Pinho-Ribeiro FA, Richardson Q, et al. The meninges host a distinct compartment of regulatory T cells that preserves brain homeostasis. Sci Immunol. 2025;10:2910 https://doi.org/10.1126/sciimmunol.adu2910.
Pasciuto E, Burton OT, Roca CP, Lagou V, Rajan WD, Theys T, et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell. 2020;182:625–e624. https://doi.org/10.1016/j.cell.2020.06.026.
Hanuscheck, N, Thalman C, Domingues M, Schmaul S, Muthuraman M, Hetsch F, et al. Interleukin-4 receptor signaling modulates neuronal network activity. J Exp Med 219 (2022). https://doi.org/10.1084/jem.20211887
Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, Kipnis J. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207:1067–80. https://doi.org/10.1084/jem.20091419.
Flood L, Korol SV, Ekselius L, Birnir B, Jin Z. Interferon-γ potentiates GABAA receptor-mediated inhibitory currents in rat hippocampal CA1 pyramidal neurons. J Neuroimmunol. 2019;337:577050 https://doi.org/10.1016/j.jneuroim.2019.577050.
Chen C, Itakura E, Nelson GM, Sheng M, Laurent P, Fenk LA, et al. IL-17 is a neuromodulator of Caenorhabditis elegans sensory responses. Nature. 2017;542:43–48. https://doi.org/10.1038/nature20818.
Lee B, Kwon JT, Jeong Y, Caris H, Oh D, Feng M, et al. Inflammatory and anti-inflammatory cytokines bidirectionally modulate amygdala circuits regulating anxiety. Cell. 2025;188:2190–.e15. https://doi.org/10.1016/j.cell.2025.03.005.
Brioschi, S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science 373 (2021). https://doi.org/10.1126/science.abf9277
Schafflick D, Wolbert J, Heming M, Thomas C, Hartlehnert M, Börsch AL, et al. Single-cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in nondiseased meninges. Nat Neurosci. 2021;24:1225–34. https://doi.org/10.1038/s41593-021-00880-y.
Wang Y, Chen D, Xu D, Huang C, Xing R, He D, Xu H. Early developing B cells undergo negative selection by central nervous system-specific antigens in the meninges. Immunity. 2021;54:2784–2794.e2786. https://doi.org/10.1016/j.immuni.2021.09.016.
Tanabe S, Yamashita T. B-1a lymphocytes promote oligodendrogenesis during brain development. Nat Neurosci. 2018;21:506–16. https://doi.org/10.1038/s41593-018-0106-4.
Utz SG, See P, Mildenberger W, Thion MS, Silvin A, Lutz M, et al. Early fate defines microglia and nonparenchymal brain macrophage development. Cell. 2020;181:557–573 e518. https://doi.org/10.1016/j.cell.2020.03.021.
Munro DAD, Movahedi K, Priller J. Macrophage compartmentalization in the brain and cerebrospinal fluid system. Sci Immunol. 2022;7:0391 https://doi.org/10.1126/sciimmunol.abk0391.
Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17:797–805. https://doi.org/10.1038/ni.3423.
Sun R, Jiang H. Border-associated macrophages in the central nervous system. J Neuroinflamm. 2024;21:67. https://doi.org/10.1186/s12974-024-03059-x.
Jordão M, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai YH, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363 (2019). https://doi.org/10.1126/science.aat7554
Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease (48, 380, 2018). Immunity. 2018;48:599–599. https://doi.org/10.1016/j.immuni.2018.02.014.
Rustenhoven J, Drieu A, Mamuladze T, de Lima KA, Dykstra T, Wall M, et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell. 2021;184:1000–1016.e1027. https://doi.org/10.1016/j.cell.2020.12.040.
Min, H, O'Neil SM, Xu L, Moseman EA, Kurtzberg J, Filiano AJ. Mural cells interact with macrophages in the dura mater to regulate CNS immune surveillance. J Exp Med 221 (2024). https://doi.org/10.1084/jem.20230326
Willis CL, Garwood CJ, Ray DE. A size selective vascular barrier in the rat area postrema formed by perivascular macrophages and the extracellular matrix. Neuroscience. 2007;150:498–509. https://doi.org/10.1016/j.neuroscience.2007.09.023.
Karam M, Janbon H, Malkinson G, Brunet I. Heterogeneity and developmental dynamics of LYVE-1 perivascular macrophages distribution in the mouse brain. J Cerebr Blood F Met. 2022;42:1797–812. https://doi.org/10.1177/0271678x221101643.
Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. 2018;48:380–395.e386. https://doi.org/10.1016/j.immuni.2018.01.011.
Barron JJ, Mroz NM, Taloma SE, Dahlgren MW, Ortiz-Carpena JF, Keefe MG, et al. Group 2 innate lymphoid cells promote inhibitory synapse development and social behavior. Science. 2024;386:1025 https://doi.org/10.1126/science.adi1025.
Peng H, Sun R, Tang L, Wei H, Tian Z. CD62L Is Critical for Maturation and Accumulation of Murine Hepatic NK Cells in Response to Viral Infection. J Immunol. 2013;190:4255–62. https://doi.org/10.4049/jimmunol.1202395.
Fung, I, Sankar P, Zhang Y, Robison LS, Zhao X, D'Souza SS, et al. Activation of group 2 innate lymphoid cells alleviates aging-associated cognitive decline. J Exp Med 217 (2020). https://doi.org/10.1084/jem.20190915
Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3:136 https://doi.org/10.3978/j.issn.2305-5839.2015.03.49.
Borlongan, CV et al. Human Astrocytes: Secretome Profiles of Cytokines and Chemokines. PLoS ONE 9 (2014). https://doi.org/10.1371/journal.pone.0092325
Lin Y, Zhang JC, Yao CY, Wu Y, Abdelgawad AF, Yao SL, Yuan SY. Critical role of astrocytic interleukin-17 A in poststroke survival and neuronal differentiation of neural precursor cells in adult mice. Cell Death Dis. 2016;7:e2273–e2273. https://doi.org/10.1038/cddis.2015.284.
Guan Y, Wang R, Li X, Zou H, Yu W, Liang Z, et al. Astrocytes constitute the major TNF-α-producing cell population in the infarct cortex in dMCAO rats receiving intravenous MSC infusion. Biomed Pharmacother. 2021;142:111971 https://doi.org/10.1016/j.biopha.2021.111971.
Canedo T, Portugal CC, Socodato R, Almeida TO, Terceiro AF, Bravo J, et al. Astrocyte-derived TNF and glutamate critically modulate microglia activation by methamphetamine. Neuropsychopharmacology. 2021;46:2358–70. https://doi.org/10.1038/s41386-021-01139-7.
Dozio V, Sanchez J-C. Profiling the proteomic inflammatory state of human astrocytes using DIA mass spectrometry. J Neuroinflamm. 2018;15:331. https://doi.org/10.1186/s12974-018-1371-6.
Savarin C, Hinton DR, Valentin-Torres A, Chen Z, Trapp BD, Bergmann CC, Stohlman SA. Astrocyte response to IFN-gamma limits IL-6-mediated microglia activation and progressive autoimmune encephalomyelitis. J Neuroinflamm. 2015;12:79. https://doi.org/10.1186/s12974-015-0293-9.
Micheau O, Tschopp J. Induction of TNF Receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. https://doi.org/10.1016/s0092-8674(03)00521-x.
Mishra A, Kim HJ, Shin AH, Thayer SA. Synapse loss induced by Interleukin-1β requires pre- and postsynaptic mechanisms. J Neuroimmune Pharm. 2012;7:571–8. https://doi.org/10.1007/s11481-012-9342-7.
Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J Neuroinflamm. 2005;2:9. https://doi.org/10.1186/1742-2094-2-9.
Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J Neuroinflamm. 2011;8:150. https://doi.org/10.1186/1742-2094-8-150.
Simpson, DSA, Oliver PL. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 9 (2020). https://doi.org/10.3390/antiox9080743
Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine Expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–30. https://doi.org/10.1523/jneurosci.23-21-07922.2003.
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/Macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–70. https://doi.org/10.1161/strokeaha.112.659656.
Dordoe C, Huang W, Bwalya C, Wang X, Shen B, Wang H, et al. The role of microglial activation on ischemic stroke: Modulation by fibroblast growth factors. Cytokine Growth Factor Rev. 2023;74:122–33. https://doi.org/10.1016/j.cytogfr.2023.07.005.
Zhang R, Wu Y, Xie F, Zhong Y, Wang Y, Xu M, et al. RGMa mediates reactive astrogliosis and glial scar formation through TGFβ1/Smad2/3 signaling after stroke. Cell Death Differ. 2018;25:1503–16. https://doi.org/10.1038/s41418-018-0058-y.
Takahashi S, Izawa Y, Suzuki N. Astroglial Pentose phosphate pathway rates in response to high-glucose environments. ASN Neuro 4 (2012). https://doi.org/10.1042/an20120002
O'shea TM, Ao Y, Wang S, Ren Y, Cheng AL, Kawaguchi R, et al. Derivation and transcriptional reprogramming of border-forming wound repair astrocytes after spinal cord injury or stroke in mice. Nat Neurosci. 2024;27:1505–21. https://doi.org/10.1038/s41593-024-01684-6.
Williamson MR, Fuertes CJA, Dunn AK, Drew MR, Jones TA. Reactive astrocytes facilitate vascular repair and remodeling after stroke. Cell Rep. 2021;35:109048 https://doi.org/10.1016/j.celrep.2021.109048.
Bouras M, Asehnoune K, Roquilly A. Immune modulation after traumatic brain injury. Front Med-Lausanne. 2022;9:995044 https://doi.org/10.3389/fmed.2022.995044.
Qin, X, Akter F, Qin L, Cheng J, Guo M, Yao S, et al. Adaptive immunity regulation and cerebral ischemia. Front Immunol 11 (2020). https://doi.org/10.3389/fimmu.2020.00689
Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 2019;565:246–50. https://doi.org/10.1038/s41586-018-0824-5.
Ma X, Qin C, Chen M, Yu HH, Chu YH, Chen TJ, et al. Regulatory T cells protect against brain damage by alleviating inflammatory response in neuromyelitis optica spectrum disorder. J Neuroinflamm. 2021;18:201. https://doi.org/10.1186/s12974-021-02266-0.
Xie L, Choudhury GR, Winters A, Yang SH, Jin K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol. 2015;45:180–91. https://doi.org/10.1002/eji.201444823.
Olsson T, Zhi WW, Höjeberg B, Kostulas V, Jiang YP, Anderson G, et al. Autoreactive Lymphocytes-T in multiple-sclerosis determined by antigen-induced secretion of Interferon-Gamma. J Clin Invest. 1990;86:981–5. https://doi.org/10.1172/Jci114800.
Häusser-Kinzel S, Weber MS. The role of B cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front Immunol. 2019;10:201 https://doi.org/10.3389/fimmu.2019.00201.
Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, et al. Microglia-mediated T-cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615:668–77. https://doi.org/10.1038/s41586-023-05788-0.
Galiano-Landeira J, Torra A, Vila M, Bové J. CD8 T-cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain. 2020;143:3717–33. https://doi.org/10.1093/brain/awaa269.
Zhang Q, Yang G, Luo Y, Jiang L, Chi H, Tian G. Neuroinflammation in Alzheimer’s disease: insights from peripheral immune cells. Immun Aging 21 (2024). https://doi.org/10.1186/s12979-024-00445-0
Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol. 2009;207:111–6. https://doi.org/10.1016/j.jneuroim.2008.12.002.
Than U, Nguyen LT, Nguyen PH, Nguyen XH, Trinh DP, Hoang DH, et al. Inflammatory mediators drive neuroinflammation in autism spectrum disorder and cerebral palsy. Sci Rep. 2023;13:22587. https://doi.org/10.1038/s41598-023-49902-8.
Fidel PL JR, Romero R, Wolf N, Cutright J, Ramirez M, Araneda H, Cotton DB. Systemic and local cytokine profiles in endotoxin-induced preterm parturition in mice. Am J Obstet Gynecol. 1994;170:1467–75. https://doi.org/10.1016/s0002-9378(94)70180-6.
Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal Influenza Infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. https://doi.org/10.1523/jneurosci.23-01-00297.2003.
Han VX, Patel S, Jones HF, Dale RC. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol. 2021;17:564–79. https://doi.org/10.1038/s41582-021-00530-8.
Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through Interleukin-6. J Neurosci. 2007;27:10695–702. https://doi.org/10.1523/jneurosci.2178-07.2007.
Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–9. https://doi.org/10.1126/science.aad0314.
Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549:528–32. https://doi.org/10.1038/nature23910.
Santisteban MM, Schaeffer S, Anfray A, Faraco G, Brea D, Wang G, et al. Meningeal interleukin-17-producing T cells mediate cognitive impairment in a mouse model of salt-sensitive hypertension. Nat Neurosci. 2023;27:63–77. https://doi.org/10.1038/s41593-023-01497-z.
Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature. 2019;577:249–53. https://doi.org/10.1038/s41586-019-1843-6.
Dong YF, Yong VW. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat Rev Neurol. 2019;15:704–17. https://doi.org/10.1038/s41582-019-0253-6.
Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of β-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci. 2009;106:1261–6. https://doi.org/10.1073/pnas.0805453106.
Schonhoff AM, Figge DA, Williams GP, Jurkuvenaite A, Gallups NJ, Childers GM, et al. Border-associated macrophages mediate the neuroinflammatory response in an alpha-synuclein model of Parkinson disease. Nat Commun. 2023;14:3754. https://doi.org/10.1038/s41467-023-39060-w.
Uekawa K, Hattori Y, Ahn SJ, Seo J, Casey N, Anfray A, et al. Border-associated macrophages promote cerebral amyloid angiopathy and cognitive impairment through vascular oxidative stress. Mol Neurodegen. 2023;18:73. https://doi.org/10.1186/s13024-023-00660-1.
Kwong B, Rua R, Gao Y, Flickinger J Jr, Wang Y, Kruhlak MJ, et al. T-bet-dependent NKp46<SUP>+</SUP> innate lymphoid cells regulate the onset of T 17-induced neuroinflammation. Nat Immunol. 2017;18:1117–27. https://doi.org/10.1038/ni.3816.
Gadani SP, Smirnov I, Smith AT, Overall CC, Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J Exp Med. 2017;214:285–96. https://doi.org/10.1084/jem.20161982.
Linares DM, Fernández M, Martín MC, Alvarez MA. Tyramine biosynthesis in Enterococcus durans is transcriptionally regulated by the extracellular pH and tyrosine concentration. Micro Biotechnol. 2009;2:625–33. https://doi.org/10.1111/j.1751-7915.2009.00117.x.
van Kessel SP, Frye AK, El-Gendy AO, Castejon M, Keshavarzian A, van Dijk G, El Aidy S. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun. 2019;10:310. https://doi.org/10.1038/s41467-019-08294-y.
Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis (vol 161, pg 264, 2015). Cell. 2015;163:258–258. https://doi.org/10.1016/j.cell.2015.09.017.
Strandwitz P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018;1693:128–33. https://doi.org/10.1016/j.brainres.2018.03.015.
Siegel, GJ, Agranoff, BW Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. (Lippincott Williams & Wilkins, 1999).
Lee B, Lee SM, Song JW, Choi JW. Gut microbiota metabolite messengers in brain function and pathology at a view of cell type-based receptor and enzyme reaction. Biomol Ther. 2024;32:403–23. https://doi.org/10.4062/biomolther.2024.009.
Silva YP, Bernardi A, Frozza RL. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol. 2020;11:25 https://doi.org/10.3389/fendo.2020.00025.
Vijay N, Morris M. Role of monocarboxylate transporters in drug delivery to the brain. Curr Pharm Des. 2014;20:1487–98. https://doi.org/10.2174/13816128113199990462.
Bachmann C, Colombo J-P, Berüter J. Short-chain fatty acids in plasma and brain: Quantitative determination by gas chromatography. Clin Chim Acta. 1979;92:153–9. https://doi.org/10.1016/0009-8981(79)90109-8.
Yang LL, Millischer V, Rodin S, MacFabe DF, Villaescusa JC, Lavebratt C. Enteric short-chain fatty acids promote proliferation of human neural progenitor cells. J Neurochem. 2019;154:635–46. https://doi.org/10.1111/jnc.14928.
Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun. 2014;5:3611. https://doi.org/10.1038/ncomms4611.
Li Z, Yi CX, Katiraei S, Kooijman S, Zhou E, Chung CK, et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut. 2018;67:1269–79. https://doi.org/10.1136/gutjnl-2017-314050.
Cook TM, Gavini CK, Jesse J, Aubert G, Gornick E, Bonomo R, et al. Vagal neuron expression of the microbiota-derived metabolite receptor, free fatty acid receptor (FFAR3), is necessary for normal feeding behavior. Mol Metab. 2021;54:101350 https://doi.org/10.1016/j.molmet.2021.101350.
Won Y-J, Lu VB, Puhl HL, Ikeda SR. β-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. J Neurosci. 2013;33:19314–25. https://doi.org/10.1523/jneurosci.3102-13.2013.
Inoue D, Kimura I, Wakabayashi M, Tsumoto H, Ozawa K, Hara T, et al. Short-chain fatty acid receptor GPR41-mediated activation of sympathetic neurons involves synapsin 2b phosphorylation. FEBS Lett. 2012;586:1547–54. https://doi.org/10.1016/j.febslet.2012.04.021.
Morland C, Frøland AS, Pettersen MN, Storm-Mathisen J, Gundersen V, Rise F, Hassel B. Propionate enters GABAergic neurons, inhibits GABA transaminase, causes GABA accumulation and lethargy in a model of propionic acidemia. Biochem J. 2018;475:749–58. https://doi.org/10.1042/bcj20170814.
Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of Histone Acetylation during memory formation in the Hippocampus. J Biol Chem. 2004;279:40545–59. https://doi.org/10.1074/jbc.M402229200.
Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium Butyrate improves memory function in an alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J Alzheimer’s Dis. 2011;26:187–97. https://doi.org/10.3233/jad-2011-110080.
Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, et al. Histone Deacetylase Inhibition by Sodium Butyrate Chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418–27. https://doi.org/10.1523/jneurosci.23-28-09418.2003.
Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, et al. Histone deacetylase inhibitors upregulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–34. https://doi.org/10.1017/s1461145708009024.
Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–98. https://doi.org/10.1111/j.1471-4159.2005.03077.x.
Zhou Z, Xu N, Matei N, McBride DW, Ding Y, Liang H, et al. Sodium butyrate attenuated neuronal apoptosis via GPR41/Gβγ/PI3K/Akt pathway after MCAO in rats. J Cereb Blood Flow Metab. 2020;41:267–81. https://doi.org/10.1177/0271678x20910533.
Chen Z, Xin L, Yang L, Xu M, Li F, Zhou M, Yan T. Butyrate promotes poststroke outcomes in aged mice via interleukin-22. Exp Neurol. 2023;363:114351 https://doi.org/10.1016/j.expneurol.2023.114351.
Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–77. https://doi.org/10.1038/nn.4030.
Soliman ML, Puig KL, Combs CK, Rosenberger TA. Acetate reduces microglia inflammatory signaling in vitro. J Neurochem. 2012;123:555–67. https://doi.org/10.1111/j.1471-4159.2012.07955.x.
Soliman ML, Combs CK, Rosenberger TA. Modulation of inflammatory cytokines and mitogen-activated protein kinases by acetate in primary astrocytes. J Neuroimmune Pharm. 2012;8:287–300. https://doi.org/10.1007/s11481-012-9426-4.
Patnala R, Arumugam TV, Gupta N, Dheen ST. HDAC inhibitor sodium butyrate-mediated epigenetic regulation enhances neuroprotective function of microglia during ischemic stroke. Mol Neurobiol. 2016;54:6391–411. https://doi.org/10.1007/s12035-016-0149-z.
Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 2012;61:349–60. https://doi.org/10.1002/glia.22437.
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4 (2013). https://doi.org/10.1038/ncomms2534
Schilling S, Chausse B, Dikmen HO, Almouhanna F, Hollnagel JO, Lewen A, Kann O. TLR2- and TLR3-activated microglia induce different levels of neuronal network dysfunction in a context-dependent manner. Brain Behav Immun. 2021;96:80–91. https://doi.org/10.1016/j.bbi.2021.05.013.
Fonken LK, Frank MG, Kitt MM, D'Angelo HM, Norden DM, Weber MD, et al. The Alarmin HMGB1 mediates age-induced neuroinflammatory priming. J Neurosci. 2016;36:7946–56. https://doi.org/10.1523/jneurosci.1161-16.2016.
Kim Y, Ryu S-H, Hyun J, Cho Y-S, Jung Y-K. TLR2 immunotherapy suppresses neuroinflammation, tau spread, and memory loss in rTg4510 mice. Brain, Behav Immun. 2024;121:291–302. https://doi.org/10.1016/j.bbi.2024.08.002.
Fernández-Arjona MDM, Grondona JM, Fernández-Llebrez P, López-Ávalos MD. Microglial activation by microbial neuraminidase through TLR2 and TLR4 receptors. J Neuroinflamm. 2019;16:245. https://doi.org/10.1186/s12974-019-1643-9.
Keene CD, Rodrigues CM, Eich T, Linehan-Stieers C, Abt A, Kren BT, et al. A bile acid protects against motor and cognitive deficits and reduces striatal degeneration in the 3-Nitropropionic acid model of Huntington’s disease. Exp Neurol. 2001;171:351–60. https://doi.org/10.1006/exnr.2001.7755.
Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc Natl Acad Sci. 2002;99:10671–6. https://doi.org/10.1073/pnas.162362299.
Rodrigues CM, Sola S, Nan Z, Castro RE, Ribeiro PS, Low WC, Steer CJ. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc Natl Acad Sci. 2003;100:6087–92. https://doi.org/10.1073/pnas.1031632100.
Shen W, Li Z, Tao Y, Zhou H, Wu H, Shi H, et al. Tauroursodeoxycholic acid mitigates depression-like behavior and hippocampal neuronal damage in a corticosterone model of female mice. Naunyn-Schmiedeberg’s Arch Pharm. 2024;398:5785–96. https://doi.org/10.1007/s00210-024-03637-z.
Li C, Wang L, Xie W, Chen E, Chen Y, Li H, et al. TGR5 deficiency in excitatory neurons ameliorates Alzheimer’s pathology by regulating APP processing. Sci Adv. 2024;10:1855 https://doi.org/10.1126/sciadv.ado1855.
Lieu T, Jayaweera G, Zhao P, Poole DP, Jensen D, Grace M, et al. The bile acid receptor TGR5 activates the TRPA1 channel to induce itch in mice. Gastroenterology. 2014;147:1417–28. https://doi.org/10.1053/j.gastro.2014.08.042.
Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, et al. The TGR5 receptor mediates bile acid–induced itch and analgesia. J Clin Invest. 2013;123:1513–30. https://doi.org/10.1172/jci64551.
Zhou L, Yan M, Luo Q, Qiu W, Guo YR, Guo XQ, et al. Elevated bile acids induce Circadian rhythm sleep disorders in chronic liver diseases. Cell Mol Gastroenterol Hepatol. 2025;19:101439 https://doi.org/10.1016/j.jcmgh.2024.101439.
McMillin M, Frampton G, Tobin R, Dusio G, Smith J, Shin H, et al. TGR5 signaling reduces neuroinflammation during hepatic encephalopathy. J Neurochem. 2015;135:565–76. https://doi.org/10.1111/jnc.13243.
Huang R, Gao Y, Chen J, Duan Q, He P, Zhang J, et al. TGR5 agonist INT-777 alleviates inflammatory neurodegeneration in Parkinson’s disease mouse model by modulating mitochondrial dynamics in microglia. Neuroscience. 2022;490:100–19. https://doi.org/10.1016/j.neuroscience.2022.02.028.
Romero-Ramírez L, García-Rama C, Wu S, Mey J. Bile acids attenuate PKM2 pathway activation in proinflammatory microglia. Sci Rep 12 (2022). https://doi.org/10.1038/s41598-022-05408-3
Zhang F, Deng Y, Wang H, Fu J, Wu G, Duan Z, et al. Gut microbiota-mediated ursodeoxycholic acids regulate the inflammation of microglia through TGR5 signaling after MCAO. Brain Behav Immun. 2024;115:667–79. https://doi.org/10.1016/j.bbi.2023.11.021.
Xu N, Bai Y, Han X, Yuan J, Wang L, He Y, et al. Taurochenodeoxycholic acid reduces astrocytic neuroinflammation and alleviates experimental autoimmune encephalomyelitis in mice. Immunobiology. 2023;228:152388 https://doi.org/10.1016/j.imbio.2023.152388.
Nunes AF, Amaral JD, Lo AC, Fonseca MB, Viana RJ, Callaerts-Vegh Z, et al. TUDCA, a bile acid, attenuates amyloid precursor protein processing and Amyloid-beta deposition in APP/PS1 Mice. Mol Neurobiol. 2012;45:440–54. https://doi.org/10.1007/s12035-012-8256-y.
Ma J, Li M, Bao Y, Huang W, He X, Hong Y, et al. Gut microbiota-brain bile acid axis orchestrates aging-related neuroinflammation and behavior impairment in mice. Pharmacol Res. 2024;208:107361 https://doi.org/10.1016/j.phrs.2024.107361.
Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood‒brain barrier permeability in mice. Sci Transl Med. 2014;6:263ra158 https://doi.org/10.1126/scitranslmed.3009759.
Romano KA, Vivas EI, Amador-Noguez D, Rey FE, Blaser MJ. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite Trimethylamine- N -Oxide. mBio. 2015;6:02481 https://doi.org/10.1128/mBio.02481-14.
Hoyles L, Pontifex MG, Rodriguez-Ramiro I, Anis-Alavi MA, Jelane KS, Snelling T, et al. Regulation of blood‒brain barrier integrity by microbiome-associated methylamines and cognition by trimethylamine N-oxide. Microbiome. 2021;9:235. https://doi.org/10.1186/s40168-021-01181-z.
Quinn M, McMillin M, Galindo C, Frampton G, Pae HY, DeMorrow S. Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Digest Liver Dis. 2014;46:527–34. https://doi.org/10.1016/j.dld.2014.01.159.
McMillin M, Frampton G, Quinn M, Ashfaq S, de los Santos M, Grant S, DeMorrow S. Bile acid signaling is involved in the neurological decline in a murine model of acute liver failure. Am J Pathol. 2016;186:312–23. https://doi.org/10.1016/j.ajpath.2015.10.005.
McMillin MA, Frampton GA, Seiwell AP, Patel NS, Jacobs AN, DeMorrow S. TGFβ1 exacerbates blood–brain barrier permeability in a mouse model of hepatic encephalopathy via upregulation of MMP9 and downregulation of claudin-5. Lab Investig. 2015;95:903–13. https://doi.org/10.1038/labinvest.2015.70.
Li Z, Zhang F, Sun M, Liu J, Zhao L, Liu S et al. The modulatory effects of gut microbes and metabolites on blood–brain barrier integrity and brain function in sepsis-associated encephalopathy. PeerJ 11 (2023). https://doi.org/10.7717/peerj.15122
Natah SS, Mouihate A, Pittman QJ, Sharkey KA. Disruption of the blood–brain barrier during TNBS colitis. Neurogastroenterol Motil. 2005;17:433–46. https://doi.org/10.1111/j.1365-2982.2005.00654.x.
Do J, Woo J. From gut to brain: Alteration in inflammation markers in the brain of Dextran sodium sulfate-induced Colitis Model Mice. Clin Psychopharmacol Neurosci. 2018;16:422–33. https://doi.org/10.9758/cpn.2018.16.4.422.
Hu S, Lin Z, Zhao S, Zhang B, Luo L, Zeng L. Pu-erh tea alleviated colitis-mediated brain dysfunction by promoting butyric acid production. Food Chem Toxicol. 2023;172:113594 https://doi.org/10.1016/j.fct.2022.113594.
Hoyles L, Snelling T, Umlai UK, Nicholson JK, Carding SR, Glen RC, McArthur S. Microbiome–host systems interactions: protective effects of propionate upon the blood–brain barrier. Microbiome. 2018;6:55. https://doi.org/10.1186/s40168-018-0439-y.
McKenna F, McLaughlin PJ, Lewis BJ, Sibbring GC, Cummerson JA, Bowen-Jones D, Moots RJ. Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J Neuroimmunol. 2002;132:34–40. https://doi.org/10.1016/s0165-5728(02)00280-1.
Leon-Ponte M, Ahern GP, O’Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood. 2007;109:3139–46. https://doi.org/10.1182/blood-2006-10-052787.
Hodo TW, de Aquino MTP, Shimamoto A, Shanker A. Critical Neurotransmitters in the Neuroimmune Network. Front Immunol. 2020;11:1869 https://doi.org/10.3389/fimmu.2020.01869.
Prud’homme GJ, Glinka Y, Wang Q. Immunological GABAergic interactions and therapeutic applications in autoimmune diseases. Autoimmun Rev. 2015;14:1048–56. https://doi.org/10.1016/j.autrev.2015.07.011.
Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M. Human T cells express a functional ionotropic glutamate receptor GluR3, and glutamate by itself triggers integrin-mediated adhesion to laminin and fibronectin and chemotactic migration. J Immunol. 2003;170:4362–72. https://doi.org/10.4049/jimmunol.170.8.4362.
Haroon E, Miller AH, Sanacora G. Inflammation, Glutamate, and Glia: A trio of trouble in mood disorders. Neuropsychopharmacology. 2017;42:193–215. https://doi.org/10.1038/npp.2016.199.
Ahern GP. 5-HT and the immune system. Curr Opin Pharm. 2011;11:29–33. https://doi.org/10.1016/j.coph.2011.02.004.
Sanidad KZ, Rager SL, Carrow HC, Ananthanarayanan A, Callaghan R, Hart LR, et al. Gut bacteria-derived serotonin promotes immune tolerance in early life. Sci Immunol. 2024;9:4775 https://doi.org/10.1126/sciimmunol.adj4775.
Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood‒brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–30. https://doi.org/10.1523/JNEUROSCI.19-24-10923.1999.
Parker A, Fonseca S, Carding SR. Gut microbes and metabolites as modulators of blood‒brain barrier integrity and brain health. Gut Microbes. 2020;11:135–57. https://doi.org/10.1080/19490976.2019.1638722.
Guedes JR, Ferreira PA, Costa J, Laranjo M, Pinto MJ, Reis T, et al. IL-4 shapes microglia-dependent pruning of the cerebellum during postnatal development. Neuron. 2023;111:3435–.e3438. https://doi.org/10.1016/j.neuron.2023.09.031.
Jiang HR, Milovanović M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol. 2012;42:1804–14. https://doi.org/10.1002/eji.201141947.
Sheean RK, McKay FC, Cretney E, Bye CR, Perera ND, Tomas D, et al. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol. 2018;75:681–9. https://doi.org/10.1001/jamaneurol.2018.0035.
Galvan-Pena S, Zhu Y, Hanna BS, Mathis D, Benoist C. A dynamic atlas of immunocyte migration from the gut. Sci Immunol. 2024;9:0672 https://doi.org/10.1126/sciimmunol.adi0672.
Sanmarco LM, Wheeler MA, Gutiérrez-Vázquez C, Polonio CM, Linnerbauer M, Pinho-Ribeiro FA, et al. Gut-licensed IFNgamma(+) NK cells drive LAMP1(+)TRAIL(+) anti-inflammatory astrocytes. Nature. 2021;590:473–9. https://doi.org/10.1038/s41586-020-03116-4.
Fitzpatrick Z, Frazer G, Ferro A, Clare S, Bouladoux N, Ferdinand J, et al. Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature. 2020;587:472–6. https://doi.org/10.1038/s41586-020-2886-4.
Rojas OL, Pröbstel AK, Porfilio EA, Wang AA, Charabati M, Sun T, et al. Recirculating Intestinal IgA-producing cells regulate neuroinflammation via IL-10. Cell. 2019;177:492–3. https://doi.org/10.1016/j.cell.2019.03.037.
Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B, et al. Microbiota Dysbiosis controls the neuroinflammatory response after stroke. J Neurosci. 2016;36:7428–40. https://doi.org/10.1523/JNEUROSCI.1114-16.2016.
Breit S, Kupferberg A, Rogler G, Hasler G. Vagus nerve as modulator of the brain–gut axis in psychiatric and inflammatory disorders. Front Psychiatry. 2018;9:44 https://doi.org/10.3389/fpsyt.2018.00044.
Jameson KG, Kazmi SA, Ohara TE, Son C, Yu KB, Mazdeyasnan D et al. Select microbial metabolites in the small intestinal lumen regulates vagal activity via receptor-mediated signaling. iScience 28 (2025). https://doi.org/10.1016/j.isci.2024.111699
Joo M-K, Shin Y-J, Kim D-H. Cefaclor causes vagus nerve-mediated depression-like symptoms with gut dysbiosis in mice. Sci Rep. 2023;13:15529. https://doi.org/10.1038/s41598-023-42690-1.
Pu Y, Tan Y, Qu Y, Chang L, Wang S, Wei Y, et al. A role of the subdiaphragmatic vagus nerve in depression-like phenotypes in mice after fecal microbiota transplantation from Chrna7 knock-out mice with depression-like phenotypes. Brain Behav Immun. 2021;94:318–26. https://doi.org/10.1016/j.bbi.2020.12.032.
Zhang J, Ma L, Chang L, Pu Y, Qu Y, Hashimoto K. A key role of the subdiaphragmatic vagus nerve in the depression-like phenotype and abnormal composition of gut microbiota in mice after lipopolysaccharide administration. Transl Psychiatry. 2020;10:186. https://doi.org/10.1038/s41398-020-00878-3.
Wang S, Ishima T, Zhang J, Qu Y, Chang L, Pu Y, et al. Ingestion of Lactobacillus intestinalis and Lactobacillus reuteri causes depression- and anhedonia-like phenotypes in antibiotic-treated mice via the vagus nerve. J Neuroinflamm. 2020;17:241. https://doi.org/10.1186/s12974-020-01916-z.
Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci USA. 2011;108:16050–5. https://doi.org/10.1073/pnas.1102999108.
Perez-Burgos A, Wang B, Mao YK, Mistry B, McVey Neufeld KA, Bienenstock J, Kunze W. Psychoactive bacteriaLactobacillus rhamnosus(JB-1) elicits rapid frequency facilitation in vagal afferents. Am J Physiol -Gastrointest Liver Physiol. 2013;304:G211–G220. https://doi.org/10.1152/ajpgi.00128.2012.
Browning KN. Role of central vagal 5-HT3 receptors in gastrointestinal physiology and pathophysiology. Front Neurosci. 2015;9:413 https://doi.org/10.3389/fnins.2015.00413.
Siopi E, Galerne M, Rivagorda M, Saha S, Moigneu C, Moriceau S, et al. Gut microbiota changes require vagus nerve integrity to promote depressive-like behaviors in mice. Mol Psychiatry. 2023;28:3002–12. https://doi.org/10.1038/s41380-023-02071-6.
Ma X, Park HS, Shin YJ, Kim JK, Hong JK, Han SW, et al. The extracellular vesicle of depressive patient-derived Escherichia fergusonii induces vagus nerve-mediated neuroinflammation in mice. J Neuroinflamm. 2024;21:224. https://doi.org/10.1186/s12974-024-03211-7.
Goswami C, Iwasaki Y, Yada T. Short-chain fatty acids suppress food intake by activating vagal afferent neurons. J Nutr Biochem. 2018;57:130–5. https://doi.org/10.1016/j.jnutbio.2018.03.009.
O'leary OF, Ogbonnaya ES, Felice D, Levone BR, C Conroy L, Fitzgerald P, et al. The vagus nerve modulates BDNF expression and neurogenesis in the hippocampus. Eur Neuropsychopharmacol. 2018;28:307–16. https://doi.org/10.1016/j.euroneuro.2017.12.004.
de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–51. https://doi.org/10.1038/ni1229.
Jin H, Li M, Jeong E, Castro-Martinez F, Zuker CS. A body–brain circuit that regulates body inflammatory responses. Nature. 2024;630:695–703. https://doi.org/10.1038/s41586-024-07469-y.
Lyu Q, Xue W, Liu R, Ma Q, Kasaragod VB, Sun S, et al. A brain-to-gut signal controls intestinal fat absorption. Nature. 2024;634:936–43. https://doi.org/10.1038/s41586-024-07929-5.
Liu J, Dai Q, Qu T, Ma J, Lv C, Wang H, Yu Y. Ameliorating effects of transcutaneous auricular vagus nerve stimulation on a mouse model of constipation-predominant irritable bowel syndrome. Neurobiol Dis. 2024;193:106440 https://doi.org/10.1016/j.nbd.2024.106440.
Wang Y, Tan Q, Pan M, Yu J, Wu S, Tu W, et al. Minimally invasive vagus nerve stimulation modulates mast cell degranulation via the microbiota-gut-brain axis to ameliorate blood‒brain barrier and intestinal barrier damage following ischemic stroke. Int Immunopharmacol. 2024;132:112030 https://doi.org/10.1016/j.intimp.2024.112030.
Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–51. https://doi.org/10.1038/nri1571.
Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–44. https://doi.org/10.1038/nm1447.
Sommershof A, Scheuermann L, Koerner J, Groettrup M. Chronic stress suppresses anti-tumor TCD8+ responses and tumor regression following cancer immunotherapy in a mouse model of melanoma. Brain Behav Immun. 2017;65:140–9. https://doi.org/10.1016/j.bbi.2017.04.021.
Leistner, C & Menke, A Hypothalamic-pituitary-adrenal axis and stress, in Sex Differences in Neurology and Psychiatry Handbook of Clinical Neurology 55-64 (2020), 175.
Muzzi C, Watanabe N, Twomey E, Meers GK, Reichardt HM, Bohnenberger H, Reichardt SD. The Glucocorticoid receptor in intestinal epithelial cells alleviates colitis and associated colorectal cancer in mice. Cell Mol Gastroenterol Hepatol. 2021;11:1505–18. https://doi.org/10.1016/j.jcmgh.2020.12.006.
You, J, Liu L, Pan X, Wu L, Tan L, Zhou C, et al. Study on the gut microbiota, HPA, and cytokine levels in infantile spasms. Front Immunol 15 (2024). https://doi.org/10.3389/fimmu.2024.1442677
Ohya T, Nagai T, Araki Y, Yanagawa T, Tanabe T, Iyoda K, et al. A pilot study on the changes in immunity after ACTH therapy in patients with West syndrome. Brain Dev. 2009;31:739–43. https://doi.org/10.1016/j.braindev.2008.11.007.
Johnson EW, Hughes TK, Smith EM. ACTH enhancement of T-Lymphocyte Cytotoxic Responses. Cell Mol Neurobiol. 2005;25:743–57. https://doi.org/10.1007/s10571-005-3972-8.
Hoadley ME, Galea J, Singh N, Hulme S, Ajao DO, Rothwell N, et al. The role of cortisol in immunosuppression in subarachnoid hemorrhage. Eur J Med Res. 2023;28:303. https://doi.org/10.1186/s40001-023-01222-3.
Petrovsky N, McNair P, Harrison LC. Diurnal rhythms of pro-inflammatory cytokines: regulation by plasma cortisol and therapeutic implications. Cytokine. 1998;10:307–12. https://doi.org/10.1006/cyto.1997.0289.
Shinkai S, Watanabe S, Asai H, Shek P. Cortisol response to exercise and post-exercise suppression of blood lymphocyte subset counts. Int J Sports Med. 2007;17:597–603. https://doi.org/10.1055/s-2007-972901.
Dong T, Zhi L, Bhayana B, Wu MX. Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury. J Neuroinflamm. 2016;13:197. https://doi.org/10.1186/s12974-016-0663-y.
Wilson BA, Jahnke JR, Roach J, Azcarate-Peril et al. Maternal precarity and HPA axis functioning shape infant gut microbiota and HPA axis development in humans. Plos One 16 (2021). https://doi.org/10.1371/journal.pone.0251782
Iino S, Nojyo Y. Muscarinic M2 acetylcholine receptor distribution in the guinea-pig gastrointestinal tract. Neuroscience. 2006;138:549–59. https://doi.org/10.1016/j.neuroscience.2005.11.021.
den Hertog A. Neural control of gastro-intestinal motility; Events following receptor activation. Vet Res Commun. 1986;10:335–40. https://doi.org/10.1007/bf02213999.
Feng C, Gao G, Wu K, Weng X. Causal relationship between gut microbiota and constipation: a bidirectional Mendelian randomization study. Front Microbiol 15 (2024). https://doi.org/10.3389/fmicb.2024.1438778
Tesfaw G, Siraj DS, Abdissa A, Jakobsen RR, Johansen ØH, Zangenberg M, et al. Gut microbiota patterns associated with duration of diarrhea in children under five years of age in Ethiopia. Nat Commun. 2024;15:7532. https://doi.org/10.1038/s41467-024-51464-w.
Ohkusa T, Koido S, Nishikawa Y, Sato N. Gut microbiota and chronic constipation: a review and update. Front Med. 2019;6:19 https://doi.org/10.3389/fmed.2019.00019.
Cooke HJ. Neurotransmitters in neuronal reflexes regulating intestinal secretion. Ann N Y Acad Sci. 2006;915:77–80. https://doi.org/10.1111/j.1749-6632.2000.tb05225.x.
Dolan B, Ermund A, Martinez-Abad B, Johansson MEV, Hansson GC. Clearance of small intestinal crypts involves goblet cell mucus secretion by intracellular granule rupture and enterocyte ion transport. Sci Signal. 2022;15:5848 https://doi.org/10.1126/scisignal.abl5848.
Specian RD, Neutra MR. Mechanism of rapid mucus secretion in goblet cells stimulated by acetylcholine. J Cell Biol. 1980;85:626–40. https://doi.org/10.1083/jcb.85.3.626.
Cornell R, Cao W, Harradine B, Godini R, Handley A, Pocock R. Neuro-intestinal acetylcholine signaling regulates the mitochondrial stress response in Caenorhabditis elegans. Nat Commun. 2024;15:6594. https://doi.org/10.1038/s41467-024-50973-y.
Ye Z, Zhu Y, Tang N, Zhao X, Jiang J, Ma J, Zhang H. α7 nicotinic acetylcholine receptor agonist GTS-21 attenuates DSS-induced intestinal colitis by improving intestinal mucosal barrier function. Mol Med. 2022;28:59 https://doi.org/10.1186/s10020-022-00485-6.
Kimura H, Imura YK, Tomiyasu H, Mihara T, Kaji N, Ohno K, et al. Neural anti-inflammatory action mediated by two types of acetylcholine receptors in the small intestine. Sci Rep. 2019;9:5887. https://doi.org/10.1038/s41598-019-41698-w.
Labed SA, Wani KA, Jagadeesan S, Hakkim A, Najibi M, Irazoqui JE. Intestinal Epithelial Wnt signaling mediates acetylcholine-triggered host defense against infection. Immunity. 2018;48:963–.e963. https://doi.org/10.1016/j.immuni.2018.04.017.
Fujita Y, Kosakamoto H, Obata F. Microbiota-derived acetylcholine can promote gut motility in Drosophila melanogaster. Philos Trans R Soc B: Biol Sci. 2024;379:20230075 https://doi.org/10.1098/rstb.2023.0075.
Iglesias-Vázquez L, Van Ginkel Riba, G, Arija V, Canals J. Composition of gut microbiota in children with autism spectrum disorder: a systematic review and meta-analysis. Nutrients 12 (2020). https://doi.org/10.3390/nu12030792
Li N, Yang J, Zhang J, Liang C, Wang Y, Chen B, et al. Correlation of gut microbiome between ASD children and mothers and potential biomarkers for risk assessment. Genomics Proteom Bioinforma. 2019;17:26–38. https://doi.org/10.1016/j.gpb.2019.01.002.
Zhao Y, Wang Y, Meng F, Chen X, Chang T, Huang H, et al. Altered gut microbiota as potential biomarkers for autism spectrum disorder in early childhood. Neuroscience. 2023;523:118–31. https://doi.org/10.1016/j.neuroscience.2023.04.029.
Gerges P, Bangarusamy DK, Bitar T, Alameddine A, Nemer G, Hleihel W. Turicibacter and Catenibacterium as potential biomarkers in autism spectrum disorders. Sci Rep. 2024;14:23184. https://doi.org/10.1038/s41598-024-73700-5.
Abuljadayel D, Alotibi A, Algothmi K, Basingab F, Alhazmi S, Almuhammadi A, et al. Gut microbiota of children with autism spectrum disorder and healthy siblings: A comparative study. Exp Ther Med. 2024;28:430 https://doi.org/10.3892/etm.2024.12719.
Wang, WJ & Fu, PC gut microbiota analysis and in silico biomarker detection of children with autism spectrum disorder across cohorts. Microorganisms 11 (2023). https://doi.org/10.3390/microorganisms11020291
Barone R, Alaimo S, Messina M, Pulvirenti A, Bastin J, MIMIC-Autism G, et al. A subset of patients with autism spectrum disorders show a distinctive metabolic profile by dried blood spot analyses. Front Psychiatry. 2018;9:636 https://doi.org/10.3389/fpsyt.2018.00636.
Orozco JS, Hertz-Picciotto I, Abbeduto L, Slupsky CM. Metabolomics analysis of children with autism, idiopathic-developmental delays, and Down syndrome. Transl Psychiatry. 2019;9:243. https://doi.org/10.1038/s41398-019-0578-3.
Leboucher A, Pisani DF, Martinez-Gili L, Chilloux J, Bermudez-Martin P, Van Dijck A, et al. The translational regulator FMRP controls lipid and glucose metabolism in mice and humans. Mol Metab. 2019;21:22–35. https://doi.org/10.1016/j.molmet.2019.01.002.
West PR, Amaral DG, Bais P, Smith AM, Egnash LA, Ross ME et al. Metabolomics as a tool for discovery of biomarkers of autism spectrum disorder in the blood plasma of children. PLoS ONE 9 (2014). https://doi.org/10.1371/journal.pone.0112445
Manghi P, Filosi M, Zolfo M, Casten LG, Garcia-Valiente A, Mattevi S, et al. Large-scale metagenomic analysis of oral microbiomes reveals markers for autism spectrum disorders. Nat Commun. 2024;15:9743. https://doi.org/10.1038/s41467-024-53934-7.
Lingampelly SS, Naviaux JC, Heuer LS, Monk JM, Li K, Wang L, et al. Metabolic network analysis of pre-ASD newborns and 5-year-old children with autism spectrum disorder. Commun Biol. 2024;7:536 https://doi.org/10.1038/s42003-024-06102-y.
Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011;25:40–45. https://doi.org/10.1016/j.bbi.2010.08.003.
Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P, et al. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol. 2006;172:198–205. https://doi.org/10.1016/j.jneuroim.2005.11.007.
Xie J, Huang L, Li X, Li H, Zhou Y, Zhu H, et al. Immunological cytokine profiling identifies TNF-α as a key molecule dysregulated in autistic children. Oncotarget. 2017;8:82390–8. https://doi.org/10.18632/oncotarget.19326.
Theoharides TC, Tsilioni I, Patel AB, Doyle R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl Psychiatry. 2016;6:e844–e844. https://doi.org/10.1038/tp.2016.77.
Siniscalco D, Schultz S, Brigida AL, Antonucci N. Inflammation and Neuro-immune dysregulations in autism spectrum disorders. Pharmaceuticals 11 (2018). https://doi.org/10.3390/ph11020056
Brister D, Rose S, Delhey L, Tippett M, Jin Y, Gu H, Frye RE. Metabolomic signatures of autism spectrum disorder. J Pers Med 12 (2022). https://doi.org/10.3390/jpm12101727
Zhang S, Li X, Yuan T, Guo X, Jin C, Jin Z, Li J. Glutamine inhibits inflammation, oxidative stress, and apoptosis and ameliorates hyperoxic lung injury. J Physiol Biochem. 2023;79:613–23. https://doi.org/10.1007/s13105-023-00961-5.
Aguayo-Cerón KA, Sánchez-Muñoz F, Gutierrez-Rojas RA, Acevedo-Villavicencio LN, Flores-Zarate AV, Huang F, et al. Glycine: the smallest anti-inflammatory micronutrient. Int J Molecular Sciences 24 (2023). https://doi.org/10.3390/ijms241411236
Niu X, Zheng S, Liu H, Li S Protective effects of taurine against inflammation, apoptosis, and oxidative stress in brain injury. Mol Med Rep (2018). https://doi.org/10.3892/mmr.2018.9465
Dan Z, Mao X, Liu Q, Guo M, Zhuang Y, Liu Z, et al. Altered gut microbial profile is associated with abnormal metabolism activity of Autism Spectrum Disorder. Gut Microbes. 2020;11:1246–67. https://doi.org/10.1080/19490976.2020.1747329.
Harber KJ, de Goede KE, Verberk S, Meinster E, de Vries HE, van Weeghel M et al. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 10 (2020). https://doi.org/10.3390/metabo10090372
Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7:13537. https://doi.org/10.1038/s41598-017-13601-y.
Zhuang Z-Q, Shen LL, Li WW, Fu X, Zeng F, Gui L, et al. Gut Microbiota is Altered in Patients with Alzheimer’s Disease. J Alzheimer’s Dis. 2018;63:1337–46. https://doi.org/10.3233/jad-180176.
Haran, JP, Bhattarai SK, Foley SE, Dutta P, Ward DV, Bucci V, McCormick BA. Alzheimer’s disease microbiome is associated with dysregulation of the anti-inflammatory P-Glycoprotein Pathway. mBio 10 (2019). https://doi.org/10.1128/mBio.00632-19
Li J, Zhu S, Wang Y, Fan M, Dai J, Zhu C, et al. Metagenomic association analysis of cognitive impairment in community-dwelling older adults. Neurobiol Dis. 2023;180:106081 https://doi.org/10.1016/j.nbd.2023.106081.
Chen Y, Yang B, Stanton C, Ross RP, Zhao J, Zhang H, Chen W. Bifidobacterium pseudocatenulatum Ameliorates DSS-Induced Colitis by maintaining intestinal mechanical barrier, blocking proinflammatory cytokines, inhibiting TLR4/NF-κB signaling, and altering gut microbiota. J Agric Food Chem. 2021;69:1496–512. https://doi.org/10.1021/acs.jafc.0c06329.
Grabrucker S, Marizzoni M, Silajdžić E, Lopizzo N, Mombelli E, Nicolas S, et al. Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis. Brain. 2023;146:4916–34. https://doi.org/10.1093/brain/awad303.
Lukiw WJ. Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s Disease. Front Microbiol. 2016;7:1544 https://doi.org/10.3389/fmicb.2016.01544.
Colombo AV, Sadler RK, Llovera G, Singh V, Roth S, Heindl S et al. Microbiota-derived short-chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 10 (2021). https://doi.org/10.7554/eLife.59826
Chen C, Zhang D, Wu D, Chen F, Li Z, Hu Y. Gut microbiome, and immune cells mediated effect on depression: A two-step, two-sample Mendelian randomization analysis. Exp Gerontol. 2024;195:112530 https://doi.org/10.1016/j.exger.2024.112530.
Liu P, Liu Z, Wang J, Wang J, Gao M, Zhang Y et al. Immunoregulatory role of the gut microbiota in inflammatory depression. Nat Commun 15 (2024). https://doi.org/10.1038/s41467-024-47273-w
Zhu X, Sakamoto S, Ishii C, Smith MD, Ito K, Obayashi M, et al. Dectin-1 signaling on colonic γδ T cells promotes psychosocial stress responses. Nat Immunol. 2023;24:625–36. https://doi.org/10.1038/s41590-023-01447-8.
Iadecola C, Buckwalter MS, Anrather J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J Clin Invest. 2020;130:2777–88. https://doi.org/10.1172/JCI135530.
Veltkamp R, Gill D. Clinical trials of immunomodulation in ischemic stroke. Neurotherapeutics. 2016;13:791–800. https://doi.org/10.1007/s13311-016-0458-y.
Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci USA. 2014;111:18315–20. https://doi.org/10.1073/pnas.1416166111.
Smith CJ, Hulme S, Vail A, Heal C, Parry-Jones AR, Scarth S, et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor antagonist in ischemic stroke): A randomized controlled Phase 2 Trial. Stroke. 2018;49:1210–6. https://doi.org/10.1161/STROKEAHA.118.020750.
Yuan C, Shi L, Sun Z, Xu F, Wang C, Shan J, et al. Regulatory T-cell expansion promotes white matter repair after stroke. Neurobiol Dis. 2023;179:106063 https://doi.org/10.1016/j.nbd.2023.106063.
Yshii L, Pasciuto E, Bielefeld P, Mascali L, Lemaitre P, Marino M, et al. Astrocyte-targeted gene delivery of interleukin 2 specifically increases brain-resident regulatory T-cell numbers and protects against pathological neuroinflammation. Nat Immunol. 2022;23:878–91. https://doi.org/10.1038/s41590-022-01208-z.
Shi L, Sun Z, Su W, Xu F, Xie D, Zhang Q, et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity. 2021;54:1527–42 e1528. https://doi.org/10.1016/j.immuni.2021.04.022.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9. https://doi.org/10.1073/pnas.82.12.4245.
Tartaglia MC, Ingelsson M. Molecular therapeutics in development to treat Alzheimer’s Disease (Sept, 10.1007/s40291-024-00738-6, 2024). Mol Diagn Ther. 2025;29:143–143. https://doi.org/10.1007/s40291-024-00752-8.
Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, et al. Donanemab in Early symptomatic Alzheimer disease The TRAILBLAZER-ALZ 2 randomized clinical trial. J Am Med Assoc. 2023;330:512–27. https://doi.org/10.1001/jama.2023.13239.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in Early Alzheimer’s disease. N Engl J Med. 2023;388:9–21. https://doi.org/10.1056/NEJMoa2212948.
Dubois B et al. Masitinib for mild-to-moderate Alzheimer’s disease: results from a randomized, placebo-controlled, phase 3, clinical trial (vol 15, 39, 2023). Alzheimers Res Ther 15 (2023). https://doi.org/10.1186/s13195-023-01230-9
Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22:135–7. https://doi.org/10.1038/nm.4022.
Rosenzweig N, Dvir-Szternfeld R, Tsitsou-Kampeli A, Keren-Shaul H, Ben-Yehuda H, Weill-Raynal P, et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat Commun. 2019;10:465. https://doi.org/10.1038/s41467-019-08352-5.
Alfaidi M, Barker RA, Kuan WL. An update on immune-based alpha-synuclein trials in Parkinson’s disease. J Neurol. 2024;272:21 https://doi.org/10.1007/s00415-024-12770-x.
Gendelman HE, Zhang Y, Santamaria P, Olson KE, Schutt CR, Bhatti D, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. Npj Parkinsons Dis. 2017;3:10 https://doi.org/10.1038/s41531-017-0013-5.
Greenland JC, Cutting E, Kadyan S, Bond S, Chhabra A, Williams-Gray CH. Azathioprine immunosuppression and disease modification in Parkinson’s disease (AZA-PD): a randomized double-blind placebo-controlled phase II trial protocol. BMJ Open. 2020;10:e040527 https://doi.org/10.1136/bmjopen-2020-040527.
Powell G, Derry-Sumner H, Rajenderkumar D, Rushton SK, Sumner P. Rationale and design of a randomized, double-blind, Placebo-controlled study to evaluate the efficacy and safety of AKST4290 in subjects with Parkinson’s disease on stable Dopaminergic treatment. Neurology. 2020;94:1929.
Shiihara T, Miyashita M, Yoshizumi M, Watanabe M, Yamada Y, Kato M. Peripheral lymphocyte subset and serum cytokine profiles of patients with West syndrome. Brain Dev -Jpn. 2010;32:695–702. https://doi.org/10.1016/j.braindev.2009.11.001.
Wang X, Huang Q, Wu L, Yang Y, Ye X, Yang B. Application of peripheral blood cytokine and immunoglobulin detection in ACTH therapy for the treatment of infantile spasms. Front Pediatr 12 (2024). https://doi.org/10.3389/fped.2024.1365917
Jun JS, Lee ST, Kim R, Chu K, Lee SK. Tocilizumab treatment for new onset refractory status epilepticus. Ann Neurol. 2018;84:940–5. https://doi.org/10.1002/ana.25374.
Stredny CM, Case S, Sansevere AJ, Son M, Henderson L, Gorman MP. Interleukin-6 blockade with Tocilizumab in Anakinra-refractory febrile infection-related Epilepsy Syndrome (FIRES). Child Neurol Open. 2020;7:2329048 https://doi.org/10.1177/2329048X20979253.
French JA, Cole AJ, Faught E, Theodore WH, Vezzani A, Liow K, et al. Safety and efficacy of Natalizumab as adjunctive therapy for people with drug-resistant epilepsy A Phase 2 study. Neurology. 2021;97:E1757–E1767. https://doi.org/10.1212/Wnl.0000000000012766.
Bittner S, Simon OJ, Göbel K, Bien CG, Meuth SG, Wiendl H. Rasmussen Encephalitis treated with Natalizumab. Neurology. 2013;81:395–7. https://doi.org/10.1212/WNL.0b013e31829c5ceb.
Ellul P, Rosenzwajg M, Peyre H, Fourcade G, Mariotti-Ferrandiz E, Trebossen V, et al. Regulatory T lymphocytes/Th17 lymphocytes imbalance in autism spectrum disorders: evidence from a meta-analysis. Mol Autism. 2021;12:68. https://doi.org/10.1186/s13229-021-00472-4.
Nie ZQ, Han D, Zhang K, Li M, Kwon HK, Im SH, et al. TH1/Treg ratio may be a marker of autism in children with immune dysfunction. Res Autism Spect Dis. 2023;101:102085 https://doi.org/10.1016/j.rasd.2022.102085.
Li M et al. Therapeutic efficacy of low-dose IL-2 in an 8-year-old autistic child with immune imbalance: a case report. Available at SSRN 4974758
Liu S, Xi H, Xue X, Sun X, Huang H, Fu D, et al. Clostridium butyricum regulates intestinal barrier function via trek1 to improve behavioral abnormalities in mice with autism spectrum disorder. Cell Biosci. 2024;14:95. https://doi.org/10.1186/s13578-024-01278-6.
Alsubaiei, S, Alfawaz HA, Almubarak AY, Alabdali NA, Ben Bacha A, El-Ansary A. Independent and combined effects of probiotics and prebiotics as supplements or food-rich diets on a propionic-acid-induced rodent model of autism spectrum disorder. Metabolites 13 (2022). https://doi.org/10.3390/metabo13010050
Abuaish S, Al-Otaibi NM, Aabed K, Abujamel TS, Alzahrani SA, Alotaibi SM, et al. The efficacy of fecal transplantation and bifidobacterium supplementation in ameliorating propionic acid-induced behavioral and biochemical autistic features in juvenile male rats. J Mol Neurosci. 2022;72:372–81. https://doi.org/10.1007/s12031-021-01959-8.
Lin T-L, Lu CC, Chen TW, Huang CW, Lu JJ, Lai WF, et al. Amelioration of Maternal Immune Activation-Induced Autism Relevant Behaviors by Gut Commensal Parabacteroides goldsteinii. Int J Mol Sci. 2022;23:13070. https://doi.org/10.3390/ijms232113070.
Prince N, Peralta Marzal LN, Markidi A, Ahmed S, Adolfs Y, Pasterkamp RJ, et al. Prebiotic diet normalizes aberrant immune and behavioral phenotypes in a mouse model of autism spectrum disorder. Acta Pharmacol Sin. 2024;45:1591–603. https://doi.org/10.1038/s41401-024-01268-x.
Prajapati SK, Wang S, Mishra SP, Jain S, Yadav H. Protection of Alzheimer’s disease progression by a human-origin probiotics cocktail. Sci Rep. 2025;15:1589. https://doi.org/10.1038/s41598-024-84780-8.
Medeiros D, McMurry K, Pfeiffer M, Newsome K, Testerman T, Graf J, et al. Slowing Alzheimer’s disease progression through probiotic supplementation. Front Neurosci. 2024;18:1309075. https://doi.org/10.3389/fnins.2024.1309075.
Andreozzi V, Cuoco S, Balestrieri M, Fierro F, Ferrara N, Erro R, et al. Synbiotic supplementation may globally improve nonmotor symptoms in patients with stable Parkinson’s disease: results from an open label single-arm study. Sci Rep. 2024;14:23095. https://doi.org/10.1038/s41598-024-74400-w.
Aktas B, Aslim B, Ozdemir DA. A neurotherapeutic approach with Lacticaseibacillus rhamnosus E9 on gut microbiota and intestinal barrier in MPTP-induced mouse model of Parkinson’s disease. Sci Rep. 2024;14:15460. https://doi.org/10.1038/s41598-024-65061-w.
Leta V, Mandal G, Zinzalias P, Staunton J, Batzu L, Trivedi D et al. Effects of a four-strain probiotic on inflammatory markers in Parkinson’s disease: a multicenter randomized controlled trial (the SymPD study) (N5.001). Neurology 102 (2024). https://doi.org/10.1212/wnl.0000000000205490
Magistrelli L, Amoruso A, Mogna L, Graziano T, Cantello R, Pane M, Comi C. Probiotics may have beneficial effects in Parkinson’s disease: in vitro evidence. Front Immunol. 2019;10:969 https://doi.org/10.3389/fimmu.2019.00969.
Westfall S, Caracci F, Estill M, Frolinger T, Shen L, Pasinetti GM. Chronic Stress-induced Depression And Anxiety Priming Modulated By Gut-brain-axis Immunity. Front Immunol. 2021;12:670500. https://doi.org/10.3389/fimmu.2021.670500.
Murray E, Sharma R, Smith KB, Mar KD, Barve R, Lukasik M, et al. Probiotic consumption during puberty mitigates LPS-induced immune responses and protects against stress-induced depression- and anxiety-like behaviors in adulthood in a sex-specific manner. Brain Behav Immun. 2019;81:198–212. https://doi.org/10.1016/j.bbi.2019.06.016.
Yang LL et al. Effects of a Synbiotic On Plasma Immune Activity Markers And Short-chain Fatty Acids In Children And Adults with ADHD—A randomized controlled trial. Nutrients 15 (2023). https://doi.org/10.3390/nu15051293
Skott E, Yang LL, Stiernborg M, Söderström Å, Rȕegg J, Schalling M, et al. Effects of a synbiotic on symptoms, and daily functioning in attention deficit hyperactivity disorder – A double-blind randomized controlled trial. Brain Behav Immun. 2020;89:9–19. https://doi.org/10.1016/j.bbi.2020.05.056.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5. https://doi.org/10.1126/science.1194637.
Sharon G, Cruz NJ, Kang DW, Gandal MJ, Wang B, Kim YM, et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell. 2019;177:1600–1618.e1617. https://doi.org/10.1016/j.cell.2019.05.004.
Ferreiro AL, Choi J, Ryou J, Newcomer EP, Thompson R, Bollinger RM, et al. Gut microbiome composition may be an indicator of preclinical Alzheimer’s disease. Sci Transl Med. 2023;15:2984. https://doi.org/10.1126/scitranslmed.abo2984.
Acknowledgements
This research was supported by grants from the National Research Foundation of Korea funded by the Korea Ministry of Science and ICT (MSIT): L.C. and H.-K.K. were supported by RS-2024-00361620, RS-2019-NR040072, RS-2025-00561456, and RS-2024-00438443. S.-H. I was supported by RS-2024-00345575. We would like to thank MID (Medical Illustration & Design), a member of the Medical Research Support Services of Yonsei University College of Medicine, for providing excellent support with medical illustrations.
Funding
Open Access funding enabled and organized by Pohang University of Science and Technology (POSTECH).
Author information
Authors and Affiliations
Contributions
JCP and LC designed and wrote this work. H-KK and S-HI helped design the concept and outline, edited the work, and supervised the overall study.
Corresponding authors
Ethics declarations
Competing interests
SHI is the CEO of ImmunoBiome but declares no conflicts of interest for this paper. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, J.C., Chang, L., Kwon, HK. et al. Beyond the gut: decoding the gut–immune–brain axis in health and disease. Cell Mol Immunol 22, 1287–1312 (2025). https://doi.org/10.1038/s41423-025-01333-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41423-025-01333-3
Keywords
This article is cited by
-
Intergenerational effects of the microbiota on neurodevelopment: mechanisms and therapeutic perspectives
Acta Pharmacologica Sinica (2026)
-
Short-Chain Fatty Acids Mitigate Perinatal Methylmercury-Induced Cognitive Impairment Via Modulation of the Gut Microbiota Composition
Biological Trace Element Research (2026)
-
High-Fat Diet Exacerbates Behavioral and Neurochemical Deficits in BTBR Mice: Linking Intestinal Inflammation and Brain Neurotransmitter Dysregulation to Melatonin Therapeutic Potential
Molecular Neurobiology (2026)
-
A dual pathway intervention of tES and FMT enhances emotion regulation in stressed military personnel
Discover Mental Health (2026)
-
Indirect influence of global climate change, mediated by nutrition, on anxiety disorders
Discover Food (2025)
