
Abstract
The perovskite manganites AMnO3 and their doped analogues A1–x B x MnO3 (A and B = main group and lanthanide metals) are a fascinating family of magnetic oxides exhibiting a rich variety of properties. They are thus under intense investigation along multiple fronts, one of which is how their structural and physical properties are modified at the nanoscale. Here we show that the molecular compound [Ce3Mn8O8(O2CPh)18(HO2CPh)2] (CeIII 2CeIVMnIII 8; hereafter Ce3Mn8) bears a striking structural resemblance to the repeating unit seen in the perovskite manganites. Further, magnetic studies have established that Ce3Mn8 exhibits both the combination of pairwise MnIII 2 ferromagnetic and antiferromagnetic exchange interactions, and the resultant spin vector alignments that are found within the 3-D C-type antiferromagnetic perovskites. First-principles theoretical calculations reveal not only the expected nearest-neighbor MnIII 2 exchange couplings via superexchange pathways through bridging ligands but also an unusual, direct MnIII–CeIV–MnIII metal-to-metal channel involving the CeIV f orbitals.
Similar content being viewed by others

Introduction
Perovskite manganites continue to be a source of great interest in the scientific community owing to the fascinating and important physical properties they exhibit, such as colossal magnetoresistance and multiferroicity1,2,3,4,5,6. These materials have been proposed for important applications in many technological fields such as spintronics and information storage7. Detailed insights into the mechanisms by which these materials function has often been limited owing to their complex nature8. In many cases, the most direct way to understand complex systems can be to study a fragment of the larger structures, and one way to do this is to apply a molecular bottom-up approach to make 0-D species whose molecular properties can overcome many of the limitations and complexities encountered in the characterization and study of bulk 3-D materials and their nanoparticles9.
Molecules possess certain important advantages over nanoparticles, including particle monodispersity, crystallinity, true solubility, and a stabilizing shell of organic ligands that is also often capable of facile modification as desired. Given the success of molecular approaches to monodisperse nanoscale magnets in other areas such as single-molecule magnetism, the synthesis and study of molecules with a structural resemblance to the perovskites would provide an alternative and complementary approach to ultra-small perovskite nanoparticles. Such advantages of molecules over their bulk counterparts for study of known physical phenomena, and discovery of new ones, are well documented in the field of single-molecule magnets (SMMs)10, 11. These are 0-D molecular nanomagnets that possess a combination of uniaxial anisotropy and a high ground state spin, and consequently function as superparamagnets below their blocking temperatures, T B 12,13,14,15,16. In a similar fashion, attainment of molecular species that combine the structural and physical properties of the perovskite manganites would provide an ideal and alternative route forward for understanding the origins of the properties of this important class of materials, including ultimately those with multiferroic behavior.
In the present work, we prepare and study a molecular compound [Ce3Mn8O8(O2CPh)18(HO2CPh)2] (CeIII 2CeIVMnIII 8; hereafter Ce3Mn8) that is structurally reminiscent of the repeating unit in perovskite manganites. Magnetic characterization shows that the spin vector alignments in this molecule are the same as in the 3-D C-type antiferromagnetic perovskites. First-principles theoretical calculations reveal an unusual and direct MnIII–CeIV–MnIII metal-to-metal magnetic exchange channel involving the CeIV f orbitals, besides the conventional nearest-neighbor MnIII 2 exchange couplings via superexchange pathways through bridging ligands. An excellent agreement between theory and experiment for the magnetic susceptibility curve is reached along with the establishment of an unprecedentedly rich physical picture of magnetic interaction. The work demonstrates the feasibility of a bottom-up molecular approach to gaining insights into the structural and physical properties of ultra-small nanoscale perovskite materials, as well as surface units on larger particles where structural relaxation effects and O vacancies are present.
Results
Synthesis and structure characteristics
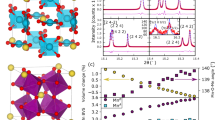
3-D perovskites are prepared at high temperatures in the solid state, whereas 0-D molecules are typically prepared at or near ambient temperatures in solution. The results reported here were thus crucially dependent on the successful development of an experimental procedure for the synthesis of pure, crystalline Ce3Mn8 (see “Methods”). The structure (Fig. 1) was determined by single-crystal X-ray diffractometry (Supplementary Methods and Supplementary Tables 1–3) and consists of a {CeIVMnIII 8(μ 3-O)8}12+ unit comprising a Mn8 distorted cube with a CeIV at its center held together by four μ 3-O2− and four μ 4-O2− ions, with the latter connecting to two external CeIII ions attached on opposite faces of the cube. The organic ligation consists of two μ 4-, four μ 3-, and twelve μ 2-benzoate groups, as well as two terminal benzoic acid groups on the CeIII ions. All MnIII atoms are six-coordinate with distorted octahedral geometries and exhibit Jahn–Teller (JT) distortion (elongation) axes. The latter are localized, meeting in pairs at the O atoms of the μ 4-benzoate groups (Fig. 1b). The CeIII and CeIV ions are nine-coordinate and eight-coordinate, respectively.

The molecular structure of Ce3Mn8 from experimental single-crystal X-ray diffractometry data. a The complete structure with H atoms omitted for clarity. The thicker black lines are guides to the eye to emphasize the distorted Mn8 cube. b The partially labeled central fragment with the MnIII JT axes indicated as thicker Mn–O bonds. Color scheme: CeIV purple; CeIII and LnIII orange; MnIII green; O red; C gray. c Comparison of the core from (a) of Ce3Mn8 (left) with a repeating unit of an ideal ABO3 cubic perovskite plus two A ions from adjacent repeating units (right). d As for (c) but showing the MnO6 polyhedra
The core of Ce3Mn8 is overall similar to a fragment of the cubic perovskite structure, shown in Fig. 1c for comparison, where it can be seen to comprise a LnMnIII 8O x (Ln = lanthanide) unit plus two additional Ln ions that in a 3-D perovskite array would be in the adjacent repeating units. It can thus be described as one complete and two partial repeating units of the perovskite structure. Also notable is that it exhibits distinct distortions from cubic symmetry and that some of these are similar to those seen in the LnMnIIIO3 manganites. This is particularly interesting as Ce3Mn8 is a discrete molecular complex buffered by the organic ligation from external influences, whereas many distortions within the more rigid 3-D perovskite structure are cooperative in nature. In Ce3Mn8: firstly, the cube undergoes a tetragonal distortion (stretching) along one direction due to the JT axis locations and the external Ce ions, giving short and long Mn…Mn edge separations of 3.16–3.33 Å and 4.84–4.94 Å, respectively; secondly, the MnO6 octahedra are tilted, allowing the bridging O2− ions to move off the Mn–Mn edges and toward the center of the cube to optimize Ce–O bond lengths; and thirdly, the long Mn...Mn edges are bridged by longer PhCO2 −groups rather than shorter O2− bridges, so that the corresponding core is {CeMn8O8(O2CPh)4} vs {CeMn8O12} for the perovskite. The first point is reminiscent of, although of greater magnitude than the tetragonal perovskite structures, and the second point is reminiscent of the orthorhombic structure of the perovskite LnMnO3 manganites, where a distortion involving tilting of the MnO6 octahedra (by an amount dependent on the Ln ionic radius) away from the ideal cubic structure similarly arises from the combined effect of MnIII JT distortions and a mismatch of the Ln–O and Mn–O bond lengths, moving the oxide ions off the Mn–Mn edges17. Interestingly, the above three points together suggest that Ce3Mn8 may best be considered a model for surface-repeating units of perovskite nanoparticles, where structural relaxation effects and oxide ion vacancies are present. Further comparison between Ce3Mn8 and the corresponding Ln = Ce manganite CeMnO3 is precluded by the absence of crystallographic information for this material18, which appears to be difficult to synthesize pure due to the multiple oxidation states accessible to both cerium and manganese.
Magnetic structure analysis
The location and orientation of the MnIII Jahn–Teller axes in Ce3Mn8 (Fig. 1b and Supplementary Table 3) represent the local Mn z axes and thus the location of the singly occupied σ-symmetry d z 2 orbitals. When two d z 2 (d σ ) orbitals meet at a bridging O atom with the acute Mn–O–Mn angles (83–88°) in Ce3Mn8, the empirical Goodenough–Kanamori (GK) rules19,20,21 predict ferromagnetic (FM) interactions for the Mn1/Mn4, Mn2/Mn6, Mn3/Mn8, and Mn5/Mn7 pairs (Fig. 1b). Exchange couplings for the other MnIII pairs are expected to be dominated by overlap of d π magnetic orbitals, and thus to be weakly antiferromagnetic (AF) according to the GK rules. For perovskite manganites, competition among spin, charge, and orbital degrees of freedom determine the magnetic properties, and the MnIII JT axes are intimately involved22. Depending on the precise orbital ordering and the associated superexchange interactions in a given manganite, a particular combination of FM and AF interactions emerges23. The common types of spin ordering that result (ferromagnetic or FM, and three types of AF: A-AF, C-AF, and G-AF) for this structure are illustrated in the left panel of Fig. 2. Below we describe the experimental magnetic data for Ce3Mn8 and then we will apply first-principles electronic structure calculations to reveal the microscopic mechanics of magnetic coupling in the Ce3Mn8 molecule, thereby providing a unique angle for understanding the exchange interactions in perovskite manganites.

Magnetic properties of Ce3Mn8 and perovskites. a Four known spin-ordering configurations in a perovskite unit cell. b Experimental χ M T vs T plot for Ce3Mn8 in a 1 kG applied dc field (+), with the small contributions from the two external CeIII ions subtracted. Solid and dashed lines are theoretical results from a multi-spin Heisenberg model using either exchange coupling J’s and anisotropy term D from fits to the experimental data (solid line) or calculated from first-principles DFT (dashed line). c The multi-spin Heisenberg model showing the magnetic exchange coupling paths labeled as J 1 to J 4; colored lines indicate symmetry-equivalent sets. Other possible paths (unlabeled) correspond to exchange coupling strengths at least an order of magnitude smaller than the ones labeled in the figure
Experimental solid-state magnetic susceptibility (χ Μ ) data on Ce3Mn8 were collected on a microcrystalline sample, restrained in eicosane to prevent torquing, in a 1 kG (0.1 T) field in the 5–300 K range. The obtained χ M T vs T plot (middle panel in Fig. 2) exhibits two regimes, increasing from 26.80 cm3 Kmol−1 at 300 K to a maximum of 32.68 cm3 Kmol−1 at 30 K, and then steeply decreasing to 12.41 cm3 Kmol−1 at 5 K. The value at 300 K is greater than the 25.62 cm3 Kmol−1 calculated for eight MnIII (S = 2, χ M T = 3 cm3 Kmol−1 with g = 2) and two CeIII (f 1, S = ½, L = 3, 2 F 5/2, χ M T = 0.81 cm3 Kmol−1)24 non-interacting ions, indicating the increasing χ M T with decreasing T to be due to dominant FM pairwise MnIIIMnIII interactions within the molecule. The steep decrease below 30 K is assigned to weaker AF interactions that begin to pair-up spins at lower temperatures. χ M T is clearly heading for a small value at ~0 K, indicating a ground state spin description for Ce3Mn8 as an AF S = 0 Mn8 cube, and two essentially non-interacting CeIII ions as expected from the very weak exchange couplings exhibited by LnIII ions25. This description is supported by the in-phase ac susceptibility (χ′ M T) vs T plot down to 1.8 K (Supplementary Fig. 1), which extrapolates to a small non-zero value consistent with the expected χ M T = 1.62 cm3 Kmol−1 for two independent CeIII ions.
First-principles energetics
To characterize the origin of the AF ground state of Ce3Mn8, the magnetic properties of Ce3Mn8 were calculated within the framework of Kohn–Sham density functional theory (DFT), using atomic coordinates for the molecule taken from the crystal structure. DFT based methods have been widely used for studying molecular magnets with Mn centers26,27,28,29,30,31. First-principles calculations were performed for eight different spin configurations of Ce3Mn8 molecules, corresponding to FM, A-AF, C-AF, and G-AF spin alignments (Supplementary Methods), with all three orientations of the ferromagnetic planes (A-AF) or axes (C-AF) considered and neglecting any noncolinearity (the easy axis is assumed to be along the global z axis). The results (Table 1) show that spin state C-AF-I has the lowest total energy. The absolute magnetization of each Mn ion is ~3.5 μ B, as expected for MnIII with some induced magnetization at the O sites. Each of the two outer CeIII sites has a magnetic moment of 1 μ B, which is included in the core region of the charge density. In the FM state, the central CeIV site has a small magnetic moment (0.28 μ B) and, as expected, no net spin density is found on the CeIV sites for any of the S = 0 AF states. The C-AF-I ground state corresponds to FM coupling in the b direction (i.e., the Mn1/Mn4, Mn2/Mn6, Mn3/Mn8, and Mn5/Mn7 pairs of Fig. 1b) and net AF coupling along the a and c directions, as suggested by the qualitative predictions from the structural parameters and χ M T vs T data. The weak net AF coupling is supported by the low-lying A-AF-II and A-AF-III excited states, corresponding to different relative alignments of the four FM pairs. The energy difference ΔE between the ground state and the FM state is about 19 meV, corresponding to a switching magnetic field of B = ΔE/gμ BΔM ≈ 5 T (to switch from C-AF-I to FM), where ΔM is the magnetic moment difference, μ B = 0.058 meV/T, and g = 2. A plot of M/Nμ B vs applied magnetic field (Supplementary Fig. 2) steadily increases and appears to be heading for saturation in the expected M/Nμ B range of gS = 32 for S = 16, or in fact a little less as g < 2 slightly for MnIII.
Theoretical analysis of the magnetic properties
The DFT results were further analyzed using a multi-spin model, defined by the spin Hamiltonian of Eq. (1) in Supplementary Methods, to estimate the various pairwise Mn/Mn exchange coupling parameters (J). Four spin coupling paths (denoted by J 1 through J 4 in the right panel of Fig. 2) were found to have significant contributions to the total energy. J 1 appears to involve a conventional superexchange mechanism via two monoatomically bridging oxygens. This was predicted above to be strongly FM by the GK rules. J 2 appears to involve two parallel superexchange pathways: one via a single monoatomically bridging oxygen and the other through a carboxylate group (Mn–O–CR–O–Mn, R = phenyl). Both J 3 and J 4 appear at first glance to involve mostly superexchange through carboxylate groups; the Mn–Mn distances are 4.6 Å and 4.9 Å for the J 3 and J 4 pathways, respectively (compared to 3.16–3.33 Å for J 1 and J 2). Initial calculations using the symmetry broken DFT method and incorporating J 1–J 4 showed that J 4 was, consistent with the structure, much weaker than J 1–J 3 and AF. In fitting experimental data, we therefore omit J 4 but include the axial anisotropy parameter (D) as even with current supercomputers it is not feasible to include both D and four J′s due to the excessive memory requirement. Our calculations show that the anisotropy easy axis is along the global z direction, perpendicular to the Mn1–Mn4–Mn2–Mn6 and Mn3–Mn5–Mn7–Mn8 planes. The calculated coupling strengths (positive for FM, negative for AF) and D were J 1 = +1.61 meV (+13 cm−1), J 2 = +0.72 (+5.8), J 3 = −1.10 (−8.87), and D = −0.07 (−0.56).
The χ M T vs T plot generated using the calculated DFT parameters is only in fair agreement with the experimental data (Fig. 2, dashed line in middle panel), missing the continuing rise in χ M T below 100 K and the peak at 30 K, before the steep decrease at lower temperatures. Therefore, the J 1–J 3 and D values were refined by fitting the experimental χ M T vs T data directly to the multi-spin Heisenberg model using the calculated DFT parameters as input values. An excellent fit was now obtained (Fig. 2, solid line) with J 1 = +1.26 meV (+10.2 cm−1), J 2 = +0.69 (+5.6), J 3 = −0.74 (−6.0), and D = −0.05 (−0.40), again resulting in a C-AF-I ground state.
Discussion
The above results present a consistent picture of a dominating J 1 FM interaction and a C-AF ground state spin configuration. In fact, as J 2 is also FM, the C-AF ground state is driven by the AF J 3 coupling, the diagonal Mn2 interaction. It is comparable in magnitude to J 2 and there is competition (spin frustration) between the J 1, J 2, and J 3 couplings in the Mn3 triangles within each Mn4 square at the top and bottom of the molecule (Fig. 2). J 1 dominates giving four FM Mn2 pairs, but the relative alignment of these pairs to give the C-AF-I ground state is caused by the slightly stronger J 3 vs J 2, i.e., |J 3| > |J 2|, and the AF J 4. If |J 3| < |J 2|, however, then the ground state would have been A-AF, the A-AF-II state in Supplementary Methods. The Ce3Mn8 ground state is thus dependent on the relative values of J 2 vs J 3: the former is clearly mediated by the superexchange pathways through the O2− and RCO2 − bridges but is opposite of the expected weakly AF character according to GK rules. Thus, we sought further insight into this unexpected result as well as the origin of the diagonal J 3.
The DFT calculations allow us to analyze the microscopic electronic and magnetic processes responsible for the magnetic couplings. The sign of J 2 suggests that the unoccupied CeIV-4f orbitals residing at the upper edge of the energy gap may have an unexpectedly important role in magnetic couplings. To reveal its role, we carried out an additional DFT calculation by replacing the Ce ions in Ce3Mn8 with La ions, and made the La3Mn8 molecule negatively charged to preserve the MnIII valence state. In contrast to Ce3Mn8, the La-4f orbitals in [La3Mn8]− are ~2.5–3.5 eV above the energy gap (Supplementary Discussion), and thus are not expected to contribute to magnetic couplings. J 1 to J 4 in the [La3Mn8]− molecule are more AF by ~0.1–0.8 meV (0.8–6.5 cm−1) than those in Ce3Mn8 (Supplementary Discussion). The comparison between Ce3Mn8 and La3Mn8 indicates that the contribution of CeIV-4f orbitals in Ce3Mn8 is FM in nature, as we further explain below. The magnetic coupling among MnIII ions in these molecules can be decomposed into two contributions from different physical origins: the AF superexchange coupling via bridging O2− ions and/or carboxylate groups, and a FM direct exchange coupling enhanced by the CeIV ion. The strength of the superexchange coupling is determined by the effective hopping integral between Mn-d z 2 states of two Mn ions. The integral can be calculated by downfolding the DFT Hamiltonian onto Mn-d z 2 orbitals using the maximally localized Wannier function method32. The calculated effective hopping integrals in Ce3Mn8 molecule are almost the same as those in [La3Mn8]− (Supplementary Discussion). From this, we conclude that the same AF superexchange couplings exist in both molecules, and rule out a possible AF superexchange coupling via CeIV-4f orbitals. The FM direct exchange depends on the shape of Mn-3d orbitals. The presence of CeIV-4f orbitals near the energy gap enhances the direct exchange coupling between neighboring Mn pairs by pulling the Mn-d z 2 orbitals closer to each other through hybridization with the CeIV-4f orbitals and forming an itinerant electron path between two Mn ions. Indeed, the shape of Mn-d z 2 Wannier orbitals shows considerable difference between Ce3Mn8 and [La3Mn8]− (Supplementary Discussion). The overall picture that emerges is an unexpected FM contribution to the Mn2 exchange couplings from a direct Mn–Ce–Mn pathway, making J 2 FM and comparable in absolute magnitude to J 3, leading to a C-AF ground state but with low-lying A-AF excited states. This suggests that small structural distortions (e.g., from applied pressure, changes in the identity of the ligands, etc.) could alter the J 2:J 3 ratio and switch the ground state to A-AF.
Overall, a synthetic method has been developed to a Ce3Mn8 cluster that shows strong structural similarity to the repeating unit of perovskite manganites. It exhibits two structural distortions common in bulk ABO3 perovskites, namely tilting of the BO6 octahedra and a tetragonal distortion driven by JT elongation of the B cations, and it also exhibits the same spin ordering as C-type antiferromagnetic perovskites. This demonstrates that these unusual structural and spin-ordering effects can be reproduced even at the level of a single repeating unit. We propose Ce3Mn8 may also be particularly relevant to surface units of nanoscale perovskites. First-principles-based investigations reveal the microscopic mechanism of the magnetic couplings inside the Ce3Mn8 molecule. The unoccupied CeIV-4f orbitals have considerable contribution to the direct exchange coupling, which in turn has a pivotal role in the competition (spin frustration) between FM and AF interactions in this molecule and the resulting ground state C-AF spin configuration with low-lying A-AF excited states. These results suggest analogous effects may be important in the magnetic couplings within the extended lattices of CeIV-containing perovskites or similar compounds in which 4f orbitals can be brought close to the Fermi energy. Attempts to complete one or both partial cubes at each end of Ce3Mn8 with additional MnIII ions to yield a molecule representing two or three face-fused repeating perovskite units are in progress, as is the synthesis of Ce3Mn8 analogues with various other lanthanide or main group metal ions to expand the experimental database. New molecules of this type, analogues to the BiMnO3 or TbMnO3 systems, may even show true multiferroic bistability and further shed light on the complex mechanisms involved in these systems.
Methods
Experiments
The comproportionation reaction of Mn(O2CPh)2·2H2O, Ce(NO3)3·6H2O, NBun 4MnO4 and PhCO2H in a 4:4:1:16 molar ratio in MeNO2 at ~80 °C gave a dark brown solution from which was isolated [Ce3Mn8O8(O2CPh)18(HO2CPh)2] (Ce3Mn8) as black crystals in ~55% yield based on Mn (Supplementary Methods). Single-crystal X-ray diffraction studies at 100 K were performed on a Bruker DUO diffractometer using MoK α (λ = 0.71073 Å) or CuK α (λ = 1.54178 Å) radiation (from an ImuS power source), and an APEXII CCD area detector. The metal oxidation states and the oxygen protonation levels were confirmed by charge considerations and bond valence sum calculations (Supplementary Tables 1, 2)33, 34.
Computations
Electronic and magnetic properties of the Ce3Mn8 molecule were calculated within the framework of Kohn–Sham DFT35 using the spin-polarized Perder–Burke–Ernzehof36 exchange correlation functional and project-augmented wave37, 38 pseudopotentials in conjuction with the plan-wave basis as implemented in the Vienna Ab-initio Simulation Package39, 40. The plane-wave cutoff energy was 500 eV, and the energy threshold for self-consistency was 10−5 eV. Owing to the strong localization of the Ce f electron, the GGA + U method was applied with U = 2 eV41 for the Ce f orbitals. Spin–orbit interactions were also included.
Data availability
The crystallographic information file (CIF) for [CeMn8O8(O2CPh)18(HO2CPh)2]·x(solvent) (Ce3Mn8) has been deposited at the Cambridge Crystallographic Data Centre with deposition code CCDC 1533475.
References
Cheong, S.-W. & Mostovoy, M. Multiferroics: a magnetic twist for ferroelectricity. Nat. Mater. 6, 13–20 (2007).
Maignan, A., Martin, C., Hebert, S. & Hardy, V. Colossal magnetoresistance manganites: importance of the coorperative phenomena. J. Mater. Chem. 17, 5023–5031 (2007).
Harris, A. B., Kenzelmann, M., Aharony, A. & Entin-Wohlman, O. Effect of inversion symmetry on the incommensurate order in multiferroic RMn2O5 (R=rate earth). Phys. Rev. B 78, 014407 (2008).
Dong, S. & Liu, J.-M. Recent progress of multiferroic perovskite manganites. Mod. Phys. Lett. B 26, 1230004 (2012).
Salamon, M. & Jaime, M. The physics of manganites: structure and transport. Rev. Mod. Phys. 73, 583–628 (2001).
Schmid, H. Multi-ferroic magnetoelectics. Ferroelectrics 162, 317–338 (1994).
Picozzi, S. & Ederer, C. First principles studies of multiferroic materials. J. Phys. Condens. Matter. 21, 303201 (2009).
Eerenstein, W., Mathur, N. D. & Scott, J. F. Multiferroic and magnetoelectric materials. Nature 442, 759–765 (2006).
Hendrickson, D. N. et al. Half-integer spin molecular nanomagnets. MRS Proc. 746, Q1.1 (2002).
Bogani, L. & Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 7, 179–186 (2008).
Friedman, J. R. & Sarachik, M. P. Single-molecule nanomagnets. Ann. Rev. Condens. Matter Phys. 1, 109–128 (2010).
Sessoli, R. et al. High-spin molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 115, 1804–1816 (1993).
Christou, G., Gatteschi, D., Hendrickson, D. N. & Sessoli, R. Single-molecule magnets. MRS Bull. 25, 66–71 (2000).
Christou, G. Single-molecule magnets: a molecular approach to nanoscale magnetic materials. Polyhedron 24, 2065–2075 (2005).
Sessoli, R., Gatteschi, D., Caneschi, A. & Novak, M. A. Magnetic bistability in a metal-ion cluster. Nature 365, 141–143 (1993).
Wernsdorfer, W., Aliaga-Alcalde, N., Hendrickson, D. N. & Christou, G. Exchange-biased quantum tunneling in a supramolecular dimer of single-molecule magnets. Nature 416, 406–409 (2002).
Tachibana, M., Shimoyama, T., Kawaji, H., Atake, T. & Takayama-Muromachi, E. Jahn–Teller distortion and magnetic transitions in perovskite RMnO3 (R = Ho, Er, Tm, Yb, and Lu). Phys. Rev. B 75, 144425 (2007).
Ambrunaz-Quezel, S. Neutron-diffraction-determined parameters of manganates of the rare earths of perovskite type and magnetic structures in praseodymium manganate(III) (PrMnO3) and neodymium manganate(III) (NdMnO3). Bull. Soc. Fr. Mineral. Cristallogr. 91, 339 (1968).
Goodenough, J. B. Theory of the role of covalence in the perovskite-type manganites [La,M]MnO3. Phys. Rev. 100, 564–573 (1955).
Goodenough, J. B. An interpretation of the magnetic properties of the perovksite-type mixed crystals La1−x Sr x CoO3. J. Phys. Chem. Solids 6, 287–297 (1958).
Kanamori, J. Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 10, 87–98 (1959).
Koppel, H., Yarkony, D. R. & Barentzen, H. The Jahn–Teller Effect (Springer, 2009).
Hotta, T., Yunoki, S., Mayr, M. & Gagotto, E. A-type antiferromagnetic and C-type orbital-ordered states in LaMnO3 using cooperative Jahn–Teller phonons. Phys. Rev. B 60, R15009–R15012 (1999).
Kahn, O. Molecular Magnetism (Wiley-VCH, 1993).
Sessoli, R. & Powell, A. K. Strategies towards single molecule magnets based on lanthanide ions. Coord. Chem. Rev. 253, 2328–2341 (2009).
Pederson, M. R. & Khanna, S. N. Magnetic anisotropy barrier for spin tunneling in Mn12O12 molecules. Phys. Rev. B 60, 9566–9572 (1999).
Park, K., Pederson, M. R., Richardson, S. L., Aliaga-Alcalde, N. & Christou, G. Density-functional theory calculation of the intermolecular exchange interaction in the magnetic Mn4 dimer. Phys. Rev. B 68, 020405 (2003).
Postnikov, A. V., Kortus, J. & Pederson, M. R. Density functional studies of molecular magnets. Phys. Stat. Sol. 243, 2533–2572 (2006).
Ruiz, E. et al. Magnetic structure of the large-spin Mn10 and Mn19 complexes: a theoretical complement to an experimental milestone. J. Am. Chem. Soc. 130, 7420–7426 (2008).
Wu, Y.-N., Zhang, X.-G. & Cheng, H.-P. Giant molecular magnetocapacitance. Phys. Rev. Lett. 110, 217205 (2013).
Li, X.-G., Fry, J. N. & Cheng, H.-P. Single-molecule magnet Mn12 on graphene. Phys. Rev. B 90, 125447 (2014).
Marzari, M., Mostofi, A. A., Yates, J. R., Souza, I. & Vanderbilt, D. Maximally localized Wannier functions: theory and applications. Rev. Mod. Phys. 84, 1419–1475 (2012).
Liu, W. & Thorp, H. H. Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. 2. Refined distance and other enzymes. Inorg. Chem. 32, 4102–4105 (1993).
Roulhac, P. L. & Palenik, G. J. Bond valence sums in coordination chemistry. The calculation of the oxidation state of cerium in complexes containing cerium bonded only to oxygen. Inorg. Chem. 42, 118–121 (2003).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, J. From ultrasoft pseudopotentials to the projector augmented wave method. Phys. Rev. B 59, 1758–1775 (1999).
Kresse, G. & Furthmuller, J. Efficiency of an-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Loschen, C., Carrasco, J., Neyman, K. M. & Illas, F. First-principles LDA + U and GGA + U study of cerium oxides: dependence on the effective U parameter. Phys. Rev. B 75, 035115 (2007).
Acknowledgements
This work was supported by the US National Science Foundation DMREF program under Grant No. CHE-1534401, and partially supported by Grant CHE-1565664. Calculations were performed at the National Energy Research Scientific Computing Center and at UF Research Computing.
Author information
Authors and Affiliations
Contributions
This project was designed and directed by H.-P.C., G.C., and X.-G.Z. A.E.T. and K.A.A. performed the experiments and analyzed the experimental data. X.-G.L. and Y.-P.W. performed the first-principles and numerical calculations, and analyzed the experimental data. All authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thuijs, A.E., Li, XG., Wang, YP. et al. Molecular analogue of the perovskite repeating unit and evidence for direct MnIII-CeIV-MnIII exchange coupling pathway. Nat Commun 8, 500 (2017). https://doi.org/10.1038/s41467-017-00642-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-00642-0
