
Abstract
Alkynes are amongst the most valuable functional groups in organic chemistry and widely used in chemical biology, pharmacy, and materials science. However, the preparation of alkyl-substituted alkynes still remains elusive. Here, we show a nickel-catalyzed deaminative Sonogashira coupling of alkylpyridinium salts. Key to the success of this coupling is the development of an easily accessible and bench-stable amide-type pincer ligand. This ligand allows naturally abundant alkyl amines as alkylating agents in Sonogashira reactions, and produces diverse alkynes in excellent yields under mild conditions. Salient merits of this chemistry include broad substrate scope and functional group tolerance, gram-scale synthesis, one-pot transformation, versatile late-stage derivatizations as well as the use of inexpensive pre-catalyst and readily available substrates. The high efficiency and strong practicability bode well for the widespread applications of this strategy in constructing functional molecules, materials, and fine chemicals.
Similar content being viewed by others
Introduction
Alkynes are one of the most valuable functional groups in organic chemistry because they are not only served as versatile synthetic building blocks for diversified chemical transformations, but also common structural motifs in a wide range of natural products, bioactive molecules and organic materials1,2,3. For example, the introduction of an alkyne into a drug molecule could provide remarkable benefits in its biological activity, such as enhanced lipophilicity, bioavailability, and metabolic stability (Fig. 1a). In addition to the widely used as functional tags in biochemistry for bioconjugation based on “alkyne-azide click chemistry”4, recent researches also indicated that alkynes have a privileged application in Raman imaging due to their unique and strong Raman scattering peaks in a cellular silent region that is free of interference from most endogenous molecules (Fig. 1b)5,6,7,8,9. Therefore, lots of efforts have been made to develop efficient methods for the construction of alkynes. Among these available transformations, the transition-metal-catalyzed Sonogashira coupling of aryl/vinyl electrophiles with terminal alkynes has proven to be one of the most powerful approaches for C(sp2)–C(sp) bond formation10,11. However, the incorporation of nonactivated, β-H-containing alkyl electrophiles in Sonogashira reaction to construct C(sp3)–C(sp) bond still remains a formidable challenge, presumably due to the following issues (Fig. 1c): (1) the reluctance of alkyl electrophiles to undergo oxidative addition with a metal catalyst, (2) the propensity of the resulting alkylmetal intermediates to undergo intramolecular β-hydride elimination, (3) the poor nucleophilicity of the sp- hybridized carbon in alkynes, and (4) the low concentration of the transmetalating species generated in situ in the reaction medium. Moreover, the facile cyclotrimerization and/or oligomerization of terminal alkynes under the catalysis of low-valent metal is another obstacle that renders such coupling a more intractable objective12,13. In a pioneering study, Fu and co-workers realized Pd/Cu-cocatalyzed Sonogashira coupling of nonactivated primary alkyl iodides and bromides by the use of an N-heterocyclic carbene (NHC) ligand14. Later on, a few elegant strategies for this transformation were developed based on the discovery of different catalytic systems including Pd/bisoxazoline-derived NHC ligand15, Ni/NN2 pincer ligand16,17, Ni/pyridine bisoxazoline (pybox) system18, and NHC pincer nickel(II) complex19 (Fig. 1d). Furthermore, Lalic et al. discovered an excellent protocol of photoinduced copper-catalyzed alkylation of terminal alkyne with alkyl iodides as the alkyl source20. More recently, Wang and co-workers further expanded the scope of alkyl to the cheap and easily accessible carboxylic acid derivatives by the utilization of redox-active esters21. Despite these significant advances, the scope for alkyl-Sonogashira-type reactions is still relatively limited. Particularly, the electrophilic partners in such reactions are largely limited to alkyl halides22, and the need of copper(I) salt as cocatalyst might also cause some detrimental effects to the reaction, such as the undesired Glaser coupling of terminal alkynes and the complicated procedure in workup23. Thus, developing simple approaches to access such coupling with more alternatives especially in copper-free conditions is highly important and appealing.

a Representative alkynyl-containing drugs (ethynylestradiol, hormone drug for estrogen medication; efavirenz, an antiretroviral drug for HIV/AIDS; dynemicin A, an antitumor drug for cancer treatments; alfaprostol, a veterinary drug for breeding control). b Important applications of alkynes in bioconjugation and molecular imaging. c Challenges in Sonogashira coupling of nonactivated alkyl electrophiles. d State-of-the-art catalytic systems for Sonogashira coupling of nonactivated alkyl electrophiles. e This work: deaminative Sonogashira coupling of alkyl amines catalyzed by nickel and amide-type pincer ligand (L4).
Alkyl amines are naturally abundant and readily available feedstock chemicals, and the prevalence of amino groups in numerous bioactive molecules, pharmaceuticals, and natural products provide expedient opportunities for late-stage functionalization and bioconjugation24,25. In this context, using alkyl amines as alkylating agents in organic synthesis would have many privileged advantages when compared to the traditional platforms using alkyl halides. However, such a promising transformation is still underexploited owing to the high bond dissociation energy of C(sp3)–N bond26,27,28. In a seminal work, Watson et al. demonstrated that pyridinium salts29, also known as “Katritzky salts” which are easily formed from primary amines and pyrylium salt, could be used as alkyl radical precursors in cross-coupling with arylboronic acids30. Since then, many elegant approaches based on the utilization of these redox-active amines for deaminative functionalization31,32,33,34, such as arylation35,36,37,38,39,40, borylation41,42,43, alkenylation44,45,46, allylation47, alkyl-Heck-type reaction48,49, carbonylation50,51,52,53,54, alkylation55,56,57,58, difluoromethylation59, and C-heteroatom bond-forming reactions60,61,62 have been established. However, the deaminative alkynylation of alkyl amines to form the C(sp3)–C(sp) bond still remains elusive. Recently, the Gryko group developed a nice protocol to access such transformation by visible-light-mediated desulfonylative alkynylation of secondary alkyl- and benzylpyridinium salts with alkynyl sulfones63. Han and co-workers reported an efficient nickel-catalyzed reductive cross-electrophile coupling of Katritzky salts with triisopropylsilyl (TIPS)-substituted bromoethyne to achieve the challenging C(sp3)–C(sp) bond64. Nevertheless, these methods rely mainly upon the use of preformed and activated alkynyl sulfones or bromides as alkynylating reagents. In addition, the limited substrate scope and the utilization of largely excess reductants (e.g. zinc flake) further disfavored their wide applications in organic synthesis. Therefore, the direct coupling of terminal alkynes with alkylpyridinium salts in a redox-neutral fashion for the synthesis of important alkynes would be highly desirable in terms of both atom-economy and practical application. To the best of our knowledge, however, such a straightforward and practical protocol has not been achieved.
Following our keen interest in nickel-catalyzed cross-coupling reactions65,66, herein, we report the general and efficient nickel-catalyzed Sonogashira coupling of alkylpyridinium salts via C–N bond activation under Cu-free conditions (Fig. 1e). The easily accessible and bench-stable amide-type NN2 pincer ligand (6-methyl-N-(quinolin-8-yl)picolinamide L4) is found to be crucial for this transformation, allowing the coupling to occur under mild reaction conditions with excellent yields and high functional group tolerance.
Results
Optimization study
Initially, the coupling of phenethylpyridinium salt 1a and phenylacetylene 2a was selected as the model reaction for optimization (Table 1). To realize such transformation, we envisaged that a pincer ligand might be feasible due to its strong and tridentate bonding mode to the metal center, thereby possibly stabilizing the alkylnickel intermediate67 and suppressing the undesired Glaser coupling68,69. Thus, the ligands were firstly screened by using 10 mol% NiCl2(glyme) as a catalyst, K3PO4 as a base in tetrahydrofuran (THF) at 80 °C. When pybox, the most efficient ligand in Liu’s work18, was applied to this reaction, the desired product 3a was obtained in 4% yield and the main product was 1,4-diphenylbutadiyne derived from the homocoupling of 2a (entry 1). While the use of a more electron-rich and bulky 4,4′,4″-tri-tert-butyl terpyridine (ttbtpy) in this process, the yield of 3a was improved to 53% (entry 2). Much to our delight, the yields of 3a could be further improved to 87% and 83%, respectively, when amide-type pincer ligand (e.g. N-(pyridin-2-ylmethyl)picolinamide (L1) or N-(quinolin-8-yl)picolinamide (L2)) was used (entries 3–4), though they were seldom used as ligands in transition metal-catalyzed cross-coupling reactions70,71,72,73. This discovery encouraged us to synthesize two sterically more hindered methylated derivatives L3 and L4 as ligands. Gratifyingly, the yield was significantly improved to 96% by employing L4 (entry 6). The reasons for the high efficiency of L4 are still unclear at present but probably related to its steric hindrance and rigidity. Screening of nickel catalysts revealed that Ni(acac)2 was ineffective (entry 7), whereas the inexpensive, air-stable and moisture-stable NiCl2·6H2O gave the best result (entry 8). Subsequently, the effect of the base was examined. K2CO3 resulted in a slightly diminished yield (entry 9). However, the reaction completely shut down by using Et3N, a frequently used base in palladium-catalyzed Sonogashira coupling of aryl halides (entry 10)11. Lowering the amount of catalyst or reaction temperature led to a reduced yield to different extent (entries 11–12). Control experiments indicated that NiCl2·6H2O, L4, and K3PO4 were all essential for achieving the transformation (entries 13–15) (For a detailed optimization study, see Supplementary Tables 1–5).
Substrate scope
With the optimized coupling conditions in hand, the scope of alkynes was first evaluated using 1a as the coupling partner. For some cases that the products were unseparated from the excess terminal alkynes, p-methoxylphenethylpyridinium salt 1b was used instead of 1a. As shown in Fig. 2, the arynes bearing both electron-donating and electron-withdrawing groups could participate in this transformation delivering the products (3b–3k) in excellent yields. Various synthetically important functional groups including methoxyl, arylhalide, ester, acetyl, trifluoromethyl, formyl, and free amino were all perfectly accommodated. Particularly noteworthy was that aryl chlorides and bromides, popular electrophilic partners in Sonogashira reactions10, remained inert under our optimized reaction conditions, highlighting the exquisite chemoselectivity of this transformation. Additionally, the presence of an ortho formyl did not hamper the reaction. Strikingly, terminal alkyne (2l) containing a boronate ester group was also successfully engaged in this transformation with its C–B bond intact, thus allowing for further diversification. Heteroaromatic rings such as pyridine and thiophene might deactivate a metal catalyst by coordination, and 1-ethynylcyclohexene could also smoothly undergo the transformation giving the corresponding products (3m–3o) in excellent yields. More importantly, aliphatic alkynes (2p–2t) could also be coupled in high efficiency. The functional groups such as Cl, NHBoc, and OH were well tolerated, affording the products (3r–3t) in high to excellent yields with excellent selectivity. Finally, TIPS- and trimethylsilyl-capped alkynes were also suitable substrates to obtain the products (3u–3v) in high yields.

Reaction conditions: 1a (0.3 mmol), 2 (0.45 mmol), NiCl2·6H2O (10 mol%), L4 (10 mol%), K3PO4 (1.3 equiv), THF (1.5 mL), 80 °C. Isolated yields. For 3g, 3h, 3j, 3k, 3l, 3s and 3t, reactions were conducted using p-methoxyphenethylpyridinium salt 1b instead of 1a. PMP p-methoxyphenyl.
Next, the generality of alkyl amines was evaluated as shown in Fig. 3. Various primary alkyl (1b–1l) and benzylpyridinium salts (1m–1o) were all suitable substrates for this transformation, and the desired products (4b–4o) could be obtained in high to excellent yields. However, the secondary alkylpyridinium salts (e.g. 1t) exhibited a dramatic drop in reaction efficiency (4t, 74%) under the optimized conditions. Then reoptimization of secondary alkylpyridinium salts was conducted by exploring various reaction parameters. Gratifyingly, 98% yield of 4t could be obtained by changing the solvent to DMF. Under the slightly modified conditions, diverse secondary alkylpyridinium salts underwent this coupling smoothly to give the desired products (4p–4w) in high to excellent yields. Similarly, good functional group tolerance was observed, as exemplified by the well compatible with methoxyl, trifluoromethoxyl, bromide, indole NH, alkenyl, tert-amine, acetal, hydroxyl, and chloride. More importantly, heterocyclic units such as thiophene (1f), pyridine (1g), indole (1h), tetrahydropyran (1s), and piperidine (1t) which are prevalent in medicinally relevant molecules were competent substrates. In addition, benzylpyridinium salts especially electron-rich benzylic salts which are not suitable in Gryko’s work63 could be coupled with high efficiency (4m–4n), emphasizing the robustness of our strategy in synthetic applications. It is worth noting that both cyclic (1p–1u) and acyclic secondary amines (1v–1w) could be readily applied to this protocol with high to excellent yields. Moreover, γ-amino acid-derived pyridinium salt (1x) proceeded well under the standard conditions. Notably, a quaternary carbon center could be successfully constructed by using a tertiary amine derivative (1y), albeit in a 44% yield. However, when phenylalanine (1z) and dipeptide (1aa) were employed in this reaction, complex products distributions were observed, and none of the desired deaminative alkynylation products were obtained.

a Scope of primary alkylpyridinium salts. b Scope of benzylpyridinium salts. c Scope of secondary alkylpyridinium salts. d Scope of amino acid and peptide-derived pyridinium salts. Reaction conditions: 1 (0.3 mmol), 2a (0.45 mmol), NiCl2·6H2O (10 mol%), L4 (10 mol%), K3PO4 (1.3 equiv), THF (1.5 mL), 80 °C. Isolated yields. For 4g, 4h, 4p, 4q, 4r, 4s, 4t, 4u, 4v, 4w, 4y, 4z, and 4aa, reactions were conducted in DMF (1.5 mL). For 4m, 4n, and 4o, reactions were conducted at 50 °C.
It is worth highlighting that this protocol was amenable to a one-pot transformation in which pyrylium salt, alkyl amine and the cross-coupling reagents were added simultaneously in a single step, and 78% yield of the product 3a could be obtained without further reoptimizing the reaction conditions (Fig. 4a). Additionally, a gram-scale reaction was successfully performed using 1a and 2c under the optimized conditions producing 3c in 87% yield, exemplifying the practicability and scalability of this process (Fig. 4b).

a One-pot transformation. b Gram-scale study.
Late-stage derivatizations
To further demonstrate the broad applicability of this method, late-stage functionalization of natural products and medicinally relevant molecules were conducted (Fig. 5). A series of pyridinium salts and alkynes derived from drugs and bioactive compounds underwent this transformation with good to excellent yields (5–20). This general protocol could be successfully applied for the rapid construction of alkyne-labeled derivatives of biomolecules (5–9). The readily attached alkynyl group is expected to serve as a labeling tool to facilitate further chemical biology studies and as a handle for rapid entry to complex derivatives. Likewise, this versatile method can be also applied in the further functionalization of alkynyl-containing bioactive molecules or intermediates (10–14). Notably, the virtues of the current method were further illustrated by the successful coupling of two drug molecules for assembling their drug-like hybrids 15–20, highlighting the potential applications of this chemistry in the discovery of pharmaceutical candidates.

Reaction conditions: pyridinium salt (0.3 mmol), alkyne (0.45 mmol), NiCl2·6H2O (10 mol%), L4 (10 mol%), K3PO4 (1.3 equiv), THF (1.5 mL), 80 oC. Isolated yields. For 7, 9, 15, 16, 17, 18, 19 and 20, reactions were conducted in DMF (1.5 mL).
Mechanistic studies
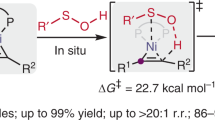
To understand the reaction mechanism, a series of experiments were performed. When the radical trapping reagent TEMPO was added to the reaction mixture, the only a trace of 3a was obtained with the concurrent formation of TEMPO-adduct 21 in 16% yield (Fig. 6a). In addition, a radical-clock experiment was also conducted by employing cyclopropylmethyl pyridinium salt 1ab. Instead of normal cross-coupling product 4ab, a ring-opened product 22 was achieved in high yield (Fig. 6b). These results suggest that an alkyl radical may be involved in this transformation. To further elucidate the role of the nickel catalyst in this reaction, a Ni complex Int-1 was synthesized by simple exposure of NiCl2·6H2O and L4 in THF at room temperature and characterized by X-ray crystallography. Gratifyingly, a high yield of 3a was obtained when Int-1 was applied to this catalytic transformation (Fig. 6c). However, when Ni(cod)2 was used as the catalyst, a remarkable decrease in efficiency was observed and only a moderate yield of 3a was achieved (Fig. 6d). These results indicate that the six-coordinate Ni complex Int-1 also exhibits an excellent catalytic activity, while a Ni(0) species is not likely involved in this chemistry. To gain more insight into the mechanism of this reaction, a Ni-alkynyl complex A1 was formed by the reaction of NiCl2(glyme), L4, and K3PO4 with p-methoxyphenylethyne in DMF, and its structure was confirmed by X-ray diffraction. Employing 10 mol% complex A1 as the catalyst, the reaction of 1a with p-methoxyphenylethyne delivered the product 3c in 93% yield, which was similar to the result obtained using NiCl2·6H2O and L4 as the catalyst (Fig. 6e). However, the stoichiometric reaction of complex A1 with 1a did not give 3c in any detectable yield (Fig. 6f). Interestingly, when this reaction was conducted in the presence of 1.0 equiv of p-methoxyphenylethyne and 1.3 equiv of K3PO4, an almost identical yield of 3c to that of the catalysis could be obtained (Fig. 6g). These results show that complex A1 itself could not react with alkyl pyridinium salt, and it needs to be activated by an additional alkynyl anion before reaction with alkyl pyridinium salt. To probe the role of alkyne on the activation of complex A1, a crossover experiment between complex A1 and p-methylphenylacetylene with 1a was performed in the presence of K3PO4. Surprisingly, both 3b and 3c were achieved in comparable yields, with an overall yield of 89% (Fig. 6h). This result implies that the two alkynyl fragments in the active Ni-species are equivalent and/or exchangeable. To further investigate whether a fast alkynyl exchange process occurred on the complex A1, the reaction of complex A1 with equal amounts of p-methylphenylacetylene was carried out under catalytically relevant conditions (Fig. 6i). A nickel complex A2 was immediately observed by NMR analysis of the reaction mixture (For details, see Supplementary Fig. 1). Then it quickly reached an equilibrium with complex A1 in a roughly 1:1 ratio, which means that the alkynyl ligated on complex A1 is exchangeable. Although the real active species for this deaminative alkynylation reaction is still unclear at present, it might be tentatively assigned to the Ni bis(acetylide) intermediate considering the outcomes achieved in Fig. 6e–i. Moreover, similar results were also observed by Hu et al. 17 further supporting the possibility of Ni bis(acetylide) intermediate as the active species for this coupling reaction.

a Radical trap experiment. b Radical clock experiment. c Catalytic transformation using Int-1 as catalyst. d Catalytic transformation using Ni(cod)2 as catalyst. e Catalytic transformation using complex A1 as catalyst. f Stoichiometric reaction of complex A1. g Stoichiometric reaction of complex A1 in the presence of terminal alkyne and base. h Crossover experiment of complex A1. i Alkyne exchange experiment of complex A1. PMP p-methoxyphenyl, Tol p-methylphenyl.
Discussion
Although a detailed mechanism awaits further studies, a plausible mechanism is depicted in Fig. 7 based on the above results and Ni/pincer-ligand system catalyzed cross-coupling of alkyl electrophiles16,17,19,74,75,76,77. Initially, coordination of L4 to the Ni center followed by base promoted transmetalation with terminal alkyne to form a complex A. However, this species possesses no reactivity toward pyridinium 1 as demonstrated by Fig. 6f. Moreover, the cyclic voltammogram of complex A1 showing an oxidation wave at 1.19 V in DMF further indicates that the direct coupling of complex A with 1 (Ered = −0.90 V vs. SCE in DMF) is not possible. (For cyclic voltammogram of pyridinium 1a and complex A1, see Supplementary Figs. 2 and 3.) Thus, a further transmetalation of complex A with alkyne was needed to generate a more electron-rich anionic species B, which is thermodynamically unstable and could rapidly disassociate an alkynyl anionic to reach an equilibrium with the dormant complex A. At this stage, the K(I) ion in the Ni bis(acetylide) intermediate is probably coordinated to the triple bond of alkyne, similar to that of binding a copper reported by Hartwig78. Then, the more active species B might undergo oxidative addition with 1 to give intermediate C, during which a radical process is likely involved based on the results obtained from Fig. 6a, b. Finally, reductive elimination from C delivers the C(sp3)–C(sp) coupling product and regenerates complex A for the next catalytic cycle. The reasons for the high selectivity of cross-coupling products are unclear at now, but probably related to the fast alkyl–alkynyl reductive elimination promoted by the NN2 pincer ligand17,76. Additionally, the oxidation state of Ni in intermediate C seems to be a NiIV, but it might also be described as a NiIII–ligand radical complex when considering the redox-active of NN2 pincer ligand75,79. Therefore, the current catalytic cycle is not in contradiction with the proposed mechanism in Ni catalysis.

A plausible mechanism involving a more electron-rich Ni bis(acetylide) species is tentatively proposed.
In summary, we have achieved a highly efficient and general Sonogashira coupling of alkylpyridinium salts by the development of a Ni/NN2 pincer ligand catalytic system. Noteworthy was the realization of the coupling of terminal alkynes with naturally abundant alkyl amines, expanding the substrate scopes used in Sonogashira reaction. The virtues of this reaction are illustrated by the broad substrate scope, well functional group tolerance in both coupling partners as well as the efficient diversification of natural products and medicinally relevant molecules. Further mechanism investigation and application of this catalytic system for the cross-coupling with other electrophiles are currently ongoing in our laboratories.
Methods
General procedure 1
In a nitrogen-filled glovebox, NiCl2·6H2O (0.03 mmol, 7.1 mg), L4 (0.03 mmol, 7.9 mg), anhydrous K3PO4 (0.39 mmol, 82.8 mg), primary alkylpyridinium salt (0.3 mmol), and THF (1.5 mL) were successively added to an oven-dried sealable Schlenk tube (10.0 mL) followed by addition of terminal alkyne (0.45 mmol) via microliter syringe (If terminal alkyne is solid, it was added before the solvent). Then the tube was securely sealed and taken outside the glovebox. And it was immersed into an oil bath preheated at 80 or 50 °C. After stirring for 24 h, the reaction mixture was cooled to room temperature and filtered through a short pad of silica gel. Then the filter cake was washed with dichloromethane or ethyl acetate. The resulting solution was concentrated under vacuum and the residue was purified by column chromatography on silica gel to afford the corresponding product.
General procedure 2
In a nitrogen-filled glovebox, NiCl2·6H2O (0.03 mmol, 7.1 mg), L4 (0.03 mmol, 7.9 mg), anhydrous K3PO4 (0.39 mmol, 82.8 mg), secondary alkylpyridinium salt (0.3 mmol), and N,N-dimethylformamide (1.5 mL) were successively added to an oven-dried sealable Schlenk tube (10.0 mL) followed by addition of phenylacetylene (0.45 mmol, 46.0 mg) via microliter syringe. Then the tube was securely sealed and taken outside the glovebox. And it was immersed into an oil bath preheated at 80 °C. After stirring for 24 h, the reaction mixture was cooled to room temperature and quenched with water. Then it was extracted with ethyl acetate or diethyl ether, washed with water and brine, and dried over anhydrous Na2SO4. The resulting solution was concentrated under vacuum and the residue was purified by column chromatography on silica gel to afford the corresponding product.
Data availability
Detailed experimental procedures and characterization of all new compounds can be found in the Supplementary Information. The authors declare that all the data supporting the findings of this study are available within the article and Supplementary Information files, and are also available from the corresponding authors upon reasonable request. CCDC 2035475 (Int-1) and 2055846 (complex A1) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Lam, J., Breteler, H., Arnason, T. & Hansen, L. Chemistry and Biology of Naturally-Occurring Acetylenes and Related Compounds (Elsevier, Amsterdam, 1988).
Patai, S. Chemistry of Triple-Bonded Functional Groups (Wiley, New York, 1994).
Diederich, F., Stang, P. J. & Tykwinski, R. R. Acetylene Chemistry: Chemistry, Biology and Material Science (Wiley-VCH, Weinheim, Germany, 2005).
Thirumurugan, P., Matosiuk, D. & Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 113, 4905–4979 (2013).
Yamakoshi, H. et al. Imaging of Edu, an alkyne-tagged cell proliferation probe, by Raman microscopy. J. Am. Chem. Soc. 133, 6102–6105 (2011).
Yamakoshi, H. et al. Alkyne-tag Raman imaging for visualization of mobile small molecules in live cells. J. Am. Chem. Soc. 134, 20681–20689 (2012).
Song, Z.-L. et al. Alkyne-functionalized superstable graphitic silver nanoparticles for Raman imaging. J. Am. Chem. Soc. 136, 13558–13561 (2014).
Wei, L. et al. Live-cell imaging of alkyne-tagged small biomolecules by stimulated Raman scattering. Nat. Methods 11, 410–412 (2014).
Li, Y., Wang, Z., Mu, X., Ma, A. & Guo, S. Raman tags: novel optical probes for intracellular sensing and imaging. Biotechnol. Adv. 35, 168–177 (2017).
Negishi, E.-I. & Sonogashira, K. In Handbook of Organopalladium Chemistry for Organic Synthesis (ed. Negishi, E.-I.) 493–529 (Wiley-Interscience, 2002).
Chinchilla, R. & Nájera, C. The Sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem. Rev. 107, 874–922 (2007).
Chopade, P. R. & Louie, J. [2+2+2] Cycloaddition reactions catalyzed by transition metal complexes. Adv. Synth. Catal. 348, 2307–2327 (2006).
Galan, B. R. & Rovis, T. Beyond Reppe: building substituted arenes by [2+2+2] cycloadditions of alkynes. Angew. Chem. Int. Ed. 48, 2830–2834 (2009).
Eckhardt, M. & Fu, G. C. The first applications of carbene ligands in cross-couplings of alkyl electrophiles: sonogashira reactions of unactivated alkyl bromides and iodides. J. Am. Chem. Soc. 125, 13642–13643 (2003).
Altenhoff, G., Würtz, S. & Glorius, F. The first palladium-catalyzed Sonogashira coupling of unactivated secondary alkyl bromides. Tetrahedron Lett. 47, 2925–2928 (2006).
Vechorkin, O., Barmaz, D., Proust, V. & Hu, X. Ni-catalyzed Sonogashira coupling of nonactivated alkyl halides: orthogonal functionalization of alkyl iodides, bromides, and chlorides. J. Am. Chem. Soc. 131, 12078–12079 (2009).
García, P. M. P., Ren, P., Scopelliti, R. & Hu, X. Nickel-catalyzed direct alkylation of terminal alkynes at room temperature: a hemilabile pincer ligand enhances catalytic activity. ACS Catal. 5, 1164–1171 (2015).
Yi, J., Lu, X., Sun, Y.-Y., Xiao, B. & Liu, L. Nickel-catalyzed Sonogashira reactions of non-activated secondary alkyl bromides and iodides. Angew. Chem. Int. Ed. 52, 12409–12413 (2013).
Wang, Z. et al. Sonogashira reactions of alkyl halides catalyzed by NHC [CNN] pincer nickel(II) complexes. N. J. Chem. 42, 11465–11470 (2018).
Hazra, A., Lee, M. T., Chiu, J. F. & Lalic, G. Photoinduced copper-catalyzed coupling of terminal alkynes and alkyl iodides. Angew. Chem., Int. Ed. 57, 5492–5496 (2018).
Mao, Y. et al. Copper-catalysed photoinduced decarboxylative alkynylation: a combined experimental and computational study. Chem. Sci. 11, 4939–4947 (2020).
Jin, L. et al. N-heterocyclic carbene copper-catalyzed direct alkylation of terminal alkynes with non-activated alkyl triflates. Chem. Commun. 53, 4124–4127 (2017).
Gelman, D. & Buchwald, S. L. Efficient palladium-catalyzed coupling of aryl chlorides and tosylates with terminal alkynes: use of a copper cocatalyst inhibits the reaction. Angew. Chem. Int. Ed. 42, 5993–5996 (2003).
Ruiz-Castillo, P. & Buchwald, S. L. Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev. 116, 12564–12649 (2016).
Liu, Y. & Ge, H. Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat. Chem. 9, 26–32 (2017).
Blanksby, S. J. & Ellison, G. B. Bond dissociation energies of organic molecules. Acc. Chem. Res. 36, 255–263 (2003).
Ouyang, K., Hao, W., Zhang, W. X. & Xi, Z. Transition-metal-catalyzed cleavage of C–N single bonds. Chem. Rev. 115, 12045–12090 (2015).
Wang, Q., Su, Y., Li, L. & Huang, H. Transition-metal catalysed C–N bond activation. Chem. Soc. Rev. 45, 1257–1272 (2016).
Katritzky, A. R., Gruntz, U., Kenny, D. H., Rezende, M. C. & Sheikh, H. Heterocycles in organic synthesis. Part 10. Conversion of amines into esters. J. Chem. Soc. Perkin Trans. 1, 430–432 (1979).
Basch, C. H., Liao, J., Xu, J., Piane, J. J. & Watson, M. P. Harnessing alkyl amines as electrophiles for nickel-catalyzed cross couplings via C–N bond activation. J. Am. Chem. Soc. 139, 5313–5316 (2017).
Kong, D., Moon, P. J. & Lundgren, R. J. Radical coupling from alkyl amines. Nat. Catal. 2, 473–476 (2019).
Sowmiah, S., Esperanca, J. M. S. S., Rebelo, L. P. N. & Afonso, C. A. M. Pyridinium salts: from synthesis to reactivity and applications. Org. Chem. Front. 5, 453–493 (2018).
He, F.-S., Ye, S. & Wu, J. Recent advances in pyridinium salts as radical reservoirs in organic synthesis. ACS Catal. 9, 8943–8960 (2019).
Peng, Y., Moser, D. & Cornella, J. Pyridinium salts: selectivity reagents for the activation of primary amino groups in organic synthesis. Synthesis 52, 489–503 (2020).
Klauck, F. J. R., James, M. J. & Glorius, F. Deaminative strategy for the visible-light-mediated generation of alkyl radicals. Angew. Chem., Int. Ed. 56, 12336–12339 (2017).
Yue, H. et al. Nickel-catalyzed C–N bond activation: activated primary amines as alkylating reagents in reductive cross-coupling. Chem. Sci. 10, 4430–4435 (2019).
Hoerrner, M. E., Baker, K. M., Basch, C. H., Bampo, E. M. & Watson, M. P. Deaminative arylation of amino-acid derived pyridinium salts. Org. Lett. 21, 7356–7360 (2019).
Liao, J. et al. Deaminative reductive cross-electrophile couplings of alkylpyridinium salts and aryl bromides. Org. Lett. 21, 2941–2946 (2019).
Martin-Montero, R., Yatham, V. R., Yin, H., Davies, J. & Martin, R. Ni-catalyzed reductive deaminative arylation at sp3 carbon centers. Org. Lett. 21, 2947–2951 (2019).
Yi, J., Badir, S. O., Kammer, L. M., Ribagorda, M. & Molander, G. A. Deaminative reductive arylation enabled by nickel/photoredox dual catalysis. Org. Lett. 21, 3346–3351 (2019).
Wu, J., He, L., Noble, A. & Aggarwal, V. K. Photoinduced deaminative borylation of alkylamines. J. Am. Chem. Soc. 140, 10700–10704 (2018).
Hu, J., Wang, G., Li, S. & Shi, Z. Selective C-N borylation of alkyl amines promoted by Lewis base. Angew. Chem. Int. Ed. 57, 15227–15231 (2018).
Sandfort, F., Strieth-Kalthoff, F., Klauck, F. J. R., James, M. J. & Glorius, F. Deaminative borylation of aliphatic amines enabled by visible light excitation of an electron donor-acceptor complex. Chem. Eur. J. 24, 17210–17214 (2018).
Zhu, Z., Tu, J. & Liu, F. Ni-catalyzed deaminative hydroalkylation of internal alkynes. Chem. Commun. 55, 11478–11481 (2019).
Hu, J., Cheng, B., Yang, X. & Loh, T.-P. Transition-metal-free deaminative vinylation of alkylamines. Adv. Synth. Catal. 361, 4902–4908 (2019).
Baker, K. M., Baca, D. L., Plunkett, S., Daneker, M. E. & Watson, M. P. Engaging alkenes and alkynes in deaminative alkyl-alkyl and alkyl-vinyl cross-couplings of alkylpyridinium salts. Org. Lett. 21, 9738–9741 (2019).
Zhang, M. & Liu, F. Visible-light-mediated allylation of alkyl radicals with allylic sulfones via a deaminative strategy. Org. Chem. Front. 5, 3443–3446 (2018).
Jiang, X., Zhang, M.-M., Xiong, W., Lu, L.-Q. & Xiao, W.-J. Deaminative (carbonylative) alkyl-Heck-type reactions enabled by photocatalytic C–N bond activation. Angew. Chem. Int. Ed. 58, 2402–2406 (2019).
Yang, Z., Xu, N., Wang, C. & Uchiyama, M. Photoinduced C(sp3)–N bond cleavage leading to the stereoselective syntheses of alkenes. Chem. Eur. J. 25, 5433–5439 (2019).
Li, C., Jiang, X., Lu, L., Xiao, W. & Wu, X. Cobalt(II)-catalyzed alkoxycarbonylation of aliphatic amines via C–N bond activation. Org. Lett. 21, 6919–6923 (2019).
Yu, C. & Matsuo, Y. Nickel-catalyzed deaminative acylation of activated aliphatic amines with aromatic amides via C–N bond activation. Org. Lett. 22, 950–955 (2020).
Wang, J., Hoerrner, M. E., Watson, M. P. & Weix, D. J. Nickel-catalyzed synthesis of dialkyl ketones from the coupling of N-alkyl pyridinium salts with activated carboxylic acids. Angew. Chem. Int. Ed. 59, 13484–13489 (2020).
Pulikottil, F. T., Pilli, R., Suku, R. V. & Rasappan, R. Nickel-catalyzed cross-coupling of alkyl carboxylic acid derivatives with pyridinium salts via C–N bond cleavage. Org. Lett. 22, 2902–2907 (2020).
Kim, I., Im, H., Lee, H. & Hong, S. N-heterocyclic carbene-catalyzed deaminative cross-coupling of aldehydes with Katritzky pyridinium salts. Chem. Sci. 11, 3192–3197 (2020).
Plunkett, S., Basch, C. H., Santana, S. O. & Watson, M. P. Harnessing alkylpyridinium salts as electrophiles in deaminative alkyl–alkyl cross-couplings. J. Am. Chem. Soc. 141, 2257–2262 (2019).
Sun, S.-Z., Romano, C. & Martin, R. Site-selective catalytic deaminative alkylation of unactivated olefins. J. Am. Chem. Soc. 141, 16197–16201 (2019).
Wu, J., Grant, P. S., Li, X., Noble, A. & Aggarwal, V. K. Catalyst-free deaminative functionalizations of primary amines by photoinduced single-electron transfer. Angew. Chem. Int. Ed. 58, 5697–5701 (2019).
Wang, C. et al. Visible-light-promoted C(sp3)–H alkylation by intermolecular charge transfer: preparation of unnatural α-amino acids and late-stage modification of peptides. Angew. Chem. Int. Ed. 59, 7461–7466 (2020).
Zeng, X. et al. Copper-catalyzed deaminative difluoromethylation. Angew. Chem. Int. Ed. 59, 16398–16403 (2020).
Yang, M., Cao, T., Xu, T. & Liao, S. Visible-light-induced deaminative thioesterification of amino acid derived Katritzky salts via electron donor–acceptor complex formation. Org. Lett. 21, 8673–8678 (2019).
Li, Z. et al. Manganese-mediated reductive functionalization of activated aliphatic acids and primary amines. Nat. Commun. 11, 5036 (2020).
Wang, X., Kuang, Y., Ye, S. & Wu, J. Photoredox-catalyzed synthesis of sulfones through deaminative insertion of sulfur dioxide. Chem. Commun. 55, 14962–14964 (2020).
Ociepa, M., Turkowska, J. & Gryko, D. Redox-activated amines in C(sp3)–C(sp) and C(sp3)–C(sp2) bond formation enabled by metal-free photoredox catalysis. ACS Catal. 8, 11362–11367 (2018).
Ni, S. et al. Ni-catalyzed deaminative cross-electrophile coupling of Katritzky salts with halides via C–N bond activation. Sci. Adv. 5, No. eaaw9516 (2019).
Zhang, X., Xie, X. & Liu, Y. Nickel-catalyzed highly regioselective hydrocyanation of terminal alkynes with Zn(CN)2 using water as the hydrogen source. J. Am. Chem. Soc. 140, 7385–7389 (2018).
Zhang, X., Xia, A., Chen, H. & Liu, Y. General and mild nickel-catalyzed cyanation of aryl/heteroaryl chlorides with Zn(CN)2: key roles of DMAP. Org. Lett. 19, 2118–2121 (2017).
Jones, G. D. et al. Ligand redox effects in the synthesis, electronic structure, and reactivity of an alkyl–alkyl cross-coupling catalyst. J. Am. Chem. Soc. 128, 13175–13183 (2006).
Leophairatana, P., Samanata, S., De Silva, C. C. & Koberstein, J. T. Preventing alkyne–alkyne (i.e., Glaser) coupling associated with the ATRP synthesis of alkyne-functional polymers/macromonomers and for alkynes under click (i.e., CuAAC) reaction conditions. J. Am. Chem. Soc. 139, 3756–3766 (2017).
Dong, X.-Y. et al. A general asymmetric copper-catalysed Sonogashira C(sp3)–C(sp) coupling. Nat. Chem. 11, 1158–1166 (2019).
Soni, V., Jagtap, R. A., Gonnade, R. G. & Punji, B. Unified strategy for nickel-catalyzed C-2 alkylation of indoles through chelation assistance. ACS Catal. 6, 5666–5672 (2016).
Andersen, T. L., Donslund, A. S., Neumann, K. T. & Skrydstrup, T. Carbonylative coupling of alkyl zinc reagents with benzyl bromides catalyzed by an NN2 pincer ligand nickel complex. Angew. Chem. Int. Ed. 57, 800–804 (2018).
Donslund, A. S. et al. Access to β-ketonitriles through nickel-catalyzed carbonylative coupling of α-bromonitriles with alkylzinc reagents. Chem. Eur. J. 25, 9856–9860 (2019).
Donslund, A. S. et al. Direct access to isotopically labeled aliphatic ketones mediated by nickel(I) activation. Angew. Chem. Int. Ed. 59, 8099–8103 (2020).
Shi, R., Zhang, Z. & Hu, X. Nickamine and analogous nickel pincer catalysts for cross-coupling of alkyl halides and hydrosilylation of alkenes. Acc. Chem. Res. 52, 1471–1483 (2019).
Vechorkin, O., Proust, V. & Hu, X. Functional group tolerant Kumada–Corriu–Tamao coupling of nonactivated alkyl halides with aryl and heteroaryl nucleophiles: catalysis by a nickel pincer complex permits the coupling of functionalized Grignard reagents. J. Am. Chem. Soc. 131, 9756–9766 (2009).
Vechorkin, O., Godinat, A., Scopelliti, R. & Hu, X. Cross-coupling of nonactivated alkyl halides with alkynyl Grignard reagents: a nickel pincer complex as the catalyst. Angew. Chem. Int. Ed. 50, 11777–11781 (2011).
Hu, X. Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem. Sci. 2, 1867–1886 (2011).
Gallego, D. et al. From bis(silylene) and bis(germylene) pincer-type nickel(II) complexes to isolable intermediates of the nickel-catalyzed Sonogashira cross-coupling reaction. J. Am. Chem. Soc. 135, 15617–15626 (2013).
Adhikari, D. et al. Structural, spectroscopic, and theoretical elucidation of a redox-active pincer-type ancillary applied in catalysis. J. Am. Chem. Soc. 130, 3676–3682 (2008).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21877206 and U1604285 to G.Z.), Zhongyuan Qianren Jihua (ZYQ201912132 to G.Z.), China Postdoctoral Science Foundation (2019M660173 to X.Z.), Henan Normal University Doctoral Initiation Fund (qd18011 to X.L.; qd18016 to X.Z.).
Author information
Authors and Affiliations
Contributions
X.Z., and G.Z. conceived the idea and guided the project. X.Z., D.Q., C.J., and X.L. performed the experiments and analyzed the results. X.Z., X.L., and G.Z. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Qi, D., Jiao, C. et al. Nickel-catalyzed deaminative Sonogashira coupling of alkylpyridinium salts enabled by NN2 pincer ligand. Nat Commun 12, 4904 (2021). https://doi.org/10.1038/s41467-021-25222-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-021-25222-1
This article is cited by
-
Recent Progress in Fragmentation of Katritzky Salts Enabling Formation of C–C, C–B, and C–S Bonds
Topics in Current Chemistry (2022)
