
Abstract
The melanocortin receptor 4 (MC4R) belongs to the melanocortin receptor family of G-protein coupled receptors and is a key switch in the leptin-melanocortin molecular axis that controls hunger and satiety. Brain-produced hormones such as α-melanocyte-stimulating hormone (agonist) and agouti-related peptide (inverse agonist) regulate the molecular communication of the MC4R axis but are promiscuous for melanocortin receptor subtypes and induce a wide array of biological effects. Here, we use a chimeric construct of conformation-selective, nanobody-based binding domain (a ConfoBody Cb80) and active state-stabilized MC4R-β2AR hybrid for efficient de novo discovery of a sequence diverse panel of MC4R-specific, potent and full agonistic nanobodies. We solve the active state MC4R structure in complex with the full agonistic nanobody pN162 at 3.4 Å resolution. The structure shows a distinct interaction with pN162 binding deeply in the orthosteric pocket. MC4R peptide agonists, such as the marketed setmelanotide, lack receptor selectivity and show off-target effects. In contrast, the agonistic nanobody is highly specific and hence can be a more suitable agent for anti-obesity therapeutic intervention via MC4R.
Similar content being viewed by others

Introduction
Based on pharmacologic and human genetic evidence, the hypothalamic melanocortin-4 receptor (MC4R) appears as a clinically validated key modulator of energy expenditure, satiety, and thus body weight. Human subjects carrying loss-of-function MC4R mutations or showing deficiency in proopiomelanocortin (POMC), the prohormone processed into bio-active melanocortin receptor peptide agonists, often show hyperphagia and severe (early-onset) obesity1,2. Conversely, subjects carrying gain-of-function MC4R mutations3 show increased satiety and reduced food uptake, resulting in lower body weight (ref. 1). Therapeutically intervening with MC4R agonists similarly increases satiety and reduces food uptake. MC4R is a rhodopsin-like class A peptide G-protein coupled receptor (GPCR) and belongs to the melanocortin receptor family which comprises 5 receptor subtypes (MC1R to MC5R). The main biological effects of melanocortin receptor stimulation are skin melanocyte pigmentation and immune regulation (MC1R)4,5, adrenal gland steroidogenesis (MC2R)6, fat mass regulation, control of growth and puberty (MC3R)7 and regulation of energy homeostasis and food uptake (MC4R)8,9. The role of MC5R is less elucidated but seems to manage immune reaction and inflammatory response, thermoregulation, and exocrine secretion10. Except for MC2R which is solely activated by the (endogenous) adrenocorticotropic hormone (ACTH), endogenous signal transduction is induced mainly by melanocyte-stimulating hormone isoforms (α-, β- and γ-MSH). MC1R, MC2R, and MC5R can also be stimulated by ACTH. MC3R, MC4R, and MC5R signaling is blocked by the naturally occurring agouti-related peptide (AGRP). All peptide agonists to MC1R, MC3R, MC4R, and MC5R share the amino acid motif HxRW, which presents key interaction residues required to induce endogenous melanocortin signaling. MC4R primarily couples the stimulatory G-protein transducer (Gs) to activate adenylyl cyclase (AC), resulting in intracellular cyclic adenosine monophosphate (cAMP) production and downstream protein kinase A (PKA) activation. Besides Gs stimulation, MC4R has been shown to recruit other G-protein dependent cytosolic signal transducers such as Gi and Gq/11. In addition, MC4R also couples to the G-protein independent transducer protein β-arrestin11 or the ion channel Kir7.112. Setmelanotide (also known as RM-493 or under its brand name Imcivree®) is a cyclic, eight amino acid peptide MC4R agonist that contains the HxRW motif (HFRW) and activates MC3R, MC1R13, and, albeit with lower potency, MC5R (see ref. 14). While setmelanotide can induce Gs signaling11, the clinical efficacy of setmelanotide on body weight regulation is hypothesized to be driven by its Gq/11—phospholipase C pathway bias15. Blood pressure and heart rate elevation observed as treatment-limiting side effects by other MC4R agonists are hypothesized to be the consequence of the activation of the Gs signaling pathway in the central nervous system (CNS). Setmelanotide was approved in 2020 for rare genetic obesity disorders after positive phase 3 clinical studies reported significant body weight loss and reduced hunger score16 in leptin receptor (LEPR)- and pro-opiomelanocortin (POMC)-deficient patients. Patients carrying such genetic mutations also show impaired MC4R signaling. While setmelanotide therapy showed a positive clinical outcome, undesired side effects were still demonstrated due to a lack of specificity towards other melanocortin receptors. The most reported off-target effect of setmelanotide is skin and hair hyperpigmentation due to the activation of MC1R17. Because of these off-target effects, there is still keen interest in the discovery of MC4R specific agonist ligands as potentially safer anti-obesity therapeutics.
Antibodies are paradigm ligands when aiming for antigen specificity. Despite an increasing reagent toolbox to facilitate de novo GPCR antibody discovery, the identification of antibodies against native GPCRs remains challenging18. In addition, the vast majority of reported antibodies that pharmacologically modulate native GPCRs via extracellular epitopes are functionally neutral or are antagonists, thus blocking GPCR signaling. The de novo discovery of antibodies that activate signaling with a similar efficacy as the endogenous agonist ligand is notoriously difficult. Indeed, despite multiple reports describing positive allosteric modulating nanobodies that increase agonist affinity or signaling potency or efficacy at low ligand tonus19,20,21,22,23, only a few are reported to agonize the GPCR in the absence of any ligand. Some of the reported agonistic antibodies have been designed by ligand tethering (genetically fusing the agonist ligand motifs to the C- or N-terminus of the fusion partner24,25 or grafting agonist ligand motifs into the antibody hypervariable complementarity determining region (CDR))26 or by structure-informed engineering27. De novo identified agonistic antibodies towards class A GPCRs are scarce and have only been reported for β1AR (bivalent IgG Mab1)28, ChemR23 (bivalent IgG)29 and APJ (monovalent nanobody JN300)30. No discovery details are reported for the agonistic ChemR23 antibody. The monovalent Fab derivative of the full agonist antibody Mab1 does not induce β1AR signaling anymore, indicating receptor cross-linking as the mode of interaction for obtaining agonist pharmacology. Whether the ChemR23 mAb or APJ antibodies are full agonists and whether they interact with allosteric or orthosteric epitopes of the GPCR is not reported. Any structural information on how these agonistic antibodies induce signal transduction is lacking.
The current study describes the use of a ConfoBody-stabilized active state GPCR conformation31,32 for efficient de novo identification of highly potent and full agonist MC4R nanobodies that do not bind to the other human melanocortin receptor subtypes. A ConfoBody (Cb) is an immunoglobulin single variable domain (such as a VHH or nanobody) that selectively stabilizes desired conformational states of conformationally dynamic (membrane) proteins such as GPCRs (for example, by interacting with the cytosolic domain of a receptor either by direct interaction with the intracellular epitopes of a GPCR, or by indirectly interacting with a signaling transducer coupled to a receptor33). We use camelid single domain antibody fragments (nanobodies) to obtain MC4R-specific agonistic antibodies as these typically interact with discontinuous three-dimensional epitopes34,35. We mined an immune library obtained after immunization with a Cb-stabilized active state MC4R. The identified MC4R specific pN162 agonist is exploited as a structural chaperone and provides structural evidence of an orthosterically interacting agonistic nanobody towards a class A GPCR. Moreover, it does not appear to rely on Ca2+ cofactor for receptor stimulation as described for other previously described agonists. Such MC4R-specific molecules can be used as tools to deconvolute MC4R signaling pathway(s) to help inform clinical translational activities for the treatment of obesity.
Results
MC4R-stabilized active state for de novo agonist nanobody discovery
In vivo matured nanobody repertoires were induced by the genetic immunization of llamas with an engineered active state MC4R conformation. The MC4R active state conformation was attained by the genetic fusion of a ConfoBody36 to the C-terminus of a MC4R-β2AR hybrid GPCR37. The β2AR G-protein mimetic Nb8035 ConfoBody was used (referred from hereon as Cb80). The MC4R chimera was synthesized by grafting the C-terminus and intracellular loops (ICLs) of β2AR onto MC4R38. The in vivo matured nanobody repertoires were enriched by phage display on custom virus-like particles (VLPs) presenting active state stabilized MC4R (Supplementary Fig. 1a). Similar to the immunogen, active state-stabilized MC4R-harboring VLPs were generated using an expression cassette containing the MC4R-β2AR chimera-Cb80 genetic fusion. Periplasmic extracts were prepared of E. coli-expressed monoclonal nanobodies picked from phage outputs that represented the different panning conditions. The antibody fragments were assessed for MC4R-specificity, the ability to recruit cytosolic signal transducers (Supplementary Fig. 1b) or to induce signaling (Supplementary Fig. 1c, d). MC4R specificity was evaluated by homogeneous time resolved fluorescence (HTRF). HTRF on HEK293T cells transiently expressing the SNAP-tagged MC4R chimera genetically fused to Cb80 showed a 33.8 % hit rate of MC4R-specific binders (227 out of 672 tested VHHs with an HTRF ratio >2 over the signal obtained by a mock nanobody; Supplementary Fig. 1b). Sequence analysis of the selected MC4R binders discriminated 73 different nanobody sequence clusters based upon complementary determining region 3 (CDR3) amino acid sequence analysis. To explore whether the MC4R binders were able to induce a conformational change that leads to signal transduction, a ConfoSensor assay was established that monitors nanobody-induced recruitment of G-protein mimetic Cb80 to the MC4R-β2AR chimera as described38,39. Out of the 73 cluster variants tested as periplasmic extract, 19 (26.0%) were able to recruit Cb80 (Supplementary Fig. 1a, c). Similar recruitment to the MC4R-β2AR chimera was detected for the MC4R clinical benchmark agonist setmelanotide (Supplementary Fig. 1c). To confirm signaling, sixteen nanobodies representing different sequence clusters (Supplementary Fig. 2a) that showed the most pronounced Cb80 recruitment were subsequently tested at a single dilution in a cAMP signaling assay (Lance Ultra HTRF; Supplementary Fig. 1d) on double Twin Strep tag-modified wild type human MC4R-overexpressing HEK293 cells. A panel of sixteen Cb80-recruiting nanobodies were selected (based on Cb80 recruitment signals and initial purification yields). All tested nanobodies were confirmed to induce signaling by producing secondary messenger cAMP and they were subsequently purified for further in vitro characterization.
To confirm MC4R activation, dose-dependent nanobody-induced cAMP signaling was assessed (Lance Ultra HTRF). All 16 nanobodies assessed showed MC4R-mediated dose-dependent cAMP accumulation with potencies ranging between 0.7 (pN162 & pN152) and 393 nM (pN161; Supplementary Fig. 2b, c). All 16 nanobodies behaved as full agonists when compared to the maximum efficacy obtained with α-MSH. In order to rank them for potency, the agonistic nanobodies were assessed for real-time cAMP monitoring in Flag-tagged human wild-type MC4R-expressing HEK293T cells (GloSensor®). pN162 was identified as the most potent full agonist nanobody with a similar EC50 as the endogenous ligand (average EC50 pN162 12.5 ± 5.8 nM; average EC50 α-MSH 14.1 ± 7.7 nM; Fig. 1a and Table 1). Binding of pN162 to the human melanocortin receptor subtypes MC1R, MC2R, MC3R, MC4R or MC5R transiently expressed as Flag-tagged receptors in HEK293T cells was assessed by flow cytometry. While receptor presence was confirmed via anti-Flag detection (an example of receptor expression is shown in Fig. 1b for MC4R), binding of 1 µM pN162 was only demonstrated for MC4R (Fig. 1c, d). No binding could be detected to any other melanocortin receptor paralogue, demonstrating that pN162 specifically interacts with MC4R (Fig. 1d). For epitope binning, a radioligand displacement assay was executed. pN162 fully displaces antagonist radioligand SHU9119 on CHO-K1 cells stably expressing human MC4R chimera (Fig. 1e) with an average potency of 6.8 ± 3.0 nM (n = 3), suggesting an interaction of pN162 to MC4R’s orthosteric pocket or an overlapping epitope. For a head-to-head comparison of receptor subtype activation, pN162, setmelanotide and α-MSH were evaluated in the GloSensor cAMP assay using HEK293 cells transiently transfected with human wild type MC1R, MC3R, MC4R or MC5R (MC2R was not included as pN162 did not interact with MC2R assessed via flow cytometry; Fig. 1f–h). Contrary to setmelanotide and α-MSH, which showed activity against all melanocortin receptors tested (Fig. 1a, f–h), pN162 was confirmed to be a specific MC4R agonist as it only induced detectable Gs-mediated signaling on MC4R (Fig. 1a; Table 1). The clinical benchmark setmelanotide is a non-specific MC4R agonist as it also activates MC1R, MC3R, and to a lesser extent MC5R (Fig. 1f–h; Table 1). The setmelanotide-induced average signaling potencies (EC50) to the distinct melanocortin subtype receptors tested in cAMP GloSensor assays were 2.0, 11.5, 2.1, 451.0 nM against human MC4R, MC1R, MC3R, and MC5R, respectively (Fig. 1a, f–h and Table 1). Compared to setmelanotide, pN162 showed a 6.6-fold reduced potency in cAMP signaling via human MC4R (GloSensor; Table 1). Interestingly, the conserved HxRW motif of melanocortin peptide agonists and antagonists was not present in any of the hypervariable gene segments (including CDR3) of the agonist nanobody pN162 (Supplementary Fig. 2d).

a Dose-dependent human MC4R -induced cAMP signaling (GloSensor) of pN162 and control ligands (endogenous agonist α-MSH and clinical benchmark peptide agonist Setmelanotide). Each data point represents the mean of two replicates. Experiments were performed minimally twice (the number of experiment repeats is indicated in Table 1). Exemplary graphs are shown. Table 1 provides the average EC50s of pN162, α-MSH, and setmelanotide. b Cell surface expression of human MC4R measured by flow cytometry. Overlays of fluorescence histograms obtained by anti-Flag mAb or isotype IgG control detection on Flag-tagged MC4R or mock GPCR transiently transfected HEK293T cells. Cell surface binding of 100 nM c-Myc-tagged pN162 to human MC4R (c) or melanocortin receptor subtypes (d) measured by flow cytometry. Overlays of fluorescence histogram obtained by pN162 or mock nanobody detection on human MC4R or mock GPCR transfected HEK293T cells (c). pN162 specific melanocortin receptor subtype staining, normalized for MC4R expressing HEK293 cells (d). Only intact cells were gated following Topro staining. MC2R was expressed in absence and presence of its expression chaperone MRAPα. e Dose dependent 125I-SHU9119 radioligand displacement with pN162. Within an experiment, each data point is an average of 2 repeats. Experiments were performed three times, and a representative graph is shown. Dose-dependent human MC1R (f), MC3R (g), and MC5R (h) induced cAMP signaling (GloSensor) of pN162 and control ligands (endogenous agonist α-MSH and clinical benchmark peptide agonist setmelanotide). Each data point represents the mean of two replicates. Experiments were performed minimally twice (the number of experiment repeats is indicated in Table 1). Exemplary graphs are shown. Table 1 provides the average EC50s of pN162, α-MSH, and setmelanotide.
To understand whether the primary MC4R signaling pathways are induced by pN162, recruitment of Gs and β-arrestin (Gs being the primary G-protein transducers recruited by α-MSH) were assessed in N-terminally Flag-tag modified WT human MC4R-expressing HEK293T cells. Like α-MSH and setmelanotide, pN162 signals via both pathways (Fig. 2a, b). pN162 dose-dependently recruits Gs and β-arrestin2. In the presence of cofactor Ca2+, in both assays, setmelanotide shows a higher potency compared to pN162 and α-MSH (Fig. 2a, b; Table 2). Strikingly, in both recruitment assays, pN162-induced recruitment seemed to be independent of Ca2+ ions, contrary to the recruitment induced by α-MSH and setmelanotide. To monitor whether pN162 induced the Gq signaling pathway, the MC4R-mediated production of secondary messenger IP1 was measured in human MC4R-expressing HEK293T cells. The Gq signaling potencies (EC50) of α-MSH, setmelanotide, and pN162 are 428.5, 9.9, and 121.3 nM, respectively (Fig. 2c; Table 3). Compared to setmelanotide, pN162 showed a 12.5-fold reduced potency in Gq signaling via human MC4R (Table 3).

Dose-dependent Gs ConfoSensor (a) or β-arrestin recruitment (b) for agonists pN162, α-MSH, and setmelanotide in the absence (dashed lines) or presence (full lines) of 1 mM Ca2+. c Dose-dependent Gq signaling (production of secondary messenger IP1) of pN162, α-MSH, and setmelanotide. Data are depicted as the mean. of two replicates. Experiments were performed twice, and exemplary graphs are shown. Tables 2 and 3 provide the average EC50s of pN162, α-MSH, and setmelanotide.
Active state MC4R structure with an agonist nanobody
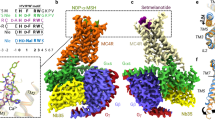
To gain insights in the interaction mode between pN162 and MC4R, a cryo-EM structure was determined of pN162-bound human MC4R in complex with Gs at a nominal map resolution of 3.41 Å. The monodisperse pN162-MC4R-Gαs-Gβ-Gγ-Cb35 complex obtained via an affinity and a size exclusion chromatography (SEC) step remained stable after a freeze-thaw cycle as assessed by analytical SEC (Supplementary Fig. 3). The complex was stabilized by Nb3532,40, also termed Cb3533. The cryo-EM map (Fig. 3a) revealed densities for all complex components, the receptor-G protein heterotrimer association mode, and, critically, the orientation of the CDR3 of the agonistic nanobody pN162 within the orthosteric binding pocket (Fig. 3b). Local densities of transmembrane regions (TMs) allowed to place most sidechains of TM2-7 residues, while TM1 showed the weakest density (Supplementary Fig. 5a). However, after final structure refinement, only the sidechains of residues showing weak or lack of density in protein-protein interfaces were removed to avoid misinterpretation of the interaction among the different subunits (Supplementary Table 1). The quality of fit of CDR3 residues only into the cryoEM density is depicted in Supplementary Fig. 5b, and a close-up view of key interactions between pN162 and MC4R in a map-to-model fit is provided in Supplementary Fig. 7. Although only one of the MC4R-characteristic disulfide bridges (C271-C277) could be identified in the density, the second disulfide bridge between C40-C279 was kept for the purpose of structure refinement (Supplementary Fig. 5c). Finally, Helix 8 had to be partially excluded from the final model due to a lack of density. Other components known for their high flexibility, such as the receptor N- and C-termini, the intracellular loops (ICLs) 1 and 3, and the extracellular loop (ECL) 1, as well as the alpha-helical domain of Gαs, were not or only partially built into the final model (Supplementary Table 1). The core of the agonistic nanobody pN162, including CDR3, showed a local resolution of ~3.5 Å. However, the peripheral portions of the nanobody showed significantly lower resolution (4.5–6.5 Å) which together with the lack of density or lower resolution of the N-terminus, ECLs, and EC portion of the TMs of the receptor allowed only limited analysis of pN162-MC4R interactions beyond CDR3 (see below).

a Cryo-EM density map (sharpened with DeepEMhancer, 8QJ2, and EMD_18442). b Final model submitted to the Protein Data Bank (PDB ID 8QJ2). The color code for the proteins is light blue for pN162, purple for MC4R, salmon pink for DNGαs (Gαs), light green for Gβ, orange for Gγ, and magenta for Cb35.
Superposition of the receptor with previously published MC4R active state structures41 and comparison with its inactive state structure42 indicate a similar activation mechanism upon pN162 binding as described for the peptide or small molecule agonists, as well as an identical conformation revealing key hallmarks of receptor activation (Fig. 4 and Supplementary Fig. 6), namely: (i) outward movement of 13.3 Å of TM6 as measured at the Cα atom M2416.31 (Fig. 4a, c, d; superscript numbers according to the Ballesteros-Weinstein numbering convention43), (ii) the CWxP6.50 toggle switch motif in MC4R, which relies on L1333.36 sitting in the upward conformation and forces W2586.48 (of motif CWxP) to adopt an inward rotation conformation that, along with the rotation of F2546.54 leads to the characteristic TM6 outward movement (Fig. 4e), (iii) the canonical discontinuous M2045.50I1373.40F2546.54 triad (MIF motif) side chain rearrangement (Fig. 4e), (iv) the class A GPCR active state TM7 NPxxY motif (D2987.49P2997.50xxY3027.53 in MC4R), which shows similar rearrangements as in other MC4R active state structures, where Y3027.53 adopts an inwards conformation towards Y2125.58 that helps stabilize the active state orientation of the conserved DRY motif (D1463.49 R1473.50 Y1483.51, Fig. 4f), and (v) the upward conformation of R1473.50 (of the DRY motif) at the top of the G-protein binding vestibule (Fig. 4f).

a, b Side views of the overlay (MC4R receptor only) of the SHU9119-bound (dark blue), inactive state MC4R structure (gray, PDB ID 6W25) with the setmelanotide- (orange, 7PIU) or pN162-bound (cyan, 8QJ2) active state MC4R structures (receptor in pink and purple, respectively). c Bottom view of the same overlay showing the hallmark TM6 outward and the TM5 inward movement upon receptor activation (indicated with arrows). d Top view of the same 6W25-7PIU-8JQ2 overlay. e MIF motif rearrangement and impact on W2586.48 of the CWxP toggle switch motif and L1333.36. f D/NPxxY and ionic lock DRY motif rearrangements upon activation and its impact on residues Y2125.58 and Y3027.53. Side chain rearrangements between inactive and active states are indicated with arrows.
As described previously, MC4R and the Gαs protein interactions occur mainly between TM3, TM5, and ICL2 of the receptor and the α5 helix and Cterm-cap loop of the Gαs protein with TM7. The G-protein interaction crevice in the pN162-MC4R structure mostly overlaps with that of the reported active state MC4R structures and shows the typical class A GPCR activation sidechain re-arrangement in the TM3 DRY motif as compared to the inactive state structure (Fig. 4f and Supplementary Fig. 6a). Comparison of the G-protein binding vestibule of MC4R with those enabled by other agonists (α-MSH-7F53 and setmelanotide-7PIU)41,44 reveals a similar interaction network between MC4R and the Gαs subunit, with minor differences (Fig. 5). While some side chains were not modeled as no clear density was seen to unambiguously assign a conformation, most of the residues previously described to participate in Gαs-coupling were visible, including the additional H-bond between Y391H5.23 (numbering of Gαs according to 8QJ2 and 7F53) and T1503.53, peculiar to MC4R active state35,41,44,45. It is known that ICL2 can adopt agonist specific conformations, and we do observe a similar behavior as the pN162-enabled structure adopts a different conformation than in the α-MSH or setmelanotide-enabled MC4R structures (Fig. 5). The α-MSH-enabled active state structure (Fig. 5a, d) shows interactions of the residue Q35H.N52, A39hns1.03 and H41S1.02 with ICL2, while in the setmelanotide-MC4R active state structure (Fig. 5b, e) R38hns1.02, A39 hns1.03 and H41S1.02 interact with ICL2. In the pN162 structure, due to a different conformation of ICL2 and the N-terminus of Gαs, neither R38hns1.02, A39hns1.03 nor H41S1.02 are within contact distance of MC4R residues. The sidechain of Q35HN.52 could not be modeled due to lack of density. Both previously published α-MSH-(7F53) and setmelanotide-MC4R structures (7PIU) revealed water networks stabilizing the Gαs-MC4R interactions (Fig. 5a, b, d, e). Giving the orientation of involved residues Y157ICL2, H158ICL2, and H387H5.19 in the pN162-MC4R structure it is still expected that water molecules are present to bridge this interaction in the between ICL2 and α5 of Gαs. However, due to the absence of visible density at our map’s lower resolution, this could not be confirmed. Overall, the analysis of the G-protein vestibule shows that pN162 can stabilize the receptor in a conformation like those induced by the previously described peptide agonists while having a different mode of binding in the orthosteric site.

Two different views of the interacting residues between G-protein (green) and MC4R in complex with either α-MSH (a, d, light blue, 7F53), setmelanotide (b, e, pink, 7PIU) or pN162 (c, f, purple, 8QJ2) are shown. Red dots represent water molecules. Black dashed lines indicate H-bond interactions. Where highlighted, individual atom colors follow the standard CPK rules (red = oxygen, blue = nitrogen). All sidechains of residues ≤4 Å distance in the MC4R-Gαs interface are depicted.
Structural insights of the pN162 MC4R interaction vestibule
Orthosteric sites of GPCRs can be buried or obstructed by the presence of large extracellular loops (ECLs). It has been shown that the MC4R orthosteric ligand binding pocket for peptide agonists and antagonists is an open cavity as the ECL2 is only one residue long, in stark contrast with other GPCRs (but observed through the melanocortin family). Comparing the structure of MC4R with pN162 to that of setmelanotide, α-MSH, and antagonist SHU9119 shows that its CDR3 occupies the same site as the endogenous ligand and its cyclic synthetic analogs (Fig. 4a, b, d, Fig. 6 and Supplementary Fig. 6a). All ligands of MC4R described bear the HxRW motif. When looking at the pose and sequence of pN162, no such motif is present. The nanobody activates the receptor by a distinct set of interactions compared to those previously described41,44,45. One of the key residues for receptor activation is the F7 or D-F5 of α-MSH or setmelanotide, respectively. It allows L1333.36 to adopt an upward conformation that forces W2586.48 to pivot and trigger the TM6 outward movement. The F7 or D-F5 pocket is partially filled by the hydrophobic residue V103 of pN162. R104 of pN162 shows a similar interaction network to TM2 and TM7 residues as the histidine residue in setmelanotide and α-MSH belonging to the critical HxRW motif for MC4R activation. Finally, a key component of the MC4R active structures described to date is the presence of a calcium ion in the orthosteric site that functions as a cofactor. It is coordinated by E1002.60, D1223.25, and D1263.29 as well as some backbone residues of the ligands (see PDB deposited structures with accession numbers 7F5341, 6W2542, 7PIU44). The MC4R-pN162 structure reveals that the Ca2+ ion is replaced by R101 and forms a salt bridge interaction with E1002.60 and D1263.29 (Fig. 6g, k, Supplementary Fig. 6b and Supplementary Fig. 7a). Although the sidechain of the third Ca2+ coordination residue described in literature (D1223.25) could not be modeled, the Cβ atom is in proximity (<4 Å) to pN162 CDR1 residue F29 (Supplementary Fig. 7b). Interestingly, in our comparison of the interaction network of pN162 (Supplementary Fig. 9a) with the one of the other agonistic ligands α-MSH (7F53) and setmelanotide (7PIU) (Fig. 6), three MC4R residues appear to exclusively interact with pN162: V1032.63 with CDR1 residue F29 (Supplementary Fig. 7b and Supplementary Fig. 9b), F1844.60 with I102 (Supplementary Fig. 7c and Supplementary Fig. 9b), and Q273ECL3 with framework residue Y37 (Supplementary Fig. 7d and Supplementary Fig. 9b). Of the remaining CDR3 residues, R98 forms a cation-pi stacking with Y2686.58 while P105 is in a hydrophobic patch composed of Y2686.58 and F2616.51 and forming a hydrogen bond through its backbone with H2646.54, all three residues known to interact with setmelanotide and α-MSH (Fig. 6).

a–d Sphere representations of setmelanotide (green, 7PIU), α-MSH (orange, 7F53), pN162 (cyan, 8QJ2), and SHU9119 (light blue, 6W25) in their respective MC4R binding sites (pink, blue, purple and gray, respectively). The horizontal dashed blue line across panels a-d shows the maximum depth reached by all ligands. e–h Side view of the main interactions of each ligand with their respective receptor. Only residue sidechains within 4 Å distance in the respective MC4R-ligand interfaces are shown; dashed lines represent Ca2+ coordination and hydrogen bonds. The blue dashed line represents the same height as in (a–d). The orange shaded circle in (d, h) highlights the deep interaction of D-Nal4 from the antagonist SHU9119 that forces L1333.36 into the inactive state conformation (see active-inactive state overlay Supplementary Fig. 6a). Top view of the key interactions involved in Ca2+ binding (i, j, l) and the position of pN162 CDR3 R101 residue sidechain (k) occupying the Ca2+ space in the binding pocket. For pN162 only CDR3 interactions are shown. For CDR1-TM7 and framework interaction with ECL3, see Supplementary Fig. 7b, d, respectively. Where highlighted, individual atom colors follow the standard CPK rules (red = oxygen, blue = nitrogen, yellow = sulfur).
Extensive experimental evidence (mutagenesis data shown in Supplementary Fig. 8 and the differential effect of Ca2+ on agonist induced signaling pathways (Fig. 2)) supports a distinct receptor activation mechanism by pN162. In the case of pN162, unlike α-MSH and setmelanotide, Ca2+ has no effect on receptor signaling (Fig. 2). Single point mutations in the CDR3 region of pN162 were generated to identify critical CDR3 residues required to induce MC4R signaling. Each residue was mutated into 16 other amino acid (AA) residues, except for M, C, and P which were not assessed (P105 was mutated into all other 17 residues, excluding M and C). Two out of the eleven residues were shown to be key for signaling: R101 and R104. Any mutation evaluated at these positions completely abolished or strongly reduced (≥than 75%) cAMP production compared to the parent pN162 (Supplementary Fig. 8). The strong impact of R101 mutations on signaling again points to the importance of that residue in mimicking Ca2+, required to trigger the activation cascade of the receptor. It is also interesting to note that the R101K mutant is the only one that led to minimal signaling (~10%), underlining the importance of a charged residue in that position for receptor activation. For two other residues (I102 and V103), 13 out of 16 mutations strongly reduced cAMP production compared to the parent pN162. For I102, two semi-conserved mutations to V or L showed little to no impact on signaling (Supplementary Fig. 8), while a mutation of that position to F or W showed ~75% reduced signaling. For V103, mutation to T or I led to a small reduction in signaling, while replacement by a smaller side chain (A, S) had a bigger impact. Larger side chains (F, L, Y, W) completely abolished signaling which might be due to steric hindrance or interaction with L1333.36 in a similar fashion as observed for the antagonist SHU9119 (Supplementary Fig. 8). Although less pronounced compared to R101 or R104, the majority of the R98 mutants show a clear reduction of signaling (lower than 50%; Supplementary Fig. 8). For position P105, only small, hydrophobic side chains permutations are permitted (Supplementary Fig. 8). While the above described pN162 CDR3 AA residues mutations impact receptor signaling, it is not clear whether these mutations still allow binding to or block of the signaling of MC4R, as this was not assessed. For all other residues of pN162 CDR3 (T99, G100, L106, D107, and Y108), the impact of introducing point mutations on signaling is less pronounced. In addition to flow cytrometry (Fig. 1c) and signaling data to melanocortin receptor subtypes (Fig. 1a, f–h), structural data presented here increase our understanding of pN162 specificity: out of 16 MC4R residues identified as interacting with pN162 (Supplementary Fig. 9a, CDR3-only residues, see also Supplementary Fig. 2a), only 5 are fully conserved in the MCxR family (E1002.60, D1263.29, F2616.51, H2646.54and F2847.35, Supplementary Fig. 9b), while the remaining 11 are either partially conserved or non-conserved (T992.56, N972.57, I1293.32, V1032.63, C1303.33, F1844.60, I1854.61, I1945.40, Y2686.58, Q273ECL3 and N2857.36).
Discussion
This study reports the identification of a sequence diverse panel of potent agonistic nanobodies to human MC4R with one of them, pN162, behaving as an MC4R-specific ligand showing full agonist pharmacology. In addition, we provide structural and experimental evidence that reveals how pN162 interacts with the orthosteric extracellular binding pocket and does not require Ca2+ as a cofactor for MC4R signaling. De novo discovery of full agonistic antibodies from immune or synthetic repertoires to class A GPCRs is notoriously difficult. Only one clear example of an antibody with full agonistic pharmacology for a GPCR has been reported in peer-reviewed literature28. The agonistic nanobodies described in this study, including pN162, are not obtained by structure-informed design27 or (repertoire) engineering24,25,26 but are enriched by phage display from an in vivo matured repertoire. The immune repertoires were induced by genetic immunization of camelids with an engineered active state MC4R conformation37,38. The active state conformation was obtained by a genetic fusion of Cb80, a β2AR G-protein mimicking ConfoBody33,35 to the C-terminus of a MC4R chimera. The MC4R chimera was synthesized by grafting the C-terminus and ICLs of β2AR on MC4R. The approach was successful for the de novo identification of large biologics, i.e., agonistic nanobodies towards MC4R. The followed discovery strategy, while independent of purified protein, enabled efficient identification of a large panel of sequence-diverse MC4R-specific, full agonistic nanobodies, some of which had signaling potencies similar to the endogenous agonist α-MSH. CDR3 amino acid sequence variability within the agonistic nanobody panel suggests that different nanobodies can stimulate MC4R signaling through different residue interactions and potentially different binding interfaces. These results strongly suggest that the native extracellular vestibule is preserved in the designed active state MC4R-β2AR chimera-Cb80 fusion. The most potent agonist, nanobody pN162, is the first reported melanocortin receptor subtype ligand that is specific to MC4R. It does not bind to nor induce signaling by any other human melanocortin receptor subtype. Given the absence in pN162 of the conserved MC4R agonist peptide HxRW motif, our structural data reveal a different amino acid repertoire which form an interaction network with MC4R than the one from known peptide agonists. CDR3 residues R101 to R104 and P105 are the critical amino acids responsible for MC4R signaling, experimentally supported by CDR3 mutagenesis analysis. The CDR3 of pN162 occupies the same orthosteric binding pocket as the endogenous ligand α-MSH, setmelanotide and the antagonist SHU9119. The pN162-MC4R structure shows, despite the reduced resolution in the peripheral areas of the active state complex, pN162-only interactions with topological domains of MC4R: V1032.63, F1844.60 and Q273ECL3. These three residues belong to the non- or partially conserved interaction network among MCxR family members as described above and are therefore contributing to MC4R specificity. Additional MC4R residues in less or non-conserved regions (ECLs or N-terminus) that are not resolved in the structure likely contribute further to pN162’s specificity for MC4R, although additional supporting evidence (e.g., mutational analysis) beyond structure is lacking. Altogether, these data provide structural evidence for a distinct mode of interaction of an agonistic antibody with a GPCR.
Similar to α-MSH and setmelanotide, pN162 activates multiple MC4R signaling pathways including the G-protein and β-arrestin pathways. Compared to setmelanotide, α-MSH shows a 7.0-fold reduced Gs signaling potency (Table 1). α-MSH however is 43.3-fold less potent in stimulating the Gq pathway (Table 3). Similar potency reductions were reported for α-MSH compared to setmelanotide: 5.9- (Gs pathway) and 81.3-fold (Gq pathway)1. Our results show similarly reduced signaling potencies of pN162 compared to setmelanotide in Gs (6.6-fold; cAMP GloSensor) and Gq (12.5-fold; IP1 HTRF) signaling, suggesting a similar pathway activation profile for pN162 and setmelanotide. Despite the use of a ConfoBody that mimics the G-protein of a GPCR primarily signaling via Gs (β2AR) for active state stabilization of the chimera, the use of such active state stabilized chimera enables the identification of an agonistic antibody fragment that also signals via Gq.
The pN162-MC4R-Gs-Cb35 complex structure shows the canonical active state structural hallmarks: a TM6 outward movement, the presence of the “active-like” toggle switch, and canonical arrangements of the MIF, NPxxY, and DRY motifs, equivalent to those obtained in complex with all other reported agonist peptide or small molecule MC4R active state structures (α-MSH41, setmelanotide44,45, NDP-α-MSH44, afamelanotide41, bremelanotide41, THIQ41). The pN162-MC4R model provides a possible explanation for how pN162 stabilizes the active but not the inactive MC4R conformation. For example, in contrast to MC4R antagonist SHU9119, pN162 has no direct interaction with MC4R’s L1333.36, the switch that controls receptor conformation (by interacting with W2586.48). The unnatural alanine analog D-naphthylalanine (D-Nal4) of SHU9119 does interact with L1333.36 and prevents the formation of the active conformational state of the receptor. Furthermore, in contrast to all described MC4R peptide and small molecule agonists, pN162-induced MC4R signaling is independent of Ca2+ (Fig. 2). The Ca2+-independent pN162 MC4R signaling is supported by the structure model of the pN162-MC4R interaction (Fig. 6g, e, f and Supplementary Fig. 6). In previously published MC4R structures the Ca2+ ion is coordinated by E1002.60, D1223.25 and D1263.29 in structure 7F53 (α-MSH) and 6W25 (SHU9119), and by E1002.60 and D1263.29 in structure 7PIU (setmelanotide; Fig. 6). In the pN162-MC4R density map, the Ca2+ space is occupied by the sidechain of pN162 CDR3 residue R101 by making salt-bridge interactions with MC4R residues E1002.60 and D1263.29 (Supplementary Fig. 6b). Since the sidechain of D1223.25 could not be modeled, we could not determine its potential interaction with pN162.
Despite orthosteric binding site homology between melanocortin receptor family members, agonistic nanobody pN162 only interacts with and induces signaling of MC4R. The scientific interest of such MC4R-specific agonistic nanobodies that bind the orthosteric pocket (confirmed by structural and radioligand displacement data) goes beyond their use as chaperone for structure determination. pN162 can be used to deconvolute the specific role of MC4R in the melanocortin receptor signaling pathways that interfere with satiety and energy homeostasis. Knowing that selective MC4R agonists oftentimes lead to unwanted adverse effects in the clinic, such an MC4R-specific antibody fragment can be further explored to understand the contribution of MC4R stimulation to the in vivo efficacy of selective MC4R agonists for treatment of (genetic) obesity and might open novel therapeutic avenues. In case such studies motivate clinical development of MC4R-specific agonistic ligands, like nanobody pN162, for monogenic anti-obesity therapeutic intervention, the pN162-MC4R active state structure can provide the basis for structure-based strategies to further improve pN162 signaling potency.
Methods
Our research complies with all relevant ethical regulations.
Mammalian cell culturing and cell transformation
HEK293T (ATCC CRL-3216) cells were routinely maintained at 37 °C and 5% CO2, under humidified atmosphere in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS). To prepare transient transfection-ready HEK293T, cells were split and grown to confluency levels of up to 70%. Transient transfections were performed with XtremeGene HP (Roche) according to the manufacturer’s instructions. CHO-K1 cells (ATCC CCL-61) were maintained under the same conditions but cultured in Ham’s F-12K medium (Thermo Fisher Scientific). The stable CHO-K1 cell line expressing Flag-tagged MC4R chimera-Cb80 was cultured in HamF12/DMEM (1:1) supplemented with 5% FBS, 8 mM Glu, and under 0.5 mg/ml G418 geneticin selection.
Detection antibodies and MC4R ligands
For primary and secondary detection in flow cytometry, anti-cMyc antibody 9E10 (Thermo Fisher Scientific, MA1-980), monoclonal anti-Flag M2 antibody (Sigma Aldrich, F3165), and goat anti-mouse IgG phycoerythrin (PE) conjugate antibody (Jackson ImmunoResearch, 115-115-164) were used. TO-PRO™−3 Iodide dye to discriminate permeable from intact cell populations was supplied by Thermo Fisher Scientific. MC4R agonist peptides α-MSH and setmelanotide (RM-493) were purchased from Tocris and ChemScene, respectively. I-SHU9119 radioligand was purchased from Perkin Elmer.
Constructs for nanobody discovery
To induce a humoral seroconversion in camelids by genetic vaccination, a mammalian expression construct was used that expresses active state human MC4R (UniprotKB P32245)37. Supplementary Table 2 provides an overview of the melanocortin receptor expression cassettes and their application. To recombinantly express active state MC4R, a GPCR chimera was designed: the human β2AR intracellular loops (ICLs) were grafted on the extracellular loops (ECLs) and transmembrane α-helical domains (TMs) of human MC4R. In addition, the MC4R chimera was fused at its C-terminus to β2AR active state stabilizing Cb8031,35,37 separated by a flexible GS linker (GSL) and additional His8-Rho1D4 tags. For translocation to and cell surface detection, a hemagglutinin signal sequence (SPHA) and Flag tag encoding sequence was added at the N-terminus of MC4R resulting in SPHA-Flag-hMC4R-hβ2AR chimera-GSL-Cb80-His8-Rho1D437. To generate custom virus-like particles (Integral Molecular), the active state MC4R chimera expression cassette was used for immunization and a similar cassette containing tagged wild type full length human MC4R (V5-hMC4R-His6) were used. To assess binding of monoclonal nanobody to MC4R via homogenous time resolved fluorescence, a SNAP-tagged active state human MC4R chimera was generated in pcDNA3.1(+) by inserting a SNAP tag after the Flag tag sequence of the immunization construct resulting in SPHA-Flag-SNAP-hMC4R-hβ2AR chimera-GSL-Cb80-His8-Rho1D4. To assess competition of monoclonal nanobody with radioligand to MC4R chimera, the expression cassette of the immunization construct was used to generate a custom stable CHO-K1 cell line (InSCREENex GmbH). To assess binding of monoclonal nanobody to human melanocortin receptor subtypes MC1R to MC5R by flow cytometry, SPHA-Flag-MCxR expression constructs cloned in pcDNA3.1(+) were used for transient transfection. For the MC1R and MC5R expression constructs, a C-terminal LgBiT tag was present. To assess binding to MC2R, transient transfection was performed in absence and presence of human MRAPα (UniprotKB Q8TCY5-1). The nucleotide sequences that encode for the human melanocortin receptors were derived from the Uniprot database: MC4R (UniprotKB P32245), MC1R (UniprotKB Q01726), MC2R (UniprotKB Q01718) MC3R (UniprotKB P41968) and MC5R (UniprotKB P33032). To monitor ligand induced recruitment of the active state stabilizing Cb80 to the hβ2AR intracellular domains of the MC4R chimera, Cb80 was genetically fused at its C-terminus to the luciferase SmBiT, while the luciferase complementary LgBiT domain was fused at the C-terminus of the MC4R chimera39 and cloned in pCDNA3.1(+) or pBiT vector. To assess ligand induced recruitment of the G-protein to human MC4R chimera, Cb35 was genetically fused at its C-terminus to SmBiT, while the LgBiT domain was fused at the C-terminus of MC4R and cloned in pBiT. To assess ligand induced recruitment of β-arrestin to human MC4R chimera, β-arrestin2 was genetically fused at its N-terminus to SmBiT and cloned in pBiT. To monitor cAMP accumulation, a custom human MC4R inducible stable HEK293T cell line (Theranyx) harboring an expression cassette 2xTwinStrep-hMC4R (AA 1-332)-TEV-GFP-His10 was used for Lance Ultra HTRF. To monitor cAMP signaling by GloSensor or IP1 production by HTRF, Flag-tagged human MC4R and human MC receptor paralogs MC1R, MC3R, and MC5R of the format SPHA-Flag-hMC4R (AA 2-end) were cloned in pcDNA3.1(+).
Llama immunization and nanobody repertoire cloning
To generate an immune response to active state human MC4R, llamas were genetically vaccinated with MC4R chimera. The vaccination experiment was executed in accordance with the European legislation (EU directive 2010/63/EC) and the Belgian Royal Decree of 29 May 2013 concerning the protection of laboratory animals (with the exception that the animals are purpose-bred). All llamas are housed in a licensed Belgian facility (accreditation number LA 1700601) staffed with appropriately trained veterinaries and animal caretakers. All animals experiments have been approved by the authorized local Animal Ethics Review board (study protocol number ECD 15-xxx-1). Animals were administered with cDNA immunogen immediately followed by an in vivo electroporation46. The in vivo matured nanobody repertoire was cloned in a phagemid vector allowing protein III phage display and expression of soluble nanobody as C-terminally His6-c-Myc tagged recombinant protein47. Custom produced virus-like particles presenting MC4R chimera-Cb80 fusions (Integral Molecular Inc.) were used for panning to enrich for MC4R binders according to standard methods47. Monoclonal nanobodies picked as E. coli TG1 re-infected phage outputs were cultured in 96-well deepwell plates (Thermo Fisher Scientific) to prepare periplasmic extracts47 for further characterization.
Nanobody sequence determination
The nucleotide sequence of a nanobody was determined via Sanger sequencing applying nanobody specific sequencing primers as described47. CDRs were delineated according to the AbM annotation48. Nanobodies were clustered based on amino acid CDR3 similarity (identical length and 85% sequence identity) using Qiagen’s CLC Main Workbench version 23.0.2 or PipeBio software suite (PipeBio). Cluster representative nanobody clones were further characterized for MC4R specificity, receptor subtype and species cross-reactivity and pharmacology.
Cell binding assays—homogeneous time resolved fluorescence (HTRF), flow cytometry, and radioligand binding
Binding of monoclonal nanobodies as E. coli periplasmic extracts to transiently transfected HEK293T cells expressing SNAP tagged MC4R chimera was assessed by HTRF in white 384-well Optiplates (Perkin Elmer). The SNAP tag was Terbium (Tb)-labeled by incubating MC4R transfected cells with 50 nM SNAP Lumi4-Terbium substrate in TagLite buffer (Cisbio) for 1 h at 37 °C, 5% CO2. Excess of label was removed with cell washing buffer (20 mM Hepes, 100 mM NaCl, 3 mM MgCl2, 0.2% BSA). Harvested cells were washed with cell washing buffer and stored at −80 °C till further use. Five thousand cells were incubated with 5-fold diluted periplasmic extract and 50 µg/mL anti-cMyc-d2 acceptor (Cisbio) in TagLite buffer supplemented with 0.2% BSA. After 1h30 incubation at room temperature (RT), HTRF ratios were detected at 665 and 620 nm on an Infinite M1000 plate reader (Tecan). Normalized HTRF ratios (665 nm/620 nm × 1E + 04) were analyzed using GraphPad Prism 8.0. Nanobody clones with a normalized HTRF ratio ≥ 2 over a mock nanobody were retained for further analysis. To assess binding of pN162 to melanocortin receptor subtypes (MC1-5R), binding of purified pN162 to transiently transfected HEK293T cells or mock GPCR transfected HEK293T cells was assessed via flow cytometry. All incubations and wash steps were performed on ice or at 4 °C in FACS buffer (PBS supplemented with 10% FBS) unless otherwise mentioned. Cells were washed in FACS buffer and resuspended at a density of 1E + 06 cells/mL. 1E + 05 cells were transferred to 96-well V-bottom culture plates (Greiner). After an additional wash step, cells were resuspended and dilutions of purified c-Myc-tagged pN162 were added. After at least 30 min of incubation, cells were washed and incubated with 5 µg/mL µg of primary anti-cMyc 9E10 detection antibody. After 1 h of incubation, cells were washed twice and incubated for 30 min in the dark with 2.5 µg/mL µg PE-labeled goat anti-mouse IgG secondary detection antibody. Immediately before data acquisition, cells were washed and resuspended in 1 µM of Topro-3 supplemented FACS buffer to gate intact from permeable cell populations. As a control for cell surface expression of Flag-tagged MC4R and mock GPCR, cells were stained with 5 µg/mL mouse anti-Flag M2 antibody followed by PE-labeled goat anti-mouse IgG secondary detection antibody. The cell fluorescence of minimally 5E + 03 gated events was measured using a MACSQuantX cytometer (Miltenyi Biotec) and data were analyzed using Flowlogic software (Miltenyi Biotec). To understand whether pN162 binds a similar epitope as orthosteric MC4R ligands, an antagonist radioligand displacement assay was performed with SHU9199. Membranes were prepared from hMC4R-β2AR-Cb80 CHO cells. Membrane extracts were resuspended in 20 mM Hepes HCl pH7.4, 0.1 mM MgSO4, and 1 mM CaCl2 supplemented with 0.2% Bovine Serum Albumin (BSA) as binding buffer. A final concentration of 0.25 nM [125I] labeled antagonist SHU9119 was added to the membranes together with a dilution series of purified pN162 or controls and the reaction mix was incubated for 1 h at room temperature. Membrane bound [125I]-SHU9119 was separated from unbound radioligand on a 96-well FilterMate harvester (Perkin Elmer) by passing all samples over a Whatman GF/C filter. Filters were then washed with cold 25 mM Hepes pH 7.4, 0.1 mM MgSO4, and 1 mM CaCl2 buffer and dried for 1 h at 50 °C. Radioligand binding was measured after adding MicroScint™-O scintillation fluid in a Wallac MicroBeta TriLux scintillation counter. The affinity (IC50) of the ligand was determined by nonlinear regression analysis and fitting for a competitive binding model using GraphPad Prism 8.0.
ConfoSensor and β-arrestin recruitment assays
A luminescence-based biosensor assay using a conformation sensing ConfoBody39 was deployed based on Promega’s NanoBiT technology (principle described in Supplementary Fig. 10a). NanoBiT luciferase complementation occurred between the MC4R-β2AR chimera fused at its C-terminus to the large fragment of Nanoluciferase (LgBiT) and β2AR active state G-protein mimetic antibody Cb80 C-terminally fused to Nanoluciferase small fragment (SmBiT)38. An alternative ConfoBody recruitment assay was established that assessed luciferase complementation between the human WT MC4R fused at its C-terminus to LgBiT and G-protein stabilizing Cb35 C-terminally fused to SmBiT39 (principle depicted in Supplementary Fig. 10b). When a ligand favorizes the receptor chimera’s active state, the ConfoBody is recruited and upon substrate addition, the kinetics of a luminescence signal is measured. 1E + 06 adherent cells HEK293T cells were transfected with the constructs encoding the NanoBiT complementation partners. Twenty-four hours post transfection, cells were harvested, washed and resuspended in OptiMEM medium (Gibco). 1,50E + 04 transfected cells were distributed into the wells of half area 96-well white plates (Greiner) and were incubated for 1 h at 37 °C. After adding Nano-Glo Live Cell reagent (Promega), background luminescence level was recorded after 30 min at RT using the EnVision 2104 Multilabel Plate Reader (Perkin Elmer). Following addition of control ligands, periplasmic extracts harboring nanobodies or purified nanobodies (diluted in OptiMEM supplemented with Tween-20 at a final concentration of 0.0016%), luminescence was recorded at 30 min and 60 min at RT (Supplementary Fig. 10c). Data were analyzed using GraphPad Prism 8.0 by applying nonlinear regression (curve fit). For the β-arrestin recruitment assay, a similar procedure was followed as for Cb80 recruitment, except that SmBiT-β-arrestin2 was used instead of Cb80-SmBiT. For both the ConfoSensor and β-arrestin recruitment assays, exogenous Ca2+ at a final concentration of 1 mM was added in all assay buffers when appropriate.
cAMP detection assays
To monitor ligand induced melanocortin receptor Gs signaling, production of secondary messenger cAMP was quantified via two assays: Promega’s GloSensor cAMP assay or the LANCE Ultra cAMP detection kit (Perkin Elmer). For GloSensor, HEK293T cells were co-transfected with pGloSensor-22F plasmid and melanocortin receptor and seeded in poly-L-lysine coated 96-well white plates (Corning). Twenty-four hours post transfection, cell culture medium was replaced with OptiMEM supplemented with 10% FBS and 2% GloSensor cAMP reagent (Promega). For clarity, the Ca2+ concentration in OptiMEM is 0.89 mM. Cells were incubated for 2 h at room temperature. Background luminescence was recorded using the EnVision 2104 Multilabel Plate Reader (Perkin Elmer). After addition of (dilution series) of control ligands or purified nanobody in OptiMEM supplemented with 10% FBS and 2% GloSensor cAMP reagent, luminescence was immediately recorded in a kinetic mode on the EnVision. Data were analyzed using Prism 9.5.0 by applying nonlinear regression (curve fit). For LANCE Ultra cAMP, the assay was executed in conditions without calcium ions according to manufacturer’s instructions with minor modifications. In brief, stable MC4R inducible HEK293T cells were pelleted, washed in stimulation buffer (Hank’s balanced salt solution (HBSS) without Ca2+ and Mg2+ supplemented with 20 mM Hepes pH7.4) and resuspended in stimulation buffer additionally supplemented with 0.5 mM IBMX. 1E + 04 cells/well were seeded in half area 96-well plates and incubated for 30 min at 37 °C. Serial dilutions of test samples in cAMP detection buffer were added and allowed to incubate for 1 h at room temperature. After addition of Eu-cAMP and ULight-anti-cAMP detection reagents according to manufacturer’s instructions and another incubation for 1 h at room temperature, TR-FRET emission signal at 665 nm was recorded with the EnVision. Graphs were prepared in Graphpad Prism 8.0 by applying nonlinear regression (curve fit).
IP1 detection assay
To monitor ligand induced melanocortin receptor Gq signaling, production of secondary messenger IP1 was quantified via the IPOne Gq HTRF kit (Cisbio). Transient human MC4R transfected HEK293T cells were pelleted, washed and resuspended in stimulation buffer (LANCE Ultra cAMP assay) supplemented with 0.1% BSA according to the manufacturer’s instructions. 2,5E + 04 cells/well were seeded in 384-well Optiplates (Revvity). Serial dilutions of test samples in stimulation buffer were added and allowed to incubate for 2 h at 37 °C. After addition of IP1 d2 reagent Antibody and IP1 Tb Cryptate Antibody according to manufacturer’s instruction and another incubation for 1 h at room temperature, TR-FRET emission signal at 665 nm and 620 nm were recorded with the EnVision. Graphs were prepared in Graphpad Prism 8.0 by applying nonlinear regression (curve fit).
Nanobody purification
cMyc-His6 tagged pN162 and His6-EPEA tagged Cb3532,40 were expressed as described47 in the periplasm of E. coli strain Rosetta (DE3). In brief, cells were grown at 37 °C in Terrific Broth medium supplemented with 0.1% glucose and 100 µg/ml carbenicillin till OD600nm 0.5 – 1.0. For induction, IPTG was added to a final concentration of 1 mM and cells were incubated for another 4 h at 37 °C or overnight at 28 °C. After centrifugation, cells were frozen at −20 °C. Periplasmic extract was made by thawing the frozen cell pellet in PBS and subsequent head-over-head incubation for 1 h at 4 °C. After centrifugation at 20,000 g for 10 min at 4 °C, the supernatant was filtered through a 0.45 µm filter (Sartorius). The nanobody was purified by immobilized metal affinity chromatography (IMAC), using Talon beads (Clontech) according to the manufacturer’s protocol. Optionally, a polishing step by size exclusion to remove contaminants was performed. The buffer switch of the purified protein to PBS was done with PD-10 desalting columns (Cytiva) according to manufacturer’s instructions and spin concentrated. For all nanobody productions, the purity was ≥ 95% as judged by SDS-PAGE and subsequent Coomassie staining. For 96-well scale nanobody purifications (to assess MC4R signaling potency of single amino acid pN162 CDR3 mutants), a single chromatography purification step was performed on E. coli periplasmic extract using PhyTip columns containing IMAC resin (Biotage) according to the manufacturer’s instructions.
Expression of recombinant MC4R and Gs protein
For cryo-EM studies, the full-length human wild-type MC4R (UniprotKB P32245) was cloned into pFastBac1 vector. At the N-terminus, the GPCR was genetically fused to the hemagglutinin signal peptide (SPHA) followed by the Flag tag and thermostabilized E. coli apocytochrome b562 (BRIL) separated by a 3 C PreScission protease cleavage sequence. At the C-terminus, MC4R was followed by the 3C sequence, mCherry, His10, the Avi and EPEA tags. To prepare recombinant baculovirus, Sf9 cells were cultured in ESF921 medium (Expression Systems) at 27 °C according to standard protocols (Bac-to-Bac Baculovirus expression system, Thermo Fisher Scientific). To generate MC4R expressing cells, 1 L Trichoplusia ni (Tni) cell cultures were infected at starting density of 4E + 06/mL of 500-fold diluted P2 baculovirus. Cell cultures were grown for 72 h, harvested by centrifugation, and stored at −80 °C for further use. To express the heterotrimeric Gs protein, recombinant baculovirus prepared from two separate constructs was used. The dominant negative human Gαs (DNGs) subunits carrying the 8 mutations to enhance GPCR active state complexes49 was cloned into the pFastBac1 vector. The N-terminally His6-tagged Gβ1 and Gγ2 subunits were cloned in opposite orientations into the pFastBac Dual vector (Thermo Fisher Scientific). Following virus generation DNGs and Gβ1-Gγ2 were co-expressed by addition of an optimized P2 virus ratio to Tni cell cultures sat a starting density of 2E6/mL. After 72 h of incubation, cells were harvested by centrifugation and stored at −80 °C for further use.
Purification of active state pN162-MC4R-Gs-Cb35 complex
All steps were performed at 4 °C unless explicitly stated. To purify the complex, a lysate of Tni insect cell pellets expressing MC4R (1 L) and DNGs-Gβ1-Gγ2 (5 L) was prepared by douncing in buffer A (50 mM Hepes pH 7.4, 150 mM NaCl, 2 mM MgCl2, 1% LMNG, 0.06% CHS, benzonase, EDTA free Complete protease inhibitor (Roche) and apyrase). In addition, structural chaperone nanobodies were added: 1 µM pN162 and 6 mg Cb35. The lysate was stirred for 1 h at room temperature and centrifuged at 125,000 × g for 30 mins at 4 °C. The supernatant was incubated with Flag affinity resin (Thermo Scientific Pierce) for 1 h at RT and transferred to an Econo-Column (BioRad). The column was extensively washed with buffer B (50 mM Hepes pH 7.4, 150 mM NaCl, 2 mM MgCl2, 0.01% LMNG, 0.006% CHS, and 100 nM pN162) before eluting MC4R-pN162 active state complex in buffer C (buffer B supplemented with 0.5 mg/mL Flag peptide). The collected fractions were concentrated using a 100 kDa molecular weight cut-off (MWCO) concentrator (Millipore) and the solution was passed through a 0.2 µm filter. To purify the monodisperse complex, subsequent polishing was performed by size exclusion chromatography (SEC) in buffer B on a Superose 6 Increase 10/300 GL column (Cytiva). Fractions that contained the complex were collected, concentrated with a 100 kDa MWCO concentrator to 10.4 mg/mL, and snap-frozen in liquid nitrogen.
Cryo-EM sample preparation, data acquisition, and processing
Snap frozen purified pN162-MC4R-Gs-Cb35 protein complex was thawed and 3 µL sample was applied to glow-discharged Quantifoil r1.2/1.3 Cu 200 mesh grids for 15 s and then blotted away for 10–14 s before plunge-freezing in liquid ethane. Plunge-freezing was performed at 4 °C and 100% humidity using a Vitrobot Mark IV (Thermo Fisher Scientific). Cryo-EM data were collected on a 200 kV Glacios 1 instrument equipped with a Falcon 4 electron detector. Images were recorded at nominal magnification of 150,000× corresponding to a pixel size of 0.95 Å/pix and then re-scaled to 1.425 Å/pix for data processing. In total, 19,531 movies were recorded and processed using cryoSPARC v4.3.1 (Supplementary Fig. 4). In short, these movies were motion corrected and dose-weighted using Patch Motion Correction job type. The contrast transfer function (CTF) parameters were estimated using Patch CTF job type. Micrographs were curated based on their maximum resolution estimation resulting in 18,738 movie selection. These images were subjected to template-based particle picking and 29,039,961 particles were picked. Two rounds of 2D classification were performed and 2D classes showing GPCR-G-protein complex features were selected resulting in a subset of 4,103,134 particles. These particles underwent multiple rounds of heterogeneous refinement, ab initio modeling, and 2D classification. This process led to selection of a subset of 220 k particles which were refined to a 3.42 Å resolution using Non-uniform Refinement job type. A final sharpening step using DeepEMhancer50 was added to improve density connectivity especially in the periphery of the active state structure model.
Model building and refinement
The initial model docked into the electron density map with Phenix software51 version 1.20.1-4487 was the structure of α-MSH-bound MC4R-Gαs-Gß-Gγ-Cb35 (PDB 7F53) and included an initial rigid-body refinement and simulated annealing. The pN162 nanobody search model was built by homology by docking the backbone of Cb35 from the GαsGβGγ–Cb35 complex of the β2-adrenergic receptor–Gs structure40 (PDB ID 3SN6) into the remaining density, with the previously docked MC4R-G-protein heterotrimer as fixed model. Initial placing of the entire complex was followed by iterative manual building and local refinement in Coot52 version 0.9.6.2-pre, and validation and global real space refinement with Phenix. After every round of manual refinement and for the final round, Real-space refinement was performed with Phenix using geometric restraints, a global minimalization protocol, and B-factor refinement (rotamer restraints; Ramachandran restraints; NCS restraints) to evaluate overall geometry. Secondary structure restraints for helices, beta-sheets, and disulfide bonds were used based on the high-resolution structures 7F53 and 3SN6. The final refinement parameters are provided in Supplementary Table 3. The residues which could not be modeled due to lack of density and the sidechains not modeled to avoid misinterpretation of the pN162-MC4R interactions are listed in Supplementary Table 1. Final structure validation was performed with Phenix and wwPDB validation service. Molecular graphics representations in this work were created using USCF ChimeraX version 1.6.1, PyMol Molecular Graphics System Version 1.3 (Schrödinger, LLC, New York, NY), and Maestro (Schrödinger, LLC, New York, NY). To allow further analysis of the binding epitope, placement of side chains and short minimization of the Cryo-EM structure of MC4R with pN162 was performed using Schrödinger’s protein preparation tools as well as Prime energy minimization software for the minimization of the structure in the presence of an implicit membrane (Software release 2022-3).
Statistical analysis
Statistical analysis of data sets was generated via Prism 9.5.0 (Graphpad) and presents the mean ± s.e.m. from repeated experiments with the number of repeats provided in the (Supplementary) Figures or Tables. Dose dependent graphs were calculated with Prism 9.5.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The model coordinates of the pN162 active state MC4R structure is deposited in the Protein Data Bank under accession number 8QJ2. Source data are provided with this paper.
References
Clément, K. et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med. 24, 551–555 (2018).
Kühnen, P. et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N. Engl. J. Med. 375, 240–246 (2016).
Lotta, L. A. et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell 177, 597–607 (2019).
Mountjoy, K. G. et al. The cloning of a family of genes that encode the melanocortin receptors. Science 257, 1248–1251 (1992).
Maaser, C. et al. Role of the melanocortin system in inflammation. Ann. N. Y. Acad. Sci. 1072, 123–134 (2006). 2006.
Chida, D. et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc. Natl Acad. Sci. USA 104, 18205–18210 (2007).
Yanik, T. & Durhan, S. T. Specific functions of melanocortin 3 receptor (MC3R). J. Clin. Res. Pediatr. Endocrinol. 15, 1–6 (2023).
Cone, R. D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 8, 571–578 (2005).
Gautron, L., Elmquist, J. K. & Williams, K. W. Neural control of energy balance: translating circuits to therapies. Cell 161, 133–145 (2015).
Xu, Y., Guan, X., Zhou, R. & Gong, R. Melanocortin 5 receptor signaling pathway in health and disease. Cell. Mol. life Sci. 77, 3831–3840 (2020).
Kühnen, P., Krude, H. & Biebermann, H. Melanocortin-4 receptor signaling: importance for weight regulation and obesity treatment. Trends Mol. Med. 25, 136–148 (2019).
Ghamari-Langroudi, M. et al. G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature 520, 94–98 (2015).
Collet, T. H. et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab. 6, 1321–1329 (2017).
Yeo, G. S. H. et al. The melanocortin pathway and energy homeostasis: from discovery to obesity therapy. Mol. Metab. 48, 101206 (2021).
Liu, Z. & Hruby, V. J. MC4R biased signaling and the conformational basis of biological function selections. J. Cell. Mol. Med. 26, 4125–4136 (2022).
Clément, K. et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 8, 960–970 (2020).
Kanti, V. et al. A melanocortin-4 receptor agonist induces skin and hair pigmentation in patients with monogenic mutations in the leptin-melanocortin pathway. Ski. Pharmacol. Physiol. 34, 307–316 (2021).
Hutchings, C. J. A review of antibody-based therapeutics targeting G protein-coupled receptors: an update. Expert Opin. Biol. Ther. 20, 925–935 (2020).
Kruse, A. C. et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013).
Scholler, P. et al. Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat. Commun. 8, 1967 (2017).
Koehl, A. et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84 (2019).
Hong, C. et al. Structures of active-state orexin receptor 2 rationalize peptide and small-molecule agonist recognition and receptor activation. Nat. Commun. 12, 815 (2021).
Toyoda, Y. et al. Structural basis of α1A-adrenergic receptor activation and recognition by an extracellular nanobody. Nat. Commun. 14, 3655 (2023).
Cheloha, R. W. et al. Improved GPCR ligands from nanobody tethering. Nat. Commun. 11, 2087 (2020).
Pan, H. et al. Everestmab, a novel long-acting GLP-1/anti GLP-1R nanobody fusion protein, exerts potent anti-diabetic effects. Artif. Cells Nanomed. Biotechnol. 48, 854–866 (2020).
Liu, Q. et al. Functional GLP-1R antibodies identified from a synthetic GPCR-focused library demonstrate potent blood glucose control. mAbs 13, 1893425 (2021).
Ma, Y. et al. Structure-guided discovery of a single-domain antibody agonist against human apelin receptor. Sci. Adv. 6, eaax7379 (2020).
Hutchings, C. J. et al. Monoclonal anti-β1-adrenergic receptor antibodies activate G protein signaling in the absence of β-arrestin recruitment. mAbs 6, 246–261 (2014).
Trilleaud, C. et al. Agonist anti-ChemR23 mAb reduces tissue neutrophil accumulation and triggers chronic inflammation resolution. Sci. Adv. 7, eabd1453 (2021).
Ren, H. et al. Function-based high-throughput screening for antibody antagonists and agonists against G protein-coupled receptors. Commun. Biol. 3, 146 (2020).
Steyaert, J. et al. Protein binding domains stabilizing functional conformational states of GPCRs and uses thereof. International Patent Application WO 2012/007593 filed on July 18, 2011, and published on January 19, 2021; applicants: vib vzw; Vrije Universiteit Brussel; the board of trustees of the Leland Stanford Junior University; the regents of the University of Michigan.
Steyaert, J. et al. Binding domains directed against GPCR: G protein complexes and uses derived thereof. International Patent Application WO 2012/175643 filed on June 21, 2012, and published on December 27, 2012; applicants: vib vzw; Vrije Universiteit Brussel; the board of trustees of the leland Stanford junior University; the regents of the University of Michigan.
Laeremans, T. et al. Accelerating GPCR drug discovery with conformation-stabilizing VHHs. Front. Mol. Biosci. 9, 863099 (2022).
De Genst, E. et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl Acad. Sci. USA 103, 4586–4591 (2006).
Rasmussen, S. G. et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469, 175–180 (2011).
Steyaert, J., Laeremans, T. & Pardon, E. Novel chimeric polypeptides for screening and drug discovery purposes. International Patent Application WO 2014/118297 filed on January 30, 2014, and published on August 7, 2014; applicants: vib vzw; Vrije Universiteit Brussel.
Laeremans, T. Methods for generating antibodies and antibody fragments and libraries comprising same. International Patent Application by Confo Therapeutics WO 2021/140205 filed on January 8, 2021, and published on July 15, 2021; Applicants: Confo Therapeutics.
De Blieck, A., Claes, P., Ververken, C., Menet, C., & Dekeyzer, L. Chimeric proteins and methods to screen for compounds and ligands binding to GPCRs. International Patent Application WO WO2020/221768 filed on April 28, 2020, and published on November 5, 2020; Applicant: Confo Therapeutics.
Menet, C., Dekeyzer, L., Martini, M. & Skieterska, K. Screening methods and assays for use with transmembrane proteins, in particular with GPCRs. International Patent Application WO WO2020/221769 filed on April 28, 2020, and published on November 5, 2020; Applicant: Confo Therapuetics.
Rasmussen, S. G. et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Zhang, H. et al. Structural insights into ligand recognition and activation of the melanocortin-4 receptor. Cell Res. 31, 1163–1175 (2021).
Yu, J. et al. Determination of the melanocortin-4 receptor structure identifies Ca2+ as a cofactor for ligand binding. Science 368, 428–433 (2020).
Ballesteros, J. A. & Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 25, 366–428 (1995).
Heyder, N. A. et al. Structures of active melanocortin-4 receptor-Gs-protein complexes with NDP-α-MSH and setmelanotide. Cell Res. 31, 1176–1189 (2021).
Israeli, H. et al. Structure reveals the activation mechanism of the MC4 receptor to initiate satiation signaling. Science 372, 808–814 (2021).
van der Woning, B. et al. DNA immunization combined with scFv phage display identifies antagonistic GCGR specific antibodies and reveals new epitopes on the small extracellular loops. mAbs 8, 1126–1135 (2016).
Pardon, E. et al. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 9, 674–693 (2014).
Abhinandan, K. R. & Martin, A. C. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol. Immunol. 45, 3832–3839 (2008).
Liang, Y. L. et al. Dominant negative G proteins enhance formation and purification of agonist-GPCR-G protein complexes for structure determination. ACS Pharmacol. Transl. Sci. 1, 12–20 (2018).
Sanchez-García, R. et al. DeepEMhancer: a deep learning solution for Cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Acknowledgements
Part of this work has been performed with support of the Flemish government “Agentschap Innoveren en Ondernemen” Vlaio [HBC_2020.2042] and the Brussels capital region government Innoviris [2016-R-49]. We thank Jimmy Ollivain for his assistance in cell culturing, Mariska Reniers for her assistance with signaling assays, and Erik Soons for the execution of the HTRF nanobody binding experiments. Nanobody and Nanobodies are trademarks for Ablynx N.V. ConfoBody and ConfoBodies are registered trademarks of Confo Therapeutics N.V.
Author information
Authors and Affiliations
Contributions
T.F. executed the nanobody discovery and in vitro characterization under supervision of S.T. and T.L. who designed the discovery strategy; N.S. designed the expression constructs, V.-P.J. and Y.-L.L. designed the protein generation for structural biology and supervised the cryo-EM part. R.V. assisted in cell pellet production and Y.-L.L. purified the complex; S.M. performed the cryo-EM data collection and processed the data to obtain the 3D electron density map; V.-P.J. and A.B. built and refined the model and reviewed the manuscript; N.L. purified the nanobodies; L.D. supervised the signaling experiments performed by C.V., M.D., L.P., K.S. and M.M; Z.S., S.D.C. and A.B. analyzed the ligand and G-protein binding vestibules and redacted the structural biology part of the manuscript; S.S. supervised and R.B. designed and performed the mutational analysis; T.L., A.B. and S.D.C. conceptually designed and wrote the manuscript; M.V.R. and C.M. supervised the overall project and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
T.F., S.T., L.D., N.S., A.B., T.L., S.D.C., M.D., C.V., K.S., S.S., R.V., N.L., M.V.R., and C.M. are current and L.P., V.-P.J., Z.S., M.M., Y.-L.L., J.O., and R.B. are former employees of Confo Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ross Cheloha, Anastasiia Gusach, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fontaine, T., Busch, A., Laeremans, T. et al. Structure elucidation of a human melanocortin-4 receptor specific orthosteric nanobody agonist. Nat Commun 15, 7029 (2024). https://doi.org/10.1038/s41467-024-50827-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-50827-7
