
Abstract
The construction of atropisomers with 1,2-diaxes, while maintaining high enantiocontrol, presents a significant challenge due to the dynamic nature of steric hindrance at ortho-aryl substituents. Although various catalytic asymmetric methods have been developed for accessing axially chiral arylpyrroles, the synthesis of axially chiral arylpyrroles with 1,2-diaxes in a catalytic asymmetric manner has remained rare. Herein, the authors report the synthesis of diverse axially chiral arylpyrroles with 1,2-diaxes, and C–C and C–N axes through copper-catalysed asymmetirc [4 + 1] annulation of yne-allylic esters with arylamines via a remote stereocontrol strategy. This approach provides facile access to a broad range of heterobiaryl atropisomers (67 examples) in excellent enantioselectivities, each bearing one or two C–C/C–N axes, demonstrating its versatility and efficiency. The utility of this methodology is further highlighted by the transformation of the product into chiral phosphine ligand, and chiral thioureas for the use in asymmetric catalysis.
Similar content being viewed by others
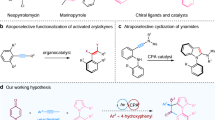
Introduction
The enantioselective synthesis of atropisomers with diaxes has emerged as a prominent research focus in organic synthesis over the past few decades. This trend is driven by their extensive applications in asymmetric catalysis, drug discovery, and material science1,2,3,4,5,6,7. However, most reported studies have concentrated on the preparing atropisomers with remote diaxes, such as 1,4 or 1,3-diaxes8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24. The construction of atropisomers with 1,2-diaxes, while maintaining high enantiocontrol, presents a significant challenge due to the dynamic nature of steric hindrance at ortho-aryl substituents (Fig. 1a)25,26,27,28,29,30. Consequently, there are relatively few reports addressing the diastereo- and enantioselective control of both axes. Pioneering works by Shibata, Sparr, Wencel-Delord, and others have employed intramolecular [2 + 2 + 2] cycloaddition or aldol condensation processes to produce ortho-terphenyls and oligo−1,2-naphthylenes with good to excellent diastereoselectivity25,31,32,33,34,35,36,37,38. In 2021, Zhou reported the modular and convergent synthesis of atropisomeric o-terphenyls with 1,2-diaxes via palladium/chiral norbornene cooperative catalysis at 110 °C39. Despite these impressive advancements, axially chiral arylpyrroles with 1,2-diaxes, which exhibit significant structural diversity, have not yet been reported.

a classes of atropisomers with diaxes. b the representive examples of synthesis axially chiral arylpyrroles. c divergent synthesis of three kinds of axially chiral arylpyrroles.
The synthesis of stereoenriched axially chiral skeletons based on pyrroles has recently emerged as a significant focus of research40,41,42,43,44,45,46,47,48,49. Various successful strategies have been reported, including the de novo synthesis of pyrroles via Paal-Knorr, Attanasi, and Barton-Zard reactions, which yield N1-arylpyrroles and C2- or C3- axially chiral arylpyrroles, respectively (Fig. 1b)50,51,52,53,54,55,56,57,58. Additionally, organocatalysed or metal- catalysed cyclization/[3 + 2] cycloaddition reactions have been proven to be reliable methods for preparing aryl or heteroaryl fused pyrroles (such as indoles or indole-like compounds). These approaches utilize well-developed central-to-axial chirality conversion techniques to successfully create an axis59,60,61,62,63,64,65. Despite these advances, the converting of a single stereocenter into two chiral axes in the synthesis of axially chiral arylpyrroles remains unexplored. Furthermore, each existing method typically yields only one type of axially chiral arylpyrroles. Consequently, there is a high demand for developing, modular approaches that can synthesize three types of axially chiral arylpyrroles from readily available starting materials under mild reaction conditions.
The chemical reactivity of yne-allylic esters has opened numerous synthetic avenues, especially in annulation and asymmetric substitution reactions. However, their application in the synthesis of axially chiral frameworks remains unexplored66,67,68,69,70,71. To address the inherent challenges, we propose a reaction of yne-allylic esters with aryl amines, proceeding via a remote substitution/annulation/aromatization pathway. This approach aims to efficiently synthesize axially chiral arylpyrroles in a single step. Herein, we report a copper-catalysed reaction of yne-allylic esters with aryl amines, achieving the synthesis of axially chiral arylpyrroles under simple and mild reaction conditions. This robust catalytic system offers a straightforward protocol for accessing three classes of axially chiral arylpyrroles, including 1,2-diaxes and those with C–C and C–N axial chirality. Using this method, more than 65 axially chiral arylpyrroles were rapidly synthesized (Fig. 1c).
Results
Reaction development
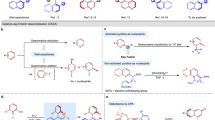
To construct axially chiral arylpyrrole, yne-allylic ester 1a was designed and applied in a reaction with 4-methoxy-1-aminobenzene 2a to explore the feasibility of forming atropisomers with a C–C axis (Table 1). However, initial attempts to employ privileged chiral Segphos L1 or tBu-Box L2 combination with Cu(CH3CN)4BF4 as catalysts gave very poor results (Table 1, entries 1 and 2). To our delight, employing iPr-Pybox L3 as the ligand resulted in the formation of the desired C–C axially chiral arylpyrrole 3a in 28% yield with 67:33 er (Table 1, entry 3). Encouragingly, the er of 3a improved as the steric hindrance of the ligands decreased. Ultimately, Me-Pybox L5 emerged as the best ligand for synthesizing axially chiral arylpyrrole, giving the desired product in 65% yield with 80:20 er (Table 1, entry 5). Further modification of the substituents at the C-4 position of the pyridyl ring significantly influenced chiral induction. With the prepared CF3-, I-, and NMe2-substituted Me-Pybox L6, L7, and L872,73,74, both yield and er showed slight improvements (Table 1, entries 6–8). Lowering the reaction temperature from 60 °C to 25 °C resulted in a substantial decrease in the reaction efficiency. A reasonable yield and an increased er were obtained when the reaction time was prolonged (Table 1, entry 9). Furthermore, replacing acetate with pentafluorobenzoate as the leaving group reduced, the reaction time to 1.5 h, delivering desired product 3a in 83% yield with 88.5:11.5 er (Table 1, entry 10). Lowering the reaction temperature further proved beneficial (Table 1, entries 11 and 12). It was observed that bases significantly affected reaction efficiency and enantioselectivity (Table 1, entries 13 and 14). Unexpectedly, the reaction of 1b and 2a proceeded smoothly without any base, providing the desired product 3a in 98% yield with 97.5:2.5 er (Table 1, entry 15). The optimal results were obtained with Cu(CH3CN)4BF4 (10 mol%) in the presence of the chiral ligand L7 (12 mol%) in MeOH at −10 °C.
Substrate scope
With the optimized reaction conditions in hand, we investigated the substrate scope of C–C axially chiral arylpyrroles with L7 as the optimal ligand (Fig. 2a). Employing 4-aminophenol as the bis-nucleophile, we exclusively obtained the desired axially chiral arylpyrrole 3b in 97% yield with 97.5:2.5 er, while the hydroxyl group remained inert throughout the transformation. This [4 + 1] annulation exhibited compatibility with a series of ortho-substituted phenyl groups of yne-allylic esters, furnishing C–C axially chiral arylpyrroles 3c–3h in 75–97% yields with 93.5:6.5–96:4 er. Notably, the alkenyl substituents of the yne-allylic esters were well tolerated, highlighting the potential for further transformations (3i). Subsequently, we investigated the influence of substituents on another benzene ring in the yne-allylic esters. By introducing various groups at different positions, we were able to achieve excellent yields and enantioselectivities in the annulated products (3j–3l). Then, we completed the scoping survey by examining different substituents on the aryl amines. As anticipated, electron-donating, electron-withdrawing groups on the phenyl amines and aniline without substituent were accommodated, yielding axially chiral arylpyrrole products 3m–3u with excellent yields and enantioselectivities. Importantly, valuable functional groups, such as terminal alkenyl (3v) and terminal alkynyl (3w) on the phenylamines were well tolerated, highlighting the potential for late-stage diversification. Remarkably, various heteroaryl amines, including indolyl and pyrazoyl group, were efficiently transformed into the corresponding highly enantioenriched C–C axially chiral arylpyrrole products 3x and 3y.

a C–C/C–N axially chiral arylpyrroles, b C–N axially chiral arylpyrroles. Reaction conditions: 1 (0.15 mmol), 2 (2.0 equiv., 0.3 mmol), Cu(CH3CN)4BF4 (10 mol%), L7 (12 mol%), MeOH (0.15 M), −10 °C. b −20 °C. c 1 (0.15 mmol), 2 (1.3 equiv., 0.195 mmol), Cu(CH3CN)4BF4 (10 mol%), L8 (12 mol%), DIPEA (1.5 equiv., 0.225 mmol), MeOH (0.15 M), −5 °C.
To our delight, the C–N atropisomers could also be accessed using L8 as the optimal ligand with DIPEA as an additive. It is noteworthy that the use of DIPEA as an additive enhances the nucleophilicity of sterically hindered amines, thereby increasing yield and improving enantioselectivity of the C–N axially chiral arylpyrrole (experimental details in Supplementary Table 4). A diverse array of aromatic substituents was investigated, and all these aryl-substituted yne-allylic esters underwent smooth [4 + 1] annulation with 8-aminonaphthalen-2-ol 2p, yielding the desired C–N axially chiral arylpyrroles in excellent yields and with high-to-excellent enantioselectivities (Fig. 2b). Various meta- and para-substituted aryl yne-allylic esters were readily transformed into the corresponding C–N atropisomers 4a–4j in 58–98% yields with 92:8–97:3 er. Notably, aliphatic substituted yne-allylic esters were compatible with this copper-catalysed [4 + 1] annulation, furnishing the desired axially chiral arylpyrroles products 4k and 4l in moderate to excellent yields and excellent enantioselectivities. Further exploration of aryl amines under the standard reaction conditions revealed the efficiency of 1-naphthylamines, bearing alkyl, chloro, or bromo substituents in delivering axially chiral arylpyrrole in excellent yields with excellent enantioselectivities (4m–4p). It was found that the hydroxyl group in 8-aminonaphthalen-2-ol is critical for controlling the enantioselectivity of the reaction. We hypothesize that hydrogen bonding plays a significant role in this process (experimental details in Supplementary Fig. 28). To our delight, the phenylamine bearing a bulky group was efficiently converted to the expected product 4q in 67% yield with 92:8 er. Naphthalen-1-amine also proved to be compatible, providing the axially chiral arylpyrrole product 4r in 72% yield with 91:9 er.
The success of the C–C and C–N axially chiral arylpyrrole synthesis encouraged us to investigate the synthesis of axially chiral arylpyrrole with 1,2-diaxes atropoisomerically (Fig. 3). To our delight, the reaction of yne-allylic ester 1b with 2p was completed in 48 h at 0°C with L5 as ligand, yielding the axially chiral arylpyrrole with 1,2-diaxes pure product 5a in 92% yield with 99.5:0.5 er and a dr of 17:1. Next, we evaluated the substrate scope of axially chiral arylpyrrole with 1,2-diaxes. Yne-allylic esters bearing diverse ortho-substituents (including iPr, Me, Et, and Bn) reacted smoothly with aryl amine 2p, affording the corresponding atropoisomers 5a–5f with high efficiency. Remarkably, phenyl-substituted yne-allylic esters containing alkenyl and chloro groups produced the desired highly functionalized axially chiral arylpyrrole with 1,2-diaxes products 5g–5 h with excellent enantioselectivities, highlighting a notable potential for further transformations. Moreover, the reaction was applicable to a naphthyl-substituted yne-allylic ester (5i). Introducing various substituents (Br, and OMe) at the C6–C8 positions in the 1,2-dihydronaphthalene backbone did not influence the reaction efficiency or stereoselectivity (5j–5l).

1 (0.1 mmol), 2 (2.0 equiv., 0.2 mmol), Cu(CH3CN)4BF4 (10 mol%), L5 (12 mol%), MeOH (0.1 M), 0 °C.
Subsequently, the reaction scope was extended to ortho-substituted aryl amines, demonstrating compatibility with various functional groups (OH, NH2), and axially chiral arylpyrrole with 1,2-diaxes products 5m–5o in excellent yields and enantioselectivities. Various protecting groups (NHPh, NHCOMe, and OMe) at different positions on the phenylamines also afforded the axially chiral arylpyrrole with 1,2-diaxes products 5p–5s in excellent yields and enantioselectivities. The use of an amine bearing an ester group afforded the desired product 5t in low yield, probably due to the reduced nucleophilicity of the amine. However, the reaction efficiency dramatically increased when a phenylamine bearing an additional hydroxyl group at the para position (5u) was used. A substrate bearing an internal alkyne motif was well tolerated, resulting in good yields and enantioselectivitiy (5w). Naphthalen-1-amine proved to be a suitable substrate, providing the desired product in 99% yield with 97.5:2.5 er and > 20:1 dr (5x). The absolute configuration of products 3a and 5a were unambiguously confirmed by X-ray crystallographic analysis, and the configurations of all other related compounds were assigned by analogy.
Product Derivatization
To further evaluate the scalability and practicality of this asymmetric protocol, gram-scale syntheses of axially chiral arylpyrroles 4j and 5m were performed without any loss of yield or enantioselectivity (Fig. 4a). Moreover, the synthetic utility of these axially chiral arylpyrroles in the syntheses of chiral ligands and catalysts was demonstrated (Fig. 4b). C-P coupling followed by protection from axially chiral arylpyrroles 5v furnished desired phosphine 6 in a total yield of 63% with 99.5:0.5 er after recrystallization. Notably, axially chiral arylpyrroles 5m could be readily protected and oxidized to chiral aldehyde 7 with cerium (IV) ammonium nitrate (CAN) as the oxidizing reagent. Subsequently, the free amine of 5o reacted with isothiocyanates 8a and 8b, leading to the corresponding thioureas 9 and 10 respectively, in moderate to excellent yields with excellent enantioselectivities. In addition, 5o could react with CSCl2 to generate an isothiocyanate intermediate, which then spontaneously reacted with chiral diamine 8c to generate the bifunctional molecule 11 in 56% yield for over two steps. Next, the potential of chiral phosphine 6 as a ligand and chiral thiourea 10 as an organocatalyst in enantioselective transformations was explored (Fig. 4c). Remarkably, phosphine 6 served as a chiral ligand, enabling enantioselective palladium-catalysed allylic alkylation in excellent yield with good er75,76. It was found that our ligand 6 exhibits better enantiocontrol compared to the biaryl-based phosphine ligands. Additionally, ligand 6 catalysed the reaction with a significantly higher yield than the ferrocene-derived P, N ligand L22. In addition, chiral thiourea 10 was applied as an organocatalyst in the enantioselective [2 + 4] cycloaddition of homophthalic anhydride 15 with 2-benzothiazolimine 16 to afford product 17 in 63% yield with 97.5:2.5 er77.

a Gram-scale reaction. b Transformations. c Applications. d Mechanistic studies. e Proposed mechanism.
Mechanistic studies
We carried out a series of reactions to elucidate the reaction mechanism. Yne-allylic ester 1b reacted smoothly with 4-chloroaniline 2i, delivering formal allylic amination intermediate 18 in 66% yield over 2 h at r.t. in the presence of the copper/Pybox L9 catalyst (Fig. 4d). Subsequently, this intermediate was converted into the corresponding pyrrole in 50% yield under standard reaction conditions. A deuterated yne-allylic ester 1ae was prepared and reacted with amine 2p. However, the target pyrrole product was isolated in 79% yield without any deuteration, indicating that the aromatization process did not involve a 1,5-H shift process. Based on these experimental observations, the reaction of yne-allylic esters with aryl amines likely proceeds in a stepwise manner including remote amination, cyclization, and aromatization (Fig. 4e). Initially, copper acetylide intermediate I, forms from the coordination and deprotonation of the terminal alkyne and copper salt, which then eliminates the leaving group to form the copper-vinylallenylidene species II. Subsequently, II undergoes remote amination to form III bearing a stereogenic chiral carbon center, which can spontaneously cyclize to afford V. Subsequently, a stereomemory aromatization sequence forms the final chiral axially arylpyrrole product 3. The central-to-axial chirality transfer during the aromatization process results in complete conversion, thereby maintaining excellent atropisomeric selectivities. Notably, the cyclic substrate design could potentially shorten the distance of the reactive site and the nucleophilic reagent, thereby improving the conversion efficiency of the intramolecular cyclization process and enhancing stereocontrol in the transfer of central to axial chirality (experimental details in Supplementary Fig. 27).
Discussion
In summary, we have designed and successfully developed an enantioselective copper-catalysed [4 + 1] annulations of yne-allylic esters with aryl amines. This protocol exhibits a broad substrate scope and excellent functional group tolerance, providing straightforward access to various axially chiral arylpyrroles with diaxes, as well as C–C, C–N axes under mild reaction conditions. The utility of this methodology is demonstrated by the transformation of the chiral products into versatile chiral ligands and organocatalysts with excellent enantiomeric purity. We anticipate that the established method will have broad application in the atroposelective synthesis of pyrrole derivatives, asymmetric catalysis, and beyond.
Methods
General procedure for the preparation of the axially chiral arylpyrroles
Cu(CH3CN)4BF4 (4.7 mg, 0.015 mmol) and ligand L7 (6.7 mg, 0.018 mmol) were placed in an oven-dried sealed tube. The tube was evacuated and refilled with nitrogen for three cycles. Anhydrous MeOH (0.5 mL) was then added, and the resulting mixture was stirred at room temperature for 1 h. Yne-allylic esters (1, 0.15 mmol), aryl amines (2, 0.3 mmol), were subsequently added to the mixture, followed by another portion of anhydrous MeOH (0.5 mL) under nitrogen at -10 °C. The reaction mixture was stirred for 48 h. After completion, the solvents were removed under reduced pressure and the obtained residue was purified by column chromatography on silica gel using a petroleum ether/ethyl acetate eluent system, yielding the desired chiral products 3.
Cu(CH3CN)4BF4 (4.7 mg, 0.015 mmol) and ligand L8 (5.2 mg, 0.018 mmol) were placed in an oven-dried sealed tube. The tube was evacuated and refilled with nitrogen for three cycles. Anhydrous MeOH (0.5 mL) was added, and the resulting mixture was stirred at room temperature for 1 h. Subsequently, yne-allylic esters (1, 0.15 mmol), aryl amines (2, 0.195 mmol), an additional portion of anhydrous MeOH (0.5 mL), and DIPEA (41 µL, 0.225 mmol) were added successively to the mixture under nitrogen at −5 °C and stirred for 10 h. After completion, the solvents were removed under reduced pressure and the resulting residue was purified by column chromatography on silica gel using a petroleum ether/ethyl acetate eluent system, affording the desired chiral products 4.
Cu(CH3CN)4BF4 (3.2 mg, 0.01 mmol) and ligand L5 (2.9 mg, 0.012 mmol) were placed in an oven-dried sealed tube. The tube was evacuated and refilled with nitrogen for three cycles. Anhydrous MeOH (0.5 mL) was added, and the resulting mixture was stirred at room temperature for 1 h. Subsequently, yne-allylic esters (1, 0.1 mmol), aryl amines (2, 0.2 mmol), and an additional portion of anhydrous MeOH (0.5 mL) were added successively to the mixture under nitrogen at 0 °C. The reaction mixture was stirred for 48 h. The mixture was passed through a short pad of silica gel using ethyl acetate as the eluent. The dr ratio was determined by 1H NMR analysis of the crude reaction mixture. The solvents were removed under reduced pressure and the obtained residue was then purified by column chromatography on silica gel using a petroleum ether/ethyl acetate eluent system, affording the desired chiral products 5.
Data availability
The X-ray crystallographic coordinates for structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2261705 and CCDC 2260916. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. Supplementary information and chemical compound information are available in the online version of the paper. For NMR analysis and HPLC traces of the compounds in this article, see Supplementary Figs. Correspondence and requests for materials should be addressed to H. X. All data are available from the corresponding author upon request.
References
Matsunaga, S. et al. Catalytic enantioselective meso-epoxide ring opening reaction with phenolic oxygen nucleophile eromoted by gallium heterobimetallic multifunctional complexes. J. Am. Chem. Soc. 122, 2252–2260 (2000).
LaPlante, S. R. et al. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 54, 7005–7022 (2011).
Zhu, Y.-Y., Wu, X.-D., Gu, S.-X. & Pu, L. Free amino acid recognition: a bisbinaphthyl-based fluorescent probe with high enantioselectivity. J. Am. Chem. Soc. 141, 175–181 (2019).
Fayez, S. et al. A near-complete series of four atropisomeric Jozimine A2-type naphthylisoquinoline dimers with antiplasmodial and cytotoxic activities and related alkaloids from Ancistrocladus abbreviatus. J. Nat. Prod. 82, 3033–3046 (2019).
Takaishi, K., Hinoide, S., Matsumoto, T. & Ema, T. Axially chiral peri-xanthenoxanthenes as a circularly polarized luminophore. J. Am. Chem. Soc. 141, 11852–11857 (2019).
Takaishi, K., Iwachido, K., Takehana, R., Uchiyama, M. & Ema, T. Evolving fluorophores into circularly polarized luminophores with a chiral naphthalene tetramer: proposal of excimer chirality rule for circularly polarized luminescence. J. Am. Chem. Soc. 141, 6185–6190 (2019).
Liu, C.-X. et al. Rhodium-catalyzed asymmetric C–H functionalization reactions. Chem. Rev. 123, 10079–10134 (2023).
Bao, X., Rodriguez, J. & Bonne, D. Enantioselective synthesis of atropisomers with multiple stereogenic axes. Angew. Chem. Int. Ed. 59, 12623–12634 (2020).
Cheng, J. K., Xiang, S.-H., Li, S., Ye, L. & Tan, B. Recent advances in catalytic asymmetric construction of atropisomers. Chem. Rev. 121, 4805–4902 (2021).
Schmidt, T. A. & Sparr, C. Catalyst control over twofold and higher-order stereogenicity by atroposelective arene formation. Acc. Chem. Res. 54, 2764–2774 (2021).
Zhang, Z.-X., Zhai, T.-Y. & Ye, L.-W. Synthesis of axially chiral compounds through catalytic asymmetric reactions of alkynes. Chem Catal. 1, 1378–1412 (2021).
Hayashi, T., Hayashizaki, K. & Ito, Y. Asymmetric synthesis of axially chiral 1,1′:5′,1″- and 1,1′:4′,1″- ternaphthalenes by asymmetric cross-coupling with a chiral ferrocenylphosphine-nickel catalyst. Tetrahedron. Lett. 30, 215–218 (1989).
Shibata, T., Fujimoto, T., Yokota, K. & Takagi, K. Iridium complex-catalyzed highly enantio- and diastereoselective [2+2+2] cycloaddition for the synthesis of axially chiral teraryl compounds. J. Am. Chem. Soc. 126, 8382–8383 (2004).
Tanaka, K., Suda, T., Noguchi, K. & Hirano, M. Catalytic [2+2+2] and thermal [4+2] cycloaddition of 1,2-bis(arylpropiolyl)benzenes. J. Org. Chem. 72, 2243–2246 (2007).
Xu, G., Fu, W., Liu, G., Senanayake, C. H. & Tang, W. Efficient syntheses of Korupensamines A, B and Michellamine B by asymmetric Suzuki–Miyaura coupling reactions. J. Am. Chem. Soc. 136, 570–573 (2014).
Tan, Y. et al. Enantioselective construction of vicinal diaxial styrenes and multiaxis system via organocatalysis. J. Am. Chem. Soc. 140, 16893–16898 (2018).
Bisag, G. D. et al. Central‐to‐axial chirality conversion approach designed on organocatalytic enantioselective povarov cycloadditions: first access to configurationally stable indole-quinoline atropisomers. Chem. Eur. J. 25, 15694–15701 (2019).
Shen, D., Xu, Y. & Shi, S.-L. A bulky chiral N-heterocyclic carbene palladium catalyst enables highly enantioselective Suzuki–Miyaura cross-coupling reactions for the synthesis of biaryl atropisomers. J. Am. Chem. Soc. 141, 14938–14945 (2019).
Bao, X., Rodriguez, J. & Bonne, D. Bidirectional enantioselective synthesis of bis-benzofuran atropisomeric oligoarenes featuring two distal C–C stereogenic axes. Chem. Sci. 11, 403–408 (2020).
Beleh, O. M., Miller, E., Toste, F. D. & Miller, S. J. Catalytic dynamic kinetic resolutions in tandem to construct two-axis terphenyl atropisomers. J. Am. Chem. Soc. 142, 16461–16470 (2020).
Nguyen, Q.-H., Guo, S.-M., Royal, T., Baudoin, O. & Cramer, N. Intermolecular palladium(0)-catalyzed atropo-enantioselective C–H arylation of heteroarenes. J. Am. Chem. Soc. 142, 2161–2167 (2020).
Xia, W. et al. Chiral phosphoric acid catalyzed atroposelective C–H amination of arenes. Angew. Chem. Int. Ed. 59, 6775–6779 (2020).
Liao, G. et al. Experimental and computational studies on the directing ability of chalcogenoethers in palladium-catalyzed atroposelective C–H olefination and allylation. Angew. Chem. Int. Ed. 61, e202115221 (2022).
Wang, Y. et al. Rhodium-catalyzed enantioselective and diastereodivergent access to diaxially chiral heterocycles. Nat. Commun. 14, 4661 (2023).
Shibata, T., Tsuchikama, K. & Otsuka, M. Enantioselective intramolecular [2+2+2] cycloaddition of triynes for the synthesis of atropisomeric chiral ortho-diarylbenzene derivatives. Tetrahedron: Asymmetry 17, 614–619 (2006).
Barrett, K. T., Metrano, A. J., Rablen, P. R. & Miller, S. J. Spontaneous transfer of chirality in an atropisomerically enriched two-axis system. Nature 509, 71–75 (2014).
Moser, D. & Sparr, C. Synthesis of atropisomeric two-axis systems by the catalyst-controlled syn- and anti-selective arene-forming aldol condensation. Angew. Chem. Int. Ed. 61, e202202548 (2022).
Zhang, X.-L. et al. Stepwise asymmetric allylic substitution-isomerization enabled mimetic synthesis of axially chiral B, N‐heterocycles. Angew. Chem. Int. Ed. 61, e202210456 (2022).
Han, T.-J. et al. Catalytic atroposelective synthesis of heterobiaryls with vicinal C–C and N–N diaxes via dynamic kinetic resolution. iScience 26, 107978 (2023).
Zhang, S.-C. et al. Enantioselective access to triaryl-2-pyrones with monoaxial or contiguous C–C diaxes via oxidative NHC catalysis. ACS Catal. 13, 2565–2575 (2023).
Oppenheimer, J., Hsung, R. P., Figueroa, R. & Johnson, W. L. Stereochemical control of both C–C and C–N axial chirality in the synthesis of chiral N,O-biaryls. Org. Lett. 9, 3969–3972 (2007).
Lotter, D., Neuburger, M., Rickhaus, M., Häussinger, D. & Sparr, C. Stereoselective arene-forming aldol condensation: synthesis of configurationally stable oligo-1,2-naphthylenes. Angew. Chem. Int. Ed. 55, 2920–2923 (2016).
Dherbassy, Q., Djukic, J. P., Wencel-Delord, J. & Colobert, F. Two stereoinduction events in one C–H activation step: a route towards terphenyl ligands with two atropisomeric axes. Angew. Chem. Int. Ed. 57, 4668–4672 (2018).
Hu, Y.-L. et al. Conversion of two stereocenters to one or two chiral axes: atroposelective synthesis of 2,3–diarylbenzoindoles. Chem. Sci. 10, 6777–6784 (2019).
Wang, B.-J. et al. Single-step synthesis of atropisomers with vicinal C–C and C–N diaxes by cobalt-catalyzed atroposelective C–H annulation. Angew. Chem. Int. Ed. 61, e202208912 (2022).
Zhang, S. et al. Atroposelective synthesis of triaryl α-pyranones with 1,2-diaxes by N-heterocyclic carbene organocatalysis. Angew. Chem. Int. Ed. 61, e202212005 (2022).
Luc, A. et al. Double cobalt-catalyzed atroposelective C–H activation: one-step synthesis of atropisomeric indoles bearing vicinal C–C and C–N diaxes. Chem. Catal. 3, 100765 (2023).
Yang, K. et al. Construction of C–B axial chirality via dynamic kinetic asymmetric cross-coupling mediated by tetracoordinate boron. Nat. Commun. 14, 4438 (2023).
Gao, Q. et al. Catalytic synthesis of atropisomeric o-terphenyls with 1,2-diaxes via axial-to-axial diastereoinduction. J. Am. Chem. Soc. 143, 7253–7260 (2021).
Wang, Y.-B. & Tan, B. Construction of axially chiral compounds via asymmetric organocatalysis. Acc. Chem. Res. 51, 534–547 (2018).
Kitagawa, O. Chiral Pd-catalyzed enantioselective syntheses of various N–C axially chiral compounds and their synthetic applications. Acc. Chem. Res. 54, 719–730 (2021).
Wu, Y.-J., Liao, G. & Shi, B.-F. Stereoselective construction of atropisomers featuring a C–N chiral axis. Green Synth. Catal. 3, 117–136 (2022).
Cai, W.-Y., Ding, Q.-N., Zhou, L. & Chen, J. Asymmetric synthesis of axially chiral molecules via organocatalytic cycloaddition and cyclization reactions. Molecules 28, 4306 (2023).
Chen, Y.-B., Yang, Y.-N., Huo, X.-Z., Ye, L.-W. & Zhou, B. Recent advances in the construction of axially chiral arylpyrroles. Sci. China Chem. 66, 2480–2491 (2023).
Faisca Phillips, A. M. & Pombeiro, A. J. L. Atropselective organocatalytic synthesis of chiral compounds containing nitrogen along the axis of chirality. Symmetry 15, 1261 (2023).
Zhang, L. et al. Phosphoric acid-catalyzed atroposelective construction of axially chiral arylpyrroles. Nat. Commun. 10, 566 (2019).
Zhang, S. et al. Enantioselective synthesis of atropisomers featuring pentatomic heteroaromatics by Pd-catalyzed C–H alkynylation. ACS Catal 9, 1956–1961 (2019).
Wang, X.-M. et al. Enantioselective synthesis of nitrogen-nitrogen biaryl atropisomers via copper-catalyzed Friedel–Crafts alkylation reaction. J. Am. Chem. Soc. 143, 15005–15010 (2021).
Yin, S.-Y., Zhou, Q., Liu, C.-X., Gu, Q. & You, S.-L. Enantioselective synthesis of N–N biaryl atropisomers through iridium(I)-catalyzed C–H alkylation with acrylates. Angew. Chem. Int. Ed. 62, e202305067 (2023).
Zhang, L., Zhang, J., Ma, J., Cheng, D.-J. & Tan, B. Highly atroposelective synthesis of arylpyrroles by catalytic asymmetric Paal–Knorr reaction. J. Am. Chem. Soc. 139, 1714–1717 (2017).
He, X.-L. et al. Asymmetric Barton–Zard reaction to access 3-pyrrole-containing axially chiral skeletons. ACS Catal 9, 4374–4381 (2019).
Wang, Y.-B. et al. Asymmetric construction of axially chiral 2-arylpyrroles by chirality transfer of atropisomeric alkenes. Angew. Chem. Int. Ed. 58, 13443–13447 (2019).
Zheng, S.-C., Wang, Q. & Zhu, J. Catalytic kinetic resolution by enantioselective aromatization: conversion of racemic intermediates of the Barton–Zard reaction into enantioenriched 3-arylpyrroles. Angew. Chem. Int. Ed. 58, 9215–9219 (2019).
Xu, Q. et al. Cu(I)-catalyzed asymmetric arylation of pyrroles with diaryliodonium salts toward the synthesis of N−N atropisomers. Org. Lett. 24, 3138–3143 (2022).
Han, T.-J., Zhang, Z.-X., Wang, M.-C., Xu, L.-P. & Mei, G.-J. The rational design and atroposelective synthesis of axially chiral C2-arylpyrrole-derived amino alcohols. Angew. Chem. Int. Ed. 61, e202207517 (2022).
Tan, C. X. A. et al. Synthesis of axially chiral CF3-substituted 2-arylpyrroles by sequential phosphine-catalyzed asymmetric [3+2] annulation and oxidative central-to-axial chirality transfer. Angew. Chem. Int. Ed. 61, e202209494 (2022).
Zhao, Y., Liu, N., Zhong, S., Wen, Z. & Wang, T. A central-to-axial chirality conversion strategy for the synthesis of C-N axially chiral N-arylpyrroles. Org. Lett. 24, 2842–2846 (2022).
Chen, Y.-B. et al. Construction of axially chiral arylpyrroles via atroposelective diyne cyclization. Angew. Chem. Int. Ed. 62, e202303670 (2023).
Man, N. et al. Organocatalytic atroposelective construction of axially chiral N-aryl benzimidazoles involving carbon-carbon bond cleavage. Org. Lett. 22, 6382–6387 (2020).
An, Q.-J. et al. Nitrosobenzene-enabled chiral phosphoric acid catalyzed enantioselective construction of atropisomeric N-arylbenzimidazoles. Angew. Chem. Int. Ed. 60, 24888–24893 (2021).
Sun, L. et al. Rhodium-catalyzed atroposelective construction of indoles via C–H bond activation. Angew. Chem. Int. Ed. 60, 8391–8395 (2021).
Yang, G. et al. Organocatalytic higher-order [8+2] cycloaddition for the assembly of atropoenantiomeric 3-arylindolizines. Org. Lett. 23, 8109–8113 (2021).
Wang, Y. et al. Construction of axially chiral indoles by cycloaddition-isomerization via atroposelective phosphoric acid and silver sequential catalysis. ACS Catal 12, 8094–8103 (2022).
Wang, Z.-S. et al. Synthesis of axially chiral N-arylindoles via atroposelective cyclization of ynamides catalyzed by chiral brønsted acids. Angew. Chem. Int. Ed. 61, e202201436 (2022).
Zhang, P. et al. Enantioselective synthesis of N–N bisindole atropisomers. Angew. Chem. Int. 61, e202212101 (2022).
Thies, N. & Haak, E. Ruthenium-catalyzed synthesis of 2,3-cyclo[3]dendralenes and complex polycycles from propargyl alcohols. Angew. Chem. Int. Ed. 54, 4097–4101 (2015).
Kong, H.-H. et al. Remote enantioselective [4+1] annulation with copper-vinylvinylidene intermediates. J. Am. Chem. Soc. 144, 21347–21355 (2022).
Ma, J.-S. et al. Copper-catalysed convergent regio- and enantioselective alkynylallylic substitution. Nat. Synth. 2, 37–48 (2022).
Niu, S. et al. Copper-catalyzed yne-allylic substitutions using stabilized nucleophiles. ACS Catal 12, 6840–6850 (2022).
Li, M.-D. et al. Copper-catalyzed remote enantioselective sulfonylation of yne-allylic esters with sodium sulfinates. Angew. Chem. Int. Ed. 62, e202313911 (2023).
Luo, D. et al. Copper-catalyzed asymmetric yne-allylic substitution using electron-rich arenes. ACS Catal 14, 2746–2757 (2024).
Sun, Y.-Z. et al. Asymmetric substitution by alkynyl copper driven dearomatization and rearomatization. Angew. Chem. Int. Ed. 62, e202314517 (2023).
Qian, H.-D. et al. Remote copper-catalyzed enantioselective substitution of yne-thiophene carbonates. Sci. China Chem. 67, 1175–1180 (2024).
Zhang, Z. et al. Enantioselective propargylic amination and related tandem sequences to α-tertiary ethynylamines and azacycles. Nat. Chem. 16, 521–532 (2024).
Trost, B. M., Machacek, M. R. & Aponick, A. Predicting the stereochemistry of diphenylphosphino benzoic acid (DPPBA)-based palladium-catalyzed asymmetric allylic alkylation reactions: a working model. Acc. Chem. Res. 39, 747–760 (2006).
Cheng, Q. et al. Iridium-catalyzed asymmetric allylic substitution reactions. Chem. Rev. 119, 1855–1969 (2019).
Liu, S.-J. et al. Rational design of axially chiral styrene-based organocatalysts and their application in catalytic asymmetric (2+4) cyclizations. Angew. Chem. Int. Ed. 61, e202112226 (2022).
Acknowledgements
Generous financial support from the National Natural Science Foundation of China (21801087 for H. X.) and the Fundamental Research Funds for the Central Universities (CCNU24JCPT013 for H. X.).
Author information
Authors and Affiliations
Contributions
C. Y., D.-R. L. and H.-M. X. performed experiments. D.-R. L. and H.-M. X. contributed equally. S.-J. L. took part in the initial reaction development. Y. L., Z. W., T. Y., J. W., K. F. participated in substrate preparation and helped with characterizing new compounds. H. X. conceived and directed the project. H. X. and C. Z. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bo Zhou and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yao, C., Li, DR., Xiang, HM. et al. Copper-catalysed asymmetric annulation of yne-allylic esters with amines to access axially chiral arylpyrroles. Nat Commun 15, 6848 (2024). https://doi.org/10.1038/s41467-024-50896-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50896-8
This article is cited by
-
Copper-catalyzed Larock-type cyclization/debenzylation of yne-allylic esters with aliphatic amines
Science China Chemistry (2025)
