
Abstract
The positive electrode|electrolyte interface plays an important role in all-solid-state Li batteries (ASSLBs) based on garnet-type solid-state electrolytes (SSEs) like Li6.4La3Zr1.4Ta0.6O12 (LLZTO). However, the trade-off between solid-solid contact and chemical stability leads to a poor positive electrode|electrolyte interface and cycle performance. In this study, we achieve thermodynamic compatibility and adequate physical contact between high-entropy cationic disordered rock salt positive electrodes (HE-DRXs) and LLZTO through ultrafast high-temperature sintering (UHS). This approach constructs a highly stable positive electrode|electrolyte interface, reducing the interface resistance to 31.6 Ω·cm2 at 25 °C, making a 700 times reduction compared to the LiCoO2 | LLZTO interface. Moreover, the conformal and tight HE-DRX | LLZTO solid-state interface avoids the transition metal migration issue observed with HE-DRX in liquid electrolytes. At 150 °C, HE-DRXs in ASSLBs (Li|LLZTO | HE-DRXs) exhibit an average specific capacity of 239.7 ± 2 mAh/g at 25 mA/g, with a capacity retention of 95% after 100 cycles relative to the initial cycle—a stark contrast to the 76% retention after 20 cycles at 25 °C in conventional liquid batteries. Our strategy, which considers the principles of thermodynamics and kinetics, may open avenues for tackling the positive electrode|electrolyte interface issue in ASSLBs based on garnet-type SSEs.
Similar content being viewed by others
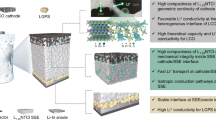

Introduction
All-solid-state Li batteries (ASSLBs) based on garnet-type solid-state electrolytes (SSEs), such as Li6.4La3Zr1.4Ta0.6O12 (LLZTO)1,2,3, are considered safer alternatives to conventional batteries due to their nonflammability4,5,6. However, ASSLBs suffer from poor interfacial stability between the positive electrode and SSEs, stemming from the inherent trade-off between chemical stability and wettability7,8,9,10. Normally, a high-temperature (>900 °C) co-sintering process between the positive electrode and SSE is required to achieve sufficient interfacial contact11. Previous studies have demonstrated that traditional positive electrode materials (e.g., LiFePO4, LiMn2O4, and layered LiCoO2) can react with LLZTO at 500 °C12,13,14,15. This indicates that the chemically stable temperature (Tstable) of the traditional positive electrode material and LLZTO is significantly lower than the wetting temperature (Twetting) of the positive electrode material, leading to unstable interfaces and poor cycling performance16. Although the use of low melting point solder as a buffer layer has been explored to improve positive electrode-electrolyte contact17,18,19, it inevitably reduces the capacity due to the limited active material content in composite positive electrodes. Therefore, novel strategies are required to enhance the interfacial stability and capacity of ASSLBs based on garnet-type SSEs.
The ultrafast high-temperature sintering (UHS) with Joule heating has been verified to partially inhibit element cross-diffusion through fast heating and cooling processes when the two phases are sintered together at high temperatures20. Unfortunately, the utilization of traditional positive electrode materials in ASSLBs still falls short of resolving the inherent trade-off dilemma between chemical stability and wettability21. High-entropy materials, comprised of multiple components, have received significant attention as an emerging class of materials22,23. By exploiting the synergistic effects of multiple elements, high-entropy materials maximize configurational entropy and form highly stable structures24,25. Consequently, they exhibit high thermal stability26,27 and require low sintering temperature28,29. As positive electrode materials, high-entropy cationic disordered rock salt positive electrodes (HE-DRXs) have shown excellent lithium storage properties28. Due to the unique entropy effects30,31,32, high-entropy materials have the potential to achieve a balance between chemical stability (high Tstable) and wettability (low Twetting).
In this work, we present a strategy that tactfully combines thermodynamics and kinetics to construct a high-stability positive electrode-electrolyte interface. By employing UHS technology, the precursor of HE-DRXs undergoes a rapid in situ phase formation reaction on dense ceramic sheets of LLZTO with a conformal interface (Fig. 1). The rapid heating and cooling process effectively curbs elemental cross-diffusion and increases the Tstable-HE-DRXs|LLZTO to 1100 °C, due to the short annealing time of ~3 s. This integration of the entropy stabilization effect and rapid reaction kinetics achieves a favorable combination of chemical stability and wettability, fostering the development of a durable conformal interface. As a result, the interface resistance of HE-DRXs|LLZTO is reduced to 31.6 Ω·cm2, representing a 700-fold decrease compared to the LiCoO2 | LLZTO interface. HE-DRXs (1.3 mg/cm2) in ASSLBs exhibit an average specific capacity of 239.7 ± 2 mAh/g at 25 mA/g with 95% capacity retention after 100 cycles relative to the initial cycle at 150 °C, far beyond the cycling performance in liquid electrolytes which retain only 76% capacity after 20 cycles at 25 °C.

Schematic of the construction of the highly stable HE-DRX | LLZTO interface using the UHS process: the high reaction temperature of the UHS technology enables the rapid synthesis of the HE-DRX positive electrode on the LLZTO surface, while the short sintering time and the stability of the HE-DRXs ensure a conformal positive electrode interface without any side reactions.
Results and discussion
Synthesis and characterization of HE-DRXs
Our study employed the UHS method to achieve a rapid synthesis of HE-DRXs with six cations, specifically Li1.3Mn2+0.1Co2+0.1Mn3+0.1Cr3+0.1Ti0.1Nb0.2O1.7F0.3 (TM6). A pressed green pellet of the TM6 precursor powders was placed between two Joule-heated carbon strips, and the rapid synthesizing reaction was achieved at 1100 °C for 3 s (Fig. 2a). The temperature rise curves are shown in Supplementary Fig. 1. X-ray Diffraction (XRD) analysis comparison of different synthesis temperatures shows that the precursor powders are completely converted to a single phase without any obvious impurity peaks28 when the reaction temperature is between 1100 °C and 1300 °C (Supplementary Fig. 2). Five peaks attributed to the (111), (200), (220), (311), and (222) planes were observed, confirming that the TM6 synthesized by UHS can be indexed to a disordered rock salt phase (Fm-3 m) (Fig. 2b)28,33. TM6 maintains a stable single-phase structure within the temperature range of 1100 to 1300 °C, indicative of excellent thermal stability. As the temperature rises to 1400 °C, the transition metal elements in the high oxidation states undergo reduction to the metallic elements due to the influence of the reducing atmosphere (Supplementary Figs. 2, 3).

a Rapid synthesis of TM6 with UHS at 1100 °C in 3 s. b XRD pattern of TM6 synthesized by UHS. c TEM-EDS mapping of the TM6 particle. d XPS spectra of Mn 2p. SEM images of TM2 (e), TM4 (f) and TM6 (g) sintered at 1150 °C for 10 s. h Comparison of synthesis temperature and sintering temperature of TM2, TM4, and TM6.
The TM6 was characterized using transmission electron microscopy (TEM)/energy-dispersive spectroscopy (EDS) to analyze the distribution of various elements. The uniform elemental distribution suggests fast reaction kinetics and rapid mixing of the precursors at 1100 °C (Fig. 2c). X-ray photoelectron spectroscopy (XPS) was conducted to determine the valence states of the metal elements (i.e., Mn, Co, Cr, Ti, and Nb). Mn has valence states of both +2 and +3 (Fig. 2d). Co is in the +2 valence state, while Cr is in the +3 valence state (Supplementary Fig. 4). Ti and Nb, utilized to stabilize the positive electrode framework, exhibit oxidation states of +4 and +5, respectively (Supplementary Fig. 4). Notably, the transition metal elements are not reduced during the rapid synthesis process and form a stable high-entropy rock salt structure with the designed valence states due to the short sintering time of only 3 s. Elemental analysis confirms that the metal ratios in the as-synthesized materials closely align with the target compositions (Supplementary Table 1). Therefore, UHS technology is an effective way for the rapid synthesis of TM6, and its excellent thermal stability gives TM6 the potential to co-sinter with electrolytes at high temperatures.
To verify the impact of the increasing number of cations on the synthesis and sintering temperature of rock salt positive electrodes (DRXs), precursors with varying cation compositions (TM2, TM4, TM6) were subjected to thermogravimetric analysis (TGA). The precursor mass of TM6 stabilized at 680 °C, whereas TM4 (Li1.3Mn2+0.2Mn3+0.2Ti0.1Nb0.2O1.7F0.3) and TM2 (Li1.3Mn3+0.4Ti0.3O1.7F0.3) reached stability only at 930 °C and 960 °C, respectively. This indicates that the synthesis temperatures of TM6 were 250 °C and 280 °C lower than those of TM4 and TM2 (Supplementary Fig. 5). Furthermore, we investigated the sintering behavior of positive electrode materials using UHS technology. TM2, TM4, and TM6 underwent one-step reaction sintering at 1150 °C for 10 s, resulting in successful phase formation (Supplementary Fig. 6). Scanning Electron Microscopy (SEM) analysis indicated that the grain size increased with an increasing number of cations during sintering at 1150 °C for 10 s. Specifically, TM6 exhibited a densification phenomenon in grain size, while the grain size of TM2 and TM4 appeared more dispersed (Fig. 2e–g). The densification of TM2 and TM4 occurred only when the temperature was raised to 1250 °C for 10 s (Supplementary Fig. 7), suggesting that the densification temperature of TM6 is 100 °C lower than that of TM4 and TM2 (Fig. 2h).
Chemical stability of TM6 and LLZTO
To improve the interface contact between the positive electrode and electrolyte, a high co-sintering temperature is necessary, requiring a highly chemically stable temperature (Tstable). Therefore, we analyzed the chemical stability between TM6 and LLZTO by heating to 1000 °C at a rate of 10 °C/min. In situ XRD shows that no reaction between TM6 and LLZTO occurs when the temperature exceeds 800 °C (Fig. 3a). Compared to traditional positive electrode materials, TM6 exhibits superior chemical stability when co-sintered with LLZTO12,13,34,35,36. However, the impurity phase LaMnO3 appears from 800 °C (Supplementary Fig. 8), which is unfavorable for interface stability and Li+ transport. The differential scanning calorimetry (DSC) data revealed that as the temperature increased, TM2 reacted with LLZTO first at 598 °C, followed by TM4 at 605 °C (Fig. 3b). In contrast, a small peak only appeared at 835 °C for the TM6 and LLZTO system, indicating that the chemical stability of the DRX positive electrode and LLZTO gradually increases with an increasing number of cations.

a In situ XRD patterns of LLZTO and TM6. b DSC of TM2 and LLZTO, TM4 and LLZTO, and TM6 and LLZTO. EDS mapping (c) and Line profiles (d) of the TM6 | LLZTO interface. e TEM images of TM6 | LLZTO interface. f XRD patterns of LLZTO and TM6 heated by UHS. g Comparison of chemical stability between different positive electrodes and LLZTO. h Each individual component of TM6 vs LLZTO pseudobinary phase diagram. The most likely reaction products are shown in Supplementary Table 2.
By employing UHS technology, the nonequilibrium rapid heat treatment conditions further extend the stable temperature range for LLZTO and TM6. Unprocessed TM6 and LLZTO particles exhibited a dispersed distribution, whereas a notable improvement in bonding was observed after heating treatment at 1100 °C for 3 s (Supplementary Fig. 9). Following heating at 1100 °C for 3 s, no detectable side reaction or cross-diffusion between LLZTO and TM6 (Fig. 3c, d, Supplementary Fig. 10). The TEM image (Fig. 3e) reveals that the TM6 | LLZTO interface exhibits a robust binding at the nanometer scale. On the right side, the lattice spacing measures 0.1635 nm, corresponding to the (222) plane of TM6 crystallization, while on the left side, the lattice spacing measures 0.5207 nm, consistent with the (211) plane of LLZTO crystallization. Similarly, the XRD patterns and Raman spectra also show that LLZTO and TM6 remain stable at 1100 °C for 3 s without any side reactions (Fig. 3f, Supplementary Fig. 11). The XPS results show that all transition metal elements maintain the designed valence states after co-sintering (Supplementary Fig. 12). However, after heating treatment at 1100 °C for 30 s, interdiffusion between the elements of TM6 and LLZTO is observed (Supplementary Fig. 9), with the emergence of the impurity phase LaMnO3 (Fig. 3f), highlighting the effectiveness of short sintering durations in inhibiting side reactions. Compared to conventional furnace treatment, UHS treatment generally increases the chemical stability temperature of the positive electrode materials and LLZTO (Fig. 3g and Supplementary Figs. 13–15). This further suggests that the rapid nonequilibrium heat treatment process effectively suppresses side reactions and cross-diffusion through kinetic control.
TM6 and LLZTO exhibit higher chemical stability compared to other positive electrode materials, demonstrating the significance of the entropy stabilization effect. As a result, we study the chemical stability between each individual component of TM6 and LLZTO. First-principles calculations and XRD patterns reveal that rock salt oxides (MnO, CoO) exhibit higher reaction energy and chemical stability with LLZTO compared to non-rock salt oxides (Fig. 3h, Supplementary Figs. 16–21). Interestingly, XRD patterns of powders obtained by mixing LLZTO and the precursor powders of TM6 do not have secondary peaks after co-sintering at 1100 °C for 3 s (Supplementary Fig. 22). This result indicates that the large configurational entropy and rapid heating process facilitate fast reaction kinetics, enabling non-rock salt oxides to integrate into stable rock salt structured oxides, while the short reaction time ensures chemical stability between TM6 and LLZTO.
TM6 | LLZTO interface characterization
The in situ reactive sintering of the positive electrode on the SSE substrate is beneficial in achieving a robust interface. Leveraging the rapid reaction kinetics and favorable chemical stability between HE-DRXs and LLZTO provides an opportunity for in situ reactive sintering. Using UHS technology initiates a rapid reaction among precursor powders, resulting in the formation of pure HE-DRXs and a bilayer structure (Fig. 4a). The heating and cooling curve of this process is shown in Supplementary Fig. 1. The cross-section SEM of the bilayer structure reveals the uniform distribution of HE-DRXs on LLZTO (Fig. 4b), with HE-DRX particles robustly growing on LLZTO (Fig. 4c, d). The short reaction duration of merely 3 s effectively suppresses element diffusion and delays the occurrence of side reactions between HE-DRXs and LLZTO (Fig. 4e, Supplementary Fig. 23). XRD patterns further confirm the absence of second phases during the in situ synthesis (Fig. 4f). XPS analysis provides additional confirmation that the transition metal elements remain unaltered during the rapid synthesis process, forming a stable high-entropy rock salt structure (Supplementary Fig. 24).

a Schematic of the in situ synthesis of HE-DRXs on dense LLZTO. SEM images (b–d) and SEM/EDS mapping (e) of TM6 | LLZTO interface. f XRD of the in situ synthesized TM6. g Zoomed in EIS of TM6 symmetric cells. h EIS of LiCoO2 symmetric cells.
The tight solid‒solid contact and excellent chemical stability are favorable to reduced interface impedance. We assembled TM6 and LiCoO2 (LCO) symmetric cells (Au|TM6 | LLZTO | TM6|Au and Au|LiCoO2 | LLZTO|LiCoO2 | Au) to quantify the interface impedance and performed electrochemical impedance spectroscopy (EIS) measurements. By analyzing the EIS fitting curves of symmetric cells (Supplementary Fig. 25), we calculated that the area-specific resistance (ASR) at the interface of TM6 | LLZTO is 31.6 Ω·cm2 (Fig. 4g, Supplementary Fig. 26 and Supplementary Table 3), whereas that of the LiCoO2 | LLZTO interface is 23,175.5 Ω·cm2 (Fig. 4h, Supplementary Table 4). Notably, the ASR of the TM6 | LLZTO interface is approximately 700 times lower than that of the LiCoO2 | LLZTO interface, indicating a substantial reduction in resistance at the TM6 | LLZTO interface compared to the LiCoO2 | LLZTO interface. As the temperature increases, the quasi-semicircle arc gradually disappears, and the total ASR of the TM6 | LLZTO interface and LLZTO drops to less than 10 Ω·cm2 at 150 °C (Supplementary Fig. 27), showing promising potential for enhancing the electrochemical performance of ASSLBs.
Electrochemical performance
We assembled all-solid-state Li |LLZTO | TM6 batteries (TM6-ASSLBs) featuring a highly stable HE-DRXs|LLZTO interface. To improve Li-ion conductivity, a small quantity of LLZTO was added to the positive electrode side at a mass ratio of 1:5 (LLZTO: HE-DRXs), while the load of active material was 1.3 mg/cm2. The electrochemical performance of the TM6-ASSLBs was evaluated via galvanostatic cycling. Voltage profiles exhibit characteristic plateaus consistent with TM6 behavior (Fig. 5a), further demonstrating the successful in situ synthesis of TM6 using the UHS technique. After two repeated experiments, cycling between 1.7 and 4.7 V at a specific current of 25 mA/g yielded a specific capacity of 239.7 ± 2 mAh/g at 150 °C. Even under a higher specific current of 100 mA/g, the battery retains a high capacity of 114.5 mAh/g, showing remarkable rate capability (Supplementary Fig. 28).

a Charge/Discharge profiles of the all-solid-state battery at different rates from 25 mA/g to 100 mA/g. XRD patterns (b) and SEM/ EDS images of the HE-DRX-ASSLBs c before and d after cycling. e Cycling performance and f Coulombic efficiency of the HE-DRX-ASSLBs at 25 mA/g. All electrochemical measurements were performed at 150 °C without organic electrolyte and external pressure.
The XRD patterns of the TM6 | LLZTO interface exhibit no secondary peaks after cycling, indicating excellent electrochemical stability between TM6 and LLZTO (Fig. 5b). Despite slight volume expansion of the HE-DRXs following charge/discharge tests, the TM6 | LLZTO interface remains robust, showing no signs of interface cracking, and no migration of transition metals is observed (Fig. 5c, d). While the utilization of Mn-based DRX positive electrodes (including HE-DRX) in liquid batteries often results in poor cycle stability due to side reactions between the positive electrode and electrolyte (Supplementary Fig. 29), the construction of a highly stable HE-DRX | LLZTO interface effectively addresses these issues. TM6 demonstrates superior cycling performance in ASSLBs compared to liquid batteries (Supplementary Fig. 29)28,37,38,39,40,41,42. This stable interface also leads to the excellent cycle stability of ASSLBs. The HE-DRX-ASSLB was cycled 100 times at 25 mA/g, maintaining a high reversible capacity of 224.43 mAh/g with a capacity retention rate of 95% (Fig. 5e, f). We provide a summary of recent publications reporting on the performance of ASSLBs based on garnet (Supplementary Table 5). The results indicate that the HE-DRXs-ASSLBs significantly improve electrochemical performances in terms of the specific capacity, cycle life, and rate capability.
We have demonstrated the efficacy of combining entropy stabilization effects with fast reaction kinetics to achieve a favorable balance of chemical stability and wettability. The high reaction temperature of the UHS technology enables the rapid synthesis of the TM6 positive electrode on the LLZTO surface, while the short sintering time and the excellent stability of the HE-DRXs ensure a conformal positive electrode interface without any side reactions. The stable TM6 | LLZTO solid-state interface also mitigates the issue of transition metal migration and interfacial side reactions typically observed with HE-DRXs in liquid electrolytes. Specifically, TM6-ASSLBs demonstrate an average specific capacity of 239.7 ± 2 mAh/g at 25 mA/g and exhibit a 95% capacity retention after 100 cycles at 150 °C.
Methods
Synthesis
Positive electrode precursors, including Li2CO3 (98%, Sinopharm Chemical Reagent Co. Ltd), CoO (98%, Aladdin), Cr2O3 (98%, Sinopharm Chemical Reagent Co. Ltd), Nb2O5 (98%, Sinopharm Chemical Reagent Co. Ltd), Mn2O3 (98%, Aladdin), MnO (99.0%, Sigma‒Aldrich) and TiO2 (98%, Sinopharm Chemical Reagent Co. Ltd), were mixed in a stoichiometric ratio. To compensate for the loss of Li, 10 wt % excess Li2CO3 was added. Following 6 h of ball milling with ethanol and ZrO2 balls, the suspension was removed for in situ synthesis, while the other precursors were dried and pelletized in an oven at 80 °C overnight. A green pellet of the HE-DRX precursor was prepared via cold isostatic pressing at a pressure of 230 MPa. Dense LLZTO pellets: Li6.4La3Zr1.4Ta0.6O12 powder was pressed into pellets under cold isostatic (DJYP-100TZ) pressure at 230 MPa, followed by sintering for 10 s using UHS technology to obtain dense LLZTO sheets20. The high temperature of the carbon heater was measured by a thermal infrared imager (628CH-L25).
Materials characterization
The crystal phases were examined by U1timaIV (Rigaku Corporation, Japan) XRD and scanned between 15° and 80° using Cu Kα radiation. To investigate the interface between the in situ-formed positive electrode and LLZTO, as well as the distribution of elements at the interface, SEM (FEI Apreo) and EDS spectra were employed at 20 kV. X-ray photoelectron spectroscopy (XPS) measurements were performed on an ESCALAB 250Xi (Thermo Scientific, UK) equipped with mono-chromated Al K alpha (energy 1486.68 eV). Raman spectra of the HE-DRXs and LLZTO were obtained by a Horiba LabRAM HR Evolution Raman spectrometer with a laser wavelength of 532 nm. TEM/EDS (FEI Talos F200X) was used to analyze the distribution of various elements in the TM6 particle and TM6 | LLZTO interface.
Preparation of the TM6-ASSLBs
The symmetric cells were configured as Au|TM6 | LLZTO | TM6|Au and Au|LCO | LLZTO | LCO|Au, with a positive electrode loading of 1 mg/cm2, and annealed at 1100 °C for 3 s using the UHS method. All preparation processes were conducted under an argon atmosphere inside a glove box. The battery assembly involved generating a layer of TM6 on dense LLZTO in situ via UHS. The LLZTO ink was then prepared by dispersing the LLZTO powder in ethanol at a concentration of 20 mg/ml for spray printing. LLZTO (LLZTO: TM6 = 1:5) was sprayed onto the bilayer structure using a solution-based printing process. UHS was then rapidly annealed at 900 °C to enhance contact and Li+ conduction. A 10 μm Li metal electrode was coated on the LLZTO surface at 260 °C. The negative electrode was coated with molten lithium at 260 °C on dense LLZTO. To coat the positive electrode particles with a current collector, carbon nanotube (CNT) solution in C5H9NO (NMP) (3 mg/ml) was dropped on the surface of the in situ positive electrode. The solution was dried at 150 °C, resulting in a 0.1 mg/cm2 coating of CNTs on the positive particles.
Electrochemical measurements
The EIS spectra of the symmetric Au|TM6 | LLZTO | TM6|Au and Au|LCO | LLZTO | LCO|Au cells were tested using Bio-Logic (SP-200) with a frequency range of 7 MHz to 100 mHz and a temperature range of 25–150 °C. EIS spectra of TM6-ASSLBs were investigated using Bio-Logic with a frequency range of 100 mHz to 1 MHz and a voltage of 30 mV. The galvanostatic charge/discharge of the TM6-ASSLBs was tested on a NEWARE (CT-4008Tn-5V10-mA-164) battery test system at 150 °C with current densities of 25, 50, and 100 mA/g without any organic electrolyte.
First-principles computation
To evaluate the trend of the reaction between each individual component in TM6 and LLZTO, a pseudobinary phase diagram of the interface of a series of precursors|LLZTO using the pymatgen package was constructed and determined the equilibrium thermodynamic driving force of the interfacial reaction by searching for components on the tie line between the electrode and the electrolyte component43,44,45,46. All density functional theory (DFT) calculations were performed using the Perdew−Burke−Ernzerhof (PBE) generalized-gradient approximation (GGA)47, implemented in the Vienna Ab initio Simulation Package (VASP)48. The projector augmented-wave (PAW)49 method was used to represent the core states. An energy cutoff of 520 eV and a k-point density of at least 1000/ (number of atoms in the unit cell) were used for all computations.
Data availability
The experiment data that support the findings of this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Miara, L. J. et al. Effect of Rb and Ta doping on the ionic conductivity and stability of the garnet Li7+2x-y(La3-xRbx)(Zr2-yTay)O12 (0 ≤ x ≤ 0.375, 0 ≤ y ≤ 1) Superionic conductor: a first principles investigation. Chem. Mater. 25, 3048–3055 (2013).
Gao, Z. et al. Promises, challenges, and recent progress of inorganic solid-state electrolytes for all-solid-state lithium batteries. Adv. Mater. 30, 1–27 (2018).
Jin, Y. et al. An intermediate temperature garnet-type solid electrolyte-based molten lithium battery for grid energy storage. Nat. Energy 3, 732–738 (2018).
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Janek, J. & Zeier, W. G. A solid future for battery development. Nat. Energy 1, 1–4 (2016).
Xu, D. et al. In situ generated fireproof gel polymer electrolyte with Li6.4Ga0.2La3Zr2O12 as initiator and ion-conductive filler. Adv. Energy Mater. 9, 1–12 (2019).
Xu, L. et al. Interfaces in solid-state lithium batteries. Joule 2, 1991–2015 (2018).
Sun, C., Liu, J., Gong, Y., Wilkinson, D. P. & Zhang, J. Recent advances in all-solid-state rechargeable lithium batteries. Nano Energy 33, 363–386 (2017).
Kim, Y., Waluyo, I., Hunt, A. & Yildiz, B. Avoiding CO2 improves thermal stability at the interface of Li7La3Zr2O12 electrolyte with layered oxide cathodes. Adv. Energy Mater. 12, 2102741 (2022).
Xiao, Y. et al. Understanding interface stability in solid-state batteries. Nat. Rev. Mater. 5, 105–126 (2020).
Kim, K. J. & Rupp, J. L. M. All ceramic cathode composite design and manufacturing towards low interfacial resistance for garnet-based solid-state lithium batteries. Energy Environ. Sci. 13, 4930–4945 (2020).
Ren, Y., Liu, T., Shen, Y., Lin, Y. & Nan, C. W. Chemical compatibility between garnet-like solid state electrolyte Li6.75La3Zr1.75Ta0.25O12 and major commercial lithium battery cathode materials. J. Materiomics 2, 256–264 (2016).
Vardar, G. et al. Structure, chemistry, and charge transfer resistance of the interface between Li7La3Zr2O12 electrolyte and LiCoO2 cathode. Chem. Mater. 30, 6259–6276 (2018).
Miara, L. et al. About the compatibility between high voltage spinel cathode materials and solid oxide electrolytes as a function of temperature. ACS Appl. Mater. Interfaces 8, 26842–26850 (2016).
Miara, L. J., Richards, W. D., Wang, Y. E. & Ceder, G. First-principles studies on cation dopants and electrolyte|cathode interphases for lithium garnets. Chem. Mater. 27, 4040–4047 (2015).
Ihrig, M. et al. Thermal recovery of the electrochemically degraded LiCoO2/Li7La3Zr2O12:Al,Ta interface in an all-solid-state lithium battery. ACS Appl. Mater. Interfaces 15, 4101–4112 (2023).
Ramkumar, B., So-young, K., Chan-woo, N., Aravindan, V. & Yun-Sung, L. LiBO2-modified LiCoO2 as an efficient cathode with garnet framework Li6.75La3Zr1.75Nb0.25O12 electrolyte toward building all-solid-state lithium battery for high-temperature operation. Electrochim. Acta 359, 136955 (2020).
Han, F. et al. Interphase engineering enabled all-ceramic lithium battery. Joule 2, 497–508 (2018).
Liu, T. et al. Achieving high capacity in bulk-type solid-state lithium ion battery based on Li6.75La3Zr1.75Ta0.25O12 electrolyte: Interfacial resistance. J. Power Sources 324, 349–357 (2016).
Wang, C. et al. A general method to synthesize and sinter bulk ceramics in seconds. Science 368, 521–526 (2020).
Chen, S. et al. Ultrafast sintering for ceramic-based all-solid-state lithium-metal batteries. Adv. Mater. 34, 1–10 (2022).
Zhang, R. Z. & Reece, M. J. Review of high entropy ceramics: design, synthesis, structure and properties. J. Mater. Chem. A 7, 22148–22162 (2019).
Oses, C., Toher, C. & Curtarolo, S. High-entropy ceramics. Nat. Rev. Mater. 5, 295–309 (2020).
Yeh, J.-W. et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004).
Akrami, S., Edalati, P., Fuji, M. & Edalati, K. High-entropy ceramics: review of principles, production and applications. Mater. Sci. Eng. R: Rep. 146, 100644 (2021).
Sarkar, A. et al. High-entropy oxides: fundamental aspects and electrochemical properties. Adv. Mater. 31, e1806236 (2019).
Cai, Z. et al. Thermodynamically driven synthetic optimization for cation-disordered rock salt cathodes. Adv. Energy Mater. 12, 1–8 (2022).
Lun, Z. et al. Cation-disordered rocksalt-type high-entropy cathodes for Li-ion batteries. Nat. Mater. 20, 214–221 (2021).
Kuo, C. H. et al. A novel garnet-type high-entropy oxide as air-stable solid electrolyte for Li-ion batteries. APL Mater. 10, 121104 (2022).
Bérardan, D., Franger, S., Meena, A. K. & Dragoe, N. Room temperature lithium superionic conductivity in high entropy oxides. J. Mater. Chem. A 4, 9536–9541 (2016).
Duan, C. Q. et al. New spinel high-entropy oxides (FeCoNiCrMnXLi)3O4 (X = Cu, Mg, Zn) as the anode material for lithium-ion batteries. Ceram. Int. 47, 32025–32032 (2021).
Ma, Y. et al. High-entropy energy materials: challenges and new opportunities. Energy Environ. Sci. 14, 2883–2905 (2021).
Huang, J. et al. Non-topotactic reactions enable high rate capability in Li-rich cathode materials. Nat. Energy 6, 706–714 (2021).
Tsai, C. L. et al. A garnet structure-based all-solid-state Li battery without interface modification: Resolving incompatibility issues on positive electrodes. Sustain. Energy Fuels 3, 280–291 (2019).
Uhlenbruck, S. et al. Cathode-electrolyte material interactions during manufacturing of inorganic solid-state lithium batteries. J. Electroceram. 38, 197–206 (2017).
Rosen, M., Finsterbusch, M., Guillon, O. & Fattakhova-Rohlfing, D. Free standing dual phase cathode tapes-scalable fabrication and microstructure optimization of garnet-based ceramic cathodes. J. Mater. Chem. A 10, 2320–2326 (2022).
Lun, Z. et al. Design principles for high-capacity Mn-based cation-disordered rocksalt cathodes. Chem. 6, 153–168 (2020).
House, R. A. et al. Lithium manganese oxyfluoride as a new cathode material exhibiting oxygen redox. Energy Environ. Sci. 11, 926–932 (2018).
Kan, W. H. et al. Evolution of local structural ordering and chemical distribution upon delithiation of a rock salt–structured Li1.3Ta0.3Mn0.4O2 cathode. Adv. Funct. Mater. 29, 1–11 (2019).
Wang, R. et al. Research on the kinetic properties of the cation disordered rock-salt Li-excess Li1.25Nb0.25Mn0.5O2 material. Solid State Ion. 339, 114999 (2019).
Wang, M. et al. Research progress in lithium-excess disordered rock-salt oxides cathode. Energy Environ. Mater. 5, 1139–1154 (2022).
Kitchaev, D. A. et al. Design principles for high transition metal capacity in disordered rocksalt Li-ion cathodes. Energy Environ. Sci. 11, 2159–2171 (2018).
Ong, S. P. et al. Python Materials Genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Richards, W. D., Miara, L. J., Wang, Y., Kim, J. C. & Ceder, G. Interface stability in solid-state batteries. Chem. Mater. 28, 266–273 (2016).
Ong, S. P., Wang, L., Kang, B., Ceder, G. & Li, - Fe - P - O2 phase diagram from first principles calculations. Chem. Mater. 20, 1798–1807 (2008).
Ong, S. P., Jain, A., Hautier, G., Kang, B. & Ceder, G. Thermal stabilities of delithiated olivine MPO4 (M = Fe, Mn) cathodes investigated using first principles calculations. Electrochem. Commun. 12, 427–430 (2010).
Perdew, J. P. et al. Rationale for mixing exact exchange with density functional approximations rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982–9985 (2010).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B – Condens. Matter Mater. Phys. 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0450401) and the National Natural Science Foundation of China (22209165). The numerical calculations in this paper were performed on the supercomputing system in the Supercomputing Center of the University of Science and Technology of China. Characterizations were partially carried out at the Instruments Center for Physical Science, USTC.
Author information
Authors and Affiliations
Contributions
C.W. and X.K. designed and conducted the experimental work. Z.J. and R.P. carried out the computational study. X.K., R.G., and C.L. carried out the co-sintering experiments, electrochemical measurements, and SEM imaging. W.X. and X.L. created the schematics. H.H. and Y.X. helped prepare the samples and conduct the XRD measurements. C.W. and X.K. analyzed the electrochemical data. K.Z. measured the in situ X-ray diffraction. C.W., X.K., L.Z., and C.Z. wrote and revised the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Swapna Ganapathy, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kong, X., Gu, R., Jin, Z. et al. Maximizing interface stability in all-solid-state lithium batteries through entropy stabilization and fast kinetics. Nat Commun 15, 7247 (2024). https://doi.org/10.1038/s41467-024-51123-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51123-0
