
Abstract
The electrocatalytic conversion of NO offers a promising technology for not only removing the air pollutant but also synthesizing valuable chemicals. We design an integrated-electrocatalysis cell featuring metal organic framework (MOF)-modified gas diffusion electrodes for simultaneous capture of NO and generation of NH4NO3 under low-concentration NO flow conditions. Using 2% NO gas, the modified cathode exhibits a higher NH4+ yield and Faradaic efficiency than an unmodified cathode. Notably, the modified cathode shows a twofold increase in NH4+ production with 20 ppm NO gas supply. Theoretical calculations predict favorable transfer of adsorbed NO from the adsorption layer to the catalyst layer, which is experimentally confirmed by enhanced NO mass transfer from gas to electrolyte across the modified electrode. The adsorption layer-modified anode also exhibits a higher NO3− yield for NO electro-oxidation compared to the unmodified electrode under low NO concentration flow. Among various integrated-cell configurations, a single-chamber setup produces a higher NH4NO3 yield than a double-chamber setup. Furthermore, a higher NO utilization efficiency is obtained with a single-gasline operation mode, where the NO-containing gas flows sequentially from the cathode to the anode.
Similar content being viewed by others

Introduction
As a major air pollutant, nitric oxide (NO) causes environmental issues such as photochemical smog, acid rain, particulate matter and ozone generation1,2,3. To control NO emission, the selective catalytic reduction (SCR) process has been widely utilized to convert NO to harmless dinitrogen4,5. However, such SCR process requires large amount of ammonia (NH3) or hydrogen (H2) gas as a reductant for NO conversion6,7,8,9. Meanwhile, NH3 is an indispensable component of nitrogen fertilizer, a vital chemical for both agriculture and chemical industry, and an important hydrogen-carrier10,11,12. Therefore, using valuable NH3 to treat NO-containing waste gas should be replaced by a more resource-efficient method. Owing to an intermediate valence state of nitrogen, NO would be a promising reactant for generating valuable nitrogenous products such as ammonia, nitric acid, and ammonium nitrate through either reduction or oxidation. At present, the Haber-Bosch process and the Ostwald process are widely used in the industrial production of ammonia and nitric acid, respectively13,14,15. However, both processes consume a large amount of energy and emit substantial CO2 and other pollutants.
The recent development of the electrochemical conversion of nitrogenous species should provide an alternative solution and its synergistic coupling with CO2 reduction provides a promising strategy for the generation of ammonia and valuable nitrogen-containing organic compounds16,17,18,19,20,21. While the catalytic conversion of N2 is highly restricted due to the presence of the strong triple bond (N ≡ N, 948 kJ/mol), the N = O bond (204 kJ/mol) in NO with a much lower binding energy than the N ≡ N triple bond can be more easily activated to be converted to ammonia and nitric acid22,23. In addition, utilizing NO in waste gas should alleviate the environmental problems associated with NO emission24,25. Electrocatalysis is being intensively investigated as an environment-friendly conversion method and a series of electrocatalysts and electrodes have been applied to NO electro-conversion via reduction or oxidation26,27,28,29,30,31,32. In addition, the development of highly efficient single-atom catalysts further propelled the advancement in the field of NO electrocatalytic conversion33,34,35. To date, most of NO electrocatalysis conversion studies have used pure NO gas as the reactant to generate ammonia36,37,38,39 whereas the concentrations of NO in various emission sources and waste gas are very low. In addition, the low solubility of NO in aqueous solution causes low mass transfer efficiency40, which further limits the NO electrocatalytic conversion. To overcome the NO mass transfer limitation at the reaction interface, the configuration of the electrode design could be referenced from the oxygen reduction reaction (ORR) and carbon dioxide reduction reaction (CO2RR) that uses gas diffusion electrodes (GDEs)41,42,43,44. However, the utilization of low-concentration NO gas still remains a great challenge for electrocatalytic NO conversion. Even with the application of GDEs, it is difficult to concentrate NO from gas phase onto the reactive interface. In this context, metal-organic frameworks (MOFs) offer a promising solution for enriching low-concentration gases. With their porous structure and large surface area, MOFs are widely applied to gas adsorption, separation and purification45,46,47,48. Some MOFs such as UIO-66, HKUST, MOF-74 in which unsaturated metal sites play a key role, were reported to have a high capacity for NO capture49,50,51,52. Accordingly, combining MOFs with GDEs can be proposed as a feasible strategy for enhancing the electrocatalytic conversion of low-concentration NO.
In this work, an integrated electrocatalytic conversion of NO is proposed for the production of ammonium nitrate by directly using dilute NO containing waste gas, which couples both NO electro-reduction reaction (NORR, Eq. 1) and electro-oxidation reaction (NOOR, Eq. 2) in one cell. To improve the NO mass transfer from the gas phase to the electrolyte in a low NO concentration gas flow, a MOFs-modified hybrid GDE (MOFs-GDE) was prepared for NO capture and electro-conversion. A series of NORR and NOOR tests confirmed that MOFs-GDE loaded with Cu (for NORR) and Ni/NiO (for NOOR) catalysts showed higher NH4+ and NO3− generation yields and higher Faradaic efficiencies (FE) than the bare GDE in both 2% NO and 20 ppm gas flow. The experimental and theoretical analysis of NO adsorption properties supported that NO can transfer from the gas phase to the electrolyte through the MOFs-GDE layer. The integrated NO electrolysis cell with the bifunctional MOF-GDEs provides a promising solution for NO removal with the concurrent production of ammonium nitrate as a valuable resource (Eq. 3). While the typical SCR process for NO removal consumes ammonia as a reagent, the present integrated electrochemical conversion for NO removal generates ammonium nitrate as a value-added product on the contrary. This is a representative example of waste-to-resource conversion for circular economy.
Results
Cathode GDEs for NO electro-reduction
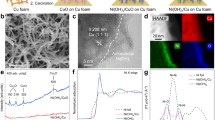
Figure 1a illustrates the hybrid GDE combined with the catalyst layer (copper nanowire, Cu NWs) and the MOFs layer on the carbon paper (CP), which was designed for the capture and the subsequent electro-conversion of low-concentration NO gas. In this hybrid structure, the MOFs layer on carbon paper captures and adsorbs NO from waste gas, and the catalyst layer converts NO. Figure 1b, the XRD patterns for CP/Cu NWs display that the broad peak around 25° corresponds to the commercial carbon paper ((002) face for graphitic carbon53) and other three diffraction peaks at 43.3°, 50.4° and 74.0° are indexed as the (111), (200) and (220) facet of Cu NWs (PDF#65-9026). The XRD spectra of the pristine Cu NWs and Cu NPs exhibit the similar diffraction peaks, indicating the same crystal facet for both Cu NPs and Cu NWs (Supplementary Fig. 1a). Compared with the CP/Cu NWs, some additional diffraction peaks appeared in the range of 2.5° to 50° for UIO-66/CP/Cu NWs, ZIF-8/CP/Cu NWs and ZIF-67/CP/Cu NWs, which are ascribed to UIO-66, ZIF-8 and ZIF-67. The XRD patterns of CP, pure MOFs, and the MOFs-loaded CP are also compared (Supplementary Fig. 1b). The XRD patterns for UIO-66, ZIF-8 and ZIF-67 are consistent with the literature data54,55,56 and those for UIO-66/CP, ZIF-8/CP and ZIF-67/CP match the diffraction peaks of UIO-66, ZIF-8, ZIF-67, respectively.

a The scheme for the preparation of MOFs-modified GDE; b the XRD patterns of CP/Cu NWs, UIO-66/CP/Cu NWs, ZIF-8/CP/Cu NWs and ZIF-67/CP/Cu NWs; the SEM images of c Cu NWs and d UIO-66; the cross-section SEM images of e CP and f UIO-66/CP/Cu NWs (hybrid GDE); the EDS elemental mapping images of g C, h Zr, and i Cu. Source data are provided as a Source Data file.
The SEM images show that Cu NWs have a nanowire morphology with an average diameter of 100 nm and an average length of 20 µm (Fig. 1c) while Cu NPs showed a nanosphere morphology with an average particle size of 300 nm (Supplementary Fig. 2a). UIO-66 displayed an octahedral morphology with an average size of 200 nm (Fig. 1d). ZIF-8 and ZIF-67 show a cubic structure with an average size of 300 nm (Supplementary Fig. 2b, c). The cross-sectional SEM image of commercial carbon paper shows that CP is composed of bundled fibers (Fig. 1e), while that of the prepared UIO-66/CP/Cu NWs GDE exhibits a hybrid structure mixed with UIO-66 and Cu NWs (Fig. 1f). The EDS elemental mapping of the hybrid UIO-66/CP/Cu NWs GDE shows that the C and Zr elements are evenly distributed over the whole GDE layer whereas the Cu element is mainly distributed on the surface of GDE (Fig. 1g–i). The cross-section SEM image and EDS elemental mapping of ZIF-8/CP/Cu NWs (Supplementary Fig. 3) and ZIF-67/CP/Cu NWs (Supplementary Fig. 4) also exhibit the similar structure and elemental distribution as UIO-66/CP/Cu NWs.
Figure 2a and Supplementary Fig. 5 illustrates the flow cell reactor setup for the electrocatalytic NORR that employs the MOFs-modified hybrid GDE through which the NO-containing gas passes. To find an optimized condition for NORR, linear sweep voltammetry (LSV) measurements for NORR were conducted on CP/Cu NPs GDE under different electrolytes (0.5 M K2SO4, KCl, and KHCO3) and different concentrations of NO gas (0%, 2%, and 10%). (Supplementary Fig. 6) The cathodic current progressively increased with increasing the NO concentration from 0% to 2% and 10%, which indicates that the NORR on the GDE is mass-transfer limited. As the highest cathodic current was obtained in 0.5 M K2SO4, K2SO4 was employed as the supporting electrolyte in this study. As for the optimal morphology of Cu catalyst, Cu NPs and Cu NWs were compared for NORR. Cu NWs exhibited consistently higher performances than Cu NPs for the NORR current, the NH4+ generation rate, and NH4+ FEs in both 10% and 2% NO gas flow (Supplementary Fig. 7 and Supplementary Fig. 8). This indicates that one-dimensional Cu NWs are more favorable for NORR than Cu NPs. Therefore, Cu NWs were chosen as the catalyst to be loaded on GDE in this study.

a Schematic illustration of the electrocatalytic NORR in a flow cell; b The LSV curves for NORR on CP/Cu NWs and UIO-66/CP/Cu NWs in 0.5 M K2SO4 (iR compensation was not applied); c the NH4+ generation rate and d the Faradaic efficiencies for NORR with 2% NO; e the NH4+ generation rate in 20 ppm NO; f the time-depended NH4+ generation rate for NORR at -0.8 V (vs. RHE) in 20 ppm NO; g the stability test for UIO-66/CP/Cu NWs at −0.5 V (vs. RHE) in 2% NO gas flow. Source data are provided as a Source Data file.
The most critical challenge in NORR is that the NO concentration from various emission sources is typically in the ppm range under which NORR should be highly limited by mass transfer. To address the mass transfer limitation, MOF materials were incorporated onto carbon paper to enhance NO adsorption capacity and facilitate the subsequent charge transfer to NO on the GDE. Figure 2b compares the LSV measurements on UIO-66/CP/Cu NWs in 2% NO and 20 ppm NO. The UIO-66/CP/Cu NWs demonstrated higher reduction currents compared to the CP/Cu NWs in both high (2%) and low (20 ppm) NO concentrations. The cathodic current on UIO-66/CP/Cu NWs was 15.7 mA/cm2 higher (in 2% NO gas flow) and 13.8 mA/cm2 higher (in 20 ppm NO gas flow) than on CP/Cu NWs at -1.2 V (vs. RHE). This validated that the MOFs loading on the CP GDE promoted the mass transfer of the low-concentration NO gas onto the Cu catalytic sites on the GDE. It is noted that there was an optimal loading of MOFs over which the overall NORR efficiency decreases. The LSV results show that loading 10 mg UIO-66 on GDE (1 cm2) achieved the highest NORR current, NH4+ generation rate, and NH4+ FEs in 2% NO gas flow whereas loading 20 mg UIO-66 reduced them (Supplementary Fig. 9). This suggests that excessive MOFs loading hinders the NORR because the excessive loading of MOFs increases the holding capacity of NO beyond the optimum to retard its subsequent transfer to the Cu catalyst. The MOFs-induced enhancement in the reduction current (compared with CP/Cu NWs) were also observed for other kind of MOFs as in ZIF-8/CP/Cu NWs and ZIF-67/CP/Cu NWs (with 10 mg loading of ZIF-8 and ZIF-67) (Supplementary Fig. 10). All three MOFs loaded on GDE exhibited higher NH4+ yield than CP/Cu NWs in 2% and 20 ppm NO and the MOFs enhancement effect is more prominent in low NO concentration flow condition (see Fig. 2c, e).
In 2% NO gas flow, the MOFs enhancement effect was also reflected in the NH4+ FE (Fig. 2d). In particular, the NH4+ generation rate on ZIF-8/CP/Cu NWs and UIO-66/CP/Cu NWs were almost two times higher than on bare CP/Cu NWs at -0.9 and -1.0 V (vs. RHE) under 20 ppm NO gas flow (Fig. 2e). MOFs is more efficient in facilitating the NORR under the low concentration condition. Incidentally, the production of NH4+ was insignificant in the absence of Cu NWs on the bare CP, ZIF-8/CP, UIO-66/CP, and ZIF-67/CP, which confirmed the role of Cu as the active sites (Supplementary Fig. 11). Figure 2f, the continuously generated NH4+ on these MOFs modified GDEs displayed a higher NH4+ yield than the bare CP/Cu NWs, these results suggested that MOFs modified GDEs could capture more NO molecular from low-concentration gases. The time profiles of NORR shows that the concentration of NH4+ is maintained consistently higher over UIO-66/CP/Cu NWs than CP/Cu NWs throughout the reaction time under both 2% and 20 ppm NO gas flow conditions (Supplementary Fig. 12), which confirms the role of MOFs in facilitating NORR in both high and low concentration conditions. Furthermore, UIO-66/CP/Cu NWs exhibited a stable NORR performance. The reduction current density (around 20 mA/cm2) was maintained for 24 h and the corresponding NH4+ FE decreased by 14% only in 24 h under the continuous flow of 2% NO gas (Fig. 2g).
The enhanced NORR performance of the MOF-modified GDE confirms the role of MOF in capturing low-concentration NO and subsequently transferring the adsorbed NO to the catalytic sites (Fig. 3a). The N2 adsorption/desorption isotherms (Fig. 3b, Supplementary Fig. 13) and the Brunauer-Emmett-Teller (BET) surface areas (Supplementary Tables 1, 2) of CP, MOFs, UIO-66/CP, ZIF-8/CP and ZIF-67/CP were measured to evaluate the adsorption property. With the loading of MOFs, the surface areas of the modified GDEs were significantly increased. In Fig. 3b, all the prepared MOFs-modified GDEs showed type I isotherms, suggesting the presence of micropores in which NO molecules should be captured. The NO adsorption capability of MOFs-GDEs was examined using NO temperature-programmed desorption (NO-TPD). According to the NO-TPD results (Fig. 3c), CP has weak NO adsorption capacity but the MOFs-GDEs exhibit highly enhanced NO adsorption performance (both higher peak intensities and temperatures), suggesting more adsorption sites and stronger adsorption energy. The desorption peak at 148 °C for ZIF-8/CP and ZIF-67/CP is ascribed to the physisorbed NO, while the peaks at higher temperature are assigned to chemisorbed NO. The higher NO adsorption capacity on the prepared MOFs-GDEs enhances the NO concentration at the gas-solid-liquid interface. However, if NO is too strongly bound in MOFs-GDEs, its subsequent desorption and transfer to the catalytic sites would be retarded, which may results in a lower electro-conversion efficiency. To estimate the NO transfer efficiency, the NO molecules that are successfully transferred from the gas phase to the electrolyte solution through the MOFs-GDEs (without catalyst) were quantified by immediately oxidizing the transferred NO to nitrate ion by H2O2 (see Methods and Supplementary Fig. 14). Table 1 shows the NO transfer flux and NO transfer efficiency through ZIF-8, UIO-66 and ZIF-67 modified GDEs with 2% NO gas flow are determined to be 1055.8, 1124.5 and 1185.7 µmol/cm2*h with the corresponding the transfer efficiency of 7.9%, 8.4% and 8.8%, respectively. In comparison, the NO transfer flux and transfer efficiency through the bare CP were 971.4 µmol/cm2*h and 7.2%. When NO concentration decreased to 20 ppm, both NO transfer flux and efficiency through the MOFs-GDEs were significantly reduced (Table 1), which indicates that the NO transfer through the MOFs-GDEs is clearly enhanced under higher NO concentration in the gas phase. Nevertheless, even under 20 ppm NO gas flow, the NO transfer flux and efficiency through the MOFs-GDEs were also enhanced from those of bare CP, confirming the successful role of MOFs in capturing and transferring NO from the low concentration of NO gas.

a MOFs-modified hybrid GDE for NO capture and conversion from low concentration NO gas; b the nitrogen adsorption-desorption isotherms of CP, UIO-66/CP, ZIF-8/CP and ZIF-67/CP; c the NO-TPD spectra of CP, UIO-66/CP, ZIF-8/CP and ZIF-67/CP (MS, mass spectrometry); d Gibbs free energy of adsorption of NO on CP, UIO-66, ZIF-8, ZIF-67 and Cu (111). Source data are provided as a Source Data file.
The density functional theory (DFT) computations were performed to estimate the NO adsorption energy on CP, ZIF-8, ZIF-67, UIO-66, and Cu (111) (Supplementary Fig. 15). Multilayer graphene model was used to simulate the CP. The atomic N of NO adsorbed on top of carbon atom, on the bridge site, and on the hollow site were calculated (Supplementary Fig. 16). The NO adsorption energies on ZIF-8 framework were calculated for five different NO adsorption sites, where NO is adsorbed on the top of zinc atom, in the center of 4-imidazeole rings, in the center of 6-imidazeole rings, on the top of imidazole ring, and in the center of cubic. (Supplementary Fig. 17) Owing to the similar structure with ZIF-8, the NO adsorption energies on ZIF-67 were also performed on such five sites. (Supplementary Fig. 18) The NO adsorption energies on UIO-66 were calculated on the top of Zr atom, on the top of benzene ring and in the center of framework (Supplementary Fig. 19). In addition, the NO adsorption energies on Cu (111) were also estimated (Supplementary Fig. 20). Supplementary Table 3 listed all of the NO adsorption energies for these models. Among these NO adsorption models, the most stable NO adsorption sites were observed on the top of the cobalt atom for ZIF-67, in the center of the 6-imidazole rings for ZIF-8, on the top of the benzene ring for UIO-66, on the top site of carbon, and on the top site of Cu (111). Figure 3d shows the NO adsorption energies for these most stable adsorbed species. As the NO adsorption energy on ZIF-67 is higher than on Cu (111), the effective diffusion of NO from ZIF-67 to the catalyst layer should be hindered, resulting in low NORR performance. The strong adsorption energy limits NO desorption from ZIF-6757. On the contrary, NO adsorption energies on UIO-66 and ZIF-8 are lower than that on Cu (111). The adsorption energy gradient facilitates NO transfer from MOFs to the catalyst layer, promoting the conversion of NO. Furthermore, the higher adsorption energy of UIO-66 compared to ZIF-8 allows UIO-66 to adsorb more NO molecules from the gas stream and efficiently transfer more NO to the catalytic surface. Such consideration of adsorption energetics is consistent with the highest NO transfer efficiency through UIO-66-GDEs. Similarly, the lowest NO adsorption energy on CP explained its low performance for NO capture and conversion.
Anode GDEs for NO electro-oxidation
In addition to NORR, NO electro-oxidation reaction (NOOR) leading to the generation of nitrate was investigated. Combining NORR on cathode with NOOR on anode can covert NO to ammonium nitrate with utilizing electricity more efficiently. Since nickel based catalysts have been widely used in electrocatalytic oxidation reactions, NiO and heterojunctioned Ni/NiO were prepared for anodic NOOR (Fig. 4a). These Ni based catalysts were prepared by calcining Ni(OH)2 precursors under different atmospheres (See Methods). In Fig. 4b, the XRD patterns of NiO Air500 and NiO Ar500 exhibited single crystal phase of NiO with the diffraction peaks identified to be cubic NiO (PDF#04-0835), and three main diffraction peaks at 37.3°, 43.3° and 62.9° were indexed as (111), (200) and (220) facet of NiO. XRD pattern of NiO HA500 (annealing in a H2/Ar mixture gas) showed the mixed phases of both NiO and Ni; the diffraction peaks at 44.5°, 51.8°, and 76.4° were indexed as (111), (200), and (220) facet of cubic Ni (PDF#65-2865). This suggests that Ni/NiO with heterojunction was formed after annealing in a H2/Ar mixture gas flow. The XRD patterns of the prepared NiO loaded GDEs exhibited broad peak around 25° which is identified as the carbon paper and also showed the NiO (or Ni/NiO) characteristic peaks (Supplementary Fig. 21a). UIO-66 modified GDE samples displayed the characteristic diffraction peaks for UIO-66 at 7.0° and 8.3° (Supplementary Fig. 21b). Moreover, XRD patterns for NiO HA400, NiO HA500 and NiO HA600 indicated that metallic nickel was preferentially formed from Ni(OH)2 precursors in a H2/Ar atmosphere (Supplementary Fig. 21c). The SEM image of NiO HA500 (Fig. 4c) showed a microsphere morphology assembled from nanosheet. The prepared NiO Air500 and NiO Ar500 also displayed a similar microsphere morphology (Supplementary Fig. 22). The SEM cross-section image and EDS mapping of CP/NiO HA500 GDE indicated that NiO HA500 microspheres were evenly loaded on carbon paper (Supplementary Fig. 23). The EDS mapping image for the cross-section of UIO-66/CP/NiO HA500 GDE indicated a hybrid GDE structure (Supplementary Fig. 24). Figure 4d, e, the TEM image and EDX mapping of NiO HA500 exhibited the formation of Ni/NiO heterojunction after annealing in H2/Ar mixture gas flow. The HRTEM image further proved the existence of Ni/NiO heterojunction (Fig. 4f), where the well-resolved lattice fringes with inter-planar distances of 0.203 nm and 0.208 nm were indexed as Ni (111) and NiO (200), respectively.

a The model of NiO and Ni/NiO; b The XRD patterns of NiO Air500, NiO Ar500 and NiO HA500; c the SEM image of NiO HA500; d the TEM image of NiO HA500; e the TEM EDX mapping of NiO HA500 (blue indicating Ni, pink indicating O); f the HRTEM image of NiO HA500; the XPS spectra of Ni 2p g and O 1 s h for NiO Air500, NiO Ar500 and NiO HA500. Source data are provided as a Source Data file.
XPS was employed to investigate the chemical state of the prepared samples and further confirmed the formation of Ni/NiO heterojunction. In Fig. 4g, the high-resolution Ni 2p XPS spectra of the prepare NiO samples show that the main valence state of Ni element is in the oxidized state. In Ni 2p3/2 spectra of NiO Air500, the peaks binding energy at 854.1 eV and 856.0 eV could be indexed to Ni2+ and Ni3+. The Ni 2p3/2 spectra of NiO Ar500 are similar to those of NiO Air500, suggesting that both samples are in the similar valence state. Compared with NiO Air500 and NiO Ar500, an obvious peak shift was observed in NiO HA500 with the binding energy decreasing by 0.6 eV. This binding energy red-shift indicates the formation of metallic nickel species (Ni0) at the binding energy of 853.5 eV58. The 2p3/2 spectra of NiO HA500 also show a peak located at 855.4 eV correspond to Ni2+. These results indicate that NiO HA500 sample consists of both metallic nickel and nickel oxide. Besides, the binding energy shift was also observed in the high-resolution O 1 s XPS spectra of these samples. The O binding energy in NiO Air500 was 529.5 eV, while that in NiO HA500 was 529.0 eV (Fig. 4h), which also confirms the formation of Ni/NiO heterojunction in NiO HA500.
The performance tests for NOOR were conducted on the NiO-loaded GDE using the flow cell setup (Fig. 5a). In 2% NO gas flow, the LSV results revealed that NiO HA500 exhibited the highest oxidation current among the prepared samples, indicating an optimal NOOR performance on NiO HA500 (Supplementary Fig. 25). In Fig. 5b, the production yield and FEs of NO2− and NO3− indicate that NO3− is the main product of NOOR. Among the tested anodes, CP/NiO HA500, the sample treated in a 5 wt% H2/Ar mixture gas at 500 °C, exhibited the highest NO3− yield of 165.6 µmol/h and FE of 89.4% at 1.8 V (vs. RHE). Meanwhile, the CP/NiO HA500 also showed the lowest NO2− yield and FE, suggesting a high NO3− selectivity. Increasing NO concentration to 10% resulted in an increased oxidation current, demonstrating the significance of the mass transfer process in NOOR (Supplementary Fig. 26a) and NiO HA500 still displayed the highest yield and selectivity for NO3− (Supplementary Fig. 26b). These results imply that Ni/NiO heterojunction effectively improves NOOR efficiency and the selectivity towards NO3−. Moreover, the annealing temperature (400, 500, 600 °C) for these NiO samples was optimized and CP/NiO HA500 exhibited the highest yield and selectivity for NO3− (Supplementary Fig. 27).

a The scheme for electrocatalysis NOOR in a flow cell setting; b the products yield and FEs on CP/NiO Air500, CP/NiO Ar500 and CP/NiO HA500 with 2% NO gas flow; c the free energy plot for NOOR on Ni (111), NiO (100) and Ni/NiO heterojunction calculated at 0 V (vs. RHE) (see Supplementary Fig. 28 for the structure); d the products yield and FEs on CP/NiO HA500 and UIO-66/CP/NiO HA500 with 2% NO and e 20 ppm NO gas flow; f the time profiles of nitrate and nitrite production via NOOR on CP/NiO HA500 and UIO-66/CP/NiO HA500 at 1.7 V (vs. RHE) under 20 ppm NO gas flow. Source data are provided as a Source Data file.
We modeled the Ni (111), NiO (100) and Ni/NiO (100) heterojunction as observed in the TEM image with density functional theory (DFT) to understand the active sites (Supplementary Fig. 28, Supplementary Fig. 29). To model the Ni/NiO(100) heterojunction, we considered a 13 atom two-layer Ni island on top of 4 × 4 × 4 cell of the observed NiO(100) surface. Ni island is considered instead of Ni slab connected across the periodic image to eliminate the undesired strain arising from lattice mismatch. We considered the following electrochemical oxidation reactions (Eqs. 4–10):
Figure 5c demonstrates that the theoretical limiting potential for the production of NO2−(aq) is similar for the NiO and Ni/NiO (1.17 eV vs 1.11 eV, respectively) limited by Eq. (6). However, the theoretical overpotential for the production of NO3−(aq) is 1.15 eV and 0.91 eV for NiO and Ni/NiO, respectively, thus, and NO3−(aq) production is expected to be faster, explaining the high yield and faradaic efficiency of the partially oxidized NiO of the CP/NiO HA 500. Furthermore, to elucidate the active sites for the NO oxidation process on the heterojunction surface, the free energy profiles for the NO oxidation process at various sites at the heterojunction interface site are compared (Supplementary Fig. 30). From Supplementary Fig. 31, it can be seen that the free energy change for the NO oxidation process is smallest at the heterojunction interface (where the intermediates interacting with the Ni and NiO simultaneously), and the *NO3 dissociation process is a downhill energy step, indicating that the active site for NO oxidation is located at the heterojunction interface.
As in the case of NORR, MOFs material was also introduced to improve the mass transfer process for NOOR in a low NO concentration. Due to a favorable NO adsorption ability, UIO-66 was also used as adsorption layer for NOOR. In Fig. 5d, the presence of UIO-66 on CP/NiO HA500 increased the yields of both NO2− and NO3− under 2% NO flow. The NO3− yield reached 187.5 µmol/h, and the NO2− yield reached 25.1 µmol/h, showing 24% increase in total NO oxidation (NO2− + NO3−). In 20 ppm NO gas flow, the LSV curves on UIO-66/CP/NiO HA500 showed a higher oxidation current than CP/NiO HA500 (Supplementary Fig. 32). UIO-66/CP/NiO HA500 also exhibited higher NO3− and NO2− generation yields compared to CP/NiO HA500 (Fig. 5e). Specifically, the NO3− yield on UIO-66/CP/NiO HA500 was nearly double that of CP/NiO HA500 at 1.8 V (vs. RHE). The results confirmed that UIO-66 modification also effectively improved the mass transfer process for NOOR in a low NO concentration gas. The time profiles of NO2− and NO3− generation on UIO-66/CP/NiO HA500 were also consistently higher than those on CP/NiO HA500 in 20 ppm NO gas at 1.7 V (vs. RHE). (Fig. 5f)
Integrated cell for NH4NO3 electro-generation
The above studies have focused on developing the separate half-reactions for NORR and NOOR, generating NH4+ and NO3− respectively as the main product. The concurrent production of NH4+ and NO3− can be combined for the production of NH4NO3, which is a valuable chemical in various industry and agriculture. To achieve this goal, an integrated NO electrocatalysis system was developed for NH4NO3 generation by feeding NO containing gas to both cathode and anode, separately. Figure 6a and Supplementary Fig. 33a illustrates a double chamber with double gasline (DC-DG) flow cell setting for the integrated NO electrocatalysis system, a Nafion membrane separating the anode chamber and cathode chamber. UIO-66/CP/Cu NWs (or CP/Cu NWs) serves as a working cathode and UIO-66/CP/NiO HA500 (or CP/NiO HA500) serves as a counter anode. The effect of MOF layer is clearly demonstrated when comparing the performance of the integrated cell between the bare GDEs and MOF-loaded GDEs systems under both 20 ppm NO and 2% NO (Fig. 6d vs. 6g). For example, in the DC-DG configuration with 2% NO (Fig. 6g), the integrated NO electrocatalysis generated 246 µmol/h of NH4+ and 452 µmol/h of NO3− at −0.9 V (vs. RHE) on UIO-66 modified GDEs, while it produced only 92 µmol/h of NH4+ and 119 µmol/h of NO3− on the bare GDEs. Note that the stoichiometric ratio of NO3− to NH4+ generated in the in the 2% NO DC-DG with MOF-loaded GDEs reached 1.84, which is close to the theoretical stoichiometric ratio of 1.67 (see Eq. 3). This close match of the NO3−/NH4+ ratio between the experimental and theoretical values strongly supports that the production of ammonium nitrate is generated via the separate production of nitrate and ammonium from NO conversion.

Integrated electrocatalytic NO conversion setup of a the double-chamber/double-gasline (DC-DG); b the single-chamber/double-gasline (SC-DG); c the single-chamber/single-gasline (SC-SG); d–f the yields of NH4+ and NO3- on UIO-66 loaded GDEs vs. bare GDEs under 20 ppm NO gas flow in a–c setup, respectively; g–i the yields of NH4+ and NO3- on UIO-66 loaded GDEs vs. bare GDEs under 2% NO gas flow in a–c setup, respectively. Source data are provided as a Source Data file.
A single chamber electrolysis (SC-DG) system without the membrane was also tested and compared for the generation of NH4NO3 (Fig. 6b and Supplementary Fig. 33b). The single chamber system exhibited the similar MOFs-enhancement effects, and the production of NO3− and NH4+ on the MOF-loaded GDEs was consistently higher than that on bare GDEs (Fig. 6e vs. 6h). In Fig. 6c and Supplementary Fig. 33c, another single chamber electrolysis (SC-SG) system that employs a single NO gasline (not double gasline as in Fig. 6a, b) that passes the cathode and the anode part serially is illustrated. This SC-SG setup should utilize NO more efficiently with minimizing the passing of unreacted NO. The MOF effect was verified again in both 20 ppm and 2% NO gas flow (Fig. 6f vs. 6i). In particular, under 20 ppm NO gas flow, the generation of ammonium nitrate on UIO-66 modified GDEs was twice as high as that on the bare GDE. In this single gasline cell, the NO-containing gas flowed from the cathode to the anode, allowing unreacted NO molecules from the cathode to be further utilized by the anode. As a result, the overall NO utilization efficiencies (the ratio of the combined output production of ammonium and nitrate to the input NO amount, see Methods) were markedly higher in the single gasline system than in the double gasline system (Fig. 7). Incidentally, in the single-chamber cell without membrane, NH4+ produced from NO reduction at the cathode may diffuse to the anode for oxidation, and NO3− produced from NO oxidation may diffuse to the cathode for reduction, potentially leading to the product loss. To check out this loss possibility, a control test of electrochemical degradation of NH4+ and NO3− was carried out in the SC-SG integrated cell, which allowed NH4+and NO3− to move freely between the cathode and anode. (Supplementary Fig. 34) The control electrochemical test results showed that neither NH4+ nor NO3− exhibited a notable decrease in concentration during the SC-SG integrated cell operation, confirming that the product (NH4NO3) loss via electrochemical conversion in the present experimental condition can be neglected.

The NO utilization efficiency in different cell setup employing UIO-66-loaded GDEs vs. bare GDEs under 20 ppm NO gas flow. Source data are provided as a Source Data file.
Discussion
Controlling NOx in various emission sources and air has been the important research topic in air pollution control technology. Given the widespread and frequent occurrence of NOx emissions in various fossil fuel combustion processes, significant research has been dedicated to this area. The widely adopted commercial method for NOx removal is the selective catalytic reduction (SCR) process, which relies on ammonia gas as a reagent—a resource of considerable value. An optimal and alternative strategy involves transforming NOx into valuable products, thereby offering a more sustainable solution than the conventional practice of using costly resources for NOx removal. This study introduces an electrochemical method to produce NH4+, NO3− and NH4NO3 as valuable products from dilute NO-containing gas. Owing to the low NO concentration in waste gases and its low solubility in water, NO conversion efficiency is limited by the slow mass transfer. To enhance the NO utilization efficiency, a hybrid-structured GDE was engineered to carry out the electro-reduction and electro-oxidation of NO simultaneously. This hybrid electrode features a NO-adsorbing MOF layer and a catalyst layer. In the electrocatalysis process, NO in the waste gas is initially captured by the MOF adsorption layer and then diffuses to the catalyst layer, where NO can be either reduced to NH4+ or oxidized to NO3−. Under 20 ppm of NO gas flow, the generation of NH4+ (NORR) and NO3− (NOOR) on MOF-modified GDEs was about two times higher than that on the bare GDE. The improvement of NO electro-conversion efficiency through MOFs-modified GDEs was further confirmed by a series of experimental characterizations and theoretical calculations. MOFs have shown a significant capability to adsorb NO from low-concentration gas. Importantly, a gradient in NO adsorption energy between the MOF and Cu catalysts aids in the transfer of adsorbed NO from the MOF layer to the catalytic layer, enhancing the process efficiency. An integrated NO electrocatalysis cell combining NORR and NOOR was developed for the joint-production of ammonium nitrate from dilute NO-containing gas. Following with concentration-separation and evaporation-crystallization steps, solid NH4NO3 can be obtained in the further industrial applications. This presents a promising alternative to traditional production methods, which could reduce both energy consumption and carbon emission. Among the various cell setups tested, a single chamber cell with a single gasline configuration exhibited superior performance in terms of NH4NO3 production yield and NO utilization efficiency. This innovative approach holds great promise for recycling NO-containing waste gases. Furthermore, MOF-modified GDEs offer a sustainable solution for capturing dilute NO gas and subsequently achieving waste-to-resource conversion. The MOF adsorption layers in GDEs should facilitate the direct utilization of specific gas components (e.g., NO, CO2) in waste gas and contaminated air through electrocatalytic conversion processes. This technology has the potential to serve as a valuable tool for reclaiming resources from waste gas, contributing to the realization of a circular economy.
Methods
Chemicals and reagents
All chemical reagents and gases used in this work were purchased and used as received without additional purification. NO (99.99%), N2 (99.999%), Ar (99.999%) and 20 ppm NO (N2 as balance gas) were purchased from DONGHAE GAS IND. The following chemicals were purchased from Sigma-Aldrich: K2SO4 ( ≥ 99.0%), KHCO3 ( ≥ 99.5%), KCl (≥ 99.0%), zirconium(IV) chloride (≥ 99.9%, ZrCl4), terephthalic acid (98.0%), 2-methylimidazole (99.0%), Zn(NO3)2·6H2O ( ≥ 99.0%), Co(NO3)2·6H2O ( ≥ 99.0%), ethylene glycol (≥ 98.0%, EG), Cu(OH)2, polyethylene glycol 2050 (PEG 2050), L-ascorbic acid (≥99.0%), Cu(NO3)2·3H2O (99%) ethylenediamine (≥99.5%), hydrazine (98%) EDTA-2Na (99%), NiSO4·6H2O (99%), urea (99%), sulfamic acid (≥99.0%), phosphoric acid (85 wt%), p-aminobenzenesulfonamide (≥99.0%), and N-(1-Naphthyl) ethylenediamine dihydrochloride (≥99.0%). Methanol (99.5%), ethanol (99.5%), N,N-Dimethylformamide (99.9%, DMF), NaOH (98%), KOH (95%), concentrated HCl (35% - 37%) and H2O2 (35.5%) were purchased from SAMCHUN Chemicals. Deionized water was purified through a Millipore water purification system with a resistivity of 18.2 MΩ cm (at 25 °C). Carbon paper (CP, GDS250,) was purchased from CeTech without any further treatment.
Synthesis of MOFs materials
To prepare UIO-66, 1.06 g of zirconium(IV) chloride (ZrCl4) and 0.76 g of terephthalic acid were dissolved in 60 mL DMF with stirring until a homogenous solution was formed. Then, such solution was transferred into a 100 mL Teflon autoclave and heated at 120 °C for 24 h. After cooling down, the prepared UIO-66 was collected and washed by centrifugation several times in ethanol54. To prepare ZIF-8, 6.5 g of 2-methylimidazole and 3.0 g of Zn(NO3)2·6H2O were dissolved in 80 mL and 40 mL of methanol. Then Zn(NO3)2·6H2O solution was slowly added into 2-methylimidazole solution with vigorous stirring for 24 h. The obtained product was centrifuged with methanol for several times and then dried at 60 °C under vacuum overnight55. To prepare ZIF-67, 2.5 g of 2-methylimidazole and 2.2 g Co(NO3)2·6H2O were dissolved in 60 mL and 30 mL methanol. Then Co(NO3)2·6H2O solution was slowly dropped into 2-methylimidazole solution with vigorous stirring for 24 h. After that, the obtained product was centrifuged with methanol for several times and then dried at 60 °C under vacuum overnight59,60.
Synthesis of Cu nanoparticles and Cu nanowires
Cu nanoparticles were synthesized according to the reference procedure. Typically, 25 mL ethylene glycol (EG) contained with 0.98 g Cu(OH)2 and 1.0 g PEG 2050 (polyethylene glycol, MW: 2050) heated to 80 °C under stirring for 30 min. An L-ascorbic acid (6.0 g) EG solution was heated to 80 °C under the same condition. Then the L-ascorbic acid solution was poured into the former flask, and the mixture was kept at 80 °C for 5 min. Finally, the product washed several times with ethanol via centrifugation61. In a typical synthesis process of Cu nanowires, 41.5 mL 0.1 mol/L Cu(NO3)2 aqueous solution was added into 833 mL of 10 M NaOH aqueous solution under vigorous stirring. After that, 6.225 mL ethylenediamine and 1.94 mL 35 wt% hydrazine aqueous solution was added in the above solution following with stirring. Then, such solution transferred to a fan oven under 60 °C for 2 hours. The afloat flocculent copper products washed with centrifugation several times in D.I. water, and such products stored in ascorbate acid containing ethanol solution to prevent oxidation62.
Synthesis of NiO nanoparticles
In a typical process, 0.3 g EDTA-2Na, 1.2 g of NiSO4·6H2O, and 0.3 g of urea were dissolved in 15 mL of D.I. water, respectively. Then the NiSO4 solution was added slowly to the EDTA-2Na solution with stirring, until the mixed solution color turned to deep blue. After that, the urea solution was also dropped into the above-mixed solution and stirred for 3 min. The pH value of the reaction system was adjusted to 6 by ammonia and a dilute sulfuric acid solution. The obtained solution was transferred to a 60 mL Teflon-lined autoclave and kept at 180 °C for 4 h. After the reactor cooled to room temperature, the green powder was collected by filtration and dried at 60 °C63. Subsequently, the NiO nanoparticles were obtained by annealing the above-mentioned green powders at different temperature (400, 500, and 600 °C) and different atmosphere (air, and Ar) for 2 h in a flow furnace. Those NiO HA samples came from the prepared NiO Air samples which further treated in 5% H2/Ar mixture gas flow for 10 minutes at different temperature.
Preparation of MOFs-modified GDEs
The MOFs modified GDEs were prepared using a dropping method. A carbon paper (CP) (15 mm × 15 mm) was used as the substrate for MOFs-modified GDEs. Firstly, 10 mg as-synthesized UIO-66, ZIF-8 or ZIF-67 was dispersed in 500 µL of ethanol under ultrasonic to prepare MOFs ink. Then, such MOFs ink was slowly dropped on one side of carbon paper to load MOFs. Then, the catalyst ink was prepared and dropped on another side of carbon paper which mixed 5 mg Cu NWs (or Cu NPs) or 10 mg NiO NPs, 950 µL of ethanol and 50 µL of Nafion solution (5 wt% in ethanol, Sigma-Aldrich). All control samples prepared by using the same dropping method, UIO-66/CP, ZIF-8/CP and ZIF-67/CP were prepared without dropping Cu NWs ink. CP/Cu NWs, CP/Cu NPs and CP/NiO were prepared without dropping MOFs ink.
Electrocatalysis tests and electrochemical measurements
All the electrocatalysis tests and measurements were conducted using a potentiostat (Gamry Instruments Reference 600) in a flow cell reactor with a standard three-electrode set up. In all of the electrochemical measurements, iR compensation was not applied. During NO electro-reduction, the prepared MOFs-modified GDE with Cu-based catalysts was served as the working cathode. Pt wire was served as the counter electrode and Ag/AgCl (in 3 M KCl) was used as the reference electrode. The external calibration method was used to calibrate the Ag/AgCl reference, a saturated calomel electrode (SCE) is employed for calibration. The calibration was performed by measuring the potential difference between the two electrodes. Unless otherwise specified, in general electrochemical tests, the anode and cathode were separated by a proton exchange membrane (Nafion 117, 1.5 cm × 1.5 cm). Nafion membrane need to be pretreated before use. Typically, the membrane was placed in a 1 mol/L H2SO4 solution and heated at 90 °C for 1 h. After that, using deionized water washed several times. Then, put it into a 5% H2O2 solution and treated at 90 °C for 1 h follow with deionized water wash several times. Finally, the treated membrane stored in deionized water for use. In the flow cell setting, both cathode chamber volume and anode chamber volume were 1 cm3 (1 cm × 1 cm × 1 cm), 10 mL electrolytes were circulated in both cathode and anode chamber controlled by a peristaltic pump with the flow rate of 2.0 mL/min.
The NO electro-reduction tests were carried out with varying the concentration of NO gas that flowed through the gas chamber with a flow rate of 250 mL/min controlled by a mass flow controller. As for the NO electro-oxidation setup, the prepared NiO-based GDE served as the working anode and Pt wire as the counter electrode for NO electro-oxidation. During the tests, 10 mL electrolyte (0.5 M K2SO4, pH = 6.8) was circulated into both cathode and anode chamber with the flow rate of 2.0 mL/min. Different concentration of NO gas or Ar gas flowed through the gas chamber with a flow rate of 250 mL/min. The settings for integrated NO electrocatalysis were similar to those of the single GDE system, except that both Cu-based GDEs and NiO-based GDEs were utilized in a single cell. This configuration allowed the simultaneous occurrence of both NORR and NOOR. Two gas chambers were installed on both side of the cathode and anode, and NO-containing gas passed through both gas chambers with the flow rate of 250 mL/min. A specific amount of 0.5 M K2SO4 electrolyte was circulated in the flow cell at the flow rate of 2.0 mL/min. Chronoamperometry (CA) method was used to test the performance for NORR and NOOR. The scanning rate of linear sweep voltammetry (LSV) measurements were 10 mV/s. The applied potential on the working electrode was rescaled to the reversible hydrogen electrode (RHE) reference using the following Eq. (11):
Products determination (NH4 +, NO2 − and NO3 −)
We employed the colorimetric method for analysis and detection of NH4+, NO3−, and NO2− as the high background concentration of electrolytes in the samples interfere with the ion chromatographic analysis of product ions. An ultraviolet-visible (UV–vis) spectrophotometer was utilized to detect the NORR and NOOR products of NH4+, NO2− and NO3− by recording the absorption intensity of characteristic peak, and calibration curves were used to calculate the product concentration (see Supplementary Fig. 35)64. The Nessler’s reagent as the color reagent was used to determine the concentration of NH4+65. Typically, a certain amount of cathode electrolyte for NO electro-reduction was sampled from the flow cell and diluted to 3 mL with 0.5 mol/L KOH aqueous solution, then 60 μL Nessler’s reagent was added into the diluted sample with stirring for 20 min. After that the absorption intensity at a wavelength of 420 nm was recorded for NH4+ concentration calculation. 1 M HCl and 0.8 wt% sulfamic acid solution were used for NO3− determination66. Briefly, a certain amount of anode electrolyte for NO electro-oxidation was sampled and diluted to the detection range. Then, 60 µL of 1 M HCl and 6 µL of 0.8 wt% sulfamic acid were added into 3 mL diluted samples. After 5 minutes, the absorption intensity at a wavelength of 220 nm and 275 nm were recorded. And the final absorption value was calculated by the following Eq. (12):
For the determination of NO2−, a mixture solution contained 4.0 g p-aminobenzenesulfonamide, 0.2 g N-(1-Naphthyl) ethylenediamine dihydrochloride, 50 mL D.I. water and 10 mL concentrated phosphoric acid was utilized as a color reagent67. Similar to determination NO3−, a certain amount of anode electrolyte was sampled and diluted to the detection range, firstly. Then, 60 µL of color reagent was added into the diluted sample with stirring for 20 min. After that, the absorption intensity was recorded at a wavelength of 540 nm. The calculation of these products were related to the concentration-absorbance plots. The Faradaic efficiency calculation for these products used the following Eq. (13):
Where n is the electron transfer number (NO to NH4+, NO to NO3− and NO to NO2−, the value of n = 5, 3 and 1), F is the Faraday constant (96500 C/mole e−), C is the concentration of N-containing products (mol/L), V is the electrolyte volume (L), Q is the total charge (C).
Measurements of NO transfer flux, transfer efficiency and NO utilization efficienciesh
To measure the NO transfer flux and transfer efficiency on MOFs modified GDE, NO molecular should be converted to NO3−, initially. This conversion process allowed for the quantification of the NO3− production rate, which served as an indicator of the transfer flux of NO. Then the amount of NO3− in electrolyte was analyzed by using color reagent as the same method as the determination of NO3− product for NO electro-oxidation. Hydrogen peroxide (H2O2) was used to oxidize NO molecular to NO3− according to the following chemical reaction Eq. (14):
In a flow cell setting as electrocatalysis tests without applied potential, the prepared MOFs modified GDEs (without catalyst loading) were used to separate the gas chamber and liquid chamber. In this configuration, NO containing gas flowed through the gas chamber and was adsorbed by MOFs modified GDEs (see Supplementary Fig. 14). Subsequently, the adsorbed NO diffused to liquid phase and convert to NO3− by H2O2. This configuration allowed for the investigation of NO adsorption and capture capacity from gas phase on the prepared GDEs, thereby evaluating their effectiveness as gas separation membranes. During the measurements, 10 mL mixture solution contained 0.5 M K2SO4 and 0.5 M H2O2 was circulated in the liquid chamber with a flow rate of 2.0 mL/min, and a series of NO containing gas continuously passed through the gas chamber with the flow rate of 250 mL/min. After running for 20 minutes, a certain amount of liquid were taken out and added the color reagent for NO3− determination following with the analysis by ultraviolet-visible (UV–vis) spectrophotometer. The calculations of NO3− amount were related to the concentration-absorbance plots. In this method, the concentration of NO3− equaled to the concentration of NO transfer amount. The calculation of NO transfer flux and transfer efficiency were according to the following Eqs. (15, 16):
For the integrated NO electrocatalysis section, the calculation of NO utilization efficiencies was according to the following Eq. (17):
Where the total n (NO) in double gas line system was combine both cathode gas and anode gas, and the total n (NO) in single gas line system was only one gas flow.
Characterization for GDE
The morphology of such prepared samples was characterized using a field emission scanning electron microscope (FESEM, JSM 7800 F). The X-ray diffraction (XRD) patterns for all the samples were obtained using an X-ray diffractometer (Ultima IV) with Cu-Kα radiation (λ = 1.54178 Å). The surface area was investigated by N2 adsorption measurements on Micromeritics TriStar II analyzer by the Brunauer-Emmett-Teller (BET) method. The NO-TPD measurements were performed by chemisorption analyzer (PCA-1200). Transmission electron microscopy (TEM) images were obtained using a JEM-1400 (JEOL Ltd., Japan) at 120 kV. The X-rays photo spectroscopy (XPS) of NiO samples were measured by Nexsa (ThermoFisherScientific) at a base pressure of 2.0 × 10−8 mBar with monochromated Al Kα (1486.6 eV) radiation, the C 1 s (284.8 eV) was used as the internal standard for charging correction.
Density functional theory calculation
The density functional theory (DFT) computations were performed using the Vienna ab initio simulation package (VASP)68,69. The interaction between the ionic core and valence electrons was described by the projector augmented wave method (PAW)70,71. The total energy convergence and the forces on each atom were set to be lower than 10−6 eV and 0.02 eV A−1. Energy cutoff of 400 eV for the plane wave basis set was used for the structure optimization. The construction of the MOFs models (UIO-66, ZIF-8 and ZIF-67) and carbon model utilized data from the crystallographic database. The 11 × 11 × 11 Monkhosrt-Pack generated k point mesh was used to sample the Brillouin zone for the primitive cells of bulk Cu, Ni and NiO72. A 4-atom primitive cell was built and optimized to define lattice parameters, the optimized lattice parameters of fcc Ni and NiO are 3.48 and 4.17 Å, respectively. For Cu and Ni slab, a 3 × 3 × 4 supercell was constructed from the primitive cell along the (111) direction. For NiO, a 4 × 4 × 4 supercell was built in the (100) direction. A 15 Å vacuum layer was added along the Z-direction to avoid interactions between periodic image replicas. During simulations, the bottom two layers of atoms were fixed, with the rest fully relaxed. For the slab models, 3 × 3 × 1 Monkhosrt-Pack generated k-points mesh was sampled from the Brillouin zone. We used the Gaussian Methfessel-Paxton smearing of 0.173. For ZIF-8, UIO-66, ZIF-67, gamma point in the Brillouin zone was sampled due to the large size of the cell. The Predew-Burke-Ernzerhor (PBE) functional with generalized gradient approximation was employed to describe the electron exchange and correlation energy69,74. To accurately describe systems containing Ni atoms, spin-polarized calculations were enabled for all Ni atoms. For the Ni slab model, the spin configuration was set to a ferromagnetic state; for the NiO slab model, the thermodynamically more stable Type-II antiferromagnetic phase spin arrangement was adopted75,76. Grimme’s DFT-D2 functional, was used to correct the dispersion forces. For the calculation of Ni and NiO system, we used the DFT + U correction where the U value of 6.3 eV was used for d electrons of Ni based on the previous studies75,77,78,79. For the heterojunction calculation, we considered the two layer Ni island on the NiO surface (100), as observed from the TEM images. We considered the Ni island instead of Ni slab connected across the periodic image in order to eliminate the undesired strain arising from lattice mismatch. All of the structures data for the optimized models are provided in Supplementary Data 1.
We modeled the oxidation reaction of NO(g) on Ni, NiO, and Ni/NiO with the Eqs. (4–10)80, the Eqs. (18–24) to calculate the Gibbs free energy of reaction, \(\Delta {G}_{i}\), of the above NO oxidation reaction in order are:
Where Gi indicates the Gibbs free energy of the species i, \({\mu }_{{{{{\rm{H}}}}}_{({{{\rm{aq}}}})}^{+}}\) and \({\mu }_{{e}^{-}}\) are the chemical potential of proton and electron, respectively, and \({G}_{{{{\rm{sol}}}},i}\) is the solvation energy of the species i. For the desorption reaction of anion species, NO2−(aq) and NO3−(aq), we adopted the formula from the Kamat et al80. The adsorption energies of NO(g) on carbon paper, ZIF-8, UIO-66, ZIF-67, and Cu (111) were calculated using \(\Delta {G}_{1}\). As the desorption reaction occur at the infinite dilution, we did not consider the dilution energy as it was considered in Kamat et al.80
The Gibbs free energy of species i, \({G}_{i}\), is calculated by using the following Eq. (25):
where \({E}_{{{{\rm{DFT}}}},i}\) is the DFT calculated energy, \({H}_{T,i}\) is the enthalpy of the species i from the vibrational contribution for surface species, as well as the rotational and translational contribution for the aqueous and gaseous species, T is the temperature, \({S}_{T,i}\) is the entropy of the species i from the vibrational contribution for surface species, as well as the rotational and translational contribution for the aqueous and gaseous species, and \({G}_{{{{\rm{sol}}}},i}\) is the Gibbs free energy of solvation. \({H}_{T,i}\) contains the zero-point energy (ZPE) corrections from the vibrational contribution calculated using the harmonic oscillator model. In the case of surface, or the substrate, we did not consider \({H}_{T,i}\), and \(T{S}_{T,i}\). The solvation energy was not considered for NO(g), H2(g) or, H2O(l). For the Gibbs free energy of H2O(l), we calculated the Gibbs free energy of the gaseous H2O(g) at the vapor pressure of 0.0613 atm81.
The solvation free energy of the solute i, \(\Delta {G}_{{{{\rm{sol}}}},i}\), at infinite dilution was calculated using implicit solvent method22,82,83,84. The solvation free energy is calculated as the difference between energy in the solvated environment, \({E}_{{{{\rm{DFT}}}},{{{\rm{s}}}}{{{\rm{ol}}}},i}\), and energy in the vacuum, \({E}_{{{{\rm{DFT}}}},i}\), as following Eq. (26):
The computational hydrogen electrode (CHE) was used to calculate the electrochemical proton transfer reaction. The chemical potential of proton and the electron is calculated as Eq. (27)81,85:
Where e is the elementary charge, and \({U}_{{{{\rm{RHE}}}}}\) is the applied potential with reversible hydrogen electrode.
The calculation energies, zero-point energies, entropy corrections and solvation energy corrections of each species in the free energy calculations were listed in Supplementary Table 4 and Supplementary Table 5.
Data availability
The authors declare that all data supporting the findings of this study are available within the main text and Supplementary Information files. The structures data of the optimized DFT computational models are provided as Supplementary Data 1. The source data underlying Figs. 1–7 in the main text and Supplementary Figs. 1, 6–13, 21, 25–27, 31–32, 34–35 in Supplementary information are provided as a Source data file. Source data are provided with this paper.
Change history
04 February 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-56682-4
References
Cheon, S., Kim, W. J., Kim, D. Y., Kwon, Y. & Han, J.-I. Electro-synthesis of ammonia from dilute nitric oxide on a gas diffusion electrode. ACS Energy Lett. 7, 958–965 (2022).
Lin, F. et al. Flue gas treatment with ozone oxidation: an overview on NOx, organic pollutants, and mercury. Chem. Eng. J. 382, 123030 (2020).
Deng, O. et al. Managing urban development could halve nitrogen pollution in China. Nat. Commun. 15, 401 (2024).
Han, L. et al. Selective catalytic reduction of NOx with NH3 by using novel catalysts: State of the Art and Future Prospects. Chem. Rev. 119, 10916–10976 (2019).
Zhou, J. et al. Cerium oxide-based catalysts for low-temperature selective catalytic reduction of NOx with NH3: a review. Energy Fuels 35, 2981–2998 (2021).
Shan, W., Liu, F., Yu, Y. & He, H. The use of ceria for the selective catalytic reduction of NOx with NH3. Chinese J. Catal. 35, 1251–1259 (2014).
Busca, G., Lietti, L., Ramis, G. & Berti, F. Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: a review. Appl. Catal. B 18, 1–36 (1998).
Wang, X., Liu, Y. & Wu, Z. Temperature-dependent influencing mechanism of carbon monoxide on the NH3–SCR process over ceria-based catalysts. ACS EST Engg. 1, 1131–1139 (2021).
Jiang, S., Su, Q., Yin, Y., Zhu, T. & Li, X. Extraordinary detoxification effect of arsenic on the cadmium-poisoned V2O5/TiO2 catalyst for selective catalytic reduction of NOx by NH3. ACS EST Engg. 3, 725–733 (2023).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Christensen, C. H., Johannessen, T., Sørensen, R. Z. & Nørskov, J. K. Towards an ammonia-mediated hydrogen economy? Catal. Today 111, 140–144 (2006).
Lan, R., Irvine, J. T. S. & Tao, S. Ammonia and related chemicals as potential indirect hydrogen storage materials. Int. J. Hydrog. Energy 37, 1482–1494 (2012).
Foster, S. L. et al. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 1, 490–500 (2018).
Kandemir, T., Schuster, M. E., Senyshyn, A., Behrens, M. & Schlögl, R. The Haber–Bosch process revisited: on the real structure and stability of “Ammonia Iron” under working conditions. Angew. Chem. Int. Ed. 52, 12723–12726 (2013).
Liu, H.-Y. et al. Electrocatalytic, homogeneous ammonia oxidation in water to nitrate and nitrite with a copper complex. J. Am. Chem. Soc. 144, 8449–8453 (2022).
Jiang, M. et al. Interfacial reduction nucleation of noble metal nanodots on redox-active metal–organic frameworks for high-efficiency electrocatalytic conversion of nitrate to ammonia. Nano Lett. 22, 2529–2537 (2022).
Jiang, M. et al. Quasi-phthalocyanine conjugated covalent organic frameworks with nitrogen-coordinated transition metal centers for high-efficiency electrocatalytic ammonia synthesis. Nano Lett. 22, 372–379 (2022).
Jiang, M. et al. Batch-scale synthesis of nanoparticle-agminated three-dimensional porous Cu@Cu2O microspheres for highly selective electrocatalysis of nitrate to ammonia. Environ. Sci. Technol. 56, 10299–10307 (2022).
Jiang, M. et al. Crystalline modulation engineering of Ru nanoclusters for boosting ammonia electrosynthesis from dinitrogen or nitrate. ACS Appl. Mater. Interfaces 14, 17470–17478 (2022).
Jiang, M. et al. Review on electrocatalytic coreduction of carbon dioxide and nitrogenous species for urea synthesis. ACS Nano 17, 3209–3224 (2023).
Jiang, M. et al. Review on strategies for improving the added value and expanding the scope of CO2 electroreduction products. Chem. Soc. Rev. 53, 5149–5189 (2024).
Anand, M., Abraham, C. S. & Nørskov, J. K. Electrochemical oxidation of molecular nitrogen to nitric acid – towards a molecular level understanding of the challenges. Chem. Sci. 12, 6442–6448 (2021).
Li, S. et al. Hydroxyl radical-mediated efficient photoelectrocatalytic NO oxidation with simultaneous nitrate storage using a flow photoanode reactor. Angew. Chem. Int. Ed. 62, e202305538 (2023).
Dai, W. et al. Gas-phase photoelectrocatalytic oxidation of NO via TiO2 nanorod Array/FTO photoanodes. Environ. Sci. Technol. 54, 5902–5912 (2020).
Zhao, S. et al. Selective nitric oxide electroreduction at monodispersed transition-metal sites with atomically precise coordination environment. Chem Catal. 3, 100598 (2023).
Long, J. et al. Direct electrochemical ammonia synthesis from nitric oxide. Angew. Chem. Int. Ed. 59, 9711–9718 (2020).
Tursun, M. & Wu, C. Single transition metal atoms anchored on defective MoS2 monolayers for the electrocatalytic reduction of nitric oxide into ammonia and hydroxylamine. Inorg. Chem. 61, 17448–17458 (2022).
Wu, F.-H., Zhao, G.-C. & Wei, X.-W. Electrocatalytic oxidation of nitric oxide at multi-walled carbon nanotubes modified electrode. Electrochem. Commun. 4, 690–694 (2002).
de Vooys, A. C. A., Beltramo, G. L., van Riet, B., van Veen, J. A. R. & Koper, M. T. M. Mechanisms of electrochemical reduction and oxidation of nitric oxide. Electrochim. Acta 49, 1307–1314 (2004).
Milsom, E. V., Novak, J., Oyama, M. & Marken, F. Electrocatalytic oxidation of nitric oxide at TiO2–Au nanocomposite film electrodes. Electrochem. Commun. 9, 436–442 (2007).
Kim, D. H. et al. Selective electrochemical reduction of nitric oxide to hydroxylamine by atomically dispersed iron catalyst. Nat. Commun. 12, 1856 (2021).
Sun, S. et al. Spin-related Cu-Co pair to increase electrochemical ammonia generation on high-entropy oxides. Nat. Commun. 15, 260 (2024).
Chen, K., Wang, G., Guo, Y., Ma, D. & Chu, K. Iridium single-atom catalyst for highly efficient NO electroreduction to NH3. Nano Res. 16, 8737–8742 (2023).
Chen, K. et al. Pd1Cu single-atom alloys for high-current-density and durable NO-to-NH3 electroreduction. Nano Lett. 24, 541–548 (2024).
Chen, K. et al. Reduction to NH3 on a W single-atom catalyst. Nano Lett. 23, 1735–1742 (2023).
Shao, J. et al. Electrochemical synthesis of ammonia from nitric oxide using a copper–tin alloy catalyst. Nat. Energy, 8, 1273–1283 (2023).
Wang, D. et al. Coupling electrocatalytic nitric oxide oxidation over carbon cloth with hydrogen evolution reaction for nitrate synthesis. Angew. Chem. Int. Ed. 60, 24605–24611 (2021).
Peng, X. et al. Ambient electrosynthesis of ammonia with efficient denitration. Nano Energy 78, 105321 (2020).
Zhang, L. et al. High-performance electrochemical NO reduction into NH3 by MoS2 nanosheet. Angew. Chem. Int. Ed. 60, 25263–25268 (2021).
Zacharia, I. G. & Deen, W. M. Diffusivity and solubility of nitric oxide in water and saline. Ann. Biomed. Eng. 33, 214–222, (2005).
Lees, E. W., Mowbray, B. A. W., Parlane, F. G. L. & Berlinguette, C. P. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 7, 55–64 (2022).
Rabiee, H. et al. Gas diffusion electrodes (GDEs) for electrochemical reduction of carbon dioxide, carbon monoxide, and dinitrogen to value-added products: a review. Energy Environ. Sci. 14, 1959–2008 (2021).
Fan, J. et al. Bridging the gap between highly active oxygen reduction reaction catalysts and effective catalyst layers for proton exchange membrane fuel cells. Nat. Energy 6, 475–486 (2021).
Li, H., Quispe-Cardenas, E., Yang, S., Yin, L. & Yang, Y. Electrosynthesis of >20 g/L H2O2 from Air. ACS EST Engg. 2, 242–250 (2022).
Ding, M., Flaig, R. W., Jiang, H.-L. & Yaghi, O. M. Carbon capture and conversion using metal–organic frameworks and MOF-based materials. Chem. Soc. Rev. 48, 2783–2828 (2019).
Liu, J., Thallapally, P. K., McGrail, B. P., Brown, D. R. & Liu, J. Progress in adsorption-based CO2 capture by metal–organic frameworks. Chem. Soc. Rev. 41, 2308–2322, (2012).
Yu, J. et al. CO2 capture and separations using MOFs: computational and experimental studies. Chem. Rev. 117, 9674–9754 (2017).
Qian, Q. et al. MOF-based membranes for gas separations. Chem. Rev. 120, 8161–8266 (2020).
Martínez-Ahumada, E. & Díaz-Ramírez, M. L. Capture of toxic gases in MOFs: SO2, H2S, NH3 and NOx. Chem. Sci. 12, 6772–6799 (2021).
Peterson, G. W., Mahle, J. J., DeCoste, J. B., Gordon, W. O. & Rossin, J. A. Extraordinary NO2 removal by the metal–organic framework UiO-66-NH2. Angew. Chem. Int. Ed. 55, 6235–6238, (2016).
Xiao, B. et al. High-capacity hydrogen and nitric oxide adsorption and storage in a metal−organic framework. J. Am. Chem. Soc. 129, 1203–1209 (2007).
Hu, J. et al. Bimetal NiCo-MOF-74 for highly selective NO capture from flue gas under ambient conditions. RSC Adv 12, 33716–33724 (2022).
Thomas, M. et al. Graphene oxide sheathed ZIF-8 microcrystals: engineered precursors of nitrogen-doped porous carbon for efficient oxygen reduction reaction (ORR) electrocatalysis. ACS Appl. Mater. Interfaces 8, 29373–29382 (2016).
Aghajanzadeh, M. et al. Preparation of metal–organic frameworks UiO-66 for adsorptive removal of methotrexate from aqueous solution. J. Inorg. Organomet. P. 28, 177–186 (2018).
Xiao, M. et al. Microporous framework induced synthesis of single-atom dispersed Fe-N-C acidic ORR catalyst and its in situ reduced Fe-N4 active site identification revealed by X-ray absorption spectroscopy. ACS Catal 8, 2824–2832 (2018).
Saliba, D., Ammar, M., Rammal, M., Al-Ghoul, M. & Hmadeh, M. Crystal Growth of ZIF-8, ZIF-67, and Their Mixed-Metal Derivatives. J. Am. Chem. Soc. 140, 1812–1823 (2018).
Medford, A. J. et al. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 328, 36–42 (2015).
Munir, A. et al. Ultrasmall Ni/NiO nanoclusters on thiol-functionalized and -exfoliated graphene oxide nanosheets for durable oxygen evolution reaction. ACS Appl. Energy Mater. 2, 363–371 (2019).
Zhao, J., Quan, X., Chen, S., Liu, Y. & Yu, H. Cobalt nanoparticles encapsulated in porous carbons derived from core–shell ZIF67@ZIF8 as efficient electrocatalysts for oxygen evolution reaction. ACS Appl. Mater. Interfaces 9, 28685–28694 (2017).
Lü, B., Qi, W., Luo, M., Liu, Q. & Guo, L. Fischer–tropsch synthesis: ZIF-8@ZIF-67-derived cobalt nanoparticle-embedded nanocage catalysts. Ind. Eng. Chem. Res. 59, 12352–12359 (2020).
Zhang, Y. et al. Facile preparation of monodisperse, impurity-free, and antioxidation copper nanoparticles on a large scale for application in conductive ink. ACS Appl. Mater. Interfaces 6, 560–567 (2014).
Chang, Y., Lye, M. L. & Zeng, H. C. Large-scale synthesis of high-quality ultralong copper nanowires. Langmuir 21, 3746–3748, (2005).
Ren, X. et al. Facile synthesis of hierarchical mesoporous honeycomb-like NiO for aqueous asymmetric supercapacitors. ACS Appl. Mater. Interfaces 7, 19930–19940 (2015).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Koroleff, F. Direct spectrophotometric determination of ammonia in precipitation. TELLUS A 562–565 (1966).
Singh, P., Singh, M. K., Beg, Y. R. & Nishad, G. R. A review on spectroscopic methods for determination of nitrite and nitrate in environmental samples. Talanta 191, 364–381 (2019).
El hani, O., Karrat, A., Digua, K. & Amine, A. Development of a simplified spectrophotometric method for nitrite determination in water samples. Spectrochim. Acta A 267, 120574 (2022).
Teter, M. P., Payne, M. C. & Allan, D. C. Solution of Schrödinger’s equation for large systems. Phys. Rev. B 40, 12255–12263 (1989).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Rohrbach, A. & Hafner, J. Molecular adsorption of NO on NiO(100): DFT and DFT+U calculations. Phys. Rev. B 71, 045405 (2005).
Solovyev, I. V. Exchange interactions and magnetic force theorem. Phys. Rev. B 103, 104428 (2021).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Rohrbach, A., Hafner, J. & Kresse, G. Molecular adsorption on the surface of strongly correlated transition-metal oxides: a case study for CO/NiO(100). Phys. Rev. B 69, 075413 (2004).
Kamat, G. A. et al. Acid anion electrolyte effects on platinum for oxygen and hydrogen electrocatalysis. Commun. Chem. 5, 20 (2022).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys 140, 084106 (2014).
Mathew, K., Kolluru, V. S. C., Mula, S., Steinmann, S. N. & Hennig, R. G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 151, 234101 (2019).
Rayne, S. & Forest, K. Comparative study on the gas to solution phase solvation free energies of model combustion flue gas compounds (N~2~, O~2~, CO~2~, H~2~O, SO~2~, and CO) in 178 organic solvents using the IEFPCM-UFF, CPCM, and SMD implicit solvent models at the Gaussian-4 (G4) level of theory. Nat. Preced. 5, 1 (2010).
Lamoureux, P. S., Singh, A. R. & Chan, K. pH effects on hydrogen evolution and oxidation over Pt(111): insights from first-principles. ACS Catal. 9, 6194–6201 (2019).
Acknowledgements
This work was supported by the Korea government (MSIT) through the National Research Foundation of Korea (NRF) grants of Leading Researcher Program (NRF-2020R1A3B2079953) and 2022M3C1A3092056.
Author information
Authors and Affiliations
Contributions
D.P. conducted key experiments, analyzed the data, and wrote the manuscript; M.A.P. and G.G. performed theoretical calculations; S.L. and H.-S.B. carried out some characterizations and analyses; F.H. designed the gas line system; W.C. directed the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ke Chu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, D., Austeria P, M., Lee, S. et al. Integrated electrocatalytic synthesis of ammonium nitrate from dilute NO gas on metal organic frameworks-modified gas diffusion electrodes. Nat Commun 15, 7243 (2024). https://doi.org/10.1038/s41467-024-51256-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51256-2
This article is cited by
-
C-F bond integration in polymeric carbon nitride enables oxygen-tolerant photocatalytic reduction of NO to N2
Nature Communications (2025)
-
Boosted charge and proton transfer over ternary Co/Co3O4/CoB for electrochemical nitric oxide reduction to ammonia
Nature Communications (2025)
