
Abstract
Mutations in the nuclear envelope (NE) protein lamin A/C (encoded by LMNA), cause a severe form of dilated cardiomyopathy (DCM) with early-onset life-threatening arrhythmias. However, molecular mechanisms underlying increased arrhythmogenesis in LMNA-related DCM (LMNA-DCM) remain largely unknown. Here we show that a frameshift mutation in LMNA causes abnormal Ca2+ handling, arrhythmias and disformed NE in LMNA-DCM patient-specific iPSC-derived cardiomyocytes (iPSC-CMs). Mechanistically, lamin A interacts with sirtuin 1 (SIRT1) where mutant lamin A/C accelerates degradation of SIRT1, leading to mitochondrial dysfunction and oxidative stress. Elevated reactive oxygen species (ROS) then activates the Ca2+/calmodulin-dependent protein kinase II (CaMKII)-ryanodine receptor 2 (RYR2) pathway and aggravates the accumulation of SUN1 in mutant iPSC-CMs, contributing to arrhythmias and NE deformation, respectively. Taken together, the lamin A/C deficiency-mediated ROS disorder is revealed as central to LMNA-DCM development. Manipulation of impaired SIRT1 activity and excessive oxidative stress is a potential future therapeutic strategy for LMNA-DCM.
Similar content being viewed by others


Introduction
Dilated cardiomyopathy (DCM), as characterized by left ventricular or biventricular dilation and impaired heart contraction, is the second most common cause of heart failure, having an estimated prevalence of > 0.4% in the general population1. Decades of research have revealed diverse aetiologies for DCM, including genetic mutations, infections, inflammation, autoimmune diseases, exposure to toxins, and endocrine or neuromuscular causes2. Multiple genetic mutations have been associated with the development of DCM, most notably in genes encoding cytoskeletal, sarcomere, desmosome, or the nuclear envelope (NE) proteins3. Despite this, only partial understanding of the underlying pathophysiology of DCM has been achieved and the wide spectrum of potential causes of DCM make it particularly challenging to prevent irreversible damage, even at early stages. Overall, there remains substantial mortality (1-year mortality of 25–30% and 5-year survival of 50%) and among such mortalities heart failure (70%) and sudden death (25–30%) remain the most common occurences4.
With the steadily increasing number of mutations that are known to be associated with DCM, LMNA-related DCM (LMNA-DCM), accounting for 4–8% cases of DCM, has received much attention3,5. The LMNA gene encodes the A-type lamins which lie along the inner nuclear membrane as a major constituent of the nuclear lamina. They play important roles in nuclear and cytoskeletal organization, mechanical stability, chromatin organization, signaling, gene regulation, genome stability, and cell differentiation5. Mutations in LMNA are associated with more than a dozen previously defined clinical disorders, collectively known as laminopathies, such as Hutchinson–Gilford progeria syndrome (HGPS), autosomal dominant Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy, and DCM. Compared to other types of DCM, LMNA-DCM is relatively atypical and more severe6. Patients prominently present with life-threatening cardiac electrical abnormalities with early onset, such as atrioventricular blocks and ventricular tachycardia and fibrillation. The high risk of sudden cardiac death (SCD) and requirement for implantable cardioverter defibrillator (ICD) implantation are among the major clinical features that affect patient quality of life and survival1,3,5,6,7.
Though the molecular mechanisms that connect LMNA deficiency with cardiomyopathy and sudden death have been widely studied over recent years, many aspects have remained elusive. Three independent mechanisms have been hypothesized to underpin the pathogenesis of LMNA-associated cardiomyopathy, namely dysregulation of heterochromatin organization, impaired nuclear mechano-resistance, and alteration of the lamin A/C-related intracellular signaling pathway8. Relating to the first one, LMNA haploinsufficiency-induced chromatin dysregulation has recently developed into a considerable focal hotspot upon the confirmation that the activation of the platelet-derived growth factor pathway, resulting from abnormal heterochromatin organization, can induce arrhythmic phenotypes in LMNA-DCM9.
Here we demonstrated that LMNA-DCM patient-specific induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) presented with elevated diastolic [Ca2+]i, abnormal Ca2+ handling, increased burden of arrhythmias, and a deformed nuclear envelope (NE). Linking these abnormalities in mutant iPSC-CMs we found resulting accelerated degradation of lamin A-binding protein sirtuin 1 (SIRT1). This, in turn, resulted in mitochondrial dysfunction and oxidative stress. We then tested the hypothesis that elevated reactive oxygen species (ROS) contributes to the development of LMNA-DCM. On one hand, elevated ROS activated Ca2+/calmodulin-dependent protein kinase II (CaMKII). This then promoted ryanodine receptor 2 (RYR2)-mediated sarcoplasmic reticulum (SR) Ca2+ leak by RYR2-Ser2814 phosphorylation and subsequently caused elevated diastolic [Ca2+]i, abnormal Ca2+ handling, and arrhythmias in LMNA-DCM iPSC-CMs. On the other hand, excessive ROS production accelerated the accumulation of SUN1, which contributed to NE deformation in LMNA-DCM iPSC-CMs. These results provide insights into the links between lamin A/C insufficiency, ROS, arrhythmias, and abnormal nuclear structure, which may lead to promising therapeutic avenues for LMNA-DCM.
Results
Clinical characteristics
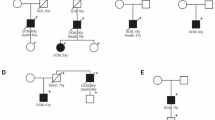
In 2016, we recruited a family pedigree with the proband (II−1) and his sister (II−2), a 57-year-old male and a 50-year-old female, respectively. The two patients were initially diagnosed with atrial fibrillation (AF), demonstrating characteristic electrocardiogram (ECG) (Supplementary Fig. 1a, b), dilated left atrium, and reserved left ventricular function by echocardiography (Supplementary Fig. 1c). Two years later, both patients developed atrioventricular blocks and a pacemaker was implanted. Premature ventricular contraction and non-sustained ventricular tachycardia were presented in the proband’s ECG. To establish an accurate diagnosis, peripheral blood samples were obtained from the proband and his relatives for genetic test, which revealed a heterozygous frameshift mutation of the LMNA gene (c.1163dupC; p.A388fs) in both patients and in one of their siblings (Fig. 1a). The pathogenicity of the variant was classified as ‘likely pathogenic’ according to the American College of Medical Genetics and Genomics (ACMG) guideline. Combined with their clinical features, the two patients were diagnosed with LMNA-DCM with predominant conduction system deficiency. During the follow-up, the proband’s sister unfortunately died of SCD and the proband’s son ultimately also developed AF.

a Pedigree of the family carrying a novel heterozygous mutation in LMNA. Individuals who were dead are marked with a fork. The proband is indicated by the red arrow. b Schematic diagram of the iPSC lines used in the study. c. Predicted effect of the LMNA A388fs mutation on the two splicing products lamin A and C. d Bar graph to compare the occurrence of arrhythmias among control (Con−1, Con-2, Con-3) iPSC-CMs, A388fs mutant (II-1, II-2) iPSC-CMs, gene-corrected (GC) (II−1-corr) iPSC-CMs, and LMNA KO (Con-3-KO) iPSC-CMs. e, f Violin graphs to compare the Ca2+ transient amplitude and diastolic [Ca2+]i among Con−1, II-1-corr, and II−1 iPSC-CMs. n = 49 (Con−1), 71 (II-1), 94 (II-1-corr) cells. g, h Violin graphs to compare the RYR2-mediated SR Ca2+ leak and SR Ca2+ load among Con-1, II-1-corr, and II-1 iPSC-CMs. n = 44 (II-1), 46 (Con-1), 60 (II-1-corr) cells. i The GO analysis identified a significant change in mitochondrial organization. j Representative graphs of mitochondria in II-1-corr and II-1 iPSC-CMs by transmission electron microscopy. Scale bar, 0.5 μm. k Analysis of oxygen consumption in Con-1, II-1-corr, and II-1 iPSC-CMs. n = 3 independently biological repeats. l Bar graphs to compare key parameters of OCR among Con-1, II-1-corr, and II-1 iPSC-CMs, including basal OCR, proton leak OCR, maximal capacity (max. cap.), ATP-linked OCR, and reserved capacity (reserved cap.). n = 3 independently biological repeats. m, n Cellular and Mitochondrial ROS levels in Con-1, II-1-corr, and II-1 iPSC-CMs measured by flow cytometry of cells stained with CellROX or MitoSOX. n = 3 independently biological repeats. Data are presented as mean ± SEM in k–n. p values were calculated by Dunnett’s multiple comparisons test in e–h, l–n and two-sided unpaired t-test in i. Figure 1b created with BioRender.com was released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license. Source data are provided as a Source Data file.
Generation and characterization of patient-specific and genome-edited iPSC-CMs
To examine the molecular and cellular properties of LMNA-DCM at the single-cell level, skin biopsies were collected from two patients (II-1, II-2) and three healthy donors (Con-1, Con-2, Con-3). Multiple control and patient-specific iPSC lines were generated using non-integrated Sendai viral method (Supplementary Table 1). Since variability among iPSC lines can profoundly influence phenotypic analysis, we generated two gene-corrected (GC) isogenic lines with corrected LMNA sequence using the patient iPSCs (II−1) through CRISPR/Cas9-mediated genome editing (Fig. 1b and Supplementary Fig. 2a). We also generated a knockout (KO) line using the control iPSCs (Con-3) by targeting exon 4 of the LMNA gene locus (Fig. 1b and Supplementary Fig. 2a). The impact of CRISPR/Cas9 on the cellular genome is primarily assessed by determining whether potential off-target sites of the single guide RNA (sgRNA) have been altered. We performed DNA sequencing on 3 predicted off-target sites of LMNA-sgRNA, and no off-target occurrences were observed in GC II-1-corr iPSCs (Supplementary Fig. 2b, c and Supplementary Table 2). The newly generated iPSC lines showed human embryonic stem cell-like morphology, expressed pluripotency markers, had normal karyotypes, and were capable of differentiating into three germ layers (Supplementary Figs. 3 and 4). These iPSCs were subsequently differentiated into cardiomyocytes using a small molecule-based monolayer differentiation protocol (Supplementary Fig. 5a). This gave rise to a yield of >95%, as indicated by fluorescence-activated cell sorting (FACS) analysis (Supplementary Fig. 5b). Single-cell patch-clamp recordings revealed that majority of the generated iPSC-CMs were ventricular-like myocytes (Supplementary Fig. 5c).
Lamin A/C haploinsufficiency causes arrhythmias and abnormal Ca2+ handling in A388fs iPSC-CMs
According to the change in base sequence, the A388fs mutation in the LMNA gene (LMNA-A388fs) can result in a premature termination codon (PTC), which could cause lamin A/C haploinsufficiency in carriers (Fig. 1c). We therefore sought to assess whether the A388fs mutation relates to the expression of LMNA and its encoding protein lamin A/C. The mRNA expression of LMNA was significantly reduced in mutant (II-1, II−2) and Con-3-KO iPSC-CMs, when compared to Con-3 and II−1-corr iPSC-CMs (Supplementary Fig. 6a and Supplementary Table 3). Similarly, Western blot analysis revealed that the protein expression levels of lamin A/C in mutant (II-1, II-2) iPSC-CMs was significantly lower than those in control (Con-1, Con-2, Con-3) and II-1-corr iPSC-CMs (Supplementary Fig. 6b, c). Moreover, the full-length or truncated lamin A/C were not detected in Con-3-KO iPSC-CMs (Supplementary Fig. 2). These results suggest that the A388fs mutation results in lamin A/C haploinsufficiency in patient-specific iPSC-CMs.
Given that the clinical features of both patients were prominently presented as arrhythmias with high risk of SCD3,5,6, we next investigated the electrophysiological properties of patient-specific iPSC-CMs carrying LMNA-A388fs by single-cell patch clamp. We observed uniform and rhythmic action potential profile in control (Con−1, Con-2, and Con-3) iPSC-CMs (Supplementary Fig. 7a). In contrast, arrhythmic waveforms were detected in a large proportion of A388fs (II−1, II-2) iPSC-CMs (67.1%, n = 73) as compared to controls (22.9%, n = 96), manifesting as delayed afterdepolarizations (DADs) (Fig. 1d and Supplementary Fig. 7a, b). Notably, we observed that II-1-corr iPSC-CMs demonstrated dramatic reduction of arrhythmic incidence (25.9%, n = 27), to the level that they resembled the action potentials recorded from controls (Fig. 1d and Supplementary Fig. 7a, b). We also observed DAD arrhythmias in a larger proportion of Con-3-KO iPSC-CMs (79.2%, n = 21) than controls, recapitulating the arrhythmic phenotype in A388fs iPSC-CMs (Fig. 1d and Supplementary Fig. 7a, b).
As DADs are related to intracellular Ca2+ mishandling in cardiomyocytes10, we next assessed Ca2+ transients from iPSC-CMs by Fura-2 ratiometric Ca2+ imaging (Supplementary Fig. 7c). The Ca2+ transient amplitude was reduced by 16.6% in II−1 iPSC-CMs, as compared to their isogenic controls (II−1-corr iPSC-CMs) (Fig. 1e). Moreover, we observed significantly increased diastolic intracellular Ca2+ ([Ca2+]i) in II-1 iPSC-CMs, as compared to Con−1 and II−1-corr iPSC-CMs (Fig. 1f). The maximal upstroke and recovery velocities were both significantly reduced in II−1 iPSC-CMs (Supplementary Fig. 7d, e). Sarcoplasmic reticulum (SR) Ca2+ leak through RYR2 is a key determinant of diastolic Ca2+ concentration11,12. To elucidate the cause of elevated diastolic [Ca2+]i, total RYR2-mediated SR Ca2+ leak and SR Ca2+ load were assessed using the standard tetracaine protocol in iPSC-CMs (Supplementary Fig. 7f). Notably, we observed that total RYR2-mediated SR Ca2+ leak, as quantified by tetracaine-induced Ca2+ shift, was significantly increased in II-1 iPSC-CMs compared to Con−1 and II−1-corr iPSC-CMs (Fig. 1g). The SR Ca2+ load, as indicated by the amplitude of caffeine-evoked Ca2+ transients, was significantly lower in II−1 iPSC-CMs than in Con-1 and II−1-corr iPSC-CMs (Fig. 1h). Similar Ca2+ handling abnormalities were also observed in II-2 and Con-3-KO iPSC-CMs as compared to Con-3 iPSC-CMs, demonstrating decreased Ca2+ transient amplitude, increased diastolic [Ca2+]i, increased RYR2-mediated SR Ca2+ leak, and decreased SR Ca2+ load (Supplementary Fig. 8). Collectively, these results suggest that lamin A/C haploinsufficiency causes arrhythmias and abnormal Ca2+ handling in A388fs iPSC-CMs.
A388fs iPSC-CMs exhibit a pathogenic signature of mitochondrial dysfunction and ROS elevation
To explore the underlying mechanisms by which deficiency of lamin A/C, a NE protein, causes arrhythmias and Ca2+ handling abnormalities, Tandem Mass Tag-Based proteomic analysis was performed to detect protein expression changes between II−1-corr and II-1 iPSC-CMs. 446 differentially expressed proteins were detected in our study (Supplementary Fig. 9a, b). Analysis using the Kyoto encyclopedia of genes and genomes (KEGG) indicated a DCM phenotype (Supplementary Fig. 9c), and Gene ontology (GO) analysis revealed a significant change in mitochondrial organization (Fig. 1i and Supplementary Fig. 9d). These results suggest that mitochondrial dysfunction may exist in A388fs iPSC-CMs. Transmission electron microscopy revealed that the mitochondria were swollen in II−1 iPSC-CMs as compared to II−1-corr iPSC-CMs (Fig. 1j and Supplementary Fig. 9e). The mitochondrial DNA (mtDNA) copy number was significantly lower in mutant (II-1, II-2) iPSC-CMs than in Con−1 and II−1-corr iPSC-CMs (Supplementary Fig. 9f). To further evaluate the mitochondrial respiratory function, we measured oxygen consumption rate (OCR) of iPSC-CMs using a Seahorse bioscience XF-96 extracellular flux analyzer. The levels of maximal capacity and reserved capacity were significantly reduced in II−1 iPSC-CMs as compared to Con-1 and II-1-corr iPSC-CMs (Fig. 1k, l), suggesting impairment of mitochondrial bioenergetics.
Considering that mitochondrial dysfunction is often associated with increased ROS production and that mitochondria are one of the major sources of ROS production13, we next assessed cellular ROS production by flow cytometry using a fluorescent CellROX probe. The ROS level was significantly higher in II-1 iPSC-CMs than in Con-1 and II-1-corr iPSC-CMs (Fig. 1m). Using a mitochondrial superoxide probe (MitoSOX), we showed that the level of mitochondrial ROS was greatly elevated in II-1 iPSC-CMs compared to Con-1 and II-1-corr iPSC-CMs (Fig. 1n). Taken together, these results demonstrate a pathogenic signature of mitochondrial dysfunction and ROS elevation in A388fs iPSC-CMs.
Accelerated SIRT1 degradation contributes to mitochondrial dysfunction and excessive ROS level in A388fs iPSC-CMs
SIRT1 is a class III histone deacetylase that acts as a sensor of redox changes and shapes the mitochondrial metabolism and inflammatory response in the host14. One previous study reported that lamin A interacts with SIRT115. We next sought to investigate whether the observed phenotypes of mitochondrial dysfunction and ROS elevation in A388fs iPSC-CMs are associated with SIRT1. Our immunofluorescence analysis demonstrated consistent colocalization of lamin A and SIRT1 in both control (Con-1) and A388fs (II-1, II−2) iPSC-CMs (Fig. 2a). The fluorescence intensity of SIRT1 was significantly decreased in A388fs iPSC-CMs as compared to controls (Fig. 2b). Moreover, co-immunoprecipitation (co-IP) results also revealed interaction between lamin A and SIRT1 in Con-1 iPSC-CMs (Fig. 2c). Western blot analysis revealed that the protein expression of SIRT1 was significantly reduced in A388fs (II-1, II-2) and Con-3-KO iPSC-CMs as compared to control (Con−1, Con-2, Con-3) and II-1-corr iPSC-CMs (Fig. 2d, e and Supplementary Fig. 10), whereas the mRNA expression levels of SIRT1 were slightly upregulated in II-1 iPSC-CMs compared to their isogenic controls (Fig. 2f and Supplementary Table 3).

a. Representative graphs of staining of SIRT1 (red) and lamin A/C (green) in Con-1 and mutant (II-1, II-2) iPSC-CMs. DAPI indicates nuclear staining (blue). Scale bar, 60 μm. b Bar graph to compare the SIRT1 intensity among different groups in a. n = 21 (Con-1), 38 (II-1), 41 (II-2) cells. c Western blot analysis revealed that lamin A was detected in anti-SIRT1 immunoprecipitates, and SIRT1 was detected in anti-lamin A/C immunoprecipitates. n = 4 independently biological repeats. d, e Western blot analysis of protein expression of SIRT1 in control (Con-1, Con-2, Con-3), II-1-corr, and mutant (II-1, II-2) iPSC-CMs. n = 3 independently biological repeats. f Bar graph to compare the mRNA expression of SIRT1 between II-1-corr and II-1 iPSC-CMs. n = 5 independently biological repeats. g Western blot analysis of SIRT1 expression in Con-3, Con-3-KO, II−1-corr, and II-1 iPSC-CMs after CHX treatment. h Line graph to compare the degradation rate of SIRT1 expression among different groups in g. n = 3 (Con-3, II-1-corr, II-1), 4 (Con-3-KO) independently biological repeats. i Bar graphs to compare the mRNA expression of TFAM, SIRT3, and SOD2 between II-1-corr and II-1 iPSC-CMs. n = 3 (TFAM-Con-3), 4 (others), and 8 (TFAM-II-1) independently biological repeats. j Analysis of oxygen consumption in II-1-corr and II-1 iPSC-CMs with or without treatment of SRT1720 (SRT). n = 3 independently biological repeats. k Bar graphs to compare key parameters of OCR among different groups in j. n = 3 independently biological repeats. l Mitochondrial ROS levels in Con-1, II-1-corr, and II-1 iPSC-CMs treated with DMSO, and II-1 iPSC-CMs treated with SRT. n = 3 independently biological repeats. m Cellular ROS levels in Con-1, II-1-corr, and II-1 iPSC-CMs treated with DMSO, II-1 iPSC-CMs treated with MT or SRT. n = 3 independently biological repeats. Data are presented as mean ± SEM in b, e, f, h–m. p values were calculated by Dunnett’s multiple comparisons test in b, i, l, m, Nested t-test in e, and two-sided unpaired t-test in f, h, k. Source data are provided as a Source Data file.
Considering the interaction between lamin A and SIRT1, and given the noted lamin A/C haploinsufficiency in A388fs iPSC-CMs, we hypothesized that lamin A may play an important role in SIRT1 turnover, thus protecting SIRT1 proteins from degradation. To test this, we compared SIRT1 degradation rates between Con-3, Con-3-KO, II-1-corr, and II-1 iPSC-CMs by cycloheximide (CHX) treatment. In this, II-1 and Con-3-KO iPSC-CMs exhibited a greatly rapid rate of SIRT1 degradation in comparison with Con-3 and II-1-corr iPSC-CMs (Fig. 2g,h). The nuclear expression of SIRT1 was significantly lower in II-1 iPSC-CMs than that in their isogenic controls, suggesting an impaired SIRT1 activity (Supplementary Fig. 11a, b). Treatment of chloroquine (CQ) but not MG-132 significantly rescued the CHX-induced SIRT1 protein degradation in Con-1 iPSC-CMs, suggesting that SIRT1 was degraded through lysosome pathway in iPSC-CMs (Supplementary Fig. 11c, d). The SIRT1 activity was significantly lower in II-1 iPSC-CMs than that in their isogenic controls, which was greatly restored by the treatment of SRT1720 (SRT, 1 μM, 24 h), a selective SIRT1 activator (Supplementary Fig. 11e). Moreover, the mRNA expression levels of TFAM, SIRT3 and SOD2, downstream genes of SIRT1, were significantly lower in II-1 iPSC-CMs than in Con-3 and II-1-corr iPSC-CMs (Fig. 2i and Supplementary Table 3).
To investigate if expression changes of SIRT1 gave rise to functional consequences, SRT was applied in both II-1-corr and II-1 iPSC-CMs to activate SIRT1. Seahorse assays demonstrated that the impaired mitochondrial bioenergetics in II-1 iPSC-CMs was rescued upon treatment with SRT, as evidenced by improved maximal capacity and reserved capacity (Fig. 2j, k). In addition, treatment of SRT in II-1 iPSC-CMs significantly reduced the mitochondrial ROS (Fig. 2l). The level of cellular ROS in II-1 iPSC-CMs was also significantly reduced by the treatment of SRT or mitochondria-targeted antioxidant MitoTEMPO (MT, 5 μM, 3 days) (Fig. 2m). Taken together, these results suggest that lamin A/C haploinsufficiency leads to accelerated SIRT1 degradation, which contributes to mitochondrial dysfunction and excessive ROS levels in A388fs iPSC-CMs.
Activated ROS-CAMKII-RYR2 pathway links down-regulated SIRT1 and arrhythmic phenotype in A388fs iPSC-CMs
The ROS-CAMKII-RYR2 pathway has been reported to be an important participant to manipulate intracellular Ca2+ homeostasis11,12. Elevated ROS-induced CaMKII activation can modulate the activity of multiple Ca2+ handling proteins, ultimately leading to abnormal Ca2+ handling16,17,18. Our prior data indicated an aberrant SR Ca2+ release in A388fs iPSC-CMs. We hypothesized that elevated mitochondrial ROS activates the CaMKII-RYR2 pathway in A388fs iPSC-CMs, leading to the abnormal Ca2+ handling and arrhythmic phenotype. To test this hypothesis, we assessed the CaMKII activity in control and A388fs iPSC-CMs using antibodies that detect phosphorylated CaMKII (phosphorylated threonine 286; p-CaMKII), oxidized CaMKII (M281-M282 oxidation; ox-CaMKII), or total CaMKII. We found that both oxidation and phosphorylation levels of CaMKII were significantly higher in A388fs iPSC-CMs than in controls (Fig. 3a–d). Intriguingly, CaMKII-dependent phosphorylation of RYR2 at Serine 2814 (p-RYR2) was dramatically enhanced in II-1 iPSC-CMs as compared to Con-1 and II-1-corr iPSC-CMs, whereas total expression of RYR2 remained unchanged (Fig. 3a, b). Similar observations were made in Con-3-KO iPSC-CMs, showing significantly higher levels of ox-CaMKII, p-CaMKII, p-RYR2, and cellular ROS, when compared to Con-3 iPSC-CMs (Supplementary Fig. 12).

a, b Western blot analysis of total CaMKII, p-CaMKII (CaMKII-T286), total RYR2, and p-RYR2 (RYR2−2814S) expression in Con−1, II-1-corr, and II-1 iPSC-CMs. n = 4 independently biological repeats. c, d Western blot analysis of protein expression of total CaMKII and oxidized CaMKII (M281-M282 oxidation; ox-CaMKII) in Con-1, II-1-corr, and II-1 iPSC-CMs. n = 4 independently biological repeats. e, f Western blot analysis of total CaMKII, p-CaMKII, total RYR2, and p-RYR2 expression in II-1 iPSC-CMs treated with DMSO, SRT, or MT, respectively. n = 4 independently biological repeats. g, h Western blot analysis of protein expression of total and oxidized CaMKII in II-1 iPSC-CMs treated with DMSO, SRT or mitoTEMPO (MT). n = 4 independently biological repeats. i Representative Ca2+ transient traces recorded from II-1 iPSC-CMs treated with DMSO, SRT, MT or KN93, respectively. j, k Violin graphs to compare the Ca2+ transient amplitude and diastolic [Ca2+]i among different groups in i. n = 37 (II-1 + KN93), 74 (II-1 + SRT), 76 (II-1 + MT), 80 (II-1 + DMSO) cells. l Representative traces of cytosolic Ca2+ fluorescence in II-1 iPSC-CMs treated with DMSO, SRT, MT, or KN93 in NT solution and exposed to 0 Na+, 0 Ca2+ solution containing tetracaine (Tet) and caffeine (Caff). m–n Violin graphs to compare the RYR2-mediated SR Ca2+ leak and SR Ca2+ load among different groups in l. n = 49 (II-1 + DMSO), 54 (II-1 + KN93), 55 (II-1 + SRT), and 58 (II-1 + MT) cells. o Representative electrophysiological measurements of spontaneous action potentials in II-1 iPSC-CMs treated with DMSO, resveratrol (resver), SRT, NAC, MT, or KN93, respectively. p Bar graph to compare the occurrence of arrhythmias among different groups in o. Data are presented as mean ± SEM in b, d, f, h. p values were calculated by Dunnett’s multiple comparisons test in b, d, f, h, j, k, m, n. Source data are provided as a Source Data file.
In line with our hypothesis, treatment with either SRT or MT significantly reduced the levels of ox-CaMKII, p-CaMKII, and p-RYR2 in II-1 iPSC-CMs (Fig. 3e–h). Meanwhile, the p-RYR2 level was reduced in II-1 iPSC-CMs after inhibition of CaMKII with KN93 (1 μM, 24 h), when compared to II-1 iPSC-CMs treated with the inactive analog KN92 (1 μM, 24 h) (Supplementary Fig. 13a, b). These data suggested that elevated mitochondrial ROS causes activated CaMKII and subsequent hyperactivation of RYR2 in A388fs iPSC-CMs. Moreover, treatment with the SIRT1 activator (SRT), the ROS scavenger (MT), or the CaMKII inhibitor (KN93), largely mitigated the Ca2+ handling abnormalities observed in II-1 iPSC-CMs, as evidenced by reduced RYR2-mediated SR Ca2+ leak and increased SR Ca2+ leak load (Fig. 3i–n and Supplementary Fig. 13c–h).
We next examined the antiarrhythmic effects of multiple drugs with different targets by patch clamp. Treatment with the SIRT activator SRT or resveratrol (10 μM, 24 h) in II-1 iPSC-CMs effectively rescued the arrhythmic phenotype (Fig. 3o, p). Moreover, MT or N-acetyl-l-cysteine (NAC, 5 mM, 4 h), a well-established antioxidant, exerted an antiarrhythmic effect in II-1 iPSC-CMs (Fig. 3o, p). Inhibition of CaMKII with KN93 also greatly reversed the arrhythmic phenotype found in II-1 iPSC-CMs (Fig. 3o, p and Supplementary Fig. 13i, j). Altogether, these results suggest that the down-regulation of SIRT1 activates the ROS-CAMKII-RYR2 pathway, resulting in abnormal Ca2+ handling and arrhythmias in A388fs iPSC-CMs.
ROS-mediated protein accumulation of SUN1 confers abnormal NE structure in A388fs iPSC-CMs
Mammalian A-type lamins are essential for the mechanical stiffness of the nucleus as a prerequisite for nuclear maintainence5,19. We therefore investigated whether LMNA-A388fs was associated with changes in NE structure. iPSC-CMs were co-stained with TNNT2 and lamin A/C, indicating cardiomyocytes and the NE, respectively. A significantly large subfraction of A388fs (II-1, II-2) iPSC-CMs (77.4%, n = 16), as compared to control (Con-1, Con-2, Con-3) (19.4%, n = 24), and II−1-corr (17.9%, n = 8) iPSC-CMs, were observed to exhibit a wrinkled NE structure (Fig. 4a and Supplementary Fig. 14). It has been previously reported that the accumulation of SUN1, an inner NE protein, is pathogenic in progeric and dystrophic laminopathies20,21. Interestingly, Western blot analysis revealed significantly higher protein expression levels of SUN1 in A388fs (II-1, II−2) iPSC-CMs than in Con−1 iPSC-CMs (Fig. 4b, c). When treated with hydrogen peroxide (H2O2), expression of SUN1 was significantly increased in both Con-1 and II-1 iPSC-CMs, suggesting that elevated ROS resulted in enhanced SUN1 expression in iPSC-CMs (Fig. 4d, e). Moreover, treatment of SRT or MT significantly reduced SUN1 expression in II-1 iPSC-CMs, as evidenced by immunofluorescence and Western blot analysis (Fig. 4f–h and Supplementary Fig. 15a, b). Importantly, the NE deformation in II-1 iPSC-CMs was partially rescued by the treatment of SRT or MT (Fig. 4i and Supplementary Fig. 15c, d).

a. Representative graphs of staining of lamin A/C (green) and cardiac-specific marker TNNT2 (red) in control (Con-1, Con-2, and Con-3), II-1-corr, and mutant (II−1, II-2) iPSC-CMs. DAPI indicates nuclear staining (blue). Scale bar, 20 μm. The experiment was repeated four times with similar results. b, c Western blot analysis of SUN1 expression in Con−1, II-1-corr, and mutant (II−1, II-2) iPSC-CMs. n = 4 independently biological repeats. d, e Western blot analysis of SUN1 expression in Con−1 and II-1 iPSC-CMs with or without the treatment of H2O2. n = 4 independently biological repeats. f, g Western blot analysis of SUN1 expression in II−1 iPSC-CMs treated with DMSO, SRT or MT. n = 4 independently biological repeats. h Representative graphs of staining of SUN1 (green) and cardiac-specific marker TNNT2 (red) in II−1 iPSC-CMs treated with DMSO or MT. DAPI indicates nuclear staining (blue). Scale bar, 60 μm. The experiment was repeated three times with similar results. i Representative graphs of staining of lamin A/C (green) and cardiac-specific marker TNNT2 (red) in II−1 iPSC-CMs treated with DMSO or MT. DAPI indicates nuclear staining (blue). Scale bar, 20 μm. The experiment was repeated three times with similar results. Data are presented as mean ± SEM in c, e, g. p values were calculated by Nested t-test in c, two-sided unpaired t-test in e, and Dunnett’s multiple comparisons test in g. Source data are provided as a Source Data file.
We also examined the rescuing effects in SUN1 knockdown (KD) cardiomyocytes by small interfering RNA (siRNA) in II-1 iPSC-CMs (Supplementary Fig. 16a, b). We observed no significant changes in expression levels of CaMKII (total, oxidized, phosphorylated) and RYR2 (total, phosphorylated) between scrambled siRNA- and SUN1 siRNA-treated II-1 iPSC-CMs (Supplementary Fig. 16a, b). In addition, SUN1 KD had no antiarrhythmic effect in II-1 iPSC-CMs (Supplementary Fig. 16c,d). Notably, the NE structural abnormality in II-1 iPSC-CMs was significantly alleviated by SUN1 KD (Supplementary Fig. 16e, f). Collectively, these results suggest that elevated ROS causes protein accumulation of SUN1 and abnormal NE structure in A388fs iPSC-CMs.
Genetic inhibition of SIRT1 in isogenic control iPSC-CMs activates ROS-CAMKII-RYR2 pathway and recapitulates pathogenic phenotypes of A388fs iPSC-CMs
The accelerated SIRT1 degradation in A388fs iPSC-CMs contributes to the excessive ROS, which might be responsible for the arrhythmic phenotype and abnormal NE structure. To further confirm the relationship between SIRT1 and pathogenic phenotypes, we, therefore, genetically inhibited the expression of SIRT1 by siRNA in II-1-corr iPSC-CMs. The KD efficiency was validated by Western blot analysis. Along with downregulation of SIRT1 expression, KD of SIRT1 in II-1-corr iPSC-CMs elicited significantly increased levels of ox-CaMKII, p-CaMKII, p-RYR2, and SUN1 (Fig. 5a, b). Moreover, SIRT1 siRNA-treated II-1-corr iPSC-CMs exhibited significantly elevated ROS level (Fig. 5c), abnormal Ca2+ handling (Fig. 5d–i), increased arrhythmic burden (Fig. 5j, k), and abnormal NE structure (Fig. 5l, m), when compared to scrambled siRNA-treated II-1-corr iPSC-CMs. Meanwhile, SIRT1 KD-induced protein expression changes and pathogenic phenotypes in II-1-corr iPSC-CMs were significantly rescued by SRT treatment (Fig. 5a–m). These results suggest that genetic inhibition of SIRT1 in isogenic control iPSC-CMs activates ROS-CAMKII-RYR2 pathway and recapitulates pathogenic phenotypes of A388fs iPSC-CMs.

a, b Western blot analysis of SIRT1, total CaMKII, ox-CaMKII, p-CaMKII, total RYR2, p-RYR2, and SUN1 expression in II-1-corr iPSC-CMs treated with scrambled siRNA (NC), SIRT1 siRNA (KD), or SIRT1 siRNA and SRT (KD + SRT), respectively. n = 4 independently biological repeats. c Cellular ROS levels in II−1-corr iPSC-CMs treated with scrambled siRNA, SIRT1 siRNA, or SIRT1 siRNA and SRT, respectively. n = 3 independently biological repeats. d Representative Ca2+ transient traces recorded from II−1-corr iPSC-CMs treated with scrambled siRNA, SIRT1 siRNA, or SIRT1 siRNA and SRT, respectively. e, f Bar graphs to compare the Ca2+ transient amplitude and diastolic [Ca2+]i among different groups in d. n = 19 (KD), 21 (NC, KD + SRT) cells. g Representative traces of cytosolic Ca2+ fluorescence in II−1-corr iPSC-CMs treated with scrambled siRNA, SIRT1 siRNA, or SIRT1 siRNA and SRT, respectively in NT solution and exposed to 0 Na+, 0 Ca2+ solution containing Tet and Caff. h, i Bar graphs to compare the RYR2-mediated SR Ca2+ leak and SR Ca2+ load among different groups in g. n = 24 (KD), 26 (KD + SRT), 30 (NC) cells. j Representative electrophysiological measurements of spontaneous action potentials in II-1-corr iPSC-CMs treated with scrambled siRNA, SIRT1 siRNA, or SIRT1 siRNA and SRT, respectively. k Bar graph to compare the occurrence of arrhythmias among different groups in j. l Representative graphs of staining of lamin A/C (green) and cardiac-specific marker TNNT2 (red) in II−1-corr iPSC-CMs treated with scrambled siRNA, SIRT1 siRNA, or SIRT1 siRNA and SRT, respectively. DAPI indicates nuclear staining (blue). Scale bar, 20 μm. m Bar graph to compare the occurrence of abnormal NE structure among different groups in l. n = 6 views from 3 independent experiments. Data are presented as mean ± SEM in b, c, e, f, h, i, m. p values were calculated by Dunnett’s multiple comparisons test in b, c, e, f, h, i, m. Source data are provided as a Source Data file.
Overexpression of SIRT1 rescues the pathogenic phenotypes in A388fs iPSC-CMs through suppressing ROS-CAMKII-RYR2 pathway
Our prior data indicated that pharmacological activation of SIRT1 could rescue the pathogenic phenotypes in A388fs iPSC-CMs. To provide the direct link between SIRT1 and ROS-CAMKII-RYR2 pathway, we overexpressed human WT SIRT1 plasmid in mutant II-1 iPSC-CMs. Overexpression (OE) of SIRT1 gave rise to significantly increased expression of SIRT1, along with significantly lower levels of ox-CaMKII, p-CaMKII, p-RYR2, and SUN1 (Fig. 6a, b). The level of cellular ROS was significantly reduced in II-1 iPSC-CMs overexpressing SIRT1 (Fig. 6c). In addition, the Ca2+ handling abnormalities observed in II-1 iPSC-CMs were markedly alleviated by SIRT1 OE, as evidenced by increased Ca2+ transient amplitude (Fig. 6d, e), decreased diastolic [Ca2+]i (Fig. 6d, e), decreased RYR2-mediated SR Ca2+ leak (Fig. 6f, g), and increased SR Ca2+ load (Fig. 6f, g). Importantly, SIRT1 OE greatly rescued the arrhythmic phenotype in II-1 iPSC-CMs, showing a normal action potential profile similar to control iPSC-CMs (Fig. 6h, i). At the same time, the phenotype of NE deformation in II-1 iPSC-CMs was partially rescued by SIRT1 OE (Fig. 6j, k). Taken together, these results suggest that SIRT1 OE rescues abnormal Ca2+ handling and arrhythmias in A388fs iPSC-CMs through suppressing ROS-CAMKII-RYR2 pathway.

a, b Western blot analysis of SIRT1, total CaMKII, ox-CaMKII, CaMKII-T286, total RYR2, p-RYR2, and SUN1 expression in II−1 iPSC-CMs overexpressed vector only (negative control, NC), or II-1 iPSC-CMs with SIRT1 overexpression (SIRT1 OE). n = 3 (total CaMKII, ox-CaMKII), 6 (SIRT1, CaMKII-T286, total RYR2, p-RYR2, SUN1) independently biological repeats. c Cellular ROS levels in NC and SIRT1 OE. n = 3 independently biological repeats. d Representative Ca2+ transient traces recorded from NC and SIRT1 OE. e Bar graphs to compare the Ca2+ transient amplitude and diastolic [Ca2+]i between the two groups in d. n = 20 (II−1-OE), 22 (II−1-NC) cells. f Representative traces of cytosolic Ca2+ fluorescence in NC and SIRT1 OE in NT solution and exposed to 0 Na+, 0 Ca2+ solution containing Tet and Caff. g Bar graphs to compare the RYR2-mediated SR Ca2+ leak and SR Ca2+ load between the two groups in f. n = 25 (II-1-OE), 28(II−1-NC) cells. h Representative electrophysiological measurements of spontaneous action potentials in NC and SIRT1 OE. i Bar graph to compare the occurrence of arrhythmias between the two groups in h. j Representative graphs of staining of lamin A/C (green) and cardiac-specific marker TNNT2 (red) in NC and SIRT1 OE. DAPI indicates nuclear staining (blue). Scale bar, 20 μm. k Bar graph to compare the occurrence of abnormal NE structure between the two groups in j. n = 6 views from 3 independent experiments. Data are presented as mean ± SEM in b, c, e, g, k. p values were calculated by two-sided unpaired t-test in b, c, e, g, k. Source data are provided as a Source Data file.
Manipulation of SIRT1 or CaMKII activity improves the function of patient-specific engineered heart tissues
To further evaluate the rescuing effects by manipulating SIRT1 or CaMKII activity at the tissue level, we generated the human-based engineered heart tissue (EHT) model. Both Con-1 and II-1 EHTs were treated with autocamtide-2-related inhibitory peptide (AIP) (CaMKII inhibitor, 25 nM) or SRT (1 μM) for 3 days. We observed that treatment of AIP or SRT in II-1 EHTs gave rise to a more compacted structure, resembling the morphology observed in Con-1 EHTs (Supplementary Fig. 17a). Furthermore, we used a customized contractile force measurement system to analyze the contraction of EHTs in stepped raising stretching length (stretching ratio 0–12%). Meanwhile, we detected the contraction status in stepped pacing frequency (0.5–3.5 Hz). We found that the optimum response frequency for the EHT bundles is 1.5 Hz in our study (Supplementary Fig. 17b), and most of the bundles can be efficiently paced at this frequency. As the frequency increased, the efficiency of pacing decreased rapidly in II-1 EHTs at 12% stretching ratio with or without isoproterenol stimulation, which can be effectively rescued by treatment of AIP or SRT (Supplementary Fig. 17b). The contraction status at 0% and 12% stretching ratio (with or without isoproterenol stimulation) was also recorded from EHTs. We found that the contractile force was significantly increased at 12% stretching ratio in AIP- or SRT-treated II−1 EHTs, when compared to DMSO-treated II−1 EHTs (Supplementary Fig. 17c). These results suggest that manipulation of SIRT1 or CaMKII activity can improve the electrical contraction coupling ability at human-based tissue level. Redressing abnormal SIRT1/CaMKII activity in LMNA-DCM cardiomyocytes could be a promising strategy to restore the electrical and contractile dysfunction.
Discussion
The high risk of SCD in association with LMNA-DCM highlights the importance of understanding how arrhythmia is provoked by the deficiency of lamin A/C1,3,5,6,7. Our study established a patient-derived cellular model of LMNA-DCM with significant proarrhythmic activity and disrupted NE structure. We then demonstrated that patient-specific iPSC-CMs with lamin A/C deficiency exhibited pathogenic signature of mitochondrial dysfunction and ROS elevation, resulting from the accelerated degradation of SIRT1. Excessive oxidative stress, known to be detrimental for both cardiomyocyte structure and function17, then led to the predisposition towards arrhythmias and exacerbation of NE disorganization. These lines of evidence indicate that the molecular pathway of lamin A/C-SIRT1-ROS plays an essential role in maintaining normal cardiac rhythm and NE structure (Fig. 7).

The novel mutation A388fs in the LMNA gene leads to lamin A/C haploinsufficiency. This causes abnormal Ca2+ handling, arrhythmias, and a disformed nuclear envelope (NE) in mutant iPSC-CMs. The reduced expression of lamin A/C in A388fs iPSC-CMs leads to accelerated degradation of the lamin A-binding protein sirtuin 1 (SIRT1), resulting in mitochondrial dysfunction and oxidative stress. On one hand, elevated ROS activates Ca2+/calmodulin-dependent protein kinase II (CaMKII). This then promotes ryanodine receptor 2 (RYR2)-mediated sarcoplasmic reticulum (SR) Ca2+ leak and subsequently causes elevated diastolic [Ca2+]i, abnormal Ca2+ handling, and arrhythmias in A388fs iPSC-CMs. Rescue of these phenotypes can be achieved via the application of the CaMKII inhibitor KN93. On the other hand, excessive ROS production accelerates the accumulation of the nuclear protein SUN1, which then contributes to NE deformation in A388fs iPSC-CMs. In this case, both arrhythmias and nuclear defects can be rescued via the application of SIRT1 activators (either SRT1720 or resveratrol) or ROS scavengers (either NAC or mitoTEMPO). Figure 7 created with BioRender.com was released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license.
Mitochondria provide the bulk of the energy required for cardiac construction22,23,24, where impaired mitochondrial metabolism increases sensitivity to stress. As observed in various cardiovascular diseases, the failing heart exhibits severe impairment of mitochondrial biogenesis and function, leading to excessive oxidative stress. In our study, a pathogenic signature of mitochondrial dysfunction and ROS elevation in LMNA-DCM iPSC-CMs was identified. While a recent study demonstrated similar phenotypes in mouse embryo fibroblasts with HGPS25, the relationship between lamin A/C and mitochondrial dysfunction remained undetermined. In this study, via elucidating the underlying mechanisms and demonstrating the rescue of mitochondrial function, we finally reveal a promising therapeutic target for LMNA-DCM.
As a major component of the nuclear peripheral meshwork, lamin A/C is known to bind with an increasing number of proteins that are implicated in numerous fundamental cellular mechanisms including nuclear and cytoskeletal organization, mechanical stability, chromatin organization, signaling, gene regulation, genome stability, and cell differentiation19. Mutations in LMNA may potentially cause alteration or disruption to any of these interactions, and lead to deleterious effects. SIRT1 is a crucial regulator for mitochondrial function and has been reported to interact with lamin A15. The interaction between SIRT1 and lamin A was confirmed in our iPSC-CM model system, where lamin A was seemingly protective for SIRT1. In A388fs and LMNA-KO iPSC-CMs, the degradation rate of SIRT1 was significantly accelerated compared to controls. Lamin A/C haploinsufficiency may lead to both impaired interaction between SIRT1 and lamin A and accelerated degradation rate of SIRT1, thus resulting in SIRT1 deficiency. With the treatment of SIRT1 activator, both the mitochondrial dysfunction and oxidative stress were significantly mitigated in A388fs iPSC-CMs, indicating that lamin A/C deficiency-related down-regulation of SIRT1 causes impaired mitochondrial function and ROS elevation in LMNA-DCM.
Oxidative stress is a hallmark of various cardiac pathologies. Overloaded oxidative stress is an important driving force for changes in the cellular cytoskeleton, cardiac dysfunction, and arrhythmias17,26. The ROS-CaMKII-RYR2 pathway has been well established in multiple studies to exacerbate cardiac dysfunction and lead to predisposition to arrhythmias11,12. Under excessive oxidative stress, the phosphorylation and oxidation levels of CaMKII become significantly elevated. As a key mediator, activated CaMKII phosphorylates the Ca2+ handling protein RYR2, which then mediates the Ca2+ leak of SR. Increased SR Ca2+ leak through RYR2 disturbs the cytoplasmic diastolic Ca2+ concentration and ultimately leads to arrhythmic activity. The treatment outcomes of the SIRT1 activators or ROS scavengers are impressive in our study. Both the Ca2+ handling abnormalities and arrhythmias in A388fs iPSC-CMs were reversed by the treatment of SRT or MT. Interestingly, we also found that the elevated ROS due to mitochondrial dysfunction accelerated the accumulation of SUN1.
SUN1 is an important pathogenic factor for nuclear deformation21. Disruption of the function of SUN1 protects cardiomyocytes from contraction-induced stress and preserves the nuclear structure. Under the treatment of the SIRT1 activator or ROS scavenger, as accompanied by reduced oxidative stress, the expression of SUN1 was significantly decreased and the NE defects were partially corrected in A388fs iPSC-CMs.
One limitation in our study is the absence of in vivo data. Further investigations in the genetic mouse model will be conducted to confirm our findings in the future. Moreover, in spite of the confirmed interaction between lamin A and SIRT1, further studies are needed to identify the exact binding site of such interaction.
In conclusion, our study identifies SIRT1 deficiency-mediated oxidative stress as a cause of the pathogenic progression in both electrophysiology and nuclear structure in LMNA-DCM. Our results also provide promising therapeutic strategies for LMNA-DCM by manipulating impaired SIRT1 activity and excessive oxidative stress.
Methods
Ethical approval
This study conformed to the principles in the Declaration of Helsinki. Our research complied with all relevant ethical regulations. 3-mm skin punch biopsies were taken from the LMNA-DCM patients and healthy donors following a written informed consent. Generation of iPSC lines was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine (No. 2020-813). The authors affirm that human research participants or their parents/legal guardians provided written informed consent for the publication of the potentially identifiable medical data included in this article.
Genetic testing
Peripheral blood samples were drawn from the LMNA-DCM patients and sent to PrecissionMD Inc (Beijing, China) for the isolation of genomic DNA and commercial genetic testing. Genomic DNA underwent panel sequencing to screen 7469 genes. DNA was fragmented to generate paired-end library. Targets were captured using xGen Lockdown Probes (Precision MD, Beijing, China). The captured libraries were sequenced using the Illumina hiseq2500 Analyzers following the manufacturer’s instructions (Illumina, San Diego, USA). Single-nucleotide variants and indels were called with SOAPsnp (The Beijing Genomics Institute in-house software) and Samtools. All single-nucleotide variants and indels were filtered via multiple databases. Pathogenic variants were assessed under the protocol issued by the ACMG. The mutation was verified in the proband and his family members by Sanger sequencing.
Culture and maintenance of skin fibroblasts
Freshly isolated skin biopsies were rinsed with Dulbecco’s Phosphate-Buffered Saline (DPBS) (Gibco, C14190500BT) and transferred into a 1.5-ml tube. Tissue was minced in collagenase I (1 mg/mL in Dulbecco’s modified Eagle medium (DMEM), Gibco, C11995500BT) and allowed to digest for 6 h at 37 °C. Dissociated skin fibroblasts were plated and maintained with DMEM containing 10% fetal bovine serum (FBS) (Gibco, 10091148) and Penicillin-Streptomycin (Gibco, 15140122) at 37 °C, 95% air and 5% CO2 in a humidified incubator. All skin fibroblasts were used for reprogramming within 5 passages.
Generation of iPSC lines
Somatic reprogramming was used to generate iPSC lines from skin fibroblasts using a CytoTune-iPS 2.0 Sendai Reprogramming Kit following the manufacturer’s instructions (Invitrogen, A16517)10.
Culture and maintenance of iPSCs
The iPSCs were cultured in feeder-free mTeSR1 (STEMCELL Technologies, 85850) media on matrigel (Corning, 354277)-coated plates at 37 °C with 5% (vol/vol) CO2. The media were changed daily and iPSCs were passaged every 3-4 days using Accutase (STEMCELL Technologies, 07920), and resuspended and seeded in mTeSR1 containing 10 μM Y27632 ROCK inhibitor (Selleck, S1049)10.
Karyotyping
Chromosome analysis by G-banding was achieved using iPSCs at passage 20 by the Prenatal Diagnosis Center of Hangzhou Women’s Hospital. At least 20 metaphase cells were analyzed at 300–400 band level.
Alkaline phosphatase staining
Alkaline phosphatase (ALP) staining was performed using the VECTOR Blue Alkaline Phosphatase Substrate Kit (Vector Laboratories, SK-5300) following the manufacturer’s instructions.
Gene correction of the A388fs mutation in the LMNA gene by CRISPR/Cas9
The exon 9 of LMNA was selected for guide RNA (gRNA) design and the gRNA was designed according to the CRISPR online design tool: http://crispor.tefor.net. The sequences of a pair of oligos for targeting site were listed as followed: Forward, 5’-CACCGCCCTACCGACCTGGTGTGGA-3’; Reverse, 5’-AAAC TCCACACCAGGTCGGTAGGGC-3’. The oligos were annealed and ligated into linearized lentiCRISPRv2 vector for generating a gRNA-expressing plasmid. Correction of the mutation (c.1163dupC; p.A388fs) in II-1 iPSCs via homologous recombination was carried out. 1 × 105 II−1 iPSCs were seeded in 12-well plates to 80% confluence. The recombinant plasmid and single-stranded oligo deoxynucleotide (ssODN) were transfected as repair templates into II−1 iPSCs using Lipofectamine 3000 Transfection Reagent (Invitrogen, L3000-015) according to the manufacturer’s instructions. Cells were placed in mTeSR1 supplemented with puromycin (Gibco, A1113803) at a concentration of 4 μg/ml. After 2–3 days, puromycin-resistant clones were selected and then verified by genomic PCR and DNA sequencing (Sangon Biotech).
Generation of LMNA knockout iPSC lines by CRISPR/Cas9
The exon 4 of LMNA was selected for the construction of guide RNA (gRNA). gRNA was then designed according to the CRISPR online design tool: http://crispor.tefor.net. The sequences of a pair of oligos for the targeting site were listed as followed: Forward, 5′-CACCGAACAGGCTGCAGACCATGA-3′; Reverse, 5′-AAACTCATGGTCTGCAGCCTGTTC-3′. The oligos were annealed and ligated into a linearized lentiCRISPRv2 vector for generating a gRNA-expressing plasmid. 1×105 Con-3 iPSCs were then seeded in 12-well plates to an 80% confluence. The recombinant plasmid was transfected into Con-3 iPSCs using Lipofectamine 3000. The cells were then placed in mTeSR1 supplemented with puromycin at a concentration of 4 μg/ml. After 2–3 days, puromycin-resistant clones were picked and verified by genomic PCR and DNA sequencing (Sangon Biotech).
Cardiac differentiation
The iPSC-CMs were generated using a 2D monolayer differentiation protocol. Briefly, ~105 undifferentiated iPSCs were dissociated and replated into matrigel-coated 6-well plates. The iPSCs were cultured, expanded to 85% confluence, and then treated for 2 days with 6 μM CHIR99021 (Axon Medchem, 1386) in RPMI 1640 (Gibco, C11875500BT) with B27 supplement minus insulin (Gibco, A1895601) (RPMI + B27-Insulin) to activate the Wnt signaling pathway. On day 2, cells were placed in RPMI + B27-Insulin with CHIR99021 removal. On days 3-4, cells were treated with 5 μM IWR-1 (Millipore, 681669) to inhibit the Wnt signaling pathway. On days 5–6, cells were removed from IWR−1 treatment and placed in RPMI + B27-Insulin. From day 7 onwards, cells were cultured in RPMI 1640 and B27 supplement with insulin (Gibco, 17504044) (RPMI + B27+Insulin) until beating was observed. Cells were then glucose-starved for 3 days with RPMI + B27+Insulin for purification. Cardiomyocytes of days 30–40 after cardiac differentiation were utilized for downstream functional assays.
Immunofluorescent staining
Cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X−100 (Sangon Biotech, A110694) for 5 min, and blocked with 3% bovine serum albumin (Sigma-Aldrich, A1933) for 1 h. Cells were then stained with appropriate primary antibodies and AlexaFluor conjugated secondary antibodies. Primary antibodies included OCT4 (Cell Signaling Technology, 2750 S, 1:200), NANOG (Santa Cruz Biotechnology, sc-33759, 1:200), SSEA-4 (Abcam, ab16287, 1:200), SOX2 (Abcam, ab171380, 1:200), TNNT2 (Abcam, ab8295, 1:500), α-actinin (Cell Signaling Technology, 6487 P, 1:100), lamin A/C (Abcam, ab108595, 1:500), SIRT1 (Santa Cruz Biotechnology, sc-74465, 1:200) and SUN1 (Abcam, ab103021, 1:500). Secondary antibodies included AlexaFluor® 647 (Abcam, ab150079, 1:200), AlexaFluor® 594 (Abcam, ab150080, 1:500; Abcam, ab150108, 1:500) and AlexaFluor® 488 (Abcam, ab150113, 1:500; Invitrogen, A11008, 1:500). Nuclei were stained with DAPI (Roche Diagnostics, 1023276001, 1 μg/ml). Pictures were taken with 60× objective on a confocal microscope (Nikon, A1) using NIS-Elements AR software (Nikon).
Transmission electron microscopy
The iPSC-CMs were dissociated with Tripsin-EDTA (Gibco, 25200072), scrapped into a 1.5-ml microcentrifuge tube, centrifuged, and then fixed with 2.5% glutaraldehyde (Sigma-Aldrich, G5882) in 0.1 M phosphate buffer overnight at 4 °C. The specimens were post-fixed with 1% OsO4 in phosphate buffer and dehydrated by a graded series of ethyl-alcohol for 15–20 min at each step, then transferred to absolute acetone for 20 min. Next, the specimens were placed in 1:1 mixture of absolute acetone and final spur resin mixture for 1 h at room temperature, transferred to 1:3 mixture of absolute acetone and final spur resin mixture for 3 h, and then transferred to final spur resin mixture overnight. The specimens were then placed in 1.5-ml tube contained spur resin, heated at 70 °C for more than 9 h, and sectioned using a LEICA EM UC7 ultratome. Sections were then stained with uranyl acetate and alkaline lead citrate for 5−10 min. Pictures were observed using a transmission electron microscopy (Hitachi, Model H-7650).
Measurements of cellular ROS
For cellular ROS detection, iPSC-CMs were incubated with 5 µM CellROX Green (Life Technologies, C10444) at 37 °C for 30 min and then washed 3 times before analysis using a CytoFLEX LX flow cytometer. Data were analyzed using FlowJo by measuring the mean fluorescence intensity of each sample.
Detection of mitochondrial ROS
For mitochondrial ROS detection, mitochondria isolated using the Mitochondrial Isolation Kit (Beyotime, C3601) were treated with 5 µM MitoSOX (Life Technologies, M36008) at 37 °C for 15 min, then washed 3 times and analyzed using a CytoFLEX LX flow cytometer. Data were analyzed using FlowJo by measuring the mean fluorescence intensity of each sample.
Mitochondrial stress analysis
Real-time oxygen consumption rates (OCR) were measured using the XFe96 extracellular flux analyser (Seahorse Bioscience). Cells were seeded at 8 × 104 cells per well in XFe96 cell culture microplates (Seahorse Bioscience) and incubated for 5 to 7 days. The OCRs were measured in Seahorse assay medium (10 mM glucose, 10 mMpyruvate, at pH7.4). Different mitochondrial respiratory states were investigated through sequential addition of diverse compounds that modulate or inhibit mitochondrial respiration differently. Firstly, ‘basal OCR’ was measured to indicate cellular respiration under normal physiological conditions. Secondly, oligomycin (1.5 μM) was added to obtain information about mitochondrial ‘proton leak’ and ‘ATP-linked OCR’. Following, the uncoupling agent carbonylcyanidep-trifluoromethoxyphenylhydrazone (FCCP; 3 μM) was added to acquire the maximum respiratory rate. The difference between maximum capacity and basal OCR indicates the ‘reserve capacity’ of the electron transport chain. Finally, a combination of rotenone (0.5 μM) and antimycin-A (Anti A; 0.5 μM) was added to inhibit the activity of complexes III and I respectively, in order to obtain non-mitochondrial OCR (‘NM OCR’) (to enable correction for non-mitochondrial sources of OCR). Specifically, the OCR parameters are calculated as follows: Basal OCR = OCR before drugs added - NM OCR; Protein leak = OCR after oligomycin - NM OCR; Maximum Capacity = OCR after FCCP – NM OCR; ATP linked OCR = OCR before drugs added – OCR after oligomycin; reserve capacity = OCR after FCCP - OCR before drugs added.
Mitochondrial copy number quantification
Mitochondrial and genomic DNAs were extracted using TIANamp Genomic DNA kit (Tiangen) according to the manufactures’ instruction. The copy numbers of mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) were quantified using TB Green® Premix EX TaqTM (Takara) on an ABI Prism 7500 system. Primers specific for ND1, ND5, SLCO2B1, and SERPINA1 were used to quantify mtDNA and nDNA (Takara,7246), respectively. The determination of the difference in the 2ΔCt values for the ND1/SLCO2B1 pair and ND5/SERPINA1 pair was conducted in the same manner using the average of the 2 values as the mtDNA copy number.
Quantitative real-time PCR
Total RNA isolation was performed using TRIzol®Reagent (Life Technologies, T9424). RNA concentration was measured using UV spectrophotometry at 260 nm (Nanodrop 2000, Thermo Scientific). cDNA was obtained using the 4× EZscript Reverse Transcription Mix II (EZB, RT2G). Quantitative real-time PCR was conducted using SYBR Green PCR Master Mix (Takara). Primer sequences used in this study were listed in Supplementary Table 3. Each reaction was run in quadruplicate using an Applied Biosystems Viia7 Dx (ThermoFisher Scientific). Gene expression values were normalized to the housekeeping gene GAPDH.
Western blot
The iPSC-CMs were detached with TrypLE and then pelleted at 94 g for 5 min at 4 °C. After washing with PBS, the pellets were re-suspended in 50-100 μl lysis buffer. Lysates were placed on ice for 30 min and then the supernatants were collected after centrifuging at 13523 g for 15 min. Protein concentration was measured using a BCA kit (Pierce, 23227). Western blot was performed using the standard protocol with the following antibodies: lamin A/C (Abcam, ab108595, 1:1000; Santa Cruz Biotechnology, sc-7292, 1:200), SIRT1 (Santa Cruz Biotechnology, sc-74465, 1:200), total CaMKII (Abcam, ab181052, 1:1000), phosphorylated CaMKII-T286 (Abcam, ab32678, 1:1000), total RYR2 (Abcam, ab2827, 1:500), phosphorylated RYR2 (Badrilla, Aolo-3/AP, 1:500), SUN1 (Abcam, ab103021,1:500) and GAPDH (Abmart, M20006M, 1:5000). Intensity values for each band were determined as the integrated density (sum of pixel values) within a fixed area using Quantity One software (Biorad).
Separation of cytoplasmic and nuclear extracts
Cytoplasmic and nuclear extracts were separated using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, 78833). The first two reagents caused cell membrane disruption and release of cytoplasmic contents. The intact nuclei were separated from the cytoplasmic extract by centrifugation. The nuclear proteins were extracted with the third reagent. Protein lysates were then followed by immunoblotting with antibodies as mentioned above.
Detection of SIRT1 activity
The SIRT1 activity was detected using the Epigenase Universal SIRT Activity/Inhibition Assay Kit (Epigentek, P-4037-48) following the manufactures’ instruction. The nuclear extracts were prepared as mentioned above. Active SIRTs bond to the acetylated histone SIRT substrate and removed acetyl groups from the substrate. The SIRT-acetylated products can be recognized with a specific antibody. The ratio of deacetylated products was measured by reading the fluorescence in a fluorescence microplate reader. The activity of the SIRT enzyme is proportional to the relative fluorescent units measured.
Co-IP
Immunoprecipitation and co-immunoprecipitation experiments were conducted utilizing Pierce Classic Magnetic IP/Co-IP Kit (Thermo Scientific, 88804). Cell lysates were incubated with a specific primary antibody at room temperature for 2 h. The immune complex was incubated with A/G magnetic beads for 1 h. The magnetic beads were then washed twice with IP buffer, followed by a washing of the antigen/antibody complex once with pure water. Elution Buffer was added to the tube and incubated with mixing for 10 min to separate the beads and the target antigen. Protein lysates were then followed by immunoblotting with antibodies as mentioned above.
Patch clamp recordings from iPSC-CMs
The iPSC-CMs were mechanically and enzymatically dissociated to obtain single cells. These were seeded on matrigel-coated glass coverslips (Warner Instruments). Cells with spontaneous beatings were selected and action potentials were recorded using an EPC−10 patch clamp amplifier (HEKA). Continuous extracellular solution perfusion was achieved using a rapid solution exchanger (Bio-logic Science Instruments, RSC-200). All signals were acquired using PatchMaster software (HEKA) and filtered at 1 kHz and digitized at 10 kHz. Data analyses were performed using Igor Pro (Wavemetrics) and GraphPad Prism (GraphPad Software). A TC-344C dual channel heating system (Warner Instruments) was used to maintain the temperature at 35.5-37 °C. Tyrode’s solution was used as the external solution containing 140 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, 1.8 mM CaCl2, 1.0 mM Na-Pyruvate and 10 mM HEPES (pH 7.4 with NaOH). The internal solution contained 140 mM KCl, 5.0 mM NaCl, 10 mM HEPES, 5 mM Mg-ATP and 5 mM EGTA (pH 7.2 with KOH). Ventricular-like iPSC-CMs were distinguished based on the action potential morphology and action potential parameters which exhibit a clear plateau phase, larger APA and Vmax values, and more negative MDP values, with APD30-40/APD70–80 > 1.5 and APD90/ APD50 ≤ 1.3.
Ca2+ imaging
The iPSC-CMs grown on coverslips were loaded with RPMI 1640 medium without phenol red (Gibco, 11835030) and supplemented with 5 μM Fura-2 AM (Invitrogen, F14185) for 30 min in the dark at room temperature. After washing twice with pre-warmed DPBS and RPMI 1640, the iPSC-CMs were immersed in imaging buffer for 30 min before imaging experiments. For imaging, iPSC-CMs were placed in a chamber equipped with a temperature-controller under constant perfusion of 37 °C imaging buffer. Ca2+ signaling was made by recording the fluorescence of cells using an Ultra High Speed Wavelength Switcher (Lambda DG-4, Sutter Instruments) with a CCD camera (Zyla, Andor) mounted on an inverted microscope (Eclipse Ti, Nikon). Fluorescent signals were obtained upon excitation at 340 nm (F340) and 380 nm (F380). The amplitude of a Ca2+ transient was defined as the ratio of F340/F380.
Assessment of RYR2-mediated diastolic Ca2+ leak and SR Ca2+ load
RYR2-mediated diastolic Ca2+ leak and SR Ca2+ load was assayed using Fluo-4 fluorescence. The iPSC-CMs were incubated for 15 min at room temperature in phenol red-free RPMI 1640 (Gibco, 11835030) containing 5 μM of the cytosolic Ca2+ dye Fluo-4 AM (Invitrogen, F23917) to load the indicator into the cytosol. Following incubation, the indicator-containing medium was removed. iPSC-CMs were washed with phenol red-free RPMI 1640 three times and immersed in imaging buffer for an additional 30-45 min at room temperature to allow for de-esterification of the indicator. Recordings were captured using an epifluorescence microscope. The iPSC-CMs were field-stimulated at 1 Hz in normal Tyrode’s solution containing 1.8 mM Ca2+ until achieving a steady-state intracellular Ca2+ content. The stimulation was then turned off, and the bath solution was rapidly changed to 0 Na+, 0 Ca2+ Tyrode’s solution (140 mM LiCl, 5.4 mM KCl, 1 mM MgCl2, 5 mM HEPES, 10 mM glucose, 10 mM EGTA, pH 7.4 with LiOH) to eliminate transsarcolemmal Ca2+ fluxes. A pulse of tetracaine (1 mM) was added to inhibit RYR2. The observed shift (Ca2+ from cytosol to SR) is then proportional to the SR Ca2+ leak. The solution was then rapidly switched to caffeine (10 mM) to deplete SR Ca2+ stores. Lastly, bath solution was switched from 0 Na+, 0 Ca2+ Tyrode’s solution back to normal Tyrode’s solution. Data was acquired using NIS-Elements software (Nikon Instruments Inc), and analysis was performed using Igor Pro (Wavemetrics) and GraphPad Prism (GraphPad Software).
Intracellular Ca2+ transients recording by IonOptix system
The iPSC-CMs was plated in confocal dish for 3–5 days before experiments. Fura-2 AM was loaded on iPSC-CMs for 30 min in the dark at room temperature. After washing twice with pre-warmed DPBS and RPMI 1640, the iPSC-CMs were immersed in imaging buffer for 30 min prior to fluorescence intensity recordings. Fluorescence emissions were detected using a fluorescence photomultiplier tube (IonOptix, Milton, MA). Fluorescent signals were obtained upon excitation at 340 nm (F340) and 380 nm (F380). The amplitude of Ca2+ transient was defined as the ratio of F340/F380.
Quantitative proteomics by multiplexed tandem mass tag MS
20 mg samples of iPSC-CMs (II−1-corr and II−1, n = 3 biological repeats) were lysed with lysis buffer (8 M urea, 100 mM triethylammonium bicarbonate (TEAB), pH 8.5), followed by being centrifuged at 12,000 × g for 15 min to collect supernatant. The extracted protein lysates were treated with 1 M DTT for 1 h at 56 °C and alkylated with iodoacetamide for 1 h at room temperature. After being qualified and quantified by Bradford protein quantitative kit, protein samples were digested with trypsin (1:20 trypsin to protein), and desalted by a Strata X C18 SPE column desalting column (Phenomenex, Torrance, CA, USA) and vacuum-dried. Prepared peptides were reconstituted with 0.1 M TEAB buffer, and subsequently labeled with tandem mass tag (TMT) labeling reagent according to the manufacture’s protocol. The TMT-labeled peptides were fractionated and pooled into 10 fractions on a Rigol L3000 HPLC system, followed by being dried under vacuum. UHPLC-MS/MS analyses were performed using an EASY-nLC 1200 UHPLC system (Thermo Fisher, Germany) coupled with Q ExactiveTM HF-X (Thermo Fisher, Germany). The resulting spectra were analyzed in UniProt database (homo_sapiens_uniprot_UP000005640 (82493 sequences)) using Proteome Discoverer (PD, Thermo, HFX, and 480). The search parameters were set as follows: mass tolerance for precursor ion at 10 ppm and mass tolerance for product ion at 0.02 Da. To improve the quality of analysis results, the PD software was used to further filter the retrieval results with Peptide Spectrum Matches (PSMs) with a credibility of more than 99% then considered as PSMs. The identified protein contains at least 1 unique peptide. The identified PSMs and protein were retained and performed with FDR no more than 1.0%. The protein quantitation results were statistically analyzed by t-test. Differentially expressed proteins (DEP) were filtered based on the criteria p < 0.05 and fold change (FC) > 1.3 or FC < 0.77. The DEP of total proteins quantified is 11.5%. Gene Ontology (GO) and InterPro (IPR) functional analysis were conducted using the interproscan program against the non-redundant protein database (including Pfam, PRINTS, ProDom, SMART, ProSite, PANTHER), and the databases of COG (Clusters of Orthologous Groups) and KEGG (Kyoto Encyclopedia of Genes and Genomes) were used to analyze the protein family and pathway. DEPs were used for Volcanic map analysis, cluster heat map analysis, and enrichment analysis of GO, IPR, and KEGG.
Overexpression of SIRT1 in iPSC-CMs
Human SIRT1 cDNA was introduced to pCDH-CMV-MCS-EF1-copGFP-T2-Puro and pCDH-CMV-MCS-EF1-T2-Puro vector, respectively. The constructed plasmid was introduced to iPSC-CMs by Lipofectamine 3000 Transfection Reagent according to the manufacturer’s instructions. The medium was changed after 9 h and green fluorescent protein (GFP) could be detected in 80°90% cells after 24 h.
Knockdown of SIRT1 or SUN1 in iPSC-CMs
The SIRT1 or SUN1 siRNA was purchased from GenePharma. The siRNA was introduced to iPSC-CMs by RNAiMAX (Invitrogen, 13778075) system following the manufacturer’s instructions. The medium was changed after 9 h. A significant reduction of SIRT1 or SUN1 expression could be observed after 3 days culturation.
Fabrication and culture of EHTs
To generate 1.5 × 8 mm 3D human cardiac tissue bundles, polydimethylsiloxane (Dow Corning, SYLGARD 184) molds were designed and microfabricated27. Hydrogel solution (24 µL 10 mg/mL fibrinogen (Sigma, F3879), 12 µL matrigel (Corning, 354277), 24 µL 2× culture medium) were mixed with 5 × 105 iPSC-CMs in 58 µL culture medium. Following the addition of 2.4 µL 50 U/mL thrombin (Sigma, T7201), the cell/gel mixture was added to the molds and placed at 37 °C for 30 min to polymerize. Cardiac bundles were removed from the molds and cultured in 12‐well plates on a rocking platform for 7 days before being used for the drug tests. The culture medium was renewed every 2 days.
Contractile force assessment of EHTs
Cardiac bundles were treated with DMSO or drugs at the indicated concentration for 3 days. The electrical and mechanical stretch-stimulated contractile force of cardiac bundles after drug treatment were assessed using a customized force measurement setup28. To assess the force-length relationship, cardiac bundles were paced with an electrical pulse (10 V, 2 Hz) using a pair of platinum electrodes in Tyrode’s solution containing 1.8 mM Ca2+, and sequentially stretched by 0% to 12% of its culture length (with or without isoproterenol stimulation). To assess the force-frequency relationship and electrical contraction coupling ability, cardiac bundles were paced under stepped raising frequencies (0.5, 1, 1.5, 2, 2.5, 3, 3.5 Hz) at 0% and 12% stretching ratio (with or without isoproterenol stimulation). Data were analyzed by the MATLAB software (version R2020b (9.9.0.1467703)).
Compounds and solutions
All the chemicals used in the electrophysiological experiments were purchased from Sigma-Aldrich. Fluo-4 AM and Fura-2 AM were purchased from Invitrogen and stock solutions were both prepared in 1 mM in 20% Pluronic F-127 (Sangon Biotech, A600750) dissolved in DMSO (Sigma-Aldrich, D2650). Resveratrol was purchased from MedChemExpress and stock solutions were prepared in 10 mM in DMSO. SRT was purchased from MedChemExpress (HY−15145) and stock solutions were prepared in 1 mM in DMSO. KN93 was purchased from Abcam (Ab120980) and stock solutions were prepared in 1 mM in DMSO. The solutions of tetracaine (Sigma-Aldrich, T7508) and caffeine (Sigma-Aldrich, C0750) were freshly prepared when required. MT was purchased from Sigma-Aldrich and stock solutions were prepared in 10 mM in DMSO. NAC was purchased from Sigma-Aldrich and stock solutions were prepared in 500 mM in DMSO. AIP was purchased from Sigma-Aldrich (SCP0001) and stock solutions were prepared in 2 μM in ddH2O.
Statistics and reproducibility
In each quantification, a single plot point indicates a cell, view or independently biological replicate (individual culture well). The quantitative data are taken from at least n = 3 biological replicates. The value of n and what n represents is indicated in each figure legend. The experiments were repeated independently for three times in Figs. 1j, k, m, n, 2a, c, d, f, g, j, i, l, m, 3a, c, e, g, i, l, 4b, d, f, h, i, 5a, c, d, g, l, and 6a, c, d, f, j; for four times in Figs. 3o, 4a, 5j, and 6h, with similar results. Statistical significance was determined using unpaired two-tailed Student’s t-tests to compare two groups and by One-way ANOVAs to compare multiple groups. A p value of <0.05 was considered statistically significant. Data were shown as mean ± SEM and analyzed by GraphPad Prism (GraphPad Software). The investigators were blinded to group allocation during data collection and analysis for all experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The proteome data generated in this study have been deposited in the ProteomeXchange Consortium via the iProX repository with the data set identifier PXD039452 [https://www.iprox.cn/page/subproject.html?id=IPX0005778001]. The raw clinical data are protected and are not available due to data privacy laws. The completely anonymized clinical data could be shared within two weeks by writing an email to the corresponding authors (Dr. Ping Liang: pingliang@zju.edu.cn; Dr. Chenyang Jiang: cyjiang@zju.edu.cn). Uncropped and unprocessed scans of blots were supplied in the Source Data file. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Information. Source data are provided with this paper.
References
Schultheiss, H.-P. et al. Dilated cardiomyopathy. Nat. Rev. Dis. Prim. 5, 32 (2019).
Richardson, P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 93, 841–842 (1996).
Hershberger, R. E., Hedges, D. J. & Morales, A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547 (2013).
Rosenbaum, A. N., Agre, K. E. & Pereira, N. L. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat. Rev. Cardiol. 17, 286–297 (2020).
Capell, B. C. & Collins, F. S. Human laminopathies: nuclei gone genetically awry. Nat. Rev. Genet 7, 940–952 (2006).
Taylor, M. D. & Towbin, J. A. The significant arrhythmia and cardiomyopathy burden of lamin A/C mutations. J. Am. Coll. Cardiol. 68, 2308–2310 (2016).
Weintraub, R. G., Semsarian, C. & Macdonald, P. Dilated cardiomyopathy. Lancet 390, 400–414 (2017).
Bertero, A. et al. Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J. Cell Biol. 218, 2919–2944 (2019).
Lee, J. et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340 (2019).
Lan, F. et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12, 101–113 (2013).
Liu, X. et al. Increased Reactive Oxygen Species–Mediated Ca 2+ /Calmodulin-Dependent Protein Kinase II Activation Contributes to Calcium Handling Abnormalities and Impaired Contraction in Barth Syndrome. Circulation 143, 1894–1911 (2021).
Pabel, S. et al. Effects of atrial fibrillation on the human ventricle. Circ. Res 130, 994–1010 (2022).
Zorov, D. B., Juhaszova, M. & Sollott, S. J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950 (2014).
Wan, X. & Garg, N. J. Sirtuin control of mitochondrial dysfunction, oxidative stress, and inflammation in chagas disease models. Front. Cell. Infect. Microbiol. 11, 693051 (2021).
Liu, B. et al. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 16, 738–750 (2012).
Hamilton, S. et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res. Cardiol. 115, 38 (2020).
Dey, S., DeMazumder, D., Sidor, A., Foster, D. B. & O’Rourke, B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ. Res. 123, 356–371 (2018).
Madreiter-Sokolowski, C. T., Thomas, C. & Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biol. 36, 101678 (2020).
de Leeuw, R., Gruenbaum, Y. & Medalia, O. Nuclear lamins: thin filaments with major functions. Trends Cell Biol. 28, 34–45 (2018).
Chen, C.-Y. et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell 149, 565–577 (2012).
Chai, R. J. et al. Disrupting the LINC complex by AAV mediated gene transduction prevents progression of Lamin induced cardiomyopathy. Nat. Commun. 12, 4722 (2021).
Wiersma et al. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells 8, 1202 (2019).
Lesnefsky, E. J., Chen, Q. & Hoppel, C. L. Mitochondrial metabolism in aging heart. Circ. Res 118, 1593–1611 (2016).
Boyman, L., Williams, G. S. B. & Lederer, W. J. The growing importance of mitochondrial calcium in health and disease. Proc. Natl Acad. Sci. USA 112, 11150–11151 (2015).
Maynard, S. et al. Lamin A/C impairments cause mitochondrial dysfunction by attenuating PGC1α and the NAMPT-NAD+ pathway. Nucleic Acids Res. 50, 9948–9965 (2022).
Hunter, M. V., Willoughby, P. M., Bruce, A. E. E. & Fernandez-Gonzalez, R. Oxidative stress orchestrates cell polarity to promote embryonic wound healing. Dev. Cell 47, 377–387.e4 (2018).
Madden, L., Juhas, M., Kraus, W. E., Truskey, G. A. & Bursac, N. Bioengineered human myobundles mimic clinical responses of skeletal muscle to drugs. eLife 4, e04885 (2015).
Juhas, M., Engelmayr, G. C., Fontanella, A. N., Palmer, G. M. & Bursac, N. Biomimetic engineered muscle with capacity for vascular integration and functional maturation in vivo. Proc. Natl Acad. Sci. USA 111, 5508–5513 (2014).
Acknowledgements
We would like to thank the core facility of Zhejiang University Institute of Translational Medicine for assistance with confocal microscopy experiments. This work was supported by Natural Science Foundation of Zhejiang Province (LD21H020001) (P.L.), National Natural Science Foundation of China (81922006, 81870175) (P.L.), National Key R&D Program of China (2017YFA0103700) (P.L.), National Natural Science Foundation of China (81970269) (C.J.), and Key Research and Development Program of Zhejiang Province (2019C03022) (C.J.). P.L. would like to thank Natalie Liang and Michael Liang for their encouragement and consistent support.
Author information
Authors and Affiliations
Contributions
P.L. and C.J. designed and supervised the study. H.Q., Y.S., X.W., T.G., J.Su., J.Shen., J.Z., J.X., H.W., X.M., G.F., and D.Z. performed the experiments and analyzed data. P.L. and H.Q. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiu, H., Sun, Y., Wang, X. et al. Lamin A/C deficiency-mediated ROS elevation contributes to pathogenic phenotypes of dilated cardiomyopathy in iPSC model. Nat Commun 15, 7000 (2024). https://doi.org/10.1038/s41467-024-51318-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51318-5
This article is cited by
-
Clinical and metabolic consequences of a historic pathogenic lamin A/C founder variant
Scientific Reports (2025)
