
Abstract
During meiosis, nucleoprotein filaments of the strand exchange proteins RAD51 and DMC1 are crucial for repairing SPO11-generated DNA double-strand breaks (DSBs) by homologous recombination (HR). A balanced activity of positive and negative RAD51/DMC1 regulators ensures proper recombination. Fidgetin-like 1 (FIGNL1) was previously shown to negatively regulate RAD51 in human cells. However, FIGNL1’s role during meiotic recombination in mammals remains unknown. Here, we decipher the meiotic functions of FIGNL1 and FIGNL1 Interacting Regulator of Recombination and Mitosis (FIRRM) using male germline-specific conditional knock-out (cKO) mouse models. Both FIGNL1 and FIRRM are required for completing meiotic prophase in mouse spermatocytes. Despite efficient recruitment of DMC1 on ssDNA at meiotic DSB hotspots, the formation of late recombination intermediates is defective in Firrm cKO and Fignl1 cKO spermatocytes. Moreover, the FIGNL1-FIRRM complex limits RAD51 and DMC1 accumulation on intact chromatin, independently from the formation of SPO11-catalyzed DSBs. Purified human FIGNL1ΔN alters the RAD51/DMC1 nucleoprotein filament structure and inhibits strand invasion in vitro. Thus, this complex might regulate RAD51 and DMC1 association at sites of meiotic DSBs to promote proficient strand invasion and processing of recombination intermediates.
Similar content being viewed by others

Introduction
Meiosis ensures the accurate reduction of chromosome numbers in gametes during sexual reproduction. Erroneous meiosis results in sterility or fertility defects owing to aberrant gamete formation. During meiosis, homologous chromosomes (homologs) undergo pairing, synapsis, and recombination. Homologous recombination (HR) is crucial for crossover (CO) formation between homologs to ensure their balanced segregation during meiosis, and for promoting pairing and synapsis of homologs in some organisms including mammals1,2,3. HR is initiated by genome-wide SPO11-dependent DNA double-strand break (DSB) formation 4. SPO11 is subsequently released from DSB sites as SPO11-oligonucleotide complex by resection machinery giving rise to 3’ single-stranded DNA (ssDNA) overhangs5,6. The heterotrimeric complex of Replication Protein A (RPA) binds to and protects the ssDNA overhangs from nucleolytic degradation. Two eukaryotic RecA-like strand-exchange proteins, RAD51 and its meiosis-specific paralog DMC1, replace RPA on ssDNA with the help of the mediator protein BRCA27,8. Both strand exchange proteins can catalyze homology search and strand exchange through invasion on an intact template, leading to formation of a joint molecule termed displacement loop (D-loop)9. The invading end primes DNA synthesis that requires the dissociation of RAD51/DMC1 from double-strand DNA (dsDNA) within the D-loop. After D-loop formation, meiotic DSB repair can produce a non-crossover (NCO), or a CO by two alternative pathways that coexist in many organisms2. In mice, the meiosis-specific class I CO pathway generates 90% of COs and is dependent on a set of proteins referred to as ZMM proteins (including the MSH4-MSH5 complex and TEX11)10,11 and the MutL homologs MLH1-MLH3. Mouse MSH4 and MSH5 are essential to repair most if not all meiotic DSBs2,12,13,14. The class II COs (~10% of COs in the mouse) depend on structure-specific endonucleases2.
Both RAD51 and DMC1 form foci, which colocalize extensively at DSB sites15,16 and are proposed to assemble into side-by-side homo-filaments on ssDNA tails, with RAD51 at the DSB-distal region and DMC1 polymerizing on the 3′, DSB-proximal region9,17. DMC1 is likely the main catalyzer of meiotic interhomolog recombination in most eukaryotes, while RAD51 plays crucial non-catalytic accessory roles18,19,20. RAD51 is the sole strand exchange protein during mitotic recombination and also plays a strand exchange activity-independent role in the replication fork protection that might rely on its dsDNA-binding capacity21,22,23,24,25,26. Besides this specific function, inactive filaments of RAD51 and DMC1 on dsDNA are likely toxic and are actively prevented27. Members of the Swi2/Snf2-related RAD54 translocase family28 prevent the accumulation of RAD51 on dsDNA in human cells29, and of Rad51 and Dmc1 in S. cerevisiae30,31. In S. cerevisiae, Rad54 and its paralog Rdh54 promote strand invasion, and remove RAD51/DMC1 from dsDNA following D-loop formation28,32. In mouse, RAD54 and its paralog RAD54B are not essential for meiotic recombination, because the Rad54 Rad54b double mutant mice are fertile33,34. Many proteins regulate RAD51/DMC1 nucleofilament formation positively or negatively. Positive factors are required to form stable and active RAD51-ssDNA filaments. They include BRCA2 and several RAD51 paralogs, which are essential for viability in mammals7,8. The conserved Shu complex comprises in mammals the distant RAD51 paralog SWSAP1, the SWIM-domain containing SWS1 and SPIDR35,36,37,38,39. This complex promotes RAD51-mediated HR in the context of replication, and RAD51- and DMC1-mediated meiotic recombination in the mouse, but is not essential for viability36,37,40,41,42. The SWSAP1-SWS1-SPIDR complex might promote specifically the stable assembly of longer RAD51 nucleoprotein filaments involved in some HR types, especially interhomolog HR36,37.
FIGNL1 (fidgetin-like 1) forms an evolutionary conserved complex with FIRRM (FIGNL1 interacting regulator of recombination and mitosis) that interacts with RAD51 and DMC1, and regulates negatively RAD51 during HR repair in mammalian cells43,44,45,46,47,48,49,50,51. In Arabidopsis and rice meiosis, FIGNL1 and FIRRM homologs negatively regulate the dynamics of RAD51 and DMC1 foci and limit the formation of class II crossovers44,45,52,53,54. Arabidopsis figl1 (Figl1 homolog) and flip (Firrm homolog) mutants are fertile with all meiotic DSBs repaired44,52,55. Conversely, unrepaired DSBs persist in rice fignl1 and meica (Firrm homolog) mutants, leading to chromosome fragmentation and sterility53,54. The regulation of RAD51/DMC1 focus formation in Arabidopsis somatic and meiotic cells involves an antagonistic interplay between BRCA2 and FIGL1, consistent with FIGL1 acting as a negative regulator of RAD51/DMC1 filament56. In human cells, an antagonistic relationship was found between BRCA2 and FIGNL1-FIRRM48, and between the SWSAP1-SWS1-SPIDR complex and FIGNL1, which interacts with SWSAP140 and SPIDR43. Indeed, FIGNL1 depletion relieves the dependency on SWSAP1 and SWS1 for forming RAD51 repair foci40. Moreover, purified human SWSAP1 protects RAD51-ssDNA filament from dissociation promoted by FIGNL1 in vitro40. However, the role of FIGNL1 and FIRRM during meiotic recombination in mammals was unknown.
In this study, we investigated the role of the FIGNL1-FIRRM complex in meiotic recombination by analyzing germ line-specific mouse conditional knock-out models for both genes. The depletion of FIGNL1 or FIRRM in mouse spermatocytes results in meiotic DSB repair failure and no full synapsis between homologs during meiotic prophase I, leading to prophase I arrest and apoptosis. Surprisingly, Fignl1 cKO and Firrm cKO spermatocytes also show an abundant DSB-independent accumulation of RAD51 and DMC1 on chromatin and meiotic chromosome axes during premeiotic replication and early meiotic prophase stages. This indicates that the FIGNL1-FIRRM complex prevents the formation of stable inactive RAD51 and DMC1 filaments, presumably on intact dsDNA, in mouse spermatocyte nuclei.
Results
FIGNL1 and FIRRM are required for meiotic prophase completion in the mouse male germline
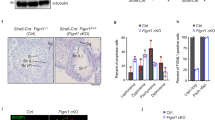
We wanted to determine the roles of FIGNL1 and its putative partner FIRRM (also called BC055324) during meiosis. As both genes are essential for mouse viability (57,58, IMPC, https://www.mousephenotype.org/), we generated cKO lines with Cre expression under the control of the Stra8 promoter59 to ablate Firrm or/and Fignl1 in the male germline shortly before meiosis onset (Firrm cKO and Fignl1 cKO, Supplementary Fig. 1a-c). Testis weight was similarly reduced in Firrm cKO, Fignl1 cKO, and Firrm-Fignl1 double cKO mice compared with wild-type controls (Fig. 1a, Supplementary Fig. 2a). Analysis of testis sections from adult Firrm cKO and Fignl1 cKO animals revealed the lack of haploid cells (spermatids) in most tubules, suggesting a prophase I arrest (Fig. 1b, Supplementary Fig. 2b-c). Some germ cell depletion was apparent on sections from older Firrm cKO and Fignl1 cKO animals, suggesting that FIGNL1-FIRRM depletion affects the viability or the proliferative capacity of spermatogonia in adults (Supplementary Fig. 2b-c). Nevertheless, the density of Sertoli cells, spermatogonia and early prophase spermatocytes appeared normal in seminiferous tubules from 4-week-old Firrm cKO animals (Fig. 1b), and no obvious defect was detected in spermatogonia by cytological analysis of juvenile testes, suggesting that the defects observed in Firrm cKO and Fignl1 cKO spermatocytes do not result from defects in spermatogonia prior to meiosis induction. The presence of some tubules with a small number of round spermatids and of few tubules with many round and elongated spermatids suggested incomplete or delayed Cre-mediated excision or protein depletion, as described in other conditional mouse mutants obtained with this Stra8-Cre transgene60,61,62. Consistent with delayed excision or protein depletion, no sperm was found in cauda epididymis from 4-month-old mice (Supplementary Fig. 2d), and CRE-mediated Firrm excision was 100% efficient, as assessed by genotyping the progeny of Firrmflox/+ Stra8-CreTg males crossed with wild-type females (Methods). In testes from 12-day post-partum (12 dpp) Firrm cKO and Fignl1 cKO mice, FIRRM and FIGNL1 protein levels in the cytoplasmic and nuclear fractions were greatly and similarly reduced compared to controls (Fig. 1c, Supplementary Fig. 2e). This suggests that they reciprocally regulate their stability, which is consistent with forming a complex. The residual protein level might result from expression in non-meiotic cells (spermatogonia or somatic cells) and/or from incomplete Cre-induced gene deletion in a fraction of spermatocytes. Conversely, RAD51 expression in the nuclear fraction was increased in Firrm cKO and Fignl1 cKO testes (Fig. 1c, Supplementary Fig. 2e), suggesting that the FIGNL1-FIRRM complex might be implicated in limiting directly or indirectly nuclear accumulation of RAD51 (but not of DMC1). This might have significant consequences, because RAD51 nuclear level is suggested to play a role in HR regulation63.

a Testis weight relative to body weight in control (gray), Firrm cKO (red), Fignl1 cKO (orange) and Firrm cKO Fignl1 cKO (purple) adult mice (30 to 154 days post-partum, dpp; kinetics is shown in Supplementary Fig. 2a). The mean ± SD is shown. Unpaired t-tests, two-sided. b Periodic acid-Schiff-stained testis sections from 4-week-old control and Firrm cKO mice. Spg, spermatogonia; Spc, spermatocytes; rSpt, round spermatids; eSpt, elongated spermatids. Scale bar, 50 µm. c Western blot analysis of cytoplasmic (80 µg) and nuclear (100 µg) fractions from testes of 12 dpp mice of the indicated genotypes. d Chromosome axes (SYCP3, red) and synaptonemal complex (SYCP1, green) were detected in spread spermatocyte nuclei from control, Firrm cKO (25 dpp) and Fignl1 cKO (56 dpp) mice. Scale bar, 10 µm. e Distribution of spermatocytes at different meiotic prophase substages in juvenile Firrm cKO mice (indicated age) and in adult (8-week-old) Firrm cKO and Fignl1 cKO mice (1 mouse per condition). Two-sided chi-square tests. For all figures: ns, non-significant; *0.01 < p ≤ 0.05; **0.001 < p ≤ 0.01; ***0.0001 < p ≤ 0.001; ****p ≤ 0.0001, exact p-values are provided as Source Data. Source data are provided as a Source Data file.
The synaptonemal complex (SC), a tripartite proteinaceous structure, links the axes of homologous chromosomes during meiotic prophase. To analyze if synapsis was impaired in Firrm cKO and Fignl1 cKO, we stained surface-spread spermatocyte nuclei with antibodies against SYCP3, a component of meiotic chromosome axes, and SYCP1, a protein of the SC central element (Fig. 1d). Firrm cKO and Fignl1 cKO spermatocytes formed apparently normal meiotic chromosome axes (leptotene stage), suggesting a normal meiotic prophase entry. However, most nuclei showed unsynapsed or partially synapsed axes, indicating the accumulation of zygotene-like cells. The small fraction of Fignl1 cKO and Firrm cKO spermatocytes that progressed toward normal-looking pachytene and diplotene might be explained by incomplete Cre-mediated excision in these cells. We followed prophase I progression during the first wave of meiosis in Firrm cKO, from 12 dpp to 18 dpp (Fig. 1e). We detected a deficit in more advanced stages already in 12 dpp Firrm cKO spermatocytes. At 16 and 18 dpp, most nuclei were arrested at a zygotene-like stage, and the percentage of nuclei at the pachytene and diplotene stages was strongly reduced (at 18 dpp, 78% of control versus 15% of Firrm cKO nuclei). Approximately 30% of Firrm cKO prophase I nuclei displayed an abnormal zygotene/pachytene-like pattern, with non-homologous synapsis and only few synapsed chromosome axes (Fig. 1d). These findings in 12 dpp to 18 dpp spermatocytes are indicative of an arrest in early pachytene and a defect in homologous synapsis. Adult Firrm cKO and Fignl1 cKO animals displayed a similar deficit in pachytene-diplotene spermatocytes (Fig. 1e), consistent with the hypothesis that FIGNL1 and FIRRM act together.
The formation and initial processing of meiotic DSBs are unaffected in Firrm cKO and Fignl1 cKO spermatocytes
A synapsis defect associated with a mid-prophase arrest might result from defective recombination initiation (e.g., Spo11−/−64,65) or defective repair of meiotic DSBs (e.g., Dmc1-/-3,66,67). To determine whether DSB formation was altered by FIRRM or FIGNL1 depletion, we quantified phosphorylated H2AX (γH2AX) that decorates chromatin in a DSB-specific manner at leptotene68. The γH2AX signal intensity in the nucleus was not different in control and Firrm cKO spermatocytes from pre-leptotene (stage of pre-meiotic replication) to mid-late leptotene (Fig. 2a, b). RPA2, a subunit of the ssDNA-binding protein complex Replication Protein A (RPA), which is involved in DNA replication and HR, forms multiple foci at replication forks in preleptotene spermatocytes, and along chromosome axes at sites of recombination intermediates from leptotene to pachytene stage60,69,70. RPA2 foci displayed a similar kinetics in Firrm cKO, Fignl1 cKO, and control spermatocytes (Fig. 2c, d). An increase in zygotene spermatocytes (by 1.3-fold for both Firrm cKO and Fignl1 cKO in early zygotene stage) might result from a different kinetics of DSB formation, from a difference in the formation or stability of HR intermediates involving detectable RPA, or from a difference in staging the nuclei, due to less or delayed synapsis initiation in mutant nuclei. Thus, the first steps of meiotic recombination (DSB formation and RPA recruitment on resected ssDNA ends) were not affected by the absence of the FIGNL1-FIRRM complex.

a Spread nuclei of spermatocytes from 12 dpp control and Firrm cKO mice stained for SYCP3 and γH2AX. Scale bar, 20 µm. b Total nuclear γH2AX signal intensity in 12 dpp control (gray) and Firrm cKO (red) spermatocytes (n = 2 mice per genotype). In cytological analyses, the black bar represents the median and two-sided Dunn’s multiple comparison tests were performed unless stated otherwise. c Spread spermatocyte nuclei from 12 dpp control and Firrm cKO mice stained for SYCP3 and RPA2. Scale bar, 10 µm. d Number of on-axis RPA2 foci in control (gray), Firrm cKO (red) and Fignl1 cKO (orange) spermatocytes. control, n = 6 (4 × 12 dpp, 1 × 16 dpp, 1 × 17 dpp), Firrm cKO, n = 4 (12 dpp) and Fignl1 cKO, n = 3 (1 × 16 dpp, 2 × 17 dpp) mice. Source data are provided as a Source Data file.
The recruitment of RAD51 and DMC1 on meiotic chromatin strongly increases in the absence of FIGNL1 or FIRRM
In mouse spermatocytes, RAD51 and DMC1 foci extensively colocalize on meiotic chromosome axes (on-axis foci) from leptotene to pachytene stage, particularly in zygotene15,16,69. Compared with controls, RAD51 and DMC1 signal intensity and foci pattern and kinetics were strikingly different in Firrm cKO and Fignl1 cKO spermatocytes (Fig. 3a-c; Supplementary Fig. 3a). First, RAD51 (but not DMC1) formed many foci at preleptotene, during premeiotic replication. Second, the mean number of RAD51 and DMC1 on-axis foci was significantly higher in Firrm cKO and Fignl1 cKO than in control spermatocytes at every stage, from early leptotene to zygotene. Third, in cKO spermatocytes, many RAD51 and DMC1 foci were located away from the chromosome axes (off-axis foci). The number of off-axis foci was highest during leptotene and progressively decreased during zygotene. Fourth, in cKO spermatocytes, RAD51 and DMC1 staining formed continuous lines, at our resolution limit, along the synaptonemal complex segments in zygotene-like nuclei. This did not allow counting RAD51 and DMC1 foci in late zygotene-like nuclei with extensive synapsis. Overall, these observations are consistent with the role of FIRRM and FIGNL1 in limiting RAD51 and DMC1 loading on chromatin in spermatocyte nuclei. We describe these different features in the following sections.

a Zygotene spermatocyte spreads from control and Firrm cKO mice stained for SYCP3, RAD51 and DMC1. Scale bar, 5 µm. Numbers of RAD51 (b) and DMC1 (c) foci in control (gray), Firrm cKO (red) and Fignl1 cKO (orange) spermatocytes (c). n = 2 mice per genotype. d Spreads of zygotene spermatocytes from 16 dpp control, Firrm cKO and Spo11YF/YF mice stained with SYCP3, SYCP1 and MSH4. Scale bar, 10 µm. e MSH4 focus density along SYCP1-marked synaptonemal complex fragments in control (gray), Firrm cKO (red), Spo11YF/YF Firrm cKO (purple), and Spo11YF/YF (blue) zygotene/zygotene-like spermatocytes. n = 3 mice per genotype. f Preleptotene spermatocyte spreads from control and Firrm cKO mice stained for SYCP3 (gray), DNA (DAPI, blue), RPA2 (red) and RAD51 (green). Scale bar, 10 µm. g. STED images of preleptotene spermatocyte spreads from control and Firrm cKO mice stained for RAD51 (STAR ORANGE, green) and RPA2 (STAR RED, red). Scale bar, 1 µm. Number of RAD51 foci that colocalized with RPA2 foci (h) and of RPA2 foci that colocalized with RAD51 (i) in spreads of preleptotene control (gray) and Fignl1 cKO (orange) spermatocyte nuclei. n = 2 (control) or n = 3 mice (Fignl1 cKO). The observed (obs) and expected by chance (random) numbers of colocalized foci are shown. Wilcoxon two-tailed test. Source data are provided as a Source Data file.
Post-strand invasion recombination foci are strongly reduced in the absence of FIRRM
The efficient recruitment of RAD51 and DMC1 prompted us to examine MSH4 and TEX11, two meiotic ZMM proteins, which stabilize post-strand invasion recombination intermediates11 and form foci on SC from zygotene to mid-pachytene3,69,71. The number of MSH4 and TEX11 foci was strongly reduced in late zygotene-like Firrm cKO nuclei compared with the control (Fig. 3d, Supplementary Fig. 3b). To normalize differences in SC extension among genotypes, we measured the density of MSH4 foci per µm of SC length. MSH4 focus density was reduced by 2.5-fold in Firrm cKO compared with control spermatocytes (Fig. 3e, Supplementary Fig. 3c), although the number of MSH4 foci was higher than in Spo11YF/YF nuclei (without DSB-inducing activity). Thus, FIRRM is required for TEX11 and MSH4 focus formation during mouse meiotic recombination. The residual MSH4 foci might result from MSH4 binding to a small fraction of normal or aberrant recombination intermediates formed in the absence of the FIGNL1-FIRRM complex. Alternatively, we cannot exclude the persistence of a small amount of FIRRM protein in Firrm cKO spermatocytes, sufficient for recruiting MSH4 to few recombination intermediates. Thus, despite the increased recruitment of RAD51 and DMC1 on chromosome axes, the processing of recombination intermediates was defective in Firrm cKO spermatocytes, suggesting a function of FIGNL1-FIRRM at a step likely before recombination intermediate stabilization by MSH4-MSH5.
RAD51 accumulates on chromatin during premeiotic replication in Firrm cKO and Fignl1 cKO spermatocytes
RPA2 forms many foci at ongoing replication forks in preleptotene nuclei (Fig. 3f, g). The kinetics of RPA2 focus formation was similar in control, Firrm cKO and Fignl1 cKO spermatocytes, and few foci remained in early leptotene stage. This suggests that premeiotic replication was completed without major alteration (Fig. 2d; Supplementary Fig. 3d). As RAD51 is involved in protecting stalled replication forks72, we hypothesized that RAD51 might colocalize with RPA during premeiotic replication in Firrm cKO and Fignl1 cKO spermatocytes. We measured the colocalization of RAD51 and RPA2 in preleptotene spermatocytes and compared these data with the colocalization of randomly distributed foci obtained from simulations (see Methods; Fig. 3h, i; Supplementary Fig. 3e). In Fignl1 cKO, 17% of RAD51 foci colocalized with RPA2 foci compared with 9% of randomly generated RAD51 foci (p < 0.0001; two-tailed Wilcoxon test), suggesting that a fraction of RAD51 foci localizes at replication forks. However, the majority of identified RAD51 foci did not colocalize with RPA2 foci, suggesting that a larger fraction of RAD51 foci may not localize at replication forks. We cannot exclude that both RAD51 and RPA localize at forks in an exclusive manner, and that RAD51 binding excludes RPA. However, because of the large number of RAD51 foci that persisted at the end of premeiotic replication and the absence of obvious gross replication defects, we hypothesize that RAD51 colocalizes transiently with RPA at replication forks. It then remains in place, possibly on intact DNA, while the forks progress and move away. DMC1 foci were rare in most Firrm cKO and Fignl1 cKO preleptotene spermatocytes (Fig. 3c), likely because meiosis-specific DMC1 production is still low at preleptotene stage.
RAD51 and DMC1 form parallel linear structures along the synaptonemal complex in the absence of FIRRM
To refine the characterization of RAD51 and DMC1 distribution in Firrm cKO spermatocytes, we visualized RAD51, DMC1 and SYCP3 using super-resolution stimulated emission depletion (STED) microscopy (Fig. 4a, b). In leptotene and zygotene control spermatocytes, RAD51 and DMC1 formed partially overlapping co-foci along the unsynapsed axes and SC segments. RAD51 was more often closer to the chromosome axis than DMC1, as described previously20,73. In Firrm cKO spermatocytes, the patterns of RAD51 and DMC1 staining were more heterogeneous. A first type of RAD51-DMC1 co-foci was similar to control foci, but RAD51 signal tended to be more extended compared with DMC1 (Fig. 4a, compare insets i–iv, control, with insets v–vi, Firrm cKO). Consistent with this observation, the intensity of RAD51 foci was higher in Firrm cKO than in control spermatocytes, whereas DMC1 focus intensity was unchanged or even lower (Supplementary Fig. 4a, b). Second, some RAD51-DMC1 co-foci formed longer structures anchored to the chromosome axis, a pattern expected if they were extending along chromatin fibers (Fig. 4a, inset vii). The localization patterns of these two types of foci are compatible with RAD51/DMC1 filaments bound to chromatin fibers at DSB sites or/and dsDNA. In addition, at some sites, RAD51 and DMC1 followed the unsynapsed axes, often filling gaps with little or no SYCP3 signal between more heavily SYCP3-stained axis segments (Fig. 4a, insets viii-ix). Lastly, in Firrm cKO zygotene-like nuclei with partial synapsis, RAD51 and DMC1 formed two parallel lines separated by ~100 nm along SC segments, between the lateral elements (axes) visualized by ~210 nm apart SYCP3 signal lines (Fig. 4b, c). The intensity of these lines was irregular with interruptions, and interspersed with more intense foci. These observations indicate a highly aberrant patterning of RAD51 and DMC1 on meiotic chromatin and on meiotic chromosome axes in the absence of FIGNL1 and FIRRM activity.

a STED images of spreads of leptotene spermatocyte nuclei stained for SYCP3 (STAR GREEN, gray), RAD51 (STAR ORANGE, green), and DMC1 (guinea-pig primary antibody, STAR RED, red). b STED images of spreads of zygotene/zygotene-like spermatocyte nuclei with extensive synaptonemal complexes, stained for SYCP3 (STAR 460 L, white), RAD51 (STAR RED, red) and DMC1 (mouse primary antibody, STAR ORANGE, green). Scale bars, 1 µm (main panels) or 200 nm (insets). c Relative intensity of SYCP3 (black), RAD51 (red) cd and DMC1 (green) signal across the synaptonemal complex in control (across RAD51-DMC1 mixed foci) and Firrm cKO (outside regions of stronger focus-like RAD51-DMC1 staining). The mean of 12 sections from STED images of 3 different nuclei.
Accumulation of RAD51 and DMC1 foci in Firrm cKO spermatocytes is meiotic DSB-independent
In Firrm cKO and Fignl1 cKO spermatocytes, RAD51 and DMC1 displayed an unusual pattern that included an increased number of foci, many off-axis foci, and linear staining along chromosome axes. This was different from the expected discrete DSB repair foci on chromosome axes16,69, raising the question of whether in these cKO models, RAD51 and DMC1 recruitment requires SPO11-generated DSBs. Thus, we generated Spo11YF/YF Firrm cKO double mutants in which SPO11 is catalytically dead74. The low early prophase γH2AX staining and the absence of RPA2 foci confirmed the absence of DSBs in these animals (Supplementary Fig. 4c, d). Strikingly, we detected large numbers of on-axis and off-axis RAD51 and DMC1 foci in Firrm cKO and in Spo11YF/YF Firrm cKO spermatocytes, and only background signal in Spo11YF/YF spermatocytes (as expected) (Fig. 5a). Overall, the pattern of RAD51 and DMC1 in Firrm cKO and Spo11YF/YF Firrm cKO were quite similar: a large number of on-axis foci detected from early prophase that persisted through zygotene, RAD51 foci formed during preleptotene stage, and both RAD51 and DMC1 off-axis foci progressively disappeared from leptotene to zygotene (Fig. 5b, c; Supplementary Fig. 4e-f). RAD51 and DMC1 foci were consistently more numerous in single Firrm cKO than in Spo11YF/YF Firrm cKO in early leptotene and zygotene, although the difference was on the merge of significance. In zygotene, the recruitment of RAD51 and DMC1 to SPO11-catalyzed DSB sites may explain this difference. It is more difficult to interpret in early leptotene, a stage with very few processed DSBs in Firrm cKO spermatocytes, based on the low number of RPA2 foci (Fig. 2d). Altogether, these observations demonstrate that the accumulation of RAD51 and DMC1 Firrm cKO spermatocytes is largely independent of SPO11-catalyzed DSBs.

a Spreads of representative control, Firrm cKO, Spo11YF/YF Firrm cKO, and Spo11YF/YF early zygotene spermatocytes stained for SYCP3 (blue), DMC1 (red) and RAD51 (green). Scale bar, 10 µm. Counts of on-axis RAD51 (b) and DMC1 (c) foci in spreads from control (gray), Firrm cKO (red), Spo11YF/YF Firrm cKO (purple), and Spo11YF/YF (blue) spermatocytes from 12 dpp mice. n = 2 mice per genotype. Percentage (corrected for random colocalization, see Methods) of on-axis RAD51 foci colocalized with on-axis DMC1 foci (d) and vice-versa (e), in spreads from control and Firrm cKO spermatocytes from 12 dpp mice. Source data are provided as a Source Data file.
In wild-type meiosis, RAD51 and DMC1 colocalization throughout the meiotic prophase reflects their cooperation at resected DSB ends15,16,17,20,73. To determine their level of colocalization in Firrm cKO spermatocytes where RAD51 and DMC1 foci form in the absence of SPO11-catalyzed DSBs, we examined their colocalization from early leptotene to mid-zygotene stage, in nuclei containing at least 10 foci for each protein (Fig. 5d, e; Supplementary Fig. 5). As expected, on-axis RAD51 foci, the number of which was higher, colocalized less frequently with DMC1 foci in Firrm cKO than in control spermatocytes, especially at earlier stages (Fig. 5d). Conversely, on-axis DMC1 foci colocalized at least as frequently with on-axis RAD51 foci in Firrm cKO and Spo11YF/YF Firrm cKO than in control spermatocytes at every stage, with a maximum in early leptotene (~70%; Fig. 5e). Off-axis foci displayed the same trend, with a very high percentage of DMC1 foci that colocalized with RAD51 foci at earlier stages in Firrm cKO (Supplementary Fig. 5c-e). Moreover, RAD51 and DMC1 colocalization levels were similar in Firrm cKO and Spo11YF/YF Firrm cKO, indicating that their association is DNA damage-independent. Altogether, these observations indicate that in the absence of FIRRM, off-and on-axis RAD51 foci assemble independently of DMC1 foci in preleptotene and early prophase spermatocytes. Detectable DMC1 foci might form by joining pre-existing RAD51 foci, or by co-assembling de novo RAD51-DMC1 foci in Firrm cKO spermatocytes.
DMC1 is recruited to DSB sites in the absence of the FIGNL1-FIRRM complex
The abundance of DSB-independent RAD51 and DMC1 foci raises the question of whether these recombinases are recruited at meiotic DSB sites at all in the absence of FIRRM or FIGNL1. Therefore, we determined the colocalization of on-axis RAD51 or DMC1 foci with RPA2 foci, used as a marker of a subset of the DSBs, in Firrm cKO and Fignl1 cKO spermatocytes. The number of on-axis RAD51-RPA2 and DMC1-RPA2 co-foci (corrected for random colocalization) in spermatocytes followed the kinetics of RPA2 foci in all genotypes (Supplementary Fig. 6a-c, compare with Fig. 2d). In Firrm cKO and Fignl1 cKO spermatocytes, the percentage of on-axis RPA2 foci that colocalized with RAD51 or DMC1 was similar to control spermatocytes in mid-late leptotene, and was even higher in mid-zygotene, maybe indicative of the accumulation of unrepaired HR intermediates (Fig. 6a; Supplementary Fig. 6d). The lower percentage of on-axis RAD51 or DMC1 foci that colocalized with RPA2 in cKO spermatocytes compared with control is likely explained by the excess of DSB-independent RAD51 and DMC1 foci (Fig. 6b; Supplementary Fig. 6e). These findings suggest that the efficiency of RAD51 and DMC1 recruitment at sites of meiotic DSBs is not affected by FIRRM and FIGNL1 absence.

Percentages of on-axis RPA2 foci colocalized with on-axis DMC1 foci (a), and vice-versa (b) in spreads from mid-late leptotene to mid-zygotene/zygotene-like spermatocyte nuclei from 12-dpp control (gray), Firrm cKO (red) and Fignl1 cKO (orange) mice. n = 2 mice per genotype. c Numbers of and shared hotspots identified by DMC1-SSDS in spermatocytes from 12 dpp control and Firrm cKO mice. d DMC1-SSDS signal correlation between control and Firrm cKO mice at hotspots identified in both genotypes. The Spearman rho and associated p-value (two-sided) are shown. Red and green dots indicate hotspots that were significantly over- and under-represented in Firrm cKO compared with control spermatocytes (DESeq2, p-value < 0.1, log2FC > 0 and log2FC < 0, respectively). Unchanged autosomal hotspots are represented in gray and chromosome X hotspots by black circled diamonds. e Average plots (top) and corresponding heatmaps (bottom) of DMC1-SSDS intensity (fragments per million, FPM) in control (left) and Firrm cKO mice (right) for hotspots that overlap with SPO11-oligo hotspots77 detected in both genotypes (common peaks), in control only (control-specific), or in Firrm cKO only (Firrm cKO-specific)(see Supplementary Fig. 7a–c). The center of intervals is defined as the center of SPO11-oligo peaks detected in B6 mice, as defined in ref. 77. f Normalized average distribution of ssDNA type 1 fragments (see Methods) originating from forward (fwd) and reverse strands (rev) at common peaks, defined in (e), for control (dark red, orange) and Firrm cKO (dark blue, light blue). The SSDS signal was normalized to have the same cumulated amount of normalized average signal over common peaks (on a 5-kb window) for both genotypes. Source data are provided as a Source Data file.
To assess directly DMC1 recruitment at SPO11-dependent DSB hotspots, we investigated the genome-wide distribution of DMC1-bound ssDNA by chromatin-immunoprecipitation (ChIP), followed by ssDNA enrichment (DMC1-Single Strand DNA Sequencing, SSDS)75 in testes from 12-dpp control and Firrm cKO mice. In control mice, the regions enriched in DMC1-bound ssDNA are the ssDNA 3’overhangs that result from DSB resection at meiotic DSB hotspots76. We detected 9907 peaks in control and 7397 peaks in Firrm cKO spermatocytes (Fig. 6c). Most of these peaks (6614) were shared. Peaks called specifically in one genotype or the other were most likely shared weakly active hotspots, as inferred from their weak enrichment in both genotypes (Supplementary Fig. 7b). Most of the detected DMC1-SSDS peaks (9297 out of 10,690) overlapped with previously identified meiotic SPO11-oligonucleotide DSB hotspots (SPO11-oligo hotspots, Supplementary Fig. 7a)77. Moreover, the DMC1-SSDS signal enrichment within peaks was highly correlated in control and Firrm cKO samples (Spearman’s rho = 0.92; Fig. 6d) with the exception of X chromosome hotspots, which were relatively less enriched in Firrm cKO than in control samples (Supplementary Fig. 7d). One possible explanation for this could be a genome-wide accumulation of HR intermediates in Firrm cKO that would erase the X chromosome-specific higher DMC1-SSDS enrichment due to delayed DSB repair20,77,78. Overall, this indicates that the recruitment of DMC1 on ssDNA at SPO11-dependent DSB hotspots was efficient, with relative hotspot intensities comparable to wild-type meiosis. The lack of Firrm cKO-specific peaks and the efficiency of hotspot detection suggest that DMC1 is not massively associated with ssDNA in non-hotspot regions in Firrm cKO testes. The recent finding by Ito and col. that no RAD51-SSDS peaks were detected in Spo11 KO Fignl1 cKO testes supports this conclusion79.
We then asked whether FIRRM depletion alters DMC1 extension on resected DSB ends at DSB hotspots. To characterize precisely the DMC1-SSDS signal distribution across DSB hotspots, we defined the center of overlapping SPO11-oligo hotspots as the center of DMC1-SSDS peaks77. This improved significantly the quality of the average DMC1-SSDS signal profile, revealing a non-identical distribution in control and Firrm cKO (Fig. 6e; Supplementary Fig. 7c). To facilitate the comparison between control and Firrm cKO DMC1-SSDS signal profiles, we normalized the overall signal intensity within common peaks and plotted the resulting normalized strand-specific profiles (Fig. 6f). The distribution of DMC1-SSDS signal in control samples was consistent with previous characterization in wild-type spermatocytes. It is characterized by a spike adjacent to the center of SPO11-oligo hotspots, followed by a shoulder, which is thought to reflect the regulation of the length of resected ssDNA tails covered by DMC1 that measure at least 300 bases and 850–900 bases on average20,77. In Firrm cKO samples, the DSB-proximal spike was present. However, the density of DMC1-SSDS signal started to decrease right next to the spike, without forming a shoulder. This suggests that the regulation of the length of DMC1-coated ssDNA is altered in the absence of FIGNL1-FIRRM. This alteration of DMC1-SSDS profile was not dependent on hotspot strength (Supplementary Fig. 7e), and was also detected at X chromosome hotspots, suggesting that this is not a consequence of delayed DSB repair (Supplementary Fig. 7d). The presence of the DSB-proximal spike suggests that DMC1 recruitment at DSB sites remains efficient on a short DSB-proximal interval close to the 3′ end of ssDNA tails independently of FIRRM. Conversely, the loss of the shoulder indicates that the mechanism controlling the DMC1 filament length requires FIRRM for full efficiency. One possible scenario is that the FIGNL1-FIRRM complex controls the balance between DMC1 and RAD51 loading on ssDNA. Alternatively, we cannot exclude that the extent of DSB resection is altered.
Firrm cKO is epistatic to Swsap1 for controlling RAD51 and DMC1 loading
In mouse meiosis, the Shu complex components SWSAP1, SWS1 and SPIDR are required for assembling normal numbers of RAD51 and DMC1 foci, which are 2- to 3-fold fewer in Swsap1−/− than in wild-type leptotene-zygotene spermatocytes36,40,41,42. FIGNL1 depletion suppresses the defect of human SWSAP1-depleted cells in forming DNA damage-induced RAD51 foci, suggesting that SWSAP1 antagonizes the anti-RAD51 activity of FIGNL140. We generated Swsap1−/− Firrm cKO and Swsap1−/− Fignl1 cKO double mutant mice to determine if the defect in forming meiotic RAD51 and DMC1 foci in Swsap1−/− spermatocytes depends on FIGNL1-FIRRM. We found that synapsis was defective and meiosis did not progress further than the zygotene-like stage with partial, partly non-homologous synapses in Swsap1−/− Firrm cKO and Swsap1−/− Fignl1 cKO spermatocytes, like in Firrm cKO and Fignl1 cKO single mutants. A small subset of nuclei progressed to pachynema, as observed for Swsap1−/− spermatocytes, most likely due to incomplete Firrm or Fignl1 deletion. Whereas the number of DMC1 foci was strongly reduced in Swsap1−/− spermatocytes, double mutant spermatocytes accumulated DMC1, like Firrm cKO and Fignl1 cKO spermatocytes (mean, 123, 30, 247 and 311 DMC1 foci in control, Swsap1−/−, Fignl1 cKO and Swsap1−/− Fignl1 cKO early zygotene spermatocytes, respectively; Fig. 7a-c, Supplementary Fig. 8a, b). Because DMC1 accumulation in Firrm cKO and Fignl1 cKO spermatocytes was essentially DSB-independent (see above), this observation indicates that the formation of DSB-independent DMC1 foci does not require SWSAP1 in the absence of FIGNL1-FIRRM.

a Spreads of control, Firrm cKO, Swsap1−/− Firrm cKO and Swsap1−/− early zygotene spermatocytes stained for SYCP3 (gray), RAD51 (yellow) and RPA2 (magenta). Scale bar, 10 µm. Total numbers of on-axis RPA2 (b) or DMC1 (c) foci, or number of on-axis RPA2 foci colocalized with on-axis DMC1 foci (corrected for the numbers expected by chance) (d) in spreads from control (gray, n = 3 mice), Fignl1 cKO (orange, n = 1), Swsap1−/− Fignl1 cKO (pink, n = 1), Swsap1−/− Firrm cKO (purple, n = 1) and Swsap1−/− (blue, n = 2) spermatocytes from 17 dpp mice. Source data are provided as a Source Data file.
We then asked specifically whether SWSAP1 is required for recruiting DMC1 at meiotic DSB sites in the absence of FIRRM or FIGNL1, by examining on-axis colocalized DMC1 and RPA2 foci, used as a proxy for a subset of meiotic DSB sites as described above (Fig. 6a, b; Supplementary Fig. 6a, b). In Swsap1−/− spermatocytes, only a small fraction of RPA2 foci colocalized with DMC1, in keeping with the reduced number of detectable DMC1 foci. In contrast, the number of RPA2 foci that colocalized with a DMC1 focus was as high in Swsap1−/− Firrm cKO and Swsap1−/− Fignl1 cKO double mutants as in Firrm cKO and Fignl1 cKO single mutants and in control spermatocytes (Figs. 6a, 7d; Supplementary Fig. 8c–e). This suggests that the formation of a normal number of detectable DSB-associated DMC1 nucleofilaments does not require SWSAP1 when FIGNL1 and FIRRM are absent, and is consistent with the hypothesis that SWSAP1 and FIGNL1-FIRRM have antagonistic roles in regulating the formation of stable RAD51/DMC1 nucleofilaments. However, it remains unknown whether these nucleofilaments, which are assembled without counterbalanced regulations by the SWSAP1-containing complex and FIGNL1-FIRRM, are functional for HR.
FIGNL1 perturbs the architecture of RAD51/DMC1 nucleoprotein filaments and inhibits RAD51- and DMC1-mediated D-loop formation in vitro
To determine the HR step(s) in which FIGNL1-FIRRM might be involved, we examined in vitro the effect of adding FIGNL1 on the assembly and stability of RAD51 and DMC1 nucleofilaments, and on their subsequent strand invasion activity. We incubated pre-formed RAD51 or DMC1 filaments assembled on a 400 nucleotide (nt) ssDNA or a 400 bp dsDNA with purified human FIGNL1ΔN, in which N-terminal 284 aa were deleted40 (Supplementary Figs. 1b, 9a). FIGNL1ΔN did not promote RAD51 and DMC1 displacement from DNA (electrophoretic mobility shift assay in Fig. 8a, b, pre-formed nucleofilament), but induced the formation of a higher molecular weight complex, suggesting that FIGNL1ΔN binds to RAD51/DMC1-DNA filaments. When we mixed FIGNL1ΔN with RAD51 or DMC1 before addition to the DNA substrate, we observed a slight increase in the fraction of free dsDNA (but not ssDNA) that was not complexed with RAD51 or DMC1 (Fig. 8a–c, no pre-formed nucleofilament). Whereas this increase was not significant, it might suggest that the presence of FIGNL1ΔN restricts RAD51 and DMC1 binding to DNA and the subsequent filament elongation. We then used transmission electron microscopy (TEM) to analyze the effect on RAD51 filament formation and architecture upon addition of FIGNL1ΔN at same time as RAD51 to a 400 nt ssDNA (Fig. 8d, e). Addition of FIGNL1ΔN induced the formation of super-complexes that contained several bridged or interwoven filaments. Simultaneously, we observed that individual RAD51 filaments not included in the super-complexes were significantly shorter than RAD51 filaments in controls (mean length of 135 versus 175 nm, respectively; Fig. 8f, Supplementary Fig. 9b). We also detected the formation of some very long filaments (more than 450 nm and up to 3–4 µm). Their length was not compatible with the length of the used DNA substrate, suggesting a DNA-independent polymerization in the presence of FIGNL1ΔN. Consistent with this hypothesis, similar structures were detected by incubating RAD51 with FIGNL1ΔN without adding any DNA (Supplementary Fig. 9b, c). Similarly, the mean length of RAD51 filaments assembled on a 400 bp dsDNA decreased from 194 nm in control to 137 nm in the presence of FIGNL1ΔN (Fig. 8f). The architecture of DMC1 filament assembled both on ssDNA and on dsDNA displayed qualitatively similar alteration (Supplementary Fig. 9d). Altogether, these results show that FIGNL1ΔN limits RAD51/DMC1 assembly on ssDNA and also dsDNA, and affect the filament architecture. We then tested whether these filaments could pair with homologous donor dsDNA (pUC19 plasmid) in a D-loop assay. Pre-formed RAD51, DMC1, and mixed RAD51-DMC1 filaments mediated the formation of 34%, 27%, and 22% of D-loop products, respectively. Addition of FIGNL1ΔN during filament assembly led to a decrease in the D-loop yield (Fig. 8g, h). When we titrated FIGNL1ΔN in the D-loop reaction, the yield decreased linearly and significantly (Fig. 8h). This showed that the contacting and pairing with homologous DNA of filaments assembled in the presence of FIGNL1ΔN might be affected. This indicates that by limiting the assembly of RAD51 and/or DMC1 on DNA, FIGNL1 could negatively regulate the next strand invasion step required for HR.

Electrophoretic Mobility Shift Assay (EMSA). Human RAD51 or DMC1 was incubated with a Cy5-labeled 400-nt ssDNA (a) or 200-bp dsDNA (b) fragment with or without human FIGNL1ΔN. RAD51 or DMC1 was incubated with DNA for 5 min before (pre-formed nucleofilament) or concomitantly (no pre-formed filament) to FIGNL1ΔN addition. c Quantification of free dsDNA in the EMSA in (b) (n = 2). Representative TEM images in positive (d) and negative staining (e) and length distribution (f) of RAD51 filaments assembled on 400-nt ssDNA fragments without (ss400-RAD51) or with human FIGNL1ΔN (ss400-RAD51 + FIGNL1ΔN). Scale bar, 200 nm (main) or 50 nm (detail). Median, quartiles, minima, and maxima are represented. Sidak’s multiple comparison tests. Two replicates were made with 35-40 filaments each. >450 nm-long filaments formed with FIGNL1ΔN (d) were not included in the quantification in (f) (see Supplementary Fig. 10b). FIGNL1ΔN inhibits the formation of a D-loop by RAD51 and DMC1 in vitro. Representative gel (RAD51 with 0.0 to 1.6 µM of FIGNL1ΔN) (g) and titration of FIGNL1ΔN (h) in the D-loop assay. Average of 2 replicates ±SD. i Model for possible roles of the FIGNL1-FIRRM complex in regulating RAD51 and DMC1 in mouse spermatocytes. i FIGNL1-FIRRM might limit the nuclear RAD51 level by sequestering a cytoplasmic RAD51 pool. (ii) FIGNL1-FIRRM might prevent the stabilization of transient dsDNA-RAD51 associations (and ensuing DMC1 recruitment) at replication forks during premeiotic replication. (iii) During meiotic recombination, FIGNL1-FIRRM might prevent stable RAD51 loading on the 3′ region of DSB ssDNA tails, promoting indirectly the 5’ to 3′ polymerization of an uninterrupted DMC1 filament (arrows) up to the 3′ ends. A factor (e.g., the SWSAP1-SWS1-SPIDR complex, SWS) might protect the RAD51 filament located in dsDNA-proximal regions from FIGNL1-FIRRM-dependent dissociation. RAD51/DMC1 filaments formed in the absence of FIGL1-FIRRM might not be fully functional for homology search, strand invasion and D-loop stabilization. Post-strand invasion, FIGNL1-FIRRM might contribute to remove RAD51/DMC1 from invading ends involved in intersister and/or interhomolog interactions. Source data are provided as a Source Data file.
Discussion
The AAA-ATPase FIGNL1 (which plant ortholog is FIGL1) and its partner FIRRM (FLIP or MEICA in plants) were identified recently as negative regulators of meiotic COs in plants44,52,53,54, and as negative regulators of RAD51 in human cells40,43,47,48,49,50,57,80. However, their role in mammalian meiosis remained unknown, although a role for Fignl1 was hypothesized in mouse male meiosis81. Here, by characterizing male germ line-specific Fignl1 and Firrm cKO mouse models, we uncovered two roles of the FIGNL1-FIRRM complex in male meiosis. First, FIGNL1 and FIRRM are required for meiotic DSB repair and for homologous chromosome synapsis during meiotic prophase I, and thus are essential for male mouse meiosis. Second, the FIGNL1-FIRRM complex prevents DNA damage-independent accumulation of RAD51 and DMC1 on chromatin and chromosome axes in spermatocyte nuclei during premeiotic replication and meiotic prophase I. Similar conclusions were reached in recent studies by characterizing the Fignl1 cKO79 and the Firrm cKO57 mice.
Our data show that FIGNL1 and FIRRM act as negative regulators of RAD51 and DMC1 during meiotic recombination, a function evolutionarily conserved from plants to mammals. However, the role of FIGNL1-FIRRM is much more crucial in mouse spermatogenesis than in A. thaliana and rice meiosis where homologous chromosome synapsis and formation of ZMM-dependent type I COs are almost normal in FIGNL1 and FIRRM mutants44,52,53,54,55. Plants and mammals show differences in their requirement of specific HR pathways for meiotic DSB repair, homologous chromosome synapsis and progression through meiotic cell cycle. These processes require DMC1, MSH4 and MSH5 in the mouse12,13,14,66,67. Conversely, in A. thaliana and rice, meiotic DSBs are repaired by RAD51-dependent intersister HR in dmc1 mutants, homologous chromosome synapsis does not depend on MSH4-MSH5, and dmc1, msh4 and msh5 mutant cells progress through meiotic prophase (reviewed in ref. 82). The difference between mice and plants in FIGNL1-FIRRM requirement might be functionally related to these other differences. However, mouse Fignl1 cKO and Firrm cKO spermatocytes also displayed defects not seen in plants, especially a massive, DNA damage-independent RAD51 and DMC1 accumulation and defects in MSH4 focus formation. This suggests that the FIGNL1-FIRRM complex has additional functions in the mouse within the shared framework of RAD51 and DMC1 negative regulation.
We found that in Fignl1 cKO and Firrm cKO spermatocytes, MSH4 focus formation and meiotic DSB repair were impaired, RAD51 and DMC1 foci accumulated at unrepaired DSB sites, and homologous synapsis was defective. These defects have been described in mutants in which strand invasion is impaired (e.g., Dmc1−/− mice that accumulate only RAD51, Hop2−/−, Mnd1−/− mice)5,6,66,67,83,84,85 and in mutants in which strand invasion might be preserved but the HR intermediates are not efficiently stabilized (e.g., Hrob−/−, Mcm8−/−, Mcmd2−/−, Msh4−/−, Msh5−/− mice)12,13,14,86,87,88,89. By altering the stability or architecture of the nucleoprotein filament formed by RAD51/DMC1 on ssDNA and/or dsDNA, the FIGNL1-FIRRM complex might play roles before and/or after strand invasion. In the case of a post-strand invasion role, this complex might favor RAD51/DMC1 dissociation from dsDNA in the D-loop, a step required for initiating DNA synthesis to extend the invading strand. In S. cerevisiae, the motor protein Rad54 and its paralog Rdh54 are involved in removing RAD51/DMC1 from dsDNA following D-loop formation28,32. In the mouse, the meiotic function of RAD54 and its paralog RAD54B is not crucial because Rad54 Rad54b double mutant mice are fertile, although they display persistent RAD51 foci during meiotic prophase33,34. Thus, the FIGNL1-FIRRM complex might be one additional factor that can disassemble RAD51 and DMC1 from dsDNA after D-loop formation. In our in vitro assay, human FIGNL1ΔN could not dissociate pre-formed RAD51/DMC1 filaments; however, the full-length FIGNL1-FIRRM complex might possess a stronger activity sufficient to dissociate RAD51/DMC1 efficiently. Alternatively, FIGNL1-FIRRM complex-dependent RAD51/DMC1 filament alteration might render it sensitive to dismantling by other factors. In addition to normal HR intermediate processing, the FIGNL1-FIRRM complex might also dissociate unproductive or potentially toxic post-synaptic RAD51/DMC1 filaments, such as multiple strand invasion or invasion on non-allelic repeated sequences90,91.
In Firrm cKO spermatocytes, the average DMC1-SSDS signal profile at meiotic DSB hotspots was altered in a way that suggests that FIRRM is involved in regulating the length of DMC1-ssDNA filaments before strand invasion. In wild-type mouse spermatocytes, the profile of DMC1-SSDS coverage at DSB hotspots and super-resolution microscopy observations indicate that DMC1 typically occupies the DSB-proximal two-third of the DSB 3′ ssDNA end, and RAD51 the DSB-distal third of the same DSB 3’ ssDNA end5,6,16,20,73,77. Because interhomolog recombination is thought to rely solely on DMC1 catalytic activity during meiosis18,19,20, defects in regulating the length or the continuity of the active DMC1 filament may affect the efficiency of interhomolog search, the formation of a D-loop that can be stabilized by MSH4-MSH5, and homologous chromosome synapsis. Several non-exclusive hypotheses can explain how the FIGNL1-FIRRM complex regulates the DMC1 filament on DSB 3’ ssDNA tails. First, RAD51 nuclear fraction was increased in Fignl1 cKO and Firrm cKO testes (Fig. 1c; Supplementary Fig. 2e), thus RAD51 might outcompete DMC1 on ssDNA tails in these mutants. Mechanistically, a balance between FIGNL1-FIRRM and BRCA2 might regulate the retention of RAD51 in the cytoplasm and contribute to fine-tune RAD51 nuclear level (Fig. 8i, (i)). Indeed, we found that purified human FIGNL1ΔN favors the formation of DNA-independent RAD51 filaments in vitro (Supplementary Fig. 9c), while it has been proposed that BRCA2 promotes RAD51 nuclear import by limiting the formation of cytoplasmic RAD51 polymers, which cannot be mobilized92. However, the FIGNL1-FIRRM complex might also play a more direct role in controlling the formation of RAD51 and DMC1 filaments at DSB ssDNA overhangs, based on a model according to which DMC1 filament is seeded at the 3′ end of a RAD51 filament (Fig. 8i, (iii))9. In vitro, RAD51 nucleates randomly on ssDNA tracts, whereas DMC1 prefers to seed at a ds/ssDNA junctions or on a RAD51 patch, and polymerizes in the 5′ to 3′ direction93. We thus hypothesize that the FIGNL1-FIRRM complex may disassemble nascent RAD51-ssDNA patches that would otherwise hamper DMC1 filament extension toward the 3’ end of ssDNA tails. In the absence of FIGNL1-FIRRM, the presence of RAD51 patches is predicted to reduce DMC1 occupancy. Accessory factors might protect RAD51 from the FIGNL1-FIRRM complex on the DSB-distal part of ssDNA tails, permitting preferential RAD51 occupancy specifically in these intervals. One candidate for protecting RAD51 from FIGNL1 is the SWSAP1-SWS1-SPIDR complex36,40,41,42. Indeed, FIGNL1 interacts with SWSAP1 and SPIDR37,40,43, and SWSAP1 protects RAD51 filaments from FIGNL1 in vivo and in vitro40. Our observation that SWSAP1 is not required for forming DMC1-RPA2 co-foci in Swsap1−/− Fignl1/Firrm cKO (Fig. 7d) is consistent with the idea that SWSAP1 supports RAD51/DMC1 focus formation by protecting the nucleofilament from FIGNL1-FIRRM. Interestingly, it was recently reported that in human cells, SWSAP1-SWS1 interacts with the cohesin regulatory protein PDS5B, which localizes to chromosome axes during meiotic prophase37,94. As generally RAD51 localizes closer to the chromosome axis than DMC1 in mouse meiotic prophase (20,73 and Fig. 4a), this interaction, if present in meiotic prophase, might provide an anchor that favors preferential RAD51 protection on the DSB-distal part of DSB ssDNA tails. Alternatively, we cannot exclude that DMC1-SSDS profile alteration results from accumulating HR intermediates with a biased DMC1-ssDNA distribution. For example, longer DMC1 filaments might be more frequently engaged in strand invasion, therefore bound on dsDNA and undetectable by ChIP-SSDS, compared with shorter filaments.
In Fignl1 cKO and Firrm cKO spermatocytes, we observed meiotic DSB-independent accumulation of RAD51 foci on chromatin during premeiotic replication that persisted and was accompanied by DMC1 accumulation during meiotic prophase. DNA damage-independent RAD51 foci accumulate in human cells upon RAD51 overexpression29, thus higher RAD51 nuclear concentration in the absence of FIRRM or FIGNL1 might contribute to favor DNA damage-independent RAD51 and DMC1 binding on intact chromatin (Fig. 8i, (i)). In addition, RAD51 and DMC1 DNA damage-independent accumulation is observed in budding yeast and human cells after depletion of RAD54 family DNA translocases29,30,31. By analogy, the FIGNL1-FIRRM complex might prevent the stabilization of normally transient nascent RAD51-dsDNA filaments at replication forks (Fig. 8i, (ii)). This hypothesis is consistent with our finding that purified human FIGNL1ΔN might reduce RAD51 and DMC1 association with dsDNA in vitro (Fig. 8b, c), and with a recent study in human cells showing FIGNL1-FIRRM association with ongoing replication forks in unchallenging conditions80.
The linear RAD51/DMC1 staining detected between SYCP3 synapsed axes suggests that RAD51/DMC1 can associate stably with chromosome axis components, in either a DNA-dependent or DNA-independent manner, in the absence of the FIGNL1-FIRRM complex. DSB-independent RAD51 (but not DMC1) staining along unsynapsed chromosome axes has been previously described in late prophase mouse oocytes74,95,96; however, these structures associating RAD51 and DMC1 along synapsed axes in Fignl1 cKO and Firrm cKO spermatocytes are unusual. RAD51 interacts with several components of meiotic chromosomes, including the axis component SYCP315, the SC central element component SYCE297,98 and the cohesion regulator PDS5A/B99,100 that interacts also with SWSAP1-SWS137. Interestingly, it has been observed by super-resolution microscopy that several cohesin subunits and HORMAD1/2 coat the outside of SYCP3 axis cores73,101, a localization resembling that of RAD51/DMC1 staining between synapsed SYCP3-positive axes. RAD51/DMC1 interactions with components of meiotic chromosome axes might facilitate the accurate HR repair of meiotic DSBs (and incidental DNA damages). In this context, a function of the FIGNL1-FIRRM complex might be to prevent the stabilization of these interactions, other than at DNA damage sites.
Meiotic cells must face the challenge of repairing hundreds of programmed DSBs through several HR pathways, while restricting inappropriate repair that may involve similar HR intermediates. In this study, we started deciphering the functions of the conserved FIGNL1-FIRRM complex in mouse meiosis. We showed that the RAD51/DMC1 filament-destabilizing activity of FIGNL1 and FIRRM is implicated in regulating meiotic recombination and restricting inappropriate formation of stable RAD51/DMC1 filaments. Interestingly, although FIGNL1 alters RAD51 and DMC1 filament similarly in vitro, it is not clear whether FIGNL1 or FIRRM absence affects DMC1 directly or indirectly through RAD51. The elucidation of the several possible functions of the FIGNL1- FIRRM complex during mouse meiosis will need more in vitro and in vivo analyses of their functional interactions with other RAD51 and DMC1 regulators.
Methods
Mice
All mice used in the study were in the C57BL/6J background. Firrmfl/+ mice (allele BC055324tm1c(EUCOMM)Hmgu, MGI:5692863) were obtained from the International Knockout Mouse Consortium (IKMC). Fignl1fl/+ mice (allele Fignl1tm1c(EUCOMM)Hmgu) were generated by Phenomin-Institut Clinique de la Souris (ICS) using the plasmid containing the Fignl1tm1a(EUCOMM)Hmgu allele (MGI:5287847) obtained from Helmholtz Zentrum München GmbH. Firrmfl/fl mice were mated with mice that express Cre under the control of the CMV promoter (C57BL/6 Tg(CMV-cre)1Cgn)102 to generate Firrm-deleted heterozygous mice (Firrm+/-). Firrm+/− mice were mated with Tg(Stra8-icre)1Reb/J (Stra8-CreTg) mice59 to generate Firrm+/-;Stra8-CreTg mice. This transgene expresses Cre specifically in male germ cells, from undifferentiated spermatogonia to preleptotene spermatocytes59. By crossing Firrmfl/fl mice with Firrm+/-;Stra8-CreTg mice, Firrmfl/-;Stra8-CreTg (Firrm cKO) and Firrmfl/+, Firrmfl/+ Stra8-CreTg or Firrmfl/- (Firrm control) males were obtained. Fignl1fl/-;Stra8-CreTg (Fignl1 cKO) males were generated using the same strategy as for Firrm cKO mice. We assessed the efficiency of germline CRE-driven excision of the Firrmfl allele by crossing Firrmfl/+ Stra8-CreTg males with wild-type females. Among 97 pups, 52 were Firrm+/+ and 45 were Firrm+/-, with no detection of the Firrmfl allele in any, indicating an 100% efficiency of transmission of the excised allele to 45 pups. The Spo11YF/YF74 and Swsap1-/-42 mouse lines were described previously. Primers used for genotyping are listed in Supplementary Table 1. All animal experiments were carried out according to the CNRS guidelines. Mice housing conditions were temperature 22 °C, unregulated humidity, 12-h light/12-h dark cycle.
Histology
Mouse testes were fixed in Bouin’s solution for periodic acid-Schiff (PAS) staining at room temperature, overnight. Testes were then embedded in paraffin and 3µm-thick slices were cut. PAS-stained sections were scanned using the automated tissue slide-scanning tool of a Hamamatsu NanoZoomer Digital Pathology system.
Spermatocyte chromosome spreads
Spermatocyte spreads were prepared with the dry-down technique103. Briefly, a suspension of testis cells was prepared in PBS, and then incubated in a hypotonic solution for 8 min at room temperature. Cells were centrifuged, resuspended in 66 mM sucrose solution and spread on slides or coverslips (1.5H, high precision) with 1% paraformaldehyde, 0.05% Triton X-100. Slides/coverslips were dried in a humid chamber for 1–2 h, washed in 0.24% Photoflo200 (Kodak), air-dried, and used for immunostaining or stored at −80 °C.
Immunofluorescence staining
Immunostaining was done as described104. After incubation with a milk-based blocking buffer (5% milk, 5% donkey serum in PBS), spermatocyte spreads were incubated with primary antibodies at room temperature overnight, followed by secondary antibodies (37 °C for 1 h). The antibodies used are listed in Supplementary Table 2. Nuclei were stained with 4′−6-diamidino-2-phenylindole (DAPI, 2 μg/ml) in the final washing step.
For immunostaining with the anti-DMC1 antibody, a specific blocking buffer (0.5% BSA, 0.5% powder milk, 0.5% donkey serum in PBS) was used prior to incubation with the primary antibody. Primary antibody incubation was performed in 10% BSA in PBS. Immunostaining of spermatocyte spreads on coverslips for STED microscopy was done with specific secondary antibodies (Supplementary Table 2), and DAPI was omitted.
Widefield fluorescent imaging
Widefield images were acquired using one of the following microscopes: Zeiss Axioimager Apotome with 100X Plan Apochromat 1.46 oil DIC objective and 1 ANDOR sCMOS ZYLA 4.2 MP monochrome camera (2048 × 2048 pixels, 6.5 µm pixel size) or Zeiss Axioimager 100X Plan Apochromat 1.4 NA oil objective and 1 Zeiss CCD Axiocam Mrm 1.4 MP monochrome camera (1388 × 1040 pixels, 6.45 µm pixel size).
Stimulated emission depletion (STED) super-resolution imaging
Super-resolution images were acquired using an STED microscope (Abberior Instruments, Germany) equipped with a PlanSuperApo 100×/1.40 oil immersion objective (Olympus, Japan). For 3-color STED imaging, immunolabeling was performed using one of the following combinations of secondary antibodies: STAR 460 L, STAR ORANGE, STAR RED or STAR GREEN, STAR ORANGE, STAR RED (Supplementary Table 2). STAR 460 L and STAR 488 were excited at 485 nm, STAR ORANGE at 561 nm, and STAR RED at 640 nm. Excitation was done with a dwell time of 10 µs. STED was performed at 595 nm for STAR 488 and at 775 nm for all other dyes. Images were collected in line accumulation mode with detection set at 571–625 nm for STAR 460 L and STAR ORANGE, 500-580 nm for STAR GREEN, and 650–750 nm for STAR RED.
Image analysis
For quantification and colocalization analyses, images were deconvolved using Huygens Professional version 22.10 (Scientific Volume Imaging).
All image analyses were performed using Fiji/ImageJ 1.53t105, with the “MeiQuant” set of tools106 available on github (https://github.com/MontpellierRessourcesImagerie/meiosis_bar).
Single nuclei were cropped manually or using an automatic DAPI signal threshold. Nuclei were sorted into meiotic prophase substages following the criteria described below.
Foci were detected using the Find Maxima function. Focus intensity was determined as the intensity of the maximum for every focus within the chosen category (e.g., on-axis RAD51 foci colocalized with an on-axis DMC1 focus). On-axis and off-axis foci were distinguished on the basis of their localization within (or outside) a binary mask. This ROI was drawn using an automatic SYCP3 axis protein staining threshold (SYCP1 staining was used for MSH4 and TEX11 foci). Because there was no SYCP3 staining-defined axis structure at preleptotene stage, all foci were considered as off-axis foci at this stage.
For two-color focus colocalization, the distance of a given channel focus to the closest second color focus was calculated. Foci were considered as colocalized when this distance was below the minimum resolution distance (0.3 µm for widefield images), as in ref. 107. The level of random colocalization of foci in channel A (foci A) with foci in channel B (foci B) in any given nucleus was estimated by simulating the random localization of the actual number of foci A within the considered area (on-axis or off-axis as defined above, or within the whole nucleus in preleptotene), and by determining the number of random foci A colocalized with actual foci B. The mean number of colocalizations from 100 simulations was taken as the number of foci A colocalized with foci B by chance in the nucleus (nrandom, “random” on figures), and this was repeated for every nucleus. Reciprocally, the level of random colocalization of foci B with foci A resulted from random simulations of foci B localizations.
In every nucleus, the number of colocalized foci A was corrected for random colocalization by considering that (1) the observed number of colocalized foci A (nobs) is composed of one subset of biologically meaningful colocalized foci (“truly” colocalized foci A, ncol) and one subset of foci A colocalized by chance; (2) the ratio nrandom /nT (where nrandom is estimated as described above and nT is the total number of foci A in the nucleus) estimates the frequency of foci A colocalizing by chance among the population of foci A not “truly” colocalized, thus the number of foci A colocalized by chance is (nT – ncol) ∗ nrandom /nT, by excluding the truly colocalized foci A from random colocalization.(3) Finally, the estimated number of colocalized foci corrected for random colocalization (ncol,) was obtained from the formula ncol = (nobs-nrandom)/(nT-nrandom), where nT was the total number of foci counted, nobs the observed number of colocalized foci and nrandom the mean number of colocalization from 100 simulations as described above. The percentage of corrected colocalization estimate was the ratio of the corrected number of colocalized foci ncol over the total number of foci in the same nucleus, ncol / nT.
For γH2AX quantification, nuclei were cropped manually and the integrated intensity of the γH2AX channel in the cropped region was measured.
Prophase spermatocytes were staged using the following criteria, based on SYCP3 staining. Preleptotene nuclei had patchy weak SYCP3 signal throughout the nucleus. Early leptotene nuclei had focus-like well-defined very short stretches of SYCP3 staining. Mid-late leptotene nuclei had short stretches of SYCP3 fragments. Early zygotene nuclei had longer SYCP3 stretches as the chromosome axes continued to elongate. Mid-zygotene nuclei had very long or full SYCP3 axes, but no or relatively few synapses marked by thicker SYCP3 stretches. Late zygotene had full SYCP3 axes with extensive synapsis marked by thicker SCP3 signal.
DMC1 chromatin immuno-precipitation, followed by single-strand DNA sequencing (DMC1-SSDS)
DMC1 ChIP-SSDS and library preparation were performed as described in ref. 108 using a goat anti-DMC1 antibody (0.5 mg/ml; Santa Cruz, reference C-20). Ten testes from 12 dpp Firrmfl/+;Stra8-CreTg (control) and from Firrmfl/+;Stra8-CreTg (Firrm cKO) mice were used in each biological replicate. Sequencing was performed on a NovaSeq 6000 PE150 platform in paired end mode (2 × 150bp).
Detection of DMC1 ChIP-SSDS peaks
Raw reads were processed using the SSDS-DMC1 Nextflow pipeline109, available on github (https://github.com/jajclement/ssdsnextflowpipeline). Briefly, the main steps of the pipeline included raw read quality control and trimming (removal of adapter sequences, low-quality reads and extra bases) and mapping to the UCSC mouse genome assembly build GRCm38/mm10. Single-stranded derived fragments were then identified from mapped reads using a previously published method75,110, and peaks were detected in Type-1 fragments (high confidence ssDNA). To control reproducibility and assess replicate consistency, the Irreproducible Discovery Rate (IDR) method111 was used, following the ENCODE procedure (https://github.com/ENCODE-DCC/chip-seq-pipeline2). The “regionPeak” peak type parameter and default p value thresholds were used. Briefly, this method performs relaxed peak calling for each of the two replicates (truerep), the pooled dataset (poolrep), and pseudo-replicates that are artificially generated by randomly sampling half of the reads twice, for each replicate and the pooled dataset. Both control and Firrm cKO datasets passed the IDR statistics criteria for the two scores (well below 2). By default, the pipeline gave the poolrep as primary output, but for this study the truerep peak sets were considered. Lastly, peak centering and strength calculation were computed using a previously published method75.
The list of SPO11-oligo hotspots from B6 mice and the coordinates (genome build GRCm38/mm10) of their center were from77.
The overlaps between intervals was determined with bedtools112 Intersect on the Galaxy France web interface. For determining overlaps between control and Firrm cKO peaks, a minimum overlap of 10%, and reciprocally, was required. The overlap between DMC1-SSDS peaks and the center of SPO11-oligo hotspots77 was considered positive if at least 1 bp of the DMC1-hotspot contained the coordinate of the center of one SPO11-oligo hotspot.
Heatmaps and average plot profiles were generated with deeptools (computeMatrix, plotHeatmap and PlotProfile) on Galaxy France server (https://usegalaxy.fr/).
Preparation of mouse testis protein extracts and western blotting
Cytoplasmic and nuclear extracts were prepared from 12 dpp control, Firrm cKO and Fignl1 cKO mice. Testes were homogenized in hypotonic buffer (10 mM Hepes, pH 7.4, 320 mM sucrose, 0.2 mM PMSF, 1x Complete protease inhibitor cocktail, EDTA-free (Roche), 0.07% beta-mercaptoethanol) in a Dounce homogenizer. After centrifugation (1000 × g at 4 °C for 10 min), the supernatant was collected and used as cytoplasmic fraction. The pellet was resuspended in half nuclear-packed volume of low salt buffer (20 mM Tris-HCl pH7.3, 12.5% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 20 mM KCl, 1× Complete protease inhibitor cocktail, EDTA-free (Roche), 0.07% beta-mercaptoethanol). Then half nuclear-packed volume of high salt buffer (same, but 1.2 M KCl) was added drop by drop, incubated at 4 °C for 30 min with agitation and centrifuged (14,000 × g at 4 °C for 30 min). The supernatant was collected as nuclear fraction. Cytoplasmic and nuclear fractions were analyzed by western blotting with rabbit anti-FIGNL1 (1/500, Proteintech, 17604-1-AP), rabbit anti-FIRRM (1/500, Abcam, ab121774), rabbit anti-beta tubulin (1/3000, Abcam, ab6046) and guinea pig anti-SYCP3 (1/3000104) antibodies. HRP-conjugated secondary antibodies were anti-rabbit IgG-HRP (1:5000; Cell Signaling Technology) and donkey anti-guinea pig IgG-HRP (1/10,000; Jackson Immuno Research, 706-035-148).
Protein purification
Human RAD51 was purified by the CiGEX Platform (CEA, Fontenay-aux-Roses) as follows. His-SUMO-RAD51 was expressed in the E. coli strain BRL (DE3) pLys. All protein purification steps were carried out at 4 °C. Cells from a 3-l culture that was induced with 0.5 mM isopropyl-1-thio-ß-D-galactopyranoside (IPTG) at 20 °C overnight were resuspended in 1x PBS, 350 mM NaCl, 20 mM imidazole, 10% glycerol, 0.5 mg/ml lysozyme, Compete Protease Inhibitor (Roche), 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF). Cells were lysed by sonication and the insoluble material was removed by centrifugation at 150,000 × g for 1 h. The supernatant was incubated with 5 ml of Ni-NTA resin (Qiagen) for 2 h. The mixture was poured into an Econo-Column Chromatography Column (BIO-RAD) and beads were washed first with 80 ml W1 buffer (20 mM Tris HCl pH 8, 500 mM NaCl, 20 mM imidazole, 10% glycerol, 0.5% NP40), followed by 80 ml of W2 buffer (20 mM Tris HCl pH 8, 100 mM NaCl, 20 mM imidazole, 10% glycerol, 1 mM DTT). Then, His-SUMO-RAD51 bound to the beads was resuspended in 8 ml of W2 buffer and incubated with SUMO protease at a 1/80 ratio (w/w) for 16 h. RAD51 without the His-SUMO tag was then recovered into the flow through and directly loaded onto a HiTrap heparin column (GE Healthcare). The column was washed with W2 buffer and then a 0.1-1 M NaCl gradient was applied. Fractions containing purified RAD51 were concentrated and dialyzed against storage buffer (20 mM Tris HCl pH 8, 50 mM KCl, 0.5 mM EDTA, 10% glycerol, 1 mM DTT, 0.5 mM AEBSF) and stored at −80 °C. Human RPA was purified by the CiGEX Platform (CEA, Fontenay-aux-Roses) as previously described113.
For human FIGNL1 purification, FIGNL1∆N without the region encoding the N-terminal 284 aa, allowing the production of soluble protein40, was inserted into the pET15 vector (Novagene), and the protein was overexpressed in E. coli BL21(DE3) cells upon addition of 0.2 mM IPTG at 37 °C for 3 h. Cell pellets were resuspended in buffer A (50 mM Tris-HCl pH7.4, 500 mM NaCl, 5% glycerol, 5 mM MgCl2, 5 mM β-mercaptoethanol, 1 mM PMSF, 0.1% NP40, 20 mM imidazole, cOmplete Protease Inhibitor Cocktail), disrupted by French press (6 bar) and cleared by centrifugation following incubation with the benzonase nuclease (Sigma) at 4 °C for 30 min. The supernatant was loaded on a 1 ml HisTrap Fast-Flow column (GE healthcare) and equilibrated with buffer A on an ÄKTA pure system. After a washing step, proteins were eluted with buffer A supplemented with 300 mM imidazole. FIGNL1∆N was further purified by size exclusion chromatography using a HiLoad 16/600 Superdex 200 column (GE Healthcare) in buffer B (50 mM Tris-HCl pH7.4, 200 mM NaCl, 10% glycerol, 5 mM MgCl2, 5 mM β-mercaptoethanol). The peak fractions were concentrated with Amicon Ultra 30 K (Millipore) and stored at −80 °C.
RAD51 and DMC1 filament electromobility shift assay (EMSA)
RAD51 and DMC1 filaments were formed by incubating 3 μM (nucleotide concentration) of 400 nt ssDNA or dsDNA labeled with Cy5 with 1 μM RAD51 (1 protein per 3 nt) or 1.5 μM DMC1 (>1 protein per 3 nt to obtain fully covered DNA) in a buffer containing 10 mM Tris-HCl pH7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 2 mM ATP, and 1 mM DTT at 37 °C for 5 min. Then, 1.6 μM of FIGLN1ΔN was added to the reaction and the incubation was prolongated for 15 min to test its effect on filament assembly and architecture (pre-formed filament). Control experiments, in absence of FIGLN1ΔN, were realized by adding the same volume of FIGLN1ΔN storage buffer to ensure that the detected effect is not due to the FIGLN1ΔN buffer components. Alternatively, RAD51 or DMC1 was added concomitantly with FIGLN1ΔN to the reaction and incubated for 20 min (no pre-formed filament). Protein-DNA complexes were fixed in 0.01% glutaraldehyde at room temperature for 5 min. Then, the reaction products were analyzed using 1% agarose gel in 0.5x Tris-acetate/EDTA at 4 °C. Images were acquired using a Typhoon imager (GE Healthcare Life Science).
Transmission electron microscopy (TEM) analysis of RAD51 and DMC1 filaments
RAD51 and DMC1 filaments were formed by incubating 7.5 μM (nucleotide concentration) of 400 nt long ssDNA and dsDNA with 2.5 μM RAD51 (1 protein per 3 nt) or 3.5 μM DMC1 in a buffer containing 10 mM Tris-HCl pH7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 2 mM ATP and 1 mM DTT at 37 °C for 20 min. Then, 1.6 μM of FIGLN1ΔN was added to the reaction at the same time as RAD51/DMC1. Again, control experiments, in the absence of FIGLN1ΔN, were realized by adding the same volume of FIGLN1ΔN storage buffer. For filament length analysis, positive staining combined with a TEM dark-field imaging mode was used: 1 μL of the reaction was quickly diluted 20 times in a buffer containing 10 mM Tris-HCl pH 7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM Cacl2. During one minute, a 5 μL drop of the dilution was deposited on a 600-mesh copper grid previously covered with a thin carbon film and pre-activated by glow-discharge in the presence of amylamine (Sigma-Aldrich, France)114,115. Grids were rinsed and positively stained with aqueous 2% (w/v) uranyl acetate, dried carefully with a filter paper. To better observe FIGLN1ΔN effect on the filament architecture, samples were also spread using negative staining and observed in bright-field mode. For this, a drop of the reaction was directly deposited on a carboned copper grid pre-activated with glow discharge (plasma).
TEM grids were observed in the annular dark-field mode in zero-loss filtered imaging or in canonical bright-field imaging using a Zeiss 902 transmission electron microscope. Images were captured at a magnification of 85,000× with a Veleta CCD camera and analyzed with the iTEM software (both Olympus Soft Imaging Solution). For quantification, the filament length was measured in at least two independent experiments with a total of at least 75 molecules measured.
D-loop in vitro assay
RAD51 and DMC1 filaments were formed in the same conditions as for the EMSA analysis. The same incubation conditions and buffer were used to assemble mixed RAD51/DMC1 filaments by incubating 3 μM (nucleotide concentration) of 400 nt ssDNA substrates with 1.25 μM RAD51 plus 0.75 μM DMC1. In the second step, 15 nM in molecules of homologous dsDNA donor (pUC19 plasmid purified on MiniQ ion exchange chromatography column) was introduced in the reaction and in the case of DMC1 filaments, 4 mM more CaCl2 was added, and then the mixture was incubated at 37 °C for 30 min. The reaction was stopped with 0.5 mg/mL proteinase K, 1% SDS, 12.5 mM EDTA at 37 °C for 30 min and separated on 1% TAE agarose gels (80 V, for 30 min).
Statistical analysis and reproducibility
The statistical analyses of cytological observations were done with GraphPad Prism 9. A contingency chi-square test was used to compare stage distributions. The nonparametric Dunn’s multiple comparison test was used to compare focus counts, colocalized focus counts and fractions, and γH2AX intensity among genotypes. The nonparametric Wilcoxon signed-ranks test was used to compare true colocalization versus random colocalization of foci. All tests, sample size, and p values (n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) are provided in the corresponding legends and/or figures. If not otherwise stated, at least two animals/genotype were analyzed and similar results were obtained.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DMC1-SSDS raw and processed data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI and are available through the project identifier PRJEB62127. Source data are provided with this paper.
Code availability
The “MeiQuant” set of tools106 for image analysis is available on Github at https://github.com/MontpellierRessourcesImagerie/meiosis_bar. The SSDS-DMC1 Nextflow pipeline109 for analysis of DMC1 ChIP-single-strand DNA sequencing data is available on Github at https://github.com/jajclement/ssdsnextflowpipeline.
References
Gerton, J. L. & Hawley, R. S. Homologous chromosome interactions in meiosis: diversity amidst conservation. Nat. Rev. Genet. 6, 477–487 (2005).
Hunter, N. Meiotic recombination: the essence of heredity. Cold Spring Harb. Perspect. Biol. 7, a016618 (2015).
Baudat, F., Imai, Y. & de Massy, B. Meiotic recombination in mammals: localization and regulation. Nat. Rev. Genet. 14, 794–806 (2013).
Robert, T., Vrielynck, N., Mézard, C., de Massy, B. & Grelon, M. A new light on the meiotic DSB catalytic complex. Semin. Cell Dev. Biol. 54, 165–176 (2016).
Paiano, J. et al. ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis. Nat. Commun. 11, 857 (2020).
Yamada, S. et al. Molecular structures and mechanisms of DNA break processing in mouse meiosis. Genes Dev. 34, 806–818 (2020).
Prakash, R., Zhang, Y., Feng, W. & Jasin, M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Perspect. Biol. 1–29. https://doi.org/10.1101/cshperspect.a016600 (2015).
Zelensky, A., Kanaar, R. & Wyman, C. Mediators of Homologous DNA Pairing. Cold Spring Harb. Perspect. Biol. 6, a016451 (2014).
Brown, M. S. & Bishop, D. K. DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb. Perspect. Biol. 7, a016659 (2014).
Börner, G. V., Kleckner, N. & Hunter, N. Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117, 29–45 (2004).
Pyatnitskaya, A., Borde, V. & De Muyt, A. Crossing and zipping: molecular duties of the ZMM proteins in meiosis. Chromosoma 128, 181–198 (2019).
Kneitz, B. et al. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 14, 1085–1097 (2000).
Edelmann, W. et al. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat. Genet. 21, 123–127 (1999).
de Vries, S. S. et al. Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes Dev. 13, 523–531 (1999).
Tarsounas, M., Morita, T., Pearlman, R. E. & Moens, P. B. Rad51 and Dmc1 form mixed complexes associated with mouse meiotic chromosome cores and synaptonemal complexes. J. Cell Biol. 147, 207–220 (1999).
Slotman, J. A. et al. Super-resolution imaging of RAD51 and DMC1 in DNA repair foci reveals dynamic distribution patterns in meiotic prophase. PLOS Genet. 16, e1008595 (2020).
Brown, M. S., Grubb, J., Zhang, A., Rust, M. J. & Bishop, D. K. Small Rad51 and Dmc1 complexes often co-occupy both ends of a meiotic DNA double strand break. PLOS Genet. 11, e1005653 (2015).
Cloud, V., Chan, Y.-L., Grubb, J., Budke, B. & Bishop, D. K. Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science 337, 1222–1225 (2012).
Da Ines, O. et al. Meiotic recombination in arabidopsis is catalysed by DMC1, with RAD51 playing a supporting role. PLoS Genet. 9, e1003787 (2013).
Hinch, A. G. et al. The configuration of RPA, RAD51, and DMC1 binding in meiosis reveals the nature of critical recombination intermediates. Mol. Cell. https://doi.org/10.1016/j.molcel.2020.06.015 (2020).
Halder, S. et al. Double-stranded DNA binding function of RAD51 in DNA protection and its regulation by BRCA2. Mol. Cell 82, 3553–3565.e5 (2022).
Mason, J. M., Chan, Y.-L., Weichselbaum, R. W. & Bishop, D. K. Non-enzymatic roles of human RAD51 at stalled replication forks. Nat. Commun. 10, 4410 (2019).
Schlacher, K. et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011).
Berti, M., Cortez, D. & Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 21, 633–651 (2020).
Kolinjivadi, A. M. et al. Moonlighting at replication forks – a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 591, 1083–1100 (2017).
Hashimoto, Y., Ray Chaudhuri, A., Lopes, M. & Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 17, 1305–1311 (2010).
Reitz, D., Chan, Y.-L. & Bishop, D. K. How strand exchange protein function benefits from ATP hydrolysis. Curr. Opin. Genet Dev. 71, 120–128 (2021).
Ceballos, S. J. & Heyer, W.-D. Functions of the Snf2/Swi2 family Rad54 motor protein in homologous recombination. Biochim. Biophys. Acta (BBA) - Gene Regul. Mech. 1809, 509–523 (2011).
Mason, J. M. et al. RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic Acids Res. 43, 3180–3196 (2015).
Holzen, T. M., Shah, P. P., Olivares, H. A. & Bishop, D. K. Tid1/Rdh54 promotes dissociation of Dmc1 from nonrecombinogenic sites on meiotic chromatin. Genes Dev. 20, 2593–2604 (2006).
Shah, P. P. et al. Swi2/Snf2-related translocases prevent accumulation of toxic Rad51 complexes during mitotic growth. Mol. Cell 39, 862–872 (2010).
Li, X. & Heyer, W.-D. RAD54 controls access to the invading 3′-OH end after RAD51-mediated DNA strand invasion in homologous recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 37, 638–646 (2009).
Wesoly, J. et al. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol. Cell Biol. 26, 976–989 (2006).
Russo, A., Cordelli, E., Salvitti, T., Palumbo, E. & Pacchierotti, F. Rad54/Rad54B deficiency is associated to increased chromosome breakage in mouse spermatocytes. Mutagenesis 33, 323–332 (2018).
Liu, T., Wan, L., Wu, Y., Chen, J. & Huang, J. hSWS1·SWSAP1 is an evolutionarily conserved complex required for efficient homologous recombination repair. J. Biol. Chem. 286, 41758–41766 (2011).
Prakash, R. et al. Distinct pathways of homologous recombination controlled by the SWS1–SWSAP1–SPIDR complex. Nat. Commun. 12, 4255 (2021).
Martino, J. et al. The human Shu complex functions with PDS5B and SPIDR to promote homologous recombination. Nucleic Acids Res. 47, 10151–10165 (2019).
Godin, S. K. et al. Evolutionary and functional analysis of the invariant SWIM domain in the conserved Shu2/SWS1 protein family from saccharomyces cerevisiae to homo sapiens. Genetics 199, 1023–1033 (2015).
Martín, V. et al. Sws1 is a conserved regulator of homologous recombination in eukaryotic cells. EMBO J. 25, 2564–2574 (2006).
Matsuzaki, K., Kondo, S., Ishikawa, T. & Shinohara, A. Human RAD51 paralogue SWSAP1 fosters RAD51 filament by regulating the anti-recombinase FIGNL1 AAA+ ATPase. Nat. Commun. 10, 1407 (2019).
Huang, T. et al. SPIDR is required for homologous recombination during mammalian meiosis. Nucleic Acids Res. gkad154. https://doi.org/10.1093/nar/gkad154 (2023).
Abreu, C. M. et al. Shu complex SWS1-SWSAP1 promotes early steps in mouse meiotic recombination. Nat. Commun. 9, 3961 (2018).
Yuan, J. & Chen, J. FIGNL1-containing protein complex is required for efficient homologous recombination repair. Proc. Natl Acad. Sci. USA 110, 10640–10645 (2013).
Fernandes, J. B. et al. FIGL1 and its novel partner FLIP form a conserved complex that regulates homologous recombination. PLoS Genet. 14, e1007317 (2018).
Zhang, P. et al. The rice AAA-ATPase OsFIGNL1 is essential for male meiosis. Front. Plant Sci. 8, 1639 (2017).
Liu, X. et al. OsRAD51 plays a vital role in promoting homologous recombination in rice meiosis. Int. J. Mol. Sci. 23, 9906 (2022).
Pinedo-Carpio, E. et al. FIRRM cooperates with FIGNL1 to promote RAD51 disassembly during DNA repair. Sci. Adv. 9, eadf4082 (2023).
Zhou, Z. et al. C1orf112 teams up with FIGNL1 to facilitate RAD51 filament disassembly and DNA interstrand cross-link repair. Cell Rep. 42, 112907 (2023).
Tischler, J. D. et al. FLIP(C1orf112)-FIGNL1 complex regulates RAD51 chromatin association to promote viability after replication stress. Nat. Commun. 15, 866 (2024).
Matsuzaki, K., Shinohara, A. & Shinohara, M. Human AAA+ ATPase FIGNL1 suppresses RAD51-mediated ultra-fine bridge formation. Nucleic Acids Res. gkae263. https://doi.org/10.1093/nar/gkae263 (2024).
Tsaridou, S. & Vugt, M. A. T. M. van. FIRRM and FIGNL1: partners in the regulation of homologous recombination. Trends Genet. 0, (2024).
Girard, C. et al. AAA-ATPase FIDGETIN-LIKE 1 and helicase FANCM antagonize meiotic crossovers by distinct mechanisms. PLOS Genet. 11, e1005369 (2015).
Hu, Q. et al. Meiotic chromosome association 1 interacts with TOP3α and regulates meiotic recombination in rice. Plant Cell 29, 1697–1708 (2017).
Yang, S. et al. FIGNL1 inhibits non-homologous chromosome association and crossover formation. Front. Plant Sci. 13, 945893 (2022).
Li, X. et al. Regulation of interference-sensitive crossover distribution ensures crossover assurance in Arabidopsis. Proc. Natl Acad. Sci. USA 118, e2107543118 (2021).
Kumar, R., Duhamel, M., Coutant, E., Ben-Nahia, E. & Mercier, R. Antagonism between BRCA2 and FIGL1 regulates homologous recombination. Nucleic Acids Res. 47, 5170–5180 (2019).
Zhang, Q. et al. The FLIP-FIGNL1 complex regulates the dissociation of RAD51/DMC1 in homologous recombination and replication fork restart. Nucleic Acids Res. gkad596. https://doi.org/10.1093/nar/gkad596 (2023).
Mazouzi, A. et al. FIRRM/C1orf112 mediates the resolution of homologous recombination intermediates in response to DNA interstrand crosslinks. Sci. Adv. 9, eadf4409 (2023).
Sadate-Ngatchou, P. I., Payne, C. J., Dearth, A. T. & Braun, R. E. Cre recombinase activity specific to post natal, premeiotic male germ cells in transgenic mice. Genesis 46, 738–742 (2008).
Shi, B. et al. Dual functions for the ssDNA-binding protein RPA in meiotic recombination. PLOS Genet. 15, e1007952 (2019).
Imai, Y. et al. PRDM9 activity depends on HELLS and promotes local 5-hydroxymethylcytosine enrichment. Elife 9, e57117 (2020).
Bao, J., Ma, H.-Y., Schuster, A., Lin, Y.-M. & Yan, W. Incomplete cre-mediated excision leads to phenotypic differences between Stra8-iCre; Mov10l1lox/lox and Stra8-iCre; Mov10l1lox/Δ mice. Genesis 51, 481–490 (2013).
Klein, H. L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 7, 686–693 (2008).
Baudat, F., Manova, K., Yuen, J. P., Jasin, M. & Keeney, S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking spo11. Mol. Cell 6, 989–998 (2000).
Romanienko, P. J. & Camerini-Otero, R. D. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell 6, 975–987 (2000).
Pittman, D. L. et al. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol. Cell 1, 697–705 (1998).
Yoshida, K. et al. The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol. Cell 1, 707–718 (1998).
Mahadevaiah, S. K. et al. Recombinational DNA double-strand breaks in mice precede synapsis. Nat. Genet. 27, 271–276 (2001).
Moens, P. B. et al. The time course and chromosomal localization of recombination-related proteins at meiosis in the mouse are compatible with models that can resolve the early DNA-DNA interactions without reciprocal recombination. J. Cell Sci. 115, 1611–1622 (2002).
Yoon, S., Choi, E.-H., Kim, J.-W. & Kim, K. P. Structured illumination microscopy imaging reveals localization of replication protein A between chromosome lateral elements during mammalian meiosis. Exp. Mol. Med. 50, 1–12 (2018).
Yang, F. et al. Meiotic failure in male mice lacking an X-linked factor. Genes Dev. 22, 682–691 (2008).
Wassing, I. E. & Esashi, F. RAD51: beyond the break. Semin. Cell Dev. Biol. 113, 38–46 (2021).
Koornneef, L. et al. Multi-color dSTORM microscopy in Hormad1−/− spermatocytes reveals alterations in meiotic recombination intermediates and synaptonemal complex structure. PLOS Genet. 18, e1010046 (2022).
Carofiglio, F. et al. SPO11-independent DNA repair foci and their role in meiotic silencing. PLoS Genet. 9, e1003538 (2013).
Khil, P. P., Smagulova, F., Brick, K. M., Camerini-Otero, R. D. & Petukhova, G. V. Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res. 22, 957–965 (2012).
Brick, K., Smagulova, F., Khil, P., Camerini-Otero, R. D. & Petukhova, G. V. Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645 (2012).
Lange, J. et al. The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell 167, 695–708.e16 (2016).
Davies, B. et al. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171–176 (2016).
Ito, M. et al. FIGNL1 AAA+ ATPase remodels RAD51 and DMC1 filaments in pre-meiotic DNA replication and meiotic recombination. Nat. Commun. 14, 6857 (2023).
Stok, C. et al. FIRRM/C1orf112 is synthetic lethal with PICH and mediates RAD51 dynamics. Cell Rep. 42, (2023).
L’Hôte, D. et al. Fidgetin-like1 is a strong candidate for a dynamic impairment of male meiosis leading to reduced testis weight in mice. PLoS One 6, e27582 (2011).
Mercier, R., Mézard, C., Jenczewski, E., Macaisne, N. & Grelon, M. The molecular biology of meiosis in plants. Annu. Rev. Plant Biol. 66, 297–327 (2015).
Petukhova, G. V., Romanienko, P. J. & Camerini-Otero, R. D. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev. Cell 5, 927–936 (2003).
Pezza, R. J., Petukhova, G. V., Ghirlando, R. & Camerini-Otero, R. D. Molecular activities of meiosis-specific proteins Hop2, Mnd1, and the Hop2-Mnd1 complex. J. Biol. Chem. 281, 18426–18434 (2006).
Zhao, W. & Sung, P. Significance of ligand interactions involving Hop2-Mnd1 and the RAD51 and DMC1 recombinases in homologous DNA repair and XX ovarian dysgenesis. Nucleic Acids Res. 43, 4055–4066 (2015).
Finsterbusch, F. et al. Alignment of homologous chromosomes and effective repair of programmed DNA double-strand breaks during mouse meiosis require the minichromosome maintenance domain containing 2 (MCMDC2) protein. PLOS Genet. 12, e1006393 (2016).
McNairn, A. J., Rinaldi, V. D. & Schimenti, J. C. Repair of meiotic DNA breaks and homolog pairing in mouse meiosis requires a minichromosome maintenance (MCM) paralog. Genetics 205, 529–537 (2017).
Lutzmann, M. et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell 47, 523–534 (2012).
Hustedt, N. et al. Control of homologous recombination by the HROB–MCM8–MCM9 pathway. Genes Dev. 33, 1397–1415 (2019).
Cole, F. et al. Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat. Genet. 46, 1072–1080 (2014).
Piazza, A. & Heyer, W.-D. Moving forward one step back at a time: reversibility during homologous recombination. Curr. Genet. 65, 1333–1340 (2019).
Yu, D. S. et al. Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol. Cell 12, 1029–1041 (2003).
Lan, W.-H. et al. Rad51 facilitates filament assembly of meiosis-specific Dmc1 recombinase. Proc. Natl Acad. Sci. 117, 11257–11264 (2020).
Viera, A. et al. PDS5 proteins regulate the length of axial elements and telomere integrity during male mouse meiosis. EMBO Rep. 21, e49273 (2020).
Rinaldi, V. D., Bolcun-Filas, E., Kogo, H., Kurahashi, H. & Schimenti, J. C. The DNA damage checkpoint eliminates mouse oocytes with chromosome synapsis failure. Mol. Cell 67, 1026–1036.e2 (2017).
Ravindranathan, R., Raveendran, K., Papanikos, F., San-Segundo, P. A. & Tóth, A. Chromosomal synapsis defects can trigger oocyte apoptosis without elevating numbers of persistent DNA breaks above wild-type levels. Nucleic Acids Res. 50, 5617–5634 (2022).
Bolcun-Filas, E. et al. Mutation of the mouse syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLOS Genet. 5, e1000393 (2009).
Crichton, J. H., Dunce, J. M., Baarends, W. M., Davies, O. R. & Adams, I. R. Parallel recruitment pathways contribute to synaptonemal complex assembly during mammalian meiosis. 2022.04.14.488335 Preprint at https://doi.org/10.1101/2022.04.14.488335 (2022).
Couturier, A. M. et al. Roles for APRIN (PDS5B) in homologous recombination and in ovarian cancer prediction. Nucleic Acids Res. 44, 10879–10897 (2016).
Morales, C. et al. PDS5 proteins are required for proper cohesin dynamics and participate in replication fork protection. J. Biol. Chem. 295, 146–157 (2020).
Xu, H. et al. Molecular organization of mammalian meiotic chromosome axis revealed by expansion STORM microscopy. Proc. Natl Acad. Sci. 116, 18423–18428 (2019).
Schwenk, F., Baron, U. & Rajewsky, K. A cre -transgenic mouse strain for the ubiquitous deletion of loxP -flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 (1995).
Peters, A. H., Plug, A. W., van Vugt, M. J. & de Boer, P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 5, 66–68 (1997).
Grey, C., Baudat, F. & Massy, B. de. Genome-wide control of the distribution of meiotic recombination. PLOS Biol. 7, e1000035 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Cau, J., Toe, L. D., Zainu, A., Baudat, F. & Robert, T. MeiQuant’: an integrated tool for analyzing meiotic prophase I spread images. Methods Mol. Biol. 2770, 263–285 (2024).
Lachmanovich, E. et al. Co-localization analysis of complex formation among membrane proteins by computerized fluorescence microscopy: application to immunofluorescence co-patching studies. J. Microsc. 212, 122–131 (2003).
Grey, C. et al. In vivo binding of PRDM9 reveals interactions with noncanonical genomic sites. Genome Res. 27, 580–590 (2017).
Auffret, P., de Massy, B. & Clément, J. A. J. Mapping meiotic DNA breaks: two fully-automated pipelines to analyze single-strand DNA sequencing data, hotSSDS and hotSSDS-extra. In Germ Cell Development: Methods and Protocols (eds Barchi, M. & De Felici, M.) 227–261. https://doi.org/10.1007/978-1-0716-3698-5_16 (Springer, 2024).
Brick, K., Pratto, F., Sun, C.-Y., Camerini-Otero, R. D. & Petukhova, G. Analysis of meiotic double-strand break initiation in mammals. In Methods in Enzymology (eds Spies, M. & Malkova, A.) vol. 601, 391–418 (Academic Press, 2018).
Li, Q., Brown, J. B., Huang, H. & Bickel, P. J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 5, 1752–1779 (2011).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Gomes, X. V., Henricksen, L. A. & Wold, M. S. Proteolytic mapping of human replication protein A: evidence for multiple structural domains and a conformational change upon interaction with single-stranded DNA. Biochemistry 35, 5586–5595 (1996).
Veaute, X. et al. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423, 309–312 (2003).
Dubochet, J., Ducommun, M., Zollinger, M. & Kellenberger, E. A new preparation method for dark-field electron microscopy of biomacromolecules. J. Ultrastruct. Res. 35, 147–167 (1971).
Acknowledgements
We would like to thank Masaru Ito and Akira Shinohara for sharing unpublished results, Maria Jasin for Swsap1 mice, Qinghua Shi for the guinea-pig anti-DMC1 antibody. We thank Thomas Robert for critical reading of the manuscript. We thank the following Montpellier Biocampus facilities for their service: Anne Sutter and the animal facility (RAM) for animal care, Manon Leportier for managing our mouse strains, the Réseau d’Histologie Expérimentale de Montpellier (RHEM) for histology. We acknowledge the support of Marie-Pierre Blanchard for help with STED microscopy and the imaging facility MRI, member of the national infrastructure France-BioImaging infrastructure supported by the French National Research Agency (ANR-10-INBS-04, “Investments for the future”). We are grateful to Xavier Veaute from the CiGEX Platform (CEA, Fontenay-aux-Roses), and to the https://www.france-bioinformatique.fr/ and the https://www.france-bioinformatique.fr/fr/cluster for providing computing resources on Galaxy France.
This work was supported by the Agence Nationale de la Recherche ANR FIRE (ANR-17-426 CE12-0015) to F.B., P.D., J.B.C. and R.K.; by the Ligue contre le Cancer (Comité départemental de l’Hérault and Comité départemental du Gard) to F.B.; by the Ligue contre le Cancer (MB/CB/070-22) to R.K. A.Z. was funded by ANR FIRE (ANR-17-426 CE12-0015) and a Ph.D. fellowship FDT202106012805 from the Fondation pour la Recherche Médicale (F.R.M.).
Author information
Authors and Affiliations
Contributions
A.Z., P.D., R.K., R.M., and F.B. conceived and designed the experiments. RM identified FIRRM as the mouse homolog of plant FLIP. A.Z. performed most mouse experiments, S.B. and F.B. performed some mouse experiments. A.Z. and F.B. interpreted and analyzed mouse data with input from B.d.M. P.D., V.R., R.K. performed, analyzed and interpreted biochemical experiments with contribution from J.B.C. J.C. developed the method for image analysis. P.A. and J.A.J.C. developed the bioinformatic pipeline for analyzing SSDS data. J.A.J.C. and F.B. performed bioinformatic analysis of SSDS data. A.Z., P.D., R.K. and F.B. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gabriel Livera, P. Jeremy Wang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zainu, A., Dupaigne, P., Bouchouika, S. et al. FIGNL1-FIRRM is essential for meiotic recombination and prevents DNA damage-independent RAD51 and DMC1 loading. Nat Commun 15, 7015 (2024). https://doi.org/10.1038/s41467-024-51458-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51458-8
This article is cited by
-
The expanding roles of homologous recombination proteins in genome stability
The EMBO Journal (2026)
-
Insights into the role of hnRNPK in spermatogenesis via the piRNA pathway
Scientific Reports (2025)
-
PCAF-mediated acetylation regulates RAD51 dynamic localization on chromatin during HR repair
EMBO Reports (2025)
