
Abstract
By connecting old and recent notions, different spatial scales, and research domains, we introduce a novel framework on the consequences of brain injury focusing on a key role of slow waves. We argue that the long-standing finding of EEG slow waves after brain injury reflects the intrusion of sleep-like cortical dynamics during wakefulness; we illustrate how these dynamics are generated and how they can lead to functional network disruption and behavioral impairment. Finally, we outline a scenario whereby post-injury slow waves can be modulated to reawaken parts of the brain that have fallen asleep to optimize rehabilitation strategies and promote recovery.
Similar content being viewed by others
Introduction
Brain injury and network disruption
The intricate relationship between brain injuries and behavioral impairment has been a central point of discussion since the dawn of modern neurology. Paul Broca’s first description in 1861 of speech impairment after a frontal lesion prompted the notion that cognitive and behavioral deficits reflect discrete structural damage and neuronal loss in specialized brain regions. However, this localistic view soon proved insufficient. In 1875, Brown-Sequard introduced the idea that deficits could result from “an irritation or inhibition effects provoked in distant areas of the brain by the tissue surrounding the lesion site”1. In 1914, von Monakow extended this notion and formalized the concept of diaschisis2,3,4, as a series of functional alterations that would cause remote dysfunction in structurally intact regions of the brain connected to the site of the lesion.
Following a prolonged period of relative neglect, the interest in the relationships between structural lesions and large-scale functional network disruptions has been recently rekindled by modern neuroimaging techniques. Studies using resting cerebral blood flow and metabolism measurements have shown reduced activity in distant brain regions connected to the lesion site, affecting various pathways and connections5,6. These remote functional effects involve not only changes in task activation but also dynamic coupling between different brain regions or networks during resting conditions7,8,9.
Resting-state functional magnetic resonance imaging (fMRI) studies have shown that an average stroke lesion of about 10 ml causes alterations in about 20% of all brain connections10, reaching up to 50% in patients with severe injuries11. These functional network alterations are typically characterized by two features: hypersynchrony within the ipsilesional hemisphere and decreased interhemispheric coupling9. The location and extent of these network alterations explain the severity of different behavioral deficits (e.g., language, memory, attention, and motor) while their normalization over time relates to the degree of behavioral recovery9,11,12,13.
These findings underscore the relevance of von Monakow’s diaschisis hypothesis to understanding behavioral deficits after brain injuries. Moreover, the striking similarity in connectivity changes across different patients suggests a common, low-dimensional response to injury14. This may represent a tractable problem and a suitable target for novel treatment strategies. However, correcting these network alterations cannot prescind from understanding their neuronal and electrophysiological underpinnings, which, to date, remain elusive.
Brain injury and slow waves
In this context, it is interesting to recall early electrophysiological observations. Already in 1937, William Gray Walter reported an association between structural lesions and slowing of electroencephalogram (EEG), most prominent in the delta frequency range, recorded in awake subjects15. This association was so consistent that, since then, neurologists relied on EEG slow waves to localize focal lesions, deduce their etiology (e.g., ischemic, hemorrhagic, traumatic, and neoplastic), and monitor their progression over time. The intrusion of these slow waves during wakefulness was most pronounced at the site of the lesion but could also extend to the hemisphere opposite the injury. Furthermore, it could be observed beyond the acute phase persisting in the brain of patients in the chronic stage of the disease.
In the 1970s and 1980s, the introduction of non-invasive structural imaging, first with CT and later with MRI, rapidly superseded electrophysiology in the clinical workup of brain-injured patients. Consequently, from the late 1970s onward, the initial focus on EEG slow waves in brain injury diminished, with only a handful of research studies continuing to explore this issue16,17,18,19,20. Hence, the observation of slow waves infiltrating the awake brain after injury was never connected to the notion of diaschisis and to the more recent neuroimaging evidence of widespread network effects.
Integrating long-standing findings into a novel perspective
Here, by connecting old and recent evidence and research domains, we propose that slow waves play a key role in altering functional networks after brain injury. First, we review evidence collected during sleep and anesthesia showing that EEG slow waves reflect transient interruptions of firing in cortical neurons and that the latter disrupt functional networks and behavior. Then, we illustrate how similar dynamics can be generated as a consequence of structural lesions and how they can, in turn, lead to functional network interference and behavioral impairment in brain-injured patients. Finally, we consider the potential protective role of sleep-like cortical dynamics following brain injury and their reversibility, opening new perspectives for treatment and rehabilitation.
The neuronal underpinnings of slow waves
Bistability between ON- and OFF-periods in cortical neurons
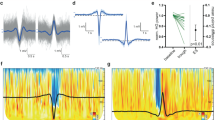
The first pillar of this framework rests on comprehending the neural basis of slow waves. Slow waves refer to a pattern of EEG activity characterized by high-amplitude oscillations in the delta (0.5–4 Hz) band. For decades, these delta waves have been observed during deep sleep, anesthesia, coma, and other neurological conditions, as well as across different species, arguably representing the most preserved physiological brain rhythm21. However, their exact neuronal mechanisms remained obscure until 1993, when Mircea Steriade and his team made a groundbreaking discovery22. Using intracellular recordings, they showed that each EEG slow wave corresponds to a period of profound membrane hyperpolarization and silence in cortical neurons, lasting for a few hundred milliseconds. These interruptions are synchronous across large populations of cortical neurons and occur every second or so, resulting in high-amplitude scalp EEG voltage fluctuations in the delta range (as illustrated in Fig. 1A).

A Simultaneous EEG and intracellular recordings. EEG slow waves correspond to the alternation between periods of depolarization and tonic firing (Up-states) sustained by synaptic activity and reverberation, and periods of deep hyperpolarization and silence (down-states) brought about by adaptation and inhibition in cortical neurons. B Neuronal Up- and Down-states can be inferred extracranially by computing the spectral power of high-frequency (>20 Hz) EEG activity. Specifically, the alternation between Up- and Down-states across large populations of cortical neurons (called ON and OFF-periods, respectively) is reflected at the scalp EEG level in the alternation between slow positive potentials crowned by fast oscillations (red color in time and spectral plots) and negative slow potentials devoid of fast frequencies (blue color in time and spectral plots), respectively. C Cortical bistability may remain a latent variable and can be best revealed by cortical perturbations. Regardless of the presence (left) or the absence (right) of EEG slow waves in ongoing traces, increasing cortical activity with direct stimulations during sleep and anesthesia readily reveals the presence of slow waves and the associated down-states.
It is now widely accepted that EEG slow waves reflect the alternation between two opposite levels of cortical activity: wakefulness-like periods of depolarization with tonic firing (known as Up-states or ON-periods) and silent periods of deep hyperpolarization (known as Down-states or OFF-periods). By virtue of this switch between two extreme states with no intermediate levels, neurophysiologists often use the term “cortical bistability” to describe the cellular underpinnings of slow waves. Following its discovery by Steriade and his colleagues, cortical bistability was soon recognized as a fundamental pattern of network activity across various conditions, species, cortical areas, and spatial scales, earning the status of “default activity pattern of cortical networks”23.
The local mechanisms of cortical bistability
Cortical bistability, due to its robust and widespread nature, appears to be governed by relatively simple intrinsic mechanisms within local cortical circuits. Accordingly, experimental data and computer models have converged on the importance of a few key dynamical elements, including activity reverberation, activity-dependent adaptation, and endogenous noise (Fig. 1A). During the ON-period, recurrent synaptic interactions enable sustained firing reverberation. At the same time, activity-dependent adaptation progressively builds up. This adaptation primarily results from hyperpolarizing Na+-dependent and Ca++-dependent K+ currents, although other activity-dependent processes, such as GABAergic inhibition, ATP-dependent K+ current, and the Na+/K+ electrogenic pump, may also play a role24,25. As adaptation reaches a critical level during the ON-period, it destabilizes synaptic reverberation, leading to the transition to the OFF-period, characterized by hyperpolarization and suppressed firing26. In the space of a few hundred milliseconds, the relaxation of adaptation together with the influence of endogenous noise, resulting from random firing27 or synaptic release28, facilitate the subsequent transition from the silent OFF-period to the next active ON-period29. In essence, a key determinant of cortical bistability is the presence of strong activity-dependent adaptation mechanisms that prevent neurons from sustaining activity and plunge them into transient silent OFF-periods.
The conditions promoting cortical bistability
How can cortical bistability be instantiated during the physiological transition from wakefulness to sleep or anesthesia? Upon falling asleep, adaptation by activity-dependent K+ currents30,31 is enhanced by decreased levels of neuromodulation from brainstem activating systems32. During non-rapid eye-movement (NREM) sleep, bistability can also be contributed by shifts in the excitation/inhibition balance due to “disfacilitation” associated with reduced synaptic input33 or to the inhibitory action of parvalbumin and somatostatin expressing neurons, which shapes the onset and duration of the OFF-period34,35. Similar mechanisms are at work during general anesthesia with different compounds influencing the relative impact of enhanced K+ currents and increased inhibition over excitation. For example, xenon largely increases adaptation by opening K+ channels, while propofol acts by strongly enhancing GABAergic tone36. An extreme version of these neuromodulation changes, leading to an activity pattern dominated by bistability and slow waves, occurs in different surgical models of cortical deafferentation, such as cortical slices, slabs, and isolated gyri16,28,37. Here, the complete severing of ascending white matter fibers totally deprives cortical circuits of the activating neuromodulatory milieu (e.g., acetylcholine or norepinephrine) that normally reduces K+ currents. At the same time, the interruption of a critical amount of long-range lateral excitatory connections may shift the excitation/inhibition balance towards the latter.
Detecting cortical bistability from scalp recordings
In humans, the occurrence of neuronal OFF-periods can be inferred by time- and frequency-domain analysis of EEG graphoelements. As shown by simultaneous EEG and intracranial measurements in animal models and humans, neuronal hyperpolarization during the OFF-period is associated with the negative polarity phase of delta waves recorded at the cortical surface or at the scalp with classic sleep montages. Further, the absence of firing and postsynaptic potentials during this phase can be detected from the same signal as a concomitant suppression of high-frequency (>20 Hz) oscillations38. Conversely, the presence of fast oscillatory activity, typically associated with the synchronous spiking activity of local neuronal populations39 characterizes the active neuronal ON-periods40. Consequently, the alternation between ON- and OFF-periods is reflected at the scalp level by slow positive waves crowned by fast oscillations, followed by negative slow waves devoid of fast frequencies (Fig. 1B). These EEG features are common to stable delta wave patterns and isolated events like spontaneous and sensory evoked K-complexes41.
Cortical bistability impairs network interactions during sleep and anesthesia
The second pillar of this Perspective is the evidence that once cortical bistability is established, it can significantly disrupt network interactions in brain circuits that are structurally intact. Functional connectivity analyses using fMRI and EEG resting-state recordings reveal network disruptions during the transition from wakefulness to states characterized by high-amplitude slow waves, such as NREM sleep and general anesthesia42. More directly, a key role of bistability in impairing large-scale neuronal communication is demonstrated by studies probing brain responsiveness to cortical stimulations. As illustrated in Box 1, perturbational approaches, whereby cortical circuits are challenged by direct stimulation, are ideally suited to reveal activity-dependent mechanisms, bistability, and their disruptive effects on network interactions.
During wakefulness, stimulating the cortex with transcranial magnetic stimulation (TMS) triggers a chain of fast recurrent waves of activity and long-range interactions, resulting in highly complex spatiotemporal network dynamics (Fig. 2A). During both deep NREM sleep and anesthesia, such distributed and rich spatiotemporal dynamics are lost, and the initial cortical activation is followed by a response that is local and stereotypical43,44,45,46. Crucially, this response is constituted by a slow negative wave47 associated with significant suppression of high-frequency activity and matches the EEG criteria for a full-blown OFF-period, the hallmark of cortical bistability (Fig. 2B). This tendency of cortical circuits to plunge into a silent period upon being activated during NREM sleep can also be assessed intracranially by combining intracortical electrical stimulation and local field potential (LFP) recordings in humans48,49.

The presence of bistability during NREM sleep impacts the ability of cortical circuits to engage in stable, reciprocal interactions and explains the breakdown of effective connectivity and network complexity. A During wakefulness, direct cortical stimulations with TMS trigger a chain of recurrent waves of activity and long-range interactions, resulting in widespread EEG spatiotemporal dynamics (left column). This is quantified by a broadband increase in power at the stimulated site, a long-lasting phase locking, and high values of PCI (right column). B During NREM sleep (and general anesthesia with various anesthetics), such distributed, rich spatiotemporal dynamics is lost (left column), and cortical activations are characterized by a local, slow response associated with a significant suppression of high-frequency power >20 Hz (matching the EEG criteria for a full-blown OFF-period), a short-lived phase locking, and low values of PCI (right column). For both panels, EEG activity is presented from six representative electrodes (yellow disks) uniformly distributed along the antero-posterior axis of the two hemispheres. For both panels, the red trace highlights the EEG recorded from the channel closest to the TMS coil.
Both TMS-EEG and studies combining intracortical electrical stimulation and LFP recordings in humans show that the disruptive effects brought about by OFF-periods go well beyond a mere interruption of activity. Indeed, as indicated by phase-locking analysis, after the OFF-period, neuronal activity resumes as a stochastic process that retains no causal relationship with the initial input. This change in the input-output properties of cortical circuits, whereby neurons hush and then forget, has a dramatic impact on the ability of cortical circuits to engage in stable, reciprocal interactions. This explains the breakdown of effective connectivity and network complexity characterizing NREM sleep and anesthesia43,50. These changes can be quantified by a synthetic measure, called the perturbational complexity index (PCI44,45; Box 1 and Fig. 2B).
The link between OFF-periods and the breakdown of neuronal interactions has been corroborated and further explored in animal models and in reduced preparations. Similar to humans, experiments employing electrical intracortical stimulation and mesoscale recordings in rodents demonstrated OFF-periods, loss of causal interactions, and disruption of network complexity (quantified by PCI) across a wide range of conditions, including NREM sleep and sevoflurane/dexmedetomidine anesthesia51 as well as propofol and isoflurane anesthesia52,53. The above measurements point to a strong correlation between the occurrence of OFF-periods and the disruption of effective network interactions; however, firmly establishing a causal role of bistability requires specific manipulations. In this vein, experiments in cortical slices have shown that the breakup of complex cortical interactions can be reverted by pharmacological manipulations that specifically reduce adaptation and the occurrence of OFF-periods54,55.
The same chain of events has been reproduced by computer simulations. At a local scale, a mean field model simulation of coupled cortical modules has shown that progressively increasing the strength of adaptation can faithfully reproduce the alteration of cortical responsiveness empirically observed in humans, including OFF-periods and the ensuing breakdown of causal interactions56. At a global level, the detrimental effects of increased adaptation on the emergence of global network dynamics have been assessed in large-scale models of brain responsiveness based on the human connectome57.
Collectively, evidence from multiple levels of investigation suggests that bistability and the associated tendency of cortical neurons to enter an OFF-period upon receiving input, can disrupt large-scale neuronal interactions. Importantly, this functional blockage occurs in structurally intact circuits and can be reversible (e.g., upon spontaneous awakening or by pharmacological modulation).
Sleep-like cortical bistability during wakefulness in brain-injured patients
In the context of the original notion of diaschisis, the evidence that cortical bistability during NREM sleep and anesthesia can lead to a dramatic impairment of neuronal interactions, and that this process is dynamic and potentially reversible, is clearly very relevant. In this section, we examine recent literature demonstrating that neuronal dynamics akin to those observed during physiological sleep can manifest during wakefulness after brain injuries with different etiologies, and that this can have important consequences for brain networks and behavior in neurological patients. This evidence will also justify the rationale for using the term “sleep-like” when referring to pathological bistability during wakefulness; the term reflects the evidence that cortical bistability after brain lesions shares the same extracellular proxies (i.e., period-amplitude and spectral features) of those observed in sleep, and that it exerts comparable effects on network interactions.
Widespread cortical bistability in the unresponsive wakefulness syndrome
One of the most dramatic consequences of brain injury is the vegetative state (VS)58, also known as unresponsive wakefulness syndrome (UWS)59, as patients lay open-eyed showing no behavioral signs of consciousness. Despite severe damage, patients with UWS can retain parts of the thalamocortical system that are structurally intact, spontaneously active, and reactive to sensory stimuli. However, their brain activity is marked by disrupted functional networks, as shown through resting-state fMRI60. Additionally, EEG studies indicate the presence of slow waves during periods of eyes opening, resembling those seen when these patients are asleep with their eyes closed61,62. Connecting these pieces of evidence within the present framework raises the question of whether the functional network disruption characterizing UWS may be due to a massive intrusion of sleep-like cortical bistability during behavioral wakefulness.
This question was addressed systematically in a series of experiments employing MRI-guided TMS-EEG in UWS. These measurements revealed a significant breakdown of long-range recurrent interactions63 and network complexity (i.e., PCI45; Fig. 3A), which was observed even when targeting portions of the cerebral cortex that were macroscopically intact as well as electrically active and reactive64. A direct comparison of cortical responses in awake patients with UWS to those observed in healthy sleeping individuals uncovered clear signs of cortical bistability65. Like sleeping subjects, open-eyed patients with UWS exhibited stereotypical slow waves and suppressed high-frequency activity indicating the occurrence of OFF-periods during the awake state. These silent periods disrupted causal interactions and prevented the emergence of complex network dynamics. Moreover, OFF-periods were invariably found in every stimulated cortical region, suggesting that cortical bistability is diffusely engaged in the residual brain of these patients (Fig. 3A). Such global intrusion of sleep-like cortical dynamics during wakefulness and the subsequent disruption of network interactions may represent an extreme form of diaschisis associated with behavioral unresponsiveness and loss of consciousness.

A TMS-EEG in unresponsive wakefulness syndrome patients shows the presence of bistability impacting the ability of cortical circuits to engage in stable, reciprocal interactions and explains the breakdown of effective connectivity and network complexity. As in NREM sleep, due to the presence of severe multifocal brain injuries (black shades), cortical responses to TMS in awake patients with UWS are characterized by a dramatic breakdown of long-range recurrent interactions and spatiotemporal dynamics (left column). Locally, these responses match the electrophysiological criteria for the detection of an OFF-period (i.e., the presence of a stereotypical slow wave, associated with a suppression of high-frequency power >20 Hz), and are invariably found in every stimulated cortical region (right column). B The clinical transition from UWS (left column) to the recovery of functional communication (right column) is characterized by a disappearance of the OFF-period, paralleled by an increase in the duration of causal interactions (as indexed by phase-locking measures) and, in turn, by a recovery of complex spatiotemporal dynamics, reflected in high values of PCI. C EEG slowing is often present in areas surrounding focal cortical lesions in awake stroke patients. D The application of TMS reveals local full-fledged signs of sleep-like cortical bistability (i.e., the presence of local stereotypical slow wave, associated with the suppression of high-frequency power >20 Hz) over perilesional areas (left and middle column) that are not detectable when stimulating the same cortical region over the contralesional hemisphere (right column). For all panels, EEG activity is presented from six representative electrodes (yellow disks in panels A, C, D) uniformly distributed along the antero-posterior axis of the two hemispheres. The red trace in panels A, B, D highlights the EEG recorded from the channel closest to the TMS coil.
Reduction of cortical bistability and recovery of consciousness
To further emphasize the clinical significance of cortical bistability, longitudinal TMS-EEG measurements revealed a gradual reduction of sleep-like dynamics in the cortex of UWS patients paralleling recovery of consciousness and behavioral responsiveness65; in these cases, the duration and depth of the OFF-period decreased progressively while the duration of causal interactions increased together with the complexity of network interactions, as measured by PCI (Fig. 3B). A similar reduction in sleep-like responses, accompanied by increased signal complexity, was observed in patients recovering consciousness after traumatic brain injury using intracranial stimulation and recordings66. Collectively, this evidence is relevant as it underscores that while widespread cortical bistability can have a dramatic impact on networks and behavior, this process is dynamic and potentially reversible.
It is noteworthy that the progressive reduction of bistability in patients who recover consciousness does not occur uniformly across the cortical areas spared by structural lesions. For example, in minimally conscious patients, cortical stimulation effectively engages complex network interactions, as indicated by the detection of high PCI values consistent with preserved consciousness45,64,67. However, mapping the reactivity of different cortical areas, especially those adjacent to lesions, often indicates the persistence of foci of bistability68. This mixed condition, characterized by the presence of stereotypical sleep-like responses alongside complex cortical dynamics resembling those recorded during typical wakefulness, aligns with the clinical presentation of patients displaying mixed behavioral signs, such as fluctuating responsiveness coupled with severe cognitive and motor impairment61. Furthermore, the piecemeal intrusion of sleep-like dynamics suggests the existence of specific topographical relationships between structural damage and cortical bistability.
Cortical bistability after focal stroke
As compared to multifocal and traumatic injuries, focal lesions provide a more suitable model for assessing the relationship between the pattern of structural injury and the spatial distribution of cortical bistability. The presence of local slow waves around focal lesions during wakefulness has been observed since the early days of EEG15(Fig. 3C). More recently, these alterations have been assessed through quantitative analysis, such as spectral analysis, singular value decomposition, and delta/alpha ratio. These investigations have revealed EEG slowing topologically consistent with the cortical areas affected by stroke in patients with either motor deficits or neglect69,70. However, the explicit link between post-stroke functional alterations and sleep-like cortical bistability was only established in 2020 by two independent TMS-EEG studies in stroke patients with unilateral focal cortico-subcortical ischemic lesions68,71.
These studies revealed the presence of full-fledged sleep-like responses to TMS in awake stroke patients, marked by a prominent slow wave and suppression of high-frequency activity over the perilesional areas (Fig. 3D). Notably, these perilesional OFF-periods were associated with the disruption of local cortico-cortical interactions68 and linked to the individual patient’s deficit71. Similar to severe brain injuries, also in the case of focal cortical injuries, the reduction of perilesional sleep-like cortical dynamics was associated with the recovery of local cortical interactions. Importantly, their reversibility was proportional to the patients’ clinical improvement72. Further, TMS-EEG measurements revealed the presence of cortical bistability even in those cases where perilesional slow waves were not immediately observable in the ongoing EEG68. This finding corroborates the notion that bistability can be a latent, albeit functionally relevant, variable that can be effectively revealed by direct cortical perturbations (Fig. 1C and Box 1) also in pathological conditions.
Besides the full-fledged signs of bistability characterizing cortical areas located in the proximity of anatomical lesions, a significant slowing of the EEG response to TMS has also been observed when targeting subsets of cortical regions at a distance, both within the ipsilesional and the contralesional hemispheres68,73. These distant, more nuanced neurophysiological alterations, resembling those found during the early stages of sleep43, suggest larger-scale functional effects. Recent findings in post-stroke delirium, characterized by fluctuating disturbances in attention, awareness, and cognition, further support this view. Post-stroke delirium patients not only exhibit diffuse EEG slowing at the scalp level but also display slower cortical responses at different cortical sites and a global impairment of network interactions, as revealed by TMS-EEG74. Given the variety in lesion size, affected hemisphere, or stroke severity in these patients, the consistent finding of such global network impairment is highly suggestive of significant functional disruption at a distance from the lesion. It is thus important to study the extent of perilesional bistability, its propagation, and its large-scale effects on neuronal activity at a finer granularity.
Perilesional cortical bistability and its intracranial percolation
The propagation of postlesional slow waves was recently investigated using mesoscale intracerebral stereo-EEG (SEEG) recordings75. This approach examined electrophysiological changes induced by controlled surgical lesions using radiofrequency-thermocoagulation (RFTC) with a radius of a few millimeters76. Overcoming methodological challenges typical of stroke studies, such as low spatial resolution in scalp EEG recording and the lack of baseline recordings before the injury, this approach provided valuable insights into perilesional cortical bistability and its effects at distant sites.
In this study75, post-lesion intracerebral activity during wakefulness featured prominent slow waves over a perilesional area extending up to a radius of 28 mm, roughly ten times larger than the structural lesion (Fig. 4). These slow waves were associated with OFF-periods and closely resembled the events recorded during NREM sleep before RFTC in the same individuals. Remarkably, similar slow waves were observed at distant cortical sites, extending beyond the perilesional area by up to 60 mm (Fig. 4). Importantly, the percolation of slow waves across remote cortical areas was not solely determined by geometrical distance but significantly correlated with individual patterns of long-range effective connectivity, as assessed through cortico-cortical responses to single-pulse electrical stimulation before RFTC. This intrusion and long-range percolation of sleep-like slow waves at specific network nodes provide direct electrophysiological support for the original hypothesis of remote functional interference in regions connected to the focal lesion3 through cortico-cortical and/or subcortico-cortical connections.

Mesoscale exploration of the effects of controlled surgical lesions performed in epileptic patients using radiofrequency-thermocoagulation (RFTC). RFTC (recorded with Stereo-EEG; SEEG) is followed by spontaneous sleep-like slow waves during wakefulness in cortical areas surrounding the lesion (black shade). This perilesional area (colored blue circles) showing prominent post-RFTC slow waves has a radius of about 30 mm, corresponding to the length of four adjacent SEEG bipolar contacts. The number and amplitude of RFTC-induced slow waves is maximal in close proximity to the lesion and rapidly decays with distance (color-coded with a deep-to-light blue gradient). Like slow waves typically found during physiological NREM sleep, these perilesional slow waves are associated with a suppression of high-frequency power >20 Hz (bottom left). Most importantly, post-RFTC slow waves can also be found at SEEG contacts (color-coded blue circles) that are distant (up to 60 mm) from the lesion but connected to it.
Towards an integrated framework on the functional consequences of brain injury
We have reviewed evidence of sleep-like cortical bistability intrusion during wakefulness following brain injury. Below, we integrate this evidence with a larger body of old and recent literature to provide the elements of a systematic framework linking structural damage to the generation of postlesional slow waves, their spread, and their effects on brain networks and behavior.
Mechanisms of postlesional slow waves
As previously discussed, the basic physiological mechanisms responsible for the engagement of cortical bistability and slow waves during sleep and anesthesia have been systematically investigated. However, the factors leading to their intrusion during wakefulness after brain injuries have not been explored. Below, we describe how different events associated with structural damage may converge to engage bistability and slow waves during wakefulness.
The first class of events involves brainstem and midbrain lesions or compressions resulting from expanding intracranial processes, impacting the ascending activating systems (Fig. 5, upper panel). This leads to decreased wake-promoting neuromodulation and increased activity-dependent adaptation mechanisms, predominantly mediated by various K+ currents in cortical neurons24,25. These effects explain the EEG slowing typically observed in clinical conditions such as unarousable coma and UWS77. Similar situations, where bistability massively affects cortical circuits in behaviorally awake (open-eyed) subjects, may occur in the case of diencephalic, typically bilateral thalamic, lesions78 or following lesions higher up in subcortical white matter. For example, diffuse axonal injury of traumatic etiology79 can interrupt a critical mass of fibers of the ascending activating systems, leading to a predominance of adaptation currents and bistability across widespread cortical areas32. Similarly, focal subcortical lesions may also interrupt ascending fibers leading to spatially restricted foci of adaptation currents and bistability in specific cortical areas80.

Many events associated with structural brain damage can converge in engaging cortical bistability during wakefulness. The first class of events involves the enhancement of activity-dependent adaptation mechanisms mediated by Ca++- and Na+-gated K+ currents in cortical neurons (top panel). These include brainstem and midbrain lesions or compressions (left), subcortical nuclei and/or white matter lesions (middle), as well as diffuse axonal injury of traumatic etiology (right). In all these instances, the influence of wake-promoting neuromodulators (e.g., cholinergic and noradrenergic) is strongly decreased, and in some cases, virtually absent. This leads to a predominance of adaptation currents in cortical areas downstream of the structural lesion. The second class of events involves a critical loss of lateral excitatory fibers, which can bring areas that are near or connected to the lesion site into a state of disfacilitation, whereby local inhibition prevails over projected excitatory influences (bottom panel). This occurs in the case of cortico-subcortical structural lesions, which can affect both local (left) and long-range (middle) cortico-cortical connections, as well as in the case of diffuse axonal injury, involving lateral fibers (right). In addition to structural disconnections, other mechanisms, such as acute edema and inflammatory processes (left panel), can lead to the generation of postlesional slow waves, via the local activation of microglia and astrocytes together with the expression of proinflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNFɑ). Finally, metabolic events induced by ischemia, such as the reduction of the ATP/ADP ratio (right panel), strongly activate K-ATP sensitive channels causing membrane hyperpolarization and suppression of activity in local neuronal populations surrounding the lesion. All these events, alone or in combination, may converge in favoring OFF-periods and slow waves during wakefulness in cortical areas topologically related to the site of injury.
The second class of events involves lesions affecting cortico-cortical connections, leading to a loss of lateral excitatory effects, creating a state of disfacilitation, with local inhibition prevailing over excitation (Fig. 5, lower panel). This mechanism aligns with the long-standing empirical evidence of sleep-like slow waves in the tissue surrounding focal cortical lesions. Since the local distribution of excitatory lateral connections is known to follow a spatial rule with exponential decay, these pathological slow waves are expected to be more prominent closer to the lesion and to decay quite steeply with distance81,82, as empirically observed by high-resolution intracranial explorations after RFTC75. The same mechanisms may account for the electrophysiological penumbra of sleep-like reactivity detected by TMS-EEG measurements in the perilesional area of stroke68,71. Similar disfacilitation effects may also occur in distant areas that are downstream to the cortical site of injury, or by the interruption of cortico-cortical fiber bundles brought about by focal white matter lesions or diffuse axonal injury. In all these cases, a critical deprivation of excitatory inputs to the disconnected area may shift the excitation/inhibition balance towards the latter, leading to cortical OFF-periods and slow waves similar to those observed in sleep34,35,83. Consistent with this view, a relative increase in tonic inhibition has been reported in a mouse model of focal ischemic stroke, and its pharmacological dampening produced an early and sustained recovery of motor function84.
These two classes of mechanisms—increased adaptation by severing of ascending activating fibers and increased inhibition by disruption of lateral connections—are not mutually exclusive and can coexist, depending on the lesion’s extent and location. Indeed, lesions in the white matter regions containing a high number of fibers (presumably, both ascending and lateral) tend to cause more numerous and more severe neurological deficits post-stroke, as well as more cortical functional connectivity abnormalities10,85.
In addition to structural disconnections, other mechanisms, such as acute edema and inflammatory processes, can facilitate the generation of postlesional slow waves (Fig. 5, left panel). For example, in animal models86,87, edema can lead to a failure of Na+/K+ electrogenic pumps and ionic imbalances that facilitate the expression of slow EEG activity88. Also, the expression of proinflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNFɑ), has been reported locally as well as in areas connected to the lesion following ischemic injury89,90. Notably, intracortical unilateral microinjections of both IL-1 and TNFɑ result in acute and lateralized increase in the number of EEG slow waves in rodents91,92. In addition, slow waves can be promoted by the activation of microglia, which has been reported to reduce norepinephrine transmission93. Consistent with the above, slow waves and EEG slowing can be observed in brain tumors15 and multiple sclerosis94, two conditions characterized by focal edema and neuroinflammation, besides structural disconnection.
Finally, the reduction of the ATP/ADP ratio following ischemia is known to strongly activate K-ATP sensitive channels, causing membrane hyperpolarization and suppression of activity in local neuronal populations surrounding the lesion (Fig. 5, right panel)95. This phenomenon, although initially neuroprotective, may have prolonged functional consequences depending on the length of the hypoxic challenge, potentially contributing to the persistence of perilesional slow waves in the sub-acute and chronic phases.
In summary, various pathological events associated with brain injury can converge to favor OFF-periods and slow waves during wakefulness in cortical areas related to the injury site. Future studies should systematically investigate the specific impact of these events, their interactions, and their relevance in both the acute and chronic stages after injury.
Propagation of postlesional slow waves
The mechanisms described above can explain the local generation of cortical bistability in areas adjacent or directly connected to the site of structural injury. However, post-lesion slow waves may also spread across brain networks as indicated by both scalp EEG20,96 and intracranial recordings75 (Fig. 4).
This phenomenon can be explained by two mechanisms. The first possibility is that remote neurons become strongly disfacilitated because they receive less excitatory input from a perilesional area engaging in frequent silent OFF-periods. In this case, a shift in the balance between reduced incoming long-range excitation and preserved local inhibition may favor bistability at distant connected sites. The second possibility is that slow waves generated in perilesional areas may actively propagate to distant connected sites. This hypothesis is supported by the evidence that, during sleep, physiological slow oscillations can behave as traveling waves through cortico-cortical and cortico-thalamocortical connections. Such long-range propagation has been shown by scalp97, intracranial98 EEG recordings in humans, in animal models99 as well as in cortical slices100. Sleep slow waves were found to travel at speeds compatible with estimates of conduction velocities derived from tractography and cortico-cortical evoked potentials in humans101, and preferentially propagate along the anterior-posterior axis through the major connectional backbone of the cortex and involving many areas of the default network102.
Both distant disfacilitation and active traveling may coexist and reinforce each other in extending cortical bistability beyond the perilesional area. In principle, the relative contribution of each mechanism could be assessed experimentally. Distant slow waves are expected to occur out-of-phase with respect to those recorded near the lesion in the case of local generation due to disfacilitation, while phase-locking and consistent time lags should be observed in the case of active traveling. Furthermore, in the case of disfacilitation, slow waves should be confined to areas strongly connected to the perilesional site, whereas active traveling would affect a larger network through polysynaptic chains.
The likelihood that perilesional slow waves may actively travel and interfere with large-scale network activity is supported by a recent study in rodents103. Chemogenetic inactivation of the prefrontal cortex, mimicking local deafferentation produced by anatomical lesions, led to the generation of prominent delta waves near the site of inactivation. These waves propagated with consistent time lags and an anterior-to-posterior direction to a broad network of directly and indirectly connected regions, resulting in the enhancement of inter-areal coherence in the low-frequency (delta) band across this distributed network.
Postlesional slow waves and alterations in fMRI networks
Remarkably, the same study also revealed interesting relationships between postlesional slow wave propagation and fMRI network alterations. Using combined electrophysiological and fMRI recordings, the authors found that the spatial distribution of slow wave propagation corresponded to functional overconnectivity in resting-state networks. This pattern strongly resembled the abnormally strong intrahemispheric correlation found in the imaging studies of stroke patients9,12.
Whether the perilesional generation and propagation of slow waves in stroke patients can explain the fMRI signal hypersynchrony typically found within the affected hemisphere and its decoupling from the contralateral hemisphere is an open question. TMS-EEG measurements have hinted at the existence of this type of network effect, with strongly asymmetric responses characterized by stereotypical slow waves around the lesion site68 and alterations in interhemispheric interactions104. On the other hand, concurrent changes in EEG and BOLD signals have already been documented in physiological conditions. For example, sleep deprivation studies have demonstrated that the intrusion of slow waves into the awake brain is linked to alterations in functional connectivity patterns105, leading to lapses in attention, impaired working memory, and reduced cognitive performance106.
Future research should investigate whether the spatial pattern of slow wave propagation matches the specific topology of fMRI network disruption in stroke patients. For example, one could expect concurrent fronto-parietal and occipital slow wave propagation and fMRI alterations in patients with unilateral spatial neglect following right perisylvian lesions11,107 or left occipito-temporal reading-specific regional propagation in the case of aphasic stroke patients108.
Postlesional slow waves and behavioral impairment
Though never explicitly connected to the topic of brain injury, a consistent body of literature has recently demonstrated that the local intrusion of bistability within the awake brain in physiological conditions can lead to selective impairments in perception, motor function, and cognition. For instance, studies in sleep-deprived rats demonstrated that isolated slow waves and cortical OFF-periods over the motor cortex predict transient failures in a pellet-reaching task109, as well as specific changes in sensory processing when triggered by sounds in early auditory cortices110. Similarly, intracranial recordings in sleep-deprived humans showed low-frequency activity and suppressed neuronal firing in the medial temporal cortex, corresponding to lapses in a simple reaction time task involving stimuli coded by those neurons111.
High-density EEG recordings have provided further evidence that the topography of local bistability during wakefulness predicts performance impairment. For example, sleep-like EEG dynamics over the frontal and parietal cortex predict poor impulse control and visuomotor performance, respectively112. In a study involving a sustained attention task during normal wakefulness, the topography of slow waves predicted the timing and type of attentional lapses, with frontal slow waves linked to a higher rate of false alarms, while posterior slow waves were associated with slower reaction times and misses. The co-occurrence of slow waves across scalp electrodes also suggest that they propagate between cortical regions along a preferred antero-posterior axis113.
The subtle behavioral effects of cortical bistability observed in healthy subjects are likely to become prominent in brain-injured patients. In such cases, OFF-periods can encompass large neuronal populations, leading to fully-fledged EEG sleep-like slow waves that propagate over distance and disrupt the complexity of neuronal interactions. The clinical consequences of these electrophysiological disturbances may vary from complete unresponsiveness to specific cognitive-motor deficits. However, to date, the intrusion of slow waves has not been explicitly connected to behavioral symptoms in neurological patients. Thus, it will be critical to systematically assess the impact of the topography and timing of these electrophysiological events on the nature and time course of post-injury cognitive-motor deficits.
Potential benefits of postlesional slow waves
Cortical bistability and slow waves during natural sleep are known to play pivotal roles in restorative and homeostatic processes114. Slow waves during sleep are associated with reduced cerebral metabolism, energy conservation115, and promote the clearance of extracellular metabolic waste products through their coupling with cerebrospinal fluid flow116. Additionally, they are strongly linked to synaptic remodeling117 and the homeostatic regulation of cortical excitability118.
Emerging evidence suggests that slow waves during NREM sleep have beneficial effects on recovery after brain injury119. In fact, reciprocal interactions between sleep and brain injury have been described at various levels120 (Box 2). However, the role of slow waves during wakefulness following brain injury remains unclear. Postlesional slow waves may lack some of the fundamental properties of sleep slow waves, such as their relationship to spindles, their thalamic control, and their tight homeostatic regulation. It is indeed uncertain whether the intrusion of sleep-like dynamics in the awake state merely reflects a regression of injured cortical circuits to a primitive default mode with detrimental network and behavioral effects or also serves some of the physiological functions of local sleep protective or restorative purposes121,122.
In future studies it will be important to investigate whether there are specific critical windows in space and time during which slow waves during wakefulness might aid in preserving or restoring the state of residual circuitry for subsequent recovery. Synchronous slow rhythms are known to be an important organizing force in the formation of new connections in the developing brain and may play a similar role during critical periods after brain injury123. Notably, in a rodent stroke model, slow waves developing during the first 3 days after injury, were found to act as a trigger for long-range axonal sprouting that was maintained over time124. A potential homeostatic role of slow waves during wakefulness has also been proposed in the areas surrounding the epileptogenic zone125. Overall, it is conceivable that sleep-like activity patterns may contribute to anatomical reorganization124, reduce metabolic costs, alleviate excitotoxicity, and mitigate extracellular compartment alterations126, particularly in perilesional areas and during the acute phase. Nonetheless, the distant propagation and chronic persistence of slow waves may result in network interference and behavioral impairment, as we have illustrated in previous sections. Understanding these intricate interactions through longitudinal measurements and specific experimental designs will be crucial for defining optimal intervention strategies. For example, this could involve controlled manipulation of cortical bistability in time and space, allowing slow waves in perilesional areas during the acute phase while blocking their propagation to distant sites and their persistence in the chronic phase.
Open questions and implications for treatment
The key tenet of the present framework is that the primary structural injury is accompanied by secondary disruptions of neuronal activity4,9 that can be dynamic, clinically relevant, but potentially reversible. Specifically, we propose that cortical sleep-like bistability is the fundamental neurophysiological underpinning for these secondary alterations, according to the following sequence of events (Fig. 6A):
-
(I)
The primary structural injury results in a critical deprivation of ascending activating influences and/or lateral excitation in perilesional areas.
-
(II)
An enhancement of adaptation mechanisms, and/or a shift in the excitation/inhibition balance (with the possible contribution of perilesional inflammation and hypoxia) lead to the local expression of sleep-like cortical bistability during wakefulness.
-
(III)
Slow waves can extend further in the brain by propagating to distant but connected circuits where they disrupt neuronal activity in a topologically specific manner.
-
(IV)
The resulting network-level interference impairs specific cognitive and behavioral domains.
-
(V)
While structural damage and disconnections may persist, bistability and its network consequences are potentially reversible.

A schematic illustration of the vision encompassing a key role of cortical bistability in the aftermath of brain injury. A The primary structural lesion (black shade) critically deprives perilesional areas of lateral excitation and/or ascending activating influences (green arrows). The ensuing shift of the excitation/inhibition balance and/or the enhancement of adaptation mechanisms (possibly facilitated by hypoperfusion and inflammation) lead to the local expression of sleep-like cortical bistability (graded blue shade) during wakefulness. The alternation between Up- and Down-states in the affected neuronal population is reflected in slow waves during wakefulness in the scalp EEG. Bistability and slow waves can then propagate either by disfacilitation of distant targets or through chains of connected circuits and disrupt neuronal activity in distant areas (blue shades of different intensity in cortical areas distant from the structural damage connected by red arrows). In this specific example, involving the network illustrated in ref. 107, the network-level intrusion of bistability during wakefulness and the resulting disruption of neuronal interactions over the right intraparietal sulcus and right frontal eye field contributes to behavioral deficits (hemispatial neglect, i.e. an attentional bias towards the right hemispace), which are not fully accounted for by the structural damage alone (located over the right temporo-parietal junction). B While structural damage and disconnections are likely to persist, bistability and its network consequences are potentially reversible, leading to a progressive recovery of function over time (t0–t1).
This sequence offers a parsimonious account for a wealth of observations made in brain-injured patients over the past century, encompassing the original evidence of diaschisis and early observations of delta waves post-injury, as well as recent TMS-EEG and fMRI findings demonstrating clinically relevant network alterations. Most importantly, this tentative framework raises new questions and serves as a starting point for further investigation and experimentation in both animal and human models. For example, it remains unknown to what extent post-injury bistability can propagate across polysynaptic chains of connected neurons potentially involving not only cortical, but also subcortical circuits. Supporting this possibility, several subcortical structures (e.g., the brainstem, the thalamus, the claustrum, the basal ganglia, and the cerebellum) are known to be involved in the synchronization127,128 as well as the propagation129,130 of physiological cortical sleep slow waves. Addressing this question, will benefit from mesoscale intracerebral recordings in humans, as well as microscale intracortical recordings and fiber tracing in animal models. Animal studies will also help systematically investigate whether the intrusion of pathological sleep-like dynamics may more likely occur in specific cell types and cortical layers similarly to what occurs during REM sleep131 and anesthesia132, and whether this may be associated with specific network and behavioral effects.
On the other hand, while animal models have provided convincing evidence of a causal link between slow wave propagation and large-scale resting-state fMRI alterations103, such a relationship has never been systematically investigated in patients with stroke. Along the same lines, although the detrimental effects of slow waves and bistability during wakefulness have been already documented in healthy animals and humans109,111,112,113, research has yet to explore whether the topology and timing of slow waves after brain injury can predict behavioral deficits with spatial and temporal specificity. For example, it would be important to investigate whether fluctuations of delta power over the right fronto-parietal cortex predict changes in the degree of attentional bias in patients with neglect (Fig. 6). Finally, systematic manipulations and longitudinal assessments in rodent models and patients are warranted to understand whether there are critical periods, in which the intrusion of slow waves during wakefulness may play a beneficial role.
Addressing these questions is vital, as the secondary disruption of functional networks after injury may influence the clinical course of countless patients, from those with specific cognitive and motor impairments following a stroke to individuals affected by post-stroke and post-traumatic delirium and disorders of consciousness. The implications are significant because cortical sleep-like dynamics and their network consequences can be spontaneously reversible, not only upon awakening from physiological sleep and anesthesia but also in certain neurological patients65,72. Whether it will be feasible to reliably manipulate post-injury slow waves and their propagation through external interventions remains an open question. Presently, evidence suggests that various tools, including drug administration, opto-pharmacology, optogenetics, transcranial electrical stimulation, and repetitive TMS, can modulate slow waves in simplified models and in vivo133,134,135. New non-invasive technologies, such as temporal interference136, are becoming available to sculpt brain rhythms with unprecedented spatial selectivity, while in silico whole-brain models of individual patients hold the promise of optimizing stimulation targets to renormalize wake-like activity after brain injury137. In parallel, future studies should assess the potential effectiveness of real-time neurofeedback techniques tailored to reducing slow waves in specific areas of interest. The exciting prospect is that rather than employing neuromodulation to induce fine-grained circuit adjustments that are currently difficult to predict and control, reducing slow waves to reawaken circuits that have fallen asleep may represent a viable and effective strategy. The stakes are high because restoring waking patterns in portions of the cortex that are functionally offline due to bistability may simultaneously improve behavioral performance and optimize the potential for targeted rehabilitation to induce specific circuit remodeling.
References
Goetz, C. G. Battle of the Titans: Charcot and Brown-Séquard on cerebral localization. Neurology 54, 1840–1847 (2000).
Carrera, E. & Tononi, G. Diaschisis: past, present, future. Brain 137, 2408–2422 (2014).
Feeney, D. M. & Baron, J. C. Diaschisis. Stroke 17, 817–830 (1986).
von Monakow, C. DIe lokalisation im grosshirn und der abbau der funktion durch kortikale herde. JAMA LXIII, 797–797 (1914).
Baldassarre, A., Ramsey, L. E., Siegel, J. S., Shulman, G. L. & Corbetta, M. Brain connectivity and neurological disorders after stroke. Curr. Opin. Neurol. 29, 706–713 (2016).
Fornito, A., Zalesky, A. & Breakspear, M. The connectomics of brain disorders. Nat. Rev. Neurosci. 16, 159–172 (2015).
Grefkes, C., Eickhoff, S. B., Nowak, D. A., Dafotakis, M. & Fink, G. R. Dynamic intra- and interhemispheric interactions during unilateral and bilateral hand movements assessed with fMRI and DCM. Neuroimage 41, 1382–1394 (2008).
Latifi, S. & Carmichael, S. T. The emergence of multiscale connectomics-based approaches in stroke recovery. Trends Neurosci. 47, 303–318 (2024).
Siegel, J. S. et al. Disruptions of network connectivity predict impairment in multiple behavioral domains after stroke. Proc. Natl Acad. Sci. USA 113, E4367–E4376 (2016).
Griffis, J. C., Metcalf, N. V., Corbetta, M. & Shulman, G. L. Structural disconnections explain brain network dysfunction after stroke. Cell Rep. 28, 2527–2540.e9 (2019).
Baldassarre, A. et al. Large-scale changes in network interactions as a physiological signature of spatial neglect. Brain 137, 3267–3283 (2014).
Siegel, J. S. et al. Re-emergence of modular brain networks in stroke recovery. Cortex 101, 44–59 (2018).
Zhu, D. et al. Changes of functional connectivity in the left frontoparietal network following aphasic stroke. Front. Behav. Neurosci. 8, 167 (2014).
Corbetta, M., Siegel, J. S. & Shulman, G. L. On the low dimensionality of behavioral deficits and alterations of brain network connectivity after focal injury. Cortex 107, 229–237 (2018).
Walter, W. G. The electro-encephalogram in cases of cerebral tumour: (Section of Neurology). Proc. R. Soc. Med 30, 579–598 (1937).
Gloor, P., Ball, G. & Schaul, N. Brain lesions that produce delta waves in the EEG. Neurology 27, 326–333 (1977).
Nuwer, M. R., Jordan, S. E. & Ahn, S. S. Evaluation of stroke using EEG frequency analysis and topographic mapping. Neurology 37, 1153–1159 (1987).
Magnus, O. & Van der Holst, M. Zeta waves: a special type of slow delta waves. Electroencephalogr. Clin. Neurophysiol. 67, 140–146 (1987).
Faught, E. Current role of electroencephalography in cerebral ischemia. Stroke 24, 609–613 (1993).
Buchkremer-Ratzmann, I., August, M., Hagemann, G. & Witte, O. W. Electrophysiological transcortical diaschisis after cortical photothrombosis in rat brain. Stroke 27, 1105–1111 (1996).
Sanchez-Vives, M. V., Massimini, M. & Mattia, M. Shaping the default activity pattern of the cortical network. Neuron 94, 993–1001 (2017).
Steriade, M., Nuñez, A. & Amzica, F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J. Neurosci. 13, 3252–3265 (1993).
Sanchez-Vives, M. V. & Mattia, M. Slow wave activity as the default mode of the cerebral cortex. Arch. Ital. Biol. 152, 147–155 (2014).
Lee, S.-H. & Dan, Y. Neuromodulation of brain states. Neuron 76, 209–222 (2012).
McCormick, D. A. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog. Neurobiol. 39, 337–388 (1992).
Compte, A., Sanchez-Vives, M. V., McCormick, D. A. & Wang, X.-J. Cellular and network mechanisms of slow oscillatory activity (<1 Hz) and wave propagations in a cortical network model. J. Neurophysiol. 89, 2707–2725 (2003).
Chauvette, S., Volgushev, M. & Timofeev, I. Origin of active states in local neocortical networks during slow sleep oscillation. Cereb. Cortex 20, 2660–2674 (2010).
Timofeev, I., Grenier, F., Bazhenov, M., Sejnowski, T. J. & Steriade, M. Origin of slow cortical oscillations in deafferented cortical slabs. Cereb. Cortex 10, 1185–1199 (2000).
Camassa, A., Galluzzi, A., Mattia, M. & Sanchez-Vives, M. V. Deterministic and stochastic components of cortical down states: dynamics and modulation. J. Neurosci. 42, 9387–9400 (2022).
Sanchez-Vives, M. V. & McCormick, D. A. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 3, 1027–1034 (2000).
Steriade, M., McCormick, D. A. & Sejnowski, T. J. Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685 (1993).
McCormick, D. A. & Williamson, A. Convergence and divergence of neurotransmitter action in human cerebral cortex. Proc. Natl Acad. Sci. USA 86, 8098–8102 (1989).
Steriade, M. Impact of network activities on neuronal properties in corticothalamic systems. J. Neurophysiol. 86, 1–39 (2001).
Funk, C. M. et al. Role of somatostatin-positive cortical interneurons in the generation of sleep slow waves. J. Neurosci. 37, 9132–9148 (2017).
Zucca, S. et al. An inhibitory gate for state transition in cortex. Elife 6, e26177 (2017).
Franks, N. P. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 9, 370–386 (2008).
Nita, D. A., Cissé, Y., Timofeev, I. & Steriade, M. Waking-sleep modulation of paroxysmal activities induced by partial cortical deafferentation. Cereb. Cortex 17, 272–283 (2007).
Mukovski, M., Chauvette, S., Timofeev, I. & Volgushev, M. Detection of active and silent states in neocortical neurons from the field potential signal during slow-wave sleep. Cereb. Cortex 17, 400–414 (2007).
Nir, Y. et al. Coupling between neuronal firing rate, gamma LFP, and BOLD fMRI is related to interneuronal correlations. Curr. Biol. 17, 1275–1285 (2007).
Ruiz-Mejias, M., Ciria-Suarez, L., Mattia, M. & Sanchez-Vives, M. V. Slow and fast rhythms generated in the cerebral cortex of the anesthetized mouse. J. Neurophysiol. 106, 2910–2921 (2011).
Cash, S. S. et al. The human K-complex represents an isolated cortical down-state. Science 324, 1084–1087 (2009).
Tagliazucchi, E. et al. Large-scale brain functional modularity is reflected in slow electroencephalographic rhythms across the human non-rapid eye movement sleep cycle. Neuroimage 70, 327–339 (2013).
Massimini, M. et al. Breakdown of cortical effective connectivity during sleep. Science 309, 2228–2232 (2005).
Sarasso, S. et al. Consciousness and complexity during unresponsiveness induced by propofol, xenon, and ketamine. Curr. Biol. 25, 3099–3105 (2015).
Casali, A. G. et al. A theoretically based index of consciousness independent of sensory processing and behavior. Sci. Transl. Med. 5, 198ra105 (2013).
Comolatti, R. et al. A fast and general method to empirically estimate the complexity of brain responses to transcranial and intracranial stimulations. Brain Stimul. 12, 1280–1289 (2019).
Massimini, M. et al. Triggering sleep slow waves by transcranial magnetic stimulation. Proc. Natl Acad. Sci. USA 104, 8496–8501 (2007).
Pigorini, A. et al. Bistability breaks-off deterministic responses to intracortical stimulation during non-REM sleep. Neuroimage 112, 105–113 (2015).
Matsumoto, R. et al. Functional connectivity in the human language system: a cortico-cortical evoked potential study. Brain 127, 2316–2330 (2004).
Ferrarelli, F. et al. Breakdown in cortical effective connectivity during midazolam-induced loss of consciousness. Proc. Natl Acad. Sci. USA 107, 2681–2686 (2010).
Cavelli, M. L. et al. Sleep/wake changes in perturbational complexity in rats and mice. iScience 26, 106186 (2023).
Arena, A., Comolatti, R., Thon, S., Casali, A. G. & Storm, J. F. General anesthesia disrupts complex cortical dynamics in response to intracranial electrical stimulation in rats. eNeuro 8, ENEURO.0343-20.2021 (2021).
Dasilva, M. et al. Modulation of cortical slow oscillations and complexity across anesthesia levels. NeuroImage 224, 117415 (2021).
D’Andola, M. et al. Bistability, causality, and complexity in cortical networks: an in vitro perturbational study. Cereb. Cortex 28, 2233–2242 (2018).
Barbero-Castillo, A. et al. Impact of GABAA and GABAB inhibition on cortical dynamics and perturbational complexity during synchronous and desynchronized states. J. Neurosci. https://doi.org/10.1523/JNEUROSCI.1837-20.2021 (2021).
Cattani, A. et al. Adaptation shapes local cortical reactivity: from bifurcation diagram and simulations to human physiological and pathological responses. eNeuro ENEURO.0435-22.2023 https://doi.org/10.1523/ENEURO.0435-22.2023 (2023).
Goldman, J. S. et al. Bridging single neuron dynamics to global brain states. Front. Syst. Neurosci. 13, 75 (2019).
Jennett, B. & Plum, F. Persistent vegetative state after brain damage. A syndrome in search of a name. Lancet 1, 734–737 (1972).
Laureys, S. et al. Unresponsive wakefulness syndrome: a new name for the vegetative state or apallic syndrome. BMC Med. 8, 68 (2010).
Demertzi, A. et al. Human consciousness is supported by dynamic complex patterns of brain signal coordination. Sci. Adv. 5, eaat7603 (2019).
Edlow, B. L., Claassen, J., Schiff, N. D. & Greer, D. M. Recovery from disorders of consciousness: mechanisms, prognosis and emerging therapies. Nat. Rev. Neurol. 17, 135–156 (2021).
Forgacs, P. B. et al. Dynamic regimes of neocortical activity linked to corticothalamic integrity correlate with outcomes in acute anoxic brain injury after cardiac arrest. Ann. Clin. Transl. Neurol. 4, 119–129 (2017).
Rosanova, M. et al. Recovery of cortical effective connectivity and recovery of consciousness in vegetative patients. Brain 135, 1308–1320 (2012).
Casarotto, S. et al. Stratification of unresponsive patients by an independently validated index of brain complexity. Ann. Neurol. 80, 718–729 (2016).
Rosanova, M. et al. Sleep-like cortical OFF-periods disrupt causality and complexity in the brain of unresponsive wakefulness syndrome patients. Nat. Commun. 9, 4427 (2018).
Mofakham, S. et al. Electrocorticography reveals thalamic control of cortical dynamics following traumatic brain injury. Commun. Biol. 4, 1210 (2021).
Sinitsyn, D. O. et al. Detecting the potential for consciousness in unresponsive patients using the perturbational complexity index. Brain Sci. 10, 917 (2020).
Sarasso, S. et al. Local sleep-like cortical reactivity in the awake brain after focal injury. Brain 143, 3672–3684 (2020).
Pirondini, E. et al. Resting-state EEG topographies: reliable and sensitive signatures of unilateral spatial neglect. Neuroimage Clin. 26, 102237 (2020).
Lanzone, J. et al. EEG spectral exponent as a synthetic index for the longitudinal assessment of stroke recovery. Clin. Neurophysiol. 137, 92–101 (2022).
Tscherpel, C. et al. Brain responsivity provides an individual readout for motor recovery after stroke. Brain 143, 1873–1888 (2020).
Sarasso, S. et al. The reduction of sleep-like perilesional cortical dynamics underlies clinical recovery in stroke. Preprint at medRXIV https://doi.org/10.1101/2024.03.16.24304272 (2024).
Pellicciari, M. C. et al. Dynamic reorganization of TMS-evoked activity in subcortical stroke patients. Neuroimage 175, 365–378 (2018).
Bai, Y. et al. Cortical reactivity to transcranial magnetic stimulation predicts risk of post-stroke delirium. Clin. Neurophysiol. https://doi.org/10.1016/j.clinph.2022.11.017 (2022).
Russo, S. et al. Focal lesions induce large-scale percolation of sleep-like intracerebral activity in awake humans. Neuroimage 234, 117964 (2021).
Cossu, M. et al. Stereoelectroencephalography-guided radiofrequency thermocoagulation in the epileptogenic zone: a retrospective study on 89 cases. J. Neurosurg. 123, 1358–1367 (2015).
Schaul, N., Gloor, P. & Gotman, J. The EEG in deep midline lesions. Neurology 31, 157–167 (1981).
Schiff, N. D. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann. N. Y Acad. Sci. 1129, 105–118 (2008).
Meythaler, J. M., Peduzzi, J. D., Eleftheriou, E. & Novack, T. A. Current concepts: diffuse axonal injury-associated traumatic brain injury. Arch. Phys. Med. Rehabil. 82, 1461–1471 (2001).
D’Ambrosio, S. et al. Detecting cortical reactivity alterations induced by structural disconnection in subcortical stroke. Clin. Neurophysiol. 156, 1–3 (2023).
Boucsein, C., Nawrot, M. P., Schnepel, P. & Aertsen, A. Beyond the cortical column: abundance and physiology of horizontal connections imply a strong role for inputs from the surround. Front. Neurosci. 5, 32 (2011).
Betzel, R. F. & Bassett, D. S. Multi-scale brain networks. Neuroimage 160, 73–83 (2017).
Zielinski, M. R. et al. Somatostatin+/nNOS+ neurons are involved in delta electroencephalogram activity and cortical-dependent recognition memory. Sleep 42, zsz143 (2019).
Clarkson, A. N., Huang, B. S., Macisaac, S. E., Mody, I. & Carmichael, S. T. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309 (2010).
Corbetta, M. et al. Common behavioral clusters and subcortical anatomy in stroke. Neuron 85, 927–941 (2015).
Clasen, R. A., Cooke, P. M., Martin, F. A., Williams, J. R. & Hass, G. M. Cerebral edema and electroencephalographic changes after local acute closed cerebral injury. AMA Arch. Neurol. Psychiatry 80, 696–707 (1958).
Schaul, N., Ball, G., Gloor, P. & Pappius, H. M. The EEG in Cerebral Edema. in Dynamics of Brain Edema (eds. Pappius, H. M. & Feindel, W.) 144–149 (Springer, 1976).
Rabiller, G., He, J.-W., Nishijima, Y., Wong, A. & Liu, J. Perturbation of brain oscillations after ischemic stroke: a potential biomarker for post-stroke function and therapy. Int. J. Mol. Sci. 16, 25605–25640 (2015).
Block, F., Dihné, M. & Loos, M. Inflammation in areas of remote changes following focal brain lesion. Prog. Neurobiol. 75, 342–365 (2005).
Gerhard, A. et al. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport 11, 2957–2960 (2000).
Yasuda, T., Yoshida, H., Garcia-Garcia, F., Kay, D. & Krueger, J. M. Interleukin-1beta has a role in cerebral cortical state-dependent electroencephalographic slow-wave activity. Sleep 28, 177–184 (2005).
Yoshida, H. et al. State-specific asymmetries in EEG slow wave activity induced by local application of TNFalpha. Brain Res. 1009, 129–136 (2004).
Ma, C. et al. Microglia regulate sleep through calcium-dependent modulation of norepinephrine transmission. Nat. Neurosci. 27, 249–258 (2024).
Kassubek, J., Sörös, P., Kober, H., Stippich, C. & Vieth, J. B. Focal slow and beta brain activity in patients with multiple sclerosis revealed by magnetoencephalography. Brain Topogr. 11, 193–200 (1999).
Sun, H. & Feng, Z. Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta Pharmacol. Sin. 34, 24–32 (2013).
Rorden, C. & Karnath, H.-O. Using human brain lesions to infer function: a relic from a past era in the fMRI age? Nat. Rev. Neurosci. 5, 812–819 (2004).
Massimini, M., Huber, R., Ferrarelli, F., Hill, S. & Tononi, G. The sleep slow oscillation as a traveling wave. J. Neurosci. 24, 6862–6870 (2004).
Nir, Y. et al. Regional slow waves and spindles in human sleep. Neuron 70, 153–169 (2011).
Volgushev, M., Chauvette, S. & Timofeev, I. Long-range correlation of the membrane potential in neocortical neurons during slow oscillation. Prog. Brain Res. 193, 181–199 (2011).
Capone, C. et al. Slow waves in cortical slices: how spontaneous activity is shaped by laminar structure. Cereb. Cortex 29, 319–335 (2019).
Lemaréchal, J.-D. et al. A brain atlas of axonal and synaptic delays based on modelling of cortico-cortical evoked potentials. Brain 145, 1653–1667 (2022).
Murphy, M. et al. Source modeling sleep slow waves. Proc. Natl Acad. Sci. USA 106, 1608–1613 (2009).
Rocchi, F. et al. Increased fMRI connectivity upon chemogenetic inhibition of the mouse prefrontal cortex. Nat. Commun. 13, 1056 (2022).
Casula, E. P. et al. Evidence for interhemispheric imbalance in stroke patients as revealed by combining transcranial magnetic stimulation and electroencephalography. Hum. Brain Mapp. 42, 1343–1358 (2021).
Chee, M. W. L. & Zhou, J. Functional connectivity and the sleep-deprived brain. Prog. Brain Res. 246, 159–176 (2019).
Ning, Y., Zheng, S., Feng, S., Li, K. & Jia, H. Altered functional connectivity and topological organization of brain networks correlate to cognitive impairments after sleep deprivation. Nat. Sci. Sleep. 14, 1285–1297 (2022).
Corbetta, M. & Shulman, G. L. Spatial neglect and attention networks. Annu. Rev. Neurosci. 34, 569–599 (2011).
Nair, V. A. et al. Functional connectivity changes in the language network during stroke recovery. Ann. Clin. Transl. Neurol. 2, 185–195 (2015).
Vyazovskiy, V. V. et al. Local sleep in awake rats. Nature 472, 443–447 (2011).
Marmelshtein, A., Eckerling, A., Hadad, B., Ben-Eliyahu, S. & Nir, Y. Sleep-like changes in neural processing emerge during sleep deprivation in early auditory cortex. Curr. Biol. 33, 2925–2940.e6 (2023).
Nir, Y. et al. Selective neuronal lapses precede human cognitive lapses following sleep deprivation. Nat. Med. 23, 1474–1480 (2017).
Bernardi, G. et al. Neural and behavioral correlates of extended training during sleep deprivation in humans: evidence for local, task-specific effects. J. Neurosci. 35, 4487–4500 (2015).
Andrillon, T., Burns, A., Mackay, T., Windt, J. & Tsuchiya, N. Predicting lapses of attention with sleep-like slow waves. Nat. Commun. 12, 3657 (2021).
Vyazovskiy, V. V. & Harris, K. D. Sleep and the single neuron: the role of global slow oscillations in individual cell rest. Nat. Rev. Neurosci. 14, 443–451 (2013).
Tüshaus, L. et al. In human non-REM sleep, more slow-wave activity leads to less blood flow in the prefrontal cortex. Sci. Rep. 7, 14993 (2017).
Fultz, N. E. et al. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science 366, 628–631 (2019).
Marshall, L., Helgadóttir, H., Mölle, M. & Born, J. Boosting slow oscillations during sleep potentiates memory. Nature 444, 610–613 (2006).
Cirelli, C. & Tononi, G. The why and how of sleep-dependent synaptic down-selection. Semin. Cell Dev. Biol. 125, 91–100 (2022).
Facchin, L. et al. Slow waves promote sleep-dependent plasticity and functional recovery after stroke. J. Neurosci. 40, 8637–8651 (2020).
Bassetti, C. L. Sleep and stroke. Semin Neurol. 25, 19–32 (2005).
Krone, L. B. & Vyazovskiy, V. V. Unresponsive or just asleep? Do local slow waves in the perilesional cortex have a function? Brain 143, 3513–3515 (2020).
Nir, Y. & de Lecea, L. Sleep and vigilance states: embracing spatiotemporal dynamics. Neuron 111, 1998–2011 (2023).
Murphy, T. H. & Corbett, D. Plasticity during stroke recovery: from synapse to behaviour. Nat. Rev. Neurosci. 10, 861–872 (2009).
Carmichael, S. T. & Chesselet, M.-F. Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J. Neurosci. 22, 6062–6070 (2002).
Sheybani, L. et al. Wake slow waves in focal human epilepsy impact network activity and cognition. Nat. Commun. 14, 7397 (2023).
Hussain, R. et al. Potentiating glymphatic drainage minimizes post-traumatic cerebral oedema. Nature 623, 992–1000 (2023).
Gent, T. C., Bandarabadi, M., Herrera, C. G. & Adamantidis, A. R. Thalamic dual control of sleep and wakefulness. Nat. Neurosci. 21, 974–984 (2018).
Narikiyo, K. et al. The claustrum coordinates cortical slow-wave activity. Nat. Neurosci. 23, 741–753 (2020).
Canto, C. B., Onuki, Y., Bruinsma, B., van der Werf, Y. D. & De Zeeuw, C. I. The sleeping cerebellum. Trends Neurosci. 40, 309–323 (2017).
Mizrahi-Kliger, A. D., Kaplan, A., Israel, Z. & Bergman, H. Desynchronization of slow oscillations in the basal ganglia during natural sleep. Proc. Natl Acad. Sci. USA 115, E4274–E4283 (2018).
Funk, C. M., Honjoh, S., Rodriguez, A. V., Cirelli, C. & Tononi, G. Local slow waves in superficial layers of primary cortical areas during REM sleep. Curr. Biol. 26, 396–403 (2016).
Bharioke, A. et al. General anesthesia globally synchronizes activity selectively in layer 5 cortical pyramidal neurons. Neuron 110, 2024–2040.e10 (2022).
Fehér, K. D. et al. Shaping the slow waves of sleep: a systematic and integrative review of sleep slow wave modulation in humans using non-invasive brain stimulation. Sleep. Med. Rev. 58, 101438 (2021).
D’Andola, M., Giulioni, M., Dante, V., Del Giudice, P. & Sanchez-Vives, M. V. Control of cortical oscillatory frequency by a closed-loop system. J. Neuroeng. Rehabil. 16, 7 (2019).
Barbero-Castillo, A. et al. Control of brain state transitions with a photoswitchable muscarinic agonist. Adv. Sci. 8, e2005027 (2021).
Grossman, N. et al. Noninvasive deep brain stimulation via temporally interfering electric fields. Cell 169, 1029–1041.e16 (2017).
Deco, G. et al. Awakening: predicting external stimulation to force transitions between different brain states. Proc. Natl Acad. Sci. USA 116, 18088–18097 (2019).
Wang, X.-J., Liu, Y., Sanchez-Vives, M. V. & McCormick, D. A. Adaptation and temporal decorrelation by single neurons in the primary visual cortex. J. Neurophysiol. 89, 3279–3293 (2003).
Hayat, H. et al. Reduced neural feedback signaling despite robust neuron and gamma auditory responses during human sleep. Nat. Neurosci. 25, 935–943 (2022).
Hermann, D. M. & Bassetti, C. L. Role of sleep-disordered breathing and sleep-wake disturbances for stroke and stroke recovery. Neurology 87, 1407–1416 (2016).
Drager, L. F., Polotsky, V. Y. & Lorenzi-Filho, G. Obstructive sleep apnea: an emerging risk factor for atherosclerosis. Chest 140, 534–542 (2011).
Giubilei, F. et al. Sleep patterns in acute ischemic stroke. Acta Neurol. Scand. 86, 567–571 (1992).
Poryazova, R. et al. Topographic sleep EEG changes in the acute and chronic stage of hemispheric stroke. J. Sleep. Res. 24, 54–65 (2015).
Vock, J. et al. Evolution of sleep and sleep EEG after hemispheric stroke. J. Sleep. Res. 11, 331–338 (2002).
Duss, S. B. et al. The role of sleep in recovery following ischemic stroke: a review of human and animal data. Neurobiol. Sleep. Circadian Rhythms 2, 94–105 (2017).
Hodor, A., Palchykova, S., Baracchi, F., Noain, D. & Bassetti, C. L. Baclofen facilitates sleep, neuroplasticity, and recovery after stroke in rats. Ann. Clin. Transl. Neurol. 1, 765–777 (2014).
Acknowledgements
The authors thank Dr. Ezequiel Mikulan, Dr. Silvia Casarotto, Dr. Andrea Pigorini, Dr. Simone Russo, and Dr. Pilleriin Sikka for their help and comments on the manuscript draft and illustrations. This work was financially supported by the following entities: ERC-2022-SYG Grant number 101071900 neurological mechanisms of injury and sleep-like cellular dynamics (NEMESIS); Italian National Recovery and Resilience Plan (NRRP), M4C2, funded by the European Union - NextGenerationEU (Project IR0000011, CUP B51E22000150006, “EBRAINS-Italy”); European Union’s Horizon 2020 Framework Program for Research and Innovation under the Specific Grant Agreement No.945539 (Human Brain Project SGA3); Tiny Blue Dot Foundation; Canadian Institute for Advanced Research (CIFAR), Canada; Italian Ministry for Universities and Research (PRIN 2022); Fondazione Regionale per la Ricerca Biomedica (Regione Lombardia), Project ERAPERMED2019–101, GA 779282; CORTICOMOD PID2020-112947RB-I00 financed by MCIN/ AEI /10.13039/501100011033; Fondazione Cassa di Risparmio di Padova e Rovigo (CARIPARO) Grant Agreement number 55403; Ministry of Health, Italy (RF-2008 −12366899) Brain connectivity measured with high-density electroencephalography: a novel neurodiagnostic tool for stroke- NEUROCONN; BIAL foundation grant (Grant Agreement number 361/18); H2020 European School of Network Neuroscience (euSNN); H2020 Visionary Nature Based Actions For Heath, Wellbeing & Resilience in Cities (VARCITIES); Ministry of Health Italy (RF-2019-12369300): Eye-movement dynamics during free viewing as biomarker for assessment of visuospatial functions and for closed-loop rehabilitation in stroke (EYEMOVINSTROKE); ERC-2023-STG: 101116748 - SleepingAwake.
Author information
Authors and Affiliations
Contributions
M.M. and S.S. wrote the original draft. M.C., M.V.S.-V., T.A., G.D., and M.R. contributed to subsections and revised the final version of the manuscript. M.R. created all the illustrations.
Corresponding author
Ethics declarations
Competing interests
Marcello Massimini is a co-founder and shareholder of Intrinsic Powers, a spin-off of the University of Milan. Simone Sarasso and Mario Rosanova are advisors of Intrinsic Powers. These affiliations in no way affect the content of this article.
Peer review
Peer review information
Nature Communications thanks Stuart Fogel, Yuval Nir, István Ulbert and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Massimini, M., Corbetta, M., Sanchez-Vives, M.V. et al. Sleep-like cortical dynamics during wakefulness and their network effects following brain injury. Nat Commun 15, 7207 (2024). https://doi.org/10.1038/s41467-024-51586-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51586-1
This article is cited by
-
Clinical significance of ADHD traits in central disorders of hypersomnolence
Journal of Clinical Sleep Medicine (2026)
-
Neural models for detection and classification of brain states and transitions
Communications Biology (2025)
