
Abstract
Liquid (organic) hydrogen carriers ([18H]-dibenzyltoluene, MeOH, formic acid, etc.) form a toolbox for the storage and transport of green hydrogen, which is crucial for the implementation of renewable energy technologies. Simple organic salts have been scarcely investigated for this purpose, despite many advantages such as low cost and minor toxicity, as well as easy handling. Here, we present a potassium formate/potassium bicarbonate hydrogen storage and release energy system, that is applicable and shows high stability (6 months). Utilizing ppm amounts of the molecularly defined Ru-5 complex, hydrogen release rates of up to 9.3 L h−1 were achieved. The same catalyst system promoted the hydrogenation of KHCO3 to HCOOK with a TON of 9650. In this way, combined hydrogen storage-release cycles can be performed for 40 times.
Similar content being viewed by others

Introduction
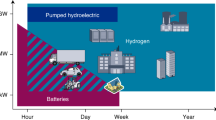
The widespread availability and use of low-cost energy has been one of the most important pillars of global economic development over the past century. It’s also the foundation for the progress of mankind regarding health and aging, food production, mobility as well as most social activities. These positive advancements are based on an increasing use of all kinds of resources. In fact, the demand of the human population statistically exceeds the amount of biological resources regenerated on earth by more than a factor of two1, mainly due to fossil-based energy generation. Since the industrial revolution, global energy consumption has increased dramatically and is expected to continue increasing2. This means that a complete change in the way we generate and use energy will have to take place over the next two decades. Accordingly, the reduction of carbon dioxide emissions by using renewable energy sources is a crucial aspect. As most forms of renewable energy, such as wind and solar power, are dynamic and not constantly available, energy storage will be a vital part of this transformation. Alongside classical engineering concepts, chemical energy storage has recently attracted increased attention3. In this respect, (green) hydrogen is a very promising energy carrier due to its availability from water via electrolysis utilizing renewable electricity4. Later on, the stored energy can be released on demand via combustion or with the aid of fuel cells providing water. Due to the physical properties and the poor volumetric energy density of hydrogen, several (organic) hydrogen carriers are considered for safe energy storage (Fig. 1)5. All these concepts share the use of an available hydrogen acceptor that can be reversibly hydrogenated and dehydrogenated using suitable catalysts. Ideally, the resulting hydrogen carrier would be liquid at ambient conditions, with the advantage that the current energy infrastructure could be used for distribution. Apart from so called liquid organic hydrogen carriers (LOHC), e.g. (hetero)arenes6, hydrogen can be converted with cheap and abandoned nitrogen or carbon dioxide to form carrier molecules like ammonia7,8, methane9, methanol10,11, or formic acid (FA)12,13. Notably, a number of these processes are currently investigated on pilot or demonstration scale14,15.

A Green hydrogen pathway from production to utilization. Excess energy from renewable sources can be used to electrolyze water. The hydrogen can then be chemically stored in so called hydrogen carriers (HC) and released using suitable catalysts. Finally, energy can be recovered via combustion or fuel cell5. B Overview of volumetric and gravimetric energy densities for various energy carriers30,31. C Chemical hazards of loaded HC compared to H2. [1] Hazardous symbols are listed for DBT.
Formic acid is a particularly interesting hydrogen carrier with its volumetric energy density of 1.77 kWh L−1 (relating to a volumetric hydrogen content of 53 g H2 L−1). Compared to other carriers it shows favorable thermodynamics for the individual hydrogenation and dehydrogenation process (Eq. (1)). Both steps can be performed under comparably mild conditions (<100 °C; <50 bar) and FA as well as its derivatives are relatively non-toxic and can be easily handled16. In fact, after the original proposal of FA for such applications17, many research groups made important contributions to this research field18,19,20. Interestingly, already in 2017, a comparative analysis of several process options applying formic acid as a hydrogen carrier, showed that the combination of aqueous, highly concentrated formate salts and their corresponding bicarbonates allows achieving the highest energy efficiency21.
A crucial issue for the use of carbon dioxide-based stationary hydrogen storage is the release of CO2 in the dehydrogenation reaction (Eq. (1)). To reintroduce the carrier molecule CO2 into the storage system, energy-intensive gas purification is required, or an excess of CO2 must be available on site. In the case of FA, under acidic conditions, the dehydrogenation step competes with dehydration yielding CO (Eq. (2)), which is a well-known poison for fuel cells and precludes many potential applications22. Both reaction pathways show minor variations in free Gibbs enthalpy23, making the selective dehydrogenation of FA difficult.
To recycle the hydrogen acceptor (CO2) and avoid a net increase in carbon dioxide emissions, we recently reported an α-amino acid-promoted “chemical hydrogen battery” based on the reversible hydrogenation of carbon dioxide to formic acid. Here, the corresponding ammonium formate is the crucial intermediate, which is formed by hydrogenation and can liberate the hydrogen. Even though a high stability and reusability of the applied manganese catalyst was achieved, a drawback of this system is the necessity to use stoichiometric amounts of the respective amino acid lysine24. To solve this problem, we became interested in the dehydrogenation of more simple formate salts instead of FA/amino acid mixtures. Obviously, dehydrogenation of metal formates in basic aqueous solution gives the corresponding bicarbonates (Eq. (3)). The equilibrium of this formate/bicarbonate cycle can be shifted easily, as the free energy change of the cycle is nearly zero at near-ambient conditions, making it an attractive reversible storage system. Although the volumetric energy density is lower compared to FA (e.g. 29.1 g H2 L−1 for HCOOK (aq) at 25 °C)25, its safety and toxicological aspects as well as sustainability make it an ideal hydrogen storage system. Formate salts are non-corrosive, non-irritating, non-toxic and compounded by earth-abundant elements, making them cheap and easy to handle26.
In general, the storage capacity of a formate/bicarbonate system is limited by the solubility27 of the respective salts. Figure 2 shows both, the mass, and the molar solubility of various formates. Although lithium presents the highest hydrogen content per weight (1.92 wt%) among the alkali salts, the low solubility of lithium formate makes it unattractive for usage. Both rubidium (0.77 wt%) and potassium (1.19 wt%) show high molar solubility at room temperature as well as at elevated temperatures. Considering the availability and price of the metals, potassium seems to be a good candidate for a hydrogen battery.

In the past decade, there have been plenty of examples, both homogeneous as well as heterogeneous, for either the hydrogenation of CO2/bicarbonate34,35 or the dehydrogenation of FA/formate36,37,38. To realize a chemical hydrogen battery, it is necessary for both, the hydrogenation and the dehydrogenation steps to be performed reversibly in the same device. This requires the catalysts to be applicable to both steps. Ideally the whole catalytic system (catalysts, additive, solvent) is applicable for both reactions. Another challenge of such a system is to avoid carbon dioxide loss through bicarbonate decomposition (Eq. 4), which would reduce the capacity of the hydrogen storage-release system after each cycle. In fact, to the best of our knowledge, no stable operating formate/bicarbonate storage system has yet been reported. Some of us have shown that an in situ formed Ru complexes enables a reversible hydrogen storage system for one cycle starting from sodium formate or sodium bicarbonate28. In the same year, Joó and Papp39,40 reported a homogeneous Ru system for formate/bicarbonate interconversion which was stable for 3 cycles in total. Soon after this work, the groups of Olah and Prakash41, as well as Laurenczy42 described improved homogeneous systems. Applying heterogeneous Pd catalysts Cao43 and Lin44 performed up to 6 cycles based on various bicarbonate salts. However, in no case more than 10 bicarbonate hydrogenation/formate dehydrogenation cycles were achieved without recharging carbon dioxide or reactivation of the catalysts45. A detailed overview of state of the art systems for reversible hydrogen storage and release based on carbon dioxide/bicarbonate and formic acid/formate is provided in Supplementary Table 3.
Here, we report an important step towards the development and practical implementation of a carbon-neutral hydrogen storage system. In this respect, the interconversion of metal bicarbonates and formates via (de)hydrogenation has been systematically studied. The presented optimal reaction system enables CO-free hydrogen generation in the presence of Ru-PiPrN(H)PiPr (Ru-5). Notable features of the concept are the excellent catalyst stability (>40 cycles within 6 months) and good productivity for both reaction steps at low temperature (90 °C).
Results
Evaluation of hydrogen generation step: dehydrogenation of alkali formates (FD)
In an ideal hydrogen storage system, the same catalyst and solvent system should be applied for both hydrogenation and dehydrogenation steps. Nevertheless, to identify most suitable conditions for a combined process, initially the hydrogenation of bicarbonate (BH) and formate dehydrogenation (FD) were investigated separately. In general, the nature of the formate/bicarbonate cation has a decisive influence on the volumetric and gravimetric energy content of the reaction system. Consequently, FD was studied utilizing Li, Na, K, Rb, and Cs formates under identical conditions (Table 1 and Supplementary Table 4). In previous works, it has been shown that the latter reaction proceeds well in aqueous DMF using [RuCl2(benzene)]2 and biphosphine ligands as in situ catalysts28.
Following this protocol, HCOOLi provided H2 in 96% yield with a TON of 3820 at 60 °C after 6 h (Table 1, entry 1). Notably, the obtained gas contained a mixture of H2/CO2 in a ratio of 65/35, demonstrating the loss of a significant amount of carbon dioxide in this system. In contrast, simple HCOONa showed high H2 productivity (98% yield; TON = 3920) and improved gas purity (H2/CO2 = 92/8) (Table 1, entry 2). Appling HCOOK as the starting material led to decreased dehydrogenation activity (45% yield and TON = 1780), but at the same time almost all carbon dioxide remained in the reaction solution (H2/CO2 = 99.5/0.5; Table 1, entry 3). HCOORb (58% yield and TON = 2320) and HCOOCs (53% yield and TON = 2120) showed comparable reactivity with a gas proportion of H2/CO2 = 90/10 and H2/CO2 = 97/3, respectively. Based on these results and considering substrate costs, molar solubility, and the composition of produced gas, HCOOK was chosen for further investigations.
Next, the impacts of different catalysts, co-solvent, and the amount of water as well as reaction temperature were studied at increased substrate concentration in more detail (Table 2, and Supplementary Tables 5–8). First, different catalysts were tested. The combinations of [RuCl2(benzene)2]/dppe (Ru-1), [RuCl2(benzene)2]/dppm (Ru-2) and RuCl3/TPPTS (Ru-3) showed low productivity with H2 yields <15% and TONs <1600. In all cases, the ratio of H2/CO2 was around 99/1 (Table 2, entries 1–3). The activity of the defined complex Ru-4 was slightly improved compared to the in situ generated system (Table 2, entry 4). Higher productivity was achieved with ruthenium pincer complexes. Using Ru-PiPrN(H)PiPr (Ru-5), Ru-MACHO (Ru-6), Ru-MACHO-BH (Ru-7) and Milstein´s Ru-catalyst (Ru-8) H2 yields up to 63%, and TONs up to 7110 were obtained (Table 2, entries 5-8). Again, the CO2 release was negligible (≤0.5%). Besides ruthenium, different Ir-pincer catalysts were tested. Ir-1 showed good results (69% H2 yield, TON 7790), but the reactivity decreased with the reaction time (Table 2, entry 9). Notably, the water-soluble Ir-Cp* complexes Ir-2 and Ir-3, which work well in formic acid dehydrogenation, showed no activity under these basic conditions (Table 2, entries 10–11). Overall, Ru-5 was selected as the catalyst for further investigations. Almost full conversion of HCOOK was obtained by increasing the reaction time from 6 h to 13 h (TON 11,260, Table 2, entry 13). Further increasing the substrate amount to 275 mmol, a TON of 17,390 and 62% H2 yield were observed (Table 2, entry 14). Additionally, increasing the reaction temperature and/or decreasing the catalyst amount (3.6 ppm) gave excellent H2 yields up to 99% and TONs up 263,200 (Table 2, entries 15–17). Noteworthy, utilizing Ru-5 for HCOONa dehydrogenation instead of HCOOK gave significantly lower H2 yield and purity (Table 2, entry 12). Among the different co-solvents tested, sulfolane, t-amylol, and triglyme allowed for significant hydrogen production rate; however, DMF performed best in FD (Supplementary Table 8). Then, the ratio of DMF and water was varied. Optimal results with respect to catalyst activity and CO2 release were obtained in DMF/water (15/10) (Table 2, entry 18 and Supplementary Table 8, entries 15–19). With regards to the activity of Ru-5 in the BH (see next chapter, Table 4), different ratios of triglyme were investigated for FD, too (Table 2, entries 18–20 and Supplementary Table 8). Using the solvent ratio of 15 mL triglyme and 10 mL water, the highest gas purity within the triglyme experiments was reached. Under the standard conditions 32% H2 yield were observed with a gas ratio of 94/6 H2/CO2 (Table 2, entry 19; for 275 mmol HCOOK: 30% H2 yield, 92/8 H2/CO2; Table 2, entry 20). Elongation of the reaction time from 6 h to 46 h increased the yield to 91% (Table 2, entry 21). Thus, FD productivity in triglyme/H2O is comparable to DMF/water (although activity is lower), and full conversion of formate in triglyme/H2O is possible.
To showcase the catalytic efficiency, the dehydrogenation of HCOOK was performed on >250 g-scale (3 mol) with only 3.3 ppm catalyst Ru-5, using DMF/H2O (150/100). Full conversion was observed with a remarkable gas release of >72 L in 25 h. This corresponds to a practically relevant catalyst turnover number (TON = 290,000), frequency (TOFmax = 38,400 h−1), maximum space-time-yield (0.76 mol h−1 L−1), and rate of hydrogen releasemax (9.3 L h−1) (Table 3 and Supplementary Table 10; for reaction in triglyme/H2O see Supplementary Table 11). The purity of the produced hydrogen was 97% and no CO was detected (detection limit <10 ppm) under these reaction conditions.
Evaluation of the hydrogen storage step: hydrogenation of potassium bicarbonate (BH)
Similar to the dehydrogenation experiments, different catalysts, solvent mixtures, and temperatures were examined for the hydrogenation step (Table 4 and Supplementary Table 12). In an ideal hydrogen storage device both H2 loading and release should be performed in the presence of the same catalyst system (catalyst, solvent). Consequently, selected well-performing catalysts in FD were investigated for their capability of BH in DMF/H2O (20/5 mL) at 90 °C (Table 4, entries 1–7). Since Ru-1, Ru-5 and Ru-7 exhibited similar activities (≤7% yield) in BH, Ru-5 was selected for further studies due to its superior performance in the dehydrogenation. Unfortunately, DMF was partially decomposed under hydrogen pressure due to catalytic reductive cleavage of the amide29. To avoid this issue, further catalytic experiments were conducted using triglyme as co-solvent. Triglyme was chosen for its anticipated stability, thermal resistance (boiling point 216 °C) and previous performance in the formate dehydrogenation vide supra. Noteworthy, applying triglyme water mixture as solvent in FD, hydrogen yields >91% were achieved (Table 2, entries 18–20; Supplementary Table 8).
Interestingly, the concentration of this co-solvent has a significant effect on the hydrogenation reaction. While a low yield (4%; Table 4, entry 8) was observed at a triglyme/H2O ratio of 20/5 mL, increasing the water concentration led to a significant increase in the formate yield up to 74% (Table 4, entries 9–12). As expected, decreasing the reaction temperature from 90 °C to 50 °C resulted in reduced formate yields (Table 4, entries 13–14 and Fig. 2).
Combination of hydrogenation and dehydrogenation steps: demonstration of H2 storage and release cycles
To demonstrate the stability of the catalysts system, we investigated the cyclability of the (de)hydrogenation over a long period with an appropriate number of cycles. Principally, the storage capacity of a hydrogen battery relies on both, FD and BH performence. Thus, a compromise of productivities achieved in FD and BH has to be made. Taken all results from the (de)hydrogenation into account, triglyme/H2O (15/10 mL) was chosen as the appropriate solvent due to its high stability and activity in BH (up to 74%) and almost full conversion for FD (up to 91%). The individual formate dehydrogenation and bicarbonate hydrogenation steps were combined using a 100 mL autoclave in the presence of KHCO3 (135 mmol), Ru-5 (10 µmol, 74 ppm). As shown in Fig. 3, 40 consecutive cycles of hydrogen storage and release were performed over a period of 6 months. While in the first two experiments, a slight decrease of the hydrogen yield was observed, from the third run onwards, highly stable activity and hydrogen yields were obtained (3rd cycle: 1195 mL; 37th cycle: 1116 mL). In 40 hydrogen storage and release cycles, nearly 50 L of hydrogen with an average purity of 99.5% were generated utilizing ppm amounts of Ru-5. In addition, the produced gas contained <0.5% carbon dioxide and less than 10 ppm carbon monoxide. As shown above, the hydrogen yield can even be increased by the elongating the reaction time. By shortening the FD reaction time, we were able to show that the optimal molecularly defined Ru complex allowes for a stable operation of the hydrogen generation for 6 months, demonstrating its potential for practical applications.

H2 storage and release cycles were started with BH: 135 mmol KHCO3 (13.5 g), 10 µmol Ru-5 (4.7 mg), triglyme/water (15 mL/10 mL), 60 bar H2, 90 °C, 18 h. After pressure release, FD was started: 90 °C, 12 h. The generated gas was measured with manual burettes. The gas was analyzed by GC. CO is undetectable in all cases (CO quantitation limit of the applied GC is <10 ppm).
Discussion
The present work describes a robust carbon neutral formate-based hydrogen storage and release system consisting only of HCOOK, KHCO3, water, an organic solvent and ppm amounts of the commercially available ruthenium complex Ru-5. Under comparably mild conditions, without the addition of a base or a Lewis acid, a noteworthy efficiency for HCOOK dehydrogenation was achieved. Performing the dehydrogenation reaction in DMF/H2O (150/100) led to a maximum hydrogen release rate of 9.3 L/h (>72 L gas in 25 h with space-time-yieldmax of 0.76 mol h−1 L−1). On the other hand, the same catalyst promoted H2 storage in HCOOK from KHCO3 using triglyme/H2O (15/10) with a TON of 9,650. The combination of the individual storage and release processes, based on the optimal reaction conditions for BH, led to a very stable system, which was tested for 6 months with 40 cycles. The average H2 purity obtained in triglyme/H2O (15/10) was 99.5% with <10 ppm CO detected in the produced gas (GC quantification limitation <10 ppm). The overall efficiency of the 40 cycles is 22%. However, elongating the reaction time for FD increases the H2 yield and consequently the potential storage efficiency. Based on the separate (de)hydrogenation reactions, a storage efficiency of up to 67% is possible. The results demonstrate the technological readiness of this methodology and pave the way for the construction of a carbon-neutral chemical hydrogen battery set-up employing HCOOK/KHCO3 salts.
Methods
General procedure for FD
Under an Ar atmosphere, 113 mmol HCOOK (9.5 g), H2O and the co-solvent (25 mL in total) were added to a 50 mL three-neck glass reactor equipped with a magnetic stir bar. The reaction solution was heated to 60 °C if not stated otherwise. Following, the reaction was started by addition of 10 µmol metal catalyst and performed for 6 h. Passed through a cooling condenser, the generated gas was collected by a manual burette and the volume was corrected by the blank volume. The gas was analyzed by GC.
General procedure for BH
Under an Ar atmosphere, 135 mmol KHCO3 (13.5 g), 10 µmol catalyst and the respective solvent mixture (25 mL in total) were added to a 100 mL autoclave. The autoclave was pressurized with 50 bar H2. The reaction solution was heated to the desired temperature and stirred for 18 h. Water (50 ml) was added, washing along the inner wall of the glass inlet and dissolving the bicarbonate as well as formate. DMSO (1 mL, 14.08 mmol) was added as an internal standard to the solution. A mixture of 0.4 mL of the reaction solution with 0.1 mL of D2O was then analyzed by 1H NMR.
Data availability
The experimental and analytical data generated in this study are provided in the Supplementary Information. All data are available from the corresponding author upon request.
References
About Earth Overshoot Day. Earth Overshoot Day https://overshoot.footprintnetwork.org/about-earth-overshoot-day/ (2024).
Ritchie, H., Rosado, P. & Roser, M. Energy Production and Consumption (Our World Data, 2024).
Khan, N., Dilshad, S., Khalid, R., Kalair, A. R. & Abas, N. Review of energy storage and transportation of energy. Energy Storage 1, e49 (2019).
Oliveira, A. M., Beswick, R. R. & Yan, Y. A green hydrogen economy for a renewable energy society. Curr. Opin. Chem. Eng. 33, 100701 (2021).
Andersson, J. & Grönkvist, S. Large-scale storage of hydrogen. Int. J. Hydrog. Energy 44, 11901–11919 (2019).
Preuster, P., Papp, C. & Wasserscheid, P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-free Hydrogen Economy. Acc. Chem. Res. 50, 74–85 (2017).
Aziz, M., Wijayanta, A. T. & Nandiyanto, A. B. D. Ammonia as Effective Hydrogen Storage: A Review on Production. Storage Utilization. Energ. 13, 3062 (2020).
Klerke, A., Christensen, C. H., Nørskov, J. K. & Vegge, T. Ammonia for hydrogen storage: challenges and opportunities. J. Mater. Chem. 18, 2304 (2008).
Saxena, S., Kumar, S. & Drozd, V. A modified steam-methane-reformation reaction for hydrogen production. Int. J. Hydrog. Energy 36, 4366–4369 (2011).
Onishi, N., Laurenczy, G., Beller, M. & Himeda, Y. Recent progress for reversible homogeneous catalytic hydrogen storage in formic acid and in methanol. Coord. Chem. Rev. 373, 317–332 (2018).
Saeidi, S., Amin, N. A. S. & Rahimpour, M. R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 5, 66–81 (2014).
Chen, Z. et al. Designing a Robust Palladium Catalyst for Formic Acid Dehydrogenation. ACS Catal. 13, 4835–4841 (2023).
Mellmann, D., Sponholz, P., Junge, H. & Beller, M. Formic acid as a hydrogen storage material – development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 45, 3954–3988 (2016).
Grasemann, M. & Laurenczy, G. Formic acid as a hydrogen source – recent developments and future trends. Energy Environ. Sci. 5, 8171 (2012).
Olah, G. A. Towards Oil Independence Through Renewable Methanol Chemistry. Angew. Chem. Int. Ed. 52, 104–107 (2013).
Dutta, I. et al. Formic Acid to Power towards Low‐Carbon Economy. Adv. Energy Mater. 12, 2103799 (2022).
Williams, R., Crandall, R. S. & Bloom, A. Use of carbon dioxide in energy storage. Appl. Phys. Lett. 33, 381–383 (1978).
Kushwaha, S., Parthiban, J. & Singh, S. K. Recent Developments in Reversible CO 2 Hydrogenation and Formic Acid Dehydrogenation over Molecular Catalysts. ACS Omega 8, 38773–38793 (2023).
Onishi, N. et al. Development of Effective Catalysts for Hydrogen Storage Technology Using Formic Acid. Adv. Energy Mater. 9, 1801275 (2019).
Sordakis, K. et al. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 118, 372–433 (2018).
Müller, K., Brooks, K. & Autrey, T. Hydrogen Storage in Formic Acid: A Comparison of Process Options. Energy Fuels 31, 12603–12611 (2017).
Baschuk, J. J. & Li, X. Carbon monoxide poisoning of proton exchange membrane fuel cells. Int. J. Energy Res. 25, 695–713 (2001).
Eppinger, J. & Huang, K.-W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2, 188–195 (2017).
Wei, D., Sang, R., Sponholz, P., Junge, H. & Beller, M. Reversible hydrogenation of carbon dioxide to formic acid using a Mn-pincer complex in the presence of lysine. Nat. Energy 7, 438–447 (2022).
Asefa, T., Koh, K. & Yoon, C. W. CO 2 ‐Mediated H 2 Storage‐Release with Nanostructured Catalysts: Recent Progresses, Challenges, and Perspectives. Adv. Energy Mater. 9, 1901158 (2019).
Calabrese, M. & Russo, D. Di Benedetto, A., Marotta, R. & Andreozzi, R. Formate/bicarbonate interconversion for safe hydrogen storage: A review. Renew. Sustain. Energy Rev. 173, 113102 (2023).
Marion, G. M. Carbonate mineral solubility at low temperatures in the Na-K-Mg-Ca-H-Cl-SO4-OH-HCO3-CO3-CO2-H2O system. Geochim. Cosmochim. Acta 65, 1883–1896 (2001).
Boddien, A. et al. CO 2 ‐“Neutral” Hydrogen Storage Based on Bicarbonates and Formates. Angew. Chem. Int. Ed. 50, 6411–6414 (2011).
Muzart, J. N,N-Dimethylformamide: much more than a solvent. Tetrahedron 65, 8313–8323 (2009).
Graetz, J. New approaches to hydrogen storage. Chem. Soc. Rev. 38, 73–82 (2009).
Niermann, M., Beckendorff, A., Kaltschmitt, M. & Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC) – Assessment based on chemical and economic properties. Int. J. Hydrog. Energy 44, 6631–6654 (2019).
CRC Handbook of Chemistry and Physics (CRC Press, 2016). https://doi.org/10.1201/9781315380476.
Balarew, C. et al. IUPAC-NIST Solubility Data Series. 73. Metal and Ammonium Formate Systems. J. Phys. Chem. Ref. Data 30, 1–163 (2001).
Ziebart, C. et al. Well-Defined Iron Catalyst for Improved Hydrogenation of Carbon Dioxide and Bicarbonate. J. Am. Chem. Soc. 134, 20701–20704 (2012).
Gunasekar, G. H., Park, K., Jung, K.-D. & Yoon, S. Recent developments in the catalytic hydrogenation of CO 2 to formic acid/formate using heterogeneous catalysts. Inorg. Chem. Front. 3, 882–895 (2016).
Marcos, R. et al. Mechanistic Studies on NaHCO 3 Hydrogenation and HCOOH Dehydrogenation Reactions Catalysed by a Fe II Linear Tetraphosphine Complex. Chem. Eur. J. 24, 5366–5372 (2018).
Wang, A. et al. Reviews on Homogeneous and Heterogeneous Catalysts for Dehydrogenation and Recycling of Formic Acid: Progress and Perspectives. Energy Fuels 37, 17075–17093 (2023).
Kushwaha, S., Parthiban, J. & Singh, S. K. Ruthenium-Catalyzed Formic Acid/Formate Dehydrogenation and Carbon Dioxide/(bi)carbonate Hydrogenation in Water. Organometallics 42, 3066–3076 (2023).
Horváth, H., Papp, G., Szabolcsi, R., Kathó, Á. & Joó, F. Water‐Soluble Iridium‐NHC‐Phosphine Complexes as Catalysts for Chemical Hydrogen Batteries Based on Formate. ChemSusChem 8, 3036–3038 (2015).
Papp, G., Csorba, J., Laurenczy, G. & Joó, F. A Charge/Discharge Device for Chemical Hydrogen Storage and Generation. Angew. Chem. 123, 10617–10619 (2011).
Kothandaraman, J. et al. Amine‐Free Reversible Hydrogen Storage in Formate Salts Catalyzed by Ruthenium Pincer Complex without pH Control or Solvent Change. ChemSusChem 8, 1442–1451 (2015).
Sordakis, K., Dalebrook, A. F. & Laurenczy, G. A Viable Hydrogen Storage and Release System Based on Cesium Formate and Bicarbonate Salts: Mechanistic Insights into the Hydrogen Release Step. ChemCatChem 7, 2332–2339 (2015).
Bi, Q. et al. An Aqueous Rechargeable Formate‐Based Hydrogen Battery Driven by Heterogeneous Pd Catalysis. Angew. Chem. Int. Ed. 53, 13583–13587 (2014).
Su, J., Yang, L., Lu, M. & Lin, H. Highly Efficient Hydrogen Storage System Based on Ammonium Bicarbonate/Formate Redox Equilibrium over Palladium Nanocatalysts. ChemSusChem 8, 813–816 (2015).
Wei, D., Sang, R., Moazezbarabadi, A., Junge, H. & Beller, M. Homogeneous Carbon Capture and Catalytic Hydrogenation: Toward a Chemical Hydrogen Battery System. JACS Au 2, 1020–1031 (2022).
Acknowledgements
The authors acknowledge financial support from the European Union and the State of Mecklenburg-Vorpommern (EFRW; project “h2cycle”). We thank the analytical team of LIKAT (Prof. Dr. Wolfgang Baumann, Susanne Schareina, Susann Buchholz, Katrin Fiedler, Dipl.-Ing. Andreas Koch, Everaldo Ferreira Krake) for their support. We thank engineers and technicians Anja Kammer and Andreas Hutter for their kind support.
Author information
Authors and Affiliations
Contributions
Conceptualization: R.S., D.W., P.R., R.J., H.J. and M.B.; catalytic experiments and results analysis: R.S., C.A.M.S., T.S., D.W., and Y.H.; discussion and experiment design: R.S., C.A.M.S., T.S., Y.H., A.L., J.M., V.T., P.S., D.W., R.J., H.J. and M.B.; funding acquisition: P.S., R.J., H.J. and M.B.; writing (original draft): C.A.M.S., R.S. and Y.H.; supervision and writing (review and editing). M.B., H.J., C.A.M.S. and R.S. All authors have read and agreed to the published version of this work. These authors contributed equally: R.S. and C.A.M.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rhett Kempe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sang, R., Stein, C.A.M., Schareina, T. et al. Development of a practical formate/bicarbonate energy system. Nat Commun 15, 7268 (2024). https://doi.org/10.1038/s41467-024-51658-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51658-2
This article is cited by
-
High cell density cultivation of Methylorubrum extorquens using a modified pH-stat supplemented with intermittent formate feeding
Biotechnology and Bioprocess Engineering (2025)
-
Liquid organic hydrogen carriers (LOHC): from definitions to recent developments
Rendiconti Lincei. Scienze Fisiche e Naturali (2025)
