
Abstract
Channelrhodopsins are popular optogenetic tools in neuroscience, but remain poorly understood mechanistically. Here we report the cryo-EM structures of channelrhodopsin-2 (ChR2) from Chlamydomonas reinhardtii and H. catenoides kalium channelrhodopsin (KCR1). We show that ChR2 recruits an endogenous N-retinylidene-PE-like molecule to a previously unidentified lateral retinal binding pocket, exhibiting a reduced light response in HEK293 cells. In contrast, H. catenoides kalium channelrhodopsin (KCR1) binds an endogenous retinal in its canonical retinal binding pocket under identical condition. However, exogenous ATR reduces the photocurrent magnitude of wild type KCR1 and also inhibits its leaky mutant C110T. Our results uncover diverse retinal chromophores with distinct binding patterns for channelrhodopsins in mammalian cells, which may further inspire next generation optogenetics for complex tasks such as cell fate control.
Similar content being viewed by others

Introduction
Microbial rhodopsins are light-gated ion channels known as channelrhodospins1. The light sensitive nature of these channels can be exploited as a valuable tool to regulate cell function by light known as optogenetics2. Since the discovery of the first cation-conducting channelrhodopsin in the chlorophyte Chlamydomonas reinhardtii in 20023, numerous cation or anion-conducting channelrhodopsin variants have been isolated and characterized4,5,6,7,8,9. Unlike these classic channelrhodopsins, the ones from H. catenoides (HcKCR1 and HcKCR2), Wobblia lunata (WiChR), Bilabrum sp. (BlChR2), and Colponema vietnamica (CovKCR1 and CovKCR2) are potassium selective10,11. While microbial rhodopsins are ion channels, ion pumps, or enzymes12, animal rhodopsins are mainly G-protein coupled receptors that act as master regulators of signal transduction cascades13. At the photoreaction level, light absorption induces photoisomerization from 11-cis to ATR resulting in movements in the transmembrane helix to form the active state in animal GPCR-type rhodopsins14. The retinal often forms a Schiff base with a lysine residue in rhodopsins, while in some vertebrate visual rhodopsins, the active state containing ATR is thermally unstable, therefore, the Schiff base can be hydrolyzed and released from opsin after rhodopsin photo-bleaching13. Many microbial rhodopsins are proposed to exhibit the canonical cyclic reaction (called photo cycle) in which the 13-cis retinal-binding form thermally returns to the initial all-trans retinal-binding form1. Several microbial rhodopsins such as histidine-kinase rhodopsins and enzyme rhodopsins are proposed to exist as bistable rhodopsins, where the thermally stable 13-cis retinal-binding form is convertible into the initial all-trans retinal-binding form by further light illumination13.
Although channelrhodopsins encode seven-helix transmembrane (7TM) like G protein-coupled receptor proteins, they form dimers or trimers instead15,16,17,18,19,20,21,22. For example, the popular algal channelrhodopsin-2 (ChR2) forms dimers22, while H. catenoides potassium-selective channelrhodopsin (KCR1) trimers18,21,23. As optogenetic tools, the microbial channelrhodopsins should adapt to the target cells, which in return possess diverse metabolic conditions to influence the retinals that regulate the channel activity24. Thus, limited understanding of the structural and functional basis of channelrhodopsins in the various cells may limit the further development of the optogenetics.
Here, we report that two channelrhodopsins, ChR2 and KCR1, exhibit distinctive light-activation behaviors with exogenous ATR or endogenous retinal in human HEK293T cells. Significantly, we identified a novel binding pocket for endogenous N-retinylidene-PE-like molecules in ChR2. We also reveal the endo-retinal in KCR1 with a noncanonical pattern, which differs from the conventional Schiff-base linked ATR. Furthermore, we show that exogenous ATR inhibits the channel activity of a leaky KCR1 mutant C110T. These findings not only uncover novel retinal binding patterns but also may enable future design of novel optogenetic tools.
Results
Distinct light activation properties of ChR2 and KCR1 with exogenous ATR
The conventional approach to test channelrodopsin activity is to express channelrodopsin in cells with exogenous ATR, as it has long been thought that ATR acts as a cofactor covalently linked to the conserved lysine residue via a protonated Schiff-base. Consequently, the protonated Schiff-base linked ATR absorbs photons and catalyzes photoisomerization to trigger structural changes and ion flow1. To see if this applies to newly identified ChR2 and KCR1, we expressed C-terminal GFP-tagged ChR2 and KCR1 in HEK293T cells with 10 μM exogenous ATR, followed by light-activated whole-cell patch clamp under near-physiological sodium versus potassium gradient conditions. Both ChR2 and KCR1 exhibited robust light-activated currents. As the reversal potentials of ChR2 and KCR1 are around +20 mV and −80 mV respectively, indicating that ChR2 is slightly sodium and KCR1 strongly potassium selective. As expected, the photocurrent magnitude of KCR1 is significantly larger than that of ChR2 (Figs. 1b, c, f, g). Unexpectedly, without exogenous ATR supplementation, the expression level of KCR1 is close to that treated with exogenous ATR for less than 6 h, but exhibiting larger photoactivated currents than that with exogenous ATR. In contrast, ChR2 is less light responsive without exogenous ATR, and its expression level has not significantly changed (Figs. 1a–h and Supplementary Fig. 1b). These results suggest that exogenous ATR enhances the light response of ChR2 but attenuates the photocurrent magnitude of KCR1 in the HEK293 cells.

a, b Representative photocurrent traces recorded from ChR2 without (a) or with (b) exogenous ATR (endoChR2) in response to 500 ms light pulses (blue box, 470 nm, 50 mW/mm2) upon 20 mV voltage increments from −10 mV to +100 mV at a holding potential of −60 mV in 150 mM NaCl bath. The blue dotted line shows the zero current level and the red trace shows the voltage at 0 mV. c Voltage versus current IV-curves as in a (black, endoChR2) and b (red, exoChR2). The Rev indicates the reversal potential of −20 mV. d Peak photoactivated current comparison of endoChR2 and exoChR2 at the voltage of −100 mV. (n = 10 independent cells for each variant, data are present as mean ± SEM). e, f Representative photocurrent traces recorded from KCR1 without (e) or with (f) exogenous ATR (endoKCR1) in response to 500 ms light pulses (blue box) upon 20 mV voltage increments from −100 mV to +100 mV at a holding potential of −60 mV in 150 mM NaCl bath. The blue dotted line shows the zero current level and the red trace shows the voltage at 0 mV. g Voltage versus current IV-curves as in e (black, endoKCR1) and f (red, exoKCR1). The Rev indicates the reversal potential of −20 mV. h Peak photoactivated current comparison of endoKCR1 and exoKCR1 at the voltage of 0 mV. Data points are the mean ± sem (n = 10 independent cells for each variant). *, p < 0.05 by Student’s t-test.
This discrepancy is of interest to us because the endogenous retinal (endo-retinal) and exogenous ATR chromophores may have different functions in ChR2 and KCR1. We use the terms endoChR2 and endoKCR1 to refer to the ChR2 and KCR1 with endogenous retinal, while exoChR2 and exoKCR1 will refer to those with exogenous ATR. We further generated GFP-tagged free ChR2 and KCR1 to see if ATR could affect the color of HEK293 cells when expressing ChR2 or KCR1. Interestingly, treatment with 10 μM exogenous ATR for more than 2 h significantly changed the visible color. Moreover, after about 24 h of treatment with 10 μM exogenous ATR, the color changes are even more significant, suggesting that exogenous ATR binds the opsin protein in a time-dependent manner and that 2 h of treatment is sufficient to influence the properties of these channelrodopsins (Supplementary Fig. 1a).
Structural determinations for exoChR2, endoChR2 and exoKCR1
To gain structural insight into these channels, we expressed and purified ChR2 with or without 10 μM exogenous ATR in a sodium environment. We then processed the purified samples with standard cryo-EM and solved the structures of exoChR2 and endoChR2 at 2.4 Å (Supplementary Fig. 2) and 2.5 Å (Supplementary Fig. 4), respectively. Since we previously determined the structure of endoKCR1 in a potassium environment without exogenous ATR25, we further solved the structure of exoKCR1 with ATR at 2.7 Å (Supplementary Fig. 6). The resulting high-quality maps enabled us to build the near-atomic models of endoChR2, exoChR2 and endoKCR1, including side chains, some water/ions, and retinal ligands (Supplementary Figs. 3, 5, and 7). The overall cryo-EM architectures of exoChR2 and endoChR2 are dimers with a Chinese chime bell shape, which is the same as the X-ray structure of ChR222. Similarly, the overall structure of trimeric exoKCR1 is identical to that of endoKCR1, with the Chinese bronze tripod shape. It is noteworthy that purified endoKCR1 and endoChR2 are slightly green due to a small amount of uncleaved GFP tag, whereas exoKCR1 and exoChR2 are lycopene and yellow, respectively (Supplementary Fig. 1c). To further determine the differences between endogenous and exogenous ATR chromophores, we applied liquid chromatogram mass spectrometry (LC-MS) analysis using a standard sample of commercial ATR. The results show that purified endoKCR1 and endoChR2 lack ATR, while exoKCR1 and exoChR2 present high peaks with the same retention time as the standard sample of ATR (Supplementary Fig. 11), confirming that the potential ligands of endoKCR1 and endoChR2 are different from ATR in exoKCR1 and exoChR2.
EndoChR2 recruits an endogenous N-retinylidene-PE-like molecule to a lateral retinal binding pocket
In exoChR2, the Schiff-base that is linked canonically to ATR is visible within the retinal binding pocket that is formed by TM3, TM4, TM5, TM6 and TM7, consistent with the X-ray structure of ChR226 (Fig. 2a). Remarkably, an apparent retinal-like density occupies the region between TM3, TM4, TM5, and TM6 in endoChR2, thereby creating another retinal binding pocket, which we have termed the lateral retinal binding pocket. Interestingly, the retinal-like density is clearly covalently linked to a lipid, so it represents the lipid-covalently linked retinal. N-retinylidene phosphatidylethanolamine (N-retinylidene PE) is the typical lipid-covalently bound retinal in the mammalian visual system26,27. The 9-cis N-retinylidene-PE is well fitted with the density (Fig. 2b), and the β-ionone ring likely overlaps with that in the canonical Schiff-base linked ATR (Figs. 2c, d). It should be noted that while 9-cis retinal fits well with the clear retinal density, the density of the β-ionone ring is somewhat flexible (Fig. 2b and Supplementary Fig. 5), indicating that endo-retinal may be analogous or post-modified, rather than the conventional all-trans or isomerized form. The endo-retinal binding pocket, created by several hydrophobic residues, competently accommodates 9-cis N-retinylidene-PE (Figs. 2e, f), suggesting that this unique retinal binding pattern is relatively stable. As no density extended to the canonical retinal binding pocket, the N-retinylidene PE-like molecule or endo-retinal is hijacked by the lateral retinal binding pocket (Supplementary Fig. 5). We mutated the F189 to alanine, which directly interacts with the lipid chain of the 9-cis N-retinylidene PE-like molecule, and show that ChR2-F189A is much less light responsive compared to wild-type ChR2 in the absence of exogenous ATR (Fig. 2g). Since the 9-cis N-retinylidene-PE-like molecule is not located in the ion permeation pathway, the conformational change caused by the photons on the 9-cis N-retinylidene-PE-like molecule may not be the same as the conventional Schiff-base linked ATR. This seems to be consistent with the electrophysiological findings that most endoChR2 remains inactive without exogenous ATR supplementation. However, since endoChR2 is not an opsin without exogenous ATR, it would be of interest to investigate the chemical formula and unique functional roles of endo-retinal in future studies.

a, b The two retinal binding pocket models of exoChR2 (a) and endoChR2 (b) were presented by Coot. The density is shown as mesh in green (exoChR2) and pink (endoChR2). Exogenous ATR was covalently linked to the conserved K257 via a canonical Schiff base in (a). Endogenous 9-cis N-retinylidene-PE-like molecule binds to another pocket without linking to the K257 residue in (b). c The two channelrhodopsin models of exoChR2 (blue) and endoChR2 (green). The pink model is the ATR, and the yellow model is the endogenous 9-cis N-retinylidene-PE-like molecule. The β-ionone ring of ATR and endogenous 9-cis N-retinylidene-PE-like molecule overlap at the same position. d Molecular formula of retinal and model comparison of 9-cis N-retinylidene-PE (9-cis N-retinylidene PE, yellow) and exoChR2 (ATR, pink). e–f Endogenous 9-cis N-retinylidene-PE-like molecule binding pocket of endoChR2. ChR2 is shown as a brown surface. The endogenous 9-cis N-retinylidene-PE-like molecule and the residues surrounding the endo-retinal are shown as cartoons in yellow and green, respectively. g Peak photoactivated current comparison of endoChR2 and endoChR2-F189A at the voltage of −100 mV. The lateral retinal binding pocket mutant endoChR2-F189A significantly reduced the light response. Data points are the mean ± sem (n = 10 independent cells for each variant). *, p < 0.05 by Student’s t-test.
Endo-retinal differs from the canonical Schiff-base linked ATR in endoKCR1
Since the retinal binding patterns of endoChR2 and exoChR2 differ so significantly, we then examined the retinal binding pockets of endoKCR1 and exoKCR1. We are confident that the retinal density in exoKCR1 can be built as Schiff-base linked ATR (Fig. 3a), which is consistent with the structure of exoKCR1 in the sodium environment and nanodiscs18,23. However, in endoKCR1, although we had previously built the retinal density as Schiff-base linked ATR25, the density fit is not as good as it could be, albeit the overall resolution is sufficiently high and the side chains of the coordinated residues are quite clear. On the one hand, the β-ionone ring of endo-retinal in endoKCR1 shows a flexibility comparable to that of endoChR2. Furthermore, the density of the endo-retinal polyene chain is also not well fitted by the polyene chain of the ATR model (Fig. 3b). Structural alignment of endoKCR1 and exoKCR1 shows that the retinal in exoKCR1 shifts towards the side chain of tyrosine 103 (Y103), resulting in an almost 70-degrees rotation of Y103 along with the phenolic plane (Fig. 3c). This suggests that the endo-retinal is different from the ATR, leading to cryptic conformational changes in the protein.

a The Schiff-base linked ATR in the retinal binding pocket model of exoKCR1 was present by Coot. The density is shown as purple mesh. Exogenous ATR is covalently linked to the conserved K233 via a canonical Schiff base. b Endo-retinal in the canonical retinal binding pocket models of endoKCR. The endo-retinal density (red box region) is not well fitted including the β-ionone ring and the polyene chain. c The retinal binding pocket models of exoKCR1 and endoKCR1 were present as cartoon. The distance between the retinal and Y103 is labeled. Y103 rotated around 70 degrees from exoKCR1 to endoKCR1 in coordination with the slight motion of the retinal.
Exo and endoKCR1 differ in ion flow
In our prior study25, we partitioned the hourglass-shaped ion permeation channel of KCR1 into six layers, denoted as L1-6. L1 is named the extracellular hydrated layer, L2 is the selectivity filter layer, L3 is the extracellular monohydrated layer, L4 is the dehydrated layer, and L5 is the intracellular hydrated layer. The process of dehydration converges on layer L4 from both the intracellular and extracellular sides of the lumen. In L4, two presumed ongoing water molecules are in contact with F195 and W199, running parallel to the dehydrated lumen25. Our analysis of the ion/water density within exoKCR1’s ion permeation pathway showed deficient ion/water densities in the assigned ongoing water sites and the dehydrated lumen, which suggests disrupted ion flow (Figs. 4a–f). The discontinuous ion flow aligns with the functional study of the attenuation of channel activity induced by exogenous ATR. This suggests that Schiff base stabilization, achieved by the addition of exogenous ATR, may cause exoKCR1 to enter a deeply closed, discontinuous ion flow state.

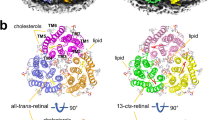
a, b Cartoon model of exoKCR1 in gray (a) and endoKCR1 in purple (b) with ion and water permeation pathway. The potential potassium ion and water are shown as purple and red spheres, respectively. The side chain of residues along the pore is labeled and color-coded. The retinal in exoKCR1 is colored in green and the retinal in endoKCR1 is colored in blue. The lumen is divided into five layers (L1–L5) according to the putative distribution and dehydration of potential potassium ion built in the pore domain: the intracellular hydrated layer (L5, E248-R244-D116), which contains the hydrated potassium; the dehydrated layer (L4), containing two water molecules (F195-W199) and two putative continuous dehydrated potassium ion; the extracellular monohydrated layer (L3, K233-D229-D105-K84), which contains a putative monohydrate potassium ion; the selectivity filter layer (L2, W102-Y222), which contains at least one partial hydrated potassium ion through π-cation interactions. the extracellular hydrated layer (L1, W100), which contains the bulky hydrated potassium ion. The exoKCR1 exhibit the continuous potassium permeation pathway while the endoKCR1 exhibit the discontinuous potassium permeation pathway. c–f The dehydrated layer of exoKCR1 (c–d) and endoKCR1 (e–f) were presented by Coot. No density is observed in exoKCR1, whereas two distinct putative water densities interact with F195 and W199, and two potassium/water densities occupy the lumen.
ATR inhibits the leaky mutant KCR1-C110T by forming Schiff-base
Since the wild-type KCR1 current is inhibited by exogenous ATR, likely due to stabilizing Schiff-base linked ATR, we carried out further experiments to investigate the effect of exogenous ATR on the leaky mutant C110T. Notably, the addition of 10 μM exogenous ATR significantly inhibited the spontaneous potassium currents of C110T in the absence of light. However, the exoC110T maintains a partially conductive state, as suggested by the current versus voltage (I–V) curve, which exhibits the background potassium current in the absence of light (Figs. 5a–d). Previously, we determined the cryo-EM structure of the endoC110T mutant in a potassium environment without exogenous ATR25. In comparison, we further acquired two classes of cryo-EM structures for exoC110T in a potassium environment, with overall resolutions of 2.6 Å and 2.6 Å, respectively (Supplementary Figs. 8–10). Interestingly, one class shows a “hydrolyzed retinal” like density with a combination of 13-cis or ATR containing state, while another class displays the conventional Schiff-base linked ATR containing state (Figs. 5e–h). These results suggest that exogenous ATR can reshape the leaky mutant retinal binding pattern and attenuate channel leaky activity, consistent with electrophysiological data showing exoC110T at a partially conducting state.

a–b Representative photocurrent traces recorded from KCR1 leaky mutant C110T without (a) or with (b) exogenous ATR (endoC110T) in response to 500 ms light pulses (blue box) upon 20 mV voltage increments from −100 mV to +100 mV at a holding potential of −60 mV in 150 mM NaCl bath. The blue dotted line shows the zero current level and the red trace shows the voltage at 0 mV. c Voltage versus current IV-curves as in a (black, endoC100T) and (b) (red, exoC110T). d Peak spontaneous dark current comparison of endoC110T and exoC110T at the voltage of 0 mV. Data points are the mean ± sem (n = 10 independent cells for each variant). *, p < 0.05 by Student’s t-test. e The retinal binding pocket model of endoC110T were present by Coot. The density is shown as green mesh. Endogenous retinal density is perched in the pocket do not well fit the canonical Schiff base covalently linked ATR. f–h Three retinal binding pocket models of exoC110T were present by Coot. ExoC110T class 1 retinal binding pocket with a canonical Schiff base linked ATR to K233 residue is shown in f. The density is present as brown mesh. In the exoC110T class 2 map, we built the mixture of the ATR or putative 13-cis retinal model. ExoC110T class 2 retinal binding pocket is with an ATR shown in g and ExoC110T class 2 retinal binding pocket is with a putative13-cis retinal shown in (h). The density is present as cyan mesh.
Discussion
Channelrhodopsins are exciting molecules with a seven-transmembrane domain opsin that generates an inner hydrophobic pocket and a photoisomerisable chromophore. In Chlamydomonas reinhardtii, ATR is the integrated chromophore where ChR2 was first identified4. ATR is naturally present in some mammalian tissues or can be supplied to animals lacking sufficient retinal.
However, our results suggest that, at least in HE293T cells, the endogenous retinal chromophore is more likely to be a retinal precursor or analog, located in the other retinal binding pocket or forming the unconventional opsin-retinal interaction, which functions differently from ATR (Figs. 6a, b). It can be summarized by the “cell color change assay”, electrophysiological studies, LC-MS and cryo-EM results, although the exact chemical formula is still not known due to the complicated nature of endo-retinal.

a For ChR2, the endogenous N-retinylidene-PE-like molecule couldn’t be sufficiently hydrolyzed and are hijacked at the endo-retinal binding pocket, resulting in photo insensitivity. b For KCR1, the endogenous N-retinylidene-PE-like molecule can probably be sufficiently hydrolyzed or directly ultilized other forms of endogenous retinal and the endogenous retinal is well located in the canonical retinal binding pocket, but cannot well form the Schiff-base linked ATR, being highly photosensitive.
It is interesting to note that although we almost cannot well detect the endo-retinal in the canonical retinal binding site in the endoChR2 from HEK293 cells, the electrophysiological results still show the light-activated currents of endoChR2, which is consistent with the widespread application of ChR2 alone for optogenetic tools28. Since in the absence of exogenous ATR, the light-activated currents are drastically reduced compared to those of exoChR2, we can assume that a tiny amount of ATR or ATR analog may still exist in the canonical retinal binding pocket, so that it cannot be detected by LC/MS and cryo-EM technology. However, since mutation of the non-canonical retinal binding pocket also has a large effect on endoChR2 light sensitivity, another explanation may be that the N-retinylidene PE can be hydrolyzed and release the ATR or ATR analogs to the canonical retinal binding pocket and then act just like endoKCR1. More interestingly, the electrophysiological behavior of endoKCR1 is opposite to that of endoChR2, further suggesting the diversity of channelrhodopsins from different species in mammalian cells without ATR.
Actually, the retinoid metabolism pathway is complicated in mamals29. Our unusual finding of this novel opsin-retinal interaction not only extends our knowledge of the mechanism of photosensitivity but also provides a new strategy for manipulating the function of channelrhodopsins in mammalian cells. Furthermore, as channelrhodopsins have great potential for different mammalian cells, it is worth noting that the different types of endogenous retinal may give rise to different function in different cell types. It is, therefore, necessary to determine the cell-specific channel properties before the intended applications.
The mechanism of channelrhodopsin activation has long been a mystery. It is thought to be a reversible Schiff-base-linked ATR-guided photoisomerization, which then triggers cellular ion flow by an unknown mechanism1. It has been reported that parallel anti- and syn-photocycles could mediate inactivation and ion selectivity changes of ChR2 during continuous illumination30. Our results imply that the retinal in the canonical retinal binding pocket not only undergo the photoisomerization but also may also generate the putative “photohydrolysis like” manner. The clues come from the two forms of the leaky mutant exoC110T (one map has Schiff-base-linked ATR and another does not). If the photohydrolysis happens in the light illumination, the light/dark adaptation could also be explained by the reversible Schiff-base formation. As each channelrhodopsin may have unique retinal binding pattern, it is possible and also exciting to investigate the activation mechanisms further with diverse arrays of channelrhodopsins in the near future.
Methods
Cloning and protein expression
The gene sequence for ChR2 from Chlamydomonas reinhardtii and KCR1 from Hyphochytrium catenoides was inserted into a customized vector based on the BacMam expression backbone. This vector contains a C-terminal PreScission protease cleavage site, a linker sequence, an enhanced green fluorescent protein (EGFP), and a FLAG tag, all of which work together to generate the ChR2/KCR1-LE-LEVLFQGP-GGK-EGFP-GSG-DYKDDDDK constructs for expression. Inverse PCR was utilized to introduce the mutations in this study. For GFP free construct, the stop coden were introduced to the C-terminal region before GFP. The KCR1 constructs and C110T mutant were transfected into DH10Bac E.coli cells, and the bacmids were generated following the manufacturer’s guidelines. The sf9 cells of Spodoptera frugiperda were cultured in sf-900 II SFM medium (Thermo Fisher Scientific) and used for subsequent steps. To produce the P1 virus, transfection of the bacmid was carried out using the Cellfectin reagent (Gibco) with adherent sf9 cells. SF9 cells were infected with P1 virus in suspension at a cell density of 1.5 million cells/mL with a multiplicity of infection (MOI) ~ 0.1. Infection was tracked using fluorescence, and P2 virus was collected 48–72 h post-infection. To create P3 virus, the same method was used. The resulting P3 viral stock was used to infect Expi-HEK293F cells for large-scale protein expression at a density of 2.5 million cells/mL with an MOI of ~2–5. The cells may require the addition of exogenous ATR at a concentration of 10 μM, following 2 days of expression. Subsequently, the cells should be collected through centrifugation at 4000 × g for 15 min, promptly frozen in liquid nitrogen, and kept in storage at −80 °C until further use.
Fluorescence-detection size-exclusion chromatography
Expi293F cells transfected by ChR2-CGFP, KCR1-CGFP and KCR1-C110T-CGFP plasmids were harvested and solubilized in Tris-buffered saline (TBS; 20 mM Tris pH 8.0 and 150 mM NaCl), 1% (w/v) Lauryl maltose neopentyl glycol (LMNG), and 0.1% (w/v) cholesteryl hemisuccinate (CHS) for 0.5 h at 4 °C, then centrifuged at 15,000 g to remove unsolubilized cells. Supernatants were centrifuged at 100,000 g in a TLA55 rotor for 30 min to remove unsolublized membrane. Supernatants were injected onto a Superose 6 increase 5/150 column (GE Healthcare), pre-equilibrated with TBS and 10 μM LMNG, and detected by a fluorescence detector (excitation 488 nm and emission 520 nm for GFP signal, excitation 280 nm and emission 335 nm for tryptophan signal).
Protein purification and EM sample preparation
Cells from one litre of culture were thawed and suspended in 100 milliliters of lysis buffer, comprising of 20 mM Tris-HCl and 150 mM NaCl (TBS) or KCl (TPS) with a pH of 8.0. Sonication was used to lyse the cells, and then the membranes were pelleted by centrifugation at 270,000 g for 40 min in a Ti45 rotor. The membrane pellets were homogenized in TBS or TPS, and then solubilised in TBS or TPS, 1% (w/v) LMNG, and 0.1% (w/v) CHS for one hour at 4 °C. Insoluble materials were removed through centrifugation at 270,000 g for 30 min in a Ti45 rotor. The resulting supernatant was then applied to anti-FLAG resin (Genscript) using gravity flow. The resin underwent subsequent washing with 10 column volumes of wash buffer (lysis buffer with 0.02% LMNG). Protein was then eluted with an added elution buffer (wash buffer comprising of 230 μg/ml FLAG peptide). To remove the C-terminal GFP tag of the recovered protein, HRV3C protease cleavage was performed for 2 h at a temperature of 4 °C. The protein was concentrated using a 100-kDa cutoff concentrator (Millipore) and subsequently loaded onto a Superose 6 Increase 10/300 column (GE Healthcare) in lysis buffer containing 0.03% digitonin. The peak fractions were then combined and concentrated to approximately 10 mg/ml to prepare the cryo-EM sample and LC/MS analysis. Before preparing the grid, the protein samples underwent centrifugation at 100,000 g for 30 min at 4 °C to achieve clearance. Newly discharged Holey Carbon, 300 mesh R1.2/1.3 gold grids (Quantifoil), were used. A 3 μl drop of protein was then dropped onto the grids. The samples were instantly frozen by dipping into ethane that had been cooled by liquid nitrogen. An FEI Vitrobot Mark IV (Thermo Fisher Scientific) was employed to automate the process, at 8 °C, 100% humidity, 3 blot force, ~5 s wait time, and 6 s blot time. It is noteworthy that all samples were prepared under dim white light conditions.
Cryo-EM data acquisition, processing, and model building
Cryo-EM data were acquired on a Titan Krios microscope (FEI) equipped with a cesium corrector operated at 300 kV. Movie stacks in super-resolution mode were automatically obtained using EPU software on a Thermo Fisher Falcon4i detector. The pixel size was 0.35 Å at the object plane, while the defocus ranged from −0.9 μm to −1.2 μm. A GIF Quantum energy filter with a 5 eV slit width was also utilized for this study. The total exposure dose was 40 e- Å−2. Data processing was conducted using the cryoSPARC v4 suite. The super-resolution image stacks were gain-normalized, binned by 2 with Fourier cropping, and patch-based Contrast Transfer Function (CTF) parameters of the dose-weighted micrographs (0.35 Å per pixel) were determined by cryoSPARC and summarized in Extended Figures. The monomers’ atomic models were constructed in Coot based on an initial model (PDB: 7SHS). The models were then manually adjusted in Coot. Trimeric models were obtained through the application of a symmetry operation on the monomer. These models were refined using Phenix.real_space_refine iteratively with secondary structure restraints and Coot. FSC curves were calculated between the EM maps and final models for validation. Coot, PyMOL and Chimera were used for figure preparation.
LC-MS analysis
The ATR detection in purified protein samples was carried by Agilent 6549 C triple quadrupole mass spectrometer connected with Agilent 1290 LC system in Westlake University, with standard sample of commercial ATR. The protein solution extracted with MTBE (methyl tert-butyl ether) by vortex and incubated for 1 h at room temperature in a shaker. The supernatants were collected and N2-drying. Extracted lipids were dissolved in 200 μl of 1:2 methanol/dichloromethane for LC-MS analysis. Chromatographic separation was achieved on an ACQUITY UPLC BEH C18 column (100 mm × 2.1 mm, 1.7 μm) at 50 °C. The mobile phase consisted of 10 mM ammonium acetate, 0.2 mM ammonium fluoride in 9:1 water/methanol (A) and 10 mM ammonium acetate, 0.2 mM ammonium fluoride in 2:3:5 acetonitrile/methanol/isopropanol (B) at a flow rate of 0.3 ml/min. The gradient of mobile phase B was 70% in 1 min, 70% to 86% in 2.5 min, held at 86% for 6.5 min, then 86% to 100% in 1 min, held at 100% for 6 min, then 100% to 70% in 0.1 min, held at 70% for 1.9 min. The sample volume injected was 3 μL. Mass spectrometer operating in positive ion mode using the following settings: Sheath gas temperature 200 °C, Sheath gas flow 11 L/min, Capliary 3.0 kV, Gas temperature 200 °C, gas flow 14 L/min, Nebulizer 20 psi. Sheath Gas Temp Compounds were measured by multiple reaction monitoring (MRM) with optimized instrumental parameters. Quantifier MRM transitions (m/z) of retinal: 285.2/91.0, 285.2/41.1. LC samples, besides 10 mg/ml endoChR2, exoChR2, endoKCR1 and exoKCR1, were the same as used for the cryo-EM study.
Whole-cell patch clamp recording from HEK293 cells
The HEK293T cells obtained from the American Type Culture Collection (ATCC) were grown on coverslips placed in a 12-well plate containing DMEM/F12 medium (Gibco) with 10% fetal bovine serum (FBS). In each well, one microgram of DNA plasmid was temporarily transfected into the cells using 3 micrograms of linear polyethyleneimine (PEI, MW 25000, Polysciences), following the instructions provided by the manufacturer. After a period of 12–0 h, the coverslips were transferred to a recording chamber filled with external solution containing 10 mM HEPES-Na pH 7.4, 150 mM NaCl, 5 mM glucose, 2 mM MgCl2, and 1 mM CaCl2. In case cells require it, they can be enriched with 10 μM of exogenous ATR prior to conducting patch clamp experiments, with supplementation occurring 2–6 h before. The borosilicate micropipettes (OD 1.5 mm, ID 0.86 mm, Sutter) were drawn out and polished to 4–6 MΩ resistance. The solution used in the pipette contained 10 mM HEPES-Na pH 7.4, 150 mM KCl and 5 mM EGTA. The bath solution included 10 mM HEPES-Na with a pH of 7.4, 150 mM NaCl, 5 mM glucose, 2 mM MgCl2 and 1 mM CaCl2. All measurements were taken in a dark environment at room temperature (~25 °C), aided by an Axopatch 700B amplifier, a Digidata 1550 digitizer, and pCLAMP software (Molecular Devices). The laser managed by pCLAMP software produced a 470 nm and 50 mW/mm2 light pulses. The membranes were held at −60 mV and monitored at a frequency of 1 kHz with a sampling frequency of 20 kHz. All current-voltage curves (IV dependencies) were corrected for liquid junction potentials (LJP), which were estimated using the LJP calculator incorporated in ClampEx. The statistical analyses were executed with ClampFit and GraphPad Prism.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-39881 (exoChR2); EMD-39880 (endoChR2); EMD-39882 (exoKCR1); EMD-39883 (exoC110T class 1); and EMD-39884 (exoC110T class2). The atomic coordinates have been deposited in the Protein Data Bank (PDB) under accession codes 8ZAN (exoChR2); 8ZAM (endoChR2); 8ZAO (exoKCR1); 8ZAP (exoC110T class 1); and 8ZAQ (exoC110T class2). Source data are provided with this paper.
References
Ernst, O. P. et al. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 (2014).
Deisseroth, K. & Hegemann, P. The form and function of channelrhodopsin. Science 357, eaan5544 (2017).
Nagel, G. et al. Channelrhodopsin-1: a light-gated proton channel in green algae. Science 296, 2395–2398 (2002).
Nagel, G. et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 100, 13940–13945 (2003).
Zhang, F. et al. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat. Neurosci. 11, 631–633 (2008).
Klapoetke, N. C. et al. Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346 (2014).
Govorunova, E. G., Sineshchekov, O. A., Janz, R., Liu, X. & Spudich, J. L. NEUROSCIENCE. Natural light-gated anion channels: A family of microbial rhodopsins for advanced optogenetics. Science 349, 647–650 (2015).
Berndt, A., Lee, S. Y., Ramakrishnan, C. & Deisseroth, K. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science 344, 420–424 (2014).
Wietek, J. et al. Conversion of channelrhodopsin into a light-gated chloride channel. Science 344, 409–412 (2014).
Govorunova, E. G. et al. Kalium channelrhodopsins are natural light-gated potassium channels that mediate optogenetic inhibition. Nat. Neurosci. 25, 967–974 (2022).
Vierock, J. et al. WiChR, a highly potassium-selective channelrhodopsin for low-light one- and two-photon inhibition of excitable cells. Sci. Adv. 8, eadd7729 (2022).
Govorunova, E. G., Sineshchekov, O. A., Li, H. & Spudich, J. L. Microbial rhodopsins: diversity, mechanisms, and optogenetic applications. Annu Rev. Biochem. 86, 845–872 (2017).
Kojima, K. & Sudo, Y. Convergent evolution of animal and microbial rhodopsins. RSC Adv. 13, 5367–5381 (2023).
Okada, T., Ernst, O. P., Palczewski, K. & Hofmann, K. P. Activation of rhodopsin: new insights from structural and biochemical studies. Trends Biochem Sci. 26, 318–324 (2001).
Kishi, K. E. et al. Structural basis for channel conduction in the pump-like channelrhodopsin ChRmine. Cell 185, 672–689 e623 (2022).
Tucker, K., Sridharan, S., Adesnik, H. & Brohawn, S. G. Cryo-EM structures of the channelrhodopsin ChRmine in lipid nanodiscs. Nat. Commun. 13, 4842 (2022).
Kato, H. E. & Nureki, O. Crystal structure of channelrhodopsin, a light-gated cation channel - all cations lead through the monomer. Biophysics (Nagoya-shi) 9, 57–61 (2013).
Morizumi, T. et al. Structures of channelrhodopsin paralogs in peptidiscs explain their contrasting K(+) and Na(+) selectivities. Nat. Commun. 14, 4365 (2023).
Kato, H. E. et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature 482, 369–374 (2012).
Kim, Y. S. et al. Crystal structure of the natural anion-conducting channelrhodopsin GtACR1. Nature 561, 343–348 (2018).
Zhang, M., Shan, Y., Xiao, L., Zhao, L. & Pei, D., Cryo-EM structures of kalium channelrhodopsins KCRs. Preprint at BioRxiv https://doi.org/10.1101/2022.11.09.515798 (2022).
Volkov, O. et al. Structural insights into ion conduction by channelrhodopsin 2. Science 358, eaan8862 (2017).
Tajima, S. et al. Structural basis for ion selectivity in potassium-selective channelrhodopsins. Cell 186, 4325–4344 (2023).
AzimiHashemi, N. et al. Synthetic retinal analogues modify the spectral and kinetic characteristics of microbial rhodopsin optogenetic tools. Nat. Commun. 5, 5810 (2014).
Zhang, M., Shan, Y., Zhao, L., Li, X. & Pei, D., Ion selectivity and activation mechanism for kalium channelrhodopsins. Preprint at BioRxiv https://doi.org/10.1101/2023.07.22.550149 (2023).
Weng, J. et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 98, 13–23 (1999).
Sun, H., Molday, R. S. & Nathans, J. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem. 274, 8269–8281 (1999).
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G. & Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 (2005).
Saari, J. C. Vitamin A metabolism in rod and cone visual cycles. Annu Rev. Nutr. 32, 125–145 (2012).
Kuhne, J. et al. Unifying photocycle model for light adaptation and temporal evolution of cation conductance in channelrhodopsin-2. Proc. Natl. Acad. Sci. USA 116, 9380–9389 (2019).
Acknowledgements
We would like to thank the Cryo-EM Facility of Westlake University for providing cryo-EM, High-Performance Computing Center of Westlake University for computation support and Wenwen Zhang and Xiaoyan Xu (Mass Spectrometry & Metabolomics Core Facility of Westlake University) for LC-MS analysis. This work was supported by National Natural Science Foundation of China (92068201) and Key R&D Program of Zhejiang (2024SSYS0029).
Author information
Authors and Affiliations
Contributions
M.F.Z. conceived the project. Y.Y.S. and M.F.Z. designed the experiments. Y.Y.S., L.P.Z., M.Y.C., and X.L., prepared the cryo-EM sample and performed the electrophysiological study. Y.Y.S. and M.F.Z. collected cryo-EM data. M.F.Z. performed image processing, built the model, analyzed data, and wrote the manuscript draft. D.Q.P. and M.F.Z. supervised the project. All authors contributed to the manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yifan Cheng, Huanghe Yang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shan, Y., Zhao, L., Chen, M. et al. Channelrhodopsins with distinct chromophores and binding patterns. Nat Commun 15, 7292 (2024). https://doi.org/10.1038/s41467-024-51811-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51811-x
This article is cited by
-
Suppression of epileptic seizures by transcranial activation of K+-selective channelrhodopsin
Nature Communications (2025)
-
Structural insights into light-gating of potassium-selective channelrhodopsin
Nature Communications (2025)
