
Abstract
DNA polymerase ζ (Pol ζ) plays an essential role in replicating damaged DNA templates but contributes to mutagenesis due to its low fidelity. Therefore, ensuring tight control of Pol ζ’s activity is critical for continuous and accurate DNA replication, yet the specific mechanisms remain unclear. This study reveals a regulation mechanism of Pol ζ activity in human cells. Under normal conditions, an autoinhibition mechanism keeps the catalytic subunit, REV3L, inactive. Upon encountering replication stress, however, ATR-mediated phosphorylation of REV3L’s S279 cluster activates REV3L and triggers its degradation via a caspase-mediated pathway. This regulation confines the activity of Pol ζ, balancing its essential role against its mutations causing potential during replication stress. Overall, our findings elucidate a control scheme that fine tunes the low-fidelity polymerase activity of Pol ζ under challenging replication scenarios.
Similar content being viewed by others


Introduction
To mitigate the detrimental effect of DNA damage on DNA replication, cells employ a mechanism known as DNA damage tolerance (DDT) to warrant continuous DNA replication across damaged templates1,2. In eukaryotes, the prominent pathway for DDT is the translesion DNA synthesis (TLS), in which DNA lesions are directly bypassed by highly specialized TLS polymerases that possess larger catalytic pockets, enabling them to accommodate DNA mismatches or covalent modifications occurring on the template DNA3,4,5. Due to the challenging nature of accurate base pairing caused by DNA lesions, there is an increased likelihood of incorporating incorrect nucleotides during TLS. In addition, TLS polymerases generally lack 3’−5’ exonuclease activity6,7, which serves as a critical proofreading mechanism for DNA polymerase. This combination significantly increases the possibility of introducing mutations or errors into the DNA sequence. As a result, TLS is considered an error-prone pathway that helps cells overcome replication blockage caused by various DNA lesions.
DNA polymerase ζ (Pol ζ) is the key TLS polymerase essential for extending the aberrant primer termini opposite DNA lesions8,9,10. Pol ζ is a multi-subunit enzyme composed of the catalytic subunit Rev3/REV3L, a regulatory subunit REV7, and two accessory subunits, Pol 31/Pol D2 and Pol 32/Pol D3, which are also integral to the replicative polymerase Pol δ. In line with the low fidelity typically seen with TLS polymerases, Rev3/REV3L inherently lacks proofreading capabilities due to the absence of 3’−5’ exonuclease activity. Given this characteristic of its catalytic subunit and its critical role in the error-prone TLS pathway, Pol ζ is responsible for the majority of DNA mutations observed in eukaryotic cells11,12,13,14,15,16. Considering the dual requirements for uninterrupted replication and high fidelity, precise regulation of Pol ζ activity during replication stress is crucial.
Previous studies have indicated that stress-induced mono-ubiquitination of proliferating cell nuclear antigen (PCNA) at lysine residue 164 is crucial for recruiting Pol ζ to the lesion sites. This recruitment process is bridged by REV1, the master orchestrator of TLS, via its interactions with both mono-ubiquitinated PCNA and two components Pol ζ complex: REV7 and Pol 32/Pol D39,17,18,19,20,21,22. However, the prolonged duration of PCNA mono-ubiquitination following stress induction raises questions about its ability to precisely control Pol ζ activity as needed23. The revelation that the active form of Pol ζ shares two accessory factors with replicative polymerase δ suggests a “polymerase switch” hypothesis in the activation of Pol ζ-mediated DNA synthesis24,25,26,27. Nevertheless, the impact of these accessory factors on the fining regulation of Pol ζ activity in response to challenged conditions remains to be determined. Furthermore, technical challenges arising from the large size of its catalytic subunit hamper efforts to unravel the activation mechanism of Pol ζ via in vitro biochemistry study. Therefore, despite extensive research over the past five decades establishing pivotal roles of Pol ζ in safeguarding DNA replication and in the development of damage-induced and spontaneous mutations, the molecular underpinnings governing its essential, yet mutagenic polymerase activity remain enigmatic.
Here, we describe an role of N70, the N-terminal cleavage product of REV3L, in providing tight “on-site” autoinhibition on REV3L. Our data further show that the stress-induced phosphorylation of the S279 cluster within N70 counteracts its inhibition and acts as a dual regulator for both activation and scavenging mechanisms of REV3L. These findings provide insights into how the low-fidelity polymerase activity of Pol ζ is precisely regulated to effectively mitigate the risks associated with both replication fork stalling and error-prone DNA synthesis during replication stress response.
Results
Pol ζ is activated by replication stress
We initially aimed to address whether Pol ζ activation responds to replication stress. However, Pol ζ-mediated DNA synthesis had not been directly monitored in mammalian cells. Thus, we first set out to establish two specific assays to evaluate the impact of replication stress on Pol ζ-mediated DNA synthesis in human cell.
First, Pol ζ has been reported to be much less sensitive to aphidicolin (APH) compared to replicative DNA polymerases α, δ, and ε28,29. Based on this knowledge and utilizing the REV3L conditional knockout cell line established in the preceding study30, we can employ the REV3L-dependent and APH-insensitive DNA synthesis as a specific indicator of Pol ζ activity. Strikingly, in comparison to DMSO-treated cells, a 24 h hydroxyurea (HU) block led to a significant increase in APH-insensitive EdU incorporation in cells expressing REV3L (Fig. 1a). Importantly, either depletion of REV3L or interference of Pol ζ recruitment at lesion sites by blocking the REV1-REV7 interaction with a small molecule compound JH-RE-0631, significantly repressed APH-insensitive EdU incorporation upon HU block (Fig. 1a, b), indicating that HU block significantly stimulates Pol ζ-mediated DNA synthesis.

Aphidicolin (APH)-insensitive EdU incorporation (a, b) and the amount of REV3L-C at active replication forks (c, d) were monitored in HCT116-R-REV3L cells upon indicated treatments. DOX, 20 ng/mL doxycycline; HU, 2 mM hydroxyurea; REV1i, 2 μM JH-RE-06. Left panel: Representative images. Scale bar, 50 μm in (a, b) 20 μm in (c, d). Right panel: Quantitative analyses results. Sample size and mean of each measurement are indicated. P values were determined using the two-sided Mann-Whitney test. Each experiment was independently replicated at least twice, yielding similar results each time. Source data are provided as a Source data file.
During its maturation, REV3L undergoes proteolytic processing into two distinct parts30. This results in a smaller 70-kDa N-terminal fragment, referred to as N70, and a larger fragment that exceeds 310-kDa at the C-terminal, designated as REV3L-C. Despite being processed, the two cleavage fragments continue to maintain their association within the Pol ζ complex30. To directly monitor the effect of replication stress on DNA polymerase activity of Pol ζ, we developed a monoclonal antibody specifically targeting REV3L-C, which contains the conserved DNA polymerase catalytic domain (Supplementary Fig. 1a,b). Strikingly, by employing an EdU-Click-coupled proximity ligation assay (PLA), we observed a robust increase in the amount of REV3L-C at actively replicating sites in cells released from a 24 h HU block compared to DMSO-treated cells (Fig. 1c), although HU treatment significantly decreased overall DNA synthesis (Supplementary Fig. 1c). The PLA signal specifically reflects Pol ζ-mediated DNA synthesis because it is substantially reduced upon either REV3L depletion or treatment with JH-RE-06 to block Pol ζ recruitment at lesion sites in HU-treated cells (Supplementary Fig. 1d and Fig. 1d).
Moderate but significant increases in both REV3L-dependent DNA synthesis and REV3L-C/EdU PLA signals were also observed after UV irradiation, indicative of a stimulating effect of UV irradiation on Pol ζ-mediated DNA synthesis (Supplementary Fig. 2). In contrast to nucleotide shortage following HU block32, UV irradiation primarily leads to the formation of cyclobutane pyrimidine dimers (CPDs) and (6-4) photoproducts33, both of which can severely impair the progression of replication forks. Taken together, these data indicate that replication stress stimulates DNA synthesis mediated by Pol ζ.
ATR-mediated REV3L phosphorylation
Intriguingly, concomitant with replication stress-induced Pol ζ activation, a high-molecular-weight smear of N70, the N-terminal cleavage product of REV3L, was detected by western blot analyses in cells exposed to a broad spectrum of replicating-blocking agents (Fig. 2a and c). Notably, the stress-induced gel mobility shift of N70 was largely eliminated by treating the lysates with calf intestinal alkaline phosphatase (CIAP) in vitro (Fig. 2b, c), indicating that REV3L-N70 undergoes post-translational phosphorylation in response to replication stress in human cells.

a–d N70 was monitored by immunoblotting. In (b) cell extracts obtained 4 h after 10 J/m2 ultraviolet (UV) irradiation were incubated with CIAP at a concentration of 0, 62.5, 125, and 250 U/mL for 30 min at 37 °C prior to analyses. In (c) the gel mobility of N70 was monitored after 4 h indicated treatments. CPT, 3 µM camptothecin; HU, 2 mM hydroxyurea; MTX, 1 µM mitoxantrone; NCS, 3 µM neocarzinostatin; Etop, 50 µM etoposide. In (d) Prior to 10 J/m2 UV irradiation, cells were treated for 1 h with caffeine (Caf, 10 mM), 2-aminopurine (2-AP, 20 mM), VE821 (VE, 4 µM), KU-60019 (KU, 20 µM) and NU7026 (NU, 10 µM). Untreated cells were included in (c, d) as control (UC). e HCT116-3F-REV3L cells were transduced by the indicated shRNA virons for 3 days prior to analyses. f The indicated cells were grown in the medium with or without 1 µg/mL doxycycline (DOX) for 5 days prior to analyses. g HEK293 cells were transfected with various REV3L expression constructs. Fourty-eight hours after transfection, the UV-induced gel mobility shift of N70 was analyzed as described in (a). h A summary of the UV-induced phosphosites within N70 revealed by IP-MS analysis (top) and alignment of the sequences flanking the S279 cluster (bottom). Asterisks mark conserved residues. Serine at SQ motifs are highlighted in red. i REV3L protein was immunoprecipitated by anti-FLAG antibody from HCT116-3F-REV3L cells under denaturing condition following the indicated treatments. Phosphorylation of the S279 cluster was examined by western blot using a phosphopeptide-specific antibody. UC, untreated control; UV, 10 J/m2, 4 h recovery; HU, 2 mM hydroxyurea, 4 h exposure; DDP, 20 µM cisplatin, 4 h exposure. j ATR inhibitor (ATRi) pretreatment (12 h) suppresses the stress-induced phosphorylation of N70 at the S279 cluster. DM, 0.1% DMSO; ATRi, 4 μM VE-821. In (a–g, i, and j) each experiment was independently replicated at least twice, yielding similar results each time. Source data are provided as a Source data file.
We utilized UV irradiation as the stimulus to compare the effect of a panel of kinase inhibitors on N70 phosphorylation. Strikingly, a marked decrease in the high-molecular-weight smear of N70 was observed in cells treated with caffeine, 2-Aminopurine (2-AP), and VE821, but not with KU-60019 and NU7026 (Fig. 2d). Since both caffeine and 2-AP are inhibitors of ATM and ATR kinases34,35, KU-60019 and NU7026 are selective inhibitors of ATM and DNA-PKcs36,37, respectively, while VE821 selectively inhibits ATR38, these results reveal a specific contribution of ATR to stress-induced N70 phosphorylation. Consistently, the depletion of ATR in shATR-virion transduced cells or the ATR-conditional knockout cells led to a concurrent decrease in N70 phosphorylation (Fig. 2e, f), further highlighting the crucial role of ATR in stress-induced phosphorylation of REV3L-N70.
ATR exhibits a preference for phosphorylating serine (S) and threonine (T) residues followed by glutamine (Q)39. There are a total of nine SQ/TQ motifs within N70. Consistent with ATR-mediated phosphorylation of N70, introducing alanine substitutions at the serine and threonine residues of all nine SQ/TQ motifs in the REV3L protein resulted in a significant reduction in the phosphorylated forms of N70 upon UV irradiation when the resulting full-length REV3L mutant, referred to as 9 A, was transiently expressed in HEK293 cells (Fig. 2g, lane 6 in comparison with lane 5). Conversely, making phosphor-mimetic aspartate substitutions at the same positions within all nine SQ/TQ motifs in the REV3L-9D mutant did not produce a similar effect on the UV-induced gel mobility shift of N70 (Fig. 2g, lane 8 in comparison with lane 7).
We next employed immunoprecipitation coupled with mass spectrometry to analyze stress-induced phosphosites on N70. The MaxQuant PTM score revealed eight potential phosphosites within N70, with S279, S282, and S286 residues (named S279 cluster) precisely situated within the SQ motifs (Fig. 2h, top panel, Supplementary Data 1). Among those, S279 and S286 show significant conservation across vertebrates (Fig. 2h, bottom panel). Based on this information, we developed the antibody specifically against a peptide carrying phosphorylated S279 and S286 residues. Taking advantage of this reagent, we verified the induction of phosphorylation at the S279 cluster in cells treated with various replicating-blocking agents (Fig. 2i). Importantly, the ATR inhibitor AZD673840 markedly suppressed the phosphorylation of REV3L S279 cluster in cells exposed to UV and HU treatments (Fig. 2j). Collectively, our data provide unequivocal evidence supporting the notion that REV3L undergoes phosphorylation at the S279 cluster upon exposure to various replicating-blocking agents, with ATR acting as a key mediator of this stress-induced phosphorylation event.
ATR is crucial for Pol ζ activation
The discovery that stress-induced Pol ζ activation is concomitant with ATR-mediated phosphorylation of REV3L raises an intriguing question: does stress-induced Pol ζ activation rely on ATR? Supporting this idea, we found that the treatment of ATR inhibitor AZD6738 results in a marked reduction in the number of REV3L-C/EdU PLA foci observed in cells 2 h after exposure to UV radiation (Fig. 3a). Considering that ATR inhibition does not negatively impact, but rather significantly enhances, overall DNA synthesis in these cells (Supplementary Fig. 3), the decrease in REV3L-C/EdU PLA foci reflects a specific reduction in Pol ζ recruitment at actively replicating sites.

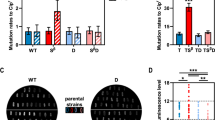
a REV3L-C/EdU PLA assay was performed in 20 ng/mL doxycycline-induced HCT116-R-3F-REV3L (R-REV3L) cells 2 h after UV irradiation, with or without 0.3 µM AZD6738 (ATRi) treatment. Scale bar, 20 μm. b, c Proteins of interest in R-REV3L cells were analyzed by western blot 24 h after a 2 h exposure to various doses of cisplatin (DDP). d HCT116-R-ATR cells were grown with (+) or without (-) 1 μg/mL doxycycline (DOX) for 5 days and subsequently exposed to cisplatin (DDP). Cellular viability was measured 48 h after cisplatin exposure. e Partial sequencing result of the RT-PCR fragment from the DOX-treated HCT116-R-3F-REV3L-3D mutant cells. f APH-insensitive EdU incorporation was assessed in the indicated cells as described in Fig. 1a. Scale bar, 50 µm. g REV3L-C/EdU PLA assays were performed under no-stressed conditions. WT, R-REV3L cells, 20 ng/mL DOX; 3D-Mut, R-REV3L-3D mutant cells,1 μg/mL DOX. Scale bar, 10 µm. h Proteins of interest were assessed as described in (b). i The phosphorylation of RPA2 and H2AX was monitored in both wild-type (WT) and mutant (3D-Mut) cells following a 20 h exposure to either DMSO or various doses of cisplatin (DDP), with or without the presence of ATRi. Mean and the standard error of the mean (SEM) from two independent experiments are presented. j, k Mitotic chromosome abnormalities (j) and cisplatin sensitivity (k) were analyzed in indicated cells 24 h after a 2 h exposure to cisplatin (DDP). Number of chromosomes (j) and cells (k) scored for each condition is indicated. Scale bar in (j) 40 m. Errors shown in quantification results in (b, d, h, j and k) represent the SEM from three independent experiments. Sample sizes are indicated in (a, f, g, j, and k). P values were calculated using the Mann–Whitney test (two-sided) for analyses (a, f and g) and using the chi-square test (two-sided) for analyses (j) and (k). Each experiment was independently replicated at least twice, yielding similar results each time. Source data are provided as a Source data file.
By utilizing the REV3L conditional knockout cell line, we established that the persistence of RPA2 phosphorylation and the accumulation of γ-H2AX following a cisplatin exposure are two biological consequences attributed to the absence of Pol ζ activity in HCT116 cells (Fig. 3b). RPA2 phosphorylation reflects the presence of replication stress41, whereas γ-H2AX is a marker of double-strand DNA breaks42. Consistent with the negative effect of ATR inhibitor on UV-induced Pol ζ activation, both RPA2 phosphorylation and γ-H2AX levels were markedly increased 24 h after a 2 h cisplatin exposure in cells where ATR, but not ATM, is inhibited or depleted (Fig. 3c, Supplementary Fig. 4). Significantly, the additional depletion of REV3L did not significantly aggravate cisplatin-induced phosphorylation of both proteins in cells treated with ATR inhibitor (Fig. 3c), indicating that ATR and Pol ζ function within the same pathway to promote cellular tolerance to cisplatin-induced stress. Consistently, cells depleted of ATR showed increased sensitivity to cisplatin (Fig. 3d), further supporting the essential role of ATR for Pol ζ to work properly in DDT pathway.
Since the S279 cluster of REV3L undergoes ATR-dependent phosphorylation in response to replication stress, we next asked whether this phosphorylation event is critical for stress-induced Pol ζ activation. To address this, we generated the HCT116-R-3F-REV3L-3D-Mut cell (REV3L-3D), in which the expression of REV3L-3D mutant carrying three phosphor-mimetic aspartate substitutions at S279, S282, and S286 residues can be induced by the addition of doxycycline (DOX) to the growth medium. A two-step genome editing strategy was employed to introduce targeted modifications at endogenous REV3L locus (Supplementary Fig. 5). Successful targeting was verified by both restriction mapping and sequencing a 657-bp region flanking the mutated nucleotides in the REV3L-3D cDNA (Supplementary Fig. 5d and Fig. 3e).
We next assessed APH-insensitive DNA synthesis in cells expressing either wild-type REV3L or the phosphor-mimicking REV3L-3D mutant. Strikingly, in the absence of external stress, the induction of REV3L-3D mutant resulted in a notable increase in APH-insensitive DNA synthesis, whereas no such effect was observed with the wild-type REV3L (Fig. 3f). Consistently, the REV3L-C/EdU PLA assay demonstrated a significantly higher presence of REV3L-C at actively replicating sites in cells expressing the REV3L-3D mutant under non-stressed conditions, compared to those expressing wild-type REV3L (Fig. 3g). Together, these data indicate that phosphor-mimetic aspartate substitutions at the S279 cluster of REV3L can circumvent the requirement for replication stress to activate Pol ζ-mediated DNA synthesis in REV3L-3D mutant cells.
Given the crucial role of Pol ζ in cellular tolerance to cisplatin, it is plausible to speculate that the constitutive activation of Pol ζ in REV3L-3D mutant cells facilitates the bypass of cisplatin-induced replication stress, thereby minimizing the adverse effects of cisplatin exposure on replication and genomic integrity. In agreement with this idea, we observed a significant decrease in the accumulation of phosphorylated RPA2 and γ-H2AX in REV3L-3D mutant cells compared to cells expressing wild-type REV3L 24 h after a 2 h exposure to cisplatin (Fig. 3h). Importantly, even though a 20 h exposure to cisplatin at low dosages (e.g., 1 and 2 μM) slightly increased the amount of γ-H2AX in REV3L-3D mutant cells, simultaneous inhibition of ATR did not result in further stimulation of phosphorylated RPA2 and γ-H2AX within these mutant cells (Fig. 3i). Conversely, when ATR was inhibited during cisplatin treatment, there was a significant increase in the levels of phosphorylated RPA and H2AX in cells expressing the wild-type REV3L. These data collectively support that aspartic acid substitutions at REV3L S279 cluster override the essential role of ATR for Pol ζ to function in tolerating cisplatin-induced stress. In line with an elevated ability to mitigate cisplatin-induced stress in 3D cells, compared to cells expressing wild-type REV3L, we observed a decrease in chromosome aberrations in 3D-mutant cells 24 h after a 2 h exposure to cisplatin at 20 μM and 40 μM, although the difference at 20 μM cisplatin treatment was not statistically significant (Fig. 3j). Consistently, a statistically significant decrease in cellular sensitivity to cisplatin treatment was also observed in cells expressing 3D mutant compared to those expressing wild-type REV3L (Fig. 3k). Taking these observations into account, we conclude that ATR is required for the activation of Pol ζ-mediated DNA synthesis.
S279 cluster phosphorylation destabilizes N70
We noticed a significant decline in the steady levels of REV3L in the REV3L-3D mutant cells compared to their parent cells that express wild-type REV3L under similar inducible conditions (Fig. 4a–c). To directly assess the impact of aspartate substitutions on N70 stability, the half-lives of N70 in wild-type and mutant cells were assessed by blocking de novo protein synthesis with cycloheximide (CHX) for different time. Indeed, N70-3D mutant is profoundly less stable than the wild-type N70 (Fig. 4d).

a–c REV3L mRNA and protein levels were analyzed by RT-PCR (a) and western blot (b) in indicated cells, with abundance quantified via ImageJ (c). Cell types: WT, HCT116-R-3F-REV3L(het), 20 ng/mL doxycycline (DOX); 3D-Mut, HCT116-R-3F-REV3L-3D-Mut (het), 1 μg/mLDOX. d, Cells pre-treated with 100 µM cycloheximide underwent western blot analysis (left panel, asterisk marks nonspecific bands) and N70 quantitative assessment (right panel, blue for WT, orange for 3D-Mut). SEM reflects the variance over three independent experiments in (c, d). e Cells were exposed to 10 µM MG132 for 2 h. Proteins of interest were examined by western blot. A non-specific band detected by anti-REV3L-C antibody is indicated by an asterisk. f Cells were treated as described in (e). 5 × 106 cells was utilized for His-pull down experiment in the presence of 6 M guanidine hydrochloride. Flag-tagged REV3L-N70 and K48-linked ubiquitin chains in whole cell extract (WCE) and pull-downed products were monitored by western blot. (g, h) Post UV or cisplatin exposure, N70 levels were measured at specified times and doses in cells with wild-type REV3L. i UV-irradiated HCT116-R-3F-REV3L cells showed dose-dependent K48-linked polyubiquitination analyzed as in (f). j, k The abundance of N70 in cells expression wild-type and the phosphor-mimicking REV3L-3D mutant was monitored via immunoblotting. In (j) R-REV3L-3D mutant cells were cultured for indicated times in the presence of either 0.3 µM AZD6738 (ATRi) alone or ATRi in combination with 10 µM MG132. In (k) after UV irradiation, cells expressing wild-type REV3L were grown into a medium with either 0.1% DMSO (DM) or ATRi for zero (R0), one (R1), and two (R2) hours. 200 μg of whole cell extract was utilized in (b) and (e) while 100 μg was utilized for (d) (h) (j) and (k). Each experiment was independently replicated at least twice, yielding similar results each time. Source data are provided as a Source data file.
Interestingly, the treatment of MG132, an inhibitor of the 26S proteasome, markedly increased N70 abundance in the mutant cells (Fig. 4e). Proteins carrying K48-linked poly-ubiquitin chains are specifically targeted for degradation by 26S proteasome43. Consistently, there was a notable increase in the amount of the K48-linkage-specific polyubiquitin chains conjugated to N70 in REV3L-3D mutant cells as compared to the parent cells expressing wild type REV3L (Fig. 4f), indicating that phosphor-mimetic aspartate substitutions at the S279 cluster of REV3L promote the ubiquitination and proteasome-mediated degradation of N70-3D mutant.
We next asked whether stress could trigger the degradation of wild-type REV3L-N70 by a similar ubiquitin-proteasome system. In agreement with this idea, UV irradiation led to a significant reduction in the steady levels of N70 in cells expressing wild-type REV3L (Fig. 4g). Importantly, analogous to N70-3D mutant, wild-type N70 was markedly stabilized by MG132 in both UV-irradiated and cisplatin-exposed cells (Fig. 4g, h). Moreover, also akin to N70-3D mutant, a dose-dependent increase in the abundance of K48-linked polyubiquitin chains conjugated to N70 was observed in wild-type cells following UV irradiation (Fig. 4i, lines 4’-6’). Additionally, while ATR inhibitor has no observable effect on N70-3D stability (Fig. 4j), it notably enhanced the stability of wild type N70 after UV irradiation (Fig. 4k). These findings collectively provide compelling evidence supporting that ATR-mediated phosphorylation of the S279 cluster stimulates the ubiquitination and proteasome-mediated degradation of REV3L-N70.
N70 negatively modulates REV3L activity
We previously demonstrated that N70 forms a post-cleavage complex (PCC) with the REV3L-C30. Surprisingly, we observed a significant reduction in the amount of co-immunoprecipitated REV3L-C with N70 from HCT116-R-3F-REV3L cells that were exposed to UV irradiation, compared to the mock-treated cells (Fig. 5a). Of note, inhibition of ATR resulted in a notable increase in the co-immunoprecipitation of REV3L-C with N70 in cells exposed to UV irradiation (Fig. 5b). Strikingly, phosphor-mimetic aspartate substitutions at the S279 cluster resulted in an almost 90% reduction in the association between the cleavage products of REV3L (Fig. 5c). Consistently, we also observed a concurrent reduction in the co-immunoprecipitation of REV1 (83.9 ± 3.8% reduction) and REV7 (80.0 ± 2.3% reduction) with N70 in 3D-mutant cells (Fig. 5c), both of which interact with REV3L PCC through its C-terminal cleavage fragment REV3L-C44,45. These findings unexpectedly reveal that phosphorylation of the S279 cluster facilitates the disassembly of REV3L PCC.

a Two hours after UV irradiation at a dose of 50 J/m2, whole cell lysates from 3 × 107 HCT116-R-3F-REV3L cells (with 20 ng/mL DOX) were subjected to co-IP analyses using anti-FLAG antibodies. The levels of indicated proteins in lysates (Input) and co-IP samples (FLAG IP) were monitored by western blot analyses. UC: untreated control; UV: 50 J/m2 UV-irradiated cells. b Cells were treated similarly to what was described in (a), except that either 0.1% DMSO (DM) or 0.3 µM AZD6738 (ATRi) was added 1 h prior to UV irradiation. Co-IP analyses were conducted as described in (a). c HCT116-R-3F-REV3L (het) (WT) and HCT116-R-3F-REV3L-3D-Mut (het) (3D) cells were grown in the presence of 2 ng/mL and 2 µg/mL of DOX, respectively. After a 4 h exposure to 10 µM MG132, cells were subjected to co-IP analyses conducted as described in (a). d Left panel: HCT116-3F-mAID-REV3L cells, with or without 50 J/m2 UV irradiation, were continuously grown in the presence of 0.9% NaCl (Buf) or 1 mM indole-3-acetic acid (IAA) for 3 h. Right Panel: After a 2 h exposure to 10 µM cisplatin (DDP), cells were continuously grown in normal medium for the indicated times prior to a 3 h treatment with 1 mM IAA. Proteins of interest in 200 μg of whole cell lysates were monitored via western blot. The co-depletion index (cDI) for REV3L-C is calculated according to the formula: cDI = [AmAID-N70/AREV3L-C]IAA / [AmAID-N70/AREV3L-C]Buf. In this equation, ‘A’ represents the amount of the proteins of interest. e After UV irradiation at 50 J/m2, HCT116-3F-mAID-REV3L cells were continuously grown in the presence of 0.9% NaCl (Buf) or 1 mM IAA for 3 h in combination with either 0.1% DMSO or 1 µM AZD6738 (ATRi). cDI for each measurement was assessed as described in (d). Each experiment was independently replicated at least twice, yielding similar results each time. Source data are provided as a Source data file.
The discovery that replication stress triggers both the degradation of REV3L-N70 and the disassembly of REV3L PCC, while simultaneously activating Pol ζ, suggests an unanticipated possibility that N70 functions as an inhibitory module of REV3L. This might occur through forming a tightly associated complex with REV3L-C, which carries the conserved catalytic active residues of DNA polymerases. To evaluate the impact of N70 on REV3L activation, we established an auxin-inducible degron (AID) system46 to rapidly degrade N70 (Supplementary Fig. 6). By utilizing CRISPR/Cas9-mediated genome-editing approach, an in-frame mini-AID (mAID) cassette was integrated at both REV3L loci immediately upstream to the start codon of the REV3L gene to generate HCT116-3F-mAID-REV3L cells. An expression cassette of TIR1, which is an auxin-responsive F-box protein derived from Oryza sativa, was subsequently introduced into HCT116-3F-mAID-REV3L cells by lentiviral transduction. In the presence of auxin, TIR1 can form functional SCF-TIR1 ubiquitin ligase with the endogenous subunits conserved in eukaryotic cells. Thus, the addition of indole-3-acetic acid (IAA), a natural auxin, to the growth medium can induce rapid degradation of N70 fused with a mAID tag.
After a 3 h treatment, steady levels of mAID-REV3L-N70 in the IAA-treated cells were reduced to ~5–15% of those observed in cells treated with the solvent alone (Supplementary Fig. 6e). Since N70 remains tightly associated with REV3L-C after TASP1-mediated cleavage, mAID-N70 degradation can simultaneously lead to an IAA-induced decline in REV3L-C abundance (Fig. 5d, lanes 2 in comparison with lane 1). Remarkably, after adjusting the steady levels of REV3L-C to match those of mAID-N70 across samples with or without IAA treatment, the co-depletion index (cDI) for REV3L-C showed a significant reduction in the IAA-induced co-depletion of REV3L-C in cells that had been treated with UV and cisplatin (Fig. 5d, lanes 4 and 6’ in comparison with 3 and 5’). These observations provide additional evidence supporting the conclusion from co-immunoprecipitation studies that genotoxic stress triggers the disassembly of the REV3L PCC. Furthermore, indicating the crucial involvement of ATR in UV-induced disassembly of REV3L PCC, treatment with an ATR inhibitor significantly enhanced the IAA-induced co-depletion of REV3L-C in cells subjected to UV irradiation, reflected in a notable increase of cDI for REV3L-C (Fig. 5e, lane 4 in comparison with lane 2).
Remarkably, in line with the idea that N70 negative regulates REV3L, both phosphorylated RPA2 and γ-H2AX showed significantly decrease in cells expressing mAID-REV3L after IAA treatment compared to those treated with solvent only, 24 h after a 3 h exposure to various doses of cisplatin (Fig. 6a, middle-top panel). Importantly, the effect of IAA on cellular tolerance to cisplatin is specifically linked to the degradation of mAID-tagged N70, as no such beneficial impact of IAA in reducing cisplatin-induced stress and DNA damages was observed in cells expressing FLAG-tagged REV3L (Fig. 6a, middle-bottom and bottom panels). Moreover, consistent with the enhanced catalytic activity of REV3L in cells treated with IAA, a notable rise in the presence of REV3L-C at active replication forks was observed using the REV3L-C/EdU PLA assay in IAA-treated cells after exposure to cisplatin (Fig. 6b). Furthermore, the ectopic over-expression of wild-type N70 significantly compromised the cellular tolerance to cisplatin in REV3L-3D mutant cells, while having no significant effect on cells expressing wild-type REV3L (Fig. 6c).

a IAA treatment reduces the accumulation of phosphorylated RPA2 and γ-H2AX induced by cisplatin (DDP) in cells expressing FLAG-mAID-tagged REV3L (mAID-REV3L), but not FLAG-tagged REV3L (FLAG-REV3L). Top panel: Experimental workflow. Middle panel: Representative images from the same blots processed in parallel. Bottom panel: Quantitative results of indicated proteins from three independent experiments. b IAA treatment significantly increases the amount of REV3L-C at active replication forks in cells expressing mAID-REV3L following cisplatin (DDP) exposure. Scale bar, 10 µm. c A HA-tagged N70 expression cassette was stably integrated into both HCT116-R-3F-REV3L (het) (WT) and HCT116-R-3F-REV3L-3D-Mut (het) (3D-Mut) cells by lentiviral transduction. Phosphorylation of RPA2 and H2AX was monitored via immunoblotting 24 h after a 2 h exposure to cisplatin (DDP) at indicated doses. Quantitative results from two independent experiments are presented. d Schematic of the targeted REV3L locus in HCT116-R-3F-REV3L-SP cells (top panel). Cells were grown in the absence of DOX for 2 days prior to applying a doxycycline (DOX) induction for the indicated time. 3F-REV3L-C, 3F-REV3L-N70, and β-actin levels were monitored by western blot (bottom-left panel). e, f APH-insensitive EdU incorporation assay and REV3L-C/EdU PLA were performed in indicated cells as described in (d) (bottom-right panel). Scale bar, 50 µm in e and 20 µm in (f). 200 μg of whole cell lysates were used in (d) whereas 50 μg of lysates were utilized in (a) and (c). In (b–e) and (f) sample size and mean of each measurement are indicated. P values were determined using the Mann-Whitney test (two-sided). Each experiment was independently replicated at least twice, yielding similar results each time. Source data are provided as a Source data file.
To directly investigate the functional activity of REV3L-C during DA synthesis, we generated a derivative of HCT116 cells, termed HCT116-R-3F-REV3L-SP (R-REV3L-SP), in which each endogenous REV3L gene is split into two separate expression cassettes (Fig. 6d, top panel and Supplementary Fig. 7). This division results in the independent transcription of the genomic DNA sequences encoding N70 and REV3L fragments, which are driven by a tet-inducible promoter (Ptet) and a constitutively active CMV promoter (PCMV), respectively. After a 2-day withdrawal of DOX, which resulted in the exclusive expression of REV3L-C in REV3L-SP cells (Fig. 6d, bottom-left panel, lane 1), measurements were taken for both APH-insensitive DNA synthesis and the REV3L-C/EdU PLA signals. Remarkably, cells expressing only REV3L-C exhibited a significant increase in these parameters under non-stressed conditions, compared to those expressing wild-type REV3L (Fig. 6e, f). This observ ation highlights the functional activity of REV3L-C when solely expressed in DNA synthesis. Significantly, a 6-h treatment with DOX led to a noticeable reduction in both APH-insensitive DNA synthesis and in the REV3L-C/EdU PLA signals in the DOX-withdrawn REV3L-SP cells (Fig. 6d–f). Taken together, these findings clearly demonstrate that REV3L-C is the catalytically active component of Pol ζ, and the presence of N70 negatively regulates its activity.
Scavenging process of activated REV3L
Despite that our data uncover a mechanism governing Pol ζ activation, there remains a gap in our understanding regarding how activated Pol ζ is deactivated once the replication stress is resolved. One possible mechanism has been suggested by the observation that the ubiquitin-proteasome mediated degradation of N70-3D is accompanied by a significant reduction in the steady levels of REV3L-C in the mutant cells (Fig. 4b, c). Considering that replication stresses stimulate N70 degradation, we asked whether exposure to such stresses could also lead to a decrease in REV3L-C abundance in wild type cells. In agreement with this idea, we found that the half-life of REV3L-C is indeed markedly reduced following UV irradiation (Fig. 7a), along with a rapid degradation of N70 (Supplementary Fig. 8). Significantly, the UV-induced REV3L-C/EdU PLA signals, which peaked at the 2 h mark after UV irradiation, rapidly declined to a level comparable to that in untreated cells by the 4 h mark (Fig. 7b), indicating that Pol ζ-mediated DNA synthesis cannot be sustained for an extended period after UV irradiation. Together, these data suggest that the compromised stability of REV3L-C contributes to the timely cessation of Pol ζ activity following the release of N70 inhibition.

a HCT116-3F-REV3L cells were treated with 100 μM cycloheximide (CHX) for indicated times. REV3L-C abundance was assessed by immunoblotting. Before CHX treatments, cells were either left untreated (UC) or exposed to 50 J/m2 UV (UV). SEM for quantitative results (right panel, orange for UC, blue for UV) reflects the variance over three independent experiments. b Cells were subjected to a REV3L-C/EdU PLA assay at one (R1), two (R2), and four (R4) hours post 50 J/m2 UV irradiation. P values were determined using the two-sided Mann–Whitney test. c Genotoxic stress induces a labile ~ 90 kD fragment (C90) recognized via anti-REV3L-C immunoblotting. Cells were grown with (+) or without (-) 10 μM MG132 for 8 h after exposure to various stresses. DMSO, 0.1% DMSO; UV, 50 J/m2 UV; DDP, 80 μM cisplatin, 2 h exposure followed by a 22 h recovery; HU, 2 mM, 24 h exposure. d The appearance of stress-induced C90 is dependent on REV3L induction. Cells were grown with (+) or without (-) 20 ng/mL doxycycline (DOX) for 2 days and subjected to treatments as described in (c). REV3L-C and C90 fragment were assessed by immunoblotting. e Inactivating caspases results in a significant reduction of stress-induced C90. Cells were treated as described in (c), except that 25 μM z-VAD-FMK (z-VAD), a pan-caspase inhibitor, was added 1 h before 10 μM MG132 treatment. f, g HCT116-R-3F-REV3L-SP (R-REV3L-SP) cells were grown with (+) or without (-) 1 µg/mL doxycycline (DOX) for 2 days and assayed for indicated mRNAs by RT-PCR (f) and proteins by immunoblotting (g). Similar results were observed in at least two individual colonies detected in parallel. h Ectopic expression of N70 stabilizes REV3L-C. An HA-tagged N70 expression cassette was stably integrated into R-REV3L-SP cells via lentiviral transduction. After being grown with (+) or without (-) 10 ng/mL DOX for 2 days, cells were assessed for REV3L-C and N70 by immunoblotting. EV, empty virion; HA-N70, virion carries HA-N70 expression cassette. Each experiment in (c–e) and (h) was independently replicated at least twice, yielding similar results each time. Source data are provided as a Source data file.
In contrast to proteasome-mediated N70 degradation, MG132 treatment did not increase the abundance of REV3L-C in cells exposed to various replication-blocking agents (Fig. 7c). However, the anti-REV3L-C antibody revealed a ~90-kDa fragment, referred to as C90 in subsequent experiments, which showed notable stabilization after MG132 treatment (Fig. 7c). Importantly, the expression of C90 was reliant on REV3L induction (Fig. 7d), suggesting that C90 is a site-specific proteolytic product of REV3L-C induced in response to replication stresses.
To further investigate the nature of this site-specific cleavage event, we conducted a small screen utilizing protease inhibitors available in the laboratory. Intriguingly, a substantial decrease in the amount of stress-induced C90 was observed when cells were exposed to genotoxic insults in the presence of z-VAD-FMK (z-VAD), a pan-caspase inhibitor (Fig. 7e).
Significantly, a decline in C90 levels coincides with a simultaneous increase in intact REV3L-C, indicating that inhibition of caspases prevents the conversion of REV3L-C into C90 following exposure to replication stresses.
Given the simultaneous induction of rapid degradation of N70 and caspase-mediated cleavage of REV3L-C upon replication stress, we sought to explore whether N70 could potentially stabilize REV3L-C by utilizing REV3L-SP cells, in which REV3L-C is constantly expressed, whereas N70 can be induced upon exposure to DOX. While the steady levels of REV3L-C mRNA did not significantly vary between cells with and without DOX treatment (Fig. 7f), a substantial decline in the abundance of REV3L-C was observed in cells where N70 was depleted upon DOX withdrawal (Fig. 7g). Notably, the ectopic expression of exogenous HA-tagged N70 augmented the steady levels of REV3L-C, even in the presence of a mild level of endogenous N70 induction (Fig. 7h, lanes 3 and 4 in comparison with lanes 1 and 2). This finding clarifies that the increase in REV3L-C abundance in DOX-treated cells is not a result of read-through from DOX-induced N70 transcription. We thus concluded that coexistence of REV3L-N70 plays a crucial role in stabilizing REV3L-C.
Discussion
In this study, we unveil a mechanism in human cells to ensure precise control over the activity of Pol ζ (Fig. 8). The key aspect of our model is the coordination between the activation and the scavenging processes of REV3L, its catalytic subunit. The molecular basis of this lies in the crucial roles of N70 in both inhibiting REV3L activation and upholding its stability, with each exerting opposing effects on Pol ζ activity.

The diagrams depict how REV3L, the catalytic subunit of Pol ζ, is repressed to unintended activation during normal growth, and how the relief of its autoinhibition and the scavenging process of activated REV3L are tightly coordinated during cellular response to genotoxic stress. I. AUTOINHIBITION: Upon synthesis, REV3L undergoes post-translational cleavage to yield two fragments, N70 and REV3L-C, which subsequently form a tightly associated post-cleavage complex (PCC). N70 serves as an autoinhibitory module, negatively modulating the catalytic activity of REV3L-C. As a result, the error-prone DNA synthesis activity of Pol ζ is stringently maintained in an inactive state. II. ACTIVATION: Autoinhibition of REV3L is unleashed by the stress-induced phosphorylation of N70 in an ATR-mediated manner. Specifically, ATR is activated in response to replication stress induced by a range of external and internal insults. Activated ATR triggers the phosphorylation of N70 at the S279 cluster, which leads to the disassembly of PCC. PCC disassembly relieves the inhibition of N70 on REV3L-C, resulting in the activation of Pol ζ. III. SCAVENGING: PCC disassembly triggers a rapid degradation of N70 through the ubiquitin-proteasome system. While N70 degradation can potentially enhance Pol ζ activation, it also causes instability in REV3L-C. Caspase(s) is involved in REV3L-C degradation. Of note, replication stress can activate caspases, potentially synergizing with the scavenging process of REV3L-C to enhance its efficiency.
Undoubtedly, auto-inhibition of REV3L via its N70 fragment can effectively prevent the deleterious impact of inappropriate REV3L activation immediately upon its synthesis. This is highly significant, when considering the error-prone nature of the DNA synthesis mediated by Pol ζ. Moreover, the localized control of the catalytic activity of REV3L guarantees a precise and rapid activation response of Pol ζ once it becomes necessary, ensuring the uninterrupted progression of replication when encountering challenges. However, this discovery initially seems paradoxical. The first 333 amino acids at the N-terminus domain (NTD) of human REV3L show homology within the B family’s replicative polymerases47, and we also observed a significant enhancement in translesion DNA synthesis activity when N70 was ectopically expressed in cells solely expressing REV3L-C in our previous investigation30. Considering the new insights from this study, the beneficial effect of N70 on REV3L-C activity observed previously can be attributed to its role in enhancing REV3L-C stability. This enhancement significantly increases the cellular capacity to alleviate stress induced by cisplatin. Through this elegant coordinating mechanism, human cells can effectively prevent the unintentional activation of Pol ζ while concurrently ensuring that sufficient REV3L is available to facilitate replication across damaged templates when an activating signal is present.
How might N70 act as an auto-inhibitory module of REV3L? The catalytic core of all DNA polymerases consists of three conserved components: the thumb, palm, and fingers domains47,48,49. The conformational switch between open and closed states in the fingers domain enables the formation of an active catalytic complex during nucleotide incorporation50,51. Unlike other B-family DNA polymerases, which typically possess two highly conserved αxA and αxB helices in their fingers domain, REV3L features an additional αF helix, similar to its yeast counterpart Rev3 (Supplementary Fig. 9a). Notably, the two conserved helices of the fingers domain (residues 2685–2736) are part of human REV3L-C, while the distinctive αF helix (residues 258–276) is located in N70. The distribution of these helices across various cleavage fragments suggests that PCC disassembly could significantly impact the conformation of the fingers domain, potentially affecting Pol ζ‘s catalytic activities. This hypothesis has been corroborated by our findings.
Positioned immediately downstream of the N-Terminal Domain (NTD), the distinctive αF helix in Rev3/REV3L may enhance the structural stability of the fingers domain through its linkage to the NTD via an embedded connecting linker. This may also endow the NTD with additional capabilities to modulate the fingers domain’s conformational switch. Interestingly, a connecting loop in yeast Rev3 exhibits inherent flexibility, as evidenced by the absence of a 7-residue segment in its cryo-EM structure52. This flexibility allows the linker to accommodate necessary conformational changes in the fingers domain during the catalytic process. However, the connecting linker is reduced from 38 residues in yeast Rev3 to 17 residues in human REV3L according to AlphaFold predictions (Supplementary Fig. 9a, top panel). Additionally, a significant portion of this linker in human REV3L forms an additional helix, contrasting with the flexible loop in yeast Rev3 (Supplementary Fig. 9a, top and middle panels). These sequence and structural disparities, developed during evolutionary processes, likely reduce the αF helix’s flexibility, potentially restricting the fingers domain to a specific conformational state. Remarkably, the fingers domain in the predicted human REV3L structure in fact resembles the inactive open conformation seen in the yeast Rev3 structure in its apo state, in contrast to the Considering this, we speculate that the increased rigidity of the connector between the NTD and the αF helix within the fingers domain in human REV3L enables N70 to act as a self-inhibitory module, negatively affecting REV3L’s catalytic activity by holding the fingers domains in an inactive conformation.
A shortened connector between the NTD and the αF helix is conserved in REV3L across metazoans (Supplementary Fig. 9b). Notably, the evolutionary development of a rigid connector is closely linked to the emergence of a unique TASP1 cleavage site located downstream of the αF helix, a feature that is absent in yeast Rev3. The auto-inhibitory function of N70, combined with the differences in the connector between the NTD and fingers domains in human REV3L versus its yeast counterpart, implies a profound evolutionary shift in the regulatory mechanisms of Pol ζ activation and highlights the critical role of TASP1-mediated cleavage in the maturation process of human REV3L, a finding that has been substantiated in our preceding study30.
An important aspect of our model is that phosphorylation of the S279 cluster serves as a dual regulator to counteract REV3L auto-inhibition and simultaneously facilitate its scavenging process by disrupting the association between N70 and REV3L-C. The dual functionality of a single phosphorylation event in launching both activating and scavenging processes of REV3L unveils a surprising yet reasonable mechanism that inherently restricts the availability of Pol ζ for DNA synthesis within a specific period following its activation. Intriguingly, our data further reveal the involvement of caspase-mediated cleavage in terminating Pol ζ activity. It is noteworthy that replication stress can concurrently lead to the activation of caspase(s)53, thereby synergizing with the disassembly of REV3L PCC to enhance the efficiency of its scavenging process. In this context, the implication that caspases could potentially be targeted to keep a tight rein on Pol ζ activity under stressed conditions, such as chemotherapy, may create new opportunities to selectively combat drug resistance during cancer chemotherapy.
Significantly, consistent with the crucial roles of Pol ζ in resolving replication issues, the phosphorylation of the S279 cluster can be specifically induced by a variety of replication-blocking agents in an ATR-mediated manner. There is extensive documentation that highlights the specific response of ATR to replication stress54,55,56. Upon activation, ATR in fact phosphorylates a range of factors that play important roles in diverse cellular responses to alleviate replication stress. Utilizing ATR-mediated phosphorylation to counteract the autoinhibition of REV3L can reinforce the coordination between lesion bypass and other stress-response mechanisms, enabling cells to tackle the detrimental instances with optimal efficiency and accuracy. In line with this concept, two recent studies have revealed that REV3L facilitates fork protection by limiting nuclease access and/or by promoting fill-in DNA synthesis5,57, supporting the impacts of Pol ζ in safeguarding DNA replication. Precise targeting of REV3L activation and/or scavenging processes holds the potential to provide deeper insights into the biological impacts of Pol ζ in cellular response to stress.
Methods
Plasmid and reagents
Constructs, antibodies, shRNAs, chemicals and oligonucleotides used in this study are described in Supplementary Tables 1–4 and Supplementary Data 2, respectively. Details of plasmid construction are available upon request. All other reagents were Sigma-Aldrich products unless noted.
Cell culture, transfection and lentivirus-based transduction
Human embryonic kidney (HEK) cell line 293 T (ATCC) was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, PAN Biotech, Germany) and 2 mM L-Glutamine (Solarbio) at 37 °C with 5% CO2. HCT116 cells (ATCC) and its derivatives were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FBS and 2 mM L-Glutamine at 37 °C with 5% CO2. Cell lines used in this study are described in Supplementary Table 5.
For transient transfection in HEK293 cells, cells were transfected using Lipofectamine 3000 reagent following the manufacturer’s instructions (Thermo Fisher), and harvested for subsequent functional analyses 36 h after transfection. Lentiviruses were prepared following the Lentiweb Protocol (https://www.addgene.org/protocols/lentivirusproduction). Virus was collected 48 h after transfection, filtered through a 0.4 μm membrane, and used directly to infect cells in the presence of 8 μg/ml polybrene.
DNA, RNA, and protein analysis
Cells (~1 x 105, cultured in a 96-well plate) were washed with PBS and lysed in 50 μL of lysis buffer [10 mM Tris-Cl (pH7.5), 10 mM NaCl, 0.5% Sarkosyl, 1 mg/ml Proteinase K]. After incubating at 50 °C for 3 h, 100 μL of NaCl/EtOH (75 mM NaCl in ethanol at -20 °C) was added to each well. Genomic DNA was precipitated after centrifuging at 2800 x g for 20 min at 4 °C.
Total RNAs were isolated following the TRIzol method (Thermo Fisher). One microgram total RNA was used to generate first-strand cDNAs in 20 μL reverse transcription reaction mixture [50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 200 U M-MLV reverse transcriptase (Beyotime), 0.5 mM dNTP, and 20 pmol random hexamer]. Primers for PCR amplification are described in Supplementary Data 2.
Cells (~3 x 106) were harvested, washed with PBS and lysed in 150 μL 1 x Laemmli loading buffer. Following brief sonication and boiling, aliquots (50–200 μg) were subjected to 10% SDS-PAGE. After transfer to nitrocellulose membrane, proteins were detected via immunoblotting. Antibodies used are described in Supplementary Table 2.
β-actin or MCM3 served as a loading control for immunoblotting analyses, while β-actin or RBM9 was used as a loading control for RT-PCR and genomic DNA PCR analyses.
CRISPR/Cas9-mediated targeting
The CRISPR/Cas9-mediated homologous recombination technique was used to integrate the donor sequences into their respective target sites. The targeting construct and the plasmid carrying the corresponding guide RNA expression cassette were transfected together into HCT116 cells using Lipofectamine 3000 reagent (Thermo Fisher). Positive colonies were first selected by their resistance to the relative antibiotics. The precise integration of the donor sequences was further verified by genomic DNA PCR, western blot or directly sequencing the genomic DNA PCR or RT-PCR products as described in the text.
To generate the N-terminal-tagged 3xFLAG-ATR conditional knockout lines in HCT116 cells, the pKS-ATR-N-tetO-target-construct and the pX330-ATR-N were co-transfected into HCT116 cells (Supplementary Fig. 4a). The cells were selected in a medium containing 300 μg/mL Puromycin for 12 days. Clones were screened by genomic DNA PCR using primers flanking the sgRNA targeted region and by western blot for 3xFLAG-ATR in whole cell lysates (Supplementary Fig. 4b–d and Fig. 2f).
HCT116-3F-REV3L-3D-Mut (het) cell line was generated by two steps (Supplementary Fig. 5a). First, the pKS-REV3L-S279-3D-target-construct and pX335-REV3L-S279 (1#-4#) were transfected together into HCT116-R-3F-REV3L (het) cells. The region starting from the 3’ half of the seventh intron to the ninth exons of the REV3L gene in the heterozygous HCT116-R-3F-REV3L cells were substituted with a fragment containing a splice acceptor site, a Puro expression cassette flanked by two loxP sites, a second splice acceptor sites and the eighth and the ninth exons of the REV3L gene bearing defined point mutations via CRISPR-Cas9n-mediated homologous recombination. The Puro expression cassette in the positive colony was subsequently excised by transient expression of Cre recombinase. Clones were first screened by genomic DNA PCR. Precise splicing of the RNAs transcribed from the mutant REV3L allele was further verified by HhaI restriction-fragment length polymorphism (RFLP) analyses and directly sequencing the RT-PCR fragments (Supplementary Fig. 5b–d and Fig. 3e).
To generate the N-terminal-tagged 3xFLAG-mAID-REV3L conditional knockout lines in HCT116 cells, the pKS-REV3L-N-mAID-target-construct and the pX330-REV3L-N were transfected together into HCT116 cells (Supplementary Fig. 6a). The cells were selected in a medium containing 300 μg/mL Puromycin for 12 days. Clones were screened by genomic DNA PCR using primers flanking the sgRNA targeted region and by western blot for 3xFLAG-mAID-REV3L in whole cell lysates (Supplementary Fig. 6b, c).
To generate HCT116-R-3F-REV3L-SP cell line, the pKS-REV3L-D525-CMV-target-construct and the pX330-REV3L-D525-2# plasmid were transfected together into HCT116-R-3F-REV3L cells (Supplementary Fig. 7a). The cells were selected in a medium containing 5 μg/mL Blasticidin for 12 days. Clones were screened by genomic DNA PCR using primers flanking the sgRNA targeted region and by western blot for 3xFLAG-REV3L-C in whole cell lysates (Supplementary Fig. 7b, c and Fig. 7g).
EdU incorporation assay
Cells were grown on glass coverslips and subjected to a EdU labeling as described in the text. Following fixation with 4% formaldehyde at room temperature, cells were permeabilized with 0.5% Triton X-100 in PBS for 10 min. To perform Click chemistry, the cells were incubated at room temperature for 30 min with a solution composed of 2 mM CuSO4, 2.5 μM azide-Alexa Fluor 488 (Thermo Fisher) and 10 mM Sodium ascorbate in PBS. After staining, cells were washed three times with PBS carrying 0.1% Tween 20. Subsequently, the cells were incubated with 1 μg/mL DAPI for 10 min to label the nuclei. Coverslips were then mounted in DABCO mounting medium and imaged using a Ni-E microscope (Nikon) equipped with a Zyla sCMOS (VDG-152V-C1E-FI) digital camera from Andor. Image acquisition and analysis were performed using NIS-Elements AR software (Nikon). A background signal, calculated as the mean of the lowest 5% of EdU signals, was subtracted from each sample to enable quantitative analysis of EdU incorporation, under the assumption that 10–40% of cells in the population do not exhibit DNA synthesis. The Mann-Whitney test (two-sided) was employed for statistical analysis. A significance threshold of p < 0.05 was applied to determine the statistical significance of observed differences.
Identification of phosphosites on REV3L-N70 through denaturing immunoprecipitation coupled with LC-MS/MS analysis
Cell pellets were lysed in one packed cell volume (PCV) of 8 M Urea lysis buffer [8 M Urea, 100 mM Tris–HCl (pH 7.5), 1% Triton X-100] and subjected to sonication at 20% output setting for 10 second. The lysate was subjected to nuclease digestion by adding two PCV of nuclease digestion buffer [50 mM Tris–HCl (pH7.5), 4 mM MgCl2] with 100 U/ml benzonase (Sigma). After a 30 min incubation at room temperature, the lysate was mixed with four PCV of dilution buffer [50 mM Tris–HCl (pH7.5), 0.75% Triton X-100, 360 mM NaCl] and incubated with anti-FLAG antibody-conjugated M2-agarose beads (Sigma) overnight at 4 °C. Immunoprecipitants were washed five times with wash buffer [50 mM Tris–HCl (pH 7.5), 1 M Urea, 500 mM NaCl, 0.5% Triton X-100, 0.2 mM EDTA], eluted with the 0.2 mg/ml 3xFLAG peptide in elution buffer [50 mM Tris–HCl (pH 7.5), 1 M Urea, 150 mM NaCl, 0.5% Triton X-100, 0.2 mM EDTA] and resolved on a 4–20% gradient SDS-PAGE gel for colloidal blue staining analysis.
Three gel blocks centered at 70-kDa, one from untreated cell and two from the UV-irradiated cells, were subjected to LC-MS/MS analysis for potential phosphorylation site identification at Beijing Protein Innovation. Specifically, the gel blocks were first extracted by 8 M Urea. A 10 µg aliquot of extracted proteins from each sample was subjected to reduction by adding 5 mM dithiothreitol (DTT) solution and incubating at 37 °C for 1 h and then was alkylated with 10 mM iodoacetamide at room temperature for 30 min in darkness. The sample was diluted 4 times by adding 25 mM ammonium bicarbonate buffer. Trypsin digestion (trypsin: protein = 1:50) was performed at 37 °C overnight.
Nanoflow LC-MS/MS analysis of tryptic peptides was conducted on a quadrupole Orbitrap mass spectrometer (Orbitrap Fusion Lumos, Thermo Fisher Scientific, Bremen, Germany), coupled to an EASY nLC 1200 ultra-high pressure system (Thermo Fisher Scientific) via a nano-electrospray ion source. A total of 500 ng of peptides were loaded onto a 25 cm column (150 μm inner diameter) packed with ReproSil-Pur C18-AQ 1.9-µm silica beads (Beijing Qinglian Biotech Co., Ltd, Beijing, China). Peptides were separated using a gradient from 8–12% buffer B over 7 min, then increased from 12–30% buffer B over 48 min, and stepped up to 40% in 10 min, followed by a 15 min wash at 95% buffer B at a flow rate of 600 nL per minute. Buffer A consisted of 0.1% formic acid in water, and buffer B was 80% acetonitrile with 0.1% formic acid in water. The total run duration was 80 min, with the column temperature maintained at 60 °C using an in-house-developed oven. The mass spectrometer operated in a ‘top-40’ data-dependent mode, collecting MS spectra in the Orbitrap mass analyzer (120,000 resolution, 340–1400 m/z range) with a maximum ion injection time of 50 ms. The most intense ions from the full scan were isolated with an isolation width of 1.6 m/z. Following higher-energy collisional dissociation (HCD) with a normalized collision energy (NCE) of 35, MS/MS spectra were collected in the Orbitrap with a maximum ion injection time of 35 ms. Precursor dynamic exclusion was enabled with a duration of 16 s.
The MS raw data were processed by using the MaxQuan suite (version 1.5.2.8) in a default setting. MS2 spectra were searched against the UniProtKB/Swiss-Prot database (2017_07 release, Homo sapiens = 20,202 entries). The Sequest HT search engine was used, and parameters were specified as follows: specific or semi-specific strict trypsin (trypsin/P) with maximum of two missed cleavages, minimum peptide length of 6, fixed carbamidomethylation of cysteine residues (+57.022 Da), variable modifications for oxidation of methionine residues (+15.995 Da), acetylation on protein N terminus (+42.016 Da), methylation of lysine and arginine residues (+14.016 Da) and phosphorylation of serine, threonine and tyrosine residues (+79.966 Da), precursor mass tolerance of 15 ppm and a fragment mass tolerance of 0.02 Da for MS2 spectra collected in the Orbitrap. Other parameters include modified score ≥ 40, delta modified score ≥ 8 and peptide length ≥ 7 amino acids. Percolator was used to filter peptide spectral matches and peptides to maintain a false discovery rate (FDR) of <1%. After these were spectrally assigned, the peptides were assembled into proteins. These proteins were then further filtered, based on the combined probabilities of their constituent peptides, to achieve a final FDR of <1%.
Analyses of ubiquitin conjugates on REV3L-N70
To monitor the ubiquitination of N70, HCT116-R-REV3L cells were pre-treated with 10 μM MG132 for 2 h, followed by denaturing His pull-down analyses of His-3F-REV3L proteins using BeaverBeads Nickel (Beaver). The cell pellet was lysed in a buffer containing 6 M guanidine hydrochloride, 100 mM Tris-HCl (pH 7.4). The lysate was then incubated with beads for 3 h at room temperature. The beads were washed with buffer containing 8 M Urea, 100 mM Tris-HCl (pH 7.4), 0.5% NP40, 1 M NaCl, and 20 mM imidazole. Ubiquitin conjugates on REV3L-N70 were analyzed by immunoblotting.
Co-immunoprecipitation (co-IP)
For co-immunoprecipitation (co-IP) of proteins associated with endogenous 3xFLAG-REV3L, cells were suspended in extraction buffer [50 mM Tris-HCl (pH7.4), 10% (V/V) glycerol, 400 mM NaCl, 2 mM MgCl2, 1 mM DTT, Protease Inhibitor Cocktail (Mei5bio)] and subjected to sonication at 20% output setting for 1 s, repeated three times, with a 10-s incubation on ice between bursts. The lysate was diluted by adding one volume of buffer containing [50 mM Tris-HCl (pH7.4), 2 mM MgCl2, 0.1% NP40, 1 mM CaCl2, 100 U/mL Benzonase, 50 μg/mL RNaseA, 1 mM DTT, Protease Inhibitor Cocktail (Mei5bio)]. After incubation at 4 °C for 30 min, the lysate was centrifuged at 16,500 x g for 10 min at 4 °C. The supernatant was collected and mixed with washed anti-FLAG antibody-conjugated M2-agarose beads (Sigma) overnight at 4 °C. Captured proteins were eluted by heating in 1x Laemmli buffer [60 mM Tris-HCl (pH 6.8), 2% SDS, 10% Glycerol] at 95 °C for 10 min and analyzed by immunoblotting.
Indirect immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for 10 min at room temperature. The cells were then permeabilized with 0.5% Triton X-100 in PBS for 10 min and blocked with 5% bovine serum albumin in PBS for 30 min. Primary antibodies were incubated for 1 h at room temperature. Secondary antibodies were incubated for 45 min at room temperature. The cells were subsequently counterstained with 40,6-diamidino-2-phenylindole (DAPI) for 10 min to stain the nucleus. Images were generated with a Nikon ECLIPSE Ni microscope (Nikon, Minato, Tokyo, Japan) and NIS-ELEMENT software (advanced research version, Nikon). A rabbit monoclonal antibody targeting REV3L-C was utilized as the primary antibody (this work, 1:1000) to monitor REV3L in cells with or without REV3L induction as described in the text.
REV3L-C/EdU Proximity Ligation Assay (PLA)
PLA technology was employed to monitor the amount of REV3L-C associated with nascent DNA in situ. After various treatments, cells were labeled with 20 μM EdU for 10 min immediately prior to harvest and were then permeabilized with Buffer I [25 mM HEPES (pH 7.0), 50 mM NaCl, 1% Triton X-100, 300 mM Sucrose, 3 mM MgCl2] for 5 min at 4 °C. Following wash with PHEM buffer [25 m MHEPES (pH 7.0), 60 mM PIPE (pH 6.9), 10 mM EGTA, 3 mM MgCl2] twice, cells were fixed in 4% formaldehyde in PBS (w/v) for 10 min at room temperature. After twice washes with PBS, coverslips were incubated with blocking solution (5% BSA, 5% Normal goat serum and 0.05% Tween 20 in PBS) for 30 min at 37 °C and subjected to a Click-iT reaction [2.5 µM Biotin-azide, 2 mM CuSO4 and 10 µM Sodium ascorbate in PBS] for 30 min at room temperature. After three washes with PBST (0.1% Tween 20 in PBS), coverslips were simultaneously incubated with the primary antibodies targeted to REV3L-C (this work, 1:1000) and Biotin (Jackson ImmunoResearch Labs, 1: 1000) for 60 min at 37 °C in a humidified chamber. Slides were then washed three times with PBS, followed by the proximity ligation assay conducted by using the Duolink In Situ Red Starter Kit Mouse/Rabbit (Sigma) according to the manufacturer’s instructions. Z-stack and max intensity projection images were generated with a Nikon ECLIPSE Ni fluorescent microscope (Nikon, Minato, Tokyo, Japan) and NIS-ELEMENT software (advanced research version, Nikon). The number of PLA foci within the nuclei was quantified, and statistical analysis was conducted using the Mann-Whitney test (two-sided). A significance threshold of p < 0.05 was applied to determine the statistical significance of observed differences.
Chromosome integrity analysis
100 ng/mL colcemid was added to the culture 45 min prior to harvesting. Cells were then harvested by trypsin digestion and subjected to a 12 min incubation in 75 mM KCl at 37 °C. After multiple washes in Carnoy’s fixative (3:1 methanol: acetic acid), cells were dropped onto slides to prepare the mitotic chromosome spread. Slides were then baked overnight at 60 °C and subjected to chromosome staining with 100 μg/mL acridine orange. Imagines were captured by Nikon ECLIPSE Ni fluorescent microscope equipped with a 100x objective. The chi-square test (two-sided) was employed for statistical analysis.
Cell survival assay
Cells were treated with the indicated dosages of cisplatin for 2 h. After releasing into normal growth medium for 24 h, an appropriate number of cells were plated into 6-well plates and cultured for nine to 14 days under normal growth conditions. The survival colonies were stained with 5 μg/mL ethidium bromide in 50% ethanol and counted. The chi-square test (two-sided) was employed for statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data of mass spectrometry analyses for potential phosphosites on REV3L-N70 are not available anymore. The parameters employed for LC-MS/MS data analyses and information about the identified phosphorylated REV3L-N70 peptides are provided as Supplementary Data 1. Source data are provided with this paper in Figshare with the identifier: https://doi.org/10.6084/m9.figshare.26097082.
References
Marians, K. J. Lesion bypass and the reactivation of stalled replication forks. Annu. Rev. Biochem. 87, 217–238 (2018).
Chang, D. J. & Cimprich, K. A. DNA damage tolerance: when it’s OK to make mistakes. Nat. Chem. Biol. 5, 82–90 (2009).
Waters, L. S. et al. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154 (2009).
Sale, J. E., Lehmann, A. R. & Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152 (2012).
Adeyemi, R. O. et al. The Protexin complex counters resection on stalled forks to promote homologous recombination and crosslink repair. Mol. Cell 81, 4440–4456 e4447 (2021).
Yang, W. Damage repair DNA polymerases Y. Curr. Opin. Struct. Biol. 13, 23–30 (2003).
Lange, S. S., Takata, K. & Wood, R. D. DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110 (2011).
Martin, S. K. & Wood, R. D. DNA polymerase ζ in DNA replication and repair. Nucleic Acids Res. 47, 8348–8361 (2019).
Makarova, A. V. & Burgers, P. M. Eukaryotic DNA polymerase zeta. DNA Repair. 29, 47–55 (2015).
Gan, G. N., Wittschieben, J. P., Wittschieben, B. & Wood, R. D. DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 18, 174183 (2008).
Lawrence, C. W. Cellular functions of DNA polymerase zeta and Rev1 protein. Adv. Protein Chem. 69, 167–203 (2004).
Lemontt, J. F. Induction of forward mutations in mutationally defective yeast. Mol. Gen. Genet 119, 27–42 (1972).
Quah, S. K., von Borstel, R. C. & Hastings, P. J. The origin of spontaneous mutation in Saccharomyces cerevisiae. Genetics 96, 819839 (1980).
Zhong, X. et al. The fidelity of DNA synthesis by yeast DNA polymerase zeta alone and with accessory proteins. Nucleic Acids Res. 34, 4731–4742 (2006).
Harfe, B. D. & Jinks-Robertson, S. DNA polymerase zeta introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Mol. Cell 6, 1491–1499 (2000).
Lawrence, C. W. & Maher, V. M. Eukaryotic mutagenesis and translesion replication dependent on DNA polymerase zeta and Rev1 protein. Biochem. Soc. Trans. 29, 187–191 (2001).
Garg, P., Stith, C. M., Majka, J. & Burgers, P. M. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase zeta. J. Biol. Chem. 280, 23446–23450 (2005).
Guo, C. et al. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell 23, 265–271 (2006).
Pustovalova, Y. et al. Interaction between the Rev1 C-terminal domain and the PolD3 subunit of Polζ suggests a mechanism of polymerase exchange upon Rev1/Polζ-dependent translesion synthesis. Biochemistry 55, 2043–2053 (2016).
D’Souza, S. & Walker, G. C. Novel role for the C terminus of Saccharomyces cerevisiae Rev1 in mediating protein-protein interactions. Mol. Cell. Biol. 26, 8173–8182 (2006).
Acharya, N., Johnson, R. E., Pagès, V., Prakash, L. & Prakash, S. Yeast Rev1 protein promotes complex formation of DNA polymerase zeta with Pol32 subunit of DNA polymerase delta. Proc. Natl Acad. Sci. USA 106, 9631–9636 (2009).
Kikuchi, S., Hara, K., Shimizu, T., Sato, M. & Hashimoto, H. Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J. Biol. Chem. 287, 3384733852 (2012).
Niimi, A. et al. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl Acad. Sci. USA 105, 16125–16130 (2008).
Johnson, R. E., Prakash, L. & Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc. Natil Acad. Sci. USA 109, 12455–12460 (2012).
Baranovskiy, A. G. et al. DNA polymerase delta and zeta switch by sharing accessory subunits of DNA polymerase delta. J. Biol. Chem. 287, 17281–17287 (2012).
Makarova, A. V., Stodola, J. L. & Burgers, P. M. A four-subunit DNA polymerase zeta complex containing pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 40, 11618–11626 (2012).
Lee, Y. S., Gregory, M. T. & Yang, W. Human Pol zeta purified with accessory subunits is active in translesion DNA synthesis and complements Pol eta in cisplatin bypass. Proc. Natl Acad Sci. USA 111, 2954–2959 (2014).
Takeuchi, R. et al. Purification of Drosophila DNA polymerase zeta by REV1 protein-affinity chromatography. Biochem. J. 382, 535–543 (2004).
Nelson, J. R., Lawrence, C. W. & Hinkle, D. C. Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science (New York, N.Y.) 272, 1646–1649 (1996).
Wang, F. et al. Site-specific proteolytic cleavage prevents ubiquitination and degradation of human REV3L, the catalytic subunit of DNA polymerase ζ. Nucleic Acids Res. 48, 3619–3637 (2020).
Wojtaszek, J. L. et al. A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178, 152–159.e111 (2019).
Bianchi, V., Pontis, E. & Reichard, P. Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J. Biol. Chem. 261, 16037–16042 (1986).
Pfeifer, G. P., You, Y. H. & Besaratinia, A. Mutations induced by ultraviolet light. Mutation Res. 571, 19–31 (2005).
Dimitrova, D. S. & Gilbert, D. M. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nature Cell Biol. 2, 686–694 (2000).
Sarkaria, J. N. et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 59, 4375–4382 (1999).
Golding, S. E. et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 8, 2894–2902 (2009).
Veuger, S. J., Curtin, N. J., Richardson, C. J., Smith, G. C. & Durkacz, B. W. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 63, 6008–6015 (2003).
Charrier, J. D. et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 54, 23202330 (2011).
Traven, A. & Heierhorst, J. SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. BioEssays News Rev. Mol. Cell. Dev. Biol. 27, 397–407 (2005).
Foote, K. M. et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J. Med. Chem. 61, 9889–9907 (2018).
Richard, D. J., Bolderson, E. & Khanna, K. K. Multiple human single-stranded DNA binding proteins function in genome maintenance: structural, biochemical and functional analysis. Crit. Rev. Biochem. Mol. Biol. 44, 98–116 (2009).
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S. & Bonner, W. M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 (1998).
Komander, D. & Rape, M. The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012).
Tomida, J. et al. REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase zeta. Nucleic Acids Res. 43, 1000–1011 (2015).
Netz, D. J. et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 8, 125–132 (2011).
Nishimura, K., Fukagawa, T., Takisawa, H., Kakimoto, T. & Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 (2009).
Yang, W. & Gao, Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu. Rev. Biochem. 87, 239–261 (2018).
Steitz, T. A. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 274, 17395–17398 (1999).
Jain, R., Aggarwal, A. K. & Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 53, 77–87 (2018).
Doublié, S., Sawaya, M. R. & Ellenberger, T. An open and closed case for all polymerases. Structure 7, R31–R35 (1999).
Joyce, C. M. & Benkovic, S. J. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry 43, 14317–14324 (2004).
Malik, R. et al. Structure and mechanism of B-family DNA polymerase ζ specialized for translesion DNA synthesis. Nat. Struct. Mol. Biol. 27, 913–924 (2020).
Gui, C. Y., Jiang, C., Xie, H. Y. & Qian, R. L. The apoptosis of HEL cells induced by hydroxyurea. Cell Res. 7, 91–97 (1997).
Flynn, R. L. & Zou, L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 36, 133–140 (2011).
Saldivar, J. C., Cortez, D. & Cimprich, K. A. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 18, 622–636 (2017).
Saxena, S. & Zou, L. Hallmarks of DNA replication stress. Mol. Cell 82, 2298–2314 (2022).
Paniagua, I. et al. MAD2L2 promotes replication fork protection and recovery in a shieldin-independent and REV3L-dependent manner. Nat. Commun. 13, 5167 (2022).
Acknowledgements
We thank Yuhua Yan, Fei Liu, Yan Zhang and Jiawei Zhu in Li lab for their assistance in generating knock-in cell lines and developing assays for monitoring Pol ζ activity. We would like to express our gratitude to Dr. Xinquan Wang (Tsinghua University, China) and Dr. Fenglin Guo (Tsinghua University, China) for help in human REV3L structure prediction using AlphaFold. We thank Dr. Wei Xiao (University of Saskatchewan, Canada) for his critical reading and discussion of the manuscript. This research was funded by the Beijing Municipal Natural Science Foundation [grant numbers 5242001 to X.L.], the Ministry of Science and Technology of the People’s Republic of China [grant number 2015CB910603 to X.L.] and the National Natural Science Foundation of China [grant number 31470780 to X.L.].
Author information
Authors and Affiliations
Contributions
X.L. designed and supervised the project, acquired funding and wrote the manuscript. C.L., S.F., P.L., Y.B., Y.W., Y.C., M.L., R.W., Y.S., Y.W., S.Z., R.W., L.G., M.L., Y.Z., F.W., S.G., S.F., J.W. and X.Q. performed investigation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kouji Hirota and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, C., Fan, S., Li, P. et al. A sophisticated mechanism governs Pol ζ activity in response to replication stress. Nat Commun 15, 7562 (2024). https://doi.org/10.1038/s41467-024-52112-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52112-z
This article is cited by
-
DNA polymerase α/primase extraction from chromatin by VCP/p97 restricts ATR activation during unperturbed DNA replication
Nature Communications (2025)
-
Human TLS DNA polymerase: saviors or threats under replication stress?
Molecular and Cellular Biochemistry (2025)
