
Abstract
Streptococcus mitis is a leading cause of infective endocarditis (IE). However, our understanding of the genomic epidemiology and pathogenicity of IE-associated S. mitis is hampered by low IE incidence. Here we use whole genome sequencing of 129 S. mitis bloodstream infection (BSI) isolates collected between 2001–2016 from clinically diagnosed IE cases in the UK to investigate genetic diversity, antimicrobial resistance, and pathogenicity. We show high genetic diversity of IE-associated S. mitis with virtually all isolates belonging to distinct lineages indicating no predominance of specific lineages. Additionally, we find a highly variable distribution of known pneumococcal virulence genes among the isolates, some of which are overrepresented in disease when compared to carriage strains. Our findings suggest that S. mitis in patients with clinically diagnosed IE is not primarily caused by specific hypervirulent or antimicrobial resistant lineages, highlighting the accidental pathogenic nature of S. mitis in patients with clinically diagnosed IE.
Similar content being viewed by others


Introduction
Infective endocarditis (IE) is a life-threatening microbial infection of the interior surface lining of the heart1, and is associated with serious multi-system complications and high mortality, which approaches 25–30% at 1 year despite optimal treatment1,2,3. Although IE is rare, with incidence estimated between 1.5 and 11.6 cases per 100,000 people per year4, hospitalisation rates in the USA have increased by 10.6% between 2000 and 20115. In Europe, the incidence has doubled over the last two decades6. In England, incidence of IE admissions rose by 86% from 26.9 cases/million in 2009–2010 to 50.0 cases/million in 2018–197. Streptococcus mitis, is a leading cause of IE8, although it is widely considered as a typical oral commensal9.
In his Gulstonian Lectures in 1885, William Olser described micrococci within the heart valve vegetations that characterise IE10. In 1931, prior to the antibiotic era, Streptococcus pneumoniae accounted for up to ~10% of IE cases11,12, frequently associated with infection elsewhere, such as pneumonia and meningitis, and was usually fatal11,13,14. However, since the introduction of penicillin and pneumococcal conjugate vaccines, S. pneumoniae now accounts for less than 1% of IE cases11. In a multivariable analysis using S. pneumoniae as a reference, S. mitis or S. oralis has been associated with a higher IE risk with an odds ratio of 31.6 (95% CI, 19.8–50.5)15. Despite the increasing clinical importance of S. mitis, little is known about the genomic epidemiology and pathogenicity of this pathogen.
S. mitis and S. pneumoniae are closely related genetically and belong to the same species complex, yet have strikingly different pathogenic potential16. S. pneumoniae invasiveness is strongly associated with the presence of the polysaccharide capsule, pneumolysin and other virulence factors17, however, it is unclear whether pneumococcal virulence factors shared with S. mitis also facilitate S. mitis disease18,19. It is noteworthy that although largely commensal, S. mitis genomes may possess a range of pneumococcal virulence genes, including those encoding the capsule, IgA1 protease, pneumolysin, and autolysin16. The polysaccharide capsule facilitates adherence, and diffusion of molecules through to the cell surface, provides resistance to specific and non-specific host immune responses, and prevents desiccation20. S. mitis strains have been demonstrated to have capsular polysaccharide synthesis (cps) loci typically resembling that found in the pneumococcus, which sometimes results in the expression of identical capsules such as those corresponding to capsule serotypes 1, 5, and 19A21,22. It has also been suggested that S. mitis isolates harbouring the lytA gene, encoding an autolysin protein, are more likely to be associated with invasive disease as this enzyme facilitates the release of the potent pore-forming exotoxin pneumolysin and inflammatory peptidoglycan and teichoic acids from lysed bacterial cells23,24,25. However, it is unclear how widely these pneumococcal virulence factors are distributed among invasive S. mitis isolates. Killian and colleagues proposed that S. pneumoniae, S. pseudopneumoniae and S. mitis lineages have evolved from a pathogenic pneumococcus-like progenitor26,27, and that commensal streptococci, including S. mitis, have subsequently lost the majority of their disease-causing virulence genes as they adapt to their upper respiratory tract niche. We and others have shown that S. pneumoniae and S. mitis continue to exchange genetic material, particularly antimicrobial resistance (AMR) genes28. It remains unknown whether the presence or absence of specific virulence-associated genes and genetic backgrounds or lineages increases the pathogenicity of certain S. mitis strains.
We therefore hypothesised that S. mitis bloodstream infection (BSI) in patients with clinically diagnosed IE is associated with specific independently acquired pathogenicity loci, or lineages which have acquired particular virulence factors or AMR profiles. However, testing of this hypothesis has so far been problematic for several reasons. Firstly, the Viridans group streptococci (VGS), which includes S. mitis, are highly heterogeneous, consisting of over 50 species that are often difficult to differentiate using traditional microbiology, molecular, and biochemical techniques29, frequently leading to misidentification30. Second, and probably the most important reason, is the limited availability of publicly available S. mitis whole-genome sequencing (WGS) data in nucleotide sequence repositories. This is partly driven by the lack of focus on S. mitis compared to the other more virulent members of the Streptococcus species, for example, S. pneumoniae, which causes life-threatening infections, including pneumonia, sepsis, and meningitis25,31.
Here, we sought to expand our understanding of the genomic epidemiology of S. mitis invasive disease by undertaking WGS of a large and unique collection of 129 well-characterised IE-associated S. mitis BSI isolates collected between 2001–2016 from national bacteraemia surveillance programmes led by the British Society of Antimicrobial Chemotherapy (BSAC) and UK Health Security Agency (UKHSA). We exploited our recently developed molecular typing schemes32 and phylogenetic analysis to investigate the population structure, and the distribution of virulence and AMR genes among the IE-associated S. mitis isolates. Our genomic dataset expands the availability of S. mitis WGS data in publicly available sequence repositories by over two-fold, including the provision of nearly all the genomes from IE-associated invasive disease isolates. We show that S. mitis BSI in patients with clinically diagnosed IE is not dominated by specific hypervirulent or AMR lineages, but the pathogenicity and virulence of these strains may be enhanced by the acquisition of virulence-promoting genes through horizontal gene transfer (HGT). Our findings provide further evidence supporting the hypothesis that all S. mitis lineages may similarly cause BSIs highlighting the opportunistic nature of S. mitis infections.
Results
Characteristics of the BSAC and UKHSA IE BSI S. mitis isolates
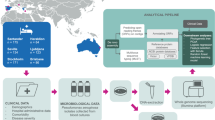
To characterise the genetic diversity, AMR gene profiles, and identify virulence genes associated with invasive S. mitis infection, we obtained and performed WGS of 217 presumed S. mitis isolates from patients with BSI and clinically diagnosed IE from the UK and Ireland between 2001 and 2016 (Fig. 1 and Supplementary Fig. 1). These isolates were collected as part of BSAC’s Resistance Surveillance Project (n = 172), and UKHSA’s voluntary identification service (n = 45). While the retrospective nature of our analysis has preluded the rigorous application of the modified Duke/European Society of Cardiology (ESC) 2023 diagnostic criteria for IE33, the isolates were all from patients where IE had been clinically diagnosed (and given the BSI, likely fulfilling the “definite” or “possible” category), and were referred to the reference laboratory for species confirmation and further antibiotic sensitivity testing. Two of 172 BSAC and three of 45 UKHSA isolates were not viable after bacterial culture of transport swabs and were, therefore, not sequenced. One UKHSA isolate subsequently failed WGS quality control due to low DNA. Accurate species determination among the VGS has been a challenge using conventional and molecular approaches34, therefore, we used a bioinformatic approach for species confirmation among the presumed S. mitis isolates obtained from BSAC and UKHSA.

The S. mitis BSI isolates analysed were collected from clinically diagnosed IE cases, between 2001 and 2016, submitted to BSAC and UKHSA. The analysis involved WGS, population structure analysis, AMR genotyping and phenotyping, and identification of virulence genes using genotyping and bacterial genome-wide association analysis approaches to identify hypervirulent and dominant S. mitis lineages. The map of the UK and Ireland was generated by the authors in R software using the maps v4.0.0 package (https://cran.r-project.org/web/packages/maps/). The confirmed S. mitis isolates are described in Supplementary Data 1 and 2.
Overall, we confirmed S. mitis by WGS in 106 of 170 (62.4%) and 23 of 41 (56.1%) of the viable BSAC and UKHSA isolates, respectively (Fig. 1, Supplementary Data 1 and 2). We found that S. mitis was most frequently misidentified as S. oralis and S. infantis for 29 of 211 (13.7%) and 27 of 211 (12.8%) sequenced isolates, respectively (Supplementary Table 1). The average nucleotide identity (ANI) is a measure of nucleotide level similarity among orthologous genes shared between two genomes and it offers robust resolution between strains of the same or closely related species35. Pairwise ANI values calculated among the 129 S. mitis isolates ranged from 91.2 to 99.6% (Median = 93.4%) (Supplementary Fig. 2). Some of these ANI values fell below the 94–96% range generally been accepted to demarcate species boundaries36,37. We speculated that such low ANI values of up to 91% observed in S. mitis genomes reflected the fact that these strains form a species complex consisting of a continuum of related but genetically diverse lineages sufficient to be considered separate sister Streptococcal species38. Additionally, we constructed a phylogenetic tree of the 129 S. mitis isolates, in the context of 188 global S. mitis isolates obtained from public nucleotide sequence repositories (Supplementary Data 3) and 5 reference genomes (Supplementary Data 4), to visualise and compare the multiple speciation approaches (Supplementary Fig. 3). Our approach of taxonomic assignment based on several approaches (see methods) resolved known species inconsistencies resulting from less discriminatory phenotypic approaches and the use of different databases when using genotypic data. Altogether, this highlights the importance of WGS and complementary genotypic-based approaches for species confirmation among the VGS34. The S. mitis isolates from patients with IE were from patients of all ages (0–99 years), with the age range 50–59 years having the highest frequency of S. mitis isolates (20.2%; 26/129) (Supplementary Table 2). Of the 129 confirmed S. mitis isolates, 70 (54.3%) were collected from men and 58 (45.0%) from women (Supplementary Table 2).
Population genetic diversity of IE-associated S. mitis
S. mitis strains are known to be highly diverse genetically16,26,32,39,40,41,42, but accurately assessing S. mitis population structure has been a challenge due to limited S. mitis genome sequences and species-specific molecular genotyping tools. Because of this, it remains unclear whether specific lineages predominantly cause BSIs associated with S. mitis in localised populations and over time. To address this, we therefore quantified the genetic diversity of the IE-associated S. mitis isolates collected from the UK and Ireland, and we determined if population-level genetic diversity changed over time during the 16-year surveillance period. We calculated the ANI values and number of single nucleotide polymorphisms (SNPs) between all the pairs of isolates to quantify the genetic diversity. The number of non-ambiguous SNPs between the pairs of isolates ranged from ~45,000 to 55,000 bp out of a total of 2,146,613 nucleotide bases from mapped sequencing reads (Supplementary Fig. 4) and varied significantly over time (Kruskal–Wallis test, p < 0.001) across the 16-year period. Similarly, the ANI values ranged from 91.8–97.1% (Supplementary Fig. 5) and varied significantly over time (Kruskal–Wallis test, p < 0.001). The observed high number of SNPs distinguishing pairs of isolates sampled in the same year and the wide range of ANI values suggested a high genetic diversity of the IE-associated S. mitis strains during the surveillance period.
Next, we confirmed the high genetic diversity of the IE-associated S. mitis isolates by the identification of long internal and terminal branches separating the isolates in the constructed maximum-likelihood phylogenetic tree (Fig. 2). The long phylogenetic branches indicated the existence of several distinct lineages. To confirm the presence of multiple distinct lineages, we then used our recently developed S. mitis MLST scheme available on the PubMLST website (https://pubmlst.org/smitis) and a complementary whole-genome-based sequence clustering approach using the PopPUNK framework (https://www.bacpop.org/poppunk/)43. This analysis identified 127 (98.4%) unique sequence types (STs) based on the S. mitis MLST scheme and an equal number of the PopPUNK lineages, that we defined as Global Sequence Clusters (GSCs), among the 129 IE-associated S. mitis isolates (Fig. 2). Two isolates isolated in 2014 and 2016, which belonged to a single ST (ST30) and lineage (GSC27), differed from each other by 3024 SNPs, had a pairwise ANI value of 99.2%, and the isolated pair had the same penicillin susceptibility profile. Similarly, two ST36 isolates collected in 2007 and 2015 belonged to lineage GSC28 and differed from each other by 6413 SNPs, had a pairwise ANI value of 99.6%, but differed by their penicillin susceptibility profiles (Fig. 2). Since we did not have access to patient-identifiable data from BSAC or UKHSA, we could not exclude the possibility that S. mitis isolates of the same STs represented recurrent infection of the same patient. Nonetheless, considering that 127 out of 129 isolates (98.4%) belonged to different STs and lineages, and the large range in pairwise distances (3024–62,803 SNPs) and ANI values (91.8–97.1%), these observations suggest that BSI and IE-associated S. mitis in the UK and Ireland is not predominantly caused by a select few dominant lineages.

Maximum-likelihood phylogenetic tree of 129 genotypically-confirmed IE S. mitis isolates. The phylogeny is built using 281,737 SNPs out of a total of 2,146,613 nucleotide bases from mapped sequencing reads, and is displayed next to metadata that includes GSC, ST, year of isolation, phenotypic susceptibility, and the presence or absence of acquired antibiotic resistance genes. Source data are provided as a Source Data file. The strain name in the same order as the phylogeny is shown in the source data file. The phylogenetic tree shows high genetic diversity of the IE-associated BSI S. mitis isolates, such that 98.4% of the isolates belong to different STs and GSCs. A limited number of resistance genes were explored as the analysis was limited to genes present in the curated public AMR databases89. Unlike S. pneumoniae, no genomic tools and public databases are available for exploring resistance in S. mitis, to antibiotics such as beta-lactam antibiotics, that are determined through point mutations89,92. Phenotypic and genotypic non-susceptibility to multiple antibiotic classes was observed for several isolates throughout the surveillance period and across the phylogeny. There was good phenotypic to genotypic resistance concordance, as 96.9% (31/32) of phenotypically tetracycline-resistant isolates had a tet(M) gene, and 94.9% (74/78) of phenotypically macrolide-resistant isolates had a mef(A) or erm(B) gene. Source data are provided as a Source Data file.
Temporal trends in AMR among the IE-associated S. mitis isolates
Due to the limited focus, misdiagnosis, and low incidence of S. mitis BSIs, temporal AMR trends for S. mitis associated with IE have not been well described. We, therefore, assessed the phenotypic susceptibility of the IE-associated S. mitis isolates against commonly used antibiotics to treat suspected S. mitis IE (penicillin, amoxicillin, gentamicin, and vancomycin)44. We found the distribution of penicillin and amoxicillin non-susceptible isolates across the entire phylogeny, not restricted to only specific phylogenetic branches containing closely related isolates (Fig. 2). Among isolates with phenotypic MIC data, 23.3% (30/129) and 6.2% (6/97) of the isolates were non-susceptible to penicillin and amoxicillin, respectively (Supplementary Data 1 and 2, antibiotic abbreviations used by BSAC and UKHSA are explained in Supplementary Table 3). We show that all 30 penicillin non-susceptible isolates belonged to different STs and GSCs (Fig. 2). We observed non-susceptibility to penicillin among isolates across the surveillance period (Fig. 2), which would impact the use of penicillin as a first-line antibiotic for Streptococcal IE44. All the isolates had low-level gentamicin resistance (MIC ≤ 128), which would not impact its use as a synergistic antibiotic in IE management44. Additionally, all isolates showed full susceptibility to vancomycin. Resistance to erythromycin and tetracycline, antibiotics not used for S. mitis IE treatment33, was observed throughout the surveillance period. S. mitis is a known reservoir of resistance genes45, and carriage of macrolide resistance isolates among 51% of healthy individuals, with a significant correlation with tetracycline co-resistance, has been described45. Macrolide and tetracycline resistance genes are often co-carried on mobile genetic elements46,47, self-transmissible DNA sequences that can move between and within species. Therefore, S. mitis, regardless of isolation source may be a reservoir of transmissible AMR for other Streptococcus species. Our AMR findings support the use of the current antibiotic treatment regimens for the management of IE; however, our data emphasises that continued surveillance remains critical for monitoring AMR trends, particularly for penicillin.
To further assess the temporal changes in antimicrobial susceptibility, we aggregated phenotypic MICs for several antibiotics into four 3-year intervals, namely 2001–2004, 2005–2008, 2009–2012, and 2013–2016. Due to the small number of phenotyped isolates per year, these 3-year intervals ensured the derivation of more robust estimates for the phenotypic MIC trends based on a sufficient number of isolates. Using this approach, we found no statistically significant differences in the median MICs across all four time intervals for seven out of eight antibiotics (Table 1 and Supplementary Fig. 6). Conversely, the median MICs for gentamicin showed a statistically significant decrease over time (Kruskal–Wallis test, p = 0.001). Despite the variability of the MIC changes over time due to the limited number of phenotyped isolates, our findings provide baseline data for the genomic surveillance of AMR in S. mitis-associated BSIs, including IE, in the UK and Ireland, regionally and globally.
Other Streptococcus species, including S. pneumoniae, S. agalactiae or Group B Streptococcus (GBS), and S. pyogenes or Group A Streptococcus (GAS), are characterised by lineages that are more likely to cause invasive diseases in humans48,49,50,51. The identification and tracking of lineages have facilitated genomic surveillance and guided clinical interventions against these species52, an approach that could be equally valuable for monitoring the epidemiology of invasive S. mitis strains. We, therefore, analysed the 129 S. mitis isolates from patients with clinically diagnosed IE in the context of globally sampled strains to better understand the global genetic diversity and the distribution of AMR and virulence genes amongst invasive S. mitis. We compiled a total of 322 confirmed whole-genome sequenced S. mitis isolates, from the present study and publicly available genomic sequence repositories, representing 258 PopPUNK lineages and 259 STs (Fig. 3a, b). Analysis of the metadata for our sequenced isolates and the contextual publicly available sequences revealed that 158 out of 322 isolates (49.1%) were from carriage, 152 (47.20%) were from invasive disease, and 12 (3.7%) were from unknown sources (Supplementary Data 3). Of the invasive isolates, 138 (42.9%) were from patients with IE (129 of these were from this study), 13 (4.0%) were from bacteraemia, and 1 (0.3%) was from pneumonia. Overall, there were no shared lineages between the S. mitis isolates obtained from patients in the UK and Ireland with clinically diagnosed IE and other global strains from carriage or invasive disease (Fig. 3c). However, the global strains that shared STs and GSCs were part of a previous carriage study that sampled the same individuals, such that the same strain was sampled multiple times32. Therefore, S. mitis isolates from asymptomatic carriage and invasive disease are distributed across the entire phylogeny of the global isolates, indicating the potential for all, rather than a select few lineages, to cause IE. Furthermore, we found no clustering of isolates based on the AMR genes (Fig. 3c), or major virulence genes associated with pneumococcal pathogenicity. Two of the virulence genes, encoded within the cps locus region and pneumolysin (ply), were found in distinct positions on the global S. mitis phylogeny (Fig. 3c). Pneumococcal adherence and virulence protein A (pavA) and pneumococcal surface adhesin A (psaA) genes were present among all 322 isolates (100%) (Supplementary Data 5). Together, these findings demonstrate that even when viewed from the global context, invasive S. mitis strains are not predominantly associated with a single or limited number of lineages.

a Frequency plot of the STs identified across the combined IE and global S. mitis dataset. The plot shows that the UK IE S. mitis belonged to unique STs, however, two isolates isolated in 2014 and 2016 belonged to ST30, and another two isolates collected in 2007 and 2015 were assigned to ST36. Multiple carriage isolates belonging to the same ST were largely from a previous study that sampled multiple isolates from individuals32. ST190 had the highest frequency of 23 isolates and was also likely due to sampling from the same individual. b Frequency plot of the GSCs identified across the combined IE and global S. mitis dataset. The plot shows that the UK IE S. mitis belonged to unique GSCs, however, two isolates isolated in 2014 and 2016 belonged to GSC27, and another two isolates collected in 2007 and 2015 were assigned to lineage GSC28. Multiple carriage isolates belonging to the same GSC were largely from a previous study that sampled multiple isolates from individuals32. GSC1 had the highest frequency of 23 isolates and was also likely due to sampling from the same individual. c Maximum-likelihood phylogeny of IE and global S. mitis is built using 473,175 SNPs out of 1,237,113 core nucleotide bases. The coloured tips of the phylogeny and the first horizontal metadata bar show the isolation condition of the S. mitis isolates. From the second to fifteenth horizontal bars, the isolate metadata shows the IE UK S. mitis isolates, the GSC lineage, and virulence genes. The virulence gene matrix has 7 capsule genes (cps4A – cps4F), autolysins (lytA and lytC), and pneumolysin (ply) genes. The phylogeny shows clustering of capsule genes among isolates in one region of the phylogeny, predominantly UK IE isolates. The last four horizontal bars show antimicrobial resistance (AMR) gene matrices. From the first to fourth matrix bar are the presence or absence of chloramphenicol (cat), macrolide (ermB and mefA), and tetracycline (tetM) genes. Source data are provided as a Source Data file.
Differential abundance of putative pathogenicity enhancing genes
Previous studies have suggested that HGT between S. mitis and other more virulent members of the Streptococcus genus, specifically S. pneumoniae, drives the spread of AMR and virulence between these species26,32,53. Additionally, S. mitis is known to harbour pneumococcal virulence genes, including those involved in the biosynthesis of serotype 1 and 5 capsules21,22 and homologues of other pathogenicity-associated genes, including Zinc metalloproteases (zmpC, zmpC, and zmpD)54, neuraminidases (nanA and nanB), pneumolysin (ply), immunoglobulin A protease (iga), and autolysins (lytA-C)18 and glucan binding protein B (gbpB or pcsB)55. However, no systematic analyses to assess the abundance of all the functionally characterised (or known) and hypothetical genes in the S. mitis pan-genome between the invasive and carriage isolates have been conducted to date, mostly due to the limited availability of genomic data. Since S. mitis is widely regarded as a source of virulence factors which enhance the pathogenicity of pneumococcal strains26, we, therefore, speculated that the S. mitis strains associated with BSIs may show a higher abundance of virulence genes compared to the isolates sampled from the asymptomatic carriage. To address this, we employed a bacterial genome-wide association study (GWAS)-type approach (Fig. 4), increasingly used to identify genomic loci associated with bacterial phenotypes56,57,58,59,60. Our null hypothesis was that no gene influenced the pathogenicity of S. mitis. Therefore, we expected the distribution of any gene would be similar among the BSI and carriage isolates due to the inclusion of phylogenetically similar but phenotypically distinct pairs of isolates. However, we found fifteen orthologous gene clusters, whose identifiers were arbitrarily defined with the prefix “SCLS”, for sequence cluster locus sequence, were differentially overrepresented among either IE-associated or carriage isolates (Table 2 and Supplementary Fig. 7). Among these genes were TP-binding cassette (ABC) transporters, competence-specific and Hca operon transcription regulators, phage-associated proteins, autolysin (a known pneumococcal virulence factor25,61,62,63), and several uncharacterised hypothetical proteins. Twelve of the genes were overrepresented in the carriage isolates when compared to invasive disease isolates, while three genes showed the opposite association.

The phylogeny of global confirmed S. mitis whole-genome sequences was built using 473,175 SNPs out of 1,237,113 nucleotide bases and annotated with disease status that was pruned to select for pairs of genetically closest carriage and invasive disease isolates. Source data are provided as a Source Data file. This phylogenetic-based approach provided an approximate matching of the isolates for the bacterial genome-wide association analysis. Source data are provided as a Source Data file.
As the pneumococcus is a close relative of S. mitis, belonging to the same species complex, we screened 493 invasive pneumococcal isolates obtained from blood from patients with bacteraemia for the presence of these 15 overrepresented genes in S. mitis (Fig. 5). The pneumococcal isolates were obtained from the Global Pneumococcal Sequencing Project and were part of the Centers for Disease Control and Prevention’s (CDC) active bacterial core surveillance (Supplementary Data 6). We identified the presence of 6 out of 15 overrepresented orthologous S. mitis gene clusters that were also prevalent among the pneumococcal isolates. Among these gene clusters, the pneumococci had a high prevalence of 2 out of 3 genes that were more overrepresented in the invasive S. mitis when compared carriage isolates, a transcriptional regulator (gene cluster SCLS3) and hypothetical gene (gene cluster SCLS7). However, gene cluster SCLS5, a hypothetical gene, was absent in all the pneumococcal strains. In future work, targeted mutagenesis experiments would aid in understanding the function of these hypothetical genes in S. mitis as well as pneumococcus. Together, these findings suggest that these identified genes may potentially modulate the pathogenicity of S. mitis, facilitating a potential rare transition from a typical commensal to a pathogenic lifestyle64,65.

Maximum-likelihood phylogeny of invasive bacteraemia S. pneumoniae isolates is built using 141,880 SNPs out of 1,520,986 core nucleotide bases. The coloured tips of the phylogeny and the first metadata row shows the Global Pneumococcal Sequence Cluster (GPSC), while the second row shows the serotype. The subsequent 12 rows show the presence or absence of the orthologous gene clusters (with prefix SCLS for sequence cluster locus sequence) that were overrepresented in invasive or carriage S. mitis. The phylogeny shows there are a few gene clusters that are also prevalent among the invasive pneumococcal isolates, however, most gene clusters are absent among the pneumococcal isolates. The largely absent S. mitis lytA gene was further investigated among the pneumococci using the virulence finder database90, which identified the presence of pneumococcal lytA among 448 out of 493 (90.9%) isolates using pneumococcal specific lytA reference sequences in the virulence finder database90. Source data are provided as a Source Data file.
Discussion
Feared by clinicians for the potential for a missed diagnosis, IE has been a puzzle in medical science, including diagnostic microbiology, since before the time of William Osler10. In this study, we undertook WGS of a large and unique collection of clinically diagnosed IE-associated S. mitis isolates, expanding the number of publicly available sequenced genomes for this poorly studied opportunistic pathogen by nearly two-fold. Taking into consideration the rarity of IE4, this dataset represents one of the largest IE-associated S. mitis datasets to date. Contrary to our hypothesis, we did not find dominant hypervirulent lineages or populations characterised by the presence of unique virulence genes or AMR across the surveillance period. These data suggest that all S. mitis isolates have the potential to cause IE-associated BSI, further supporting the notion that S. mitis is likely an accidental pathogen. Our analysis suggests that the presence of pneumococcal virulence genes and phenotypic AMR has not led to a selective advantage in the carriage population whereby increased risk for causing disease is a consequence. However, our phylogeny-based GWAS suggest that there are genes that may potentially modulate the pathogenicity of S. mitis, facilitating the rare switch from a typical commensal to a pathogenic lifestyle. The identification of several S. mitis populations with different virulence profiles similar to S. pneumoniae supports the hypothesis that the commensalism of S. mitis evolved through genome reduction and loss of virulence genes to attain a commensal lifestyle16,26. Additionally, our chronologically sampled S. mitis dataset provides the foundation on which to monitor changes in the population structure and AMR of IE-associated S. mitis.
We have demonstrated that WGS and a combination of appropriate bioinformatic analytical techniques are crucial for the accurate identification of S. mitis species from related VGS strains, which have implications for understanding S. mitis disease and epidemiology. Here we show that WGS more accurately distinguishes S. mitis isolates from closely related species compared to phenotypic testing and matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF) methods. Although MALDI-TOF is increasingly used by reference laboratories to rapidly identify bacterial species66, the method fails to accurately differentiate species of the Mitis group67. WGS, therefore, is a potential adjunct for accurately differentiating S. mitis and other VGS, which avoids false diagnosis at the patient level and incorrect ascertainment of the contribution of S. mitis to IE relative to related VGS species. Indeed, the false species ascertainment at diagnostic laboratories may have contributed to the low numbers of S. mitis isolates obtained across the 16-year period, and it is likely that other VGS-causing IE may be S. mitis. However, WGS is not routinely carried out in diagnostic laboratories, and the option for sequencing all archived VGS from UKHSA and BSAC laboratories to increase the S. mitis sample size in this study was resource-intensive and not typically feasible.
S. mitis that have been isolated from patients with clinically diagnosed IE between 2001 and 2016 in the UK and Ireland have remained susceptible to antibiotics commonly used as first-line treatment44. We did not identify high-level vancomycin or gentamicin resistance among the IE-associated isolates, suggesting that gentamicin used synergistically with penicillin or vancomycin is also likely to remain effective against S. mitis. Penicillin MICs have remained stable throughout the surveillance period, which is consistent with data from the USA between 2010 and 202068. However, as penicillin non-susceptible isolates were identified across the surveillance period, continued surveillance to monitor the AMR trends among IE-associated S. mitis isolates remains important alongside WGS, as species misidentification can skew AMR trends and distribution as species-specific differences in AMR among the VGS have been identified69.
We have shown extensive genetic diversity among IE S. mitis, marked by the association of each isolate with a unique lineage and a variable distribution of pneumococcal virulence genes. Similar to models proposed for Staphylococcus epidermidis opportunistic infection70, our findings suggest that S. mitis is a true commensal with accidental pathogenicity, such that multiple genetically divergent clones are found to cause IE-associated BSI and have virulence determinants equally distributed among isolates from carriage and disease. Therefore, as previously highlighted elsewhere19, the presence of pneumococcal virulence genes alone may not necessarily be enough to determine S. mitis invasiveness. For example, the polysaccharide capsule is a major virulence factor for the pneumococcus20, however, transformation experiments have shown that serotype 4 capsule acquisition by S. mitis does not increase resistance against early clearance in a mouse model to levels similar to S. pneumoniae serotype 4 capsule wild-type19. It is possible that a more complex combination of genes related to virulence, metabolism, and other functions, in addition to the host immunity, may determine the potential to cause IE.
PavA and PsaA adhesion genes are known to play a role in the pathogenesis of pneumococcal disease20,71, and the genes may also potentially contribute to the adhesion properties among S. mitis8,18. We established that both genes were not exclusive to the IE S. mitis isolates, but PavA and PsaA were present among all carriage and disease S. mitis isolates. It remains unclear whether there are differences in the presence of other adhesion genes among carriage and disease isolates. Targeted mutagenesis is potentially a valuable approach in identifying endovascular IE virulence factors, but there is considerable redundancy in the streptococcal virulence factors that may mediate disease. In the context of such extensive genome variation, a bacterial GWAS-type approach offers the opportunity to identify overrepresented genes associated with disease isolates72. Here we show, using a pilot phylogenetic-based GWAS-type approach, that some genes may be overrepresented in disease when compared to carriage isolates, which may potentially influence the pathogenicity of S. mitis and include a transcription regulator and uncharacterised hypothetical proteins. Although larger genome datasets are required to validate these pilot GWAS analyses, this will require the systematic collection of many hundreds to thousands of S. mitis isolates from individuals with asymptomatic carriage and disease to achieve sufficient statistical power to unravel potential associations.
Our finding of multiple populations with distinct virulence profiles is concordant with the current theory that S. mitis has transitioned from a more pathogenic species to adopt a commensal lifestyle. S. mitis and S. pneumoniae are very closely related yet have strikingly different pathogenic potentials16. Both species have been suggested to have evolved from a common ancestor with all properties associated with virulence16, however, S. mitis evolved through reductive evolution to become 15% smaller in genome size compared to the pneumococcus and adopt a more commensal lifestyle27. The mosaic pattern of virulence gene presence or absence among S. mitis lineages supports the suggestion that gene loss has occurred and may still be ongoing in the species16. Although this theory has been described as linear, it is possible that multiple selective pressures at various time points may have facilitated the emergence of several populations of S. mitis as we observed in our analysis.
S. mitis is known to be highly transformable, therefore, can acquire genetic material through HGT73. It is, therefore, also possible that S. mitis evolution through both gene loss and gain, via HGT and homologous recombination processes continuously shapes the genetic diversity of this species. We, therefore, hypothesise that S. mitis lineages with increased invasiveness could potentially emerge through acquiring a combination of virulence genes and other determinants from closely related pathogenic species such as the pneumococcus. Therefore, continued surveillance of IE-associated S. mitis infections is important for monitoring the changing population structure and virulence of S. mitis lineages.
Our study has some limitations. First, although the present study relies on 129 newly sequenced genomes, which equates to 40.1% (129/322) of the total publicly available S. mitis genomes, our sample size is still limited. Although the study was restricted to one geographical region, we utilised a valuable resource of archived IE-associated isolates with phenotypic data obtained through robust surveillance systems not readily found elsewhere. A prospective study would be advantageous, however, due to the rarity of S. mitis-associated invasive disease, such a study would be possible but challenging as it would take many years to achieve even a modest sample size. While the sample size requirement could be partly resolved by conducting a multi-site study involving several countries, such a study is likely to be costly and difficult to justify considering the overall low incidence of S. mitis invasive disease compared to diseases caused by other bacterial pathogens. In contrast, retrospective studies of already collected isolates from several countries through international collaborations, as evidenced by consortiums such as the Global Pneumococcal Sequencing (GPS) project74, may provide additional insights on S. mitis diversity and pathogenicity. However, considering the low incidence of S. mitis-associated IE, our dataset represents a unique and the largest collection of IE-associated S. mitis isolates to date, which will provide much-needed baseline genomic data for further comparative studies of S. mitis diversity and pathogenicity. Secondly, as highlighted, we were not able to retrospectively ascertain whether these S. mitis BSI in patients with clinically diagnosed IE fulfilled the modified Duke/ESC 2023 diagnostic criteria for IE75. While this potential imprecision may have affected our ability to identify rare hypervirulent lineages, given the BSI it is likely that these patients fulfilled the “definite” or “possible” IE categories, it is very unlikely that misclassification materially biased the conclusions of our genomic analysis. Thirdly, the S. mitis isolates were not collected systematically from all regions of the UK and Ireland as submission of isolates for bacterial surveillance was voluntary, possibly introducing bias in the samples submitted by the hospital laboratories to BSAC and UKHSA. Lastly, due to the retrospective nature of the study, we did not have access to S. mitis carriage isolates from the same geographical region and across a similar time frame, which would have helped to contextualise the IE-associated S. mitis isolates.
In conclusion, using a rare collection of IE-associated S. mitis isolates from the UK and Ireland, we have shown that S. mitis isolates from patients with suspected IE are highly diverse, with a wide distribution of AMR genes. We have shown that suspected S. mitis associated IE disease is not predominantly caused by a select few dominant lineages and that the presence of known virulence genes from S. pneumoniae does not noticeably influence invasiveness. However, our pilot GWAS-type approach of phylogenetically paired invasive and carriage S. mitis isolates suggest that some genes may be differentially abundant among invasive and carriage S. mitis strains and thus may likely modulate pathogenicity. These findings, therefore, provide further evidence for opportunistic and accidental pathogenicity and expand on the existing theory of the commensal lifestyle of S. mitis. While our AMR findings support the use of the current antibiotic treatment regimens for the management of IE, continued surveillance remains critical for monitoring the AMR trends particularly for penicillin. Our unique dataset expands the publicly available genomic dataset S. mitis by over two-fold and constitutes nearly all invasive IE-associated S. mitis genomic data available to date, which will therefore provide critical baseline data to inform further in-depth investigations of the epidemiology and biology of IE and other BSIs caused by this accidental pathogen.
Methods
Ethical approval
This study complies with ethical regulations applied in public health surveillance. UKHSA holds approvals to process patient-identifiable data for the purposes of infectious disease surveillance, in accordance with Section 60 of the Health and Social Care Act 2001. All isolates were anonymised to the key researcher, and patient-identifiable information was not included in the study. Isolates submitted to BSAC were collected as part of routine clinical investigations and were processed as such by the original laboratory.
Bacteraemia surveillance isolates and sample selection
The isolates included in the analysis were from BSI surveillance. The first batch of isolates were obtained from the British Society of Antimicrobial Chemotherapy (BSAC) Resistance Surveillance Project, which is a long-term study that aims to monitor AMR among bacterial isolates from lower respiratory tract infections and from BSI among patients in the UK and Ireland (https://bsac.org.uk/)76. The bacterial isolates are re-identified by the BSAC Central Testing Laboratory using matrix-assisted laser desorption ionisation-time of flight mass spectrometry (MALDI-TOF MS) to confirm the species. The second batch of isolates were obtained from the UKHSA voluntary surveillance, which includes streptococcal isolates obtained from patients with BSI. Species identification was confirmed by UKHSA using phenotypic analytical profile index (API) testing.
All available isolates that were collected, archived, and identified as S. mitis from suspected IE cases by BSAC and UKHSA from 2001–2016 were included in the study. However, well-phenotyped isolates from more recent years were not available. The diagnosis of IE assigned to the UKHSA and BSAC S. mitis isolates was made by the referring clinical teams. The modified Duke/ESC 2023 diagnostic criteria for IE33 were not available. However, in view of the BSI, and the referral of the isolates for species confirmation and antibiotic sensitivity testing for the management of IE, the patients likely fulfilled the “definite” or “possible” modified Duke/ESC 2023 diagnostic categories. The geographical locations of individual isolates were not available, however, BSAC surveillance covered the UK and Ireland76, while UKHSA surveillance covered England, Wales, and Northern Ireland. Age ranges for the patients were available from both surveillance programmes.
Bacterial culture and antimicrobial susceptibility testing
Bacterial transport swabs obtained from BSAC and UKHSA were inoculated on Columbia agar plates supplemented with 5% horse blood (CBA) (Thermo Scientific, UK). CBA plates were incubated at 37 °C in 5% CO2 for 18 hr, then a single colony was sub-cultured to obtain a plate with pure growth. Pure presumed S. mitis colonies were picked and suspended in cryovials with 1 ml of Todd Hewitt Broth with yeast extract (THY) (Merck, Germany) and 20% glycerol (Merck, Germany), then stored at −80 °C for downstream processing.
Amoxicillin, cefotaxime, clindamycin, erythromycin, gentamicin, penicillin, tetracycline, and vancomycin MICs were previously determined for the presumed S. mitis isolates by the reference laboratory using the agar dilution method. Where data was not available, we derived penicillin MICs for presumed S. mitis isolates, the first-line antibiotic option for S. mitis IE44,75, using the E-test® (bioMérieux, UK). The American Type Culture Collection (ATCC) 49619 S. pneumoniae strain was used as an internal control for all Antimicrobial Susceptibility Testing (AST). Although not consistently determined for all isolates, MICs for additional antibiotics are included in Supplementary Data 1 and 2.
Decreased phenotypic susceptibility (intermediate and resistant) was defined by MIC breakpoints established by the Clinical and Laboratory Standards Institute (CLSI) (https://clsi.org) for penicillin (≥0.25 μg/mL), cefotaxime (≥2 μg/mL), vancomycin (>1 μg/mL), erythromycin (≥0.5 μg/mL), tetracycline (≥4 μg/mL), and clindamycin (≥0.5 μg /mL). In the absence of CLSI guidelines, the European Committee on Antimicrobial Susceptibility Testing (EUCAST) breakpoints (https://www.eucast.org/) for amoxicillin (>2 μg/mL) and gentamicin were used. Gentamicin MIC of ≤128 μg/mL is associated with low-level intrinsic resistance, and isolates with gentamicin MIC of >128 μg/mL are associated with high-level resistance.
Bacterial DNA extraction, library preparation, and WGS
Genomic DNA for WGS was extracted from S. mitis colonies using the Qiagen DNeasy Blood & Tissue Kit (Qiagen, Germany) according to the manufacturer’s instructions. A pre-lysis step for Gram-positive bacteria using a solution consisting of 30 mg/ml of lysozyme (Merck, Germany) and 50 U/ml mutanolysin (Merck, Germany) dissolved in 1× TE buffer (Promega, UK) was included. The quality of extracted DNA was assessed by agarose gel electrophoresis (0.7%), and by measuring 260/280 and 260/230 ratios on a Nanodrop machine (Thermo Scientific, UK). DNA samples were stored at −20 °C prior to dispatch for WGS. DNA quantification, genomic library preparation, and WGS was done by University College London Pathogen Genomics Unit (PGU). The NEBNext Ultra II DNA Library Prep Kit for Illumina was used for library preparation (New England Biolabs, Ipswich, MA, USA), and the Illumina NextSeq platform (Illumina, San Diego, CA, USA) was used for WGS, which generated paired-end sequence reads of 150 bp in length and 50–100x coverage.
Post-sequencing quality control, genome assembly and speciation
Illumina sequencing reads of presumed IE S. mitis, from our previous work32, were checked for quality using FastQC (version 0.11.9) (https://github.com/s-andrews/FastQC), and trimmed using Trimmomatic77 (version 0.39) and a phred score of at least 33 per read was used as the minimum quality score threshold. De novo genome assembly was performed using default parameters in SPAdes78 (version 3.12), and genome quality was determined using the quality assessment tool for genome assemblies (QUAST version 5.0.2) with default parameters79 (Supplementary Data 7). Taxonomic classification of the sequenced S. mitis, that formed our curated dataset of 322 S. mitis genomes32, was firstly done using KRAKEN80 (version 1.0) against the MiniKraken DB_8GB database, and KRAKEN80 (version 2.0) against the minikraken2_v2_8GB_201904 database using default parameters (Supplementary Fig. 8). Genomes assigned as S. mitis were further screened by applying the online PathogenWatch Speciator tool (https://pathogen.watch/), where the in-house species identification tool applied MASH81 to search a curated NCBI RefSeq database82. Genomes that were not assigned as S. mitis by both KRAKEN versions and PathogenWatch were excluded. In this current analysis, we reanalysed the curated 322 S. mitis genome dataset using the speciation methods described above, and as an additional screening step, average nucleotide identity (ANI) values were calculated using fastANI35 (version 1.32). All strain pairs were tested against each other and against a list of complete S. mitis genomes using the “many to many” method and by using the “–matrix” option. Previous studies have suggested that ANI values of 94–96 % are generally accepted as a species boundary36,37, however, S. mitis has been shown to have lower ANI values of up to 91% as the group consists of a continuum of lineages38. Therefore, a relaxed approach using a 90% ANI threshold was used. Lastly, an S. mitis phylogeny was generated using the methods described below, and species assignment methods were assessed together to confirm the species.
Global S. mitis genomes were used to contextualise locally obtained isolates in a broader perspective. All publicly available S. mitis genome assemblies used in this project were downloaded from The National Center for Biotechnology Information (NCBI) genome database (https://www.ncbi.nlm.nih.gov/) and were from carriage, invasive disease, and unknown conditions (Supplementary Data 3).
Pairwise-SNP distance, phylogeny, and population structure analysis
Snippy (version 4.6.0) (https://github.com/tseemann/snippy) was used to map confirmed UK IE S. mitis sequence reads to the S. mitis B6 reference genome (GenBank Accession: GCA_000027165.1) to obtain SNPs, determine genetic diversity, and the alignment was used to construct maximum-likelihood phylogenies using fasttree83 (version 2.1.10). We used the generalised time-reversible model of nucleotide evolution to generate the phylogenies, which were visualised and annotated using the online Interactive Tree of Life (iToL) software84 (version 3.0) and microreact85 (version 240). Isolates were clustered into GSC using PopPUNK43 (version 2.4.0), and STs were defined using a novel multi-locus sequence typing (MLST) scheme (https://pubmlst.org/organisms/streptococcus-mitis)32.
To obtain a core-genome alignment using global S. mitis, genome assemblies were first annotated using Prokka86 (version 1.13.4), and a core-genome analysis was conducted using Panaroo87 (version 1.2 .9) to obtain a core-genome alignment. An alignment of SNPs was generated from the core-genome alignment using Snp-Sites88 (version 2.5.1), and phylogenies were constructed as described above. Acquired AMR and virulence genes were identified among the streptococci using Abricate (version 0.9.8) (https://github.com/tseemann/abricate). The ResFinder89 and virulence finder90 databases were used as references for AMR genes and virulence genes, respectively. Since very few S. mitis genomes have been sequenced and studied, genotypic resistance was used to determine concordance with phenotypic data.
Bacterial GWAS
We undertook a pilot bacterial genome-wide association study to identify specific genetic changes overrepresented in IE-associated S. mitis isolates when compared to those collected from nasopharyngeal carriage. Due to the high genetic diversity, and the modest dataset size, we only investigated the relative abundance of genes or gene clusters identified from the pan-genome analysis using Panaroo. Because of the extremely high within-species genetic diversity of S. mitis and the challenges of collecting matched isolates from invasive disease and carriage from the same setting and time frame, we performed a two-stage bacterial GWAS analysis. First, we generated a maximum-likelihood phylogenetic tree of recently sequenced and publicly available confirmed S. mitis whole-genome sequences. We annotated the phylogenetic tree with the disease status of the isolates based on the body isolation site, i.e., blood as an ‘IE-associate BSI’ and oropharynx or nasopharynx as ‘asymptomatic carriage’. Second, we selected pairs of genetically closest carriage and invasive disease isolates that shared the most recent ancestors regardless of their genetic divergence. We then pruned the initial phylogenetic tree of all the isolates to remain with a subtree with an equal number of invasive diseases and carriage S. mitis isolates, where each pair of carriage and invasive disease isolates formed monophyletic clades. This approach provided an approximate matching of the isolates, albeit with higher divergence than seen with similar analyses in other bacterial species, such as Staphylococcus aureus56, Staphylococcus epidermidis70, and Mycobacterium tuberculosis91, to allow for a robust assessment of the genes potentially enriched in the carriage and invasive disease isolates. Due to the phylogenetic matching or pairing of the S. mitis isolates, we used the exact McNemar’s test to identify genes or gene clusters overrepresented in IE or carriage-associated isolates. We used the function “mcnemar.test” in the stats (version 4.0.3) R package to perform the exact McNemar’s test. Genes or gene clusters with P value < 0.05 were considered to be statistically significant. Overrepresented genes identified as hypothetical genes were further checked using the online NCBI BLAST tool, its databases, and default parameters to determine any known gene functions. NCBI BLAST matches with the highest total score, sequence coverage, and sequence identity were used to assign potential gene function.
Invasive Streptococcus pneumoniae genomes used for screening of overrepresented S. mitis genes were obtained from the Global Pneumococcal Sequencing Project (Supplementary Data 6). The GPS was screened to identify S. pneumoniae genomes obtained via blood from patients with bacteremia74. The largest collection of S. pneumoniae genomes collected through bacteraemia surveillance was therefore used and is part of the CDC active bacterial core surveillance. Abricate (version 1.0.1) was used with default settings and the overrepresented S. mitis genes as the database to screen invasive pneumococcal genomes for the presence of these genes.
Statistical analysis
Statistical tests and associated diagrams were generated in R (version 2.11.1) (R Core Team 2014; https://www.R-project.org/), GraphPad Prism (version 8.0) (GraphPad Software, San Diego, California, USA), and edited in Inkscape version 1.0.0. Parametric data collected included the age group of the IE cases and were presented as frequencies. Non-parametric data, which included antibiotic minimum inhibitory concentrations (MICs) and pairwise-SNP distances, are presented as individual data points and median values. The Kruskal–Wallis test was used to compare median MICs among isolates grouped by year, and the test was also used to compare population-level genetic diversity by pairwise SNPs and ANI values across the 16-year surveillance period. Statistical significance was defined as p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genomes sequenced in this study have been deposited in the US National Center for Biotechnology Information (NCBI) database under BioProject accession code PRJEB55310. Publicly available genomes used in this project are under BioProjects PRJNA480039, PRJEB42564, PRJEB42963, and PRJEB53188. All genomes used in this study are also shared under genome assembly accessions listed in Supplementary Data 3, 4, and 6. Source data are provided with this paper Source data are provided with this paper.
Code availability
We have described all the tools and methods used for the analysis in the Material and Methods sections.
References
Holland, T. L. et al. Infective endocarditis. Nat. Rev. Dis. Primer 2, 1–22 (2016).
Prendergast, B. D. The changing face of infective endocarditis. Heart 92, 879–885 (2006).
Murdoch, D. R. et al. Clinical presentation, etiology and outcome of infective endocarditis in the 21st century: the international collaboration on endocarditis-prospective cohort study. Arch. Intern. Med. 169, 463–473 (2009).
Bin Abdulhak, A. A. et al. Global and regional burden of infective endocarditis, 1990–2010: a systematic review of the literature. Glob. Heart 9, 131–143 (2014).
Pant, S. et al. Trends in infective endocarditis incidence, microbiology, and valve replacement in the United States from 2000 to 2011. J. Am. Coll. Cardiol. 65, 2070–2076 (2015).
Talha, K. M. et al. Escalating incidence of infective endocarditis in Europe in the 21st century. Open Heart 8, e001846 (2021).
Thornhill, M. H. et al. An alarming rise in incidence of infective endocarditis in England since 2009: why? Lancet 395, 1325–1327 (2020).
Mitchell, J. Streptococcus mitis: Walking the line between commensalism and pathogenesis. Mol. Oral Microbiol. 26, 89–98 (2011).
Smith, D. J., Anderson, J. M., King, W. F., van Houte, J. & Taubman, M. A. Oral streptococcal colonization of infants. Oral Microbiol. Immunol. 8, 1–4 (1993).
Osler, W. Culstonian lectures on malignant endocarditis. Lancet 125, 505–508 (1885).
Taylor, S. N. & Sanders, C. V. Unusual manifestations of invasive pneumococcal infection. Am. J. Med. 107, 12–27 (1999).
Thayer, W. S. Bacterial or infective endocarditis. The Gibson lectures for 1930: Lecture I. Edinb. Med. J. 38, 237 (1931).
Austrian, R. The syndrome of pneumococcal endocarditis, meningitis and rupture of the aortic valve. Trans. Am. Clin. Climatol. Assoc. 68, 40–50 (1957).
Ruegsegger, J. M. Pneumococcal endocarditis. Am. Heart J. 56, 867–877 (1958).
Chamat-Hedemand Sandra et al. Prevalence of infective endocarditis in Streptococcal bloodstream infections is dependent on streptococcal species. Circulation 142, 720–730 (2020).
Kilian, M. et al. Evolution of Streptococcus pneumoniae and its close commensal relatives. PloS One 3, e2683 (2008).
Rai, P., He, F., Kwang, J., Engelward, B. P. & Chow, V. T. K. Pneumococcal pneumolysin induces DNA damage and cell cycle arrest. Sci. Rep. 6, 22972 (2016).
Rasmussen, L. H. et al. In silico assessment of virulence factors in strains of Streptococcus oralis and Streptococcus mitis isolated from patients with Infective Endocarditis. J. Med. Microbiol. 66, 1316–1323 (2017).
Rukke, H. V. et al. Protective role of the capsule and impact of serotype 4 switching on Streptococcus mitis. Infect. Immun. 82, 3790–3801 (2014).
Mitchell, A. M. & Mitchell, T. J. Streptococcus pneumoniae: virulence factors and variation. Clin. Microbiol. Infect. 16, 411–418 (2010).
Pimenta, F. et al. Streptococcus infantis, Streptococcus mitis, and Streptococcus oralis strains with highly similar cps5 loci and antigenic relatedness to serotype 5 pneumococci. Front. Microbiol. 9, 3199 (2018).
Lessa, F. C. et al. Streptococcus mitis expressing pneumococcal serotype 1 capsule. Sci. Rep. 8, 17959 (2018).
Whatmore, A. M. et al. Genetic relationships between clinical isolates of Streptococcus pneumoniae, Streptococcus oralis, and Streptococcus mitis: Characterization of “Atypical” Pneumococci and Organisms Allied to S. mitis Harboring S. pneumoniae Virulence Factor-Encoding Genes. Infect. Immun. 68, 1374–1382 (2000).
Johnston, C. et al. Detection of large numbers of Pneumococcal virulence genes in Streptococci of the Mitis group. J. Clin. Microbiol. 48, 2762–2769 (2010).
Kadioglu, A., Weiser, J. N., Paton, J. C. & Andrew, P. W. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6, 288–301 (2008).
Kilian, M., Riley, D. R., Jensen, A., Brüggemann, H. & Tettelin, H. Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. mBio 5, e01490-01414 (2014).
Kilian, M. & Tettelin, H. Identification of virulence-associated properties by comparative genome analysis of Streptococcus pneumoniae, S. pseudopneumoniae, S. mitis, Three S. oralis Subspecies, and S. infantis. mBio 10, e01985-19 (2019).
Kalizang’oma, A. et al. Streptococcus pneumoniae serotypes that frequently colonise the human nasopharynx are common recipients of penicillin-binding protein gene fragments from Streptococcus mitis. Microb. Genomics 7, 000622 (2021).
Doern, C. D. & Burnham, C.-A. D. It’s not easy being green: the viridans group Streptococci, with a focus on pediatric clinical manifestations. J. Clin. Microbiol. 48, 3829–3835 (2010).
Pearce, C. et al. Identification of pioneer viridans streptococci in the oral cavity of human neonates. J. Med. Microbiol. 42, 67–72 (1995).
Weiser, J. N., Ferreira, D. M. & Paton, J. C. Streptococcus pneumoniae: transmission, colonization and invasion. Nat. Rev. Microbiol. 16, 355–367 (2018).
Kalizang’oma, A. et al. Novel multilocus sequence typing and global sequence clustering schemes for characterizing the population diversity of Streptococcus mitis. J. Clin. Microbiol. 0, e00802-22 (2022).
Delgado, V. et al. 2023 ESC Guidelines for the management of endocarditis: Developed by the task force on the management of endocarditis of the European Society of Cardiology (ESC) Endorsed by the European Association for Cardio-Thoracic Surgery (EACTS) and the European Association of Nuclear Medicine (EANM). Eur. Heart J. 44, 3948–4042 (2023).
Sadowy, E. & Hryniewicz, W. Identification of Streptococcus pneumoniae and other Mitis streptococci: importance of molecular methods. Eur. J. Clin. Microbiol. Infect. Dis. 39, 2247–2256 (2020).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Konstantinidis, K. T. & Tiedje, J. M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. 102, 2567–2572 (2005).
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. 106, 19126–19131 (2009).
Jensen, A., Scholz, C. F. P. & Kilian, M. Re-evaluation of the taxonomy of the Mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. Int. J. Syst. Evol. Microbiol. 66, 4803–4820 (2016).
Bek-Thomsen, M., Tettelin, H., Hance, I., Nelson, K. E. & Kilian, M. Population diversity and dynamics of Streptococcus mitis, Streptococcus oralis, and Streptococcus infantis in the upper respiratory tracts of adults, determined by a nonculture strategy. Infect. Immun. 76, 1889–1896 (2008).
Kirchherr, J. L. et al. Clonal diversity and turnover of Streptococcus mitis bv. 1 on shedding and nonshedding oral surfaces of human infants during the first year of life. Clin. Diagn. Lab. Immunol. 12, 1184–1190 (2005).
Hohwy, J., Reinholdt, J. & Kilian, M. Population dynamics of streptococcus mitis in its natural habitat. Infect. Immun. 69, 6055–6063 (2001).
Fitzsimmons, S. et al. Clonal diversity of Streptococcus mitis biovar 1 isolates from the oral cavity of human neonates. Clin. Diagn. Lab. Immunol. 3, 517–522 (1996).
Lees, J. A. et al. Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 29, 304–316 (2019).
Habib, G. et al. 2015 ESC Guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC) Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur. Heart J. 36, 3075–3128 (2015).
Malhotra-Kumar, S. et al. Oropharyngeal carriage of macrolide-resistant viridans group streptococci: a prevalence study among healthy adults in Belgium. J. Antimicrob. Chemother. 53, 271–276 (2004).
Santoro, F., Vianna, M. E. & Roberts, A. P. Variation on a theme; an overview of the Tn916/Tn1545 family of mobile genetic elements in the oral and nasopharyngeal streptococci. Front. Microbiol. 5, 535 (2014).
Chancey, S. T. et al. Composite mobile genetic elements disseminating macrolide resistance in Streptococcus pneumoniae. Front. Microbiol. 6, 26 (2015).
Turner, C. E. et al. The emergence of successful streptococcus pyogenes lineages through convergent pathways of capsule loss and recombination directing high toxin expression. mBio 10, e02521-19 (2019).
Zhi, X. et al. Emerging invasive group A streptococcus M1UK lineage detected by allele-specific PCR, England, 20201. Emerg. Infect. Dis. 29, 1007–1010 (2023).
Lo, S. W. et al. Emergence of a multidrug-resistant and virulent Streptococcus pneumoniae lineage mediates serotype replacement after PCV13: an international whole-genome sequencing study. Lancet Microbe 3, e735–e743 (2022).
Jamrozy, D. et al. Increasing incidence of group B streptococcus neonatal infections in the Netherlands is associated with clonal expansion of CC17 and CC23. Sci. Rep. 10, 9539 (2020).
Lo, S. W. et al. Pneumococcal lineages associated with serotype replacement and antibiotic resistance in childhood invasive pneumococcal disease in the post-PCV13 era: an international whole-genome sequencing study. Lancet Infect. Dis. 19, 759–769 (2019).
Coffey, T. J., Dowson, C. G., Daniels, M. & Spratt, B. G. Horizontal spread of an altered penicillin-binding protein 2B gene between Streptococcus pneumoniae and Streptococcus oralis. FEMS Microbiol. Lett. 110, 335–339 (1993).
Bek-Thomsen, M., Poulsen, K. & Kilian, M. Occurrence and evolution of the paralogous zinc metalloproteases IgA1 protease, ZmpB, ZmpC, and ZmpD in streptococcus pneumoniae and related commensal species. mBio 3, e00303-12 (2012).
Harth-Chu, E. N. et al. PcsB expression diversity influences on streptococcus mitis phenotypes associated with host persistence and virulence. Front. Microbiol. 10, 2567 (2019).
Chaguza, C. et al. Prophage-encoded immune evasion factors are critical for Staphylococcus aureus host infection, switching, and adaptation. Cell Genomics 2, 100194 (2022).
Power, R. A., Parkhill, J. & de Oliveira, T. Microbial genome-wide association studies: lessons from human GWAS. Nat. Rev. Genet. 18, 41–50 (2017).
Wee, B. A. et al. Population analysis of Legionella pneumophila reveals a basis for resistance to complement-mediated killing. Nat. Commun. 12, 7165 (2021).
Chewapreecha, C. et al. Comprehensive identification of single nucleotide polymorphisms associated with beta-lactam resistance within pneumococcal mosaic genes. PLoS Genet. 10, e1004547 (2014).
Farhat, M. R. et al. GWAS for quantitative resistance phenotypes in Mycobacterium tuberculosis reveals resistance genes and regulatory regions. Nat. Commun. 10, 2128 (2019).
Berry, A. M., Lock, R. A., Hansman, D. & Paton, J. C. Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect. Immun. 57, 2324–2330 (1989).
Whatmore, A. M. et al. Molecular characterization of equine isolates of streptococcus pneumoniae: natural disruption of genes encoding the virulence factors pneumolysin and autolysin. Infect. Immun. 67, 2776–2782 (1999).
Canvin, J. R. et al. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J. Infect. Dis. 172, 119–123 (1995).
Sitkiewicz, I. How to become a killer, or is it all accidental? Virulence strategies in oral streptococci. Mol. Oral Microbiol. 33, 1–12 (2018).
Catto, B. A., Jacobs, M. R. & Shlaes, D. M. Streptococcus mitis. A cause of serious infection in adults. Arch. Intern. Med. 147, 885–888 (1987).
Welker, M., Van Belkum, A., Girard, V., Charrier, J.-P. & Pincus, D. An update on the routine application of MALDI-TOF MS in clinical microbiology. Expert Rev. Proteomics 16, 695–710 (2019).
Isaksson, J. et al. Comparison of species identification of endocarditis associated viridans streptococci using rnpB genotyping and 2 MALDI-TOF systems. Diagn. Microbiol. Infect. Dis. 81, 240–245 (2015).
Singh, N. et al. Antibiotic susceptibility patterns of viridans group streptococci isolates in the United States from 2010 to 2020. JAC-Antimicrob. Resist. 4, dlac049 (2022).
Chun, S., Huh, H. J. & Lee, N. Y. Species-specific difference in antimicrobial susceptibility among viridans group streptococci. Ann. Lab. Med. 35, 205–211 (2015).
Méric, G. et al. Disease-associated genotypes of the commensal skin bacterium Staphylococcus epidermidis. Nat. Commun. 9, 5034 (2018).
Holmes, A. R. et al. The pavA gene of Streptococcus pneumoniae encodes a fibronectin-binding protein that is essential for virulence. Mol. Microbiol. 41, 1395–1408 (2001).
Gori, A. et al. Pan-GWAS of streptococcus agalactiae highlights lineage-specific genes associated with virulence and niche adaptation. mBio 11, e00728-20 (2020).
Salvadori, G., Junges, R., Morrison, D. A. & Petersen, F. C. Competence in Streptococcus pneumoniae and close commensal relatives: mechanisms and implications. Front. Cell. Infect. Microbiol. 9, 94 (2019).
Gladstone, R. A. et al. International genomic definition of pneumococcal lineages, to contextualise disease, antibiotic resistance and vaccine impact. EBioMedicine 43, 338–346 (2019).
Delgado, V. et al. 2023 ESC Guidelines for the management of endocarditis. Eur. Heart J. 44, 3948–4042 (2023).
White, A. R., On behalf of the BSAC Working Parties on Resistance Surveillance. The British Society for Antimicrobial Chemotherapy Resistance Surveillance Project: a successful collaborative model. J. Antimicrob. Chemother. 62, ii3–ii14 (2008).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Lam, M. M. C. et al. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12, 4188 (2021).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Argimón, S. et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom. 2, e000093 (2016).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Tonkin-Hill, G. et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21, 180 (2020).
Page, A. J. et al. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genomics 2, e000056 (2016).
Bortolaia, V. et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. https://doi.org/10.1093/jac/dkaa345 (2020).
Chen, L., Zheng, D., Liu, B., Yang, J. & Jin, Q. VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44, D694–D697 (2016).
Farhat, M. R., Shapiro, B. J., Sheppard, S. K., Colijn, C. & Murray, M. A phylogeny-based sampling strategy and power calculator informs genome-wide associations study design for microbial pathogens. Genome Med. 6, 101 (2014).
Li, Y. et al. Penicillin-binding protein transpeptidase signatures for tracking and predicting β-lactam resistance levels in Streptococcus pneumoniae. mBio 7, e00756-16 (2016).
Acknowledgements
This research was funded by the NIHR project reference 16/136/46 (R.S.H.) and NIHR200652 grant using UK aid from the UK Government to support global health research. R.S.H. is a NIHR Senior Investigator. We thank the British Society for Antimicrobial Chemotherapy (BSAC) for allowing the use of isolates from the BSAC Resistance Surveillance Programme. We are grateful to the Microbiology laboratory staff for the referral of isolates, the AMR and Healthcare-Associated Infections (AMRHAI) Reference Unit for antimicrobial susceptibility testing, and members of the Respiratory and vaccine-preventable bacteria reference unit (RVPBRU) for assistance in isolate identification and UKHSA for funding. We thank Andrea Gori for his expert input into the analysis. The funders had no role in study design, data collection and analysis, decision to publish, and preparation of the manuscript. Views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR, the UK Department of Health and Social Care, UKHSA, or BSAC.
Author information
Authors and Affiliations
Contributions
A.K., C.C., and R.S.H. conceived the study. J.C., K.B., B.P., K.L.H., and V.C. provided the IE isolates through the UKHSA. K.L.H. conducted phenotypic antibiotic susceptibility testing. S.B. contributed additional genomes for analysis. A.K. performed culture and DNA extractions for WGS. D.R. accessed and verified the raw data post-sequencing, while S.D.B. facilitated access to computing clusters and genomic pipelines at the Wellcome Sanger Institute. A.K. and C.C. led and conducted the analyses, with B.K., S.D.B., and R.S.H. offering guidance and suggestions on alternative approaches that were adopted. R.S.H. and C.C. supervised the study. A.K. wrote the first draft of the manuscript, and all authors reviewed and edited it. R.S.H. secured the funding. All authors approved the final manuscript and had ultimate responsibility for the decision to submit it for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks François Vandenesch, who co-reviewed with Coralie Bouchiat and the other anonymous reviewer(s), for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kalizang’oma, A., Richard, D., Kwambana-Adams, B. et al. Population genomics of Streptococcus mitis in UK and Ireland bloodstream infection and infective endocarditis cases. Nat Commun 15, 7812 (2024). https://doi.org/10.1038/s41467-024-52120-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52120-z
This article is cited by
-
Genomic identification and characterization of Streptococcus oralis group that causes intraamniotic infection
European Journal of Clinical Microbiology & Infectious Diseases (2025)
