
Abstract
The artificial photocatalytic synthesis based on graphitic carbon nitride (g-C3N4) for H2O2 production is evolving rapidly. However, the simultaneous production of high-value products at electron and hole sites remains a great challenge. Here, we use transformable potassium iodide to obtain semi-crystalline g-C3N4 integrated with the I-/I3- redox shuttle mediators for efficient generation of H2O2 and benzaldehyde. The system demonstrates a prominent catalytic efficiency, with a benzaldehyde yield of 0.78 mol g−1 h−1 and an H2O2 yield of 62.52 mmol g−1 h−1. Such a constructed system can achieve an impressive 96.25% catalytic selectivity for 2e- oxygen reduction, surpassing previously reported systems. The mechanism study reveals that the strong crystal electric field from iodized salt enhances photo-generated charge carrier separation. The I-/I3- redox mediators significantly boost charge migration and continuous electron and proton supply for dual-channel catalytic synthesis. This groundbreaking work in photocatalytic co-production opens neoteric avenues for high-value synthesis.
Similar content being viewed by others


Introduction
The utilization of hydrogen peroxide (H2O2) as an effective and eco-friendly energy vector in domains such as medicine, chemical industry, and biology is experiencing a consistent upsurge1. This increasing demand necessitates alternative methodologies to the traditional yet energetically intensive and waste-generating anthraquinone process2. Implementing artificial photosynthesis for H2O2 from water and oxygen, which translates inexhaustible solar energy into storable chemical forms, marks a substantial advancement in the energy transition3,4. Significant progress has been made in developing H2O2 photosynthesis, though numerous challenges impede its application. For instance, the lethargic kinetics of the water oxidation half-reaction contributed to a deficient proton supply, critically hindering the efficiency of the photocatalytic oxygen reduction reaction (ORR)5,6. Conventionally, sacrificial agents like triethanolamine and isopropanol, among other small organic molecules, have been utilized to circumvent water oxidation reaction (WOR)7,8,9. Their inclusion enhanced proton availability and mitigated the recombination of electrons and holes. While the introduction of sacrificial agents significantly accelerated H2O2 production, the generation of essentially valueless oxidative by-products led to the unproductive expenditure of photo-generated hole energy10,11.
Nonetheless, relentless global research efforts have culminated in the advent of a novel trend: the photosynthesis of H2O2 concurrent with the parallel generation of high-value organic compounds, thereby optimizing photon and atom utilization12. A key oxidation product in this process, the conversion of benzyl alcohol (BzOH) to benzaldehyde (BA), holds significant value as an intermediate in the synthesis of fragrances and pharmaceuticals13. Moreover, the biphasic system comprising BzOH and water facilitates the separation of H2O2 and other potential active substances during the reaction, safeguarding the catalyst against oxidative degradation14. With an oxidation potential lower than WOR, BzOH emerged as an advantageous organic substrate15. Consequently, the shift towards a more efficient photocatalytic coupled production approach necessitates advanced multifunctional catalyst designs.
Graphitic carbon nitride (g-C3N4, CN) has garnered considerable attention in photocatalytic synthesis, attributed to its cost-effectiveness, stability, and structural tunability16,17. Nonetheless, the intrinsic slow surface reaction kinetics and pronounced charge carrier recombination in unmodified CN substantially constrain its photocatalytic efficiency18,19. Crystal engineering, promoting the formation of a robust built-in electric field, has emerged as a potent approach to augment CN’s inherent photocatalytic properties20,21. This highly organized structural configuration enhanced the dissociation of photo-generated carriers and improved charge transfer from bulk to surface22. Moreover, strategies encompassing surface modifications or elemental doping within the ambit of crystal engineering could further escalate photocatalytic activity, thereby potentially revolutionizing photocatalytic performance.
A prevalent method for synthesizing crystalline CN involved the utilization of a LiCl/KCl eutectic salt as both a solvent and a template23,24,25. This approach notably expedited the deammoniation process, which in return diminished the interlayer spacing and fostered the migration of photo-generated charge carriers, culminating in enhanced photocatalytic activity. Prior studies have demonstrated that employing a KI/KCl mixed salt during calcination can produce semi-crystalline CN, significantly outperforming pristine CN in photocatalytic H2O2 production26. Observations indicated that various potassium salt combinations improve CN crystallinity. Notably, specific transformable salts like KI could induce the formation of a KI/KI3 mixed salt, suggesting their potential as versatile options in molten salt templates. In recent discussions, the I-/I3- redox shuttle mediator has been recognized for its stable and efficient electron transfer capabilities, particularly in applications like battery development and water splitting27,28. The integration of eutectic salts that not only enhanced CN’s crystallinity but also embed I-/I3- redox shuttle mediator into the structure represented a synergistic strategy. Thus, a combined modification approach, incorporating both crystalline engineering and the integration of the I-/I3- redox mediators, is poised to significantly advance the capabilities of CN, particularly for both ORR and selective alcohol oxidation. Simultaneously, we anticipate that incorporating various iodine species will reinforce the iodine’s stability between CN layers and consistently yield high-value products over time. Nevertheless, the facile preparation of CN with this dual-functional efficiency remains a complex challenge. Also, it is required to elucidate the impact of these dual-benefit strategies on the overall photocatalytic performance.
As anticipated, a KI/KI3 mixed eutectic salt, synthesized from post-site insertion by combining KI and I2, facilitated the creation of high-crystallinity CN (CN-I/I3) embedded with I-/I3- redox mediators through a facile two-step calcination (Fig. 1a). The implementation of this dual-modification strategy significantly augmented the photocatalytic efficiency of CN-I/I3. Notably, an impressive H2O2 generation rate of 62.52 mmol g−1 h−1 (with a high selectivity of 96.62%) could be achieved at the conduction band (CB) reduction platform and a benzaldehyde production rate of 0.78 mol g−1 h−1 (selectivity of 92.65%) on the oxidation platform of valance band (VB). Advanced diagnostic methodologies, such as in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS), femtosecond transient absorption spectroscopy (fs-TAS), Kelvin probe force microscopy (KPFM), and theoretical simulations, supported the conclusion that the increased crystallinity and the resulting robust built-in electric field significantly enhance the separation of photo-generated carriers. Incorporating I-/I3- redox mediators could effectively improve the transfer and mobility of these carriers, supplying enough electrons and protons. Notably, under natural sunlight, the concentration of H2O2 reached 158.48 mM after 8 h. CN-I/I3 also exhibited its versatility under various complex conditions. This study sets a neoteric precedent in developing efficient CN-based photocatalysts and presents a viable method for co-producing high-value chemicals, optimizing atom-economical utilization by employing electrons and holes in separate reactions.

a Schematic synthesis of CN and CN-I/I3. b, c, d, e, f, g, h, l electrostatic potential (b) two-dimensional electron density (c), surface work function (d, h, l), and electronic state density (e, f, g) of CN, CN-I, and CN-I/I3. i, j, k The H2O2 production (i), BzOH conversion rate, benzaldehyde selectivity (j), and corresponding evolution rates (k) for CN, CN-I, and CN-I/I3. The error bars represent the standard deviation of three replicate tests. m, n, o The stability evaluation of CN-I/I3 (m), the sustained recycling performance of BzOH, Inset: Corresponding yield and selectivity (n), and H2O2 production performance, (o) in different reaction solutions.
Results
Phenomenon identification—theoretical prediction—activity evaluation
Observations revealed that the decomposition of potassium iodide, mainly when used for an extended period, could occur (Supplementary Fig. 2a). Integrating these decomposed components into the g-C3N4 (CN) matrix drastically enhanced its catalytic performance (Supplementary Fig. 2b, c). A vital aspect of this improved performance was the formation of I3-, predominantly attributed to incorporating I2 (Supplementary Fig. 2a). Drawing inspiration from these findings, various amounts of I2 were introduced into the CN system. This addition aimed to simulate the metathesis process and facilitate the forming of intercalated layers comprising I-/I3- redox mediators, potentially enhancing the photocatalytic efficacy.
To elucidate the impact and functionality of I- or I3- species as redox mediators in subsequent high-value chemical reactions, this study established three distinct model systems: the original CN, CN co-polymerized with KI (termed CN-I), and CN co-polymerized with a KI/I2 mixture (CN-I/I3) (Fig. 2a–c, Supplementary Fig. 3 and Supplementary Data 1). A comprehensive investigation utilizing density of states (DOS) analyses was conducted to assess the influence of I- or I3- incorporation on the electronic band structure and band gap of CN (Fig. 1e–g). The valence band (VB) of unmodified CN predominantly consisted of N’s 2p orbitals, while its conduction band (CB) was chiefly composed of the 2p orbitals of C and N. Integration of I- the CN matrix led to the emergence of mid-gap states within the CB, causing a downward shift in the CB edge and narrowing the band gap. In contrast, introducing I3- elicited an upward shift in CN’s VB. Notably, CN co-polymerized with both I- and I3- (CN-I/I3) exhibited the most significant reduction in the band gap. This narrower band gap enhanced visible light absorption and improved the driving force for ORR.

a, b, c Optimized CN (a), CN-I (b), and CN-I/I3 (c) geometric structures with an isosurface value of 0.009 e/Bohr3. d TEM-EDS mappings of CN-I/I3. e–k XPS spectra (e, f, g), FT-IR spectra (h), XRD patterns (i), C K-edge (j), and N K-edge (k) XANES spectra of CN, CN-I, and CN-I/I3. l ESR spectra of CN and CN-I/I3. m, n, o TEM (inset), and HR-TEM images of CN (m), CN-I (n), and CN-I/I3 (o). p Relationship between H2O2 production and FWHM value of the (002) plane for samples with different CN-I-I2 ratios.
Theoretical calculations revealed surface work functions for CN, CN-I, and CN-I/I3 as 5.150, 4.254, and 4.168 eV, respectively (Fig. 1d, h, l). Incorporating I- and the combined I-/I3- systems could result in a progressive elevation of the Fermi level, easing the constraints on free electron movement and concurrently reducing the work function. These changes suggested that the addition of I- or I3- facilitated the migration of photo-generated electrons, thereby increasing surface reaction rates29. The synergistic effect was more pronounced with the simultaneous introduction of I- and I3-. Furthermore, electrostatic potential (ESP) analyses and two-dimensional electron density mapping were used to investigate the charge distribution characteristics in CN post I- or I3- (Fig. 1b, c). While electronegative atoms presented negative electrostatic potential and conjugated surfaces displayed positive potential, creating electron-rich and electron-deficient areas, the symmetric structure of pristine CN led to a uniformly distributed charge, impeding charge transfer. The inclusion of I- or I-/I3- disrupted such uniformity, promoting charge separation. Furthermore, optical property evaluations based on a six-flux model indicated that CN-I/I3 showed better characteristics compared to CN and CN-I, implying enhanced photoactivity (Supplementary Fig. 4; Supplementary Tables 1 and 2). These findings confirm that embedding I- or I3- boosted light absorption and charge separation efficiency in CN.
Taking into account the aforementioned considerations, we developed a dual high-value conversion system to validate the innovative design of the catalyst. In photocatalytic H2O2 production, benzyl alcohol (BzOH) was used as the organic substrate to evaluate the performance of a precisely engineered catalyst. This assessment was conducted within an optimized reaction system using the six-flux model (Fig. 1i and Supplementary Figs. 5–7). Incorporating KI into the catalyst notably amplified its capacity for H2O2 production. Furthermore, the subsequent addition of I2 to the CN-I framework resulted in a discernible secondary enhancement in H2O2 by CN-I production. Compared to the baseline CN and the CN-I variant, CN co-polymerized with both KI and I2 (CN-I/I3) demonstrated a stronger photocatalytic activity, achieving an apparent quantum yield of 34.61% at 400 nm. This performance largely surpassed similar reported systems for the artificial photosynthesis of H2O2 (Supplementary Fig. 8). The catalytic activity for H2O2 decomposition was not notably inhibited (Supplementary Fig. 9), suggesting that the observed improvement was likely attributable to the enhanced crystallinity and the formation of I-/I3- redox mediators within its layered structure. To optimize the yield of photocatalytically produced H2O2, a strategy involving segmented addition of the organic substrate was implemented during the reaction process (Supplementary Figs. 11–15). This approach increased H2O2 yield from 12.34 mM to 15.63 mM, indicative of more efficient utilization of photo-generated carriers. The successful H2O2 production using various organic substrates corroborates the effectiveness of this enhanced photocatalytic strategy.
In this study, the efficacy of H2O2 photo-production and concurrent oxidation for benzaldehyde (BA) synthesis was meticulously investigated (Fig. 1j, k; Supplementary Figs. 16 and 17). Co-polymerization with both KI and KI/I2 markedly improved H2O2 production. Significantly, the CN-I/I3 sample exhibited exceptional performance in H2O2 generation, reaching 62.52 mmol g−1 h−1, which was approximately 143.0 times higher than that of pristine CN (0.436 mmol g−1 h−1), and displayed optimal BzOH oxidation efficiency. The conversion rate of BzOH using CN-I/I3 w was around 17.18%, with a high BA selectivity of 92.65%, in stark contrast to the 0.94% conversion rate and 14.06% selectivity observed with unmodified CN.
Interestingly, an analysis of the functional relationship between photocatalytic rates of H2O2 and BA across different samples revealed high correlation coefficients, up to 0.970 (Supplementary Fig. 19). This strong correlation underscores a synergistic effect between photocatalytic reduction and oxidation processes, enabling dual-efficient production H2O2 and BA while fully utilizing generated electrons and holes. Additionally, the specifically engineered CN-I/I3 system demonstrated effective production of H2O2, 2,5-diformylfuran (a selective oxidation product of hydroxymethylfurfural, achieving 82.59% selectivity), and dihydroxyacetone (a selective oxidation product of glycerol, with 74.88% selectivity). This result was achieved even when using diverse reactants such as 5-hydroxymethylfurfural and glycerol. These results highlight the system’s prominent adaptability in coupling the selective oxidation of organic substrates with the photo-production of H2O2 (Supplementary Fig. 21 and Supplementary Table 3). After five cycles of reuse, CN-I/I3 retained its photocatalytic activity with minimal change (Fig. 1m). In the fifth cycle, H2O2 yield remained above 50 mmol g−1 h−1, and BA yield reached 0.77 mol g−1 h−1, indicating its excellent durability in both H2O2 generation and selective BzOH oxidation. In various complex environments, including different pH values and the presence of assorted ionic species, CN-I/I3 consistently achieved high H2O2 yields (Supplementary Fig. 22). It also maintained significant activity in continuous H2O2 production without alterations in its structural and chemical properties, as evidenced by Scanning electron microscopy (SEM), X-ray diffraction spectroscopy (XRD), X-ray photoelectron spectroscopy (XPS), and Fourier-transform infrared spectroscopy (FT-IR) analyses after repeated reaction cycles (Supplementary Figs. 23–25), further confirming its stability and resistance to interference.
Notably, cyclic utilization of BzOH increased the conversion rate from 17.18% to 95.60% after five reactions, accompanied by significantly enhanced yields of H2O2 (91.32 mmol g−1) and BA (1.72 mol g−1) (Fig. 1n), suggesting a higher potential for practical application. The effectiveness of the catalyst was further validated by substantial H2O2 production in real-world samples, including Yangtze River water (9.16 mM) and Yellow Sea seawater (7.13 mM) (Fig. 1o), highlighting its applicability.
Physicochemical characterizations of target catalysts
To verify the successful synthesis of the required catalyst, a comprehensive chemical structure analysis of the prepared catalysts was performed using XPS, X-ray absorption near-edge structure spectroscopy (XANES), FT-IR, and XRD (Fig. 2e–k; Supplementary Figs. 26–36, and Supplementary Tables 4–7). Deconvolution of high-resolution XPS spectra for C 1 s and N 1 s revealed characteristic peaks at ~284.8, 286.0, 288.0, 398.0, 399.0, and 400.0 eV, corresponding to C=C, C-NHx/C, N-C=N, C-N=C, N-C3, and C-N-H functionalities within the CN framework30 (Supplementary Figs. 28, 29, 32,33). These XPS findings, along with their fitting analyses, suggest that the co-polymerization with KI or KI/I2 successfully integrated K and I elements into the CN framework without altering its inherent chemical structure. The K 2p and I 3d3/2 spectra of CN-I and CN-I/I3 showed subtle shifts near 293.0/295.0 and 630.0 eV, respectively, compared to standard KI, affirming the incorporation of K and I into the CN lattice rather than existing as mere KI aggregates. Notably, in the I 3d3/2 spectrum of CN-I/I3, a new peak indicative of I3- emerged, contrasting with the CN/KI spectrum. This observation suggested that the addition of I2 resulted in the formation of I3- (KI + I2 → KI3), potentially acting as a redox mediator alongside I-, thereby enhancing the transport of photo-generated charge carriers during the photocatalytic process.
The electronic environments of C and N in CN, CN-I, and CN-I/I3 were then examined using XANES (Fig. 2j, k). The C-K-edge XANES spectra of these samples exhibited characteristic resonances around 290.0 eV, corresponding to π*(N-C=N) transitions. In CN-I and CN-I/I3, these π*(N-C=N) transition peaks were negatively shifted by approximately 0.08 eV compared to pristine CN, a shift attributable to the increased electron density around carbon atoms following co-polymerization with KI and KI/KI3. Additionally, a distinct shoulder peak at 289.5 eV in CN-I and CN-I/I3, absent in CN, was observed, indicative of enhanced in-plane and interlayer interactions in these modified samples. The N-K-edge XANES spectra revealed similar electron transition peaks among all samples, including π*(C-N=C), π*(N-C3), π*(C-NHx), and π*(N-C=N). The only notable difference was the upward shift of the π*(C-N=C) transition peak in CN-I/I3 by about 0.07 eV compared to CN and CN-I, suggesting that I3- incorporation increased π* electron density, thereby providing additional electrons for reduction reactions.
The XRD analyses of CN revealed characteristic diffraction peaks at 12.9° (100) and 27.7° (002), corresponding to the in-plane stacking and interfacial packing of heptazine units, respectively31. The KI or KI/I2 co-polymerization altered the crystal structure, leading to the disappearance or weakening of certain lattice plane diffraction peaks. The persistence of the (002) peak indicated the retention of heptazine units, while the absence of the (100) peak and a slight shift of the (002) peak towards higher angles suggested reduced interlayer spacing and a denser structure, generally indicative of enhanced crystallinity32,33.
Compared to CN and CN-I, CN-I/I3 exhibited smaller average pore sizes and increased thickness, confirming the denseness of their structures (Supplementary Fig. 39). As anticipated, high-resolution transmission electron microscopy (HR-TEM) images of CN-I/I3 showed lattice stripes corresponding to the (002) plane, with the quantity and quality of these stripes increasing with the amount of I2 co-polymerized (Fig. 2o and Supplementary Fig. 40). In contrast, the HR-TEM results of CN and CN-I lacked lattice fringes (Fig. 2m, n), suggesting that the mixed salt (KI/KI3) in KI/I2 might enhance crystallinity by facilitating in-plane ‘sewing’ and interlayer ‘cutting’ of CN34. The slight transition of CN-I/I3 to a more crystalline PHI phase further substantiated this perspective35,36,37 (Supplementary Fig. 38).
A functional relationship was established between the full width at half maximum (FWHM) value of the (002) plane, and the photocatalytic H2O2 production rate (Fig. 2p). A significant negative correlation between the FWHM value and H2O2 production underscored the critical role of enhanced crystallinity in improving the photocatalytic activity of CN-I/I3. This enhancement was further attributed to the potential formation of I-/I3- redox mediators induced by KI/I2 co-polymerization and the associated increase in unpaired electron density in CN-I/I3 compared to CN (Fig. 2l), facilitating more efficient separation of photo-generated charge carriers.
Therefore, the qualitative characterization results affirmatively suggest the improved crystallinity of CN-I/I3 and the potential formation of the I-/I3- redox mediators. Furthermore, the observed configurations aligned with DFT predictions corroborated the precise synthesis of the catalyst.
Electronic structure and carrier separation properties
Optical properties are pivotal in assessing the catalytic performance of photocatalysts. To this end, UV–visible diffuse reflectance spectroscopy (UV–Vis DRS), Tauc plot analysis, and Mott-Schottky (MS) assessment were utilized to investigate the effects of KI or KI/I2 co-polymerization on the electronic and optical attributes of the photocatalyst (Fig. 3a and Supplementary Figs. 41–43). Compared to CN, both CN-I and CN-I/I3 displayed a slight red shift in optical absorption spectra and a reduction in band gap width. This shift indicates that co-polymerization with KI or KI/I2 enhances the light-harvesting efficiency of the photocatalyst. Subsequent band edge position analyses revealed that this co-polymerization effectively modulated the conduction band (CB) position. The addition of I2 to CN-I/I3 resulted in a downward shift of its CB position, likely linked to increased crystallinity38. The modified CB positions (≤−0.73 eV) were strategically aligned above the reduction potentials of both single-electron (O2 + e- →·O2- + e- + 2H+ → H2O2, ENHE = −0.33 eV) and double-electron pathways (O2 + 2H+ + 2e- → H2O2, ENHE = 0.68 eV) in the ORR process. This indicates that the co-polymerization of KI or KI/I2, while facilitating the necessary reduction potential for efficient H2O2 generation, simultaneously maximized the valence band (VB) position, enhancing the oxidative capacity for selective oxidation of organic substrates. Also, by augmenting the H+ supply, the H2O2 production could be simultaneously improved.

a, b, c UV–Vis DRS spectra, Inset: The corresponding band structure diagram (a), PL spectra, Inset: TR-PL spectra (b), and EIS spectra, Inset: photocurrent response (c) of CN, CN-I, and CN-I/I3. d, e, f, g, h, i Three-dimensional contour plots of fs-TAS (d, g), transient absorption kinetics at 375 nm within 1400 ps (e, h), and transient absorption intensity decay curves (f, i) within the 500–550 nm range for CN-I and CN-I/I3.
Beyond band structure modifications, the co-polymerization of KI or KI/I2 was anticipated to influence the separation of photo-generated carriers. CN exhibited a broad and intense photoluminescence (PL) emission peak centered around 450 nm (Fig. 3b). Conversely, CN-I and CN-I/I3 markedly reduced PL intensities, with CN-I/I3 exhibiting almost negligible emission. Time-resolved PL spectra were employed to analyze the charge separation process and the average lifetimes of photo-generated charge carriers. The average lifetimes for CN, CN-I, and CN-I/I3 were 3.84 ns, 0.95 ns, and 0.9 ns, respectively. The substantial decrease in PL emission peaks and shorter carrier lifetimes in CN-I and CN-I/I3 suggest accelerated separation of electron-hole pairs. CN-I/I3, in particular, exhibited the fastest carrier separation rate, likely due to its significantly enhanced crystallinity and the formation of I-/I3- redox mediators. Additionally, CN-I/I3 exhibited the highest photocurrent response, the greatest open-circuit potential difference, the smallest electrochemical impedance spectroscopy (EIS) arc radius, and the most rapid electron accumulation (Fig. 3c and Supplementary Figs. 44–47). These findings collectively demonstrate its stronger efficiency in transferring photo-generated charge carriers compared to both CN and CN-I.
To delineate the decay kinetics of photo-generated charge carriers, femtosecond transient absorption spectroscopy (fs-TAS) was utilized to probe the absorption characteristics of intermediate species. This analysis was aimed at shedding light on the impacts of augmented crystallinity and the introduction of I-/I3- redox mediators on the oxidation and reduction reactions occurring on the photocatalyst’s surface. Upon 375 nm excitation, the 3D contour plots and absorption profiles of CN and CN-I/I3 were delineated in Fig. 3d, g. The negative absorption features observed in CN are indicative of ground-state bleaching or stimulated emission (SE), while CN-I/I3 displayed a pronounced positive absorption feature in the excited-state absorption (ESA) spectrum, implying effective retention of photo-generated electrons, holes, or electron-hole pairs39,40. Representative dynamic decay traces captured by fs-TAS are presented in Fig. 3f, i, and the kinetic curves for CN and CN-I/I3 in the 500–550 nm wavelength range were accurately modeled using bi-exponential functions. The derived time constants, τ1 and τ2 are representative of photo-generated electron-hole pair recombination and shallow electron trapping processes, respectively41,42. Compared to CN (τ1 = 4.53 ps, τ2 = 72.22 ps), CN-I/I3 (τ1 = 7.56 ps, τ2 = 198.82 ps) exhibited prolonged durations for both photo-generated electron-hole recombination and shallow electron trapping.
The observed retardation in the decay of photo-generated carriers and the amplified shallow electron trapping in CN-I/I3 suggest an increased likelihood and abundance of active electron transfers during photocatalysis. This facilitates the persistence of a substantial number of active electrons and holes, thereby enhancing the efficiency of H2O2 generation and the selective conversion of organic substrates.
To corroborate the separation process of photo-generated carriers on the catalyst surface, Kelvin probe force microscopy (KPFM) was implemented to visualize the surface potential with a nanoscale spatial resolution (Fig. 4). The contact potential difference (CPD) was assessed based on the potential differential between the Kelvin probe tip and the sample. Under illumination, the surface potentials of CN, CN-I, and CN-I/I3 shifted negatively, a phenomenon attributed to interlayer charging owing to the segregation of photo-generated charge carriers43. The CPDs of CN, CN-I, and CN-I/I3 progressively increased to 48.98 mV, 95.21 mV, and 107.42 mV, respectively, under light exposure (Fig. 4d–f). The most pronounced ∆CPD signal in CN-I/I3 indicates an accelerated separation of photo-generated electron-hole pairs, facilitated by the newly formed I-/I3- redox mediators and improved crystallinity.

a, b, c, d, e, f Surface morphology, height difference distribution, and surface potential difference distribution under illumination for CN (a, d), CN-I (b, e), and CN-I/I3 (c, f).
Overall, under the combined influences of an internally generated electric field due to heightened crystallinity and the I-/I3- redox mediators, CN-I/I3 exhibited an enhanced capability for light absorption and prolonged participation of carriers in the reaction process, compared to CN and CN-I. These empirical observations are congruent with the increased yields of H2O2 and BA demonstrated by CN-I/I3.
Synergistic photocatalytic reduction and oxidation reactions
To elucidate the fundamental mechanism of H2O2 production, a series of experiments were conducted under varying atmospheric conditions and through active species trapping (Fig. 5a). The marked contrast in H2O2 yields under N2 and O2 atmospheres irrefutably confirmed the essential role of O2 as a reactant in H2O2 synthesis. The complete inhibition of H2O2 photo-production under K2Cr2O7 shielding, coupled with a progressive decline in free charge intensity in the presence of TEMPO as an electron scavenger, decisively indicated the crucial involvement of photo-generated electrons in H2O2 formation (Fig. 5c). Notably, the negligible H2O2 yield in the presence of p-benzoquinone (pBQ) underscored the pivotal role of superoxide radicals (•O2−) as intermediates in the production process. Electron paramagnetic resonance (EPR) experiments using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin trap and nitroblue tetrazolium (NBT) as a detection probe further corroborated the presence of •O2− (Fig. 5b and Supplementary Figs. 48–50). Compared to CN, the more pronounced •O2− EPR signal and greater NBT reduction observed with CN-I/I3, suggested a more efficient H2O2 generation in CN-I/I3. These findings robustly support the hypothesis that a two-step single-electron reduction pathway of O2 (O2 → •O2− → H2O2) was the predominant mechanism for H2O2 generation.

a Photocatalytic H2O2 production using CN-I/I3 with different reaction gases, electron capture agent (K2Cr2O7), and •O2- scavenger (p-BQ). b EPR signals of superoxide radicals for CN and CN-I/I3. c EPR signals of free charges for CN-I/I3. d, e, f In situ FT-IR spectra of CN-I/I3 under different reaction conditions and corresponding peak intensity changes with the light irradiation time. g Quasi-in situ cyclic voltammetry curves of CN and CN-I/I3. h, i Polarization curves (h), corresponding H2O2 selectivity, and average electron transfer number (i) for CN, CN-I, and CN-I/I3.
In situ diffuse reflectance infrared Fourier-transform spectroscopy (in situ DRIFTS) was utilized to further delineate the mechanistic details of H2O2 production and BzOH oxidation under various testing conditions (Supplementary Fig. 51). The in situ FT-IR spectra of CN-I/I3 in both dry and BzOH aqueous solutions, as shown in Fig. 5d, e, revealed distinctive vibrational bands in the ranges of 908–1156 cm−1, 1300–1400 cm−1, 1600–1700 cm−1, and 3000–3300 cm−1. These bands were attributed to adsorbed O2, *OOH, *H2O2, and -OH groups (originating from the BzOH solution) on CN-I/I3, respectively44,45. In a dry environment, the increasing intensity of adsorbed O2 peaks over time indicated effective O2 adsorption by CN-I/I3. Conversely, in the BzOH aqueous solution, a decrease in these characteristic peaks with time was observed (Fig. 5f), suggesting that in the presence of a proton source, CN-I/I3 efficiently activates adsorbed O2 for H2O2 production. The rise in vibrational signals corresponding to *OOH and *H2O2 with prolonged exposure in the BzOH solution provided direct evidence of H2O2 formation. Additionally, the diminishing vibrational signal of hydroxyl (-OH) groups from BzOH over time confirmed the oxidative dehydrogenation of BzOH. The observed increase in the *H2O2 signal and decrease in the -OH signal indicated the concurrent occurrence of photocatalytic reduction (ORR) and oxidation (BzOH oxidation) processes. These concurrent reactions mutually facilitated each other by synchronously consuming photo-generated electrons and holes, efficiently utilizing photo-generated charge carriers.
In quasi-in situ cyclic voltammetry (CV) experiments, the electrochemical active surface area (ECSA) of CN-I/I3 demonstrated a progressive increase with the reaction time, in contrast to pristine CN. Such an increase, coupled with the emergence of distinct oxidation (I- → I3-) and reduction (I3- → I-) peaks, provided solid evidence for the formation of an I-/I3- redox mediator within the catalyst structure46 (Fig. 5g and Supplementary Fig. 54). The parallel trends in ECSA enhancement and oxidation-reduction peak profiles observed in CN-I and CN-I/I3 during the reaction indicated their mutual engagement in converting the I-/I3- redox mediator. However, CN-I/I3 displayed a notably larger ECSA both pre- and post-reaction compared to CN-I and exhibited more pronounced changes in the oxidation-reduction peaks at equivalent reaction durations. This large disparity suggests that the incorporation of I2 effectively pre-implanted the I-/I3- redox mediator within the catalyst. The pre-formation of the redox mediator facilitated more rapid oxidation-reduction cycling in the photocatalytic process, accelerating the transfer of photo-generated charge carriers and enhancing overall photocatalytic activity.
The rotating ring-disk electrode (RRDE) technique was further employed to evaluate the selectivity of O2 reduction and determine the number of electron transfers (N) during the ORR, based on the recorded reduction and oxidation currents (Fig. 5h, i and Supplementary Fig. 55). The estimated N values for CN, CN-I, and CN-I/I3 were 2.77, 2.49, and 2.06, respectively. Notably, the electron transfer characteristics of CN-I/I3 closely aligned with a 2e- O2 reduction process, favoring the selective production of H2O2 with a high selectivity of 96.62%47. This result suggests the enhanced suitability of CN-I/I3 for targeted H2O2 generation, positioning it as a potent candidate for selective photocatalytic applications.
Thorough dissection of dual-channel high-value conversion mechanisms
The pivotal contribution of KI or KI/KI3 co-crystallization to the enhancement of photocatalytic activity was further elucidated through DFT calculations. Electronic density differences for CN-I and CN-I/I3 are depicted in Fig. 6a, b and Supplementary Fig. 56, with regions of electron increase and decrease represented by green and yellow, respectively. In CN-I, charge transfer predominantly occurred from I- to the CN framework. In contrast, CN-I/I3 exhibited more extensive charge redistribution due to concurrent charge transfer from the CN framework to both I- and I3-. These multiple charge transfer pathways augmented the electron exchange efficiency of CN-I/I3 and substantiated the existence of the I-/I3- redox shuttle mediator. The electron density distributions of the lowest unoccupied molecular orbitals (LUMO) and highest occupied molecular orbitals (HOMO), along with LUMO+1 and HOMO-1, for CN, CN-I, and CN-I/I3, provided detailed insights into their charge transfer characteristics (Supplementary Fig. 57). In CN, HOMOs were predominantly located on N atoms, while LUMOs were concentrated on C atoms. This uniform distribution led to efficient charge carrier recombination in CN48. The incorporation of I- facilitated aggregation of HOMOs, creating an uneven spatial distribution of HOMOs and LUMOs. The insertion of I3- intensified these spatial charge distribution disparities, effectively reducing e--h+ pairs recombination and enhancing charge separation, thus favoring photocatalytic reactions.

a, b, c Electron density difference diagrams of CN-I (a), CN-I/I3 (b), and CN-I/I3-BzOH (c) with an isosurface value of 0.009 e/Bohr3 (Regions of electron increase and decrease represented by green and yellow, respectively). d, f Energy diagrams of H2O2 evolution reaction on CN, CN-I, and CN-I/I3. e, g Energy diagrams of BzOH oxidation reaction on CN, CN-I, and CN-I/I3. h Adsorption structures of *O2, *OOH, *H2O2, *C7H8O, *C7H7O, *C7H6O on CN-I/I3. i Schematic diagrams of the enhanced oxidation and reduction conversions.
Moreover, the Gibbs free energy changes in the ORR and BzOH oxidation processes intricately demonstrated the efficacy of KI/KI3 co-polymerization (Fig. 6d–g). The intercalation of I- or I-/I3- led to uneven charge redistribution, enhancing O2 adsorption via van der Waals interactions between the material’s fixed dipole and the induced dipole of O2 molecules. Specifically, the free energies for O2 adsorption on CN, CN-I, and CN-I/I3 were calculated to be +0.074 eV, +0.044 eV, and +0.03 eV, respectively, with corresponding O-O bond lengths post-adsorption of 1.24 Å, 1.26 Å, and 1.27 Å (Fig. 6h and Supplementary Fig. 58). Lower adsorption energies and longer O-O bond lengths implied the increased affinity for O2 adsorption and activation on CN-I/I3. Due to the synergistic effect of I- and I3-, the ORR pathway on CN-I/I3 exhibited a more favorable energy profile compared to CN and CN-I, facilitating the formation of the critical intermediate *OOH and its subsequent hydrogenation to H2O2. The concurrent selective oxidation of BzOH to BA was also analyzed. Following BzOH adsorption, the α-H atom was first removed, then the -O-H atom, leading to benzaldehyde formation. The thermodynamically rate-determining step involved the removal of the α-H atom. Notably, CN-I/I3 displayed a more pronounced uneven charge distribution, resulting in a longer C-H bond length in adsorbed BzOH (1.104 Å) compared to CN (1.099 Å) and CN-I (1.101 Å). This disparity significantly lowered the energy barrier for the rate-determining step (*C7H8O → *C7H7O). Furthermore, CN-I/I3-BzOH exhibited stronger electron transfer tendencies from BzOH to the material compared to CN-BzOH and CN-I-BzOH (Fig. 6c and Supplementary Fig. 59), indicating more pronounced charge accumulation at the adsorption site of benzyl alcohol, which facilitated charge transfer to the material, consuming more holes during the oxidation process. In summary, due to the incorporation of I-/I3- and enhanced crystallinity, both the ORR and BzOH oxidation pathways on CN-I/I3 were energetically more favorable than on CN and CN-I, significantly promoting the synergistic production of H2O2 and BA.
Based on these findings, a likely mechanism for the dual-channel photosynthesis using CN-I/I3- was proposed (Fig. 6i). The I-/I3- redox shuttle mediator played a crucial role in speeding up charge separation and ensuring a continuous carriers supply. This mechanism allowed the photo-generated electrons to quickly interact with adsorbed O2 through a two-step, single-electron pathway, converting O2 into •O2− and subsequently into H2O2. Concurrently, the holes participated in the selective oxidation of BzOH, effectively producing BA and yielding excess protons for ORR.
Does the substantial accumulation of H2O2 have a practical application potential?
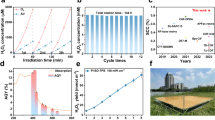
Leveraging its enhanced crystalline structure and the synergistic interaction of I-/I3- redox mediators, the CN-I/I3 exhibited exceptional photocatalytic performance, with its H2O2 yield largely surpassing those of other recently reported photocatalysts under analogous conditions (Fig. 7a, Supplementary Fig. 60 and Supplementary Tables 8–9). The photocatalytic efficacy of CN-I/I3 was further evaluated under natural sunlight. In various environments, including deionized water (H2O2, 158.48 mM, 1.58 mmol; BA, 2.55 mmol), authentic Yangtze River water (H2O2, 81.25 mM, 0.81 mmol; BA, 2.80 mmol), and Yellow Sea seawater (H2O2, 71.01 mM, 0.71 mmol; BA, 3.31 mmol), CN-I/I3 efficiently generated H2O2 and selectively produced BA over 8 h of natural sunlight exposure (Fig. 7b and Supplementary Fig. 61a). In an expanded reactor setup with a 100 mL working volume, CN-I/I3 consistently achieved a high H2O2 yield (33.16 mM).

a Comparison with other recently reported photocatalysts for the production of H2O2. b CN-I/I3 under natural light irradiation: H2O2 production, BzOH conversion, and benzaldehyde selectivity in different solutions. c H2O2 production by continuous immobilization of CN-I/I3 under natural light, Inset: Continuous production of H2O2 physical diagrams. d Removal of ATZ in H2O2/O3 combined system. e Removal of TC-Cr(VI) by CN-I/I3 under intermittent light irradiation. f Efficiency of CN-I/I3 sample in activating PI for SMX removal under natural light, and OD600 values and bacterial counts of E. coli/cells in solution at different time intervals. g Comparison of disinfection effects using different samples on E. coli/cells and corresponding bacterial counts of E. coli. The error bars represent the standard deviation of three replicate tests. h, i, j, k Scenario diagrams for the applications of CN-I/I3 system.
For practical applications, the used CN-I/I3 could be readily recovered through techniques such as centrifugation and filtration. However, implementing a continuous flow system with CN-I/I3 immobilized on a membrane (containing 5 mg CN-I/I3) simplifies this process. This system demonstrated a high H2O2 yield of 1.82 mmol under 8 h of natural sunlight at a flow rate of 0.8 mL min−1, underscoring the potential for solar panel-level H2O2 photosynthesis using CN-based photocatalysts (Fig. 7c and Supplementary Fig. 61d).
To better align with practical industrial development, we developed an integrated process system for the photocatalytic production, concentration, and separation of products (Supplementary Fig. 63). By loading the photocatalyst onto hydrophobic melamine foam, we further eliminated the need for solid-liquid separation while establishing a gas-liquid-solid catalytic interface to enhance mass transfer rates, further promoting the co-production of BA and H2O2. In a natural light photocatalytic production trial lasting up to 21 h (12 h on the first day, 9 h on the second), the conversion efficiency of BzOH reached 94.99%, with a BA yield of 6.29 mmol. The produced H2O2 and BA mixture could be easily concentrated through low-temperature (50 °C) evaporation. In the first concentration process, the H2O2 concentration increased from 47.28 mM to 97.39 mM, and in the second concentration process, it increased from 254 mM to 496.67 mM, achieving a yield of 10.93 mmol. Due to the difference in phase properties, the concentrated mixture solution was separated using a simply hydrophobically modified stainless steel mesh. The higher-density BA flowed out from beneath the hydrophobic membrane, while the H2O2 aqueous solution remained above it (Supplementary Fig. 62). The successful implementation of this integrated process system provided a promising feasibility for the practical application of photocatalytic simultaneous production of two high-value chemicals.
Also, CN-I/I3’s dual enhancement mechanism was highly effective in water purification, activating periodate (PI) under natural sunlight. The system achieved complete decolorization of 100 mg/L rhodamine B (RhB) within 14 min (Supplementary Fig. 65) and complete degradation of 20 mg/L sulfamethoxazole (SMX) in 6 min (Fig. 7f). The treated water exhibited a higher density of E. coli survival, healthier wheat growth, and normal development of zebrafish embryos, accompanied by a reduction in the toxicity of the intermediates. These results indicate that the CN-I/I3-PI system possesses strong purification and detoxification capabilities for organics-contaminated water after activation under natural sunlight (Supplementary Figs. 66–70).
The efficiently accumulated H2O2 in CN-I/I3 could be effectively utilized for in situ applications. The H2O2 generated by CN-I/I3 was harnessed for E. coli disinfection, exhibiting outstanding antibacterial efficacy. Specifically, CN-I/I3 achieved an impressive 99.38 ± 0.04% inactivation rate of E. coli within 15 min of exposure to visible light irradiation (Fig. 7g), demonstrating significant potential for practical applications in medical and health-related fields. Additionally, in the H2O2/O3 system, the combined treatment process significantly outperformed the individual processes in degrading atrazine, illustrating a prominent synergistic effect (Fig. 7d).
Considering the well-documented memory photocatalytic effect associated with CN-based materials, the post-irradiation catalytic activity of CN-I/I3 was assessed for the removal of tetracycline and hexavalent chromium (TC-Cr(VI)) (Fig. 7e and Supplementary Fig. 71a). After 40-min continuous illumination, TC-Cr(VI) was completely removed, a process attributed to the oxidative breakdown of TC-Cr(VI) bonds and the oxidation of TC by photo-generated holes. The reduction of Cr(VI) was presumably driven by photo-generated electrons. Interestingly, under intermittent light irradiation, nearly equivalent TC-Cr(VI) removal to that achieved with continuous 40-min exposure was accomplished in 20 min. The substantial removal of both TC and Cr(VI) during dark periods suggests the release of electrons even in the absence of light, underscoring the material’s effective utilization of stored energy.
Therefore, the well-designed CN-I/I3, under both natural and visible light irradiation, has demonstrated prominent potential in simulated environmental and antibacterial scenarios. These findings position CN-I/I3 as a promising and yet relatively unexplored photocatalyst with broad application prospects.
Discussion
Inspired by the commonplace phenomenon of KI decomposition and underpinned by comprehensive theoretical calculations, this study leveraged I2 to synthesize a KI/KI3 mixed salt, facilitating the formation of a highly semi-crystalline CN embedded with an I-/I3- redox mediators (CN-I/I3) by a secondary calcination process. The integration of theoretical simulations and extensive experimental characterizations revealed that the heightened crystallinity and stability were instrumental in enhancing the built-in electric field. In concert with the embedded I-/I3- redox mediators, this effect significantly improved the dissociation of photo-generated carriers. This dual-action design efficiently maximized the utilization of photo-generated electrons and holes, thus producing both high values of H2O2 and BA in a coupled photocatalytic system. Further empirical validation underscored the versatile potential of CN-I/I3 in various applications. Notably, CN-I/I3 exhibited an impressive H2O2 production of 158.48 mM (1.58 mol) under natural sunlight. This efficiency could be augmented through integrating with photothermal effects to boost catalytic performance.
Furthermore, CN-I/I3 could effectively activate oxidant for the purification and regeneration of contaminated wastewater, demonstrating capabilities in E. coli cultivation and disinfection, and highlighting its potential in combating resistant bacteria. This suggests that CN-I/I3 could be particularly effective in applications involving antibiotic-resistant genes, especially in treating medical wastewater. The potential of CN-I/I3 extended to the photocatalytic eradication of tumor cells, adding to its multifaceted utility. Interestingly, CN-I/I3 retained significant catalytic activity in dark conditions post-light exposure, hinting at potential applications in alternating light-dark reactions, which may be relevant in bio-inspired designs. Crucially, the synergistic effect observed in the industrialized H2O2/O3 system underscores the strong practical applicability of CN-I/I3. These findings position CN-I/I3 as a highly competitive yet underexplored photocatalyst in the market. This research contributes substantially to the understanding and harnessing of electrons and holes in redox reactions, aiming to orchestrate a notable advance in this field. The above results indicate that accurately engineered CN- or other semiconductor-based photocatalysts can facilitate high-value, concurrent production at both the oxidation and reduction ends of the spectrum.
Methods
Chemicals and reagents
Melamine (MA, AR), potassium iodide (KI, AR/GR), iodine (I2), potassium titanium oxalate (C4H2K2O10Ti, AR), sulfuric acid (H2SO4), sodium hydroxide (NaOH), triethylamine, isopropyl alcohol (IPA, AR), methanol (MeOH, AR), ethanol (EtOH, AR), ethylene glycol, tert-butanol (TBA, AR), sulfamethoxazole (SMX), rhodamine B, p-benzoquinone (pBQ, AR), silver sulfate (Ag2SO4, AR), potassium dichromate (K2Cr2O7, AR), sodium chloride (NaCl), potassium chloride (KCl), magnesium chloride (MgCl2), potassium sulfate (K2SO4), sodium nitrate (NaNO3), sodium carbonate (Na2CO3), and potassium hydrogen phosphate (K2HPO4) were all purchased from Sinopharm Chemical Reagent Co., China, Sigma-Aldrich Chemical Reagent Co., China, Macklin Chemical Reagent Co., China, or Shanghai Chemical Reagent Co., China. The Yangtze River water was collected from Chongqing City, China, while the seawater was collected from the Yellow Sea, China. All solutions were prepared with Milli-Q water with an 18.25 MΩ/cm resistivity.
Synthesis of photocatalysts
-
(a)
CN preparation: A homogeneous mixture of 10 g of melamine was placed in a covered alumina crucible and subjected to a calcination process within a tube furnace under an air atmosphere. This process was carried out for 4 h, employing a controlled heating rate of 2.5 °C/min. After the calcination, the resultant product was meticulously ground, thoroughly washed, and then efficiently separated and collected through centrifugation.
-
(b)
CN-I preparation: A mixture consisting of 1 g of CN and varying amounts of KI—specifically, 1 g, 2 g, and 3 g—was prepared in 80 mL of deionized water. This mixture was subjected to ultrasonic treatment for 10 min to ensure homogeneity. Subsequently, it was heated to 80 °C in an oil bath, with continuous stirring to facilitate solvent evaporation and to aid in product collection. The resultant material was then transferred to a covered alumina crucible for calcination. This calcination process was conducted in a tube furnace under an air atmosphere for 4 h, maintaining a steady heating rate of 2.5 °C/min.
After calcination, the product was thoroughly ground, washed, and then efficiently separated using centrifugation. The different samples were systematically named based on the weight of KI used in their preparation, resulting in CN-I-1, CN-I-2 (collectively referred to as CN-I), and CN-I-3.
-
(c)
CN-I/I3 preparation: Typically, 1 g of CN, 2 g of KI, and varying amounts of I2—specifically, 1 mg, 3 mg, 5 mg, and 8 mg—were added to 80 mL of deionized water. This mixture was subjected to ultrasonication for 10 min to ensure homogeneity, followed by heating at 80 °C in an oil bath. Continuous stirring was employed during this heating process to facilitate solvent evaporation and the collection of the resultant product. The gathered product was subsequently placed in a covered alumina crucible and calcinated within a tube furnace under an air atmosphere. This calcination was conducted over 4 h, with a controlled heating rate of 2.5 °C/min. Post-calcination, the obtained material was meticulously ground, washed, and separated via centrifugation. The different samples, differentiated by the weight of I2 used, were sequentially named CN-I/I3-1, CN-I/I3-3, CN-I/I3-5 (collectively as CN-I/I3), and CN-I/I3-8.
Dual-channel evaluation of H2O2 production and BzOH oxidation
The coupled photocatalytic reaction involving oxygen reduction and selective alcohol oxidation was conducted in a 100 mL double-layered glass reactor. Initially, 5 mg of the photocatalyst was ultrasonically dispersed in 10 mL of an aqueous BzOH solution for 5 min to ensure uniform distribution. This was followed by a dark stirring for 10 min to establish adsorption-desorption equilibrium. The catalytic reaction was then initiated using a 300 W xenon lamp (PLS-FX300HU, Beijing Perfectlight Technology Co., China, λ ≥ 420 nm) as the light source, maintaining a constant ambient temperature of 25 °C. During the reaction, aliquots of 1 mL were sampled at 10-min intervals. The photocatalyst was subsequently separated from these samples through filtration using a 0.22 μm membrane filter. The H2O2 concentration in the sampled aliquots was quantified employing the potassium titanium oxalate method. Post-reaction, the solution was centrifuged, and the concentrations of BzOH and BA were analyzed using high-performance liquid chromatography (HPLC). The conversion rate of BzOH and selectivity of BA were calculated according to the Eqs. 1 and 2:
where C0 is the original concentration of BzOH, CBzOH is the concentration of the residual BzOH, and CBA is the concentration of the corresponding BA after the photocatalytic reaction.
Characterizations
Near-edge X-ray absorption fine structure spectroscopy (EXAFS) analysis was conducted at the BL12B beamline of the Hefei National Synchrotron Radiation Laboratory. Kelvin probe force microscopy (KPFM) measurements were performed using a conductive probe (SCM-PIT-V2) on an Atomic Force Microscope (AFM, Bruker Multimode 8). In situ infrared spectroscopy was conducted on a spectrometer (INVENIO S, Bruker Co., Germany, equipped with a high-temperature reaction chamber and liquid nitrogen-cooled MCT). Additionally, in situ infrared spectroscopy was performed again at the synchrotron source. Ultrafast transient absorption spectroscopy (fs-TAS) analysis was carried out using the Helios pump-probe detection system (Ultrafast Systems LLC) and an amplified femtosecond laser system (Coherent). Quasi-in situ CV tests were conducted using a standard three-electrode system with pure water as the electrolyte on a CHI660E electrochemical workstation. The crystallographic phases of the synthesized samples were characterized using a Bruker D8 Advanced X-ray diffractometer (XRD), employing Cu Kα X-ray radiation at operational settings of 40 kV and 40 mA. The microstructural attributes of the various samples were meticulously examined utilizing a field emission scanning electron microscope (FESEM, Apreo 2 C, Thermo Fisher Inc., USA) and a transmission electron microscope (TEM) equipped with FEI Tecnai G20 (Hitachi Co., Japan). Elemental analyses and mapping results were conducted using energy-dispersive X-ray spectroscopy (EDS) on a JSM-7900F spectrometer. Optical properties, including light absorption profiles, were analyzed using ultraviolet-visible diffuse reflectance spectroscopy (UV–Vis DRS) on a UV3600-plus spectrometer, covering a spectral range of 250–800 nm. X-ray photoelectron spectroscopy (XPS) measurements for detailed chemical state and elemental composition analysis were performed using an ESCALAB 250Xi spectrometer (Thermo Fisher Inc., USA). Surface area determinations were carried out using the Brunauer-Emmett-Teller (BET) method, utilizing a Builder 4200 instrument (Tristar II 3020 M, Micromeritics Co., USA). The identification of surface functional groups present in the samples was conducted through Fourier-Transform Infrared Spectroscopy (FT-IR, Nicolet Is10, Thermo Fisher Inc., USA). Photoluminescence (PL) characteristics and time-resolved PL spectra were obtained using an FLS-1000 fluorescence spectrometer. To identify the active species present in the photocatalytic system, electron spin resonance (ESR) spectroscopy (ER200-SRC, Bruker Co., USA) was used, utilizing 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) and 5,5-dimethylpyrroline N-oxide (DMPO) as spin-trapping reagents. Furthermore, the acute and chronic toxicological impacts of sulfamethoxazole (SMX, antibiotics) and its degradation products were evaluated using the ecological structure-activity relationships (ECOSAR) and toxicity estimation software tool (T.E.S.T) systems. This assessment provides crucial insights into the environmental and health-related implications of SMX and its derivatives.
Analysis of optical properties of the photocatalysts
A comprehensive understanding of a photocatalyst’s optical properties is imperative for the identification and development of high-performance photocatalysts. The optical characteristics of a catalyst are fundamental in determining its photon absorption efficiency and, by extension, its photocatalytic performance49. Building upon the foundations of existing research, the current study employed a six-flux model to evaluate the light absorption capacity of the photocatalyst50,51. It is noteworthy that the six-flux model facilitates the calculation of critical optical parameters such as the total photon absorption rate (TRPA), local volume rate of photon absorption (LVRPA), and apparent optical thickness (τapp). These parameters are instrumental in enabling a detailed and comprehensive assessment of the photocatalyst’s efficiency.
At present, LVRPA is recognized as a critical metric for quantifying the local photon absorption within the reaction medium52. Accurately calculating the synthesized catalyst’s optical thickness, as delineated through Eqs. 3–7, is an essential step in determining the LVRPA.
where Ccat is the photocatalyst loading, σ* and κ* are the spectral average specific scattering and absorption coefficients, respectively, and L is the characteristic length of light extinction in the reactor, which is 80 mm. Obtained scattering coefficient (σ*), absorption coefficient (κ*), and extinction coefficient (β*) are listed in Supplementary Table 1.
The optical thickness of different materials could be calculated as follows:
The determination of the local volume rate of photon absorption (LVRPA) for the catalyst was conducted using Eqs. 8–13, as shown in Supplementary Table 2. Notably, a positive correlation was observed between the concentration of the catalysts and their respective optical thicknesses. As the concentration of the catalysts increased, there was a corresponding linear enhancement in the LVRPA at the catalyst surface, as depicted in Supplementary Fig. 2. Significantly, among the evaluated catalysts, CN-I/I3 demonstrated the steepest slope in this relationship. This finding underscores its superior optical properties, aligning well with the predictions made by theoretical calculations.
Furthermore, considering factors such as the optimal optical thickness range of the comprehensive reactor (1.8-4.4), mass transfer rate, etc., a catalyst concentration of 0.5 g/L was chosen for the photocatalytic activity evaluation.
Among a, b, ωcorr, and γ are defined below:
Degradation of sulfamethoxazole (SMX)
The direct degradation of SMX was carried out in a 100 mL double-layer glass beaker. In brief, 5 mg of CN-I/I3 was ultrasonically dispersed in 50 mL of SMX solution. The mixture was stirred in the dark for an additional 10 min to ensure adsorption-desorption equilibrium. Photocatalytic reactions were initiated by adding 1 mM sodium periodate (PI, NaIO4) under natural light exposure. At 2-min intervals, 1 mL of the solution was collected, and the concentration of SMX was determined using HPLC (1260 Infinity, Agilent Inc., USA). To further assess the detoxification capability of CN-I/I3 under natural light exposure, LB culture media with varying reaction times were prepared. These media were used to inoculate Escherichia coli (E. coli), and their OD600 values were monitored, along with the colony survival rate on agar plates.
Zebrafish cultivation for real toxicity assessment
Initially, a trio of zebrafish—one female and two males—were placed in a tank filled with a nutrient-rich solution, where they were cultured for a duration of 12 h. This was done with the aim of obtaining a substantial quantity of zebrafish embryos. Following this initial phase, the first batch of collected embryos was subjected to a washing process using deionized water. The purpose of this step was to meticulously remove any surface impurities present in the embryos. Eight-cell stage embryos were then selected under a microscope for further use. To investigate the detoxification performance of CN-I/I3 activated PI on SMX-contaminated water under natural sunlight, solutions obtained from reactions in the CN-I/I3-PI-Sunlight and CN-I/I3-sunlight systems were designated as experimental groups. Solutions containing only SMX and the nutrient solution (NC) served as control groups. These solutions were collectively referred to as exposure solutions. The selected zebrafish embryos were placed in a cell culture plate, then 1 mL of nutrient solution and 1 mL of exposure solution were added to each well. Each exposure solution was cultured five times. The development of embryos in each group was observed and recorded under a microscope every 24 h until hatching was completed in the NC group. Realistic toxicity assessment using zebrafish cultivation presented above can better reveal the response system’s processing performance and subsequent utility.
Cultivation or disinfection of E. coli
Inoculate 50 μL of E. coli (BL21, wild bacteria) freeze-dried powder into prepared LB culture medium and cultivate in a shaking incubator at 37 °C (180 rpm, Honour, HNY-200B) for 10 h to obtain activated strains. In the in situ antibacterial experiment, sterilized catalyst and benzyl alcohol were simultaneously added to a 20 mL quartz test tube containing activated strains. The investigation was conducted under the irradiation of a 300 W xenon lamp (λ ≥ 420 nm). Suspension was collected every 5 min, diluted 10,000 times with PBS solution, and spread on agar plates. The plates were then incubated at 37 °C for 18 h, and the colony count on the agar plates was calculated.
The procedure for E. coli culture was the same as above, except the LB culture medium was prepared with SMX solution of different reaction times. Before spreading on agar plates, the dilution factor was 100,000. The antibacterial efficiency calculation formula is as follows (Eq. 14):
where M refers to the initial number of colonies and N represents the number of colonies after photocatalytic sterilization. All experiments were carried out on a clean workbench (AIRTECH, SW-CJ-1FD). All the consumables used in the experiments were sterilized at 121 °C for 15 min in an autoclave (IMJ-85A, STIK Co., USA) or under UV radiation (254 nm) for 20 min.
Performance assessment of the H2O2/O3 degradation system
Performance evaluation of atrazine (ATZ, pesticide residues) degradation was carried out under natural light conditions employing the combined H2O2/O3 system. Specifically, a mixture comprising 10 mg of the catalyst and a trace amount of BzOH was ultrasonically dispersed in an aqueous solution containing ATZ. During the experiment, O3 was continuously introduced into the solution under natural light irradiation. Samples of 1 mL volume were systematically collected at 3-min intervals for analysis. The concentration of ATZ in these samples was subsequently quantified using high-performance liquid chromatography (HPLC, 1260 Infinity, Agilent Inc., USA). This methodological approach allowed for a precise and systematic evaluation of the catalyst’s efficacy in degrading ATZ in an environmentally relevant photocatalytic system.
The future development of an integrated system for H2O2 production, encompassing in situ utilization, concentration, collection, and reuse, holds substantial potential for practical applications. Such a system would not only streamline the production process of H2O2 but also optimize its use, thereby enhancing overall efficiency and sustainability. This integrated approach is auspicious for industries where H2O2 is a crucial reactant or cleaning agent, offering a more environmentally friendly and cost-effective solution.
Photoelectrochemical measurements
The obtained catalyst was electrochemically characterized using a standard three-electrode system on a CHI 760E electrochemical workstation (Shanghai Chenhua Instrument Co., China). The characterization included electrochemical impedance spectroscopy (EIS), transient photocurrent (i-t) curves, and Mott-Schottky (MS) curves. The reference electrode and counter electrode were Ag/AgCl and Pt plate electrodes, respectively. The working electrode was an FTO glass coated with the catalyst, prepared from 5 mg of the catalyst, 2 mL of ethanol, and 20 μL of Nafion solution. The electrolyte used was 0.1 M Na2SO4.
The rotating ring-disk electrode (RRDE) test was conducted in a three-electrode reaction cell to describe the oxygen reduction reaction characteristics of the obtained catalyst. The reference electrode and counter electrode were Ag/AgCl and Pt plate electrodes, respectively. The working electrode consisted of an RRDE composed of a glassy carbon disk and a platinum ring. The prepared slurry mentioned above (6 µL) was coated onto the glassy carbon disk and vacuum-dried to prepare the working electrode.
The H2O2 production selectivity is calculated according to Eq. 15:
The electron transfer number (n) is calculated according to Eq. 16:
where iR and iD are the ring and disk currents, respectively, and N is the collection efficiency of the RRDE (N = 0.25).
Details of apparent quantum yield (AQY) test
The AQY for artificial H2O2 synthesis in the CN-I/I3 system was measured using a 400 ± 15 nm band-pass filter. The calculation formula is provided in Eq. 17.
where n is the amount of H2O2 molecules (mol, 46.15 × 10−6 mol), NA is the Avogadro constant (6.022 × 1023/mol), h is the Planck constant (6.626 × 10−34 JS), c is the speed of light (3 × 108 m/s), S is the irradiation area (m2, 15.89 × 10−4 m2), P is the intensity of irradiation light (W m−2, 27.9 W m−2), t is the photoreaction time (s, 1800 s), λ is the wavelength of the monochromatic light (m, 400 × 10−9 m). Combining the above data, the AQY at 400 nm of our system was calculated to be 34.61%.
Quasi-in situ UV–Vis absorption spectroscopy test
To systematically evaluate the stability of the optimized catalytic system, quasi-in situ UV–Vis absorption spectroscopy was employed for real-time detection of potentially leached I3-. The photocatalytic reaction followed the same procedure as artificial H2O2 synthesis but without benzyl alcohol. Samples were taken at specific intervals and centrifuged, and the supernatant was collected for full-spectrum scanning on a UV–Vis spectrophotometer (TU1391901) within a wavelength range of 200–500 nm.
Catalyst immobilization procedure
The catalyst immobilization setup was similar to the working electrode preparation method described in Photoelectrochemical measurements, with the distinction that the prepared slurry was sprayed onto a 2 cm diameter circular filter membrane, achieving a loading amount of 5 mg.
Quantification of •O2 -
The generation of •O2- during the photocatalytic reaction was detected using nitroblue tetrazolium (NBT) as a probe. Specifically, 9.5 μM of NBT and 5 mg of the photocatalyst were added to 10 mL of benzyl alcohol aqueous solution. The mixture was ultrasonicated and allowed to react in the dark for 10 min to establish adsorption-desorption equilibrium. Upon initiating the light source, samples were collected at specific intervals, and centrifuged, and the supernatant’s absorbance was measured at 259 nm using a UV–Vis spectrophotometer. The yield of •O2- was calculated based on the stoichiometric ratio of the reaction between •O2- and NBT (4: 1).
Hydrophobic stainless steel mesh membrane modification procedure
Cut stainless steel mesh (400 mesh) was ultrasonically cleaned with acetone and deionized water for 20 min to remove dust and stains. Polydimethylsiloxane (PDMS, 2 g) was added to hexane (40 mL) and stirred thoroughly for 30 min, followed by the addition of silica nanoparticles (1 g) and continued stirring for another 30 min. Subsequently, silicone resin (0.2 g) was added as a curing agent. The prepared mixture was sprayed onto the stainless steel mesh from a distance of 30 cm using a spray gun at a pressure of 275 kPa. The mesh was then dried in an oven at 60 °C for 5 h to solidify. The unmodified stainless steel mesh is referred to as SS, while the modified version is named MSS.
Hydroponic experiment of wheat seeds
Hydroponic experiments with wheat seeds were conducted using water samples purified by different catalytic systems, alongside original SMX and deionized water, to evaluate the detoxification effects of various catalytic systems on contaminated water from a botanical perspective. Specifically, 20 healthy, plump wheat seeds were selected and placed in culture dishes equipped with filter paper at the bottom, to which 10 mL of different water sources were added. The growth of the seeds was recorded at set intervals, with water sources replenished as needed. After 7-d cultivation, all the wheat seedlings were collected, and differences in root length and shoot length among each group were recorded.
Details of theoretical calculations
We conducted density functional theory (DFT) calculations within the generalized gradient approximation (GGA) framework, employing the Perdew-Burke-Ernzerhof (PBE) formulation53. Brillouin zone of 2 × 2 × 1 k-points was applied for geometry optimization. Valence electrons were represented using a plane wave basis set with a kinetic energy cutoff of 400 eV. Partial occupancies of the Kohn−Sham orbitals were introduced using the Gaussian smearing method with a width of 0.05 eV. We ensured self-consistency in the electronic energy calculations, considering the energy change smaller than 10−5 eV as a convergence criterion. Moreover, geometry optimization was deemed convergent if the energy change was below 0.02 eV per Ångström (eV•Å−1). To minimize artificial interactions between periodic images, we typically included a 15 Å thick vacuum layer at the surface. For the treatment of weak interactions, the DFT + D3 method was used, implementing Grimme’s empirical correction scheme.
The photocatalytic production process of H2O2 can be represented by the following steps (Eqs. 18–21):
The standard hydrogen electrode model was employed to calculate the free energy change (Δ G) of the ORR intermediate in the aforementioned reaction pathways54,55. G(H+) is typically described as 1/2 G(H2) − kBT ln(10) × pH at non-zero pH (p = 1 bar, T = 298.15 K). For each step, the reaction free energy was calculated by Δ G = Δ E + Δ ZPE – TΔ S + Δ GU + Δ GpH, where Δ E, Δ ZPE, and TΔ S represent the total energy, zero-point energy, and entropy contribution obtained from DFT calculations, respectively. Δ GU = – eU, where e is the elementary charge, U is the electrode potential. Δ GpH is the correction of H+ free energy. The Δ G in H2O2 production can be calculated using the following formula (Eqs. 22–25).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data analyses are mainly carried out in the software Origin 2022 and XPS PEAK 41. The data that support the findings of this work are available within the manuscript, Supplementary information files, and Source Data File. Source data are provided in this paper. The X-ray crystallographic coordinates for structures (g-C3N4) reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers JCPDS 87-1526. Source data are provided in this paper.
References
Freese, T., Meijer, J. T., Feringa, B. L. & Beil, S. B. An organic perspective on photocatalytic production of hydrogen peroxide. Nat. Catal. 6, 553–558 (2023).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374–384 (2021).
Hou, H., Zeng, X. & Zhang, X. Production of hydrogen peroxide through photocatalytic processes: a critical review of recent advances. Angew. Chem. Int. Edit. 59, 17356 (2019).
Liu, J. et al. Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway. Science 347, 970–974 (2015).
Li, Q. et al. Shear stress triggers ultrathin-nanosheet carbon nitride assembly for photocatalytic H2O2 production coupled with selective alcohol oxidation. J. Am. Chem. Soc. 145, 20837–20848 (2023).
Wang, W. et al. Photothermal-enabled single-atom catalysts for high-efficiency hydrogen peroxide photosynthesis from natural seawater. Nat. Commun. 14, 2493 (2023).
Dong, W. et al. Isomeric oligo (phenylenevinylene)-based covalent organic frameworks with different orientation of imine bonds and distinct photocatalytic activities. Angew. Chem. Int. Edit. 62, e202216073 (2023).
Li, K. et al. H2S involved photocatalytic system: a novel syngas production strategy by boosting the photoreduction of CO2 while recovering hydrogen from the environmental toxicant. Adv. Funct. Mater. 32, 2113002 (2022).
Yang, J., Jing, J. & Zhu, Y. A full-spectrum porphyrin-fullerene D-a supramolecular photocatalyst with giant built-in electric field for efficient hydrogen production. Adv. Mater. 33, e2101026 (2021).
Luo, N. & Wang, F. Visible-light-driven co-production of diesel precursors and hydrogen from lignocellulose-derived methylfurans. Nat. Energy 258, 575–584 (2019).
Wu, J., Wang, Y., Zhang, S., Liu, Y. & Wang, F. Poly (dibenzothiophene-S, S-dioxide)-Fe2O3 heterojunction for photocatalytic hydrogen production coupled with selective oxidation of benzyl alcohol. Appl. Catal. B-Environ. 332, 122741 (2023).
Mennen, S. M. et al. The evolution of high-throughput experimentation in pharmaceutical development and perspectives on the future. Org. Process. Res. Dev. 23, 1213–1242 (2019).
Shi, Y. et al. Homogeneity of supported single-atom active sites boosting the selective catalytic transformations. Adv. Sci. 9, e2201520 (2022).
Zhao, W. et al. Accelerated synthesis and discovery of covalent organic framework photocatalysts for hydrogen peroxide production. J. Am. Chem. Soc. 144, 9902–9909 (2022).
Wang, H., Yang, C., Chen, F., Zheng, G. & Han, Q. A crystalline partially fluorinated triazine covalent organic framework for efficient photosynthesis of hydrogen peroxide. Angew. Chem. Int. Edit. 61, e202202328 (2022).
Cao, S., Low, J., Yu, J. & Jaroniec, M. Polymeric photocatalysts based on graphitic carbon nitride. Adv. Mater. 27, 2150–2176 (2015).
Zhang, G. et al. Electron deficient monomers that optimize nucleation and enhance the photocatalytic redox activity of carbon nitrides. Angew. Chem. Int. Edit. 58, 14950 (2019).
Liu, B. et al. Boosting O2 reduction and H2O dehydrogenation kinetics: surface N‐hydroxymethylation of g‐C3N4 photocatalysts for the efficient production of H2O2. Adv. Funct. Mater. 32, 2111125 (2021).
Zhou, G. et al. Half-metallic carbon nitride nanosheets with micro grid mode resonance structure for efficient photocatalytic hydrogen evolution. Nat. Commun. 9, 3366 (2018).
Yu, Z., Yue, X., Fan, J. & Xiang, Q. Crystalline intramolecular ternary carbon nitride homojunction for photocatalytic hydrogen evolution. ACS Catal. 12, 6345–6358 (2022).
Algara-Siller, G. et al. Triazine-based graphitic carbon nitride: a two-dimensional semiconductor. Angew. Chem. Int. Edit. 53, 7450–7455 (2014).
Zhang, J. et al. Improved charge separation in poly (heptazine‐triazine) imides with semi‐coherent interfaces for photocatalytic hydrogen evolution. Angew. Chem. Int. Edit. 61, e202210849 (2022).
Zhang, G. et al. Optimizing optical absorption, exciton dissociation, and charge transfer of a polymeric carbon nitride with ultrahigh solar hydrogen production activity. Angew. Chem. Int. Edit. 56, 13445–13449 (2017).
Li, F. et al. Understanding the unique S-scheme charge migration in triazine/heptazine crystalline carbon nitride homojunction. Nat. Commun. 14, 3901 (2023).
Zhang, Y. et al. Molecular heptazine‐triazine junction over carbon nitride frameworks for artificial photosynthesis of hydrogen peroxide. Adv. Mater. 35, 2306831 (2023).
Liu, L.-L., Chen, F., Wu, J.-H., Chen, J.-J. & Yu, H.-Q. Synergy of crystallinity modulation and intercalation engineering in carbon nitride for boosted H2O2 photosynthesis. Proc. Natl. Acad. Sci. USA 120, e2215305120 (2023).
Li, W. et al. Designing ternary hydrated eutectic electrolyte capable of four-electron conversion for advanced Zn-I2 full batteries. Energ. Environ. Sci. 16, 4502–4510 (2023).
Liu, X. et al. Integrating mixed halide perovskite photocatalytic hi splitting and electrocatalysis into a loop for efficient and robust pure water splitting. Adv. Mater. 35, 2208915 (2023).
Chen, F. et al. Enhanced full solar spectrum photocatalysis by nitrogen-doped graphene quantum dots decorated BiO2-x nanosheets: ultrafast charge transfer and molecular oxygen activation. Appl. Catal. B-Environ. 277, 119218 (2020).
Fu, H. et al. Ultrathin porous carbon nitride bundles with an adjustable energy band structure toward simultaneous solar photocatalytic water splitting and selective phenylcarbinol oxidation. Angew. Chem. Int. Edit. 60, 4951 (2021).
Xu, Y. et al. Homogeneous carbon/potassium‐incorporation strategy for synthesizing red polymeric carbon nitride capable of near‐infrared photocatalytic H2 production. Adv. Mater. 33, 2101455 (2021).
Zhang, G. et al. Ionothermal synthesis of triazine-heptazine-based copolymers with apparent quantum yields of 60% at 420 nm for solar hydrogen production from “Sea Water”. Angew. Chem. Int. Edit. 130, 9516–9520 (2018).
Ma, T. Single-crystal X-ray diffraction structures of covalent organic frameworks. Science 361, 48–52 (2018).
Zhang, G. et al. In-plane charge transport dominates the overall charge separation and photocatalytic activity in crystalline carbon nitride. ACS Catal. 12, 4648–4658 (2022).
Botari, T., Huhn, W., Lau, V. W.-h, Lotsch, B. V. & Blum, V. Thermodynamic equilibria in carbon nitride photocatalyst materials and conditions for the existence of graphitic carbon nitride g-C3N4. Chem. Mater. 29, 4445–4453 (2017).
Kessler, F. K. et al. Functional carbon nitride materials-design strategies for electrochemical devices. Nat. Rev. Mater. 2, 17030 (2017).
Wang, Y. et al. Current understanding and challenges of solar-driven hydrogen generation using polymeric photocatalysts. Nat. Energy 4, 746–760 (2019).
Zhang, G. et al. Breaking the limitation of elevated coulomb interaction in crystalline carbon nitride for visible and near‐infrared light photoactivity. Adv. Sci. 9, e2201677 (2022).
Lin, H. et al. Molecular dipole‐induced photoredox catalysis for hydrogen evolution over self‐assembled naphthalimide nanoribbons. Angew. Chem. Int. Edit. 61, e202117645 (2022).
Corp, K. L. & Schlenker, C. W. Ultrafast spectroscopy reveals electron-transfer cascade that improves hydrogen evolution with carbon nitride photocatalysts. J. Am. Chem. Soc. 139, 7904–7912 (2017).
Xu, F. et al. Step-by-step mechanism insights into the TiO2/Ce2S3 S-scheme photocatalyst for enhanced aniline production with water as a proton source. ACS Catal. 12, 164–172 (2021).
Wang, W. et al. In situ protonated-phosphorus interstitial doping induces long-lived shallow charge trapping in porous C3-N4 photocatalysts for highly efficient H2 generation. Energy Environ. Sci. 16, 460–472 (2023).
Zhang, P. et al. Heteroatom dopants promote two‐electron O2 reduction for photocatalytic production of H2O2 on polymeric carbon nitride. Angew. Chem. Int. Edit. 132, 16343–16351 (2020).
Luo, Y. et al. Sulfone‐modified covalent organic frameworks enabling efficient photocatalytic hydrogen peroxide generation via one‐step two‐electron O2 reduction. Angew. Chem. Int. Edit. 135, e202305355 (2023).
Nayak, S., McPherson, I. J. & Vincent, K. A. Adsorbed intermediates in oxygen reduction on platinum nanoparticles observed by in situ IR spectroscopy. Angew. Chem. Int. Edit. 130, 13037–13040 (2018).
Wang, X. et al. Double-photoelectrode redox desalination of seawater. Water Res. 239, 120051 (2023).
Nosaka, Y. & Nosaka, A. Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 117, 11302–11336 (2017).
Wang, Y. et al. Increasing solar absorption of atomically thin 2D carbon nitride sheets for enhanced visible‐light photocatalysis. Adv. Mater. 31, 1807540 (2019).
Tolosana-Moranchel, A., Casas, J. A., Carbajo, J., Faraldos, M. & Bahamonde, A. Influence of TiO2 optical parameters in a slurry photocatalytic reactor: kinetic modelling. Appl. Catal. B-Environ. 200, 164–173 (2017).
Grcic, I. & Puma, G. L. Six-flux absorption-scattering models for photocatalysis under wide-spectrum irradiation sources in annular and flat reactors using catalysts with different optical properties. Appl. Catal. B-Environ. 211, 222–234 (2017).
Acosta-Herazo, R., Mueses, M. A., Puma, G. L. & Machuca-Martínez, F. Impact of photocatalyst optical properties on the efficiency of solar photocatalytic reactors rationalized by the concepts of initial rate of photon absorption (IRPA) dimensionless boundary layer of photon absorption and apparent optical thickness. Chem. Eng. J. 356, 839–849 (2019).
Mena, E., Rey, A. & Beltrán, F. TiO2 photocatalytic oxidation of a mixture of emerging contaminants: a kinetic study independent of radiation absorption based on the direct-indirect model. Chem. Eng. J. 339, 369–380 (2018).
Perdew, J. P., Burke, K. & Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B. 54, 16533 (1996).
Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Origin of the electrocatalytic oxygen reduction activity of graphene-based catalysts: a roadmap to achieve the best performance. J. Am. Chem. Soc. 136, 4394–4403 (2014).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B. 108, 17886–17892 (2004).
Acknowledgements
The authors thank the National Natural Science Foundation of China (52270149 and 51908528) and the Fundamental Research Funds for the Central Universities (2021CDJQY-014) for supporting this work. The authors also thank the related testers from Shiyanjia Lab (www.shiyanjia.com) for SEM measurements.
Author information
Authors and Affiliations
Contributions
F. Chen and C.W. Bai conceived and planned the experiments, and C.W. Bai conducted the related experiments. F. Chen provided program support. P. J. Duan contributed to the DFT calculations. C.W. Bai conducted, and Z.Q. Zhang, Y.J. Sun, X.J. Chen, Q. Yang, and F. Chen assisted in collecting data and analyzing various characterizations. C.W. Bai, F. Chen, and H.Q. Yu wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hongwei Mi, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, F., Bai, CW., Duan, PJ. et al. Merging semi-crystallization and multispecies iodine intercalation at photo-redox interfaces for dual high-value synthesis. Nat Commun 15, 7783 (2024). https://doi.org/10.1038/s41467-024-52158-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52158-z
