
Abstract
Meta-omics is commonly used for large-scale analyses of microbial eukaryotes, including species or taxonomic group distribution mapping, gene catalog construction, and inference on the functional roles and activities of microbial eukaryotes in situ. Here, we explore the potential pitfalls of common approaches to taxonomic annotation of protistan meta-omic datasets. We re-analyze three environmental datasets at three levels of taxonomic hierarchy in order to illustrate the crucial importance of database completeness and curation in enabling accurate environmental interpretation. We show that taxonomic membership of sequence clusters estimates community composition more accurately than returning exact sequence labels, and overlap between clusters can address database shortcomings. Clustering approaches can be applied to diverse environments while continuing to exploit the wealth of annotation data collated in databases, and selecting and evaluating these databases is a critical part of correctly annotating protistan taxonomy in environmental datasets. We argue that ongoing curation of genetic resources is crucial in accurately annotating protists in in situ meta-omic datasets. Moreover, we propose that precise taxonomic annotation of meta-omic data is a clustering problem rather than a feasible alignment problem.
Similar content being viewed by others


Introduction
Protists (microbial eukaryotes) are ubiquitous and essential organisms that provide multifarious ecosystem services, ranging from interactions with other microbes to impact on global biogeochemical cycles1,2,3,4,5. Protists have complex ecosystem roles and morphology, and often bridge seemingly disparate scales of interactions, which makes them difficult to visually differentiate yet critical to census for a complete understanding of ecosystem ecology1,3,4.
Molecular surveys of microbial communities have allowed researchers to characterize taxonomic diversity without microscopy or imaging and their associated limitations. Computational approaches are used to assess the taxonomic composition of metagenomic or metatranscriptomic samples. However, approaches that have been available since the early days of metagenomics, like Naïve Bayes classification6,7, deep learning, and topic modeling have become less popular in recent literature in favor of more direct comparisons to databases, which are more interpretable but also minimally predictive8,9,10. Comparison approaches may include: k-mer profiling of raw reads11,12,13; direct recruitment of raw reads from the meta-omic (community-level) sequencing sample to a reference or set of references of interest (e.g., genome, transcriptome, metagenome-assembled genome (MAG), or single-amplified genome (SAG))14,15,16,17; identification and recovery of well-known marker genes (e.g., 18S rRNA) from meta-omic raw reads or from assembled contigs followed by phylogenetic alignment and within-sample quantification18,19,20,21; or sequence search of assembled contigs to a database, using match quality and percentage identity cutoffs to assign best-available level of confidence to taxonomic annotation of genes22,23,24,25,26. Computational approaches to assign taxonomic identities range in the scale over which they can be applied (Supplementary Fig. 1).
All annotation methods share a reliance on databases containing labeled sequences from past studies (“reference sequences”), some of which may carry study-specific features. Environmental microeukaryote meta-omic studies often rely on annotations from transcriptomes of cultured representatives of protists17,27,28,29,30, and are therefore representative of conditions or treatments specific to an experiment. Though transcriptomes constitute a fraction of the genome, they are more readily available than genomes due to the high time and monetary cost of sequencing the large repetitive and intergenic regions common to eukaryotes31. Because it is difficult to collect laboratory genetic data when populations are in decline and expression levels are low, microorganisms that are in an unusual or poor state of metabolism are more challenging to detect in the field using transcriptome reference databases. Moreover, reference datasets that include different cell life cycle stages and environmental conditions would be ideal to link taxonomic identity to functional role but are not always available32.
Here, we highlight three vignettes that span three scales of taxonomic hierarchy (genus, family, and phylum) and explore how alignment-based taxonomic annotation of assembled predicted proteins may be impacted by database composition. We extend the existing documentation of database annotation challenges in the literature33,34,35 with a systematic evaluation of how these issues impact eukaryotic microbiome sequence data. To demonstrate how clustering methods provide a complement to alignment-based taxonomic annotation, we applied a two-stage clustering technique that includes unsupervised clustering to a simplified metatranscriptomic use-case. We propose that clustering approaches highlight the limit of our ability to taxonomically annotate de novo assembled sequences. Our method re-poses taxonomic annotation as a clustering problem and can be used to improve characterization of community composition at multiple levels of taxonomy or to recruit potential sequences associated with some taxon for which an insufficient number of database sequences are available.
Results
Genus
Genetic differentiation between species complicates accurate identification of genus-level community composition
Species in the haptophyte genus Phaeocystis are genetically related, yet have distinctive geographic distributions and morphologies. Phaeocystis antarctica and P. pouchetii are cold-adapted and form large blooms at high latitudes, and along with globally-ubiquitous P. globosa form colonies (“colony-formers”), while P. cordata and P. jahnii are found at mid-latitudes and do not form colonies (“free-living”)36,37,38,39. We re-analyzed Tara Oceans metagenomic samples from the Mediterranean Sea and the Southern Ocean, assembling contigs and then annotating using standard lowest common ancestor (LCA) algorithm against three modified MMETSP and MarRef databases containing: 1) all Phaeocystis references (both colony-formers and free-living), 2) only the colony-formers, and 3) only the free-living; all databases contained non-Phaeocystis taxa. Given that all three databases contain Phaeocystis representatives to the genus level, our expectation was that all three databases would differentiate Phaeocystis at the genus level. In the Southern Ocean where large blooms of P. antarctica are observed, 79.0% of the total Phaeocystis sequences identified with a combined database were identified using the colony-former database, whereas only 11.3% of the Phaeocystis sequences were identified using the free-liver database (Fig. 1). In the Mediterranean Sea where free-living Phaeocystis are more abundant40, 58.8% of Phaeocystis sequences were identified using the free-liver database as compared to 39.9% with the colony-former database (Fig. 1). This implies that the presence of biogeographically distinct species ecotypes in our databases complicates reliable identification of expected taxa - ecotypes that have not been added to the database may be entirely missed.

A Abundance of metagenomic proteins in each ocean basin coassembled from the Tara Oceans dataset annotated to be Phaeocystis by a combined database of the colony-forming references (left in each group; purple), a combined database of the free-living references (middle in each group; pink), a combined database of all Phaeocystis references (right in each group; black). Each group of bars represents either the large (>20 μm) or the small size (0.8–5 μm) fraction samples. Abundance is shown via read coverage (TPM) of annotated metagenomic contigs. B Phylogenetic tree of Phaeocystis references and genomic and transcriptomic outgroups. The bars to the right of the tree show the total number of orthogroups in each species that are a, pink or lavender: shared by other members of the same ecotype (colony-former or free-liver), b, maroon: shared among multiple Phaeocystis species regardless of ecotype, or c, white: present only within one species. C Percentage of sequences from the coassembly from the Southern Ocean Tara Oceans samples annotated to be Phaeocystis by any of the databases that were annotated as Phaeocystis using (top group of two bars) a combined reference database containing all of the free-living Phaeocystis references, (middle group of bars) a combined reference database containing all of the colony-forming Phaeocystis references, (bottom group of bars) a combined reference database containing all Phaeocystis references. The top bar in each group (brown) corresponds to the smallest Tara Oceans size fraction, while the bottom bar in each group (blue) corresponds to the largest Tara Oceans size fraction. D Identical to Panel C, but for the Tara Oceans samples from the Mediterranean Sea.
Family
Database imbalance limits phylogenetic resolution in closely related diatom taxa
Taxonomic annotations are impacted when many closely related taxa have uneven database representation. When a large number of reference sequences belong to one family, but none or only a few references belong to another, this imbalanced database representation may alter annotation recovery unexpectedly. We explored this phenomenon using metatranscriptomic data from a 2012 survey28 paired with associated microscopic cell counts (University of Rhode Island Long-Term Plankton Time Series; https://web.uri.edu/gso/research/plankton/data/). We focus our analysis on diatoms, a group that is well-represented in reference databases (266 transcriptomes in MMETSP; Source Data), but has uneven representation across families (Anderson-Darling Test against uniform distribution: An=70.221; p = 1.3e-5). The diatom Dactyliosolen fragilissimus (family Rhizosoleniaceae) constituted over 38–60% of the cells counted using light microscopy in 3 of 4 sampled weeks (Fig. 2A). However, it was not consistently identified in the metatranscriptomes (<1% of species-level annotations)28,41, despite the observed species being present in the reference database (Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP))29,31,42. Four other Rhizosoleniaceae are also included in the MMETSP database (Source Data)31, yet the family constituted just 0.5–4.3% of family-level annotations and 0.1–0.7% of total sequence abundance. By contrast, the diatom family Skeletonemataceae represented as much as 95% of microscopy counts in one sample, and given the availability of isolates from Narragansett Bay in the database, it was well-annotated in the metatranscriptomes (Fig. 2A). Cerataulina pelagica (family Hemiaulaceae) was also abundant in the microscopy data. Counterintuitively, while not present within the MMETSP database, contigs in the metatranscriptome were consistently annotated as belonging to Hemiaulaceae using a single related reference (Eucampia antarctica; Fig. 2A). The outcomes of low database taxonomic resolution were incongruent between taxa: though both missing taxa of Hemiaulaceae and Rhizosoleniaceae had a member of the same family available in the database (Fig. 2B), only Hemiaulaceae yielded annotations at the expected taxonomic resolution. Critically, this implies that taxonomic coverage alone often does not lead to accurate phylogenetic labels. This vignette highlights that metatranscriptomic data should not be directly interpreted as representative of community abundance. The combined impact of bias in recovering RNA fragments from different taxa, expression differences between taxa, and taxonomic annotation database ambiguity cumulatively contribute to annotation uncertainty.

A Community composition of diatoms in Narragansett Bay based on light microscopy counts (top) compared to their metatranscriptomic activity (bottom). Lineage-conflicted refers to predicted proteins that were annotated as belonging to class Bacillariophyta, but had a conflict at the family level. “Other” refers to diatom families with associated TPM of less than 1000. Circles (top) indicate cells per L (right y-axis). B Mean percentage identity of non-self hits meeting a minimum bitscore value threshold (≥50) for diatom families represented in the MMETSP. C The bars to the right of the heatmap mean percentage identity plot indicate the total number of transcriptomes contained in the MMETSP for each family.
Phylum
Broad-rank absence from databases leads to inaccurate community composition estimates
Sequence representation across major lineages in the eukaryotic tree of life is variable1,43. We explored the impact of missing one eukaryotic lineage from a reference database on the predicted taxonomy of metatranscriptomes. Data from the North Atlantic along a transect from Woods Hole Oceanographic Institution (WHOI) to the Bermuda Atlantic Time Series (BATS) station (“BATS transect”)44 were annotated using a popular marine microeukaryote database (MMETSP)31,42 composed of diverse eukaryotic lineages, though missing key groups such as radiolarians (phylum Retaria) that are especially difficult to culture hence frequently inadequately covered in reference databases45. This is a common problem in microeukaryotic databases because limited reference sequences are available from the ocean, failing to represent the full extent of lineage diversity. This exercise left 42,736 putative radiolarian proteins unannotated and 46,283 annotated as different phyla across diverse lineages (Fig. 3A–C). Adding radiolarians (see Online “Methods” section) to the database impacted not only the total sequences labeled but also changed assigned annotations of existing taxa, highlighting how database incompleteness impairs community interpretation via both missing and incorrect annotations. Further, of 1,021,229 (8.6%) ORFs that were annotated at the domain–but not the phylum–level (“lineage-conflicted”), 95.8% were assigned a functional annotation, a higher rate than likelihood of functional annotation among all ORFs (45.8%). This suggests that highly conserved proteins will be left out of lineage-specific analysis because they tend to be taxonomically ambiguous (Fig. 3D), with distinctions in lineage-conflicted ORFs additionally noted between metaproteomes and metatranscriptomes44.

A Map of the BATS transect colored by the distance of each sample from the shore in kilometers. B Fraction of annotated scaled abundance of proteins that changed annotation before and after the radiolarian sequences were added, grouped by depth. C Among sequences that changed annotations, comparison of their annotation without radiolarian sequences (left axis) to with radiolarian sequences (right axis). In both cases the database contained the MMETSP and MarRef2 databases. While the majority category of putative Radiolarian sequences was those previously unannotated at the phylum level, some were previously classified as other phyla. Some phylum-level annotations were lost due to conflicts with added radiolarian sequences. D Comparison of the number of proteins that were taxonomically annotated (“Annotated”), taxonomically unannotated (“Unannotated”), or had conflicting taxonomy (“Conflicted”) according to whether they were also functionally annotated.
Clustering and kAAmer approaches increase the scope of taxonomic exploration in environmental -omics
Combining database expansion, targeting to taxa of interest, and unsupervised clustering can expand the reach of sequence classification for assembled sequences from meta-omic datasets. Unsupervised approaches have been developed to combat inadequate reference database coverage46,47. Current unsupervised approaches largely classify highly dissimilar fragments (e.g., separating sequences at the domain level between eukaryotes and prokaryotes) because finer scale differences are not easily inferred due to sequence overlap. We posit that leveraging large eukaryotic databases, preprocessing the database to reduce problem size and taxonomic overlap, and then training an unsupervised model on unknown sequences alongside curated databases can improve interpretability of community assessment.
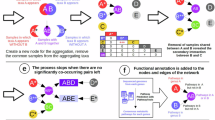
To explore this idea, we leverage existing clustering tools in a two-stage method of taxonomic assignment, an approach we have named “tax-aliquots: Assigning Lineage to Queries Over Two Steps” (Fig. 4). Proteins are first clustered according to their homology, and then hierarchically using the kAAmer (subsequences of amino acids) content of the proteins in the homology-based cluster. The advantages of this method are twofold: we reduce the computational complexity of kAAmer matching48, which is an effective tool to distinguish taxonomic groups49, and we ensure that assignment is also constrained by sequence alignment. We applied three distance thresholds for tax aliquots in the second clustering stage: a permissive, intermediate, and stringent strategy (see “Methods” section). Similar to the percent identity cutoffs used to make decisions about taxonomic level in the Least Common Ancestor (LCA) approach, the distance threshold determines how small the distance between sequences needs to be in order for them to fall into the same cluster. Unlike the LCA approach, all labels are retained in each cluster once they meet the cutoff (Supplementary Figs. 13 and 14). We envision that combining the traditional BLAST + LCA approach with clustering approaches like tax-aliquots enable rapid, global annotation of sequences (BLAST-LCA) alongside maximizing available taxonomic resolution and recovering novel content that performs poorly via a traditional alignment approach.

The workflow is intended to be used alongside the LCA algorithm to detect ambiguity in taxonomic assignment and identify possible taxonomic annotations of sequences which cannot be annotated using the short alignment method. By assessing similarity using subsequence patterns over the entire sequence length, tax-aliquots can also identify discrepancies in the taxonomic annotation selected by alignment and the LCA algorithm.
To demonstrate the utility of the tax-aliquots approach in identifying taxa of interest, we constructed a simplified mock metatranscriptomic example consisting of a single taxon, Phaeocystis pouchetii–one species from vignette 1 above. This particular taxon is known to form colonies, yet was absent from reference databases until recently. Additionally, there are several related, bloom-forming species of Phaeocystis (i.e., P. globosa and P. antarctica) available in the MMETSP and other databases. We generated a default (the UniRef90 protein database50 or the MMETSP database combined with the MarRef2 bacterial database31,51) and a Phaeocystis-only database, each with only the P. pouchetii sequences which were not being tested included, to examine the performance of (1) BLAST + LCA via EUKulele24, (2) mmseqs252, and (3) tax-aliquots in the taxonomic annotation of the P. pouchetii sequences from the mock metatranscriptome (Fig. 5A). We prefiltered putative haptophyte sequences based on their BLAST-LCA taxonomy via EUKulele24 from the mock metatranscriptome and then applied both tax-aliquots clustering and two LCA-based approaches, EUKulele24 and mmseqs2 taxonomy52 (Fig. 5). Then, we split the P. pouchetii sequences into two parts, annotating the taxonomy of one half of sequences and including the other half of sequences in the Phaeocystis database to simulate the case where only a partial transcriptome was previously sequenced and included in the database. This use-case is designed to emulate the common scenario of having sequences of an unknown or unsequenced species in a sample with some closely or distantly related relatives present in the sequencing database. Because we split the P. pouchetii sequences and left the complement of the tested sequences in the Phaeocystis database, we were testing the case where previous sequencing efforts have been insufficient, even though the taxon is technically represented in the database.

A Left panel: Workflow schematic; first, we annotated a “mock metatranscriptome” (a Phaeocystis pouchetii transcriptome) and filtered putative haptophyte sequences using EUKulele (Right panel: results of annotating the mock metatranscriptome with BLAST + LCA (EUKulele) as compared to mmseqs2). Then, we split the sequences into two parts, and annotated half of putative haptophyte sequences with a custom Phaeocystis-only reference database which excluded the half of P. pouchetii being tested (but included the other half as a simulated partial database transcriptome) using BLAST + LCA (EUKulele), mmseqs2, and tax-aliquots. B Tax-aliquots clusters using the “permissive” clustering scheme for the putative haptophyte sequences retrieved from the BLAST + LCA approach in panel B. C Comparison of the fate of the test putative haptophyte sequences between the BLAST + LCA, mmseqs2, and tax-aliquots approaches.
The initial annotation with the default database for BLAST + LCA (EUKulele) resulted in 89.8% of total sequences being annotated as haptophytes, 41.1% of which were annotated as genus Phaeocystis without a species label, and approximately 5.8% of which were annotated as a non-pouchetii species of Phaeocystis. The EUKulele default settings conservatively annotated sequences but did not retain information about lineage beyond phylum and/or genus. The mmseqs2 tool using default settings and a similar default database annotated 34.1% of sequences as haptophytes, including 25.3% as a non-pouchetii species. Additionally, 30.3% of sequences were misannotated as non-haptophytes and 35.6% were not annotated as any lineage (Fig. 5B). 89.8% (n = 57,002) of the sequences were identified as haptophyte by EUKulele to the phylum level. We split the P. pouchetii transcriptome into mock metatranscriptome sequences (n = 31,777) and retained those that were identified as haptophytes (n = 31,056), and included the remaining (n = 31,778) sequences in the reference database. We generated a custom reference database containing all non-pouchetii Phaeocystis reference sequences as well as the latter (n = 31,778) reserved P. pouchetii sequences from the split described above. We then re-annotated the putative haptophyte sequences (n = 31,056) identified from the split using mmseqs2, BLAST + LCA via EUKulele, and tax-aliquots (Fig. 5A). Using the custom Phaeocystis database, EUKulele annotated 60.6% of sequences as Phaeocystis (no species), and 31.4% as a haptophyte (no genus); the remaining 8.0% of species were labeled as a Phaeocystis species (including 1.0% labeled as Phaeocystis pouchetii using the partial transcriptome included in the database). The identical database using the mmseqs2 tool resulted in 45.1% of sequences labeled as Phaeocystis species, 32.1% unannotated, and 22.7% annotated as Phaeocystis (no species) (Fig. 5C). Hence, while EUKulele accurately returned no species label for the majority of sequences, mmseqs2 more liberally assigned conclusive species annotations (Fig. 5D).
Using the tax-aliquots approach, relationships between sequences are identified and reported, rather than returning exact taxonomic labels (i.e., LCA estimates). The tax-aliquots algorithm conservatively clusters sequences regardless of the total size of the reference database (unlike BLAST + LCA, which as shown in Fig. 2 is impacted by database composition). Thus, tax-aliquots allows the closest taxonomic relatives of the query sequences to be identified independent of database completeness. For example, 64.5% of the putative haptophyte sequences were clustered with one of the other Phaeocystis reference sequences, but the majority (63.4%) of these clusters contained multiple additional Phaeocystis species. This observed overlap between Phaeocystis species is analogous to sequences being unambiguously assigned only at the genus level using BLAST + LCA, but with the additional benefit that information is directly retained about the closest species relative to the unknown sequence. Unknown P. pouchetii sequences tended to fall into clusters only with sequences from colony-forming Phaeocystis species (45.2%), which provides insight into the probable ecology of the “unknown” species in an environmental sample (Fig. 5B). By contrast, using BLAST + LCA or mmseqs2, the nearest species lineage is discarded unless a species-level annotation is made (Fig. 5C). Some of the P. pouchetii sequences also fell into clusters with other P. pouchetii sequences–the largest such cluster contained 10 sequences–and 20.9% of P. pouchetii sequences were in clusters with two or more P. pouchetii proteins. The P. pouchetii sequences that could not be clustered would be viewed as an unknown or novel (relative to taxonomy and/or gene content) sequence in the metatranscriptomic setting. Additional information about the proposed tax-aliquots approach is included in Supplementary Note 1 for all three vignettes described in this study.
This clustering example demonstrates the utility of the approach for surveying close relatives of taxonomically-ambiguous taxa (Fig. 5) and expanding the number of sequences on which some inference can be made (Supplementary Note 1). We envision that tax-aliquots could be used in conjunction with a conventional taxonomic annotation tool to expand candidate sequences for a taxon of interest. For example, if P. pouchetii was of interest, but only a single transcriptome reference was available, an LCA-style alignment-based taxonomy tool could be used to conservatively annotate proteins as pouchetii-like, and then those sequences could be combined with the P. pouchetii reference sequences as query sequences for tax-aliquots. This combination of alignment and clustering based methods could enable more sequences with similar subsequence profiles (via kAAmer or k-mer content) to P. pouchetii proteins from the same sample to be identified and explored in-depth.
Discussion
The growth of taxonomically diverse sequence databases and the development of complementary computational analysis approaches have enabled taxonomic predictions for community assessment in meta-omics16,17,31,43,53. The overall size of available databases has expanded dramatically since the first environmental metagenome, fueled by the growing availability of genomes, new sequencing technology that can be deployed straight from the lab (e.g., Nanopore sequencing54,55,56), and the curation of resources from transcriptomes24,29,31,42,57,58,59 and metagenome-assembled genomes14 for eukaryotes15,16,17,60, which expand databases to include non-marker genes or full contigs.
Database curation plays a critical role in how sequences are taxonomically annotated, which directly impacts downstream ecological and biological data interpretation (e.g., how taxonomic identity is linked to functional role)61. All database matching is selective and implicitly biased, because only a selection of organisms have been isolated, subsequently sequenced, and added to protein reference databases. Because microeukaryotes have high average genetic differentiation62, much of our ability to annotate diversity hinges on tradeoffs inherent to building appropriate databases from an unbalanced number of available references for different phyla and orders. We demonstrated the impact of high-level database composition via the misannotation of Radiolaria transcripts in the BATS dataset, where Radiolarian references were absent in the MMETSP31 but present in the EukProt and EukZoo databases43,59. This is one example from a transect dataset, but in more remote environments such as the deep sea, where a smaller proportion of environmental sequences are expected to have been cultured and sequenced, closely related, complete database counterparts, using an entirely generative and flexible approach such as topic modeling or global hierarchical clustering may be warranted rather than a homology search, as this approach may facilitate the better identification of clusters of sequences from the same organism that lack similarity to a reference database.
While the absence of complete lineages limits our ability to accurately annotate environmental sequences, database expansion does not always remedy the annotation problem. Annotation is challenging because very highly conserved proteins often cannot be disentangled, and some unique sequences rarely have homology with others in the reference database even when coverage is relatively good. Our family-level analysis showed that even when a group had higher database representation, it was not necessarily easier to identify in community data (Fig. 2). We also showed that more than half of sequences within an abundant and ecologically significant protistan phylum (Bacillariophyta) lack non-self hits to another sequence of the same family (Table 1 and Supplementary Fig. 6). Because non-self, same-family hits appeared to be limited to a maximum value regardless of the number of available family-level relatives in the database (Supplementary Fig. 6), this observation is unlikely to be solely a consequence of database incompleteness. In some cases, the sequences lacking family overlap might be spurious, and in other cases sequences may constitute valuable variability that could enable understanding of population dynamics in protists63,64. In our analysis, the addition of genomes and transcriptomes at genus resolution in the Tara Oceans samples similarly did not necessarily increase our ability to identify a different species from that genus using typical annotation approaches. Further, percentage identity within a high-scoring alignment for protein matching is frequently an unreliable indicator of phylogenetic relatedness (e.g., Fig. 3B). Training models or selecting thresholds using a phylogeny-aware approach takes into account the patterns in sequence overlap that differentiate microorganisms (e.g., what defines distinct species at the sequence-level for one family may be different for another family).
Accurate taxonomic annotation of environmental sequences has evolved with both algorithms and the increasing size of databases. Using an unsupervised method and a clustering approach such as the tax-aliquots workflow shown here reduces bias associated with particularly rare taxonomic groups for which only a single database representative might be available. Multiple repeated hits are not weighted more heavily by clustering algorithms, allowing annotation challenges to be diagnosed. Taken together, our vignettes and the output of the tax-aliquots workflow illustrate the importance of critically evaluating the completeness and composition of the database selected. Using clustering and engaging with sequence content offers an approach to target taxa that are insufficiently covered in current databases or may be novel. Considering taxonomic annotation as a clustering problem may also be complementary to emerging approaches in leveraging protein structure information to understand proteins of unknown function61. We encourage applying clustering workflows like tax-aliquots to challenging datasets with low rates of taxonomic annotation to expand inference on groups of interest. Ultimately, critical reassessment of datasets and reevaluation of methods is a vital step towards improving taxonomic annotation and enhancing our ability to link taxonomic variability to functional potential in natural communities of ecologically essential protists.
Methods
In order to evaluate and select a sequence identity cutoff for use in taxonomic classification, we performed a bidirectional DIAMOND search65 of the MMETSP database using the blastp algorithm66. We used a cutoff of hits with bitscore of at least 50, and processed hits according to their percentage identity. We removed self-hits to the same sequence, and then recorded the percentage of sequences within each taxonomic family that had (a) hits to other sequences in the same taxonomic family and (b) hits to other sequences in different taxonomic families using eight different percentage identity cutoffs (30, 40, 50, 60, 65, 70, 80, and 90). We compared each of these percentages to the total number of transcriptomes associated with each family within the MMETSP. The results from this bidirectional search were used for the diatom family best hits displayed in Fig. 1D and for the diatom family mean percentage identity results in Fig. 2B. A similar bidirectional search which also included additional Radiolarian references was used to generate Supplementary Fig. 2E, and the same bidirectional search among the Phaeocystis references above was used to generate Supplementary Fig. 2F. We tested the uniformity of the counts of each diatom family in the MMETSP using the Anderson-Darling test against the uniform distribution generated with a count bound of zero to 10 greater than the maximum observed per-family count using the goftest package (version 1.2–3) in R67.
Genus Scale: Tara Oceans metagenomes
Metagenomic samples from the global ocean were retrieved from the Tara Oceans project68. Assemblies were previously generated in Alexander et al. (2021)17, with input sequencing reads grouped by ocean basin, depth, and size fraction; in brief, assemblies were generated by the MEGAHIT assembler69 after trimming with the Trimmomatic software70. Protein prediction was performed with Prodigal47,71. The taxonomic identity of predicted proteins was obtained using EUKulele v2.0.324, first using a combined database containing the MMETSP29,31,42, MarRef72, and additional Phaeocystis references, including the genome resources for Phaeocystis antarctica and Phaeocystis globosa73,74 available from the IMG/M (Integrated Microbial Genomes & Microbiomes) database (Phaant1 and Phaglo1, respectively), Phaeocystis cordata, Phaeocystis jahnii, and Phaeocystis globosa transcriptome resources75,76,77, and a Phaeocystis pouchetii transcriptome (Mars Brisbin et al. in prep). The contigs associated with the proteins identified to the genus Phaeocystis were quantified against the raw reads using the CoverM software in contig mode, from which we obtained estimates for total coverage in TPM as represented in Fig. 1 (v0.6.2; https://github.com/wwood/CoverM; coverm contig --min-covered-fraction 0).
Subsequently, separate EUKulele databases were created that contained the MMETSP29,31,42 with all genus Phaeocystis references removed, the MarRef72 database, and one of the ten distinct Phaeocystis genome or transcriptome references, inclusive of species Phaeocystis antarctica, Phaeocystis globosa, Phaeocystis pouchetii, Phaeocystis jahnii, Phaeocystis cordata, and Phaeocystis rex. A third set of EUKulele databases was created which contained the MMETSP29,31,42 with all genus Phaeocystis references removed, the MarRef72 database, and all of either the colony-forming Phaeocystis species or the free-living Phaeocystis species (Phaeocystis cordata, Phaeocystis jahnii, and Phaeocystis rex). Each Tara Oceans assembly was annotated with each of these databases. All databases used for the mapping are available online on Zenodo (https://zenodo.org/record/8269166).
A phylogenetic tree for the Phaeocystis references was constructed by conducting orthologous group clustering against all Phaeocystis references, a selection of Emiliania huxleyi transcriptome assemblies from the MMETSP (MMETSP0994, MMETSP0995, MMETSP0996, MMETSP0997, MMETSP1006, MMETSP1007, MMETSP1008, MMETSP1009, MMETSP1150, MMETSP1151, MMETSP1152, MMETSP1153, MMETSP1154, MMETSP1156, MMETSP1157), Gephyrocapsa oceanica transcriptome assemblies from the MMETSP (MMETSP1363, MMETSP1364, MMETSP1365, MMETSP1366), Isochrysis galbana transcriptome assemblies from the MMETSP (MMETSP0943, MMETSP00595), and three reference genomes from the JGI’s IMG/M (Integrated Microbial Genomes & Microbiomes) database73,74 - Chrysochromulina tobinii (Chrsp), Oxytricha trifallax (Oxytri1), and Guinardia theta (Guith1). Orthologous groups were created from proteins from all references using OrthoFinder (v2.5.4)78, and orthologous groups containing a single protein from all of the Phaeocystis references were used to create an alignment and phylogenetic tree. This amounted to 40 total single-copy genes shared across references which were used to build the alignment. The MAFFT tool was used for multiple sequence alignment of each of the concatenated lists of single-copy genes (one file per gene containing all gene versions across organisms in the alignment; version 7.508), followed by the removal of possible spurious sequences using trimAl79 (version 1.4.rev15), and then a secondary multiple sequence alignment using Clustal-Omega80. Sequences in the alignment were adjusted to standardize their trimmed lengths, and the subsequent alignments were concatenated and trimmed once more with trimAl. FastTree (version 2.1.11) was used to build the phylogenetic tree with 100 resamples (-boot 100)81.
Family Scale: metatranscriptomes from Narragansett Bay
The metatranscriptome assembly and annotation process for the metatranscriptomic samples from Narragansett Bay is described in full in Krinos et al. (2023)41. In brief, raw reads were trimmed and quality-assessed, and then assembled in parallel using the eukrhythmic pipeline41. Trimming was performed using Trimmomatic version 0.3970, with a minimum read length of 50 basepairs, a sliding window of length 4 and quality score 2, and a standard list of Illumina adapters (ILLUMINACLIP:<adapter-list > :2:30:7 LEADING:2 TRAILING:2 SLIDINGWINDOW:4:2 MINLEN:50). Assembly was performed using default parameters to the eukrhythmic pipeline and used MEGAHIT, rnaSPAdes, metaSPAdes, and Trinity69,82,83,84. Taxonomic annotations were assigned using the EUKulele tool24 using a combined database containing the MMETSP and MarRef2 sequences31.
Phylum Scale: metatranscriptomes from a transect between WHOI and BATS
Samples from the transect between Woods Hole Oceanographic Institution (WHOI) and the Bermuda Atlantic Time Series (BATS) stations were assembled and post-processed as described in Cohen et al. (2023)44, with assembly products available online through Zenodo (https://zenodo.org/record/8287779). EUKulele24 was used for the BLAST-LCA search against these sequences, first using the MarRef and MMETSP database31 and then adding all radiolarian references available in the EukProt and EukZoo databases34,44. These organisms included Sticholonche zanclea (EP00491), Amphilonche elongata (EP00492), Phyllostaurus siculus (EP00493), Astrolonche serrata (EP00494), Collozoum sp. 1 RS2012 (EP00495), Lithomelissa setosa (EP00496), and Spongosphaera streptacantha (EP00497). All data associated with this project are published as part of Cohen et al. (2023; in prep). Raw sequences have been deposited to the NCBI SRA database under BioProject ID PRJNA903389. Assemblies, annotations and count data are available through Zenodo (https://zenodo.org/record/7317272#.Y3Z5w-zMInV).
Hybrid partially-supervised clustering workflow
A very permissive protein clustering is performed using DIAMOND DeepClust85, followed by taxonomic profiling using hierarchical clustering on a matrix formed in parallel by calculating kAAmer overlap between sequences present in the cluster. This enables exact kAAmer overlap to be computed efficiently, and does not taxonomically annotate sequences for which an alignment is based on sequence coverage of <20-50% of the protein. Unlike other LCA-based approaches where ancestry is computed using the aligned fragment, this method profiles the short kAAmers over the entire length of the proteins which were originally clustered together on the basis of a short and potentially low sequence similarity alignment. This allows sequences with promising homology, even with low percentage identity, to be clustered based on consistency in sequence content over the entire protein length.
We ran DIAMOND DeepClust85 against the predicted proteins from the MMETSP and MarRef2 databases31 using a 50% coverage threshold for the shorter sequence in the alignment and no minimum percentage identity. First, kAAmers were identified in parallel separately for each cluster. We used the pyahocorasick package, which implements the Aho-Corasick algorithm for efficient string matching86,87. After counting all kAAmers of length 4 using this approach and the “Automaton” utility from pyahocorasick, we computed similarity between each sequence in the protein cluster according to the formula:
Where \({intersections}\left(i,\, j\right)\) is the number of intersecting kAAmers between proteins sequences \(i\) and \(j\) and \(\left({n}_{{kAAmers}}\left(i\right),\, {n}_{{kAAmers}}\left(i\right)\right)\) is the minimum number of kAAmers found in each of the two protein sequences, which is used to scale the raw number of intersections. These distance numbers were used for the downstream hierarchical clustering steps, which were conducted using the fcluster function from SciPy88.
We linked original sequences from the database to revised taxonomic annotations according to the taxonomic coherence of the cluster to which it was assigned using the two-part algorithm. We created a new taxonomy string dictionary which takes into account the taxonomic ambiguity of sequences according to their kAAmer overlap. The stringent approach used a distance threshold of 0.2, the intermediate a threshold of 0.5, and the permissive approach used a distance threshold of 0.8. We explored the utility of this approach using a “mock metatranscriptome” (the Phaeocystis pouchetii transcriptome) as a hypothetical scenario of an unknown taxon to which sequences could be recruited via clustering; for this example we used the MMETSP and MarRef combined database and a kAAmer length of 3 (Fig. 5). We conducted an initial EUKulele search with the default database containing MMETSP and the MarRef database31,51 and filtered sequences that were annotated as haptophytes for the second search with tax-aliquots and the two LCA-based tools with only Phaeocystis sequences as described below. To compare our approach to other taxonomic annotation tools, we annotated the taxonomy of the same “metatranscriptome” with the mmseqs2 taxonomy tool52, using a default UniRef90 database available with mmseqs250 as well as a custom database containing either all Phaeocystis apart from Phaeocystis pouchetii (applied to the the sequences annotated as haptophytes using EUKulele). Finally, we annotated the transcriptome using EUKulele (version 2.0.7)24 using the custom databases described above that contained the MMETSP and MarRef databases31,42,51 as well as a custom database containing all Phaeocystis but excluding Phaeocystis pouchetii (applied to the sequences annotated as haptophytes using the initial EUKulele search). We applied tax-aliquots to the filtered haptophyte sequences with the custom Phaeocystis-only database. The figures and discussion in the text refer to a less stringent 0.8 distance cutoff for the hierarchical clustering step of tax-aliquots, but we also ran tax-aliquots with a 0.3 and a 0.5 distance cutoff for demarcating sequences as part of the same cluster, corresponding to more stringent clustering.
Figures were generated in R (version 4.1) and in Python (version 3.10.1) using the ggplot2 software (including the world map dataset using the map_data function from ggplot2), ggridges package, ggUpSet package, ggmap package, and ggalluvial package89,90,91,92,93,94.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
For the Phaeocystis sequences: EUKulele databases used to annotate the Tara68 assemblies17 are available at Zenodo (https://zenodo.org/record/8269166). Raw read sequences for the Tara dataset are available from the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI; accession number PRJEB4352). For the Narragansett Bay dataset: all raw read sequences and assemblies are available as described in Alexander et al. (2015) and Krinos et al. (2023)28,41, and raw read sequences were deposited to the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA; www.ncbi.nlm.nih.gov/sra; accession no. SRP055134). For the BATS dataset: raw sequences have been deposited to the NCBI SRA database under BioProject ID PRJNA903389. Assemblies, annotations, and count data are available through Zenodo (https://zenodo.org/record/7317272#.Y3Z5w-zMInV). Source data are provided with this paper.
Code availability
Code for running the tax-aliquots clustering can be accessed at https://github.com/akrinos/tax-aliquots (also published on Open Science Framework; https://doi.org/10.17605/OSF.IO/P865G). Code for analysis and figure generation can be found at https://github.com/akrinos/2023-euk-diversity (also published on Open Science Framework; https://doi.org/10.17605/OSF.IO/PH8EC).
References
Keeling, P. J. & Campo, J. D. Marine protists are not just big bacteria. Curr. Biol. 27, R541–R549 (2017).
Cuddington, K., Byers, J.E., Wilson, W.G. & Hastings, A. Ecosystem Engineers: Plants to Protists. (Academic Press, 2011).
Caron, D. A., Countway, P. D., Jones, A. C., Kim, D. Y. & Schnetzer, A. Marine protistan diversity. Ann. Rev. Mar. Sci. 4, 467–493 (2012).
Sherr, E. B. & Sherr, B. F. Significance of predation by protists in aquatic microbial food webs. Antonie Van. Leeuwenhoek 81, 293–308 (2002).
Worden, A. Z. et al. Environmental science. Rethinking the marine carbon cycle: factoring in the multifarious lifestyles of microbes. Science 347, 1257594 (2015).
Rosen, G. L., Reichenberger, E. R. & Rosenfeld, A. M. NBC: the Naive Bayes Classification tool webserver for taxonomic classification of metagenomic reads. Bioinformatics 27, 127–129 (2011).
Rosen, G., Garbarine, E., Caseiro, D., Polikar, R. & Sokhansanj, B. Metagenome fragment classification using N-mer frequency profiles. Adv. Bioinform. 2008, 205969 (2008).
Delgado-Serrano, L., Restrepo, S., Bustos, J. R., Zambrano, M. M. & Anzola, J. M. Mycofier: a new machine learning-based classifier for fungal ITS sequences. BMC Res. Notes 9, 402 (2016).
Bouchot, J.-L. et al. Advances in machine learning for processing and comparison of metagenomic data. Comput. Syst. Biol. Mol. Mech. Dis. 295–329 (2013).
Fierst, J. L. & Murdock, D. A. Decontaminating eukaryotic genome assemblies with machine learning. BMC Bioinform. 18, 533 (2017).
Brown, C. T. & Irber, L. sourmash: a library for MinHash sketching of DNA. J. Open Source Softw. 1, 27 (2016).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Royo-Llonch, M. et al. Compendium of 530 metagenome-assembled bacterial and archaeal genomes from the polar Arctic Ocean. Nat. Microbiol. 6, 1561–1574 (2021).
Delmont, T. O. et al. Nitrogen-fixing populations of Planctomycetes and Proteobacteria are abundant in surface ocean metagenomes. Nat. Microbiol. 3, 804–813 (2018).
Delmont, T. O. et al. Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean. Cell Genom. 2, 100123 (2022).
Alexander, H. et al. Eukaryotic genomes from a global metagenomic data set illuminate trophic modes and biogeography of ocean plankton. mBio 14, e01676–23 (2023).
Massana, R. Marine Protists: Diversity and Dynamics (eds. Ohtsuka, S., Suzaki, T., Horiguchi, T., Suzuki, N. & Not, F.) 3–21 (Springer Japan, 2015).
Laso-Jadart, R., O’Malley, M., Sykulski, A. M., Ambroise, C. & Madoui, M.-A. How marine currents and environment shape plankton genomic differentiation: a mosaic view from Tara Oceans metagenomic data. bioRxiv 2021.04.29.441957 https://doi.org/10.1101/2021.04.29.441957 (2021).
Hamilton, M. et al. Spatiotemporal variations in antarctic protistan communities highlight phytoplankton diversity and seasonal dominance by a novel cryptophyte lineage. mBio 12, e0297321 (2021).
Obiol, A. et al. A metagenomic assessment of microbial eukaryotic diversity in the global ocean. Mol. Ecol. Resour. 20, 718–731 (2020).
Cohen, N. R. et al. Marine microeukaryote metatranscriptomics: sample processing and bioinformatic workflow recommendations for ecological applications. Frontiers in Marine Science 9, 867007 (2022).
Levy Karin, E., Mirdita, M. & Söding, J. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 8, 48 (2020).
Krinos, A. I., Hu, S. K., Cohen, N. R. & Alexander, H. EUKulele: Taxonomic annotation of the unsung eukaryotic microbes. JOSS 6, 2817 (2021).
Delgado, L. F. & Andersson, A. F. Evaluating metagenomic assembly approaches for biome-specific gene catalogues. Microbiome 10, 72 (2022).
Villar, E. et al. The Ocean Gene Atlas: exploring the biogeography of plankton genes online. Nucleic Acids Res. 46, W289–W295 (2018).
Groussman, R. D., Coesel, S. N., Durham, B. P. & Armbrust, E. V. Diel-regulated transcriptional cascades of microbial eukaryotes in the North Pacific subtropical gyre. Front. Microbiol. 12, 682651 (2021).
Alexander, H., Jenkins, B. D., Rynearson, T. A. & Dyhrman, S. T. Metatranscriptome analyses indicate resource partitioning between diatoms in the field. Proc. Natl Acad. Sci. USA 112, E2182–E2190 (2015).
Caron, D. A. et al. Probing the evolution, ecology and physiology of marine protists using transcriptomics. Nat. Rev. Microbiol. 15, 6–20 (2017).
Lambert, B. S. et al. The dynamic trophic architecture of open-ocean protist communities revealed through machine-guided metatranscriptomics. Proc. Natl. Acad. Sci. USA. 119, e2100916119 (2022).
Keeling, P. J. et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12, e1001889 (2014).
Anderson, O. R. Recent advances in application of transcriptomics: research on heterotrophic and autotrophic protists. Acta Protozool. 61, 47–75 (2022).
Bokulich, N. A. et al. mockrobiota: a public resource for microbiome bioinformatics benchmarking. mSystems 1, e00062-16 (2016).
Lydon, K. A. & Lipp, E. K. Taxonomic annotation errors incorrectly assign the family Pseudoalteromonadaceae to the order Vibrionales in Greengenes: implications for microbial community assessments. PeerJ 6, e5248 (2018).
Afshinnekoo, E. et al. Geospatial resolution of human and bacterial diversity with city-scale metagenomics. Cell Syst. 1, 97–97.e3 (2015).
Medlin, L. & Zingone, A. Phaeocystis, Major Link In The Biogeochemical Cycling Of Climate-relevant Elements (eds. van Leeuwe, M. A. et al.) 3–18 (Springer Netherlands, 2007).
Rousseau, V., Chrétiennot-Dinet, M.-J., Jacobsen, A., Verity, P. & Whipple, S. The life cycle of Phaeocystis: state of knowledge and presumptive role in ecology. Biogeochemistry 83, 29–47 (2007).
Decelle, J. et al. An original mode of symbiosis in open ocean plankton. Proc. Natl Acad. Sci. USA 109, 18000–18005 (2012).
Zingone, A., Chrétiennot-Dinet, M.-J., Lange, M. & Medlin, L. Morphological and genetic characterization of Phaeocystis cordata and P. jahnii (Prymnesiophyceae), two new species from the Mediterranean sea. J. Phycol. 35, 1322–1337 (1999).
Da Silva, O. et al. Genomic differentiation of three pico-phytoplankton species in the Mediterranean Sea. Environ. Microbiol. 24, 6086–6099 (2022).
Krinos, A. I., Cohen, N. R., Follows, M. J. & Alexander, H. Reverse engineering environmental metatranscriptomes clarifies best practices for eukaryotic assembly. BMC Bioinform. 24, 74 (2023).
Johnson, L. K., Alexander, H. & Brown, C. T. Re-assembly, quality evaluation, and annotation of 678 microbial eukaryotic reference transcriptomes. Gigascience 8, giy158 (2019).
Richter, D. J. et al. EukProt: A database of genome-scale predicted proteins across the diversity of eukaryotes. Peer Community J. 2, e56 (2022).
Cohen, N. R., Krinos, A. I., Kell, R. M., Chmiel, R. J., Moran, D. M., McIlvin, M. R. et al. Microeukaryote metabolism across the western North Atlantic Ocean revealed through autonomous underwater profiling. Nature Communications, 15, 7325 (2024).
Nakamura, Y. et al. DNA metabarcoding focused on difficult-to-culture protists: An effective approach to clarify biological interactions. Environ. Microbiol. 25, 3630–3638 (2023).
Karlicki, M., Antonowicz, S., & Karnkowska, A. Tiara: deep learning-based classification system for eukaryotic sequences. Bioinformatics, 38, 344–350 (2022).
West, P. T., Probst, A. J., Grigoriev, I. V., Thomas, B. C. & Banfield, J. F. Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res. 28, 569–580 (2018).
Déraspe, M., Boisvert, S., Laviolette, F., Roy, P. H. & Corbeil, J. Flexible protein database based on amino acid k-mers. Sci. Rep. 12, 9101 (2022).
Pierce-Ward, T., Botvinnik, O. B., Reiter, T. E., Irber, L. & Titus Brown, C. Amino Acid k-mers enable assembly- and alignment-free sequence analysis. Manubot (2022).
Suzek, B. E., Huang, H., McGarvey, P., Mazumder, R. & Wu, C. H. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288 (2007).
Klemetsen, T. et al. The MAR databases: development and implementation of databases specific for marine metagenomics. Nucleic Acids Res. 46, D692–D699 (2018).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Massana, R. & López-Escardó, D. Metagenome assembled genomes are for eukaryotes too. Cell Genom. 2, 100130 (2022).
Liechti, N., Schürch, N., Bruggmann, R. & Wittwer, M. Nanopore sequencing improves the draft genome of the human pathogenic amoeba Naegleria fowleri. Sci. Rep. 9, 16040 (2019).
Semmouri, I., De Schamphelaere, K. A. C., Mees, J., Janssen, C. R. & Asselman, J. Evaluating the potential of direct RNA nanopore sequencing: metatranscriptomics highlights possible seasonal differences in a marine pelagic crustacean zooplankton community. Mar. Environ. Res. 153, 104836 (2020).
d’Avila-Levy, C. M. et al. First Draft Genome of the Trypanosomatid Herpetomonas muscarum ingenoplastis through MinION Oxford Nanopore Technology and Illumina Sequencing. Trop. Med. Infect. Dis. 5, 25 (2020).
Allen, L. Z. et al. Influence of nutrients and currents on the genomic composition of microbes across an upwelling mosaic. ISME J. 6, 1403–1414 (2012).
Dupont, C. L. et al. Genomes and gene expression across light and productivity gradients in eastern subtropical Pacific microbial communities. ISME J. 9, 1076–1092 (2015).
Liu, Z., Hu, S. & Caron, D. EukZoo, an aquatic protistan protein database for meta-omics studies. [Dataset]. Zenodo. https://doi.org/10.5281/zenodo.1476236 (2018).
Cao, S. et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome 8, 47 (2020).
Robinson, S. L. Structure-guided metagenome mining to tap microbial functional diversity. Curr. Opin. Microbiol. 76, 102382 (2023).
Pernice, M. C., Logares, R., Guillou, L. & Massana, R. General patterns of diversity in major marine microeukaryote lineages. PLoS ONE 8, e57170 (2013).
Rengefors, K., Kremp, A., Reusch, T. B. H. & Wood, A. M. Genetic diversity and evolution in eukaryotic phytoplankton: revelations from population genetic studies. J. Plankton Res. 39, 165–179 (2017).
Logares, R. Population genetics: the next stop for microbial ecologists? Open Life Sci. 6, 887–892 (2011).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Faraway, J., Marsaglia, G., Marsaglia, J. & Baddeley, A. Classical Goodness-of-Fit Tests for Univariate Distributions [R package goftest version 1.2–3]. (2021).
Sunagawa, S. et al. Tara Oceans: towards global ocean ecosystems biology. Nat. Rev. Microbiol. 18, 428–445 (2020).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Hernández, L. et al. Evolutionary history of dimethylsulfoniopropionate (DMSP) demethylation enzyme DmdA in marine bacteria. PeerJ 8, e9861 (2020).
Mukherjee, S. et al. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Res. 49, D723–D733 (2021).
Chen, I.-M. A. et al. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677 (2019).
Uwizeye, C. et al. Cytoklepty in the plankton: a host strategy to optimize the bioenergetic machinery of endosymbiotic algae. Proc. Natl. Acad. Sci. USA. 118, e2025252118 (2021).
Brisbin, M. M. Characterization of Acantharea-Phaeocystis Photosymbioses: Distribution, Abundance, Specificity, Maintenance And Host-control (2020).
Mars Brisbin, M. & Mitarai, S. Differential gene expression supports a resource-intensive, defensive role for colony production in the bloom-forming haptophyte, Phaeocystis globosa. J. Eukaryot. Microbiol. 66, 788–801 (2019).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 8, giz100 (2019).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Buchfink, B., Ashkenazy, H., Reuter, K., Kennedy, J. A. & Drost, H.-G. Sensitive clustering of protein sequences at tree-of-life scale using DIAMOND DeepClust. bioRxiv https://doi.org/10.1101/2023.01.24.525373 (2023).
Muła, W. & Ombredanne, P. Pyahocorasick Library (2021).
Aho, A. V. & Corasick, M. J. Efficient string matching: an aid to bibliographic search. Commun. ACM 18, 333–340 (1975).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Bunn, A. & Korpela, M. Crossdating in dplR. https://cran.microsoft.com/snapshot/2014-09-08/web/packages/dplR/vignettes/xdate-dplR.pdf (2014).
Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. The Journal of Machine Learning Research. 12, 2825–2830 (2011).
Wilke, C. O. ggridges: ridgeline plots in ‘ggplot2’. R package version 0.5 (2024).
Ahlmann-Eltze, C. ggupset: combination matrix axis for ‘ggplot2’to Create ‘UpSet’Plots. https://CRAN.R-project.org/package=ggupset (2024).
Kahle, D., Wickham, H. & Ggmap Spatial visualization with ggplot2. R. J. 5, 144 (2013).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer Science & Business Media, 2009).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Advanced Scientific Computing Research, Department of Energy Computational Science Graduate Fellowship under Award Number DE-SC0020347 (A.I.K.). The work conducted by the US Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science user facility, is supported by the Office of Science of the US Department of Energy, operated under contract no. DE-AC02-05CH11231 (F.S. and A.I.K.). M.M.B. was supported by a Simons Foundation Postdoctoral Fellowship in Marine Microbiology (award #874439 to M.M.B.). H.A. is supported by a Simons Foundation Early Career Investigator in Aquatic Microbial Ecology and Evolution Award (award #931886 to H.A.). A Simons Collaboration on Computational Biogeochemical Modeling of Marine Ecosystems (CBIOMES) supported M.J.F. and A.I.K. (award #549931 to M.J.F. also supporting A.I.K.).
Author information
Authors and Affiliations
Contributions
A.I.K., M.M.B., and H.A. conceived the original idea for an exploration of the influence of missing references on metatranscriptomics and metagenomic analysis of marine microbial eukaryotic communities; AIK conceived the clustering idea. F.S. contributed to development of the clustering approach and tax-aliquots. S.K.H. and N.R.C. contributed ideas to approach and analysis of data; N.R.C. contributed analysis products. T.A.R. contributed expertise on diatom communities in Narragansett Bay. M.J.F. consulted on idea development throughout the project and secured funding for the analyses. A.I.K. conducted analyses on all three datasets, generated figures, developed and tested the tax-aliquots approach, and wrote the initial manuscript draft. All authors (A.I.K., M.M.B., S.K.H., N.R.C., T.A.R., M.J.F., F.S., and H.A.) contributed to writing and editing of the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Krinos, A.I., Mars Brisbin, M., Hu, S.K. et al. Missing microbial eukaryotes and misleading meta-omic conclusions. Nat Commun 15, 9873 (2024). https://doi.org/10.1038/s41467-024-52212-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52212-w
