
Abstract
Cancer is a significant cause of death around the world, and for many varieties, treatment is not successful. Therefore, there is a need for the development of innovative, efficacious, and precisely targeted treatments. Here, we develop a series of Au(I) complexes (1-4) through rational manipulation of ligand structures, thereby achieving tumor cell specific targeting and orchestrated tumor eradication via chemo-phototherapy and induced immunogenic cell death. A comprehensive exploration based on in vitro and in vivo female mice experimentation shows that complex 4 exhibits proficiency in specific tumor imaging, endoplasmic reticulum targeting, and has robust therapeutic capabilities. Mechanistic elucidation indicates that the anticancer effect derives from the synergistic actions of thioredoxin reductase inhibition, highly efficient reactive oxygen species production and immunogenic cell death. This work presents a report on a robust Au(I) complex integrating three therapeutic modalities within a singular system. The strategy presented in this work provides a valuable reference for the development of high-performance therapeutic agents.
Similar content being viewed by others


Introduction
As a major disease threatening human health, cancer has the characteristics of a high incidence rate and high mortality. In recent years, both the incidence and mortality rates of cancer continue to grow globally1. According to data released by the International Agency for Research on Cancer (IARC) of the World Health Organization, there were ~19.29 million new cancer cases and 9.96 million cancer-related deaths globally in 20202. At present, the primary methods of cancer treatment include radiotherapy, surgical therapy and chemotherapy3,4. Among them, radiotherapy involves using radiation to eliminate local tumor lesions, but it poses risks of damaging normal tissues around the tumor and induces various side effect, such as nausea, vomiting, loss of appetite, overall fatigue and decreased bone marrow function. Surgical therapy involves tumor removal through resection, yet has many drawbacks, including reduced immune function, high risk of tumor resection in sensitive areas and susceptibility to recurrence. So far, chemotherapy is still the primary cancer treatment method inhibiting the proliferation, infiltration and metastasis of tumor cells using chemical drugs. Although widely use, chemotherapy still exist many drawbacks in clinics, such as the lack of tumor targeting, strong toxic side effects and the emergence of drug resistance due to frequent drug use5. In order to improve the efficiency of cancer therapy, experts are still continuously exploring new strategies. However, the essential shortcomings of current therapies, including targeting issues, toxicity, and drug resistance, persist as significant obstacles. Therefore, in the current research and treatment of cancer, there is an urgent need to develop new, efficient, and precise treatment strategies.
In recent years, immunotherapy has emerged as a powerful and promising method in cancer therapy6. This approach involves activating or enhancing the host’s immune response to track, kill, and eliminate cancer cells7. The unique advantage of immunotherapy lies not only in eliminating local metastatic tumors, but also in preventing tumor recurrence through long-term immune memory8. Despite its success, immunotherapy is not universally effective for all types of cancer, and it still has some shortcomings, such as high cost, development of resistance, limited applicability and effectiveness of single modality9. Therefore, researchers are exploring to synergistic approaches by combining immunotherapy with other treatment methods, aiming to achieve more optimal outcomes in cancer therapy10. Compared with traditional treatment methods, phototherapy, as an emerging and important treatment method, hold the advantages of non-invasiveness and minimal side effects11,12. In addition, due to its unique ability of inducible immunogenicity, it has attracted much attention in recent years13,14,15. Studies have demonstrated that phototherapy can induce tumor immunogenic cell death (ICD) under light irradiation, promote the release of tumor-related antigens from residual tumor cell, and further enhance the activation, proliferation and infiltration of antigen-specific T cells16,17,18. Therefore, combining phototherapy with immunotherapy have shown potential to improve the overall efficiency of anticancer therapy.
However, traditional photosensitizers used in phototherapy often show poor photostability and suffer from severe aggregation fluorescence quenching (ACQ) effect in the aggregate state19,20. Additionally, the enhanced non-radiative attenuation caused by aggregation can reduce the phototherapeutic effects of many traditional photosensitizer21. In this context, aggregation-induced emission luminogens (AIEgens) present an ideal solution for solving the ACQ problems of traditional photosensitizers in aggregate states. AIEgens exhibit enhanced emission as aggregates, possess large Stokes shift and strong photostability. More importantly, AIEgens also hold excellent fluorescence imaging abilities, allowing the integration of diagnostic and therapeutic functions into a single platform for precise imaging-guided cancer treatment22. However, current AIEgens still face challenges of low absorptivity and insufficient targeting ability, which limit their luminescence and phototherapy efficiency.
To tackle the aforementioned challenges faced by AIEgens, our group has proposed a simple yet effective strategy in our previous work, involving the introduction of Au(I) unit23. This strategy addresses the two key aspects. Firstly, it significantly enhances the luminescence of AIEgen ligand, enabling deep-tissue penetration and high-resolution in vivo imaging. The elevation of absorptivity improves the non-radiative phototherapeutic efficiency of AIEgen ligand. Secondly, the Au(I) center also achieves excellent antitumor performance through a special thioredoxin reductase (TrxR) inhibition pathway24,25,26. Therefore, the introduction of Au(I) can further achieve a synergistic outcome of chemotherapy and phototherapy, maximizing the antitumor efficacy. Although Au(I)-based AIEgens has made some progress in antitumor research27, this field is still in its early stage. And, current treatment methods mainly focused on chemotherapy and phototherapy, and there are few reports on immunotherapy. Although the latest relevant reports have expanded into the field of immunology27,28, the limited therapeutic modalities have resulted in insufficient tumor treatment effects and immune activation capabilities. Considering the above limitations of current immunotherapy and phototherapy, we thus envision the development of Au(I)-based AIE systems that integrate chemotherapy, phototherapy and immunotherapy to achieve potent antitumor effect. At present, such a powerful system has not yet been reported, as it is a challenging task.
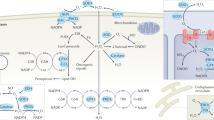
In this work, we propose an approach by integrating the NHC-Au unit and AIE ligands to design a series of complexes. Inspired by the recent work of Prof. Arambula and colleagues29, reporting that N-heterocyclic carbene (NHC)-Au complex could act as an efficacious ICD inducer to trigger ICD via redox modulation. This integration aims to harness the advantages of both components, resulting in a synergistic outcome for chemotherapy, phototherapy, and immunotherapy. Furthermore, we derived the NHC ligand into triphenylphosphine and systematically compared their biological imaging and anticancer performances to identify optimal candidates. Ultimately, it turns out that the designed NHC-Au(I) complexes 1-3 exhibit efficient anticancer treatment through combined chemotherapy, phototherapy and immunotherapy (Fig. 1). Notably, the triphenylphosphine coordinated complex 4 exhibited more prominent imaging and antitumor performance. To the best of our knowledge, this is the first report of powerful Au(I) complexes integrating three therapeutic approaches, i.e., chemotherapy, phototherapy and immunotherapy, into a single system. It will open the way for the development of high-performance theranostic agents.

Following intravenous injection, the nanoparticles of the Au(I) complex will accumulate passively at the tumor site. Once inside the tumor cells, they can target specific organelles, induce cellular oxidative stress, including endoplasmic reticulum (ER) stress, facilitate the occurrence of ICD, and enhance the release of damage-associated molecular patterns (DAMPs) from the tumor cells. These DAMPs include the translocation of calreticulin (CRT), the secretion of adenosine triphosphate (ATP), and the release of high-mobility group box 1 (HMGB1). The DAMPs released from dying cancer cells can activate a specific antitumor immune response, promote the generation of tumor-specific effector T cells, further eliminate residual or metastatic lesions, eradicate the primary tumor, and ultimately foster long-term immune memory to prevent tumor recurrence.
Results
Design, synthesis and characterization of Au(I) complexes
We conceived the idea to integrate pyridine-based AIE ligands with the NHC-Au(I) unit based on inspiration drawn from the work of Professor Lin et al.30. Until now, the reported Au(I) complexes coordinated by pyridine and NHC are rare, and the related luminescent systems has not yet been reported. Although we previously developed three multifunctional pyridine-based AIE ligands coordinated pentafluorophenyl-Au(I) complexes with demonstrated anticancer applications31,32,33, they remain neutral molecules, and their binding affinities to cells need enhancement. Recognizing this challenge, the successful integration of pyridine-based AIEgens with the NHC-Au(I) unit would represent a groundbreaking approach in molecular design, considering the extensive modifiability inherent in both AIEgens and carbene structures. Thus, we selected three classical pyridine-based AIE ligands with gradually enhanced donor (D)-accepter (A) characteristics (Fig. 2a)34,35,36, to achieve the manipulation of both the luminescence wavelength and reactive oxygen species (ROS) generation capability. Additionally, we also replaced the NHC ligand with a triphenylphosphine unit to make a comparative exploration of their photophysical and biological properties.

a Molecular structure of complexes 1-4. b Plots of normalized PL intensity of complexes 1-4 in the aggregate states. c Photoluminescence (PL) spectra of complex 3 (2.0 × 10−5 M) in DMF water mixtures, λex = 480 nm. Insert: photos of DMF solution and DMF/water mixture (fw = 90%). d Relationship diagram between the relative maximum emission peak intensity (αAIE) and fw of the DMF water mixture of complexes 1-4 (where αAIE = I/I0; I = emission intensity; I0 = emission intensity in DMF solution). e DFT calculated the molecular orbitals of HOMO and LUMO, while TD-DFT calculated the energy levels (B3LYP/Def2-TZVP), major orbital configurations, and ISC channels of complexes 1-4. The lowest singlet excited state (S1), the lowest triplet excited state (T1), and high-level triplet excited states (Tn) are denoted as different electronic states. Possible intersystem crossing (ISC) channels from the singlet state (S1) to its higher- or lower-lying triplet states (Tn) are indicated by red and blue dashed arrows, respectively. The symbols H and L represent HOMO and LUMO orbitals, respectively. ΔEST represents the energy gap between the S1 state and the T1 state.
According to the above design strategy, we systematically synthesized Au(I) complexes 1-4 via the proposed synthetic route (Supplementary Fig. 1). Comprehensive characterization through NMR and high-resolution mass spectrometry provided satisfactory results for all intermediates and final complexes (Supplementary Figs. 2–14). The powders of the target complexes display high stability under ambient conditions and show good solubility in common organic solvents, including tetrahydrofuran (THF), chloroform, dichloromethane, and dimethylformamide (DMF), etc.
Then we systematically measured the photophysical properties of complexes 1-4. As observed, they all exhibited typical AIE properties and both the absorption and the maximum luminescence wavelengths of their aggregate states were gradually redshifted from complex 1 to complex 2 and then to complex 3 (Fig. 2b–d and Supplementary Figs. 15–19). Comparatively, the emission wavelength of complex 3 to complex 4 were close due to the existence of the same AIE ligands. The overall capabilities of complexes 1-4 to produce ROS were assessed using the commercial indicator 2,7-dichlorodihydrofluorescein (H2DCFDA)37. Upon light irradiation, all complexes were able to generate ROS quickly and efficiently within 30 seconds, demonstrating significantly better production abilities compared to the commercially available photosensitizer, rose bengal (RB) (see Supplementary Fig. 20a). Complex 3 exhibited the strongest ROS generation ability, followed by complex 2 and complex 4. While the production abilities of singlet oxygen (1O2) for them showed some difference (Supplementary Fig. 21), complexes 3 and 4 presented parallel and optimal performance, followed by complex 2 and complex 1. The results indicate that the addition of thiophene and benzothiazole units to complex 1 endow complexes 2-4 with much stronger ROS generation abilities. We calculated the singlet oxygen quantum yields of complexes 1-4 and the values, determined relative to RB, are as follows: 0.98-fold for complex 1, 1.03-fold for complex 2, 5.82-fold for complex 3, and 5.50-fold for complex 4. Accordingly, the generation abilities of 1O2 of all the four complexes were much better than that of RB. Additionally, we employed dihydrorhodamine 123 (DHR123) to detect the generation of superoxide anion of the relatively better complexes 3 and 4 in an aqueous solution under white light irradiation (72 J/cm2) (Supplementary Fig. 20b). In the presence of complexes 3 or 4, the fluorescence intensities of DHR123 enhanced obviously with the increase of irradiation time, implying their production abilities of type I superoxide anion. Thus, these results demonstrate that complexes 1-4 possess the potentials of photodynamic therapy.
To explore the mechanisms of ROS generation, a thorough series of density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were carried out. The topographies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), as well as energy level diagrams for all four complexes, are shown in Fig. 2e. The calculated energy gaps between the HOMO and LUMO orbitals shed light on the sequence of maximum absorption wavelengths (λabs) among the complexes: 4 ≈ 3 > 2 > 1. Similarly, the calculated energy of the excited state S1 reveals the order of maximum emission wavelengths (λem): 4 ≈ 3 > 2 > 1. Generally, the feasibility of the singlet-to-triplet transition through intersystem crossing (ISC) is governed by the nature of the triplet state (Tn). When the transition orbital compositions of Tn are identical to those of S1, and its energy level falls within the range of ES1 ± 0.3 eV or ±0.4 eV, the singlet-to-triplet transition from S1 to Tn is regarded as an efficient channel for ISC. Even though the energy gap between S1 and T1 in complex 1 is 0.26 eV (lower than 0.3 eV), T2 lacks the same transition orbital compositions as S1, eliminating it as a potential ISC channel. For complexes 3 and 4, only one possible singlet-to-triplet transition channel satisfies both criteria for efficient ISC (S1 to T1). On the other hand, complex 1 only has one channel which meets the basic criteria of having the same transition orbital compositions, however, its significantly higher ΔEst value of 0.55 eV leads to a rather low ISC efficiency, establishing an ISC efficiency order of 3 > 4 > 1. Notably, although the energy gap between S1 and T1 in complex 2 surpasses the other complexes, an alternative ISC channel from S1 to a higher T2 state, with an energy gap notably smaller than 0.3 eV (0.16 eV), exists. While this secondary channel in complex 2 contributes to its higher ROS generation efficiency compared to complexes 1 and 4, its endothermic nature might position complex 2 behind complex 3 in ROS generation efficiency. Consequently, an ISC efficiency sequence of 3 > 2 > 4 > 1 is established, delineating the observed ROS generation capability order from experiments. We also performed additional calculations for both ground and excited state energies using various DFT functionals and basis sets. The results show that the choice of basis set has minimal impact on these calculated. Moreover, using the M062X functional, commonly used in excited states calculations, gave a similar trend to those obtained with the B3LYP functional. The article also presents calculations and analyses of spin-orbit coupling (SOC, in cm−1), energy gaps between accessible triplet states and low-lying singlet states, differences in dipole moments between ground and excited states (Δμ, in D), effective distances (DCT, in Å), and the amount of transferred charge (qCT, in A.U.) for the S1 and accessible Tm states (see Supplementary Tables 1–3 for details)38,39. The energy gaps between accessible triplet states and low-lying singlet states show that for all four complexes, triplet states can be accessible with a trend of increasing accessibility from complex 1 to 4, particularly when considering the contribution from T2 states in complex 2. The calculated results confirm that all the accessible excited states exhibit obvious intramolecular charge transfer (ICT) characteristics. In addition, the calculated emission profiles at the B3LYP/Def2-TZVP level of theory are well consistent with experimental spectra (Supplementary Fig. 22a). Furthermore, the difference in dipole moments between the excited state being examined and the ground state serves as an initial indicator of the ICT characteristics of the transitions. In nearly all accessible excited states of complexes 1 to 4, this difference in dipole moments is significant, exceeding 3D in all except the T2 state of complex 2, which still presents a considerable value of 1.55D (Supplementary Tab. 3). As a result, ICT seems to be significant in these complexes. Furthermore, the extent of charge transfer within the molecule can be assessed more precisely by calculating the DCT index. This index indicates the average separation between the hole and the electron, along with the associated fraction of transferred electrons, represented as qCT. Except for the T2 state of complex 2, the magnitude of the transferred charge and the corresponding effective distance for all accessible excited states confirm the presence of ICT. Subsequently, theoretical analysis has been conducted to examine the bond strengths between the Au(I) atoms and their respective ligands, along with the distributions of electrostatic potential (Supplementary Fig. 22b). The analyses showed that the bond energy associated with Au(I)–P is comparatively weaker than that of Au(I)–C (for carbene). This suggests that complex 4 is likely to have a greater tendency to bind with TrxR than complex 3. Moreover, the significantly less electron-rich environment surrounding the Au(I) centre in complex 4 implies a higher possibility of interaction with negatively charged membranes or organelles.
In vitro tumor visualization and therapeutic investigation
Considering the advantage of excellent luminescence in the aggregate state of AIEgens, we then focused on investigating the imaging effect of the target complexes 1-4. Different tumor cells were individually incubated with complexes 1-4. The fluorescence imaging results (Fig. 3a and Supplementary Figs. 23–25) revealed that all the complexes exhibited significant imaging performance towards tumor cells. After confirming the excellent imaging performance of complexes 1-4 in tumor cells, we proceeded to assess their imaging performance in normal cells. The results demonstrated no obvious fluorescence signals for all the complexes in normal cells (Fig. 3a and Supplementary Fig. 26), suggesting specific selectivity of complexes 1-4 towards tumor cells. This selectivity is attributed to the specific binding of Au(I) centers of them to TrxR that is over-expressed in tumor cells, as validated by our previous work23. In our previous study33, the designed neutral complex TBP-Au exhibited no selectivity between normal and tumor cells (Supplementary Fig. 27). TBP-Au primarily targeted lysosomes. This suggests that the localization within different organelles affects selectivity due to the isoforms of TrxR are predominantly expressed in cytoplasm, mitochondria, or testis23. Consequently, it implies that introducing positive charge into the Au(I) complex is also important, as it affects its binding to TrxR by influencing the organelle localization (Supplementary Fig. 28). Therefore, the rationality of our initial design strategy, involving the introduction of charge, was thus well confirmed. We analyzed the interactions between complex 4 and TrxR, building upon similar covalent docking methods from our previous study23. By comparing the calculated bond energies of Au(I)–P and Au(I)–N for complex 4 (Supplementary Fig. 29), we inferred that the pyridine-based AIE ligand should be replaced before or upon interaction with TrxR. The docking results show that Au-P and Au-S bonds have bond lengths of 2.32 Å and 2.38 Å, respectively, consistent with previous covalent docking results and thus validating our docking approach. Further analysis was conducted on the interactions of the Ph3P-Au+ fragment in the binding pocket of human thioredoxin reductase (hTrxR, PDB ID: 2ZZB), as detailed in Supplementary Fig 29. The results indicate that the bulky PPh3 group inserts into the hydrophobic cavity of hTrxR. The three benzene rings of the Ph3P group interact with their surrounding amino acids (ASN418, ASN419, and TRP407) mainly through hydrophobic and van der Waals interactions. These interactions are crucial for enhancing molecular targeting capabilities.

a Confocal fluorescence images of 4T1 cells and 3T3 cells stained with 3 μM of complexes 1, 2, 3 and 4 for 0.5 h, respectively. For complexes 1 and 2: λex = 405 nm; λem = 500–600 nm; complexes 3 and 4: λex = 488 nm; λem = 600–700 nm. b Co-localization images of 4T1 cells co-stained with complex 4 (3 μM) and ER-Tracker™ Red, Mito-Tracker™ Red, or Lyso-Tracker™ Red. Complex 4: λex = 488 nm; λem = 600–700 nm. Scale bar: 50 μm. Data represent three independent experiments. An image representative of three biologically independent samples from each group is displayed.
Furthermore, we employed siRNA reagents to ascertain the effect of TrxR on tumor cell-specific imaging. For this purpose, we established three distinct groups for each of the complexes 1-4: a control group, a non-targeting siRNA (siRNA-NT) group, and a TrxR-targeting siRNA (siRNA-TrxR) group. Our findings revealed no significant difference in imaging efficacy between the control and siRNA-NT groups for any of the complexes (Supplementary Fig. 30), with both groups exhibiting excellent capabilities for tumor cell visualization. However, there was a marked decline in the imaging performance of the siRNA-TrxR group when compared to the other two groups under identical conditions, with a near-complete loss of imaging functionality. These results underscore the importance of TrxR overexpression in tumor cells for achieving specific imaging of these cells. Subsequently, we employed a laser confocal microscope to evaluate the subcellular distribution of complexes 1-4. Co-localization analyses with various subcellular structural probes revealed their enrichment in organelles (Fig. 3b, Supplementary Figs. 31–34). It is noteworthy that complex 4 exhibited excellent targeting ability towards endoplasmic reticulum (ER) and mitochondria. To further verify the organelle targeting specificity of complex 4, we conducted immunofluorescence experiments and captured the corresponding images using a super-resolution microscope. For ER labeling, we employed the BiP/GRP78 antibody, a calcium ion-binding molecular chaperone predominantly located in the ER. The COXIV antibody, a typical mitochondrial protein marker, was utilized for mitochondrial identification. The immunofluorescence data (Supplementary Fig. 35) revealed a high co-localization index of 0.81 between complex 4 and the ER marker BiP/GRP78, and a substantial index of 0.62 with the mitochondrial marker COXIV. These findings indicate the excellent targeting affinity of complex 4 for both the ER and mitochondria. The precise organelle targeting capability of complex 4 can facilitate the damage to ER and mitochondria induced by locally generated ROS upon exposure to light irradiation40. It’s noteworthy that ER, recognized as the largest organelle in eukaryotic cells, is integral to processes such as protein synthesis, folding, and translation, and other aspects41. As the literature documented42, the delivery of photosensitizers to the ER can generate ROS within the ER calcium channel, leading to the ER calcium release and triggering ER-stress. In addition, recent studies have shown that ER-targeting PDT can effectively induce ICD43,44. Based on these inspired findings and our rational molecular design involving PDT ability, the potential ICD process should also be an important aspect of our subsequent research. Additionally, we conducted an analysis of their photostability. In comparison to commercially available probes such as RB, all the complexes exhibited more excellent photostability and imaging capabilities (Supplementary Figs. 36 and 37).
Following the above inspiration, our research extended to a comprehensive investigation of their in vitro therapeutic efficacy. Firstly, we quantitatively evaluated the biological safety of complexes 1-4 using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, a standard method (Fig. 4a and Supplementary Fig. 38). The results indicated that complex 4 exhibited the highest level of biosafety in normal cells compared to complexes 1-3, presumably due to its higher selectivity in normal and tumor cells. Prior to tumor cell toxicity validation, we initiated our assessment with MTT experiments to determine the survival rate of 4T1 cells when exposed to the complex under varying lighting conditions at multiple time intervals. The purpose is to evaluate the dark toxicity and phototoxicity of the complex on tumor cells at different durations. Our observations indicated that complexes 1-4 demonstrated minimal dark and phototoxic effects on tumor cells after 2 and 6 hours of exposure (Supplementary Figs. 39 and 44). However, an escalation in both types of toxicity was observed with prolonged exposure times, culminating in peak levels after 24 hours. A subsequent decline in toxicity was noted at the 48-hour mark, and the dark and phototoxic effects had decreased again by 72 hours, lower than the levels observed at 48 hours. Significantly, the disparity in cell survival rates between the illuminated and non-illuminated groups was most pronounced at the 24-hour time point. This suggests that the differential impact of dark toxicity and phototoxicity is most substantial after 24 hours of administration. Considering the therapeutic window and safety considerations, the experimental outcomes suggest that a 24-hour in vitro drug treatment regimen is the most suitable approach. Subsequently, we investigated their cytotoxicity against various cancer cell lines using the MTT assay. Complexes 1-4 exhibited obvious concentration-dependent cytotoxicity towards 4T1 cells (Fig. 4b and Supplementary Figs. 45 and 47). Under white light exposure, all the complexes showed more pronounced cell-killing effects. Comparatively, under the same conditions, the cellular cytotoxicity of complex 4 is the highest, followed by complex 3, complex 2 and complex 1. It is obvious that the stronger ROS generation abilities of complexes 2-4 (Supplementary Fig. 20) lead to more efficient anticancer effects. Comparatively, the performance of complex 4 outperformed complex 3, and this superiority was attributed to the weaker binding ability of triphenylphosphine to Au(I)45, favoring the competitive combination of TrxR. Subsequently, we validated the phototoxicity of 3T3 and HUVEC cells (Supplementary Fig. 48). The experimental data indicated that both 3T3 cells and HUVEC cells exhibited a significant decrease in cell survival rate after light exposure. Furthermore, compared to tumor cells under the same conditions, the survival rate of tumor cells was lower. We presumed that this outcome might be associated with the disparity in TrxR levels between normal and tumor cells (Supplementary Fig. 28). The elevated TrxR concentrations in tumor cells are likely to enhance the binding affinity to the Au(I) active center of the complexes, thereby inhibiting TrxR activity and augmenting the chemotherapeutic impact. Consequently, the complexes exhibit excellent chemotherapeutic (dark toxicity) and combined chemo-phototherapeutic (phototoxicity) efficacy in tumor cells relative to normal cells. The cLogP for complexes 1-4 were also calculated as 16.412 (complex 1), 18.985 (complex 2), 18.136 (complex 3), and 14.367 (complex 4), respectively. Comparatively, the lowest cLogP value of complex 4 suggests that it has the lowest lipophilicity, which is beneficial for its biocompatibility and aligns with the findings involving the lowest dark toxicity. These observations corroborate the conclusion that complexes 1-4 display a therapeutic selectivity that favors tumor cells over normal cells, potentially offering a targeted approach to cancer treatment. We calculated IC50 based on the MTT experimental results to guide our subsequent experiments (Supplementary Figs. 49 and 51). In addition, live/dead cell staining analysis is used to visually detect cell death. Calcein-AM with green fluorescence was used to identify live cells, while SYTOXTM Deep Red Fluorescent Nucleic Acid dye with red fluorescence stains dead cells (Fig. 4c). In contrast to the control group with bright green fluorescence, each group showed varying degrees of red fluorescence after light irradiation. Notably, complex 4 demonstrated the most significant increase in cell mortality when exposed to light irradiation, aligning with the MTT results. Clone formation experiments were further conducted to evaluate the combined therapeutic effect of complexes 1-4 under light irradiation (Supplementary Fig. 52). This experiment, crucial for assessing cell proliferation ability, invasiveness, and sensitivity to killer factors, yielded similar results. Complex 4 exhibited the most noticeable reduction in the rate of cell clone formation and effectively suppressed the proliferation of tumor cells under white light irradiation. In summary, considering all the above results, it can be concluded that the selective imaging, ER-targeting and specific anticancer abilities of complex 4 are the most excellent, highlighting the crucial role of rational molecular structural manipulation.

a Relative viability of 3T3 cells and HUVEC cells after Co incubate with complexes 1, 2, 3 and 4 at various concentrations in the dark. b Relative survival rate of 4T1 cells treated with varying concentrations of complexes 1, 2, 3, and 4, followed by irradiation with either dark or white light (72 J/cm2) for 20 minutes, was measured using the MTT assay. c Assessment of tumoricidal efficacy in 4T1 cells treated with complexes 1, 2, 3, and 4 (3 μM) using calcein-AM and SYTOX™ Deep Red Fluorescent Nucleic Acid dye. Emissions for calcein-AM and SYTOX™ Deep Red were recorded at 517 nm and 682 nm, respectively. Scale bar = 100 μm. L: Light. Data are based on three independent experiments. An image representative of three biologically independent samples from each group is provided.
In vitro antitumor mechanism
Following the evaluating of in vitro anticancer efficacy for complexes 1-4, we conducted more in-depth exploration of their mechanism. TrxR is a crucial component of the cellular antioxidant system, playing a pivotal role in mitigating cellular oxidative stress and maintaining redox homeostasis. Inhibiting TrxR induces ROS production, disrupting redox balance and initiating apoptosis46. There should be ample evidence supporting the effective hindrance of cancer cell growth by Au(I) complexes through TrxR inhibition in the redox system. Thus, we further validated the efficacy of complexes 1-4 in inhibiting TrxR activity. TrxR activity progressively decreased with the increase of their concentrations (Fig. 5a), demonstrating the effectiveness of complex 4. This result well supported our prior presumption that the weaker binding ability of triphenylphosphine to Au(I) favors the competitive combination of TrxR. Consequently, complex 4 displayed the strongest inhibition ability to TrxR, effectively countering the ROS elimination from the antioxidant system. This observation demonstrates its most potent chemotherapy effect, as indicated by the MTT assay.

a In vitro activity inhibition of TrxR by complexes 1, 2, 3 and 4 at various concentrations. b DCFH-DA fluorescence images of tumor cells after different treatments. The administered concentration for complexes 1-4 is 3 μM. Scale bar = 50 μm. Data are based on three independent experiments. An image representative of three biologically independent samples from each group is displayed. c 4T1 cells were treated in different ways (The administration concentrations of complexes 1-4 are 3 μM) and stained with Annexin V-FITC and SYTOXTM Deep Red Fluorescent Nucleic Acid dye. Detection of apoptotic cell percentage in each group by flow cytometry. Q1: necrotic or mechanically damaged cells; Q2: late-phase apoptotic cells; Q3: early-phase apoptotic cells; Q4: normal cells. d Expression level of the apoptosis-related molecules in four treatment groups. The administration concentration of complex 4 is 3 μM. Statistical analyses were performed using GraphPad Prism 9.0.0. Data are expressed as mean ± SD. Significance levels are indicated as NS (not significant), *P < 0.05, **P < 0.01, and ***P < 0.001. A two-way ANOVA was used for comparisons, followed by Tukey’s post hoc test for multiple comparisons (L: Light).
Subsequently, we explored their capacity of in situ ROS production in vitro using 2,7-dichlorofluorescein diacetate (DCFH-DA) as an indicator. The results demonstrated varying degrees of green fluorescence for complexes 1-4 (Fig. 5b and Supplementary Fig. 53). Furthermore, it was observed that all the complexes exhibited more pronounced fluorescence upon light irradiation. Overall, complex 4 exhibited the strongest ROS production ability under light irradiation. These results indicate that complexes 1-4 not only impede ROS elimination through TrxR inhibition but also acted as photosensitizers, efficiently producing ROS and enhancing synergistic anticancer performance.
Moreover, we validated the occurrence of cell apoptosis caused by the destruction of the antioxidant system in vitro by chemotherapy and phototherapy. The result of flow cytometry (Fig. 5c) exhibited varying degrees of apoptosis in each group of cells, and complex 4 led to the most pronounced tumor cell apoptosis under light exposure, further substantiating the excellent structural design of complex 4. Additionally, we performed a Western blot (WB) analysis to detect apoptosis-related proteins (Fig. 5d), and the result further confirmed the significant induction of tumor apoptosis by complex 4 under light exposure.
In vitro ICD-mediated by complex 4
The above results demonstrate that the rationally designed complex 4 has the ability to target organelles and exhibits excellent photodynamic effect. Subsequently, to capture the dynamic cellular changes during photoactivation, in situ fluorescence imaging was performed on 4T1 cells by selecting complex 4 as the representative. The imaging data (Fig. 6a) revealed a rapid fluorescence response in the 4T1 cells, indicating the swift entry and light-up of the cells by complex 4. Interestingly, under oxidative stress induced by ROS under light irradiation, it was observed that the internal cellular structures showed progressive swelling and deformation, as indicated by dashed circles. Given the fluorescence co-localization data indicating that complex 4 predominantly targets the ER and mitochondria, we presume that the extensive ER regions within the cells are also likely to undergo swelling and deformation. As we discussed above43,44, the ROS generated by complex 4 can cause damage to the ER and they are likely to induce the release of calcium ions from the ER, thereby triggering the occurrence of ER stress.

a Confocal imaging of 4T1 cells stained with complex 4 (3 μM) under continuous 488 nm laser scanning for 0–30 scans. λex = 488 nm; λem = 600–700 nm. Scale bar = 5 μm. b Immunofluorescence images showing CRT exposure in 4T1 cells treated with different ways for 24 h. The administration concentration of complex 4 is 3 μM. Scale bar = 50 μm. c Extracellular ATP levels of 4T1 cells treated with different ways by an ATP luminescence test kit (n = 3). The concentration of complex 4 is 3 μM. d Detection of ER stress-related proteins in 4T1 cells and culture supernatant after treatment. e Complex 4 induced ER stress and enhanced the release of tumor cell DAMPs under light exposure. DAMPs include CRT translocation, ATP secretion, and HMGB1 release, among others. n = 3 independent experiments. The statistical analyses were conducted using GraphPad Prism 9.0.0. All data were presented as mean ± SD. The outcomes were compared via one-way ANOVA (with Tukey’s post hoc correction for multiple comparisons).
To further validate the occurrence of ER stress, WB analysis (Fig. 6d) was conducted to monitor the expression levels of C/EBP homologous protein (CHOP) and glucose regulatory protein (GRP) 78, which are associated with ER stress47. During the PDT process mediated by complex 4, the expression patterns of CHOP and GRP78 were similar. Compared to other groups, complex 4 markedly increased the expression of GRP78 and CHOP under light irradiation, indicating a substantial response to severe ER stress47. To further verify that complex 4 can effectively induce ER stress, we conducted a more in-depth study on its mechanism of action. During ER stress occurs, the ER activates unfolded protein response (UPR)48. If the ER stress response persists or cannot be resolved, the UPR signal shifts from promoting cell survival to inducing apoptosis49. Key molecules in ER stress-induced UPR signaling include protein kinase-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6)50,51. During ER stress, PERK detaches from its chaperone protein GRP78, undergoes self-phosphorylation, and activates the α subunit of eukaryotic initiation factor 2 (eIF2α) at Serine 51, leading to reduced overall protein translation52. Phosphorylated eIF2α leads to an increase in mRNA levels of activating transcription factor 4 (ATF4)49. Once in the nucleus, ATF4 activates downstream factors, including CHOP53. The overexpression of CHOP promotes apoptosis by enhancing the expression of pro-apoptotic genes such as Bim and Bax, while inhibiting anti-apoptotic proteins like BCL-2 and GADD3454. Besides PERK, both ATF6 and IRE1 can induce CHOP transcription, but the PERK-eIF2α-ATF4 pathway is the primary mechanism for CHOP protein expression during ER stress. Additionally, the accumulation of unfolded proteins in the ER triggers IRE1 oligomerization, which activates its endonuclease activity and results in the splicing of X-box binding protein 1 (XBP-1) mRNA55. Moreover, during ER stress, released GRP78 cleaves ATF6, releasing its N-terminal domain to activate transcription of multiple chaperone molecules (PDI, GRP78, GRP94, and calcium-binding proteins)50,55. We initially assessed ER stress sensor activation using WB and reverse transcription quantitative polymerase chain reaction (RT-qPCR). The results (Supplementary Figs. 55–57) showed a significant increase in the expression of phosphorylated-PERK (p-PERK), p-IRE1, and ATF6 in the “complex 4 + L” group. In addition, the expression level of eukaryotic initiation factors (p-eIF2α) also increased (Supplementary Figs. 55, 57). These results preliminarily demonstrated the activation of UPR. We further detected its downstream molecules using RT-qPCR. The results (Supplementary Fig. 57) showed that ATF4 and CHOP, as the most important downstream effector and promoting factor of UPR induced cell apoptosis, were significantly increased in the “complex 4 + L” group. The pro-apoptotic genes Bim and Bax were also increased, while BCL-2 and GADD34 expression decreased, confirming the pro-apoptotic role of ER stress. In addition, the expression level of XBP1s in the “complex 4 + L” group decreased, indicated that complex 4 reduced the formation of XBP1s under light, impairing cellular adaptability to ER stress and promoting tumor cell apoptosis. Moreover, the elevated expression of multiple companion molecules (PDI, GRP78, GRP94, endoplasmic reticulum protein 44, 57, 72 (ERP-44, ERP-57, ERP-72) and 58 kDa inhibitor of PKR (P58ipk)) further confirmed the occurrence of ER stress (Supplementary Fig. 57). These experimental results demonstrated the existence of ER stress and the correlation between ER stress and tumor apoptosis.
Accumulating evidence has demonstrated that chemo-phototherapy induced cellular oxidative stress, including ER stress, may be effective means to induce ICD, subsequently triggering related antitumor immune responses43,44. During the ICD process, damage-associated molecular patterns (DAMPs) can serve as natural adjuvants for the body’s immune system, enhancing the immunogenicity or stress of dead cancer cells. DAMPs include surface exposed calcium reticulum protein (CRT translocation), adenosine triphosphate (ATP) secretion, high-mobility histone B1 (HMGB1), Heat shock protein 70 (HSP70) and annexin A1 (Annexin A1), which are crucial indicators of the ICD cascade. CRT translocation serves as a pivotal event of ICD, wherein, during endoplasmic reticulum stress, CRT protein migrates from the ER cavity to the cell membrane, acting as a signal to mediate antitumor immune response. We assessed the levels of membrane-bound and total CRT in cells using immunofluorescence staining and WB analysis, respectively. The results (Fig. 6b, d) demonstrate that the application of complex 4 induced the translocation of CRT when exposed to light. Despite variations in light treatment, the overall quantity of CRT protein was found to be consistent across all experimental groups. Specifically, an increase in membrane-bound CRT was observed in the “complex 4 + L” group, suggesting that PDT mediated by complex 4 can effectively promote the translocation of CRT. Furthermore, we investigated additional characteristic biomarkers of ICD, specifically ATP, high-mobility HMGB1, HSP70, and Annexin A1. PDT mediated by complex 4 resulted in a significant increase in the extracellular levels of these ICD-associated DAMPs (Fig. 6c, d, and Supplementary Fig. 58). It is particularly noteworthy that the extracellular ATP levels exhibited an early elevation at 12 hours post-treatment, preceding the rise in HMGB1, HSP70, and Annexin A1 levels observed at 24 hours. These observations imply that complex 4, has the potential to act as a type II ICD inducer. It effectively facilitates the release of DAMPs associated with ICD under light irradiation in vitro, as represented in Fig. 6e.
In vivo tumor imaging and therapeutic investigation
After confirming the exceptional imaging and therapeutic efficacy of complex 4 in vitro, we further investigated its performance in vivo. Considering the enhanced penetration ability resulting from the long-wavelength luminescence of complex 4, we speculated that it may also possess excellent imaging capability in deep tissues. Accordingly, we conducted further investigations on its imaging effect in living animals (Fig. 7a). In vivo imaging of mice bearing tumors was performed by administering intravenous injection, and the changes in fluorescence intensity were monitored over time. Four hours post-injection, robust fluorescence was detected at the tumor site, and the fluorescence signal could persist for up to 96 hours in the tumor area. These findings suggested that complex 4 holds significant potential for long-term tumor monitoring. Given its excellent in vitro anticancer property, application in imaging-guided therapy is anticipated. After 96 hours, the images (Fig. 7b) of the main organs and tumor tissues obtained by euthanizing the mice presented strong fluorescence signals at the tumor site, demonstrating the targeting effect of complex 4. In addition, kidney and liver, as metabolic organs, also displayed slight fluorescence, suggesting potential metabolism via these organs. Subsequently, the in vivo anticancer efficacy of complex 4 was evaluated using a 4T1 tumor-bearing BALB/c mouse model (Fig. 7c). After observing the treatment for 21 days, it was observed that the tumor growth in the control group and the light-alone group were barely inhibited, whereas the “complex 4 + L” group presented a complete suppression (Fig. 7d). Additionally, body weight monitoring of all the groups revealed no significant difference (Fig. 7e), indicating their steady growth trend and the excellent biological safety of complex 4. After the 21-day treatment period, the mice were euthanized, and their major organs (hearts, livers, spleens, lungs, kidneys) as well as tumor tissue were collected. tumor weight measurements further supported the conclusion of significant tumor growth suppression in the “complex 4 + L” group (Fig. 7f). To further validate the antitumor efficacy of complex 4, tumor tissue was subjected to hematoxylin-eosin (H&E), terminal deoxynucleotide transferase dUTP nick end labeling (TUNEL), and Ki-67 staining (Fig. 7g). The H&E staining results revealed that the tumor cells in the control group exhibited a tightly arranged pattern without notable damage, whereas the “complex 4 + L” group exhibited a sparser distribution of tumor cells, indicating significant structural damage to the tumor cells in this group. TUNEL staining revealed that “complex 4 + L” led to a considerably higher proportion of TUNEL-positive apoptotic cells (indicated by green fluorescence) in comparison to the control group. On the contrary, Ki-67 staining showed a significant decrease in the number of positive value-added cells (indicated by pink fluorescence) in the “complex 4 + L” group. In addition, complex 4 exhibited excellent blood compatibility in hemolysis tests (Supplementary Fig. 59), suggesting its ability to maintain stability during blood circulation. Furthermore, we performed H&E staining of major organs and blood biochemistry experiments. The H&E staining results (Supplementary Fig. 60) showed no pathological damage in the treated organs and no significant difference in morphology and structure compared to the control group. In the blood biochemistry experiment, we collected orbital blood to detect liver function abnormalities (ALT, AST, and ALP), renal function abnormalities (UA, URA, and CREA), and myocardial injury markers (CK and CK-MB). The results (Supplementary Fig. 61) indicated that biochemical indicators were within normal ranges, suggesting normal liver, kidney, and myocardial function in the treatment groups. Overall, these findings demonstrated that the histological damage caused by complex 4 to normal tissues and organs can be negligible, indicating its excellent biological safety in vivo. These preliminary results establish the suitability of complex 4-mediated combination therapy for in vivo applications.

a Continuous in vivo imaging observation was performed on 4T1 tumor-bearing mice after intravenous injection of complex 4 (3 mM, 100 μL). b Imaging of major organs and tumors excised from mice after intravenous injection with for 96 h. c This diagram illustrates the construction of animal models and the treatment process 4. Firstly, an animal tumor model was constructed by injecting 4T1 cells subcutaneously into mice. After 5 days, different treatments were administered to mice via intravenous injection, and next day, irradiated with white light (900 J/cm2) for 5 min at the primary tumor sites. The treatment process lasts for 21 days. d Tumor volume in 4T1 tumor-bearing mice treated with different ways. e Body weight of mice bearing 4T1 tumors after various treatments. f Weights of isolated tumors in after 21 days. g H&E, Tunel and Ki-67 staining of tumor sections from mice after different treatments. Statistical analyses were performed using GraphPad Prism 9.0.0, with data presented as mean ± SD. Each group comprised n = 5 independent animals. Representative images from five biologically independent samples per group are shown. Data comparisons were made using one-way ANOVA with Tukey’s post hoc test for multiple comparisons. (L: Light).
In vivo ICD and related tumor immune responses mediated by complex 4
To decipher the underlying mechanism contributing to the excellent antitumor efficacy of complex 4-mediated in vivo combination therapy, tumor tissue and main organs were collected from the tumor animal model after completion of the treatment (Fig. 8a). Immunofluorescence analysis of tumor tissue sections showed pronounced CRT exposure in the “complex 4 + L” group (Fig. 8b), further validating the induction of endoplasmic reticulum stress in vivo through the complex 4-mediated combination therapy. CRT exposure acted as a prominent signal, prompting the maturation of dendritic cells (DCs) by binding to surface receptors on these cells. This interaction enhanced the presentation of tumor antigens to T cells and triggers subsequent immune responses. Therefore, the maturation of DCs following diverse treatments was meticulously assessed using flow cytometry (CD11c+, CD80+ and CD86+), and the results revealed a significant augmentation in DCs maturation within the “complex 4 + L” group compared to the other groups (Fig. 8c). The presentation of antigens by mature DCs to T cells could instigate adaptive immune responses, marked by the generation of T lymphocytes (CD4+ T cells and CD8+ T cells) against cancer cells. Immunofluorescence staining of spleen tissue sections underscored an elevation in the count of CD4+ T cells and CD8+ T cells within the “complex 4 + L” group compared to the other three groups (Fig. 8d). This observation implied a more robust immune response following combination therapy mediated by complex 4, leading to the recruitment of a higher number of CD4+ T cells and CD8+ T cells into the spleens within this group. Further level scrutiny of CD8+ T cells, CD4+ T cells, and Foxp3+cells in tumors using immunofluorescence staining indicated an escalation in fluorescence intensity for CD8+ T cells and CD4+ T cells in the “complex 4 + L” group, concomitant with a marked reduction in the fluorescence intensity of Foxp3+cells (Fig. 8b and Supplementary Fig. 62). This suggests enhanced antitumor immune activity in the “complex 4 + L” group. Additionally, flow cytometry analysis of CD4+ T cells (CD3+ CD4+) and CD8+ T cells (CD3+ CD8+) in spleen also revealed a substantial increase in CD4+ T cells and CD8+ T cells in the “complex 4 + L” group (Fig. 8e, f), suggesting the activation of CD4+ T cells and CD8+ T cells. Collectively, these findings indicate that the combination therapy mediated by complex 4 effectively triggered ICD in vivo, thereby eliciting corresponding antitumor immune responses.

a After intravenous injection, complex 4 accumulates passively at the tumor site, inducing tumor cell apoptosis combined with chemotherapy and PDT. The apoptosis process promotes the release of DAMPs and stimulates the maturation of dendritic cells (DCs), thereby initiating T cell activation and the immune response. b Immunofluorescence staining for calreticulin (pink) and Foxp3+ T cells (pink) in tumor sections post-treatment. Scale bar = 200 μm. c Flow cytometry plots showing CD80+ and CD86+ dendritic cells (gated on CD11c+) in the spleen. d Representative immunofluorescence images of CD4+ T cells (pink) and CD8+ T cells (pink) in spleen slices after various treatments. Scale bar = 100 μm. e Flow cytometry plots for CD3+ and CD4+ T cells (gated on CD45+) in the spleen. f Flow cytometry plots for CD3+ and CD8+ T cells (gated on CD45+) in the spleen. (L: Light). n = 3 independent samples per group. Representative images from three biologically independent samples are shown.
To gain a deeper understanding of the effects of complex 4-mediated in vivo combination therapy on the activation of immune responses, we established a mouse model for immune activation validation. After 21 days of treatment as previously described in the constructed mouse tumor model (Supplementary Fig. 63), the tumor was surgically removed from the mice. Subsequently, these mice were subcutaneously inoculated with homologous 4T1 tumor cells and monitored continuously for an additional 30 days. As a result, it was found that the tumor volume in the “complex 4 + L” treatment group was markedly reduced compared to the other treatment groups (Supplementary Fig. 64). This finding suggests that the combination of complex 4 with light irradiation can potently stimulate adaptive immune responses and significantly suppress the proliferation of new tumors in vivo.
Discussion
In this study, we designed a series of complexes by integrating NHC-Au(I) and PPh3-Au(I) units with AIE ligands, and achieving a remarkably efficacious anticancer effect that combines chemotherapy, phototherapy, and immunotherapy. In vitro experiments demonstrated the specific tumor imaging, organelle targeting and robust therapeutic capabilities inherent in this series complexes. Notably, among these, complex 4, featuring a triphenylphosphine ligand, exhibited optimal performance across all evaluated aspects. A comprehensive exploration of the underlying mechanisms revealed that the potent anticancer effects of complex 4 was derived from the orchestrated actions of effective TrxR inhibition, highly efficient ROS production and cell oxidative stress-induced ICD characterized by a significant release of DAMPs, with ER stress playing an important role. Subsequent in vivo experiments substantiated the excellent tumor imaging capabilities of complex 4 and its powerful therapeutic efficiency augmented by the activation of immune responses. Therefore, our deliberate design not only imparts Au(I) complexes with exquisite molecular structures, rendering them as efficacious photosensitizers, but also underscores their formidable capacity to induce ICD in vitro and evoke immune responses in vivo. These findings accentuate the substantial advantages and promising prospects of the Au(I)-AIE complex in the realm of combination therapy, and further bolstering precise immunotherapy. Admittedly, our current systems face certain limitations. Specifically, their absorption spectrum does not extend to the more desirable 600–850 nm range and the emission wavelengths are capped at below 800 nm. As a result, the systems’ in vivo imaging capabilities, tissue penetration, and phototherapy efficiency are significantly constrained. There is ample scope for molecular refinement and performance enhancement to extend the absorption to longer wavelengths, boost efficiency, and achieve longer emission spectra, potentially tapping into the near-infrared II region. We are committed to making these advancements in our ongoing research efforts.
Methods
Ethical regulations
All experimental protocols and ethical reviews related to animal experiments have been approved by the Experimental Animal Ethics Review Committee of Southern Medical University (SMUL2022257).
Materials
The starting materials, 1,3-bis(2,6-di-isopropylphenyl) imidazol-2-ylidene Au(I) chloride and (PPh3)AuCl, were prepared using methods described in the literature. Other compounds were synthesized according to the preparation route outlined in Supplementary Fig. 1. Additional reagents were purchased and used directly from their respective suppliers. Dulbecco’s modified Eagle’s medium and RPMI 1640 culture medium were obtained from Grand Island Biological Company (USA). Fetal bovine serum (FBS) was sourced from Procell Life Science & Technology Co., Ltd. (China). ER-Tracker™ Red, Lyso-Tracker™ Red, and Mito-Tracker™ Red were acquired from Yeasen Biotechnology (Shanghai) Co., Ltd. (China). The MTT test kit was obtained from Kulaibo Technology Co., Ltd. (China). The live-dead cell staining kit and DCFH-DA were provided by Beyotime Biotechnology Co., Ltd. (China). The CheKine™ TrxR Colorimetric Assay Kit was purchased from Box Shenggong Technology Co., Ltd. (China). Primary antibodies were sourced from Proteintech Group (China).
Apparatus
NMR spectra for 1H, 13C, and 31P were acquired using a Bruker AVIII 400 MHz NMR spectrometer. Deuterated chloroform or THF served as solvents, and tetramethylsilane (TMS; δ = 0) was used as the internal reference. High-resolution mass spectrometry was conducted with a GCT Premier CAB048 mass spectrometer in MALDI-TOF mode. UV-visible spectra were obtained with a Varian CARY 50 spectrophotometer, while photoluminescence (PL) spectra were recorded using a Horiba Fluoromax 4 spectrofluorometer. Cellular fluorescence images were captured with a Nikon confocal laser scanning microscope. Optical density measurements of cell samples were carried out using a Thermo Fisher Multiskan MK3 microplate reader. Near-infrared animal imaging was performed with an IVIS Spectrum system from PerkinElmer.
Cell culture
The 4T1 cells were sourced from Procell Life Science & Technology Co., Ltd. and cultured in RPMI 1640 medium (Gibco) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin (HyClone) in a 5% CO2 atmosphere at 37 °C. Cell experiments were performed after the 4T1 cancer cells reached 80% confluence in the culture flask.
Cell imaging
5 × 104 cells were seeded in each 35 mm confocal culture dish and allowed to incubate for 24 hours. After the cell medium was replaced, the cells were treated with 3 μM AIEgens for 30 minutes. For co-staining imaging, the 4T1 cells were subsequently treated and incubated with ER-Tracker™ Red, Mito-Tracker™ Red, and Lyso-Tracker™ Red (Yeasen) individually. The cells were washed 2–3 times with phosphate-buffered saline (PBS). They were then imaged using a laser confocal microscope (Nikon A1 HD25, Japan). For the imaging parameters, 1 and 2 utilized an excitation wavelength of 405 nm with an emission filter of 500–600 nm, while 3 and 4 used an excitation wavelength of 488 nm with an emission filter of 600–700 nm.
Immunofluorescence super-resolution imaging
Placed 5 × 104 cells in each 35 mm confocal culture dish and culture for 24 hours. After replacing the cell medium, they were treated with 3 μM AIEgens. Subsequently incubated with a rabbit anti-BiP/GRP78 antibody and COXIV antibody (Proteintech). Finally, the experimental results captured by super-resolution microscope.
Cytotoxicity assay
In the cell viability assay, the MTT was utilized to assess the metabolic activity of cells. The experiment was conducted in 96-well plates, with each well initially seeded 6 × 103 cells/well. After an addition 24 hours of 24 hours of incubation, the medium in each well was replaced by a 100 μL suspension of AIEgens with varying concentrations. After incubation for 30 minutes, expose the plates to white light (72 J/cm2) for 20 minutes, while keeping the other group of plates in the dark as a control. Subsequently, the cells were incubated for an additional 24 hours. Next, add MTT reagent to each well and incubate in a 37 °C incubator. Measure the absorbance of MTT at 560 nm using a microplate reader after 2 hours (Thermo Fisher Multiskan MK3, America).
Live/dead cell staining
Placed 5 × 104 cells in each 35 mm confocal culture dish and culture for 24 hours. After replacing the cell medium, they were treated with 3 μM AIEgens for 30 minutes, and then exposed to white light (72 J/cm2, 20 min) or kept in the dark. After incubation for 24 hours, wash the cells with PBS. Cell staining was performed according to the manual of the calcein-AM (Beyotime) and SYTOXTM Deep Red Fluorescent Nucleic Acid dye (Thermo Fisher), and the stained cells were observed using a laser confocal microscope (Nikon A1 HD25, Japan).
Intracellular ROS generation
Intracellular ROS levels were measured utilizing DCFH-DA (Beyotime). Placed 5 × 104 cells in each 35 mm confocal culture dish and culture for 24 hours. After replacing the cell medium, they were treated with 3 μM AIEgens for 30 minutes, and then exposed to white light (72 J/cm2, 20 min) or kept in the dark. Subsequently, DCFH-DA (10 μM) was added and incubated for another 20 minutes. Intracellular ROS levels were visualized and captured using a laser confocal microscope (Nikon, Japan, excitation: 488 nm), and mean fluorescence intensity was quantified by Image J software.
Determination of TrxR activity
To measure the TrxR activity at the cellular level, the 4T1 cells were incubated with AIEgens for 24 hours. The concentrations used were 0.003 μM, 0.03 μM, 0.3 μM, 3 μM, and 10 μM. After 24 hours of treatment, cells from each group were collected and counted. Subsequently, 5 × 106 cells were taken from each group and detected using a thioredoxin Reductase Assay Kit (boxbio). Then use a microplate reader (Thermo Fisher Multiskan MK3, America) to measure TrxR activity (412 nm).
To verify the TrxR activity of different cells, the thioredoxin reductase assay kit (boxbio) was used to detect the TrxR activity of 4T1, HUH7, H292, 3T3 and HUVEC cells according to the provided instructions. Then use a microplate reader (Thermo Fisher Multiskan MK3, America) to measure TrxR activity (412 nm).
Cell apoptosis analysis
Placed cells in 6-well plates and culture for 24 hours and subjected to different ways. After a 24-hour incubation period, the cells were collected and stained with Annexin V-FITC (excitation: 494 nm, emission: 517 nm) and SYTOXTM Deep Red Fluorescent Nucleic Acid dye (excitation: 660 nm, emission: 682 nm). The number of apoptotic cells was measured using flow cytometry (BD LSRFortessa X-20, America) within 1 h.
Western blot analysis
Cells were seeded in 6-well culture plates and incubated for 24 hours. After treating the cells with PBS and 3 μM AIEgens for 30 minutes, they were either exposed to white light irradiation (72 J/cm2 for 20 minutes) or left without irradiation. After an additional 24 hours, both the cells and the culture supernatant were collected. To extract proteins, RIPA lysis buffer was used to lyse the cells. The concentration of the extracted proteins was measured with a BCA protein detection kit (Beyotime). The resulting proteins were then subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by transfer to a membrane (Solarbio). To minimize nonspecific binding, the membrane was blocked with 5% BSA for 1 hour. The membrane was then incubated first with the primary antibody (Proteintech) and subsequently with the secondary antibody (Bioss). Finally, the target proteins were detected using an ECL chemiluminescence detection kit (HaiGene).
Detection of CRT
We utilized immunofluorescent staining to investigate the cell surface exposure of CRT. Placed 5 × 104 cells in each 35 mm confocal culture dish and culture for 24 hours. After treating the cells with either PBS or AIEgens (3 μM), they were either irradiated with white light or left untreated. Incubate cells with rabbit anti CRT antibody (Proteintech). To stain the cell nucleus, DAPI (Solarbio) was used. Finally, the exposure of CRT was observed under a laser confocal microscope (Nikon, Japan, excitation: 652 nm; DAPI excitation: 358 nm).
ATP detections
The extracellular ATP levels were assessed using the ATP Determination Kit (Beyotime). Briefly, Placed cells in 6-well plates and culture for 24 hours. The cells were then treated with either PBS or AIEgens (3 μM) and subsequently exposed to white light irradiation or kept in the dark. After 6, 12, and 24 hours of treatment, supernatant was collected and analyzed using the ATP Determination Kit (Beyotime) following the manufacturer’s instructions.
Animals
Female Balb/c mice (6–7 weeks old) were obtained from SPF (Beijing) Biotechnology Co., Ltd. The mice were housed in a specific pathogen-free (SPF) environment at the Animal Experiment Center of Southern Medical University. All animal experiments in this study were conducted in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. The volume of the subcutaneous tumors in the mice did not exceed the ethical maximum limit of 2000 mm3.
In Vivo biodistribution and imaging studies
Establishment of tumor model in mice by subcutaneous injection of 4T1 cells. AIEgens were administered intravenously, followed by in vivo biodistribution and fluorescence imaging at various time intervals using an IVIS spectrum system (PerkinElmer, America). At the 96-hour time point after intravenous injection, mice were euthanized and dissected, and their tumors, spleens, livers, hearts, lungs, and kidneys, were collected and photographed using an IVIS spectroscopy system (PerkinElmer, America).
In vivo antitumor research
In this study, 6–7 week-old female BALB/c mice were used as animal models. A tumor model was established by subcutaneously injecting 4T1 cells into the mice. Once the tumor volume reached ~150–200 mm3, the mice were randomly divided into four groups, with five mice in each group. The groups received the following treatments via intravenous injection: (1) PBS; (2) PBS + Light; (3) AIEgens (3 mM, 100 μL); and (4) AIEgens + Light (3 mM, 100 μL). After 24 hours, the tumors were exposed to white light irradiation (900 J/cm2, 5 minutes). Tumor volume (calculated using the formula length × width2/2) and body weight were monitored and measured every 3 days throughout the treatment period. After 21 days of treatment, the mice were euthanized, and their tumors were collected for various analyses, including hematoxylin-eosin (H&E) staining, TUNEL staining, and immunofluorescence staining. Additionally, spleen samples were collected on day 21 for immunofluorescence staining. Flow cytometry (BD LSRFortessa X-20, USA) was employed to analyze DCs, CD8+ T cells, and CD4+ T cells in the splenocytes.
In vivo immune activation research
In the study, BALB/c mice (6–7 weeks old, female) were used as animal models. Establishment of tumor model in mice by subcutaneous injection of 4T1 cells. When the measured volume of the mouse tumor reaches ~150–200 mm3, the mice will be randomly assigned, with five mice in each group, for a total of four groups. The groups were treated by intravenous injection as follows: (1) PBS; (2) PBS + Light; (3) AIEgens (3 mM, 100 μL); (4) AIEgens + Light (3 mM, 100 μL). After 24 hours, the tumors were exposed to white light irradiation (900 J/cm2, 5 minutes). Throughout the entire treatment period, tumor volume (calculated using the formula length × width2/2) and body weight were observed and measured every 3 days. After 21 days of tumor model treatment, the mouse tumor was excised and subcutaneously inoculated with homologous tumor cell 4T1 for 30 days of continuous observation.
Statistical analysis
All experiments were conducted three times or more. Significant values are presented as mean ± standard deviation. A P value of <0.05 was deemed statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this article, its Supplementary information files, and source data files. All other data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
International agency for research on cancer. Latest global cancer data: Cancer burden rises to 19.3 million new cases and 10.0 million cancer deaths in 2020. IARC (2020).
Hao, Y. et al. Near-infrared responsive 5-fluorouracil and indocyanine green loaded MPEG-PCL nanoparticle integrated with dissolvable microneedle for skin cancer therapy. Bioact. Mater. 5, 542–552 (2020).
Shi, S. et al. Evolving role of biomaterials in diagnostic and therapeutic radiation oncology. Bioact. Mater. 5, 233–240 (2020).
Sahin, U., Karikó, K. & Türeci, Ö. mRNA-based therapeutics–developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Riley, R. S., June, C. H., Langer, R. & Mitchell, M. J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 18, 175–196 (2019).
Fukumura, D., Kloepper, J., Amoozgar, Z., Duda, D. G. & Jain, R. K. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 325–340 (2018).
Lin, Y. X. et al. Reactivation of the tumor suppressor PTEN by mRNA nanoparticles enhances antitumor immunity in preclinical models. Sci. Transl. Med. 13, eaba9772 (2021).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Vanneman, M. & Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 12, 237–251 (2012).
Dai, J. et al. Efficient near-infrared photosensitizer with aggregation-induced emission for imaging-guided photodynamic therapy in multiple xenograft tumor models. ACS Nano 14, 854–866 (2020).
Benson, S. et al. Photoactivatable metabolic warheads enable precise and safe ablation of target cells in vivo. Nat. Commun. 22, 2369 (2021).
Sun, Z. et al. Aggregation-induced-emission photosensitizer-loaded nano-superartificial dendritic cells with directly presenting tumor antigens and reversed immunosuppression for photodynamically boosted immunotherapy. Adv. Mater. 35, e2208555 (2023).
Dai, J. et al. Red blood cell membrane-camouflaged nanoparticles loaded with AIEgen and poly(I:C) for enhanced tumoral photodynamic-immunotherapy. Nat. Sci. Rev. 8, nwab039 (2021).
Jia, S. et al. Sonosensitized aggregation-induced emission dots with capacities of immunogenic cell death induction and multivalent blocking of programmed cell death-ligand 1 for amplified antitumor immunotherapy. CCS Chem. 4, 501–514 (2022).
Zhao, L. P. et al. Self-delivery photo-immune stimulators for photodynamic sensitized tumor immunotherapy. ACS Nano 14, 17100–17113 (2020).
Sen, S., Karoscik, K., Maier, E. & Arambula, J. F. Immunogenic cell death-inducing metal complexes: From the benchtop to the clinic. Curr. Opin. Chem. Biol. 73, 102277 (2023).
Galluzzi, L., Guilbaud, E., Schmidt, D., Kroemer, G. & Marincola, F. M. Targeting immunogenic cell stress and death for cancer therapy. Nat. Rev. Drug Discov. 23, 445–460 (2024).
Guo, W. J. et al. Visualization of supramolecular assembly by aggregation-induced emission. Aggregate 4, e297 (2023).
Ye, Z. Y., He, W., Zhang, Z. C., Qiu, Z. J., Zhao, Z. & Tang, B. Z. AIEgens for microorganism-related visualization and therapy. Interdiscip. Med. 1, e20220011 (2023).
Liao, Y., Peng, Z., Liu, X., Hu, Y. & Zhang, J. Theranostic applications of biomolecule-responsive aggregation-induced emission luminogens. Interdiscip. Med. 1, e20230024 (2023).
Wang, X. Y., Gong, J., Zou, H., Liu, S. H. & Zhang, J. Aggregation‐induced conversion from TADF to phosphorescence of gold(I) complexes with millisecond lifetimes. Aggregate 4, e252 (2023).
Zhang, J. et al. Multifunctional Au(I)-based AIEgens: manipulating molecular structures and boosting specific cancer cell imaging and theranostics. Angew. Chem. Int. Ed. 59, 7097–7105 (2020).
Yang, Z. et al. Biotin-targeted Au(I) radiosensitizer for cancer synergistic therapy by intervening with redox homeostasis and inducing ferroptosis. J. Med. Chem. 65, 8401–8415 (2022).
Long, Y., Cao, B., Xiong, X., Chan, A. S. C., Sun, R. W. & Zou, T. Bioorthogonal activation of dual catalytic and anti-cancer activities of organo Au(I) complexes in living systems. Angew. Chem. Int. Ed. 60, 4133–4141 (2021).
Zhang, C. et al. An artemisinin-derivative-(NHC)-Au(I) hybrid with enhanced cytotoxicity through inhibition of NRF2 transcriptional activity. Angew. Chem. Int. Ed. 59, 12062–12068 (2020).
Duo, Y. et al. Targeted delivery of novel Au(I)-based AIEgen via inactivated cancer cells for trimodal chemo-radio-immunotherapy and vaccination against advanced tumor. Nano Today 51, 101920 (2023).
Miao, Z. et al. Endoplasmic reticulum-targeting AIE photosensitizers to boost immunogenic cell death for immunotherapy of bladder carcinoma. ACS Appl. Mater. Interfaces 16, 245–260 (2024).
Sen, S. et al. Rationally designed redox-active Au(I) N-heterocyclic carbene: an immunogenic cell death inducer. J. Am. Chem. Soc. 142, 20536–20541 (2020).
Chiou, J. Y. Z. et al. Au(I) complexes of N-heterocyclic carbenes and pyridines. Eur. J. Inorg. Chem. 1434–1948 (2009).
Zhang, J. et al. A new strategy to elevate absorptivity of AIEgens for intensified NIR-II emission and synergized multimodality therapy. Adv. Mater. 35, e2306616 (2023).
Zhang, J. et al. Endowing AIE with extraordinary potential: a new Au(I)-containing AIEgen for bimodal bioimaging-guided multimodal synergistic cancer therapy. Adv. Funct. Mater. 32, 2108199 (2022).
Zou, H. et al. Making aggregation-induced emission luminogen more valuable by gold: enhancing anticancer efficacy by suppressing thioredoxin reductase activity. ACS Nano 15, 9176–9185 (2021).
Park, H. et al. Precise and long-term tracking of mitochondria in neurons using a bioconjugatable and photostable AIE luminogen. Chem. Sci. 13, 2965–2970 (2022).
Wang, D. et al. Highly efficient photosensitizers with far-red/near-infrared aggregation-induced emission for in vitro and in vivo cancer theranostics. Adv. Mater. 30, e1802105 (2018).
Kanwar, R., Gradzielski, M., Prevost, S., Kaur, G., Appavou, M. S. & Mehta, S. K. Physicochemical stimuli as tuning parameters to modulate the structure and stability of nanostructured lipid carriers and release kinetics of encapsulated antileprosy drugs. J. Mater. Chem. B. 7, 6539–6555 (2019).
Liu, Z. et al. Tuning organelle specificity and photodynamic therapy efficiency by molecular function design. ACS Nano 13, 11283–11293 (2019).
Karges, J. et al. Rationally designed ruthenium complexes for 1- and 2-photon photodynamic therapy. Nat. Commun. 11, 3262 (2020).
Spiegel, M. & Adamo, C. Tuning the photophysical properties of Ru(II) photosensitizers for PDT by protonation and metallation: a DFT study. J. Phys. Chem. A 127, 3625–3635 (2023).
Luo, T. et al. Metal-organic layer delivers 5-aminolevulinic acid and porphyrin for dual-organelle-targeted photodynamic therapy. Angew. Chem. 135, e202301910 (2023).
Huang, X. et al. Rationally designed heptamethine cyanine photosensitizers that amplify tumor-specific endoplasmic reticulum stress and boost antitumor immunity. Small 18, e2202728 (2022).
Dandekar, A., Mendez, R. & Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 1292, 205–214 (2015).
Ma, H. et al. ER-targeting cyanine dye as an NIR photoinducer to efficiently trigger photoimmunogenic cancer cell death. J. Am. Chem. Soc. 144, 3477–3486 (2022).
Liu, X. et al. ER-targeting PDT converts tumors into in situ therapeutic tumor vaccines. ACS Nano 16, 9240–9253 (2022).
González, J. J. et al. Luminescent Au(I) complexes of 1-pyridyl-3-anthracenylchalcone inducing apoptosis in colon carcinoma cells and antivascular effects. Inorg. Chem. 58, 12954–12963 (2019).
Lu, Y. et al. SERD-NHC-Au(I) complexes for dual targeting ER and TrxR to induce ICD in breast cancer. Pharmacol. Res. 190, 106731 (2023).
Zhuang, Z. et al. Type I photosensitizers based on phosphindole oxide for photodynamic therapy: apoptosis and autophagy induced by endoplasmic reticulum stress. Chem. Sci. 11, 3405–3417 (2020).
Liu, X. et al. Targeting LIPA independent of its lipase activity is a therapeutic strategy in solid tumors via induction of endoplasmic reticulum stress. Nat. Cancer 3, 866–884 (2022).
Xu, F. & Wang, L. Deciphering ER stress-unfolded protein response relationship by visualizing unfolded proteins in the ER. Cell Rep. 43, 114358 (2024).
Yi, X. et al. SIRT7 orchestrates melanoma progression by simultaneously promoting cell survival and immune evasion via UPR activation. Signal Transduct. Target Ther. 8, 107 (2023).
Yüksek, V., Dede, S., Çetin, S., Usta, A. & Taşpınar, M. Vitamin D may assist the UPR against sodium fluoride-induced damage by reducing RIPK1, ATG5, BECN1, oxidative stress and increasing caspase-3 in the osteoblast MC3T3-E1 cell line. J. Trace Elem. Med. Biol. 80, 127293 (2023).
Zhu, H. et al. Pifithrin-μ incorporated in gold nanoparticle amplifies pro-apoptotic unfolded protein response cascades to potentiate synergistic glioblastoma therapy. Biomaterials 232, 119677 (2020).
Rigg, N., Abu-Hijleh, F. A., Patel, V. & Mishra, R. K. Ketamine-induced neurotoxicity is mediated through endoplasmic reticulum stress in vitro in STHdhQ7/Q7 cells. Neurotoxicology 91, 321–328 (2022).
Chen, X. & Cubillos-Ruiz, J. R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 21, 71–88 (2021).
Zhang, G. et al. Redox-responsive dendrimer nanogels enable ultrasound-enhanced chemoimmunotherapy of pancreatic cancer via endoplasmic reticulum stress amplification and macrophage polarization. Adv. Sci. 10, e2301759 (2023).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22205096, 31971293, 22103062), the National Key R&D Program of China (2021YFE0202400), the National Key R&D Program of China (2021YFA1300604), the National Science Fund for Distinguished Young Scholars (82025024), the Outstanding Youths Development Scheme of Nanfang Hospital, Southern Medical University (2021J002), GuangDong Basic and Applied Basic Research Foundation (2021A1515110373 and 2023A1515011924), Shanghai Pujiang Program (No. 22PJ1402800). The schematic diagram of Fig. 1 was drawn by Figdraw.
Author information
Authors and Affiliations
Contributions
N.F and J.Z. conceived and designed the experiments. Z.P. performed the synthesis. N.F. did all the biological experiments and analyzed the data. X.Z. and L.H. conducted the calculation. X.Z. and Y.L. took part in the discussion and gave important suggestions. N.F, L.H., and J.Z. co-wrote the manuscript. L.Z. and B.Z.T. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Safacan Kölemen, Federica Maschietto, Sajal Sen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, N., Peng, Z., Zhang, X. et al. Strategically engineered Au(I) complexes for orchestrated tumor eradication via chemo-phototherapy and induced immunogenic cell death. Nat Commun 15, 8187 (2024). https://doi.org/10.1038/s41467-024-52458-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52458-4
This article is cited by
-
Fluorescent ferroptosis inducers leveraging the pharmacological activity of rhodanine to promote antitumor immunity
Science China Chemistry (2025)
