
Abstract
Continuous directed evolution of base editors (BEs) has been successful in bacteria cells, but not yet in mammalian cells. Here, we report the development of a Continuous Directed Evolution system in Mammalian cells (CDEM). CDEM enables the BE evolution in a full-length manner with Cas9 nickase. We harness CDEM to evolve the deaminases of cytosine base editor BE3 and adenine base editors, ABEmax and ABE8e. The evolved cytidine deaminase variants on BE4 architecture show not only narrowed editing windows, but also higher editing purity and low off-target activity without a trade-off in on-targeting activity. The evolved ABEmax and ABE8e variants exhibit narrowed or shifted editing windows to different extents, and lower off-target effects. The results illustrate that CDEM is a simple but powerful approach to continuously evolve BEs without size restriction in the mammalian environment, which is advantageous over continuous directed evolution system in bacteria cells.
Similar content being viewed by others

Introduction
CRISPR-Cas systems or base editors (BEs) have been widely studied to improve their performance including editing efficiency, specificity, or precision. Directed evolution has become one critical approach for this purpose. Directed evolution in unicellular context has been successfully performed to improve the specificity1 or broaden the protospacer adjacent motif (PAM) compatibility2,3 of Cas protein, and to improve the editing activity or modify the editing window of BEs including adenine base editor (ABE)4,5,6, cytidine base editor (CBE)7, and C-to-U RNA base editor8. Despite great success, the current unicellular-based evolution systems still face some challenges. The performance of evolved protein via directed evolution in unicellular context can falter or even fail to function when transferring to mammalian context. The reason could be due to different mechanisms of trafficking, compartmentalization, protein maturation, and non-native co-factors occurring in eukaryotes and prokaryotes9. Moreover, prokaryotic-based evolution systems4,10 including phage-assisted continuous and non-continuous evolution (PACE and PANCE)3,5,7,11 harnessed dead Cas9 (dCas9) instead of Cas9 nickase (Cas9n) during evolution. However, Cas9n was commonly used in BE to improve editing efficiency in mammalian cells even though more indels are introduced compared with dCas9. In addition, restricted by a payload size (~4.4 kb12) of phage, BE in a split format was used by PACE3,5,7. These variances may undermine the effectiveness of bacteria-based evolution as well as the functionality of the evolved product when transferring to mammalian cells.
Directed evolution of CRISPR-Cas systems or BEs in mammalian cells have lagged behind unicellular evolution systems. So far, only a few continuous directed evolution systems based on mammalian cells have been reported and they are all viral-based evolution systems that couple virus propagation to the function of the evolved target13,14. However, none of these systems have been reported to evolve BEs or CRISPR/Cas proteins. One reason could be the challenge to design a functional evolution circuit coupling virus propagation to gene editing. In addition, the payload size of the viral systems is a restricting factor especially for evolution target with a large size that needs to be introduced into the viral genomes, while the genes encoding Cas protein or BE are often large (e.g. BE typically is more than 5 kb). Compared with continuous directed evolution, non-continuous directed evolution or screening has a few disadvantages. First, limited mutation variants produced by in vitro mutagenesis hamper its efficiency and wide application. Second, evolutionary success depends on the total round numbers of the evolution performed15, therefore effectiveness could be dramatically enhanced through continuous evolution. At present, only a few reports on non-continuous directed evolution of Cas protein or BE in mammalian cells have been reported16,17,18. Taken together, a continuous directed evolution system in mammalian cells is needed for further improvement of CRISPR systems or BEs.
CBE and ABE enable programmable single base conversion in target DNA without relying on double strand breaks (DSBs), and have showed great therapeutic potentials in which precise editing and low off-target activity are required19,20. However, several challenges impede their therapeutic applications, one of which is that CBE and ABE can edit all targets within their editing windows and thus generate undesired bystander base editing. For example, the state-of-the-art ABE8e5 has a very wide editing window and high off-target activity on both DNA and RNA levels. Although several studies have been reported to narrow the editing window of CBE21,22,23,24 and ABE18,25,26 through structure-guided protein engineering, these window-optimized variants still suffer from product purity21,24,27, off-target effects on both DNA23,28 and RNA22,25,29 levels. To our knowledge, narrowing or shifting the editing window through directed evolution has not been reported so far.
In this study, we develop a continuous directed evolution system in mammalian cells (CDEM) by combining CRISPR-X30 for in vivo mutagenesis and antibiotic resistance for positive selection. We apply CDEM to evolve the cytidine deaminase component of BE3 and the adenosine deaminase of ABEmax and ABE8e respectively, generating CBE and ABE variants with narrowed or shifted editing windows, higher product purity and lower off-target effects.
Results
Development of a continuous directed evolution system in mammalian cells
To circumvent the challenges of the current directed evolution systems for BE, we sought to develop a non-viral directed evolution system in mammalian cells. Mutagenesis and selection are two major elements for a directed evolution system. To introduce targeted mutagenesis on deaminase, we harnessed the CRISPR-X system, which consists of a dCas9, a MS2-AID (activation-induced cytidine deaminase) fusion protein, and a guide RNA (gRNA) containing a MS2-binding loop to recruit MS2-AID30 (Fig. 1c). CRISPR-X can mutate a large variety of C/G to other bases and a low level of A/T mutation within a ~ 100 base pair (bp) window, at a rate up to ~1/500 to 1000 per bp.

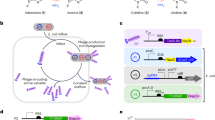
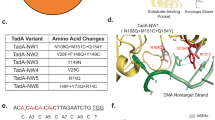
a General schematic of CDEM. b The BEs to be evolved: BE3, ABEmax and ABE8e. c Schematic of the two MPs. d Schematic of deficient SP conversion to active SP. e SP design for cytidine or adenine deaminase evolution. f Genotypes of APOBEC1 variants. See Supplementary Fig. 2 for the whole 18 evolved variants. g Genotypes of heterodimer TadA variants of ABEmax. See supplementary Fig. 8 for the 16 evolved TadA genotypes. h Genotypes of TadA variants of ABE8e. See supplementary Fig. 9 for all the 21 evolved genotypes. TadA and TadA* represent the wild type and evolved TadA, respectively. The SP-selection# refers to the SP design and the round number of selections. Source data are provided as a Source Data file.
CDEM (Fig. 1a) starts with the construction of a HEK293T cell line constitutively expressing BE (Fig. 1b) and a gRNA targeting deficient selection plasmid (SP) (Fig. 1d) (Step 1), followed by the delivery of the mutagenesis plasmids (MPs) (Fig. 1c) for mutating the deaminase (Step 2). Subsequently, cells were transfected with deficient SP followed by addition of a corresponding antibiotic for selection (Step 3). The cells with desired base editing on deficient SP would survive from antibiotic selection, whereas cells with unedited or incorrectly edited deficient SP will die (Step 4). As SP is transfected transiently and will be degraded in a few days, continuous selection can be achieved by repetitive SP transfection. The acquired mutations from evolution were determined by sequencing the deaminase gene from genomic DNA.
We first tested CDEM to evolve BE3, which is promiscuous with a relatively large editing window (C4-C12). We replaced the blasticidin start codon (ATG) with GTG to make a deficient SP (BE3 C6 design) (Fig. 1e). Base editing of C6, but not C4 nor C4C6 editing, can convert GTG back to ATG that will turn on blasticidin expression. We thus anticipated that this evolution strategy could select BE3 variants with narrowed editing windows. As the number of evolutionary rounds increased, we observed an elevated level of C to T conversion at C6 on SP, indicating the desired genotype enrichment (Supplementary Fig. 1). We stopped the evolution at round 5 at day 60. The cytidine deaminase gene from the genomic DNA was PCR-amplified and cloned back to BE3 plasmid for Sanger sequencing. 43 candidate variants were obtained after excluding the ones with wild-type (WT), synonymous and frame-shift mutations (Fig. 1f, Supplementary Fig. 2a). A clear enrichment of several amino acid mutations was observed: T5G, E24G, Y120H, V139M, T140A and E146G, indicating a result from evolution selection.
Inspired by the results from CBE evolution, we set out to evolve adenosine deaminase component of ABEmax and ABE8e. We installed ATA to replace the ATG start codon in A4 and A7 design (Fig. 1d, e). In A7 design, only a desired A7 base editing (A to G), but not A5 nor A5A7, can lead to an ATG start codon for puromycin resistance gene expression, and a similar selection strategy was also used to shift or narrow the editing window of ABEmax and ABE8e. In A4 design, only a desired A4 base editing (A to G), but not A2 nor A2A4, lead to an ATG start codon for puromycin resistance gene expression. After five rounds of evolution, the TadA genes were amplified and cloned into expression plasmid for sequencing. We obtained 16 (Fig. 1g, Supplementary Fig. 8a) and 21 (Fig. 1h, Supplementary Fig. 9a) variants from ABEmax and ABE8e evolution, respectively. Enrichment of amino acid mutations (ABEmax: Y73C, D119G, N127D; ABE8e: N38G or N38D, R39G, L68P, R107G, L121P) were observed. We also observed two mutations (R39G, Y73C) enriched for both ABEmax and ABE8e. Intriguingly, we found a large deletion in one ABEmax mutant, forming a minimized ABEmax with a TadA monomer.
Evolved CBEs with narrowed editing windows, high product purity and low off-target activity
Evolved cytidine deaminase variants on BE3 architecture were first characterized at the endogenous genomic FANCF site 1 in HEK293T cells. Compared to the original BE3, the 18 variants showed significantly varying editing activity with the majority displaying reduced activity (Supplementary Fig. 2b). However, we observed a few variants (N2-BE3, N5-BE3, N7-BE3 and N12-BE3) exhibiting comparable base editing activity at peak editing position but lower editing at other positions, implying narrowed editing windows of these variants. We also noted that N5-BE3 (4 out of 43 clones), N7-BE3 (7 out of 43 clones) and N12-BE3 (6 out of 43 clones) were highly enriched (Supplementary Fig. 2a), indicating an evolutionary selection. Thus, we chose these 4 variants for further characterization. As BE4 outperformed BE3 in editing efficiency and product purity, and they are structurally similar to each other31, we decided to characterize the evolved cytidine deaminases on BE4 architecture.
We tested these four BE4 variants (N2-BE4, N7-BE4, N5-BE4 and N12-BE4) at 16 genomic sites in parallel with WT BE4 and a previously engineered YE1-BE4 with narrowed editing window24. We observed that the four variants and YE1-BE4 showed comparable editing efficiency at the peak editing position (C5 to C7) at most sites, but lower editing activity at positions distal to the peak position compared to BE4 (Fig. 2a and Supplemental Fig. 3a). An analysis of all 16 sites showed that BE4 was active in a large window from position 4 to 12, whereas YE1-BE4 and four BE4 variants were active within a narrower editing window ranging from position 5 to 8 (Fig. 2b and Supplementary Fig. 3b). Notably, N7-BE4 showed a narrower editing window than that of YE1-BE4. Thus, the four BE4 variants displayed narrower editing windows without compromising editing activity compared to the BE4.

a The evolved CBE variants exhibited comparable editing efficiency and narrower editing windows compared with BE4; n = 3 or 4 independent experiments. b Frequencies of C converted to T across protospacer at the edited sites (PAM located at positions 21–23). Single dots represent individual data points from 3 or 4 independent replicates per site. Boxes span the interquartile range (25th to 75th percentile); horizontal line in the box indicates the median (50th percentile); and small horizontal bars mark the minimal and maximal values. c The indel decrease fold of YE1, N2 and N7 relative to BE4 in HEK293T cells. Bars represent median values; n = 16 target sites (see details in supplementary Fig. 4a). d The decrease fold of all Cs in the spacer converted to A/G for YE1, N2 and N7 relative to BE4 in HEK293T cells. The maximal fold change was set as 100. Bars represent median values; n = 16 target sites (see all variants in supplementary Fig. 4c). e The decrease fold of off-target activity for YE1, N2 and N7 compared with BE4. Bars represent median values; n = 21 off-target sites (see detailed data in supplementary Fig. 5). f Cas9-independent off-target analysis of the cumulative C edits by the orthogonal R-loop assay; n = 3 independent experiments. g RNA off-target evaluation of ABEs; n = 2 independent experiments. Data were presented as mean±s.d. in histograms.
We next examined the base editing product purity of the four BE4 variants. An analysis of the deep sequencing data of the above 16 sites showed that all four BE4 variants exhibited significantly lower frequencies of unwanted base editing alterations than that of BE4 at almost all the sites (Fig. 2c, d and Supplementary Fig. 4). All four BE4 variants generated lower level of indels than BE4 at all 16 sites, and lower or similar level of indels compared to YE1-BE4 at most sites (12 out of 16) (Fig. 2c and Supplementary Fig. 4a). Quantification of the indel frequency showed that, compared with BE4, YE1-BE4, N2-BE4, N7-BE4, N5-BE4 and N12-BE4 had a 2.8-, 3.2-, 4.8-, 4.7- and 8-fold indel decrease, respectively (Fig. 2c and Supplementary Fig. 4b). We also compared the frequency of all Cs or specific C converted to A or G on spacer (Supplementary Fig. 4c,d). While BE4 has a higher or comparable frequency of targeted specific C converted to A or G compared to YE1-BE4 and four evolved BE4 variants, the frequency of all targeted Cs converted to A or G by BE4 turned out to be significantly higher than that of the YE1-BE4 and the other variants at most sites, indicating that the evolved variants also have narrowed windows for C to A or G editing. A frequency quantification of all Cs converted to A or G on spacer illustrated that compared to BE4, YE1-BE4, N2-BE4, N7-BE4, N5-BE4 and N12-BE4 exhibited a 1.8, 1.6, 2.9, 4.5 and 12.3 median fold decrease, respectively (Fig. 2d and Supplementary Fig. 4e). Taken together, the four evolved BE4 variants, particularly the N7-BE4, showed higher product purity than that of the original BE4 and YE1-BE4.
To investigate the off-target effects of the evolved BE4 variants, we probed Cas9-dependent off-target activity at three endogenous genomic sites (EMX1, FANCF site 1and HEK site 4) as previously described24 (Fig. 2e and Supplementary Fig. 5), and characterized the Cas9-independent off-target activity by an orthogonal R-loop assay in four dSaCas9 R-loop sites (Fig. 2f, Supplementary Fig. 6). All four evolved BE4 variants showed reduced DNA editing at all three Cas9-dependent off-target sites and all four Cas9-independent sites (Supplementary Figs. 5a,c, 6). Compared to BE4, YE1-BE4, N2-BE4, N7-BE4, N5-BE4 and N12-BE4 have a 5.2-, 6.5-, 49.7-, 21.4-, 20.5-fold decreased DNA editing, respectively, at three Cas9-dependent off-target sites (Fig. 2e and Supplementary Fig. 5b). Notably, N7-BE4 showed the lowest off-target activity at both Cas9-dependent and -independent sites, outperforming YE1-BE4.
As CBE was reported to deaminate RNA cytosines22, we further evaluated the extent of cellular RNA editing by these BE4 variants. We treated HEK293T cells with plasmids encoding BE4, the above four evolved BE4 variants, YE1-BE4 or GFP together with a vector expressing gRNA targeting the endogenous PPP1R12C site 1, and then measured the substitution frequency across the transcriptome. Consistent with previous results, YE1-BE4 exhibited a significantly lower cellular RNA editing than BE428. The four evolved variants have a similar RNA editing to YE1-BE4, with N7-BE4 having the lowest RNA editing (Fig. 2g, Supplementary Fig. 7).
Evolved ABE variants with narrowed/shifted editing windows and low off-target activity
We chose 16 variants evolved from ABEmax and 21 variants evolved from ABE8e for characterization at three (HEK293 site 1/11/16) and four (HEK293 site 3/13/16/18) genomic sites, respectively (Supplementary Figs. 8, 10). We did not observe a consistent improvement on editing activity for all these variants. However, some ABEmax variants and all ABE8e variants showed narrowed editing window compared to the original ABEmax and ABE8e. Based on the editing profiles, 2 variants (M3-ABEmax, M1-ABE) evolved from ABEmax and 6 variants (E4-ABE, E18-ABE, E2-ABE, E14-ABE, E11-ABE and E21-ABE) evolved from ABE8e were chosen for a more extensive and parallel characterization at 21 genomic sites (Fig. 3a, c and Supplementary Figs. 9, 11). Note that M1-ABE has a TadA monomer while M3-ABEmax has a TadA heterodimer.

a Base editing activity of ABEmax and evolved variants M3 and M1 in HEK293T cells; n = 3 or 4 independent experiments. c Base editing activity of ABE8e and the evolved variants profiled in HEK293T cells; n = 3 or 4 independent experiments. b, d Frequencies of A-to-G editing across the protospacer from the edited sites (PAM located at positions 21–23). Single dots represent individual data points from 3 or 4 independent replicates per site. Boxes span the interquartile range (25th to 75th percentile); horizontal line in the box indicates the median (50th percentile); and small horizontal bars mark the minimal and maximal values. e Schematic of editing windows for ABEs. f The fold decrease of Cas9-dependent off-target activity of ABEs; n = 12 off-target sites (see detailed data in supplementary Fig. 12a). Bars represent median values. g Cas9-independent off-target analysis of the cumulative adenine edits by the orthogonal R-loop assay; n = 3 independent experiments. (h) RNA off-target evaluation of ABEs; n = 2 independent experiments. Data were presented as mean ± s.d. in histograms.
An analysis of the editing activity and window for the selected variants at 21 genomic sites showed that M3-ABEmax and M1-ABE have a comparable or slightly lower editing activity compared with ABEmax. While M1-ABE from A7 design has an editing window similar to ABEmax, M3-ABEmax from A4 design exhibits a slightly narrower editing window but with a lower editing at A7 position compared to ABEmax, which reflected the selection pressure of A4 design for M3-ABEmax (Fig. 3a, b). E14-ABE and E21-ABE derived from A7 design displayed comparable editing activity to ABE8e at peak editing positions, while they all exhibited a narrower editing window (A4-A8 vs. A3-A10 for ABE8e) (Fig. 3d and Supplementary Fig. 11). In addition, ABE8e was also active in the marginal positions including A2, A11 and A12, while the three variants were almost completely inactive at these positions. Different from A7 design variants, the E4-ABE and E2-ABE from A4 design exhibited comparable editing activity at peak editing positions at half of tested sites and lower activity at the other half sites. However, they both had a significantly narrower editing window (A5-A7) than ABE8e (Fig. 3c, d, Supplementary Fig. 11). Intriguingly, E4-ABE and E2-ABE had a predominant editing at A5 position but low activity at A7 position, indicating an evolution outcome from A4 design (Fig. 3c, Supplementary Fig. 11). A cross comparison of variants evolved from ABEmax and ABE8e showed that M3-ABEmax, E2-ABE and E4-ABE exhibited narrower editing windows than ABEmax. The editing window widths of E14-ABE, E11-ABE and E21-ABE were in between that of ABE8e and ABEmax (Fig. 3b, d, e and Supplementary Figs. 9, 11).
We also observed that the peak editing window position of E11-ABE shifted to the A7 (particularly at ABE site 5/8/16/25) (Fig. 3c, d), which complies with the A7 design evolution strategy. E11-ABE variant displayed a different editing window profile compared to ABEmax and ABE8e, with a peak editing position at A7 rather than A5 for ABEmax and A5-A7 for ABE8e (Fig. 3a, c).
As the editing window widths of variant E14-ABE, E11-ABE, and E21-ABE were in between ABE8e and ABEmax, this characteristic may have advantages at certain circumstances. More genomic sites (PPP1R12C site 3 and 9, HEK293 site 6 and 26, HBG site 5, EMX1 site 2, PDCD2) were included for a further characterization (Supplementary Fig. 15). The result is consistent with previous result from 21 genomic sites, variant E14-ABE, E11-ABE, and E21-ABE exhibited editing windows in between ABE8e and ABEmax. Notably, variant E14, E11, and E21 exhibited higher editing activities at position A4, A7, and A8 compared with ABEmax, but lower editing activities at position A1–3 and A9–12 compared with ABE8e. These variants may benefit certain therapeutic applications that require high editing activity at A4, A7, A8 positions but low off-target activity.
We next assessed the DNA off-target activity of these evolved ABE variants as assayed for CBE. To compare Cas9-dependent off-target activity, we plotted the off-target activity decrease fold for the evolved variants and ABEmax relative to ABE8e (Fig. 3f, Supplementary Fig. 12). For comparison of Cas9-independent off-target activity, we calculated the percentage of cumulative adenine edits by the orthogonal R-loop assay (Fig. 3g, Supplementary Fig. 13). We observed a higher median fold decrease of Cas9-dependent off-target activity for M1-ABE (89.8) and M3-ABEmax (56.4) than that for ABEmax (34.6), and lower or comparable Cas9-independent off-target activity compared with ABEmax (Fig.3f, g). For variants evolved from ABE8e, the fold decrease at Cas9-dependent off-target sites was 4.0 (E14-ABE), 15.1 (E11-ABE), 40.1 (E4-ABE), 77 (E2-ABE), 10.8 (E21-ABE) and 126.7 (E18-ABE) (Fig. 3f, Supplementary Fig. 12e) and a similar pattern was also observed at Cas9-independent off-target sites (Supplementary Fig. 13). A cross comparison of ABEmax and ABE8e clusters showed that M3-ABEmax, M1-ABE, E4-ABE, E18-ABE and E2-ABE had a lower off-target activity than that of ABEmax. Additionally, variants E14, E11 and E21 exhibited interspersed off-target activity between ABEmax and ABE8e.
We next evaluated the RNA editing activity of the evolved ABE variants. To do so, we measured the cellular RNA editing by four evolved variants relative to ABE8e and ABEmax in HEK293T cells. Consistent with previous results5, ABE8e has a significantly higher transcriptome-wide RNA editing than that of ABEmax. The two variants evolved from ABEmax have lower transcriptome-wide RNA editing than ABEmax, and similarly, four variants evolved from ABE8e showed lower transcriptome-wide RNA editing than ABE8e (Fig. 3h, and Supplementary Fig. 14).
Adenine base editing with evolved ABEs at disease-relevant loci in human cells
We finally tested the utility of the evolved variants in disease-relevant contexts. To do so, we tested ABE variants (E14-ABE, E11-ABE, E7-ABE and E44-ABE) to target pathogenic SNPs on four gene loci, including CFTR (causing cystic fibrosis), VHL (causing non-cancerous tumors), POLG (causing early childhood mitochondrial DNA depletion syndromes or later-onset syndromes), SCN1A (causing Dravet syndrome). To mimic the in situ disease-causing mutations, we generated HEK293T cell lines with the integration of a 100-base pair (bp) CFTR/VHL/POLG/SCN1A gene sequence. We then compared the ability of ABE8e/ABEmax/E14-ABE/E11-ABE to install an A•T to G•C edit at position A8 on CFTR, A4 on VHL/POLG, A5 on SCN1A (Fig. 4). For CFTR locus, we observed desired A8 editing and also unwanted A10 editing that causes a missense mutation. ABE8e and two variants (E14-ABE and E11-ABE) significantly outperformed ABEmax for A8 editing on CFTR. While ABE8e and two variants showed a comparable A8 editing, these two variants exhibited much lower A8 + A10 co-editing, with a 5- to 11-fold decrease. For VHL locus, while ABE8e showed the highest desired A4 editing, it was also accompanied with a high A4 + A12 co-editing. Both ABEmax and E14-ABE exhibited almost no co-editing, but E14-ABE mediates much more A4 editing. We observed high desired A4 editing for ABEmax, ABE8e and E14-ABE on the POLG locus, but again, ABE8e caused high undesired A4 + A12 co-editing. For SCN1A locus, ABEmax was slightly better than E4-ABE and E2-ABE for desired A5 editing, but caused an obvious stronger undesired editing. We barely observed desired A5 editing, but there was significant undesired co-editing for ABE8e, which is consistent with its wide editing spectrum. These results demonstrated that the evolved ABE variants are well suited for correcting disease-causing mutations with minimal undesired base editing relative to their parental BEs.

Comparison of correcting pathogenic mutations in four stable HEK293T cell lines. Desired A-to-G percentiles of alleles (red bar) are exhibited; Data were presented as mean ± s.d. in histograms; n = 3 independent experiments. Source data are provided as a Source Data file.
Discussion
In summary, we developed a continuous directed evolution system in mammalian cells termed CDEM that was successfully applied to evolve cytidine deaminase of CBE, and adenosine deaminases on ABEmax and ABE8e architectures, separately. Using CDEM, we generated CBE variants with narrowed editing windows compared with BE4, and evolved ABEmax and ABE8e to generate variants with narrowed or shifted editing windows (Figs. 2a, b, 3a–d). Interestingly, all evolved variants also showed lower off-target activity on both DNA and RNA levels. In addition, the evolved BE4 variants also exhibited higher product purity - lower indel ratios and lower undesired edits - than that of BE4 (Figs. 2c–g, 3e–g).
BEs can typically edit multiple bases within a certain sequence window, which often poses a big threat to therapeutic application where only a specific position requires base editing. Narrowed editing window is thus critical, which can not only improve editing precision but also lower off-target effect on both DNA and RNA level. It seems that the editing window width is correlated with off-target activity (Figs. 2, 3), which is consistent with previous reports that a narrowed window results in a low off-target activity5,24. In this study, through CDEM we generated CBE and ABE variants with narrowed editing windows of varied widths, which maximize the likelihood of achieving base editing with high efficiency and precision for a certain base alteration application (see a comparison of our selected and other reported ABE variants at Supplementary Table 2). In addition, the strategy of shifting the editing window also proved to be feasible as demonstrated by the E11 variant, indicating that such a similar circuit could be also applied to other BEs (e.g., CGBE32, AYBE33 and GYBE34) to widen their targeting spectrums. Taken together, the strategies of narrowing or shifting the editing window illustrated here can be used to enhance the editing precision of BEs, which is important in therapeutic applications.
The major advantage of CDEM is that it allows the evolution of BEs directly in a mammalian context. As the gene to be evolved depends on the host cell for signaling, maturation and function, CDEM thus should be able to evolve BEs maximally and directly yield superior BE variants that fit mammalian environment. CDEM wholly mimic the context of BEs application in mammalian cells, unlike unicellular-based evolution system mentioned. First, unlike PACE or other evolution systems in prokaryotic cells where dCas9 was used for CBE and ABE evolution5,7,11, CDEM used Cas9n. In mammalian cells, Cas9n introducing a nick can enhance the editing activity, but is accompanied with significant indel production27. Second, CDEM could directly evolve the full-length BE (deaminase-nCas9), the format that is commonly used in mammalian cells, while the deaminase and dCas9 in PACE were expressed in a split format due to the payload size limitation. The full-length and split format could lead to different evolution performances. These differences might explain the observation that all the evolved cytidine deaminase variants on BE4 architecture had significantly decreased indel ratio compared with BE4. By contrast, the evolved CBE variant through PACE had a similar or even higher indel ratio compared to the initial CBE7. Therefore, it is very likely that CDEM-evolved BEs using Cas9n introduce less indel on selection plasmids and thus provide host cells with a higher survival probability, while PACE with the use of dCas9 does not have such an evolution pressure on indel production. However, we did not observe decreased indel ratio by the evolved ABE variants (data not shown). This could be due to a fewer indel introduction of ABE compared to that of CBE, which is consistent with previous results4.
The CDEM with AID as source of mutation is highly mutagenic30,35, as also demonstrated in this study by the diverse mutation paradigm in the evolution experiments especially for ABEmax and ABE8e (Supplementary Figs. 8, 10). These diverse mutation data can be used to fine-tune a machine learning process for protein engeneering36. The mutation paradigm of CDEM seems different from that of virus-based evolution systems2,4,7,14. The variants with single or few amino acid mutations can still be frequently observed after several rounds of evolution in CDEM but seems not in virus-based evolution systems. The reason could be due to differences in selection pressure. The selection pressure of viral evolution systems depends on the growth competition for virus survival, while the selection pressure of CDEM comes from a higher antibiotic concentration than the cell tolerance threshold, which can be controlled by simply adjusting the antibiotic concentration. The high selection pressure of viral evolution system could lead to a quick convergence, however, for CDEM it is unlikely to occur rapid premature convergence, which could lead to residues being fixed prematurely and formation of local optimum15.
CDEM has more advantages due to its non-viral characteristics (Supplementary Table 3). CDEM has no length restriction on the evolution targets, unlike the viral evolution system that often has a payload size limit. In addition, CDEM does not need high biosafety level laboratories which are normally required by viral evolution systems. CDEM also does not require complex adjustments of experimental parameters as conducted for PACE12, and there is no need to perform cloning or site-directed mutagenesis when transferring between prokaryotic and eukaryotic cells. Furthermore, the evolution circuit of modifying editing window is easy to use. We thus believe that CDEM can be adapted to evolve other Cas proteins of different status (nuclease, nickase or dead), or effectors of other BEs, or the corresponding gRNA components. Likewise, CDEM could be employed to improve the targeting specificity of CRISPR systems and BEs. We also envision that evolution of other biological proteins (e.g., enzymes, binding proteins) should be possible with CDEM (Supplementary Fig. 17). For some proteins that are difficult or impossible to evolve in a prokaryotic context due to incompatibility of biological systems, CDEM provides an alternative option.
Nevertheless, CDEM still has a significant room for improvement. For mutagenesis resources, the current mutation pattern of AID prefers to mutate C or G to other bases30,35, which limits the mutagenesis diversity. This could be enhanced using other effectors such as the recently reported AYBE (editing A to other bases)33 and TGBE (editing T to other bases)37. During the evolution process, mutational tolerance (gained mutations resistant to the same gRNA targeting because of mismatch between the gRNA spacer and DNA target) could lead to lower mutagenesis ratio38, which could be ameliorated by designing more gRNAs. We also anticipate that CDEM can be improved by combining it with other approaches such as bioinformatic and protein structural analysis or artificial intelligence. In summary, we report a continuous directed evolution system in mammalian cells with several superiorities over the unicellular evolution and other eukaryotic evolution systems, and expand the growing toolbox of synthetic biology.
Methods
Construct design
Lentiviral plasmid pLenti-FNLS-P2A-Puro (Addgene, #110841) expressing BE3 was a gift from Lukas Dow39. Lentiviral plasmid expressing ABEmax (Lv-ABEmax-Cas9n-Blast) and ABE8e (Lv-ABE8e(V106W)-Cas9n_blast) were made by cloning the ABEmax and ABE8e gene sequence from pCMV-ABEmax (Addgene, #112095)40 and ABE8e(TadA-8e V106W) (Addgene, #138495)5, respectively. pCMV-BE4max-puro was constructed through the exchange of GFP with puromycin gene on plasmid pCMV-BE4max-P2A-GFP (Addgene, #112099)40. YE1-BE4max (Addgene, #138155)41 was a gift from David Liu. Plasmids N2-BE4, N7-BE4, N5-BE4, N12-BE4 were constructed through the exchange of cytidine deaminase gene on plasmid pCMV-BE4max-puro with the evolved cytidine deaminase variants. pLenti-SpBsmBI sgRNA-Hygro (Addgene, #62205)42 was a gift from Rene Maehr. pTE-dCas9-NeoR was constructed based on pTE4398 (Addgene, #74042) by substituting the LbCpf1 gene with dCas9 (lenti-dCAS-VP64-Blast, Addgene, # #61425). pGL-MS2gRNA-MS2-AID-Δ-BeoR was constructed based on pGH335-MS2-AID*Δ-Hygro (Addgene, #85406) and pGH224-sgRNA-2xMS2-Puro (Addgene, #85413)30.
DNA ligation was performed using ClonExpress® II One Step Cloning Kit (Vazyme) and all clones were transformed into home-made Stbl3 Competent E. Coli. Individual colony was grown at 37 °C (non-lentiviral plasmid) or 30 °C (lentiviral plasmid) at 220 rpm for ~16–22 h. Plasmids were purified with HiPure Plasmid Mini/midi Kit, HiPure Plasmid EF Midi Kit (Magen, #P1002-02, #P1003-02, #P1113-02). All constructs were validated by Sanger sequencing and sequence alignment was conducted with CodonCode Aligner.
Cell culture
HEK293T cells (purchased from National Collection of Authenticated Cell Cultures in China, GNHu44) were grown in a humidified 37 °C incubator with 5% CO2 using DMEM media supplemented with 100 I.U./mL penicillin and 100 µg/mL streptomycin (Thermo Fisher, #15140163).
Transfection
Transfection in the evolution assay: HEK293T cells grown in the absence of antibiotic were seeded on 24-well plates (Biofil, China). Cells were transfected at approximately 70% confluency with 6 μl Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturers’ protocol, 3 μg dCas9 plasmid and 1 μg gRNA mix.
Transfection in the function validation experiment: HEK293T cells were seeded into 24-well plates and transfected with 1,500 ng BE and 500 ng gRNA plasmids per well using EZtrans (life iLAB, China) following the manufacturers’ instructions. 24 h after transfection, the culture media were exchanged with fresh culture media supplemented with 1 µg/ml puromycin (Solarbio, China) and 10 µg/ml blasticidin (Solarbio, China). 5 days after transfection, genomic DNA were extracted for PCR amplification and the resultant PCR products were subjected to deep sequencing.
Transfection for RNA sequencing: HEK293T cells were seeded on 6-well plates and transfected at ~70% confluence with 3 μg editor and 1 μg gRNA-expressing plasmids or the control GFP plasmid using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturers’ protocol. 3 days after transfection, cells were collected for total RNA isolation (Vazyme).
Transduction
Lentiviral vectors were produced by transfection of HEK293T cells with the lentiviral plasmid and packaging plasmids (psPAX2 and pVSV-g) using Lipofectamine 2000. 6 h after transfection, the culture medium was replaced with Opti-MEM (Invitrogen). 48 h after transfection, the supernatant containing lentiviral vector was collected, centrifuged, filtered (0.45 µm), aliquoted, and stored at -80 °C until use.
Directed evolution of BE3 in mammalian cells
HEK293T cells stably expressing BE3 and gRNA targeting deficient blasticidin were made by transducing HEK293T cells with lentivirus (pLenti-FNLS-P2A-Puro) followed by puromycin (1 μg/ml) (Solarbio) selection, and subsequently by a second lentivirus (Lv-gBlast-Hygro) transduction followed by Hygromycin B (400 μg/ml) (APExBIO) selection. The MPs (pTE-dCas9-NeoR plasmid and 10 gRNA plasmids as a mixture (each gRNA with same amount) targeting the cytidine deaminase gene) were delivered into HEK293T cells by transfection, 4 days after which the cells were seeded on a new plate for transfection of SP (Lv-GTG-blast). 3 days after SP transfection, the cells were split in a 1:4 ratio and blasticidin were added starting from 1 μg/ml to 200 μg/ml. After five rounds of mutagenesis and selection, the cells were collected for genomic DNA extraction.
Directed evolution of ABEmax in mammalian cells
HEK293 T cells stably expressing ABEmax and gRNA targeting deficient puromycin were made by transducing HEK293T cells with lentivirus (Lv-ABEmax-Cas9n-Blast) followed by blasticidin (10 μg/ml) (Solarbio) selection, and by another lentivirus (Lv-gPuro1/2-Hygro) transduction followed by Hygromycin B (400 μg/ml) selection. Transient transfection of the stable 293 T cells was conducted to deliver MPs (pTE-dCas9-NeoR plasmid and 15 gRNA plasmids targeting the adenosine deaminase gene on ABEmax). 4 days after transfection, the cells were spread and SPs (A4: Lv-ATA1-Puro, A7: Lv-ATA2-Puro) were transiently transfected, 3 days after which puromycin were added starting from 1 μg/ml to ~200 μg/ml. After five rounds of mutagenesis and selection, the cells were harvested for genomic DNA extraction.
Directed evolution of ABE8e in mammalian cells
HEK293 T cells stably expressing ABE8e and gRNA targeting deficient puromycin gene were made by transducing HEK293T cells with lentivirus (Lv_ABE8e(V106W)_Cas9n_blast) followed by blasticidin (10 μg/ml) selection, and lentivirus (Lv-gPuro1/2-Hygro) followed by Hygromycin B (400 μg/ml) selections. Transient transfection was performed on the 293 T cells with MPs (pTE-dCas9-NeoR plasmid and 19 gRNA plasmids targeting the adenosine deaminase gene on ABE8e). 4 days after transfection, the cells were spread and selection plasmids (A4: Lv-ATA1-Puro, A7: Lv-ATA2-Puro) were transiently transfected, 3 days after which puromycin were added starting from 1 μg/ml to ~200 μg/ml. After five rounds of mutagenesis and selection, the cells were harvested for genomic DNA extraction.
Genomic DNA extraction and PCR amplification
Genomic DNA extraction in the evolution assay: The cells were harvested after antibiotic selection and the genomic DNA extraction was performed according to manufacturers’ instructions (Thermo Scientific).
Genomic DNA extraction in the base editing test assay: The harvested cells were mixed with 20 μl lysis buffer (10 mM Tris-HCl (pH 8.0), 50 mM KCl, 1.5 mM MgCl2, 0.5% Nonidet P-40, 0.5% Tween-20, and 100 μg/ml proteinase K (Roche)) and incubated under the following procedure: 68 °C for 30 min, 16 °C for 2 min and 98 °C for 5 min. The lysates were centrifugated at 12,000 rpm for 3 min and the supernatants were collected as template for PCR amplification under the following program: 95 °C for 5 min, 35 cycles of 95 °C for 30 s, 56 °C for 30 s, 72 °C for 20 s, and 72 °C for 5 min. The purified PCR products were subjected to Sanger sequencing or targeted deep sequencing. Sanger sequencing data were analyzed by EditR for the calculation of base editing activity. (https://moriaritylab.shinyapps.io/editr_v10/). The primers and amplified DNA sequences are listed in Supplementary Data 1–3.
Targeted deep sequencing and data analysis
PCR products were purified by the VAHTS DNA clean Beads (Vazyme) and then subjected to library construction and high-throughput sequencing on an Illumina Nova 6000 with PE150 mode (Annoroad, Beijing, China). The amplicon sequencing data were analyzed using CRISPResso2 (v.2.0.3) in the batch mode, with parameters “--base_edit --wc −8 --fastq_output --base_editor_output --write_cleaned_report --place_report_in_output_folder.” Editing efficiency was quantified from the “Quantification_window_nucleotide_percentage_table.txt” table. Indels were quantified from the “Alleles_frequency_table_around_sgRNA_*.txt” table. The results including C-to-T conversion rates, C-to-non-T (C-to-G and C-to-A) conversion rates, and indel rates were calculated.
RNA off-target analysis by RNA-seq
Raw reads were processed by fastp (version 0.20.1) to filter out bad reads and cut adapter sequences. Clean reads were aligned to human genome (hg38) with STAR (version 2.7.10b). Then, Sambamba (version 0.6.6) was employed to process the aligned bam files, including sorting the order of reads, removing duplicated reads and indexing the bam files. Strelka (version 2.9.10) was used to perform variant detection with the parameter ‘--exome’. The raw variants output from strelka with a filter label ‘PASS’ were chosen as high confidence variants for downstream analysis. The genomic information of SNVs and indels was annotated by ANNOVAR (version 2017Jul17). Downstream analysis and visualization were accomplished by custom R scripts.
Statistics and reproducibility
GraphPad Prism 9 software (version9.0) was used to analyze the data. All numerical values are presented as mean ± s.d., as noted otherwise. Data from the targeted deep sequencing and RNA-seq were corrected for between-session variation using Factor Correction43. n = 2–4 biological replicates were performed and listed in each figure. In this study, no statistical method was used to predetermine sample size, and no data were excluded from the analyzes. The cell experiments were not randomized and the Investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw high-throughput sequencing data generated in this study have been deposited in the NCBI sequence Read Archive database under PRJNA1093219. Plasmids used in this study (# 226583, #226584, # 226585, # 226586, # 226587, # 226588) are available on Addgene. Source data are provided with this paper.
References
Lee, J. K. et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nature communications 9, 3048 (2018).
Hu, J. H. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature https://doi.org/10.1038/nature26155 (2018).
Miller, S. M. et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nature biotechnology 38, 471-+ (2020).
Gaudelli, N. M. et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nature biotechnology, https://doi.org/10.1038/s41587-020-0453-z (2020).
Gaudelli, N. M. et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nature biotechnology 38, 892–900 (2020).
Thuronyi, B. W. et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nature biotechnology 37, 1070–1079 (2019).
Abudayyeh, O. O. et al. A cytosine deaminase for programmable single-base RNA editing. Science 365, 382–386 (2019).
Hendel, S. J. & Shoulders, M. D. Directed evolution in mammalian cells. Nature methods 18, 346–357 (2021).
Goldberg, G. W. et al. Engineered dual selection for directed evolution of SpCas9 PAM specificity. Nature communications 12, 349 (2021).
Neugebauer, M. E. et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nature Biotechnology 41, 673-+ (2023).
Miller, S. M., Wang, T. & Liu, D. R. Phage-assisted continuous and non-continuous evolution. Nature protocols 15, 4101–4127 (2020).
English, J. G. et al. VEGAS as a platform for facile directed evolution in mammalian cells. Cell 178, 748–761 e717 (2019).
Das, A. T. et al. Viral evolution as a tool to improve the tetracycline-regulated gene expression system. The Journal of biological chemistry 279, 18776–18782 (2004).
Voigt, C. A., Kauffman, S. & Wang, Z. G. Rational evolutionary design: the theory of in vitro protein evolution. Adv Protein Chem 55, 79–160 (2000).
Fu, J. et al. Human cell based directed evolution of adenine base editors with improved efficiency. Nature communications 12, 5897 (2021).
Xie, H. et al. High-fidelity SaCas9 identified by directional screening in human cells. PLoS biology 18, e3000747 (2020).
Tu, T. et al. A precise and efficient adenine base editor. Molecular therapy: the journal of the American Society of Gene Therapy 30, 2933–2941 (2022).
Porto, E. M., Komor, A. C., Slaymaker, I. M. & Yeo, G. W. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov 19, 839–859 (2020).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nature biotechnology 38, 824–844 (2020).
Lee, S. S. et al. Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome- and transcriptome-wide off-target effects. Science Advances 6, doi:ARTN eaba1773 (2020).
Grunewald, J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019).
Gehrke, J. M. et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nature biotechnology 36, 977–982 (2018).
Kim, Y. B. et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nature biotechnology 35, 371–376 (2017).
Grunewald, J. et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nature biotechnology 37, 1041–1048 (2019).
Chen, L. et al. Engineering a precise adenine base editor with minimal bystander editing. Nature Chemical Biology 19, 101-+ (2023).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420-+ (2016).
Zuo, E. W. et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nature methods 17, 600-+ (2020).
Zhou, C. et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature, https://doi.org/10.1038/s41586-019-1314-0 (2019).
Hess, G. T. et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nature methods 13, 1036–1042 (2016).
Komor, A. C. et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv 3, eaao4774 (2017).
Kurt, I. C. et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nature biotechnology, https://doi.org/10.1038/s41587-020-0609-x (2020).
Tong, H. et al. Programmable A-to-Y base editing by fusing an adenine base editor with an N-methylpurine DNA glycosylase. Nature biotechnology, https://doi.org/10.1038/s41587-022-01595-6 (2023).
Tong, H. et al. Programmable deaminase-free base editors for G-to-Y conversion by engineered glycosylase. Natl Sci Rev 10, nwad143 (2023).
Ma, Y. et al. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nature methods 13, 1029–1035 (2016).
Yang, K. K., Wu, Z. & Arnold, F. H. Machine-learning-guided directed evolution for protein engineering. Nature methods 16, 687–694 (2019).
Ye, L. J. et al. Glycosylase-based base editors for efficient T-to-G and C-to-G editing in mammalian cells. Nature biotechnology, https://doi.org/10.1038/s41587-023-02050-w (2024).
Wang, G., Zhao, N., Berkhout, B. & Das, A. T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Molecular therapy: the journal of the American Society of Gene Therapy 24, 522–526 (2016).
Zafra, M. P. et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nature biotechnology 36, 888–893 (2018).
Koblan, L. W. et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nature biotechnology 36, 843–846 (2018).
Doman, J. L., Raguram, A., Newby, G. A. & Liu, D. R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nature biotechnology 38, 620–628 (2020).
Pham, H., Kearns, N. A. & Maehr, R. Transcriptional regulation with CRISPR/Cas9 effectors in mammalian cells. Methods in molecular biology (Clifton, N.J.) 1358, 43–57 (2016).
Ruijter, J. M. et al. Factor correction as a tool to eliminate between-session variation in replicate experiments: application to molecular biology and retrovirology. Retrovirology 3, https://doi.org/10.1186/1742-4690-3-2 (2006).
Acknowledgements
This work was supported by National Natural Science Foundation of China (82271909) to G.W. We thank Junhao Guo (Precise Genome Engineering Center, School of Life Sciences, Guangzhou University) for the assistance in the R-loop assay.
Author information
Authors and Affiliations
Contributions
N. Z., and G.W. conceived the study. N. Z., J.Z., Z.G and G.W. designed the experiments. N. Z., J.Z. performed experiments with the assistance from Q.W., J.T., D.L., Z.G., S.W., T.T., S.G., J.L. and X.L. performed computational analysis. N. Z., J.Z., Z.G and G.W. analyzed the data with assistance from JK.Z. and J.S.O. N. Z., J.Z., Z.G. and G.W. wrote the manuscript with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
N.Z. and G.W. are co-founders of JinHua JiYin (进化基因). G.W., N.Z. and J.Z. have submitted a patent application to the China Patent & Trademark Office pertaining to the aspects of this work (202211365250.6). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, N., Zhou, J., Tao, T. et al. Evolved cytidine and adenine base editors with high precision and minimized off-target activity by a continuous directed evolution system in mammalian cells. Nat Commun 15, 8140 (2024). https://doi.org/10.1038/s41467-024-52483-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52483-3
