
Abstract
Developing highly active and durable air cathode catalysts is crucial yet challenging for rechargeable zinc-air batteries. Herein, a size-adjustable, flexible, and self-standing carbon membrane catalyst encapsulating adjacent Cu/Na dual-atom sites is prepared using a solution blow spinning technique combined with a pyrolysis strategy. The intrinsic activity of the Cu-N4 site is boosted by the neighboring Na-containing functional group, which enhances O2 adsorption and optimizes the rate-determining step of O2 activation (*O2 → *OOH) during the oxygen reduction reaction process. Meanwhile, the Cu-N4 sites are encapsulated within carbon nanofibers and anchored by the carbon matrix to form a C2-Cu-N4 configuration, thereby reinforcing the stability of the Cu centers. Moreover, the introduction of Na-containing functional groups on the carbon atoms significantly reduces the positive charge on their outer shell C atoms, rendering the carbon skeletons less susceptible to corrosion by oxygen species and further preventing the dissolution of Cu centers. Under these multi-type regulations, the zinc-air battery with Cu/Na-carbon membrane catalyst as the air cathode demonstrates long-term discharge/charge cycle stability of over 5000 h. This considerable stability improvement represents a critical step towards developing Cu-N4 active sites modified with the neighboring main-group metal-containing functional groups to overcome the durability barriers of zinc-air batteries for future practical applications.
Similar content being viewed by others

Introduction
The excessive consumption of fossil fuels and their environmental impact drive researchers to explore alternative energy carriers or devices for energy storage and conversion1,2,3. In recent years, rechargeable zinc-air batteries (ZABs) have emerged as one of the most promising technologies due to their low cost, high safety, high theoretical energy density, and environmental friendliness4. However, unsatisfactory energy conversion efficiency and limited durability are the main bottlenecks restricting the widespread application of ZABs5,6,7. These shortcomings predominantly originate from the sluggish kinetics of oxygen reduction/evolution reactions (ORR/OER) and the low stability of catalysts in air cathodes8,9,10. Although satisfactory catalytic activity can be achieved with precious metal-based catalysts (Pt, Ir, Ru, etc.), their high cost and poor durability force researchers to explore non-precious metal alternatives11. Recently, atomically dispersed M-N-C (M = non-precious metal) catalysts have attracted increasing interest as promising ORR electrocatalysts. In particular, Fe-N-C catalysts exhibit remarkable ORR activity even comparable to that of commercial Pt/C catalyst12,13,14. However, due to the Fenton effect, the Fe sites will inevitably react with the H2O2 formed during the ORR process and resulting in relatively low ORR/ZABs stability15. Thus, achieving a balance between the activity and durability of ORR catalysts is crucial for advancing the practical application of ZABs.
Inspired by nature, cytochrome c oxidase is commonly employed in animal cells to convert oxygen to water, which is composed of heme a3/CuB (heme-copper oxidases)16. This suggests that copper-based materials could serve as potential ORR catalysts. Notably, compared with Fe-N-C catalysts, Cu-N-C catalysts can effectively prevent the occurrence of the Fenton reaction. However, Cu-N-C catalysts featuring the Cu-N4 configuration usually demonstrate inferior oxygen adsorption energy and poor ORR catalytic performance due to the less accessible d-orbitals in the central Cu sites17. How to modify the Cu-N-C sites to ensure their high catalytic activity while maintaining good stability has become a bottleneck in the development of efficient ORR catalysts. In recent years, main-group metal catalysts, including Mg, Ca, Sn, and Sb, have been widely explored as an emerging class of catalysts for hydrosilylation, hydroamination, and electrochemical reactions18,19,20,21. However, due to the lack of the combination of empty and filled host orbitals, the majority of main-group metal-based catalysts encounter challenges in accelerating electron transfer processes during ORR22. Indeed, some main-group metals exhibit a suitable affinity for oxygenated species, particularly Na. It can be reasonably proposed that rationally optimizing the electronic structure of the Cu-N4 active site by introducing oxyphilic Na may improve the conversion of oxygenated species and accelerate the kinetics of the ORR. However, seldom research has been reported to study the activity effects of main-group metals on transition metal catalysts towards ORR/ZABs up to now.
While considerable attention has been directed towards developing highly active catalysts for ORR and rechargeable ZABs, the stability of these catalysts has often been overlooked. Some studies have indicated that the decline in catalyst stability may be attributed to the poisoning of active sites by intermediates or by-products generated during electrochemical reactions23. Based on this, Bao and co-workers synthesized a catalyst with Fe nanoparticles confined inside pea-pod-like carbon nanotubes. They utilized the graphitic wall to protect the active centers, and achieved an improved stability of polymer electrolyte membrane fuel cell in 201324. Since then, the concept of carbon encapsulation has emerged as an important strategy to enhance the stability of metal-based catalysts. Additionally, forming coordination bonds between the metal active sites and the substrate atoms is another effective method to improve the stability of electrocatalysts, which can also enhance the conductivity of the active sites25. In addition to the poisoning and deactivation of active sites, the stability of the carbon support is also crucial for the stability of catalysts. Carbon corrosion at high potentials represents the main cause of the migration, dissolution, and aggregation of metal active centers, and is also an important factor leading to the deterioration of catalyst stability26. Notably, when the C atoms adjacent to the metal active site become less positively charged, it will alleviate the attack by negatively charged oxygen intermediates, thereby reducing the leaching of active sites from the carbon matrix caused by the corrosion of neighboring C atoms. Therefore, modifying the C atoms around the metal active sites with functional groups to make them less positively charged is a rational strategy to prevent carbon corrosion and further enhance the stability of metal active centers. Unfortunately, limited effort has been devoted to mitigating carbon corrosion in M-N-C catalysts. In this regard, it is reasonable to expect that if one can thoroughly analyze the effects of main-group Na-containing functional groups on the Cu-N-C site and synthesize an electrocatalyst with an appropriate coordination environment for Cu atoms, combined with carbon encapsulation and substrate catalytic site fixation strategies, highly active and durable ZABs could be achieved. Nevertheless, it is undoubtedly a great challenge to obtain such a rationally designed Cu/Na dual-atom sites catalysts to meet these multi-type regulations.
Herein, we report a rationally designed dual-metal site catalyst with transition Cu atoms and main-group Na-containing functional groups encapsulated in carbon nanofibers (CuNa-CF) by using a pyrolysis-followed solution blow spinning strategy (SBS). The intrinsic activity of the Cu-N4 site in the CuNa-CF catalyst is boosted by the adjacent Na-containing functional group, which addresses the problem that the traditional Cu-N4-C catalysts have inferior oxygen adsorption energy and are not suitable for serving as ORR catalysts. Thus, our CuNa-CF catalyst exhibits an excellent ORR half-wave potential as high as 0.89 V. Meanwhile, thanks to the neighboring main-group metal-ligand effect, carbon encapsulation, and support anchoring effect, the electrocatalytic stability of the CuNa-CF catalyst is further improved, and it can maintain 97.85% relative current after 48 h of long-term ORR catalytic stability measurement. The ZAB with CuNa-CF catalyst as the air cathode demonstrates remarkable discharge/charge cycle stability for more than 5000 h without noticeable decay at the current density of 1 mA cm−2. Additionally, the CuNa-CF air cathode can even operate stably for over 950 h at a higher current density of 50 mA cm−2. The as-assembled flexible solid-state ZAB and button-type all-solid-state ZAB also show satisfactory cycling stabilities, offering new perspectives for powering wearable devices and microelectronic devices.
Results
Catalyst synthesis and structural characterization
As illustrated in Fig. 1, the CuNa-CF catalyst was synthesized by using SBS technology followed by a temperature-programmed pyrolysis process. Firstly, chlorophyllin sodium copper complex (CSCC) was used as the heteroatom metal precursor, in which, the Cu atom coordinates with four N atoms and there are three Na atoms bonded to three branches through carboxylate bonds. Polyacrylonitrile (PAN) was selected as the carbon precursor due to its high stability of carbon skeleton structure at high temperatures27. By using SBS technology, a PAN nanofiber membrane with uniform CSCC dispersion (CSCC-PAN) was first prepared. After pre-oxidation in air, the linear PAN chains in the CSCC-PAN converted into heat-resistant aromatic ladder structures, and this unique aromatic structure could stabilize CSCC by forming π-π interactions with aromatic-like structures in CSCC28. In order to protect the loading of Na atoms and the coordinating N atoms adjacent to the Cu atom, the annealing conditions during the pyrolysis process were carefully optimized. Finally, a flexible CuNa-CF catalyst membrane was successfully obtained (Supplementary Fig. 1). As shown in Supplementary Fig. 2a, the Brunauer-Emmett-Teller (BET) specific surface area of the as-prepared CuNa-CF catalyst membrane is 597.31 m²/g, and the catalyst mainly possesses micropores and mesopores. Thermogravimetric analysis (TGA) is a powerful technique for determining the formation of catalysts during the pyrolysis of catalyst precursors. The overall weight loss of CSCC was approximately 65%, indicating that apart from the loss caused by physically adsorbed water, CSCC partially decomposed during the pyrolysis process (Supplementary Fig. 2b). The pre-oxidized pure PAN membrane began to decompose at about 300 °C and maintained a mass ratio of 37.6% at 900 °C. During the carbonization process from 300 °C to 900 °C, the pre-oxidized pure PAN membrane underwent significant degradation and weight loss. Non-carbon elements such as H, O, and N in the oxidized PAN fiber were partially removed in the form of small molecular gases or liquid tar, and taking away a large amount of carbon atoms29. The pre-oxidized CSCC-PAN membrane exhibited a TGA curve similar to that of the pre-oxidized PAN membrane, which is attributed to the partial decomposition of CSCC and the partially removal of non-carbon elements generated after the pre-oxidation step. The metal contents of Cu and Na in CuNa-CF catalyst are 1.89 wt% and 0.711 wt% (the atom ratio of Cu to Na is close to 1:1) as determined by inductively coupled plasma mass spectrometry (ICP-MS, Supplementary Table 1), respectively. In order to confirm the significance of Na in the CuNa-CF catalyst, the CuPc-CF catalyst (Na-free) was prepared by using copper phthalocyanine (CuPc) as the metal precursor through the same strategy, and the loading amount of Cu in CuPc-CF catalyst is 2.60 wt% (Supplementary Fig. 3 and Supplementary Table 1).

The dual metal site catalyst with transition Cu atom and main-group Na-containing functional group encapsulated in the flexible carbon nanofibers (CuNa-CF) was synthesized by using pyrolysis followed solution blow spinning technology. Uniformly dispersed chlorophyllin sodium copper complex-polyacrylonitrile (CSCC-PAN) membrane can be scalable produced by using solution blow spinning strategy.
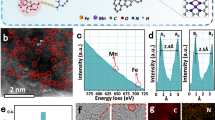
The scanning electron microscope (SEM) and transmission electron microscopy (TEM) were used to examine the morphology of the CuNa-CF catalyst. As shown in Fig. 2a, the fluffy and aligned carbon nanofibers of CuNa-CF intricately interlace with each other, forming an interconnected 3D network structure. The TEM image of CuNa-CF reveals a smooth surface, and the average diameter of the nanofibers is approximately 220 nm, also no obvious metal nanoparticles can be observed (Supplementary Fig. 4a). This agrees well with the results from X-ray diffraction (XRD) analysis, in that there are no obvious diffraction peaks corresponding to Cu or CSCC, and it only displays a broad peak and a tiny peak at 24.1° and 44.2° assigned to the (002) and (101) crystal planes of carbon (Supplementary Fig. 4b), indicating that Cu atoms are uniformly dispersed in carbon fibers without agglomeration during the pyrolysis step30. The dispersion of Cu atoms was directly monitored by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) in sub-angstrom resolution (Fig. 2b). Due to the sensitive Z-contrast of heavy elements, the homogeneously and densely dispersed bright dots (tagged by red circles) corresponding to Cu atoms could be easily observed, revealing that Cu atoms were dispersed at the atomic level in CuNa-CF31. It is worth noting that the Cu atoms are situated on the carbon lattice, further confirming that CSCC enters into the aromatic structure formed by PAN during the pyrolysis process. As shown in Fig. 2c, the AC-HAADF-STEM image and the corresponding EDS elemental mapping images illustrate the well distributions of C, N, O, Cu, and Na elements in CuNa-CF. Notably, the CuPc-CF (Na-free) catalyst synthesized with CuPc as the metal precursor also exhibits a similar Cu single-atom site on the carbon lattice as observed in the CuNa-CF catalyst (Supplementary Fig. 5). To further confirm the neighboring relationship between Cu and Na atoms in the CuNa-CF catalyst, the HADDF-STEM image and the corresponding EELS spectrum of a selected small area (0.5 nm × 0.5 nm) are shown in Fig. 2d–f, in which the signals of Cu and Na atoms were both detected, which provided strong evidence for the presence of neighboring Cu and Na atoms. Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) were also conducted to investigate the structural transformations during the pre-oxidation/pyrolysis processes, and the chemical composition of CuNa-CF and CuPc-CF catalysts. These analyses indicated that the structure and composition of the Cu complex (CSCC or CuPc) were partially retained in their final catalyst (Supplementary Note 1, Supplementary Figs. 6–9, and Supplementary Table 2). After that, the presence of the -COONa functional group in the CuNa-CF catalyst was further revealed by Fourier transform infrared spectroscopy (FT-IR) analysis. As shown in Supplementary Fig. 10, the FT-IR spectra of CSCC and CuNa-CF catalyst both exhibit an obvious peak at 1567 cm−1, which can be attributed to the -COO⁻ asymmetric vibration band32. Combined with the XPS results, the existence of the -COONa functional group in the CuNa-CF catalyst was confirmed.

a Scanning electron microscope image of the CuNa-CF membrane. b Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) image, some of Cu atoms are marked by red circles. c HAADF-STEM image of the CuNa-CF catalyst and the corresponding energy dispersive X-ray spectroscopy elemental mapping images for C, N, O, Cu, and Na. d HAADF-STEM image of the CuNa-CF catalyst and the corresponding scanning transmission electron microscopy-electron energy loss spectroscopy (STEM-EELS) mapping (e) taken from the orange boxed area in (d). f the corresponding EELS spectrum of the 0.5 nm × 0.5 nm selected small area in (e).
To further confirm the electronic structure and coordination environment of Cu atoms in CuNa-CF and CuPc-CF catalysts, X-ray absorption spectroscopy (XAS) measurements were performed33. As shown in the X-ray absorption near-edge structure (XANES), the absorption edges of CuNa-CF and CuPc-CF partially overlap, indicating that the oxidation states of Cu atoms in these two catalysts are similar (Fig. 3a and Supplementary Fig. 11). By comparing with other reference samples, the absorption-edge positions for CuNa-CF and CuPc-CF catalysts are both located between Cu foil and CuO, implying that the Cu atoms in these two catalysts are positively charged with an oxidation state between 0 and +2 (Supplementary Fig. 12). The Fourier transform (FT) extended X-ray absorption fine structure (EXAFS) spectra in the R space verify the absence of Cu aggregation in both CuNa-CF and CuPc-CF catalysts, and the Cu atoms remain in an isolated state due to the absence of Cu-Cu coordination path when compared to Cu foil (Fig. 3b and Supplementary Fig. 13). The FT-EXAFS curves of CuNa-CF and CuPc-CF display main peaks at approximately 1.44 Å and 1.50 Å, corresponding to the Cu-N bond. Also, a tiny peak at ~2.3 Å can be detected in the both two catalysts, which is attributed to the interaction between the Cu atom and the C atoms in the underlying carbon lattice. For the high resolution in R-space and k-space, the wavelet transform (WT)-EXAFS analysis was performed to justify the atomic dispersion of Cu atoms in these two catalysts. As shown in Fig. 3c, the main intensity maximum can be observed at k ~ 3.85 Å−1 in CuNa-CF and CuPc-CF catalysts responding to the Cu-N bonds, which is consistent with CSCC and CuPc complexes. By comparing with the Cu foil and CuO, no Cu-Cu signals can be detected in these two catalysts (white dashed line), confirming that the Cu atoms in CuNa-CF and CuPc-CF are atomically dispersed and coordinated with N atoms. Interestingly, both CuNa-CF and CuPc-CF show a slight intensity maximum at k ~ 6.55 Å−1 (white dot line), which differs from the Cu-C coordination at the second shell in CSCC and CuPc complexes, indicating that the Cu-C coordination in CuNa-CF and CuPc-CF is distinct from that in metal precursors. In order to gain more detailed structural understanding of CuNa-CF and CuPc-CF, the Cu quantitative coordination configurations were constructed, and Fourier-transformed EXAFS fitting curves were calculated (Fig. 3d, e and Supplementary Table 3). Based on the fitting results, the Cu atoms in both CuNa-CF and CuPc-CF catalysts are coordinated with four N atoms, which is consistent with the structures of CSCC and CuPc complexes. Additionally, two Cu-C coordination bonds are present in both catalysts, indicating that the Cu-N4 sites are embedded in the carbon matrix and there have some strong interactions between Cu atoms and the C atoms in the underlying carbon lattice matrix. These findings support the formation of the C2-Cu-N4 site in these two catalysts. Combined with the XPS and FT-IR results, the dominant active sites in CuNa-CF and CuPc-CF catalysts can be postulated as C2-Cu-N4-COONa and C2-Cu-N4, respectively (Supplementary Note 1). In order to get a specific schematic model, density functional theory (DFT) calculations were carried out (Supplementary Fig. 14 and 15)34,35,36. The ortho-C atoms in the underlying carbon lattice matrix served as anchors to stabilize the Cu atom in both CuNa-CF and CuPc-CF catalysts, and the para-C atom of Cu in the carbon matrix was used to anchor the -COONa functional group in the CuNa-CF catalyst because of the existence of the strong coordination interaction (vide infra). The corresponding schematic modularizations of CuPc-CF catalyst and CuNa-CF catalyst are illustrated in the insets of Fig. 3d, e.

a Cu K-edge X-ray absorption near-edge structure spectra. b Fourier transformed extended X-ray absorption fine structure (EXAFS) spectra in R space. c Wavelet transformed EXAFS spectra for Cu foil, CuPc-CF, CuNa-CF, CuO, CuPc, and CSCC. d, e Fourier transformed EXAFS fitting curves for CuPc-CF and CuNa-CF, respectively. Insets in d and e show schematic models of the CuPc-CF catalyst and CuNa-CF catalyst, respectively (Cu atom is green, Na atom is purple, O atoms are pink, N atoms are blue, C atoms in the layer where the Cu site is located are dark gray, and C atoms in the layer below Cu site are light gray).
Electrochemical catalytic properties
To further validate that the distinctive electrocatalytic performance of the C2-Cu-N4 site is inspired by the neighboring Na atom in the CuNa-CF catalyst, the CuPc+NaCl-CF catalyst was prepared by introducing NaCl during the synthesis of the CuPc-CF catalyst. NaCl-CF (Cu-free) and CF (Cu, Na-free) were also synthesized by using the same methods, except for the different metal precursors (Supplementary Note 2, Supplementary Figs. 16–21, and Supplementary Tables 1 and 2). The electrocatalytic ORR performance of the as-prepared catalysts was evaluated in an O2-saturated 0.1 M KOH electrolyte37. The CuNa-CF catalyst exhibited the highest activity with the most positive half-wave potential (E1/2, 0.89 V) among the as-prepared catalysts and the commercial 20 wt% Pt/C catalyst (Fig. 4a). It is worth noting that the CuNa-CF catalyst has a similar Cu coordination environment to that of CuPc-CF in the first coordination shell. The enhanced ORR performance of the CuNa-CF catalyst compared to the CuPc-CF catalyst can be attributed to the presence of Na-containing functional groups adjacent to C2-Cu-N4 sites in the CuNa-CF catalyst, which modulate the ORR catalytic activity of the C2-Cu-N4 sites. The half-wave potential of the CuPc+NaCl-CF catalyst is between that of CuNa-CF and CuPc-CF, providing further experimental evidence for the effect of the neighboring Na-containing functional group and confirming the significant role of the C2-Cu-N4-COONa site in the ORR process. During the synthesis of the CuPc+NaCl-CF catalyst, NaCl was randomly distributed in the PAN fibers, resulting in an uncontrolled coordination relationship between the Cu and Na atoms, which led to the difference in catalytic performance between the CuNa-CF and CuPc+NaCl-CF catalysts. Specifically, the ORR polarization curve of the CuNa-CF catalyst also exhibits an onset potential of 1.04 V, which is 20 mV and 30 mV higher than those of the CuPc+NaCl-CF and Pt/C catalysts, respectively, suggesting that less energy is required to generate ORR intermediates and drive the ORR on the CuNa-CF electrode. Notably, CuNa-CF catalyst displayed the highest kinetic current density (Jk = 10.64 mA cm−2) at 0.85 V, surpassing other studied catalysts and the commercial Pt/C catalyst (Fig. 4b, and the detailed data are listed in Supplementary Table 4). Moreover, the CuNa-CF catalyst also exhibited a high kinetic current density of 4.51 mA cm−2 at its half-wave potential (0.89 V), representing its high intrinsic ORR electrocatalytic activity (Supplementary Fig. 22). The Tafel slopes calculated from polarization curves are commonly used to analyze the ORR kinetics on different catalysts. The CuNa-CF catalyst displayed a lower Tafel slope compared to other catalysts, indicating reduced oxygen binding energy and accelerated ORR kinetics at the C2-Cu-N4-COONa site (Supplementary Fig. 23 and Supplementary Table 5). To further understand the catalytic processes of the CuNa-CF catalyst, linear sweep voltammetry (LSV) tests on a rotating disk at varying speeds from 400 to 1600 rpm were carried out (Fig. 4c). The limiting current density increased with the promotion of rotating speed, confirming that the ORR performance of CuNa-CF is highly dependent on the rate of oxygen diffusion, which conforms to the first-order kinetics model. By using the Koutecky-Levich equation, the electron transfer numbers at various potentials were calculated to be ~ 4 (Fig. 4d), indicating that the ORR on the CuNa-CF catalyst follows a four-electron transfer pathway from O2 to OH−. As shown in Fig. 4e, a nearly complete four-electron transfer pathway for the ORR was also observed on both the CuNa-CF and Pt/C catalysts by operating rotating ring disk electrode measurement over a wide potential range, which is consistent with the findings calculated by Koutecky-Levich equation38. Notably, the yield of H2O2 with the CuNa-CF catalyst is less than 5% in a wide potential range from 0.2 V to 0.9 V, certifying that the ORR followed a highly selective and efficient four-electron transfer pathway.

a Oxygen reduction reaction polarization curves without iR correction for CuNa-CF, CuPc+NaCl-CF, CuPc-CF, NaCl-CF, CF, and commercial 20% Pt/C catalysts in O2-saturated 0.1 M KOH solution at 1600 rpm with a scan rate of 5 mV s−1 (the resistance of the solution was 40 ± 5 Ω). b Comparison of onset potential (Eonset), half-wave potential (E1/2), and kinetic current density (Jk, at 0.85 V, V versus RHE) for different catalysts. c Linear sweep voltammetry (LSV) curves of the CuNa-CF catalyst at different rotation speeds (400, 625, 900, 1225, and 1600 rpm). d Koutecky-Levich plots and corresponding electron transfer numbers for the CuNa-CF catalyst at different potentials (0.2 V, 0.3 V, 0.4 V, 0.5 V, 0.6 V, and 0.7 V, V versus RHE). e H2O2 yield and electron transfer number of the CuNa-CF catalyst and Pt/C catalyst measured by the rotating ring-disk electrode (RRDE). f Long-term electrochemical stability of the CuNa-CF catalyst and Pt/C catalyst.
Apart from the ORR catalytic selectivity and activity, methanol tolerance and catalytic durability are also important in evaluating ORR catalysts39. As displayed in Supplementary Fig. 24, a negligible attenuation in current density was recorded of the CuNa-CF catalyst during the methanol tolerance ability tests. In sharp contrast, the current density of Pt/C catalyst suffered from a sharp drop, highlighting the outstanding methanol tolerance ability of CuNa-CF. During the long-term stability test, the ORR current density of the commercial Pt/C catalyst decreased rapidly. In contrast, no significant decrement was observed with the CuNa-CF catalyst, and its final relative current remained at 97.85% even after 48 h of the ORR electrocatalytic stability test (Fig. 4f). As compared, the CuPc-CF (Na-free) catalyst showed poor ORR stability with only 63.27% relative current retained after a 10-hour chronoamperometry test (Supplementary Fig. 25). The HAADF-STEM image reveals the aggregation of the Cu atoms after the long-term stability test (Supplementary Fig. 26). Based on the above results, it is a promising way to enhance the stability of atomically dispersed Cu catalysts by constructing a Na-containing functional group adjacent to the C2-Cu-N4 active site, which could prevent the corrosion of carbon atoms around the active site by reducing the positive charge of the carbon atoms near the copper active site, and further avoid the migration, dissolution, and aggregation of the metal active center (Supplementary Note 3 and Supplementary Figs. 27 and 28). In addition, the Cu-N4-COONa sites embedded in the carbon matrix and the strong Cu-C coordination interaction can stabilize the Cu/Na site and enhance its ORR stability. Due to the rational design of the catalytic site, the CuNa-CF catalyst is comparable to or even surpasses other reported precious metal/non-precious metal catalysts (Supplementary Table 6). To evaluate the practical application potential of this catalyst in batteries, we also examined its oxygen evolution reaction (OER) performance (including the OER catalytic activity and stability of the as-prepared catalysts). Surprisingly, this rationally designed catalytic site in the CuNa-CF catalyst also has significant facilitating effect on OER (Supplementary Note 4, Supplementary Figs. 29–32, and Supplementary Table 7).
ZABs performance and durability
Encouraged by the remarkable ORR/OER electrocatalytic activity and stability of the CuNa-CF catalyst, we assembled various types of Zn-air batteries by using CuNa-CF as the air cathode to demonstrate its potential applications in practical energy devices. Supplementary Figs. 33 and 34 reveal that the CuNa-CF air cathode liquid-state ZAB delivers a high specific capacity of 621.55 mAh g−1, corresponding to an energy density of 715.15 Wh kg−1, which surpasses that of the battery with the commercial Pt/C catalyst as the air cathode (specific capacity of 612.23 mAh g−1 and energy density of 645.73 Wh kg−1). The CuNa-CF catalyst also achieved a peak power density of 264.18 mW cm−2, which is obviously higher than that of the Pt/C cathode ZAB (Supplementary Fig. 35), illustrating its considerable potential as an alternative to precious metal cathodes. Besides, the long-term discharge/charge cycle stability of ZAB is regarded as a critical judging indicator for practical applications. For liquid-state ZABs, the cycling stabilities of CuNa-CF and Pt/C + RuO2 air cathodes were evaluated by continuous discharge and charge tests at a constant current density (Supplementary Fig. 36). As shown in Fig. 5a, the CuNa-CF cathode ZAB demonstrates remarkable stability for more than 5000 h with negligible changes in discharge/charge voltage at a constant current density of 1 mA cm−2. Such high stability has rarely been reported to date (Supplementary Table 8). After over 5000 h of ZAB operational stability testing, the CuNa-CF catalyst was characterized again (Supplementary Fig. 37a), and HAADF-STEM analysis confirmed that the Cu atoms remained homogeneously and densely dispersed in the carbon fibers without forming nanoclusters. Furthermore, in order to reveal the presence of neighboring Cu and Na atoms in the CuNa-CF catalyst after long-term discharge/charge cycle stability testing, HAADF-STEM imaging combined with corresponding EELS spectrum analysis were further conducted. A selection of 0.5 nm × 0.5 nm small area HAADF-STEM image and the corresponding EELS spectrum are shown in Supplementary Fig. 37b–d, the signals for Cu and Na atoms were both detected and provided strong evidence for the presence of neighboring Cu and Na atoms. Surprisingly, the ZAB with the CuNa-CF air cathode can also tolerate high current density discharge/charge cycles, and deliver a smooth discharge and charge cycling curve for more than 1200 h at the current density of 10 mA cm–2, indicating its outstanding rechargeable ability, which undoubtedly exceeds the performance of the commercial Pt/C + RuO2 cathode ZAB (Supplementary Figs. 38 and 39). Inspired by the high discharge/charge cycle stability of ZABs with the CuNa-CF air cathode, we further conducted the discharge/charge cycle stability test of ZAB at a higher current density of 50 mA cm−2. The catalyst loading was optimized first and the initial discharge voltage of ZAB reached its optimal value with a CuNa-CF catalyst loading of 1.0 mg cm−2 (Supplementary Fig. 40). Then, a ZAB with this optimal CuNa-CF catalyst loading on the air cathode was assembled and its discharge/charge cycle stability at a high current density of 50 mA cm−2 was tested. As shown in Supplementary Fig. 41, the ZAB with the CuNa-CF air cathode is still able to exhibit good discharge/charge cycle stability for over 950 h even at a high constant current density of 50 mA cm−2, further confirming the high electrocatalytic stability of the CuNa-CF catalyst and its application potential in rechargeable ZABs. As the CuNa-CF catalyst is a flexible membrane material, it can be directly used as the air cathode of both flexible solid-state ZAB and button-type all-solid-state ZAB. The schematic illustration of the flexible solid-state ZAB is shown in Fig. 5b, in which the CuNa-CF membrane is employed as the air cathode and a home-made flexible hydrogel serves as the electrolyte. During the stability test at a constant current density of 1 mA cm–2, although the flexible solid-state ZAB experienced folding and revert flattening states, it presented a highly stable discharge and charge profile (Fig. 5c), indicating its practical application potential in flexible energy storage devices. In response to the development of small or micro appliances and the growing demand for button-type batteries, a button-type all-solid-state ZAB with CuNa-CF membrane air cathode was also assembled (Fig. 5d). It is gratifying that the button-type all-solid-state ZAB held on a stable running for more than 1000 min at a constant current density of 1 mA cm–2 (Fig. 5e). It was also capable of powering a series of LED lights on a wristband using just two series-connected batteries (Supplementary Fig. 42), indicating the application ability of CuNa-CF membrane in miniature electrical devices.

a Discharge/charge cycling curves of CuNa-CF air cathode liquid-state zinc-air battery (ZAB). b Schematic illustration of the flexible solid-state ZAB. c Cycling stability of flexible solid-state ZAB with CuNa-CF membrane as the air cathode, and the insets are digital photos of the flexible solid-state ZAB in various states (flat/bent/revert flat). d Simplified schematic of the button-type all-solid-state ZAB. e Operating stability of button-type all-solid-state ZAB using CuNa-CF membrane as the air cathode.
Theoretical insights into the activity and stability improvement
To gain a deeper insight into the enhanced ORR electrocatalytic performance of the CuNa-CF catalyst, density functional theory (DFT) calculations were carried out40. Based on the HAADF-STEM, XAS and XPS results, the main active site in the CuNa-CF catalyst is identified as the Cu-N4-COONa site, also the ortho-C atoms in the underlying carbon lattice matrix serve as anchors for stabilizing the Cu active site. At first, the interactions between the Cu-N4-COONa active sites and the graphite adsorbates (C) were studied by analyzing the projected density of state (PDOS, Supplementary Fig. 43), and we found that the graphene substrate barely influences the electronic structure of the Cu-N4-COONa site, except that the conductivity of the Cu-N4-COONa sites is significantly enhanced thanks to the π–π stacking. Due to the longer bond length between Cu and C (already exceeds 3 angstroms from EXAFS and DFT results) for CuNa-CF and CuPc-CF, the impact on the electronic structure for axially adjacent C atoms is minimal. Further PDOS analysis reveals that the combination with graphene has almost no effect on the position of the d-band center of the Cu 3d orbitals (Supplementary Fig. 44). Thus, we primarily focus on the role of major active Cu-N4-COONa sites in CuNa-CF catalyst, and consider different atomistic configurations in further DFT calculations. As shown in Supplementary Figs. 45 and 46, with -COONa adsorption on the S1 site of Cu-N4 monolayer, the Cu-N4-COONa shows the lowest total energy and is regarded as the most stable structure. After modification with the neighboring -COONa functional group, charge transfer and more charge accumulation can be observed at the Cu-N4 site (Fig. 6a–c), forming a unique electronic structure. As shown in Supplementary Fig. 47, the introduction of the -COONa functional group increases the distribution of electronic states near the Fermi level in Cu-N4 site, and thus enhancing its conductivity. As a result, the introduction of the coordination environment (-COONa functional group) could modify the electronic states of the Cu-N4 active site, which in turn modulates its conductivity and further enhances its ORR electrocatalytic activity41,42,43,44. To further analyze the effect of the coordination environment on the catalytic activity, we further investigated the ORR catalytic process on the pure Cu-N4 site and the Cu-N4 site which modified with the -COONa functional group (Supplementary data 1 and 2). For 4e− transfer ORR process, the metal active sites will adsorb oxygen molecules in the first step and the nearly positive adsorption energies of O2 on Cu-N4 sites (−0.08 eV, Fig. 6d) reveal its inertness. By contrast, the much more negative adsorption energies after the –COONa coordination (−0.58 eV) indicate that the Cu-N4-COONa site can spontaneously activate O2 molecules and ensure the subsequent ORR process. Moreover, the ORR free energy diagrams for pure Cu-N4 and Cu-N4-COONa are investigated in Fig. 6e, f. Due to the weak protonation ability of O2 (*O2 → *OOH step), the Cu-N4 site shows a high ORR overpotential of 1.21 V. After the -COONa modification, the ORR overpotential of Cu site in the Cu-N4-COONa configuration declines to 0.83 V, suggesting that the adjacent ligand effect of -COONa is beneficial for optimizing the adsorption energies of ORR intermediates and improving the catalytic activity of Cu sites, in good agreement with our experimental results. Subsequently, according to the Bader charge transfer results (Fig. 6g), the Cu-N4-COONa site (0.57 e) can accept more charge from *OOH than the pure Cu-N4 site (0.40 e), indicating the strong anchoring of OOH* on the Cu-N4-COONa site. The PDOS analysis also corroborates that the Cu-3d orbital are more overlapped with *OOH intermediates after the -COONa functional group modification, demonstrating the better activation for O2 molecules on the Cu-N4-COONa site. In order to quantify the bonding strength in electronic state, the crystal orbital Hamilton population (COHP) analysis was conducted (Fig. 6h), in which the interaction between Cu and O atoms of adsorbed *OOH species is described by the product of their corresponding Hamiltonian matrix element and the densities of states matrix. From the COHP analysis, it can be seen that the absolute values of integrated COHP of Cu–O bonds from adsorbed *OOH hybridization up to the Fermi level on Cu-N4-COONa site is larger than that of the pure Cu-N4 site, suggesting the stronger binding strength, and thus also further optimize the binding strength of ORR intermediates and guaranteeing better electrochemical catalytic performance. After that, considering the potential ion exchange of the Na ions in the CuNa-CF catalyst and the K ions in the electrolyte, ICP-MS and DFT calculations were further conducted. It was found that even the Cu-N4 site modified with the -COOK functional group, it can also exhibit an improvement in its ORR catalytic activity (Supplementary Note 5, Supplementary Fig. 48, and Supplementary data 3). Combining the experimental and theoretical results, we believe this provides an efficient strategy and successful case for enhancing electrocatalytic activity through coordination with main-group metals containing functional groups.

Atomistic structures of (a) Cu-N4 and (b) Cu-N4-COONa configurations. c Charge density differences of Cu-N4-COONa, the isosurface levels are set to 0.002 e Å−3, where charge depletion and accumulation were depicted by cyan and yellow, respectively. It can be seen that there is charge accumulation on the C around COONa. d Adsorption energies for O2 on Cu-N4 and Cu-N4-COONa sites. The calculated Gibbs free energy evolution diagrams for ORR through a 4e− pathway on active sites of (e) Cu-N4 and (f) Cu-N4-COONa under electrode potential of U = 0 V, where the elementary reaction in red dotted line represents the potential limiting step. g Projected density of state (PDOS) analysis of Cu-3d orbital with OOH intermediates on Cu-N4 and Cu-N4-COONa. Charge density differences and Bader charge transfer are illustrated inside. h The crystal orbital Hamilton population (COHP) analysis of Cu active sites and O atoms of OOH intermediates, the integrated crystal orbital Hamilton population (ICOHP) values (in eV per bond) are listed.
Discussion
Balancing the activity and stability of oxygen electrocatalysts is crucial for their practical applications, which has driven the exploration of non-Fe-based atomically dispersed M-N-C catalysts. Drawing inspiration from nature, we have synthesized a kind of atomically dispersed Cu-N-C catalyst via a facile solution blow spinning followed by pyrolysis strategy. To enhance the intrinsic activity of the Cu-N-C catalyst, a Na-containing functional group was introduced in proximity to the Cu-N4 active centers. The results revealed that the introduction of Na-containing functional group facilitated the adsorption/activation of O2 at the Cu-N4 site, thereby enhancing its intrinsic activity. Simultaneously, the introduction of Na-containing functional group can mitigate the carbon corrosion and stabilize the Cu-N4 sites, thereby improving its stability. Consequently, the obtained CuNa-CF catalyst exhibited remarkable ORR performance, particularly demonstrating a discharge/charge cycle stability of more than 5000 h when used as the air cathode catalyst of ZAB. We believe that the concept and strategy of modulating Cu-N-C catalysts with the main-group element functional groups proposed in this work not only expand and enrich the research of main-group elements in electrocatalysis applications, but also provide guidance for the future design of non-Fe-based M-N-C oxygen electrocatalysts that balance activity and stability. Furthermore, it offers a reliable methodology for the development of high-performance non-precious metal-based air cathode catalysts for ZABs.
Methods
Materials
Polyacrylonitrile (PAN, M.W.,150,000, Sigma Aldrich Co.), N,N-Dimethylformamide (DMF, 99.8%, Damas Beta Co., Ltd.), Chlorophyllin Sodium Copper Salt (95%, Meryer Shanghai Chemical Technology Co., Ltd.), Copper (II) Phthalocyanine (CuPc, 95%, Alfa Aesar Co., Ltd.), Sodium Chloride (NaCl, 99.5% Sigma Aldrich Co.), Methanol (99.8%, Sinopharm Chemical Reagent Co. Ltd), Potassium Hydroxide (KOH, 85%, General-Reagent Co., Ltd.), Zinc Acetate (Zn(CH3COO)2, 99.99%, Aladdin), Polyvinyl Alcohol (PVA, 99%, Shanghai Aladdin Bio-Chem Technology Co., Ltd.), Commercial Pt/C (Pt, 20 wt%, Suzhou Sinero Technology Co., Ltd.), Ruthenium (IV) oxide (RuO2, 99.9% trace metals basis, Sigma-Aldrich), Nafion D-521 Dispersion (5%, Alda Aesar), and Isopropanol (99.7%, Sinopharm Chemical Reagent Co., Ltd.) were used as received. The ultrapure water was obtained from the Milli-Q System.
Catalyst synthesis
In a typical synthesis of the CuNa-CF catalyst, 0.1709 g of chlorophyllin sodium copper salt was added to 10.0 mL of DMF solution under vigorous agitation. The mixture was ultrasonicated for 30 min and stirred for 24 h to form a homogeneous solution. Then, 1.00 g of PAN was added to the above solution under vigorous agitation and stirred for 48 h to obtain a homogeneous and viscous solution. Subsequently, the solution was transferred into a 3 mL syringe with a 30 G needle and delivered at a constant flow rate of 1.00 mL/h to the surrounding high-speed air jet flow (45.00 L/min) for solution blow spinning process. The distance between the tip of nozzle and the non-woven collector was 35 cm. The obtained chlorophyllin sodium copper-PAN membrane was dried in the vacuum oven at 60 °C overnight. Then, the chlorophyllin sodium copper-PAN membrane was pre-oxidized in air at 260 °C for 3 h with a heating rate of 1 °C/min. Finally, the pre-oxidized chlorophyllin sodium copper-PAN membrane was annealed at 900 °C for 2 h under a gas flow of 100 sccm Ar with a heating rate of 5 °C/min. After cooling to 25 °C, the CuNa-CF catalyst was obtained. By replacing the chlorophyll sodium precursor with copper phthalocyanine, copper phthalocyanine and sodium chloride, sodium chloride, and no metal precursor, we also synthesized CuPc-CF catalyst, CuPc+NaCl-CF catalyst, NaCl-CF catalyst, CF catalyst, respectively.
Materials characterization
Transmission electron microscopy (TEM) images of the as-synthesized catalysts were obtained using a JEM-2100F. Field emission scanning electron microscope (FE-SEM) images were acquired with a JEOL JSM-7001F. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and energy dispersive X-ray spectroscopy (EDS) mapping images were attained on a FEI Titan 80–300 (acceleration voltage, 300 kV). Powder X-ray diffraction (XRD) patterns were collected with a D/max-2500/PC powder diffractometer using monochromatized Cu-Kα radiation (λ = 0.15418 nm, 9 kW). X-ray photoelectron spectroscopy (XPS) was performed with a Thermo Fisher ESCALAB 250Xi. Quantitative analysis of the metal loading in different catalysts was conducted using an Agilent ICP-MS 8800. The Raman spectra were collected with a Horiba LabRAM HR Evolution Raman microscope using 532 nm laser excitation. The specific surface area and porosity were measured by isothermal nitrogen adsorption-desorption analysis on an Autosorb-iQ1-MP. The specific surface areas were calculated using the multi-point Brunaure-Emmert-Teller (BET) method. The thermogravimetric analysis (TGA) was performed on a NETZSCH-TGA-X70 over the temperature range from 25 °C to 900 °C in an argon atmosphere. Fourier transform infrared spectroscopy (FT-IR) spectra were recorded using a Spotlight 400 Perkin-Elmer infrared spectrum microscope. The absorption spectra of the Cu K-edge were measured by 1W1B beamline of Beijing Synchrotron Radiation Facility (BSRF, China) in fluorescence mode. The X-ray absorption spectroscopy (XAS) data were analyzed using Athena software and the Fourier-transformed EXAFS fitting curves in R-space were obtained using Artemis module of IFEFFIT software packages. All spectra were collected under ambient conditions.
Electrochemical measurements
1 mg of the as-prepared catalyst was dispersed in 200 μL of a mixture solution containing 95 μL of isopropanol, 95 μL of ultrapure water and 10 μL of Nafion solution. The mixture was ultrasonicated for 3 h to form a homogeneous catalyst ink. Then 8 μL of the catalyst ink was dropped onto the surface of a polished glassy carbon rotating disk electrode (RDE, Pine Research Instrumentation) or a rotating ring-disk electrode (RRDE, Pine Research Instrumentation) followed by drying in the air. The resulting electrode was used as the working electrode for the oxygen reduction reaction (ORR) with a catalyst loading of approximately 0.2 mg cm−2. For oxygen evolution reaction (OER), 10 μL of the catalyst ink was loaded onto the surface of the carbon cloth (0.25 cm2) and used as the working electrode, with a catalyst loading of 0.2 mg cm−2. The commercial Pt/C catalyst and RuO2 catalyst-modified electrodes were prepared in the same way.
All electrochemical tests were conducted in a conventional three-electrode system at a temperature of 25 ± 1 °C by employing a CHI 760E electrochemical station (Shanghai Chenhua Instruments Company) equipped with a Pine Modulated Speed Rotator. A graphite rod (Shanghai Chenhua Instruments Company) and an Ag/AgCl electrode (3.5 M KCl, Shanghai Chenhua Instruments Company) were used as the counter electrode and reference electrode, respectively. The obtained potentials were normalized to the reversible hydrogen electrode (RHE) according to Nernst equation (\({E}_{{RHE}}={E}_{{Ag}/{AgCl}}+0.0591\times {pH}+0.205\)). For ORR, a rotating disk electrode or rotating ring-disk electrode coated with the as-prepared catalyst were served as the working electrodes. The electrolyte was 0.1 M KOH solution (pH = 13.0 ± 0.2), and the resistance of the solution was 40 ± 5 Ω. Prior to measurement, the O2 was purged in the electrolyte for about 30 min to keep saturated O2 in the electrolyte. The linear sweep voltammetry (LSV) experiments were performed with a scan rate of 5 mV s−1 at various disk rotation speeds of 400, 625, 900, 1225, and 1600 rpm. The number of electrons transferred (n) at different potentials were calculated by using Koutecky-Levich Eqs. (1)-(3):
Here J is the measured current density (mA cm−2), JL and JK are the diffusion-limiting current density (mA cm−2) and kinetic current density (mA cm−2), ɷ is the angular velocity of the disk (rpm), n represents the electron transfer number, F is the Faraday constant (96485 C mol−1), Co is the saturated O2 concentration (1.2×10−6 mol cm−3), Do is the diffusion coefficient of O2 in the electrolyte (1.9×10−5 cm2 s−1), V is the kinematic viscosity (0.01 cm2 s−1). RRDE test is the most effective way to test and calculate the electron transfer number and the yield of hydrogen peroxide (Eqs. 4–5):
Here Id is the disk current and Ir is the ring current, N is the H2O2 collection coefficient at the ring. Long term stability tests were carried out by measuring the current changes under an operation potential of 0.66 V (vs. RHE) for 48 h.
For OER, the electrolyte was 1.0 M KOH solution (pH = 13.8 ± 0.2), and the resistance of the solution was 6 ± 1 Ω. The OER performance of the as-prepared catalysts were obtained from LSV with a scan rate of 10 mV s−1. The long-term stability tests were carried out by conducting chronopotentiometry (V-t) test for 48 h under a constant current density of 10 mA cm−2 for each catalyst.
Rechargeable liquid-state zinc-air battery tests
A homemade liquid-state zinc-air cell was assembled to evaluate the catalytic activity of the as-prepared catalyst in practical applications. A polished zinc foil (1.5 cm×1 cm) was used as the anode, a gas diffusion layer coated with the catalyst (1 cm×1 cm) served as the air cathode. The electrolyte was a mixture solution of 6.0 M KOH and 0.2 M zinc acetate. The specific capacity and energy density were collected by galvanostatic discharge testing at a current density of 10 mA cm−2 until the zinc foil in contact with the electrolyte was fully reacted, and then normalized to the consumed mass of the Zn foil (Eq. 6).
Here, Idischarge is the discharge current density (10 mA cm−2), T is the time when reaction stops, mZn1 and mZn2 are the weights of the Zn foil before and after discharge process, respectively. The discharge/charge cycle stability tests were held at constant current densities of 1 mA cm−2, 10 mA cm−2 and 50 mA cm−2 (catalyst loading: 0.2 mg cm−2 for 1 mA cm−2 and 10 mA cm−2, 1.0 mg cm−2 for 50 mA cm−2). All tests were operated in ambient environment with the LAND testing system (LAND Electronics Ltd.).
Rechargeable flexible solid-state zinc-air battery test
For the flexible solid-state zinc-air battery, a PVA-KOH-Zn(CH3COO)2 hydrogel polymer was used as the electrolyte. The hydrogel was prepared by dissolving 2.00 g of polyvinyl alcohol (PVA) in 20 ml of deionized water and stirring for 2 h at 90 °C. Then, 2 ml of 6.0 M KOH solution with 0.2 M Zn(CH3COO)2 was added to the above mixture and stirred for 0.5 h to form a homogeneous solution. Finally, the gel was poured onto a plate and the PVA-KOH-Zn(CH3COO)2 hydrogel polymer was obtained after cooling in a freezer. The catalyst membrane (5×10 mm) and a polished zinc foil were used as the air cathode and anode, respectively. The battery testing method followed the procedure used for the liquid-state zinc-air battery (the stability test was hold at a constant current density of 1 mA cm−2).
Rechargeable button-type all-solid-state zinc-air battery test
A button-type all-solid-state zinc-air battery is also composed of an air cathode (catalyst membrane), a zinc foil anode (0.1 mm thickness) and an electrolyte hydrogel polymer (PVA-KOH-Zn(CH3COO)2 hydrogel polymer prepared using the same method as for the flexible solid-state zinc-air battery). The battery testing method followed the procedure used for the liquid-state zinc-air battery (the stability test was hold at a constant current density of 1 mA cm−2).
Theoretical calculation method
All of the calculations were performed by means of spin polarized density functional theory (DFT) methods using the Vienna Ab initio Simulation Package (VASP)45. The projector augmented wave (PAW) method was adopted to describe electron-ion interaction45. The Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional within a generalized gradient approximation (GGA) was employed, while a 520 eV cut off energy for the plane-wave basis set was used for the valence electrons. Moreover, the DFT-D3 scheme of dispersion correction was used to describe the van der Waals (vdW) interactions in molecule adsorption46. The self-consistent filed (SCF) calculations were performed with an energy and force convergence criterion of 10−5 eV and 0.02 eV Å−1, respectively. To avoid the interactions between two adjacent periodic images, the vacuum thickness was set to be 15 Å. The atomic structures were analyzed by using the VESTA code47. The free energy correction in ORR was obtained similarly by including the ZPE and entropic contributions from vibrational degrees of freedom calculated with the substrate fixed.
(1) Computational details of the Gibbs free energy for 4e− ORR. The reaction steps considered for the electrochemical reduction of 4e− ORR under base condition are generally reported to proceed as follow (Eqs. 7−11):
Here * represents either the catalytic active sites of vacant surface, or intermediate species adsorbed on the active sites. By considering the zero-point energy (ZPE) and entropy corrections, the Gibbs free energy for ORR can be calculated with the following Eq. 1248:
Here ΔE indicates the adsorption energy difference for each species adsorbed on the catalyst, ΔZPE and ΔS are the zone point energy and entropy difference between the adsorbed state and corresponding free-standing state, respectively. The calculated values of ΔZPE and ΔS are listed as follows:
Species | Pressure (bar) | Temperature (K) | Energy (eV) | ZPE (eV) | TS (eV) |
|---|---|---|---|---|---|
H2O | 0.035 | 298.15 | −14.23 | 0.57 | 0.58 |
H2 | 1 | 298.15 | −6.77 | 0.27 | 0.40 |
O2 | 1 | 298.15 | −9.86 |
The zero point energy (ZPE) is calculated by Eq. 13 49
Here \(\hslash\) is the reduced Planck constant and \({\omega }_{j}\) is the frequency associated with the harmonic mode in the gamma point. The same values for the adsorbed species are used throughout the entire text, as vibrational frequencies have been found to depend much less on the catalyst surface. The contributions (TS) values of the free molecules are taken from the NIST database50, For water, the entropy is calculated at 0.035 bar through S=S0 + kBTln(P/P0) to derive the chemical potential of liquid water, because at this pressure gas-phase water is in equilibrium with liquid water at 298.15 K.
(2) Electronic Structure Analysis. Crystal orbital Hamilton population (COHP) analysis was performed with the LOBSTER 3.2.0 package, which reconstructs the orbital-resolved wave functions via projection of the delocalized PAW to localized atomic-like basis sets51,52. Basis sets given by Koga with additional functions fitted to atomic VASP-PBE wave functions were used53,54. Atomic charges were computed using the atom-in-molecule (AIM) scheme proposed by Bader55. The electron density differences were evaluated using the formula Δρ = ρ(AB) - ρ(A) - ρ(B), then analyzed by using the VESTA code.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Information. Source data are provided with this paper.
References
Wussow, M. et al. Exploring the potential of non-residential solar to tackle energy injustice. Nat. Energy 9, 654–663 (2024).
Ueckerdt, F. et al. Potential and risks of hydrogen-based e-fuels in climate change mitigation. Nat. Clim. Chang. 11, 384–393 (2021).
Kondori, A. et al. A room temperature rechargeable Li2O-based lithium-air battery enabled by a solid electrolyte. Science 379, 499–505 (2023).
Wang, Y. et al. Accelerated deprotonation with a hydroxy-silicon alkali solid for rechargeable zinc-air batteries. Nat. Commun. 14, 6968 (2023).
Zhang, W. et al. Two-electron redox chemistry via single-atom catalyst for reversible zinc-air batteries. Nat. Sustain. 7, 463–473 (2024).
Jiang, Z. et al. Interfacial assembly of binary atomic metal-Nx sites for high-performance energy devices. Nat. Commun. 14, 1822 (2023).
Lei, X. et al. High-entropy single-atom activated carbon catalysts for sustainable oxygen electrocatalysis. Nat. Sustain. 6, 816–826 (2023).
Yuan, Y. et al. Zirconium nitride catalysts surpass platinum for oxygen reduction. Nat. Mater. 19, 282–286 (2020).
Liu, M. et al. In situ modulating coordination fields of single-atom cobalt catalyst for enhanced oxygen reduction reaction. Nat. Commun. 15, 1675 (2024).
Chen, Y., Zheng, D. J., Xu, Z. J. & Shao-Horn, Y. Best practices for oxygen electrocatalysis. Nat. Sustain. 7, 371–374 (2024).
Wu, Z. Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Zhou, Y. et al. Boosting oxygen electrocatalytic activity of Fe-N-C catalysts by phosphorus incorporation. J. Am. Chem. Soc. 145, 3647–3655 (2023).
Adabi, H. et al. High-performing commercial Fe-N-C cathode electrocatalyst for anion-exchange membrane fuel cells. Nat. Energy 6, 834–843 (2021).
Zhang, P. et al. Inter-site structural heterogeneity induction of single atom Fe catalysts for robust oxygen reduction. Nat. Commun. 15, 2062 (2024).
Guo, Y. et al. Stabilizing Fe single atom catalysts by implanting Cr atomic clusters to boost oxygen reduction reaction. Appl. Catal. B 344, 123679 (2024).
Wang, Z. et al. Dual‐atomic‐site catalysts for molecular oxygen activation in heterogeneous thermo‐/electro‐catalysis. Angew. Chem. Int. Ed, 135, e202301483 (2023).
Wang, J. et al. Rational design of the first and second coordination spheres for copper single-atom catalyst to boost highly efficient oxygen reduction. Appl. Surf. Sci. 605, 154832 (2022).
Kong, R. & Crimmin, M. Activation and functionalization of C-C σ bonds of alkylidene cyclopropanes at main group centers. J. Am. Chem. Soc. 142, 11967–11971 (2020).
Färber, C. et al. Teaming up main group metals with metallic iron to boost hydrogenation catalysis. Nat. Commun. 13, 3210 (2022).
Jiang, J. et al. Alloyed Pt-Sn nanoparticles on hierarchical nitrogen-doped carbon nanocages for advanced glycerol electrooxidation. Nano Res. 17, 4055–4061 (2024).
Fazekas, E. et al. Main group metal polymerisation catalysts. Chem. Soc. Rev. 51, 8793–8814 (2022).
Liu, S. et al. Turning main-group element magnesium into a highly active electrocatalyst for oxygen reduction reaction. Nat. Commun. 11, 938 (2020).
Choi, E. Y. et al. Cobalt nanoparticles-encapsulated holey nitrogen-doped carbon nanotubes for stable and efficient oxygen reduction and evolution reactions in rechargeable Zn-air batteries. Appl. Catal. B 325, 122386 (2023).
Deng, D. et al. Iron encapsulated within pod‐like carbon nanotubes for oxygen reduction reaction. Angew. Chem. Int. Ed. 52, 371–375 (2013).
Li, Y. et al. Dual-atom Ag2/graphene catalyst for efficient electroreduction of CO2 to CO. Appl. Catal. B 268, 118747 (2020).
Yan, Z. et al. Nitrogen-doped bimetallic carbide-graphite composite as highly active and extremely stable electrocatalyst for oxygen reduction reaction in alkaline media. Adv. Funct. Mater. 32, 2204031 (2022).
Li, Z. et al. Constructing a novel carbon skeleton to anchor Sn/SnO2 nanodots for flexible supercapacitor with excellent rate capability. Carbon 194, 197–206 (2022).
Kang, S. H. et al. Polyacrylonitrile/phosphazene composite-based heat-resistant and flame-retardant separators for safe lithium-ion batteries. ACS Appl. Energy Mater. 5, 2452–2461 (2022).
Simitzis, J. C. & Georgiou, P. C. Functional group changes of polyacrylonitrile fibres during their oxidative, carbonization and electrochemical treatment. J. Mater. Sci. 50, 4547–4564 (2015).
Wu, Q., Li, W., Liu, S. & Jin, C. Hydrothermal synthesis of N-doped spherical carbon from carboxymethylcellulose for CO2 capture. Appl. Surf. Sci. 369, 101–107 (2016).
Chen, C. et al. Engineering molecular heterostructured catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 145, 21273–21283 (2023).
Tushar, S. Y., Meena, P. & Warkar, S. G. Synthesis and characterization of novel xanthan gum-based pH-sensitive hydrogel for metformin hydrochloride release. Colloid Polym. Sci. 301, 1147–1158 (2023).
Timoshenko, J. Spectroscopy predicts catalyst functionality. Nat. Catal. 5, 469–470 (2022).
Zhao, H. et al. The role of Cu1-O3 species in single-atom Cu/ZrO2 catalyst for CO2 hydrogenation. Nat. Catal. 5, 818–831 (2022).
Zhu, E. et al. Simultaneous regulation of thermodynamic and kinetic behavior on FeN3P1 single-atom configuration by Fe2P for efficient bifunctional ORR/OER. Appl. Catal. B 347, 123796 (2024).
Huang, B. et al. Decrypting the influence of axial coordination on the electronic microenvironment of Co-N5 site for enhanced electrocatalytic reaction. CCS Chem. 5, 1876–1887 (2023).
Zhang, P. et al. A robust asymmetric diatomic electrocatalyst for oxygen reduction reaction in both acidic and alkaline media. Appl. Catal. B 344, 123645 (2024).
Zhong, X. et al. Stabilization of layered lithium-rich manganese oxide for anion exchange membrane fuel cells and water electrolysers. Nat. Catal. 7, 1–14 (2024).
Xiao, W., Yan, D., Zhao, Q., Bukhvalov, D. & Yang, X. Regulating electrocatalytic properties of oxygen reduction reaction via strong coupling effects between Co-NC sites and intermetallic Pt3Co. Appl. Catal. B 346, 123740 (2024).
Pei, Z. et al. Atomically dispersed Fe sites regulated by adjacent single Co atoms anchored on N‐P co‐doped carbon structures for highly efficient oxygen reduction reaction. Adv. Mater. 36, 2306047 (2024).
Yu, H. X. et al. DFT study on the oxygen titanium porphyrin as sustainable cyclic catalyst for water splitting. Int. J. Hydrog. Energy 44, 19920–19928 (2019).
Yang, C. et al. Electronic properties of double-atom catalysts for electrocatalytic oxygen evolution reaction in alkaline solution: a DFT study. Nanoscale 14, 187–195 (2022).
Liu, L. L., Chen, C. P., Zhao, L. S., Wang, Y. & Wang, X. C. Metal-embedded nitrogen-doped graphene for H2O molecule dissociation. Carbon 115, 773–780 (2017).
Gui, Y. Luo, P., Ji, C., Lin, Y., Chen, X. First-principles study of the gas sensing of benzene and formaldehyde by Ag2O- and CuO-modified MoSe2 nanosheets. ACS Appl. Nano Mater. 5, 12907-12914 (2022).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A. consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 132, 154104 (2010).
Momma, K. & Izumi, F. VESTA: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 41, 653–658 (2008).
Li, M., Zhang, L., Xu, Q., Niu, J. & Xia, Z. N-doped graphene as catalysts for oxygen reduction and oxygen evolution reactions: theoretical considerations. J. Catal. 314, 66–72 (2014).
Wei, C., Kong, F. T. & Gong, H. R. Phase stability and elastic property of PdH and PdCuH phases. Int. J. Hydrog. Energy 38, 16485–16494 (2013).
Govind, N., Petersen, M., Fitzgerald, G., King-Smith, D. & Andzelm, J. A generalized synchronous transit method for transition state location. Comp. Mater. Sci. 28, 250–258 (2003).
Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 115, 5461–5466 (2011).
Dronskowski, R. & Bloechl, P. E. Crystal orbital hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617–8624 (1993).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Wuttig, M. et al. The role of vacancies and local distortions in the design of new phase-change materials. Nat. Mater. 6, 122–128 (2007).
Bader, R. F. W. A quantum theory of molecular structure and its applications. Chem. Rev. 91, 893–928 (1991).
Acknowledgements
We acknowledge the support from the National Natural Science Foundation of China (Grant Nos. 52371228, 51972191 of Ruitao Lv), the National Key Research and Development Program of China (Grant Nos. 2021YFA1200800, 2021YFC2902900, 2021YFF0500503 of Ruitao Lv and Chen Chen), the National Natural Science Foundation of China (Grant Nos. 21925202, U22B2071 of Chen Chen), Yunnan Provincial Science and Technology Project at Southwest United Graduate School (Grant No. 202302AO370017 of Chen Chen), and International Joint Mission on Climate Change and Carbon Neutrality (Chen Chen).
Author information
Authors and Affiliations
Contributions
Ruitao Lv and Yifan Li conceived and designed the project. Yifan Li carried out the sample synthesis, characterizations, electrochemical measurements, and manuscript writing. Chang Chen contributed to the zinc-air batteries tests and manuscript writing. Aijian Huang and Chen Chen carried out the computational investigation and provided the analyses. Zewen Zhuang contributed to the analysis of the XAS results. Feiyu Kang contributed to the discussion and revision of the manuscript. Lingxi Zhou, Bohan Li, and Muyun Zheng helped to discuss the experimental data. Ruitao Lv is responsible for the overall supervision of the project. All the authors participated in preparing the manuscript and contributed to the discussion.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiao-Chun Wang, Yuan-Yao Li, and Ramendra Sundar Dey for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Huang, A., Zhou, L. et al. Main-group element-boosted oxygen electrocatalysis of Cu-N-C sites for zinc-air battery with cycling over 5000 h. Nat Commun 15, 8365 (2024). https://doi.org/10.1038/s41467-024-52494-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52494-0
This article is cited by
-
Ten thousand hour stable zinc air batteries via Fe and W dual atom sites
Nature Communications (2025)
