
Abstract
The P2X1 receptor is a trimeric ligand-gated ion channel that plays an important role in urogenital and immune functions, offering the potential for new drug treatments. However, progress in this area has been hindered by limited structural information and a lack of well-characterised tool compounds. In this study, we employ cryogenic electron microscopy (cryo-EM) to elucidate the structures of the P2X1 receptor in an ATP-bound desensitised state and an NF449-bound closed state. NF449, a potent P2X1 receptor antagonist, engages the receptor distinctively, while ATP, the endogenous ligand, binds in a manner consistent with other P2X receptors. To explore the molecular basis of receptor inhibition, activation, and ligand interactions, key residues involved in ligand and metal ion binding were mutated. Radioligand binding assays with [3H]-α,β-methylene ATP and intracellular calcium ion influx assays were used to evaluate the effects of these mutations. These experiments validate key ligand-receptor interactions and identify conserved and non-conserved residues critical for ligand binding or receptor modulation. This research expands our understanding of the P2X1 receptor structure at a molecular level and opens new avenues for in silico drug design targeting the P2X1 receptor.
Similar content being viewed by others

Introduction
There are seven subtypes of the P2X ligand-gated ion channel (P2X1-7) and these receptors control a vast number of biological processes that make them exciting drug targets1. P2X receptors consist of three individual subunits that can be homotrimeric or heterotrimeric to form a ligand-gated ion channel2,3. Each P2X subunit contains two transmembrane-spanning helices, a large extracellular domain, and a smaller intracellular N- and C-terminus except for the P2X7 receptor, which has an intracellular domain similar in size to the extracellular domain. The endogenous ligand, ATP, activates P2X receptors by binding to ATP binding sites located at the extracellular interface of each receptor subunit. Upon activation, P2X receptors form a non-selective pore that is permeable to cations such as potassium, calcium, and sodium4.
Research among the P2X receptor family has primarily focused on the P2X3, P2X4 and P2X7 receptors5. Experimentally determined structures exist for each of these P2X receptor subtypes alongside a selection of potent and specific small molecule compounds5,6,7,8,9,10,11. These groundbreaking studies greatly improved our understanding of the P2X receptors. However, for some of the lesser studied P2X receptors, crucial information is still lacking, particularly regarding molecular mechanisms of activation and inhibition and the relationship between structure and function, with a focus on how this knowledge could be utilised for drug discovery12. The P2X1 receptor is among these receptors, known for its high affinity for the endogenous ligand ATP and its rapid desensitisation following receptor activation5. The P2X1 receptor holds therapeutic promise, particularly in the urogenital and immune system. P2X1 receptor knockouts revealed reduced fertility in male mice, attributed to diminished contractility of the vas deferens, suggesting a P2X1 receptor antagonist could be used as a male contraceptive13,14. Additionally, the P2X1 receptor’s involvement in platelets and neutrophils contributes to thrombo-inflammatory conditions, which could potentially be targeted by a drug to suppress clot formation, thereby serving as an antithrombotic agent15,16. Recent studies even indicated the involvement of P2X1 receptors in different types of cancers and small arteries, hinting at potential medical applications yet to be discovered17,18,19.
Given the promising clinical potential, research groups employed structure-activity relationship studies with known P2X1 receptor ligands or compound screening methods to discover new P2X1 receptor ligands. The most promising P2X1 receptor antagonists were PSB-2001,20 ATA,21 MRS2159,22 and an unnamed series of P2X1 and P2X3 receptor antagonists23,24. Yet, none are as potent or P2X receptor selective as an older generation P2X1 receptor antagonist, NF44925,26. It is important to note that NF449 also exhibits several off-target effects and is a large polar molecule making it difficult to improve27. Nevertheless, NF449 could serve as a basis for developing potent and specific inhibitors for the P2X1 receptor. To date, the majority of drug discovery efforts were directed towards the generation of P2X1 receptor antagonists, and there are currently very few P2X1 receptor agonists, suggesting potential for significant improvement in this area12. As new P2X1 receptor ligands are discovered, the focus is shifting toward developing high-quality tool compounds to validate clinical opportunities. However, the absence of high-resolution P2X1 receptor structures to guide these studies is hindering development21,28,29.
To enhance our structural understanding of the P2X1 receptor, we determine the receptor's structure in both an ATP-bound desensitised state and an NF449-bound closed state. These structures guide mutagenesis studies aimed at unravelling ligand binding at a molecular level. Structural findings combined with site-directed mutagenesis and pharmacological assays are used to delineate structural features and the mechanistic basis for subtype-specific interactions of the P2X1 receptor. This work aims to support structure-based drug discovery efforts and accelerate drug development targeting the P2X1 receptor.
Results
Cryo-EM structure determination
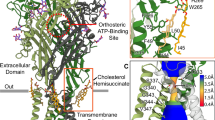
The full-length human P2X1 receptor was extracted from insect cells and reconstituted into detergent micelles for single-particle cryogenic electron microscopy (Supplementary Fig. 1). Cryo-EM imaging of the P2X1 receptor revealed severe preferred orientation of the receptor in vitreous ice. The addition of fluorinated fos-choline-8 before vitrification alleviated orientation bias and allowed for the determination of the P2X1 receptor to high resolution (Supplementary Fig. 2). In an early cryo-EM dataset of the P2X1 receptor, cryo-EM density was observed in the orthosteric pocket, corresponding to ATP, even though ATP was not added during the purification of the receptor. This finding mirrors observations from the initial crystal structures of the hP2X3 receptor10. In a subsequent sample ATP and MgCl2 were added at high concentrations to determine the ATP-bound P2X1 receptor structure (PDB: 9B73) to a resolution of 1.96 Å (Fig. 1A, B). Addition of the high affinity and competitive P2X1 receptor antagonist NF44925 was used to remove ATP-bound to detergent solubilised P2X1 receptor, (Supplementary Fig. 1D-F) and allowed for determination of the NF449-bound P2X1 receptor structure (PDB: 9B95) to a resolution of 2.61 Å (Fig. 1C, D). The P2X1 receptor models correlated well with their respective density maps and assignment of backbone, side chains, ligands, metals, glycosylation and waters were possible at the achieved resolution (Supplementary Fig. 3, 4). The structures of both the ATP-bound and NF449-bound P2X1 receptors exhibit a characteristic chalice-like shape, consistent with other experimentally determined P2X receptor structures (Fig. 1)8,9,10. The structure of the P2X1 receptor monomer can be likened to that of a dolphin, featuring a head domain, left and right flippers, dorsal fin, upper body, lower body and a fluke (Fig. 1B, D)2.

Cryo-EM maps of the (A) ATP-bound P2X1 receptor and (C) NF449-bound P2X1 receptor, with individual chains coloured and the cryo-EM ligand density depicted in violet. Enlarged depiction of Mg-ATP and NF449 fitted into the cryo-EM map. Atomic model and structural characteristics of the (B) ATP-bound P2X1 receptor and the (D) NF449-bound P2X1 receptor colourised to resemble features of a dolphin. The head domain is purple, the lower body is light blue, the dorsal fin is orange, the upper body is blue, the right flipper is red, the left flipper is yellow, and the fluke is green. The dolphin figure was created in BioRender. Bennetts, F. (2023) BioRender.com/r92x211.
Receptor gating cycle and pore region
To define the state of the P2X1 receptors, the pore region was analysed. This analysis revealed that the ATP-bound P2X1 receptor has two gates, V344 and V347 from transmembrane 2 of each monomer, which constrict the pore to its narrowest radius (Fig. 2A). Conversely, in the NF449-bound P2X1 receptor structure, S337 from transmembrane 2 of each monomer constricts the pore to its narrowest radius, with residues T333 and G336 also contributing to the gating region (Fig. 2B). Both P2X1 receptor structures were in non-pore conducting states due to the small pore radius, measuring 0.6 Å for the ATP-bound receptor and 1.0 Å for the NF449-bound receptor, making it insufficient to facilitate the transport of cations (Fig. 2C). Hence, the P2X1 receptor bound to the antagonist NF449 is in a closed state, whereas the P2X1 receptor bound by the agonist ATP is in a desensitised state.

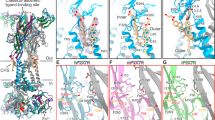
Cartoon representation of the (A) ATP-bound P2X1 receptor and (B) NF449-bound P2X1 receptor pore region, coloured by monomer. C Graph of the pore radius along the length of the pore region, with the residues responsible for constricting the pore radii highlighted. The ATP-bound P2X1 receptor is depicted in red, while the NF449-bound P2X1 receptor is represented in blue. The pore radius measured 0.6 Å for the ATP-bound receptor and 1.0 Å for the NF449-bound receptor, marked by the dotted line. D Overlay of the transmembrane domains (E) and a corresponding graph of the pore radius of the ATP-bound P2X1 receptor (red) and the desensitised state ATP-bound hP2X3 receptor (PDB: 5SVL, green). Pore for each P2X3 receptor is displayed by radius with red, white, and blue, with red indicating the area with the tightest constriction point. F Overlay of the transmembrane domains (G) and a corresponding graph of the pore radius of the of the NF449-bound P2X1 receptor (blue) and the closed state TNP-ATP-bound hP2X3 receptor (PDB: 5SVQ, yellow). For all pore volumes, the surface volume of the pore radius is highlighted in red, white, and blue, with red indicating the area with the tightest constriction point.
Structures of the P2X3 receptor were determined in all of the major ion channel states (desensitised, apo, open, closed) and the P2X3 receptor shares the fast desensitisation profile of the P2X1 receptor, making it a useful tool for comparison10. To gain deeper insights into the pore region of the P2X1 receptor, the desensitised and closed structures of the P2X3 receptor were compared to their equivalent P2X1 receptor structures (Fig. 2D, F). The desensitised P2X1 and P2X3 receptor exhibited a similar wider opening at the top of the pore, followed by a lower constriction site. This gate is controlled by two valine residues (V344 and V347) in the P2X1 receptor and one valine residue (V334) in the P2X3 receptor (Fig. 2E). The presence of the second valine residue (V347) in the P2X1 receptor results in a longer gate, while both valine residues contribute to a much smaller pore radius, of 0.6 Å, compared to 1.7 Å for the P2X3 receptor. The closed P2X1 and P2X3 receptors are similar in overall pore shape and size, with the closed P2X3 receptor having a slightly smaller constriction site radius of 0.5 Å compared to 1 Å for the P2X1 receptor (Fig. 2G). The gate in these two closed receptors is located towards the top of the pore, formed by multiple residues, resulting in a longer gate region (Fig. 2F). These subtle differences highlight the variability in the pore region between the P2X1 and P2X3 receptors, despite their similar properties. A similar observation between the P2X1 and P2X3 receptor structures was the absence of a modellable cytoplasmic cap in the desensitised and closed states, with the cytoplasmic cap only resolved in the open state structure of the P2X3 receptor10. Thus, it is likely that this region is highly dynamic and disordered in the desensitised and closed states of the P2X1 and P2X3 receptors.
NF449 is a competitive P2X1 receptor antagonist and aligning the ATP-bound P2X1 receptor and NF449-bound P2X1 receptor revealed one benzene disulfonic acid group of NF449 overlapping with bound ATP, demonstrating how NF449 competitively inhibits ATP from binding the P2X1 receptor (Fig. 3A). Initially it was difficult to visualise how a large molecule like NF449 would fit into the orthosteric binding pocket however, from the structure it was apparent that NF449 adopts a distinct orientation with unique contacts to the lower body, head domain, and dorsal fin of the P2X1 receptor (Fig. 3A). Furthermore, there were significant differences around the ligand binding sites at a structural level. The left flipper, right flipper, head domain and dorsal fin moved outward in the NF449-bound P2X1 receptor structures (Fig. 3A). The changes around the ligand binding site propagate a conformational change through the lower body to the pore region (Fig. 3B). The lower body and upper portion of the transmembrane domains moved diagonally inward resulting in the vestibule above the pore reducing size and the constriction site of the pore moving up the transmembrane domain (Fig. 3B). In the NF449-bound P2X1 receptor, transmembrane one has moved laterally across the face of the pore region, while transmembrane two has moved inward and downward, compared to the ATP-bound P2X1 receptor (Fig. 3B). Despite both structures being in non-conductive states, they adopt vastly different poses and gating regions, highlighting the significant differences between the desensitised and closed structures of the P2X1 receptor.

A Overlay of the ATP-bound P2X1 receptor (red) and NF449-bound P2X1 receptor (blue) at the orthosteric binding site, illustrating movement in secondary structure with arrows. B Overlay of the ATP-bound P2X1 receptor (red) and NF449-bound P2X1 receptor (blue) at the lower body and transmembrane domain, illustrating movement in secondary structure with arrows.
ATP binding site
ATP exhibits high affinity and potency for the P2X1 receptor, which was further confirmed in this study using a competition radioligand binding assay and a calcium ion influx assay with a more stable closely related analogue of ATP, α,β-methylene ATP. α,β-methylene ATP exhibited an affinity of 43 nM (Supplementary Fig. 5) and a potency of 99 nM (Fig. 4B) for the P2X1 receptor. The potency of α,β-methylene ATP was comparable to previous studies,30 while the affinity was previously reported at a lower value of 8.1 nM,31 with the differences likely due to different experimental conditions. The cryo-EM structure of the ATP-bound P2X1 receptor revealed a wide range of interactions between ATP and the receptor (Fig. 4A, Supplementary Table 1). Residues K68, K70, R292, and K309 formed salt bridges with the phosphate chain of ATP, with residue N290 and S286 additionally contributing a hydrogen bond in this region, collectively generating a highly polar environment around the phosphate chain of ATP. The phosphate chain of ATP formed an ionic interaction with a magnesium ion, which in turn engaged in a direct ionic interaction with residue D170, while also interacting with residue E122 through a water network, contributing to the highly polar local environment around the phosphate chain of ATP. Residues K68, K70, N290, R292, and K309 were previously studied using single alanine mutations and were shown to cause large reductions in the potency of ATP when compared to the WT-P2X1 receptor29. Another critical residue was T186, which forms hydrogen bonds with the adenine ring of ATP utilising both the backbone carbonyl and the side chain hydroxyl group (Fig. 4A). Mutation of T186 was shown to be an important component of ATP-mediated receptor activation29. A finding, possible because of the high resolution of the ATP-bound P2X1 receptor structure was a water-mediated hydrogen bond between R139 and the adenine ring of ATP (Fig. 4A). In addition, residue K140 formed a hydrogen bond with the ribose sugar of ATP, while the backbone carbonyl of K68 formed a hydrogen bond with the adenine ring of ATP. Together, residues, K68, R139, and K140 form a polar pocket around the adenine ring and ribose sugar of ATP. F188 engages in a pi-pi stacking interaction with the adenine ring, while on the opposite side of the adenine ring, K70 participates in a pi-cation interaction. Finally, hydrophobic residues, M214, and V229 form a small hydrophobic pocket around the adenine ring of ATP, with part of this region being previously studied using cysteine scanning mutagenesis and shown to be important in ATP-mediated P2X1 receptor activation32.

A Graphic of the ATP binding site, with key interactions illustrated as black dotted lines and nearby residues labelled. B, C Increasing concentrations of the agonist α,β-methylene ATP on HEK293 cells expressing WT-P2X1 or single residue mutants of the P2X1 receptor. The data is normalised to 10 µM ionomycin and subsequently adjusted so that the highest value corresponds to 100 and the lowest value to 0. A log(agonist) nonlinear regression curve using a four-parameter model is fitted to the data with the top and bottom constrained to 100 and 0, respectively. Data points are represented as mean values with error bars indicating the standard error of the mean (SEM). All P2X1 receptor variants were derived from four independent experiments conducted in triplicate. D Amino acid sequence alignment of key residues from the human P2X1 receptor (hP2X1:P51575), aligned with corresponding residues from human P2X receptor subtypes (hP2X2:Q9UBL9, hP2X3:P56373, hP2X4:Q99571, hP2X5:Q93086, hP2X6:O15547, hP2X7:Q99572). Residue categorisation based on properties: Hydrophobic (A, I, L, M, F, W, V, Y) in blue, positive charge (K, R, H) in red, negative charge (E, D) in magenta, polar (N, Q, S, T) in green. Special cases (C, G, P) in orange. Gaps represented in white.
Although most of the residues interacting with ATP in the binding site were previously studied,29 several residues, including E122, R139, K140, D170, M214, and S286 remain unexplored. Some of these residues, E122, R139, K140, and D170 show low amino acid sequence conservation across the P2X receptor family, which is uncommon in the conserved ATP binding site (Fig. 4D). To understand their role in ATP-mediated receptor activation, each of these residues were individually mutated to alanine. Subsequently, activation of the P2X1 receptor was assessed using a calcium ion influx assay. The mutants R139A, M214A, E122A, and D170A significantly decreased the potency of the P2X1 agonist α,β-methylene ATP compared to WT-P2X1 with a 3.5-fold, 7-fold, 18.5-fold, and 58-fold decrease, respectively (Table 1, Fig. 4B). In contrast, mutant S286A and K140A did not significantly change the potency of α,β-methylene ATP compared to WT-P2X1 (Table 1, Fig. 4B). To validate if these mutations were driving a change in the affinity of α,β-methylene ATP, each mutant receptor was tested in a radioligand binding experiment using [3H]-α,β-methylene ATP. The mutants S286A and K140A, which did not affect the potency of α,β-methylene ATP, exhibited binding affinity comparable to that of WT-P2X1 (Supplementary Fig. 5). Mutated residue R139A showed a 6-fold decrease in affinity for [3H]-α,β-methylene ATP and mutants M214A, E122A, and D170A exhibited reduced binding affinities that were beyond the threshold that could be accurately measured using a radioligand probe (Supplementary Fig. 5). These findings correlate with the substantial decrease in potency observed in the calcium ion activation assay for each of these mutants and establishes that the decline in potency for the mutants R139A, M214A, E122A, and D170A stems at least in part from a decrease in the agonist’s affinity to bind to the mutated P2X1 receptor. In addition to the known residues crucial for ATP-mediated P2X1 receptor activation, residues R139, M214, E122, and D170 emerge as significant contributors to P2X1 receptor activation29. Furthermore, these residues exhibit lower conservation within the P2X receptor family compared to other interactions of the ATP binding site, implying that they enhance the potency of ATP for the P2X1 receptor and could be residues to target for the design of subtype-selective agonists.
To evaluate the impact of ATP-based mutants on receptor desensitisation, data from the calcium ion influx assay were analysed to measure desensitisation rate by tracking the decrease in fluorescence response over time. To validate this assay, a previously identified slowly desensitising 2Nβ P2X1 receptor mutant33 was generated and evaluated. Compared to WT-P2X1 receptor, the 2Nβ P2X1 receptor showed a significant 12-fold decrease in the 50% decay rate, which is comparable to the 7-fold reduction reported in the original study33 (Supplementary Fig. 6A, B). To determine if any P2X1 receptor mutants used in this study affected the desensitisation rate, fluorescence was measured over time after agonist stimulation. Mutants R139A, R139P, and M214A all significantly slowed the desensitisation rate compared to the WT-P2X1 receptor (Supplementary Fig. 6C, D). Residues E122A, D170A, and F188L also exhibited a smaller increase in the time to achieve 50% decay, approximately 1.5 times higher than the WT-P2X1 receptor (Supplementary Fig. 6C, D). Of these residues R139A, R139P, M214A, E122A, and D170A were found to decrease the potency of α,β-methylene ATP, suggesting that the slowed desensitisation rate may be a result of reduced binding (Table 1). An exception was the R292A mutant P2X1 receptor, which caused a significant decrease in the potency of α,β-methylene ATP, with desensitisation rates comparable to the WT-P2X1 receptor. All other mutants generated in this study produced comparable desensitisation to WT-P2X1 (Supplementary Fig. 6C, D). These results demonstrate that mutating the orthosteric binding pocket can affect the desensitisation rate of the P2X1 receptor, which is often associated with reductions in receptor affinity and potency. While these studies examine the binding, activation, and desensitisation properties of the P2X1 receptor, they do not address ion selectivity and efficacy. These aspects may be affected by receptor mutations, so further research is needed to determine if mutations in the orthosteric binding pocket influence these receptor properties.
NF449 binding site
NF449, a large polar molecule, is recognised for its high affinity and potency for the P2X1 receptor25. In this study, the activity of NF499 was checked using a competition radioligand binding assay and a calcium ion influx assay, demonstrating a binding affinity of 6.6 nM (Fig. 5B) and an inhibitory potency of 66 nM, respectively (Fig. 5C). The decrease in inhibitory potency observed compared to the previously reported value of 0.05 nM at human WT-P2X1 receptors could be attributed to the utilisation of the voltage clamp technique on Xenopus oocytes as opposed to cell assays on HEK293 cells expressing the P2X1 receptor25. As mentioned, NF449 adopts a distinctive orientation, requiring substantial movement of residues within the binding site, and engages in numerous interactions with the P2X1 receptor within the binding site (Fig. 5A, Supplementary Table 2). The P2X1 receptor engages in several salt bridge interactions with NF449, involving residues K68, K70, K136, K215, and R292. Additionally, residues L72 (backbone carbonyl and amine), K140 (backbone amine), M214, C217 (backbone amine), and N290 form hydrogen bonds with NF449. The multitude of highly polar interactions, particularly around the sulfonic acids of NF449, likely underpins its high affinity for the P2X1 receptor (Fig. 5A). F188 engages in a pi stacking interaction with one benzene ring of NF449, while R139 participates in a cation-pi interaction at another benzene ring. Six additional residues, T186, V209, C217, L218, P228, and V229, were found near NF449, suggesting their involvement in hydrophobic interactions, typically around the benzene rings of NF449. Previous research on NF449 has identified several key residues crucial for NF449-mediated inhibition. The most significant was a selection of positively charged residues in the head domain of the P2X1 receptor (K136, K138, R139, and K140) that was shown to play a significant role in mediating NF449 inhibition28,34,35. In the NF449-bound P2X1 receptor, K136, R139, and K140 interact with NF449, which explains why mutating these residues reduced NF449’s inhibitory potency.

A Graphic of the NF449 binding site, with key interactions illustrated as black dotted lines and nearby residues labelled. B Competition radioligand binding of a submaximal concentration of [3H]-α,β-methylene ATP (50 nM) with increasing concentrations of NF449 on HEK293 cells expressing WT-P2X1 receptor. A nonlinear regression one site, fit Ki curve is fitted to the data. Data points are represented as the mean with the SEM, derived from 3 independent experiments conducted in duplicate. C, D Calcium ion influx using submaximal concentrations of α,β-methylene ATP (0.316–100 µM, 80–95% of max response per mutant) with increasing concentrations of the antagonist NF449 on HEK293 cells expressing WT-P2X1 and single residue mutants of the P2X1 receptor. Data was normalised to 10 µM ionomycin and subsequently adjusted so that the highest value corresponds to 100 and the lowest value to 0. A log(antagonist) nonlinear regression curve fit using a four-parameter model is fitted to the data with the top and bottom constrained to 100 and 0, respectively. Data points were represented as the mean with the error bars indicating the SEM. P2X1 receptor variants D170A, M214A, E122A, K140A, and S286A were derived from 3 independent experiments and WT-P2X1, R139A, K215A, K136A, E282A, R292A, K136W, R139P, F188L, and L218T were derived from 4 independent experiments, with each experimental condition conducted in triplicate. E Amino acid sequence alignment of NF449 binding site residues from the human P2X1 receptor, aligned with residues from human P2X receptor subtypes. Residue categorisation based on properties: Hydrophobic (A, I, L, M, F, W, V, Y) in blue, positive charge (K, R, H) in red, negative charge (E, D) in magenta, polar (N, Q, S, T) in green. Special cases (C, G, P) in orange. Gaps represented in white.
Given that most of the molecular studies on NF449 binding to the P2X1 receptor conducted to date did not mutate individual residues,28,34,35 and the NF449-bound P2X1 receptor revealed a selection of conserved and non-conserved residues interacting with NF449 (Fig. 5E), single alanine mutagenesis was performed on key residues interacting with NF449 to determine their importance in ligand binding. The follow P2X1 receptor residues, M214, K140, R292, K215, R139, and K136, were mutated to alanine. The mutant E282A was included as a control. Although it is located nearby, it does not interact with NF449 and was therefore not expected to reduce the potency of NF449 (Fig. 5A). To explore the molecular basis behind the high selectivity of NF449 for the P2X1 receptor,36 non-conserved residues, F188, K136, L218, R139 were mutated to their equivalent residues in the P2X7 receptor (Fig. 5E), a receptor with over 500-fold lower potency for NF44925. Initially, the potency and binding affinity of α,β-methylene ATP to each mutant were validated to ensure the mutant was functional. The single residue M214A, R139A, R139P, and R292A P2X1 receptor mutations showed reduced potency (Table 1, Fig. 4C) and affinity (Supplementary Fig. 5) for α,β-methylene ATP. Each of these residues forms interactions with ATP (Table 1). Conversely, P2X1 receptor mutants K215A, E282A, K136A, F188L, K136W and L218T, which do not make contacts with ATP, maintained similar α,β-methylene ATP potency (Table 1, Fig. 4C) and α,β-methylene ATP affinity (Supplementary Fig. 5) to that of the WT-P2X1 receptor. To ensure similar occupancy of each mutant receptor, an 80 to 95% maximal concentration of α,β-methylene ATP was used when testing increasing concentrations of NF449 in calcium ion influx assays.
Among the single residue alanine mutations in the P2X1 receptor, M214A, K215A, R139A, K136A, and K140A exhibited a significant 4-fold, 15-fold, 32-fold, 35-fold, and 83-fold decrease in the inhibitory potency of NF449, respectively (Table 2, Fig. 5C). Residue E282, which is nearby NF449, but does not directly interact with NF449, yielded no change in the potency of NF449 when mutated to alanine (Table 2, Fig. 5A). The residues K215, R139, K136, K140, and M214 in the P2X1 receptor, crucial for NF449 activity, participate in salt bridge or hydrogen bond interactions with NF449. These interactions highlight the significance of at least four sulfonic acids and a benzene ring in NF449, underscoring their crucial role in the inhibitory activity of NF449 (Fig. 5A). Furthermore, these interactions take place at various locations within NF449, indicating the accuracy of the binding mode of NF449 in the P2X1 receptor structure. The P2X1 to P2X7 single residue mutants F188L, K136W, L218T, and R139P were generated by substituting the respective residues with those matched from the P2X7 receptor sequence (Fig. 5E). These residues are relatively non-conserved across the P2X receptor family, which suggests they may play a role in driving selectivity for the P2X1 receptor (Fig. 5E). Mutants, K136W and L218T of the P2X1 receptor significantly attenuated the inhibitory activity of NF449, showing a 6.5-fold and 9.5-fold reduction in inhibitory potency compared to WT-P2X1, respectively (Table 2, Fig. 5D). In contrast, the mutants F188L and R139P exhibited an NF449 inhibitory potency comparable to that of the WT-P2X1 receptor (Table 2, Fig. 5D). Intriguingly, while the R139A mutation discussed earlier caused a reduction in the inhibitory potency of NF449, the R139P mutation did not, suggesting that R139P retained interactions with NF449. However, among the four P2X1 to P2X7 mutants tested, half of them (K136W and L218T) caused a reduction in the inhibitory potency of NF449. This suggests that these residues may contribute to the reduced affinity NF449 has for the P2X7 receptor and could be targeted for designing selective P2X1 receptor antagonists.
Metal ion binding sites
Initial cryo-EM model building of the ATP-bound P2X1 receptor revealed a large density adjacent to the gamma phosphate of ATP (Fig. 6A). A metal binding site adjacent to the gamma phosphate of ATP was identified in crystal structures of the human P2X3 receptor and the Gulf Coast Tick P2X receptor10,37,38. To validate the metal ion within the ATP binding site, metal binding parameters were calculated, including valency, occupancy, geometry, and contact distances (Fig. 6D)39,40. The metal ion site was coordinated by six contacts in an octahedral geometry, four to water molecules, one to the gamma phosphate of ATP and one to D170. Each interaction formed an ionic bond at a distance of 2.0 Å. The characteristics of these interactions suggest that this site was occupied by a magnesium ion, while other metal ions, such as calcium and sodium, do not fit well into the binding site (Fig. 6D). Both residues D170 and E122, which interact with the magnesium ion, play critical roles in ATP-mediated receptor activation and ATP receptor binding. Mutating either residue to alanine significantly reduced the potency and affinity of α,β-methylene ATP (Fig. 6E-G). Sequence alignment of P2X1 receptor residues E122 and D170 reveals negatively charged residues at the same location at the hP2X3 and hP2X4 receptors (Fig. 6C). Studies established that ATP complexed with magnesium maintains high efficacy for the P2X3 and P2X1 receptor, while P2X2 and P2X4 receptors display diminished responses to Mg-ATP,41 suggesting that only some P2X receptor subtypes likely contain this cation binding site. Building on this research, the same group established that this metal ion site can stabilise ATP interactions with the P2X3 receptor and slow ATP unbinding and receptor recovery from desensitisation37. Furthermore, alanine mutants targeting the residues responsible for binding metal ions in the P2X3 exhibited slower desensitisation. At the P2X1 receptor, these mutants exhibited a slight 1.5 to 1.6 times decrease in the time required to achieve a 50% reduction in fluorescence compared to the WT-P2X1 receptor, indicating reduced desensitisation of the receptor compared to the WT-P2X1 receptor (Supplementary Fig. 6C, D). Further studies are needed to assess this metal ion site at the P2X1 receptor for ATP unbinding and recovery from desensitisation. However, these findings indicate that the P2X1 receptor has a similar metal ion site to that of the P2X3 receptor, which plays a comparable role in ATP potency and receptor desensitisation.

A The ATP Mg2+ ion binding site modelled into the cryo-EM map of the ATP-bound P2X1 receptor, with surrounding residues, ligands, and waters. B The possible central metal ion binding site cryo-EM density of the ATP-bound P2X1 receptor, with surrounding residues. C Amino acid sequence alignment of D97, E122 and D170 from the human P2X1 receptor (hP2X1: P51575), aligned with the corresponding residue from human P2X receptor subtypes (hP2X2: Q9UBL9, hP2X3: P56373, hP2X4: Q99571, hP2X5: Q93086, hP2X6: O15547, hP2X7: Q99572). Residue colourisation based on properties: Hydrophobic: blue, positive: red, negative: magenta, polar: green, special cases: orange, gaps: white. D Validation metrics for the magnesium ion positioned adjacent to ATP, colour-coded as follows: green: acceptable, yellow: borderline, and red: poor. No metal ion could be conclusively identified at the potential central metal ion site. E Increasing concentrations of the agonist α,β-methylene ATP on HEK293 cells expressing WT-P2X1 or P2X1 receptor mutants. The data is normalised to 10 µM ionomycin and subsequently adjusted so that the highest value corresponds to 100 and the lowest value to 0. A log(agonist) nonlinear regression curve using a four-parameter model is fitted to the data with the top and bottom constrained to 100 and 0, respectively. Data points represent the mean, with error bars indicating the SEM, from four independent experiments conducted in triplicate. F Saturation radioligand binding on HEK293 cells expressing WT-P2X1 receptor or P2X1 receptor mutants. A nonlinear regression one site, fit Ki curve is fitted to the data. Data points represent the mean, with error bars indicating the SEM. P2X1 receptor variants WT-P2X1 were derived from 5 independent experiments, D170A and E122A from 3, and D97A from 2, with each condition performed in triplicate. Values that exceeded 300 nM were classified as not defined. G Calculated EC50 and Ki values from experiments conducted on WT-P2X1 or P2X1 receptor mutants. Statistically significant differences compared to WT-P2X1 were calculated using a one-way ANOVA with a Dunnett’s multiple comparison test. P values: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
The cryo-EM map of the ATP-bound and NF449-bound P2X1 receptor suggested there may be another metal ion site located centrally within the extracellular domain coordinated by the D97 residue from each monomer (Fig. 6B). In P2X receptors, P2X4, P2X5, and P2X6 receptors possess either aspartic acid or glutamic acid at the sequence-matched residue, implying the possible presence of this metal ion site in these receptors as well (Fig. 6C). Indeed, it was previously shown that P2X4 receptors contain a metal ion binding site at the same location from the closed zfP2X4 receptor structure2. To validate the presence of a possible central cation ion located within the extracellular domain, metal ion parameters were calculated and analysed. However, no metal ion could be accurately assigned. Poorer resolution in this area of the cryo-EM map may hinder resolving bound water molecules, which are needed to assign the occupancy, geometry, and interaction characteristics of a metal ion at this site. To investigate this site further, the D97 residue was mutated to alanine, and the mutated P2X1 receptor was analysed using calcium ion activation and radioligand binding assays. These assays revealed that the P2X1 D97A receptor exhibited a similar α,β-methylene ATP affinity, α,β-methylene ATP potency, and desensitisation rate to that of the WT-P2X1 receptor (Fig. 6E–G, Supplementary Fig. 6C, D). The lack of effect observed when residue D97 was mutated to alanine, rendering it unable to coordinate a metal ion, suggests that this potential metal ion site may not be functionally relevant or that the experimental conditions were not suitable for studying its role.
Discussion
This study presents high-resolution cryo-EM structures of the P2X1 receptor in a desensitised ATP-bound and closed NF449-bound state. Features such as the binding site, metal ion sites, and the gating cycle were studied in relation to other P2X receptors. Additionally, mutagenesis was used to study the interactions of ATP, NF449 and metal ions, revealing both conserved and non-conserved residues that regulate the binding of NF449 and ATP.
The cryo-EM structure of the ATP-bound P2X1 receptor revealed that ATP exhibits a remarkably similar binding mode to the ATP-bound P2X3, P2X4 and P2X7 receptor structures. Alignment of these structures shows that ATP from the P2X3, P2X4, and P2X7 receptors deviate by root mean square deviations (RMSD) of 0.70, 0.93, and 1.3 Å, respectively (Supplementary Fig. 7). Moreover, the interactions of bound ATP in the P2X1 receptor closely resemble those in the ATP-bound P2X3, P2X4, and P2X7 receptor structures (Supplementary Fig. 7). Despite these consistent interactions across ATP-bound P2X receptor structures, it is surprising that the affinity of ATP for the P2X receptor can vary between these subtypes5. The affinity of ATP for the P2X1 receptor ranks among the highest compared to other P2X receptors. Upon examining interactions of ATP at the P2X1 receptor, it became apparent that there are additional interactions, which may account for this high affinity. The residues K140, D170, E122 and R139 each interact with bound ATP in the P2X1 receptor with residue K140 forming a hydrogen bond with the ribose sugar, a feature not observed in other P2X receptors. Similarly, residues D170 and E122 interact with the adjacent magnesium ion or the water network associated with the magnesium ion, while R139 interacts with the adenine ring of ATP through a water molecule. The mutagenesis data in this study highlighted the significance of D170, E122, and R139 in receptor activation (Table 1). However, it is the combination of additional interactions that likely contributes to the increased affinity of ATP for the P2X1 receptor.
During ATP-mediated activation, P2X receptors experience a conformational change where the dorsal fin and head domain move inward, and the left flipper moves away, resulting in cleft closure10. In the NF449-bound P2X1 receptor, cleft closure is prevented due to the dorsal fin and head domain being sterically blocked by disulfonic acid benzene groups of NF449 (Fig. 3A). Importantly, NF449 forms a selection of interactions with both of these key regions (Supplementary Table 2). The residues K136, R139, and K140 are located on the head domain, while M214, K215, and L218 are positioned on the dorsal fin of the P2X1 receptor (Fig. 5A). As mentioned when each of these residues was mutated, they led to a decrease in the potency of NF449 for the P2X1 receptor (Table 2). In contrast, residues R292 and F188 are located on the upper and lower body domains, respectively, and form interactions with bound NF449 but showed no effect on NF449-mediated inhibition of the P2X1 receptor when mutated. Derivatives of NF449, including those featuring NF449 split in half on its symmetrical axis, exhibited large decreases in potency compared to NF449, suggesting that most of the NF449 molecule is required to retain activity at the P2X1 receptor26. An explanation for this effect was the loss of interactions at either the dorsal fin or the head domain, which seem to be crucial for mediating the inhibitory activity of NF449, suggesting that these two regions are key, while the lower body, upper body, and left flipper are less important in facilitating NF449’s inhibitory effect. Interestingly, previous molecular studies on NF449 identified positively charged residues of the head domain (K136, K138, R139, and K140) to be critical in mediating NF449 inhibition at the P2X1 receptor; however, introducing these residues into the P2X2 and P2X4 receptor produced at best minor improvements in the inhibitory potency of NF44928,34,35. Perhaps P2X1-specific structural movements or the structural conformation of the binding site, particularly the dorsal fin and head domain, are required for NF449 binding, which cannot be examined from mutagenesis studies alone. Further studies in this area are warranted, especially in light of the information presented in the cryo-EM NF449-bound P2X1 receptor structure, as it may pave the way for designing P2X1 specific antagonists.
NF449 is an interesting molecule as it is highly potent and specific to the P2X1 receptor, yet it is a poor drug candidate due to its off-target effects and high polarity27,36. P2X receptor structures bound to orthosteric antagonists were aligned with the NF449-bound P2X1 receptor structure to compare the binding mode of orthosteric antagonists with NF449 (Supplementary Fig. 8). The antagonists A-317491 from the hP2X3 (5SVR), TNP-ATP from the ckP2X7 (5XW6), and PPNDS from the pdP2X4 (8JV8) receptor structures all overlap with the ATP binding site and a portion of the NF449 binding site (Supplementary Fig. 8)10,42,43. From these structural comparisons, NF449 occupies a distinct part of the P2X receptor binding pocket that has was not previously explored by other P2X receptor antagonists, opening the door for designing new ligands that target this specific region of the receptor. Furthermore, some sections of NF449 do not form any interactions with the P2X1 receptor (Fig. 5A), indicating this molecule could be further optimised. Notably, A-317491 has additional interactions at the dorsal fin of the P2X1 receptor, similar to NF449 (Supplementary Fig. 8). A-317491 was designed to be a specific P2X3 receptor antagonist and does retain some weak activity for the P2X1 receptor44. These findings may be useful in identifying unique binding sites inspired by NF449 or in designing orthosteric antagonists with enhanced potency for the P2X1 receptor.
The high-resolution structures of the desensitised ATP-bound and closed NF449-bound P2X1 receptors unveil structural features and essential molecular interactions, shedding light on the binding of ATP and the distinctive binding mechanism of the potent antagonist NF449. These structural revelations open exciting opportunities for innovative structure-based discovery efforts in the realm of P2X1 receptor modulation. As the development of new therapeutics targeting the P2X1 receptor is still in its early stages, these structures are poised to guide the design of novel tool compounds, which may play a pivotal role in advancing our understanding and harnessing the therapeutic potential of the P2X1 receptor.
Methods
Construct design and generation
The full-length human P2X1 sequence (WT-P2X1) with C-terminal tag (CT) containing a 3C-cleavage site followed by muGFP (ultra stable GFP45) and an octa-histidine sequence (P2X1-CT) was synthesised from Integrated DNA Technologies (Iowa, USA) (Supplementary Fig. 1A). Empty pFastBac and pcDNA5/FRT/TO plasmid were Gibson assembled with WT-P2X1-CT and WT-P2X1 sequences, respectively. Primers were used to generate mutants of the P2X1 receptor from the pcDNA5/FRT/TO WT-P2X1 construct using polymerase chain reaction (PCR) Q5 high-fidelity polymerase. DNA constructs were transformed and purified. Each construct was sequenced, and the purity and concentration checked prior to use. DNA solutions were stored at −20 °C.
Expression and purification
Utilising the Bac-to-Bac system, WT-P2X1-CT pFastBac plasmid was transformed into a bacmid to make baculovirus and transfected into S. frugiperda cells using FuGENE HD transfection reagent. The P2X1 receptor was then expressed in S. frugiperda cells using baculovirus, with cell harvest conducted approximately 60 h post-transfection, followed by freezing at −80 °C.
Purification protocols were initially derived from existing literature reports on P2X receptor purifications and subsequently optimised specifically for the P2X1 receptor8,9,10. Cell pellets were thawed and lysed using a buffer consisting of 50 mM Tris (pH 8), 1 mM EDTA, 1 mM MgCl2, 2.5 µL/L benzonase, and a protease inhibitor cocktail containing, 0.2 mM phenylmethylsulfonyl fluoride, 5 µg/ml leupeptin, 5 µg/ml soybean trypsin inhibitor, and 0.2 mg/ml benzamidine. The resulting cell membranes were then subjected to centrifugation and subsequently resuspended in a solubilisation buffer comprising 50 mM Tris (pH 8), 15% glycerol, 750 mM NaCl, 1 mM MgCl2, 5 mM imidazole, 2.5 µL/L benzonase, 0.5% Lauryl Maltose Neopentyl Glycol (LMNG), 0.03% Cholesteryl Hemisuccinate Tris Salt (CHS), and the protease inhibitor cocktail. The cell membranes were homogenised using a dounce homogeniser with both a small and large pestle, each used six times, and then stirred for 1.5 h. Following this, the sample underwent high-speed centrifugation and was then batch-bound to pre-equilibrated Talon resin for 2 h. The sample was subsequently washed with a buffer containing 50 mM Tris (pH 8), 5% glycerol, 750 mM NaCl, 20 mM imidazole, 0.01% LMNG, and 0.0006% CHS. Upon loading onto a glass column, it was washed with 6 column volumes, and elution was performed using elution buffer comprising 50 mM Tris (pH 8), 5% glycerol, 100 mM NaCl, 200 mM imidazole, 0.01% LMNG, and 0.0006% CHS, collecting 5 column volumes. The sample underwent buffer exchange through an Amicon Ultra-15 mL 100 kDa molecular mass cut-off centrifugal filter unit, followed by cleavage with 3 C protease at a 1:1 w/w ratio overnight at 4 °C. The cleaved sample was then passed through clean Talon resin, and the flowthrough was collected. Subsequently, size exclusion chromatography (SEC) was employed for purification, utilising SEC buffer consisting of 50 mM Tris (pH 8), 100 mM NaCl, 0.01% LMNG, and 0.0006% CHS. The sample was loaded onto a pre-equilibrated Superdex 200 Increase 10/300 GL column, and fractions containing the trimeric P2X1 receptor were pooled, concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C. Initial studies also utilised a Superose 6 Increase 10/300 GL column. This purification process yields the ATP-bound desensitised P2X1 receptor as determined by cryo-EM studies.
To obtain the NF449-bound P2X1 receptor, the aforementioned protocol is modified with the inclusion of 100 µM NF449, 5 µL of apyrase, and 500 µM CaCl2 during the overnight 3 C protease cleavage at 4 °C. Additionally, 1–2 µL of apyrase is incorporated into both the elution and Ni wash buffers. To verify sample purity and homogeneity, each sample was loaded onto TGX precast (4%–15%) polyacrylamide gels alongside Precision Plus Protein Dual Colour Standards. Following electrophoresis, gels were stained with Coomassie blue and imaged with a camera. Additionally, samples were validated through negative stain electron microscopy with uranyl formate. Stained grids were visualised using a Talos L120C 120 kV microscope (Thermo Fisher Scientific, USA).
Single particle cryo-EM
To determine the structure of the ATP-bound P2X1 receptor, purified ATP-bound P2X1 receptor (19 mg/ml) was supplemented with 1 mM ATP and 1 mM MgCl2 overnight before vitrification. Additionally, 0.4 mM or 1.2 mM of fluorinated fos-choline-8 was added just 1 min prior to vitrification46. To determine the structure of the NF449-bound P2X1 receptor, purified NF449-bound P2X1 receptor (15.5 mg/ml) was supplemented with 100 µM NF449 overnight prior to vitrification. Additionally, 0.3 mM of fluorinated fos-choline-8 was added 1 min prior to vitrification. All samples were pipetted at a volume of 3 µL onto UltrAufoil R1.2/1.3 300 mesh holey grids which were glow discharged in air at 15 mA for 180 seconds using a Pelco easiGlow (Ted Pella, USA). Grids were plunge frozen in liquid ethane using a Vitrobot Mark III (Thermo Fisher Scientific, USA) operated at 4 °C and 100% humidity with 12 blot force and 2 seconds blot time. The cryo-EM data were acquired at the Ramaciotti Centre for Cryo-Electron Microscopy, on a G1 300 kV Titan Krios microscope (Thermo Fisher Scientific, USA) fitted with S-FEG, a BioQuantum energy filter and K3 detector (Gatan, Pleasanton, California, USA). The Krios microscope was operated at an accelerating voltage of 300 kV, utilising a 50 μm C2 aperture, and a 100 µm objective aperture, along with zero-loss filtering with a slit width of 10 eV. The microscope was set to an indicated magnification of 105,000 times in nanoprobe EFTEM mode. Data collection was assisted using aberration-free image shift with Thermo Fisher Scientific EPU software. Additional microscope details are listed in Supplementary Table 3.
Cryo-EM image processing and model building
Movies were collected at 0.82 Å per pixel and processed through motion correction and CTF correction using CryoSPARC v3 or v447. Template particle picking was conducted on motion-corrected images using 2D templates of the P2X1 receptor generated from a screening dataset. Following this, multiple rounds of 2D and 3D classification were conducted using CryoSPARC v3 or v4 to remove low-resolution particles, noise, and damaged particles. Bayesian polishing was applied to the ATP-bound P2X1 receptor in RELION v3.148. A CryoSPARC 3D variability analysis was conducted on a larger set of NF449-bound P2X1 receptor particles in clustered mode (2 clusters). This analysis was used to eliminate a subset of particles representing a different state (desensitised) from the desired state (closed). The final homogenous refinement for the ATP-bound receptor utilised 481,309 combined particles from two grids, resulting in a 1.96 Å map (FSC = 0.143). The NF449-bound receptor underwent non-uniform refinement, yielding a 2.61 Å map (FSC = 0.143) from 41,932 particles. The unmasked resolution estimates were 2.6 Å for the ATP-bound P2X1 receptor and 3.1 Å for the NF449-bound P2X1 receptor. Both maps were refined with C3 symmetry applied. C1 symmetry maps were also generated and, apart from a small decrease in resolution, exhibited the same shape and size as C3 symmetry maps. An initial P2X1 receptor model was obtained from the AlphaFold Protein Structure Database49,50 and rigid body fitted into the ATP-bound P2X1 receptor density using UCSF ChimeraX51. For the NF449-bound P2X1 receptor, the ATP-bound P2X1 receptor model was used as the starting point and was rigid body fitted into the NF449-bound P2X1 receptor density. Multiple rounds of manual model building and real-space refinement were conducted in Coot52 and PHENIX,53 respectively. Residues in the intracellular region of the ATP-bound P2X1 receptor (M1-G30 and L357-S399) and the NF449-bound P2X1 receptor (M1-G30 and D350-S399) were not modelled due to poor density in the cryo-EM map. Model quality was assessed using MolProbity54 before PDB deposition. Structure figures were generated using ChimeraX v1.6. Ligand interactions and metal ion analysis were performed using ChimeraX v1.6 and CheckMyMetal: metal binding site validation server,39 respectively. Pore radius calculation was conducted in Mole v2.555 and visualised in ChimeraX v1.6. Each step of the cryo-EM workflow for determining the ATP-bound and NF449-bound P2X1 receptor structures is detailed in Supplementary Fig. 2.
Stable transfections
HEK293 Flp-In T-REx cells were grown to about 80% confluency and the media was replaced with a minimal volume of Opti-MEM reduced serum medium. Cells were transfected with a pcDNA5/FRT/TO construct containing an insert of WT-P2X1 receptor or P2X1 receptor mutants. A transfection mixture composed of 250 µL Opti-MEM reduced serum medium, 0.7 µg P2X1 construct, 6.3 µg pOG44 (in a 10:1 ratio to P2X1 construct), and 28 µL FuGENE (in a 4:1 ratio to total DNA), was incubated for 30 min before addition to cells. After 48 h, selection with complete media containing 175 µg/mL of Hygromycin B was initiated until individual colonies formed. These colonies were reseeded, expanded under selection conditions, and preserved by freezing. To induce P2X1 receptor expression tetracycline was added at a concentration of 150 ng/mL two days prior to experimental testing.
Radioligand binding
Two days prior to experimentation HEK293 cells expressing WT-P2X1 or single residue mutants of the P2X1 receptor were seeded in 96-well isoplates at a density of 15,000 cells per well supplemented with 150 ng/ml of tetracycline. A binding buffer solution with the following composition: 150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2.6H2O, 10 mM D-glucose, 10 mM HEPES, 2.2 mM CaCl2.H2O at pH 7.4 was prepared, and the cells were washed twice. Subsequently, compounds of interest, ATP, and then [3H]-α,β-methylene ATP was added to wells. ATP was added in excess (10 μM) to determine non-specific binding. Plates were gently agitated for 3 h at room temperature, which radioligand association experiments determined to be appropriate for complete binding of the radioligand. Cells were then washed twice with ice-cold 0.9% NaCl buffer and briefly dried before the addition of UltimaGold liquid scintillation cocktail to each well. Data were recorded on a MicroBeta counter (PerkinElmer, Waltham, USA) as corrected counts per minute (CCPM). Data analysis was conducted using GraphPad Prism v9 (GraphPad, La Jolla, USA). Experimental conditions were repeated multiple times, with each condition run in duplicate or triplicate unless otherwise specified. [3H]-α,β-methylene ATP concentrations for each experiment were checked on Tri-Carb scintillation beta counters (PerkinElmer, Waltham, USA) as disintegrations per minute (DPM) and converted to concentration.
Saturation assays were conducted by adding increasing concentrations of [3H]-α,β-methylene ATP. Nonlinear regression - one site-specific binding curves were fit to experimental saturation binding data, with results reported in corrected counts per minute (CCPM). Each condition has the non-specific binding subtracted to calculate the specific binding. Competition assays were conducted by the addition of a KD concentration of [3H]-α,β-methylene ATP (50 nM), with increasing concentrations of the compounds of interest. Nonlinear regression one-site fit Ki curves were fit to experimental competition binding data, in relation to the concentration and affinity of [3H]-α,β-methylene ATP with results reported in corrected counts per minute (CCPM). Each condition has the non-specific binding subtracted to calculate the specific binding.
Calcium ion influx
Two days prior to experimentation HEK293 cells expressing either WT-P2X1 or single residue mutants of the P2X1 receptor were seeded into 96-well flat bottom clear plates at a density of 15,000 cells per well supplemented with 150 ng/ml of tetracycline. On the day of experimentation, a calcium buffer solution was prepared, consisting of 150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2.6H2O, 10 mM D-glucose, 10 mM HEPES, 2.2 mM CaCl2.H2O, and 2 mM probenecid at pH 7.4. All cell incubations were performed at 37 °C in a 5% CO2 humidified incubator with drug plates and tips pre-heated to 37 °C prior to use. Cells were washed once with calcium buffer, and subsequently, 1 mM Fluo-8 was added to each well under reduced light conditions. Cells were incubated for 1 h and then washed once with calcium buffer. Calcium buffer was added to each well and some wells contained P2X1 receptor antagonists or compounds of interest. Cells were incubated for an additional 30 min. Drug plates were prepared containing the calcium ionophore ionomycin, control buffer, and the P2X1 agonist α,β-methylene ATP. Data were recorded with excitation at 485 nm, emission at 525 nm, and an auto cut-off at 515 nm on a FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, California). Initial baseline readings were taken over a 20-second period, followed by stimulation, and measurements were recorded for an additional 2 min at regular intervals. For each condition, the maximum fluorescence value subtracted from the minimum fluorescence value was exported and analysed using GraphPad Prism v9 (GraphPad, La Jolla, USA). Experimental conditions were repeated multiple times, with each condition run in triplicate unless otherwise specified.
For the concentration-response data, each condition was normalised to 10 µM ionomycin, with the highest value adjusted to 100 and the lowest value to 0. Data from increasing concentrations of P2X1 receptor agonists were fitted to a log(agonist) nonlinear regression curve using a four-parameter model, with the top and bottom constrained to 100 and 0, respectively. Data from increasing concentrations of P2X1 receptor antagonists were fitted to a log(antagonist) nonlinear regression curve using a four-parameter model, with the top and bottom constrained to 100 and 0, respectively. EC50 and IC50 values were extracted from individual experiments, grouped and plotted on column graphs. These data were analysed using a one-way ANOVA with a Dunnett’s multiple comparison test to determine if there was a significant difference (<0.05) between different conditions and the control response. For the desensitisation data, a maximal concentration of α,β-methylene ATP was applied to HEK293 cells expressing WT-P2X1 or mutant P2X1 receptors, with the fluorescence trace normalised to a maximum response of 100% and a minimum response of 0%. Data readings were taken every second and plotted as a line from three independent experiments conducted in triplicate. The 50% decay rate of the P2X1 receptor and mutant P2X1 receptors was calculated based on the time taken to decrease from 100% to 50% fluorescence response. Statistical significance (<0.05) compared to WT-P2X1 was determined using a one-way ANOVA followed by Dunnett’s multiple comparison test.
Kinase-Glo ATP-based luminescent assay
To measure ATP bound to detergent purified P2X1 receptor, an ATP luminescent kinase assay was used. Detergent purified P2X1 receptor was incubated with 0.3 µL of apyrase for 20 min to remove ATP in solution and then heated at 70 °C for 5 min to release bound ATP and degrade apyrase. To establish conditions for ATP removal from detergent-purified P2X1 receptor samples, the receptor was pre-treated with different P2X1 receptor antagonists for 2 h, followed by a 20-min incubation with apyrase. Controls including ATP and ATP apyrase conditions were included in these experiments, and all incubation steps were carried out at 4 °C. Samples were plated at 20 μL total volume into solid white 384 well plates with a 1:1 ratio to Kinase-Glo reagent. Luminescence is recorded on a Lumistar at room temperature (BMG LABTECH, Germany) and graphed in GraphPad Prism v9. Measurements are recording in relative light units (RLU).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All cryo-EM maps for the human P2X1 receptor in the ATP-bound and NF449-bound states were deposited in the Electron Microscopy Data Bank (EMDB) under the accession codes: EMD-44299 (ATP-P2X1) and EMD-44370 (NF449-P2X1). The cryo-EM maps include half maps, masks, sharpened, and unsharpened maps, along with validation metrics. The corresponding atomic coordinates for the P2X1 receptor structures were deposited in Protein Data Bank under the PDB accession codes: 9B73 (ATP-P2X1) and 9B95 (NF449-P2X1). Previously published structures referenced in this study can be accessed via accession codes: 5SVL, 5SVQ, 5SVR, 8JV8, 5XW6, 5YVE, 8JV5, 5U1X, 6U9W, and 4DW1. The source data underlying Figs. 4, 5, and 6, as well as Supplementary Figs. 5 and 6, are provided as a Source Data file. Source data are provided with this paper.
References
Burnstock, G. The therapeutic potential of purinergic signalling. Biochem. Pharmacol. 151, 157–165 (2018).
Kawate, T., Michel, J. C., Birdsong, W. T. & Gouaux, E. Crystal structure of the ATP-gated P2X 4 ion channel in the closed state. Nature 460, 592–598 (2009).
Saul, A., Hausmann, R., Kless, A. & Nicke, A. Heteromeric assembly of P2X subunits. Front. Cell. Neurosci. 7, 250 (2013).
North, R. A. Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1068 (2002).
Illes, P. et al. Update of P2X receptor properties and their pharmacology: IUPHAR Review 30. Br. J. Pharmacol. 178, 489–514 (2021).
Wang, J. et al. Druggable negative allosteric site of P2X3 receptors. Proc. Natl. Acad. Sci. 115, 4939–4944 (2018).
Akira, K. & Toshimitsu, K. Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 5 (2016).
Motoyuki, H. & Eric, G. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485, 207 (2012).
McCarthy, A. E., Yoshioka, C. & Mansoor, S. E. Full-length P2X7 structures reveal how palmitoylation prevents channel desensitization. Cell 179, 659–670.e13 (2019).
Mansoor, S. E. et al. X-ray structures define human P2X3 receptor gating cycle and antagonist action. Nature 538, 66 (2016).
Shen, C. et al. Structural insights into the allosteric inhibition of P2X4 receptors. Nat. Commun. 14, 6437 (2023).
Bennetts, F. M., Mobbs, J. I., Ventura, S. & Thal, D. M. The P2X1 receptor as a therapeutic target. Purinergic Signal 18, 421 (2022).
White, C. W. et al. Male contraception via simultaneous knockout of α1A-adrenoceptors and P2X1-purinoceptors in mice. Proc. Natl. Acad. Sci. USA. 110, 20825–20830 (2013).
Mulryan, K. et al. Reduced vas deferens contraction and male infertility in mice lacking P2X1 receptors. Nature 403, 86 (2000).
Darbousset, R. et al. P2X1 expressed on polymorphonuclear neutrophils and platelets is required for thrombosis in mice. Blood 124, 2575–2585 (2014).
Oury, C. & Wéra, O. P2X1: a unique platelet receptor with a key role in thromboinflammation. Platelets 32, 902–908 (2021).
del, Carmen, Gonzalez-Montelongo, M., Meades, J. L., Fortuny-Gomez, A., Fountain, S. J. & Neuropeptide, Y. Direct vasoconstrictor and facilitatory effects on P2X1 receptor-dependent vasoconstriction in human small abdominal arteries. Vascul. Pharmacol. 151, 107192 (2023).
He, X. et al. P2X1 enhances leukemogenesis through PBX3-BCAT1 pathways. Leukemia 37, 265–275 (2023).
Ledderose, S. et al. P2X1 and P2X7 receptor overexpression is a negative predictor of survival in muscle-invasive bladder cancer. Cancers 15, 2321 (2023).
Tian, M. et al. Discovery and structure relationships of salicylanilide derivatives as potent, non-acidic P2X1 receptor antagonists. J. Med. Chem. 63, 6164–6178 (2020).
Obrecht, A. S. et al. Identification of aurintricarboxylic acid as a potent allosteric antagonist of P2X1 and P2X3 receptors. Neuropharmacology 158, 107749 (2019).
Kim, Y.-C. et al. Structure−activity relationships of pyridoxal phosphate derivatives as potent and selective antagonists of P2X1 receptors. J. Med. Chem. 44, 340–349 (2001).
Jung, K.-Y., Cho, J.-H., Lee, J. S., Kim, H. J. & Kim, Y.-C. Synthesis and structure–activity relationships of carboxylic acid derivatives of pyridoxal as P2X receptor antagonists. Bioorg. Med. Chem. 21, 2643–2650 (2013).
Jung, Y.-H. et al. Discovery of potent antiallodynic agents for neuropathic pain targeting P2X3 receptors. ACS Chem. Neurosci. 8, 1465 (2017).
Hülsmann, M. et al. NF449, a novel picomolar potency antagonist at human P2X1 receptors. Eur. J. Pharmacol. 470, 1–7 (2003).
Kassack, M. U. et al. Structure–activity relationships of analogues of NF449 confirm NF449 as the most potent and selective known P2X1 receptor antagonist. Eur. J. Med. Chem. 39, 345–357 (2004).
Hohenegger, M. et al. Gsα-selective G protein antagonists. Proc. Natl. Acad. Sci. 95, 346–351 (1998).
Farmer, L. K., Schmid, R. & Evans, R. J. Use of chimeras, point mutants, and molecular modeling to map the antagonist-binding site of 4,4′,4″,4‴-(Carbonylbis-(imino-5,1,3-benzenetriylbis(carbonylimino)))tetrakisbenzene-1,3-disulfonic Acid (NF449) at P2X1 receptors for ATP. J. Biol. Chem. 290, 1559–1569 (2015).
Chataigneau, T., Lemoine, D. & Grutter, T. Exploring the ATP-binding site of P2X receptors. Front. Cell. Neurosci. 7, 273 (2013).
Bianchi, B. R. et al. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur. J. Pharmacol. 376, 127–138 (1999).
Varani, K. et al. Binding thermodynamic characterization of human P2X1 and P2X3 purinergic receptors. Biochem. Pharmacol. 75, 1198–1208 (2008).
Roberts, J. A., Valente, M., Allsopp, R. C., Watt, D. & Evans, R. J. Contribution of the region Glu181 to Val200 of the extracellular loop of the human P2X1 receptor to agonist binding and gating revealed using cysteine scanning mutagenesis. J. Neurochem. 109, 1042–1052 (2009).
Allsopp, R. C. & Evans, R. J. The intracellular amino terminus plays a dominant role in desensitization of ATP-gated P2X receptor ion channels. J. Biol. Chem. 286, 44691–44701 (2011).
Sim, J. A., Broomhead, H. E. & North, R. A. Ectodomain lysines and suramin block of P2X1 receptors. J. Biol. Chem. 283, 29841 (2008).
El-Ajouz, S., Ray, D., Allsopp, R. & Evans, R. Molecular basis of selective antagonism of the P2X1 receptor for ATP by NF449 and suramin: contribution of basic amino acids in the cysteine-rich loop. Br. J. Pharmacol. 165, 390–400 (2012).
Catia, L. et al. Medicinal chemistry of P2X receptors: agonists and orthosteric antagonists. Curr. Med. Chem. 22, 915–928 (2015).
Li, M. et al. Molecular mechanisms of human P2X3 receptor channel activation and modulation by divalent cation bound ATP. eLife 8 (2019).
Kasuya, G. et al. Structural Insights into Divalent Cation Modulations of ATP-Gated P2X Receptor Channels. Cell Rep. 14, 932–944 (2016).
Gucwa, M. et al. CMM—An enhanced platform for interactive validation of metal binding sites. Protein Sci. Publ. Protein Soc. 32, e4525 (2023).
Zheng, H. et al. CheckMyMetal: a macromolecular metal-binding validation tool. Acta Crystallogr. D: Struct. Biol. 73, 223–233 (2017).
Li, M., Silberberg, S. D. & Swartz, K. J. Subtype-specific control of P2X receptor channel signaling by ATP and Mg2+. Proc. Natl. Acad. Sci. 110, E3455–E3463 (2013).
Sheng, D. et al. Structural insights into the orthosteric inhibition of P2X receptors by non-ATP-analog antagonists. eLife 12 (2023).
Kasuya, G. et al. Structural insights into the competitive inhibition of the ATP-gated P2X receptor channel. Nat. Commun. 8, 876 (2017).
Jarvis, M. F. et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc. Natl. Acad. Sci. USA. 99, 17179–17184 (2002).
Scott, D. J. et al. A novel ultra-stable, monomeric green fluorescent protein for direct volumetric imaging of whole organs using CLARITY. Sci. Rep. 8, 667 (2018).
Kampjut, D., Steiner, J. & Sazanov, L. A. Cryo-EM grid optimization for membrane proteins. iScience 24, 102139 (2021).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50, D439–D444 (2022).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. Publ. Protein Soc. 32, e4792 (2023).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D: Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D: Biol. Crystallogr. 66, 213–221 (2010).
Williams, C. J. et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. Publ. Protein Soc. 27, 293–315 (2018).
Pravda, L. et al. MOLEonline: a web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 46, W368–W373 (2018).
Acknowledgements
This work was funded by the National Health and Medical Research Council of Australia Investigator Grant (1196951): D.M.T. The cryo-EM imaging and sample vitrification were performed at the Monash University Ramaciotti Centre for cryo-electron microscopy. The cryo-EM data processing and data storage were done using the Monash University MASSIVE high-performance computing facility and supercomputing resources.
Author information
Authors and Affiliations
Contributions
D.M.T., S.V., and J.I.M. designed the overall research. F.M.B. designed, expressed, and purified protein samples. F.M.B performed negative-stain EM. F.M.B., J.I.M., A.G., H.V. performed sample vitrification and cryo-EM imaging. F.M.B. processed the EM data and generated and analysed atomic models. F.M.B. generated DNA constructs and performed and analysed pharmacology experiments. D.M.T., S.V., and J.I.M. provided supervision. F.M.B. wrote the manuscript with contributions and input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Toshimitsu Kawate and Mark Young for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bennetts, F.M., Venugopal, H., Glukhova, A. et al. Structural insights into the human P2X1 receptor and ligand interactions. Nat Commun 15, 8418 (2024). https://doi.org/10.1038/s41467-024-52776-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52776-7
