
Abstract
The route of oncolytic virotherapy is pivotal for immunotherapeutic efficacy in advanced cancers. In this preclinical study, an oncolytic reovirus (RC402) is orally administered to induce antitumor immunity. Oral reovirus treatment shows no gross toxicities and effectively suppresses multifocal tumor lesions. Orally administered reovirus interacts with the host immune system in the Peyer’s patch of the terminal ileum, increases IgA+ antibody-secreting cells in the lamina propria through MAdCAM-1+ blood vessels, and reshapes the gut microbiome. Oral reovirus promotes antigen presentation, type I/II interferons, and T cell activation within distant tumors, but does not reach or directly infect tumor cells beyond the gastrointestinal tract. In contrast to intratumoral reovirus injection, the presence of the gut microbiome, Batf3+ dendritic cells, type I interferons, and CD8+ T cells are indispensable for orally administered reovirus-induced antitumor immunity. Oral reovirus treatment is most effective when combined with αPD-1(L1) and/or αCTLA-4, leading to complete colon tumor regression and protective immune memory. Collectively, oral reovirus virotherapy is a feasible and effective immunotherapeutic strategy in preclinical studies.
Similar content being viewed by others


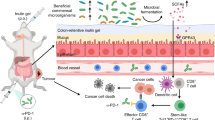
Introduction
Oncolytic viruses (OVs) are therapeutic agents that have been selected or genetically engineered to specifically infect and lyse tumor cells without affecting normal cells1,2,3,4,5. The tumor selectivity of OVs is owing to the parallel interplay of oncogenic mechanisms and viral signaling, which renders cancer cells vulnerable to viral infection6,7. Cancer cells evade immune surveillance by suppressing interferon (IFN) signaling and IFN is a critical component of antiviral defense. Therefore, cancer cells have intrinsic vulnerability to viral infection8,9. Moreover, dysregulation of RAS, TP53, and RB signaling pathways in cancer cells increases their susceptibility to viral infection10,11.
Reoviruses are a class of OVs that cause self-limited asymptomatic or mild infections of the upper respiratory and gastrointestinal (GI) tracts in humans12,13,14. They are non-enveloped viruses with 10 double-stranded RNA segments encoding proteins that allow them to evade antiviral IFN responses15,16. Reovirus σ1 binds with high affinity to JAM-1, which is overexpressed on the surface of various cancer cells, resulting in receptor-mediated endocytosis of reovirus17. Moreover, reoviruses preferentially replicate in transformed tumor cells compared with normal cells because of the suppressed antiviral defense, increased proteolytic activity, and dysregulated protein synthesis often present in cancer cells18,19. Reoviruses induce tumor cell oncolysis by triggering various cell death mechanisms20,21. Reoviral dsRNA stimulates TRAIL through RNA sensors such as RIG-1 and MDA522,23. TRAIL then binds to surface death receptors and activates apoptosis through caspase-3 and caspase-724,25. Reoviruses also promote apoptosis by inhibiting Bcl-2 and activating Bax/Bak in tumor cells26,27. Furthermore, they can induce necroptosis by type I IFN responses and autophagy by acute endoplasmic reticulum stress28,29. In addition to direct oncolysis, tumor microenvironment (TME) remodeling and induction of antitumor immunity are likely to be involved in the antitumor effects of reoviruses, yet these mechanisms remain largely unexplored30.
RC402 (ReoCure) is a clinical formulation of the unmodified reovirus T3D strain31. It selectively infects various types of cancer cells and induces tumor cell death32,33. Preclinical studies show that RC402 has strong antitumor and antiangiogenic efficacies in vivo12,34. Recently, a first-in-human phase I (1a/1b) trial of RC402 as a monotherapy or in combination with pembrolizumab was conducted in patients with advanced solid tumors and RC402 is generally well tolerated with no dose-limiting toxicities nor clinically significant adverse events (Registration Number ACTRN12620000777998)35. However, further studies are required to determine the mechanism of immunotherapeutic activity of RC402, and optimize RC402-based immunotherapy.
Intratumoral (IT) or intravenous (IV) administration of reoviruses was performed in previous clinical trials36,37. However, these administration routes have limitations for clinical application. IT injection is an invasive procedure that is operator-dependent, thus impeding patient recruitment in clinical trials38. Most IT injections are repeated at regular intervals that increases the cumulative risk of procedure-related complications such as tumor rupture or hemorrhage over time39. Moreover, while IT injections can elicit direct immune responses in the local TME, they are insufficient to induce systemic responses in distant un-treated metastases40,41. Meanwhile, IV injection reoviral therapy is a systemic treatment that allows for rapid and widespread distribution of the virus42,43. However, it can elicit non-specific systemic inflammatory responses and induce rapid viral clearance by host immunity that can compromise antitumor efficacy of reoviral therapy44,45. Moreover, IV injection can provoke very strong antiviral antibodies compared to IT injections46.
Here, we demonstrated that the oral administration of reovirus is a feasible and safe oncolytic virotherapy that maximizes immunotherapeutic efficacy in murine colon cancer and melanoma. Oral RC402 treatment effectively reduced multifocal tumor burden and enhanced immune checkpoint blockade (ICB). It mostly depended on gut microbiome remodeling and antitumor T cell immunity to exert its antitumor efficacy, but not on direct oncolysis (which is a dominant mechanism in IT injection). Overall, this study can serve as a scientific rationale for future clinical trials of oral reovirus virotherapy.
Results
Oral administration of reovirus shows potent antitumor efficacy on a multifocal tumor model
CT26 tumor cells were injected into the right flank of mice and RC402 was administered intratumorally or orally (Fig. 1A); IT RC402 showed superior tumor growth suppression compared with oral RC402 (mean −72.9% vs. −46.1%) (Fig. 1B, C). Consistently, IT RC402 suppressed MC38 colon cancer growth better than oral RC402 (mean −64.4% vs. −44.2%) (Fig. 1D).

Mice were subcutaneously implanted with CT26 or MC38 tumor cells in single (A–D) or multifocal (E–H) lesions and treated with RC402 (R) and/or αPD-1 (PD1). A Schematic diagram of the treatment schedule in single tumor-bearing mice. Arrows indicate treatments. B Comparison of tumor growth in single tumor-bearing mice treated with phosphate buffered saline (PBS, n = 5), intratumoral (IT) RC402 (n = 6), or oral RC402 (n = 6). C Comparison of tumor growth suppression in single tumor-bearing mice. Each bar indicates the % change in each tumor volume compared to the mean of control tumor volumes at the end of treatment. D Comparison of MC38 tumor growth in single tumor-bearing mice treated with PBS, IT RC402, or oral RC402 (n = 6 per group). E Schematic diagram of the treatment schedule in multifocal tumor-bearing mice. The tumor in the right upper quadrant was labeled as tumor 1, and others are labeled as 2, 3, and 4 in a counterclockwise direction. Mice were treated with PBS, IT RC402, or oral RC402, and/or αPD-1. Arrows indicate treatments. Comparison of tumor growth suppression in multifocal tumor-bearing mice treated with IT RC402 (F) or oral RC402 (G) (n = 5 per group). Each bar indicates the % change in each tumor volume compared to the mean of control tumor volumes at the end of treatment. H Comparison of the sum of all tumor volumes per mouse. Overall tumor volume was calculated by summing the volume of every tumor per mouse and normalizing it to the mean overall tumor volume of the control group. Dotter line indicates the mean of overall tumor burden in control group (n = 5 per group). Data are pooled from two independent experiments. Values are expressed as the mean ± standard deviation (SD). Two-tailed Student’s t test and ANOVA with Tukey post-hoc test were used. Source data are provided as a Source Data file.
Patients with a single tumor mass are eligible for local treatment such as surgical resection or IT treatment, but the majority of patients with advanced cancers have multifocal metastases, even hundreds, and are not eligible for such local treatment. To compare the antitumor efficacy of IT and oral treatment in an advanced cancer setting, a multifocal tumor model of CT26 colon cancer was generated by subcutaneous implantation of tumor cells into each quadrant of the dorsal skin of mice and the mice were treated with IT or oral RC402 and/or αPD-1 ICB (Fig. 1E). In the IT RC402 group, RC402 was IT-injected into tumors located in the right upper quadrant (tumor 1), and PBS was orally administered using a feeding needle (Fig. 1F). In the oral RC402 group, RC402 was orally administered and PBS was IT-injected (Fig. 1G). The total number of implanted tumor cells and the total amount of RC402 administered per mice was the same between the IT and oral treatment group. IT RC402 had the strongest anti-tumor effect in the RC402-injected tumor (tumor 1, Fig. 1F). Meanwhile, oral RC402, showed a relatively even anti-tumor effect in all tumors, whether as a monotherapy or in combination with αPD-1 ICB (Fig. 1G). Regarding the sum of all tumor volumes per mouse, oral RC402 treatment significantly reduced tumor burden, both as monotherapy and in combination with αPD-1 ICB, in contrast to IT RC402. (Fig. 1H). Taken together, oral RC402 treatment is inferior to IT treatment when there is only one tumor lesion, but is superior to IT treatment when there are multifocal tumor lesions.
Entry of oral reovirus in Peyer’s patches elicits strong adaptive immunity within the tumor
We further investigated the effects of oral administration of reovirus on tumor-bearing mice, especially on the host immune system. We examined the intestinal passage and distribution of orally administered RC402 labeled with AF647 fluorescence dye (Fig. 2A). RC402 underwent rapid gastric transit, followed by the majority of RC402 fluorescence observed in the small intestine from 3 h, and the highest level was found in the terminal ileum 12 h after oral administration; however, it was not detected in other organs (Fig. 2B, C). Histologic analyses revealed that the major interface between RC402 and the host immune system were Peyer’s patches (PP) within the terminal ileum where RC402 crossed the mucosal barrier and interacted with host immune cells within 24 h of oral administration (Fig. 2D). However, the virus itself was not observed in lymphoid organs such as mesenteric lymph nodes and spleen, and was even absent in tumor tissue (Fig. 2E, F). RC402 did not directly migrate to lymph nodes and tumor tissue; however, it induced extensive immune remodeling inside lymph nodes and tumor tissue, as well as PP (Fig. 2G). RC402 activated innate and adaptive immunity in PP, lymph nodes, and tumor tissue (Fig. 2H). Of note, innate immune activation was most pronounced in PP, while adaptive immune activation was most prominent in tumor tissues (Fig. 2H, I). RC402 induced dendritic cell markers in PP and tumor tissues, and enhanced antigen processing and presentation by inducing transporter associated with antigen processing 1 (Tap1) and major histocompatibility complex (MHC) class I genes, but not MHC class II genes in PP. Notably, RC402 strongly induced type I (Ifna1, Ifnb1) and type II (Ifng) interferons (IFNs) and Th1/Th2 genes, resulting in strong T cell activation within the tumors (Fig. 2I). Intriguingly, immune checkpoints such as Pd-1, Pd-l1, and Ctla-4 were induced within the tumor after RC402 treatment, compared with PP and lymph nodes (Fig. 2I). Moreover, we observed that the expression of genes related to tertiary lymphoid structure, such as Ltb, Light, Ccl19, and Cxcl13, increased after oral RC402 treatment (Supplementary Fig. 1A).

CT26 tumor-bearing mice were orally administered with AF647-fluorescent labeled RC402 and the reovirus distribution was serially monitored. A Schematic diagram depicting the gastrointestinal (GI) migration and distribution of orally administered RC402. B, C Representative images and quantifications of the fluorescent labeled RC402 localization in each organ over time (n = 3 per group). D Representative images showing reovirus and CD45+ leukocytes within terminal ileum and Peyer’s patch (PP). Arrows indicate RC402 infected mucosal epithelial cells. E Representative images showing the absence of RC402 within mesenteric lymph nodes (LN), spleen, and tumor tissues. F Comparison of reovirus transcripts within PP, LN, spleen, and tumor tissue after oral RC402 treatment (n = 4 per group). G Volcano plot showing transcriptome changes in reovirus-treated PP, LN, and tumor tissues compared with PBS-treated matched-control tissues after 7 days of oral RC402 treatment (n = 3 per group). Colored dots indicate significantly upregulated genes after oral RC402 treatment (one-tailed Student’s t test). Red lines denote P = 0.05. H Enrichment of Gene Ontology (GO) biological processes in each tissue after 7 days of oral RC402 treatment (n = 3 per group, one-tailed Fisher’s Exact test). I Heatmap showing the expression of genes related to innate immunity, interferons (IFNs), Th1/Th2 responses, T cell activation, immune checkpoints, and tumor microenvironment after 7 days of RC402 treatment (n = 3 per group). Values are fold changes in RC402-treated tissues compared with matched-control tissues. Data are pooled from two independent experiments. Values are expressed as the mean ± SD. Scale bars, 100 μm. Source data are provided as a Source Data file.
To validate the specificity of oral RC402 treatment, we compared oral treatment of RC402, inactivated RC402, and myxoma virus as other type of oncolytic virus (Supplementary Fig. 1B). Oral treatment with inactivated RC402 or myxoma virus, unlike conventional RC402 treatment, did not significantly suppress tumor growth (Supplementary Fig. 1C). In addition, inactivated oral RC402 or oral myxoma virus treatment partially upregulated a limited number of immune-related genes in PP and tumor tissues; however, the magnitude of these changes was considerably lower than that of conventional RC402 treatment (Supplementary Fig. 1D).
To determine whether IFN treatment was comparable to oral RC402 treatment, IFN-α was intraperitoneally administered in CT26 colon cancer-bearing mice (Supplementary Fig. 1E). IFN-α treatment showed weaker tumor growth suppression than that by oral RC402 (Supplementary Fig. 1F). Although IFN-α slightly elicited innate immunity in LNs and tumors, oral RC402 induced more widespread and robust immunologic changes in tumors (Supplementary Fig. 1D, G).
Overall, orally administered RC402 initially interact with the host immune system in PP of the terminal ileum. RC402 did not directly migrate and infect tumor cells beyond the GI tract, although it can trigger robust immune responses within the distant TME, especially inducing type I and type II IFNs and activating intratumoral T cells.
Oral RC402 reshapes the gut microbiome by increasing IgA+ antibody-secreting cells (ASCs) in the terminal ileum via MAdCAM-1
The gut microbiome resides in the GI tract and plays a critical role in regulating immune homeostasis47.
To explore the role of gut microbiome in modulating antitumor immunity during RC402 treatment, the tumor growth suppression by IT or oral RC402 treatment were compared in the presence or absence of the gut microbiome. A cocktail of antibiotics (ampicillin, neomycin, metronidazole, and vancomycin) was administered to deplete the gut microbiome following a previous protocol (Supplementary Fig. 2A)48. Although IT RC402 treatment showed antitumor efficacy in the absence of the gut microbiome, oral RC402 could not suppress colon tumor growth in the absence of the gut microbiome. (Fig. 3A). Therefore, the antitumor efficacy of oral RC402 was substantially mediated by the gut microbiome, unlike IT RC402.

Mice were subcutaneously implanted with CT26 tumor cells, orally administered with RC402 (RC), and gut microbiomes were analyzed using shotgun metagenomic sequencing (B–H). A The gut microbiome was depleted with a cocktail of antibiotics. Then, mice were implanted with CT26 tumor cells and treated with PBS, IT, or oral RC402. Comparison of tumor growth (n = 5 for IT PBS without antibiotics; n = 6 for IT RC402 without antibiotics; n = 6 for oral PBS or oral RC402 without antibiotics; n = 8 for other groups). B Principal coordinate analysis (PCoA) showing distinct clustering of gut microbiome in baseline and PBS- or RC402-treated mice (n = 3 for baseline; n = 4 for other groups). C Comparisons of gut microbiome diversity in terms of Shannon index. D PCoA plot showing Bray-Curtis dissimilarity. E Relative abundance of gut microbiota at the genus level (n = 4 per group). F Heatmap showing the relative abundance of gut microbiota at the genus level. Values are z-scores (n = 4 per group). G Abundance of specific genus in PBS- or RC402-treated mice. H Volcano plot showing differentially expressed microbial metabolic pathways and abundance of specific metabolic pathways between PBS- or RC402-treated mice (one-tailed Student’s t test). I–N CT26 tumor-bearing mice were treated with oral PBS or RC402 in the presence and absence of αMAdCAM-1 neutralization antibody. I Comparison of CT26 tumor growth. (n = 6 for RC402; n = 7 for other groups). J, K Representative images and comparisons of IgA+ ASCs within lamina propria of terminal ileum. L Comparisons of fecal IgA levels. Expression was normalized to total fecal protein. M, N Abundance of specific genus in PBS-, RC402-, and/or αMAdCAM-1-treated mice. Data are pooled from two independent experiments. Values are expressed as the mean ± SD. Unless otherwise denoted, two-tailed Student’s t test and ANOVA with Tukey post-hoc test were used. Scale bars, 100 μm. Source data are provided as a Source Data file.
Next, we performed a shotgun metagenomic analysis to evaluate RC402-induced changes in the gut microbiome composition and metabolic pathways. The principal coordinate analysis (PCoA) plot revealed distinct clustering between the RC402 and control-treated groups (Fig. 3B). The α-diversity, Shannon index, was significantly higher in RC402-treated versus control mice (Fig. 3C). When we analyzed different time points (0, 3, and 9 days after treatment) in a paired manner, the α-diversity was consistently maintained over time following RC402 treatment but markedly reduced in control mice during tumor progression (Supplementary Fig. 2B). The PCoA plot with Bray-Curtis dissimilarity showed differences in β-diversity for the gut microbiome between control- and RC402-treated mice (Fig. 3D). A compositional analysis revealed that the intestinal bacterial distribution changed after RC402 treatment (Fig. 3E and Supplementary Fig. 2C). Remarkably, at the genus level, the relative abundance of Bacteroides significantly decreased, whereas that of Ligilactobacillus increased (Fig. 3F, G). Furthermore, we examined the alterations in microbial metabolic pathways that may be associated with physiological functions in the GI tract. The results identified significant enrichment of the pathways in L-valine biosynthesis, inosine-5’-phosphate biosynthesis I, TCA cycle V, glycolysis IV, and stachyose degradation after RC402 treatment (Fig. 3H).
The homing of IgA+ ASCs to intestinal lamina propria by MAdCAM-1+ venules regulates the gut microbiome49. To investigate the mechanism of how RC402 regulates the gut microbiome, we compared the antitumor efficacy of oral RC402 in the presence and absence of MAdCAM-1. When MAdCAM-1+ venules were depleted by αMAdCAM-1 in Peyer’s patches and the terminal ileum (Supplementary Fig. 3A, B), the therapeutic efficacy of orally administered RC402 was almost completely abolished (Fig. 3I). Notably, oral RC402 treatment increased IgA+ ASCs in the lamina propria of the terminal ileum, but not after MAdCAM-1 neutralization (Fig. 3J, K). Consistent with this finding, oral RC402 treatment increased fecal IgA concentrations, but not after MAdCAM-1 neutralization (Fig. 3L). When we compared gut bacteria using metagenome shotgun sequencing, we observed that RC402-induced changes in gut bacteria were abolished upon MAdCAM-1 neutralization (Fig. 3M, N). Overall, the oral administration of RC402 increased IgA+ ASCs in the lamina propria of the terminal ileum through MAdCAM-1+ blood vessels and reshaped the gut microbiome.
Oral reovirus suppresses tumor growth by activating tumor-specific CD8+ T cell immunity
To further define the antitumor immune response by oral RC402 treatment, CT26 tumor-bearing mice were orally administrated with PBS or RC402 once daily. Histologic analyses of tumor tissues revealed that oral RC402 treatment increased tumor-infiltrating CD8+ T cells by 2.3-fold, granzyme B (GzB) expressing activated CD8+ T cells by 2.2-fold, and Casp3+ apoptotic tumor cells by 1.7-fold within tumors (Fig. 4A, B). Consistently, flow cytometric analyses showed an increase in CD8+ T cells, while there were no significant changes in CD4+ and CD4+Foxp3+CD25+ regulatory T cells (Fig. 4C–E). The AH-1 (SPSYVYHQF) tetramer assay showed that the tumor-specific CD8+ T cells were higher in RC402-treated CT26 tumor-specific T cells compared with control tumors (Fig. 4F). These findings were also consistently observed in MC38 colon cancer-bearing mice treated with oral RC402 (Supplementary Fig. 3C–I).

A–F Mice were subcutaneously implanted with CT26 tumor cells and treated orally with PBS or RC402. A, B Representative images and comparisons of CD8+ T cells, CD31+ tumor vasculatures, granzyme B (GzB)+ T cells, and caspase 3 (Casp3)+ apoptotic cells within tumor tissues (n = 5 per group). C, D Comparison of CD8+ T cells and CD4+ T cells in tumors (n = 5 per group). E Comparisons of CD4+Foxp3+CD25+ Tregs (n = 5 per group). F Comparison of the AH-1 tetramer+ CD8+ T cells (n = 4 per group, one-tailed Student’s t test). G MC38 tumor cells were subcutaneously implanted into wild-type (WT) (n = 7), Batf3 knockout (KO) (n = 6), and αIFNAR-1-treated WT mice (n = 7), and the mice were treated with PBS, IT, or oral RC402.Comparisons of tumor growth. H Nude mice were subcutaneously implanted with CT26 tumor cells and treated with PBS, IT, or oral RC402. Comparisons of tumor growth (n = 9 for IT PBS; n = 8 for IT RC402; n = 9 for oral PBS; n = 8 for oral RC402). I Mice were subcutaneously implanted with CT26 tumor cells and treated with oral RC402 and depleting antibodies for CD8+ T cells (αCD8) or CD4+ T cells (αCD4) (n = 5 per group). Comparisons of tumor growth. Data are pooled from two independent experiments. Values are expressed as the mean ± SD. Unless otherwise denoted, two-tailed Student’s t test and ANOVA with Tukey post-hoc test were used. Scale bars, 100 μm. Source data are provided as a Source Data file.
Type I IFNs and Batf3+ dendritic cells play a critical role in innate immune sensing of tumor cells and priming of CD8+ T cells50. To determine the extent to which they contribute to oral RC402-induced tumor immunity, we compared the effects of RC402 in Batf3-knockout mice and those with type I IFN signaling blockade using an anti-IFNAR1 antibody. In contrast with IT RC402 treatment, the effects of oral RC402 treatment were abolished in the absence of Batf3 or IFNAR1 signaling (Fig. 4G).
To explore the possible involvement of adaptive T cell immunity in the mode of action of RC402, the antitumor efficacies of IT RC402 and oral RC402 were compared in nude mice. Although IT RC402 showed antitumor efficacies in nude mice, the oral administration of RC402 did not inhibit tumor growth in nude mice (Fig. 4H). To identify the specific compartment of T cell immunity responsible for the anticancer effects of oral RC402, we compared the efficacy of oral RC402 after treatment with CD8 or CD4 neutralizing antibodies. Intriguingly, CD8 depletion completely abrogated the antitumor effects of RC402, but CD4 depletion did not affect the tumor growth inhibition by oral RC402 treatment (Fig. 4I).
Collectively, unlike IT RC402, Batf3+ dendritic cells, Type I IFNs, and CD8+ T cells are indispensable in oral RC402-induced antitumor immunity.
Oral reovirus synergizes with αPD-L1 and suppresses the progression of orthotopic tumors and colitis-associated colorectal cancers
Oral reovirus extensively remodeled the gut microbiome and TME; however, the antitumor efficacy of oral RC402 alone was moderate. Therefore, we further explored the combination treatment of RC402 with ICBs. Mice were orthotopically injected with CT26 colon cancer cells into the cecal wall and subsequently treated with oral RC402 daily and αPD-L1 every three days (Fig. 5A). The combination therapy with RC402 and αPD-L1 most potently suppressed orthotopic colon tumor growth (Fig. 5B, C). In addition, the overall survival was longest in mice receiving combined treatment with RC402 and αPD-L1 (Fig. 5D). The combination therapy group showed increased intratumoral infiltration of CD8+ cytotoxic T cells and decreased tumor vascular densities compared with those in the control group (Fig. 5E, F). In orthotopic colon cancers, reovirus was detected in tumors following oral treatment with RC402 (Supplementary Fig. 3J).

A–F Mice were orthotopically implanted with CT26 colon and treated with oral RC402 (RC) and/or intraperitoneal αPD-L1 (PDL1). A Schematic diagram showing the treatment schedule for orthotopic CT26 colon cancer. B, C Representative images and comparisons of orthotopic colon cancer growth at the end of treatment (n = 6 per group). D Kaplan−Meier curves for overall survival (n = 4 for vehicle; n = 5 for RC402; n = 6 for RC402 and αPD-L1). E, F Representative images and comparison of CD8+ T cells and CD31+ tumor vasculatures within tumor tissues (n = 6 per group). G–K Mice with colitis-associated colorectal cancers were treated with oral RC402 (R) and/or intraperitoneal αPD-L1 (P) for a longer duration. G Schematic diagram showing the treatment schedule. After the initial administration of azoxymethane (AOM), mice were orally administered with drinking water containing 3% dextran sodium sulfate (DSS) for a week every three-week interval. Oral RC402 and αPD-L1 were administered at the indicated time points. H Comparison of body weight changes from baseline (n = 4 for control; n = 8 for other groups). I Comparison of the disease activity index score (n = 4 for control; n = 8 for other groups). J Comparisons of the colon length and tumor nodules. K Kaplan−Meier curves for overall survival (n = 7 per group). L–N Mice were subcutaneously implanted with B16F10 melanoma cells and treated with oral RC402 and/or αPD-L1. L Schematic diagram showing the treatment schedule. M Comparison of B16F10 tumor growth in mice (n = 15 for vehicle; n = 10 for other groups). Mean tumor growth curves over time. N Kaplan−Meier curves for overall survival (n = 10 per group). Data are pooled from two independent experiments. Values are expressed as the mean ± SD. Two-tailed Student’s t test, ANOVA with Tukey post-hoc test, and log-rank test were used. Scale bars, 1 cm. Source data are provided as a Source Data file.
We employed the azoxymethane/dextran sulfate sodium (AOM/DSS) model of inflammatory colon cancer to confirm the long-term efficacy and safety of oral reovirus treatment on tumorigenesis51. Oral reovirus and/or αPD-L1 were administered from week 3 to week 13 from the day of AOM administration (Fig. 5G). There were no significant differences in body weight among vehicle-treated mice and RC402- or αPD-L1-treated mice (Fig. 5H). The disease activity of DSS-induced colitis significantly improved at week 12 in mice treated with the combination of RC402 and αPD-L1 compared with vehicle-treated mice (Fig. 5I). In mice administered with αPD-L1 alone, colon length was not restored and the number and size of intestinal tumor nodules showed only moderate changes. On the other hand, treatment with RC402 alone or in combination with αPD-L1 resulted in intestinal length recovery and a remarkably reduced number of tumor nodules (Fig. 5J). Furthermore, all mice treated with RC402 and αPD-L1 combination therapy survived up to 28 weeks, whereas only 40% of the vehicle-treated mice survived (Fig. 5K).
The efficacy of combination therapy was also examined in the B16F10 melanoma model (Fig. 5L). Combination immunotherapy of RC402 and αPD-L1 showed the most potent tumor suppression and the longest overall survival in B16F10 melanoma-bearing mice (Fig. 5M, N).
Triple combination immunotherapy of oral reovirus with αPD-1 and/or αCTLA-4 leads to complete tumor regression and establishes protective immune memory
We further evaluated the potential of oral RC402-based combination immunotherapy. CT26 tumor cells were inoculated on the flank of BALB/c mice and treated with oral reovirus and intraperitoneal αPD-1 and/or αCTLA-4 (Fig. 6A). RC402 or αPD-1 monotherapy could not induce CT26 tumor regression, while complete tumor regression was noted in 33.3% of the dual combination group (RC402 and αPD-1) and 66.7% of the triple combination therapy group (RC402, αPD-1, and αCTLA-4) without gross toxicities (Fig. 6B and Supplementary Fig. 3K). When the gut microbiome was depleted by a cocktail of antibiotics, the anti-cancer efficacy of the triple combo was reduced to a complete regression rate of 25%. Thus, the effectiveness of RC402-based triple combination immunotherapy was dependent on the presence of the gut microbiome (Fig. 6B). Moreover, mice in the triple combination immunotherapy group revealed an extensive infiltration of CD8+ cytotoxic T cells into the tumor and a reduction in tumor vascular densities within the tumor (Fig. 6C, D). GzB+ activated T cells and Casp3+ apoptotic tumor cells were most highly increased in tumors treated with triple combination immunotherapy compared with control tumors (Fig. 6C, D). Collectively, oral RC402 combined with αPD-1 and αCTLA-4 ICB showed the most potent immunotherapeutic efficacy that can completely eradicate established tumor nodules.

A–D Mice were subcutaneously implanted with CT26 tumor cells and treated with RC402 (RC), αPD-1 (P), αCTLA-4 (C) and/or a cocktail of antibiotics. A Schematic diagram of the treatment schedule. B Comparison of CT26 tumor growth. The number of tumor-free mice is indicated. C, D Representative images and comparisons of CD8+ T cells (n = 7 for control; n = 6 for other groups), CD31+ tumor vasculatures (n = 7 for control; n = 6 for other groups), GzB+ activated CD8+ T cells (n = 5 per group), and Casp3+ apoptotic cells (n = 5 per group) within tumors. E–K CT26 tumor-bearing mice treated with the triple combination therapy of oral RC402, αPD-1, and αCTLA-4. Mice experiencing complete regression (CR) of tumors were rechallenged with CT26 tumor cells (n = 5 for naïve; n = 4 for CR). E Scheme of the triple combination immunotherapy and tumor rechallenge. F Comparison of tumor growth in naïve mice or mice experiencing CR with combination immunotherapy. G Representative plot showing the tumor-specific AH-1 tetramer+ fraction of CD8+ T cells in the spleen. H, I Comparison of CD8+ T cell and AH-1 tetramer+ CD8+ T cell fractions in the spleen. J, K Representative plot and comparison of the naïve, central memory (CM, CD44+CD62L+), and effector memory (EM, CD44+CD62L−) T cells among CD8+ T cells within the spleen. Data are pooled from two independent experiments. Values are expressed as the mean ± SD. Two-tailed Student’s t test was used. Scale bars, 100 μm. Source data are provided as a Source Data file.
We reinjected CT26 tumor cells to mice that experienced complete regression of CT26 tumors (CR mice) four weeks after RC402 combination immunotherapy and monitored the tumor growth to confirm whether tumor-specific immune memory is elicited with oral RC402-based immunotherapy (Fig. 6E). Naïve mice showed robust tumor growth, while CR mice did not show tumor regrowth and remained tumor-free for over six months (Fig. 6F). Moreover, CR mice had increased CD8+ T cells that can specifically bind to AH-1 peptide, the major tumor rejection antigen of CT26 colon cancer (Fig. 6G–I). Furthermore, CR mice had more central and effector memory T cells in CD8+ T cells compared with naïve mice (Fig. 6J, K). Collectively, combination immunotherapy of RC402 and ICBs induced durable and protective antitumor immunity in colon cancer.
Discussion
Oncolytic viruses are promising antitumor agents that selectively infect and lyse cancer cells and elicit antitumor immunity4. Reovirus is an OV that was evaluated in a number of preclinical and clinical trials for various malignancies. To date, most preferred routes of reovirus administration in clinical trials are IT or IV injection. However, these routes are limited to induce optimal efficacy of reovirus therapy. Therefore, it is critical to provide an optimal route of administration that can maximize the immune regulatory efficacy of reovirus within the TME.
Here, we employed orally available reovirus (RC402) to trigger robust antitumor immunity and enhance ICB therapy. Oral RC402 showed several mechanistic features to establish antitumor immunity. Orally administered RC402 interacted with the host immune system in PP of the terminal ileum, increased IgA+ ASCs in the lamina propria of the terminal ileum through MAdCAM-1+ blood vessels, and reshaped the gut microbiome. Intriguingly, the antitumor efficacy of oral RC402 was substantially dependent on the gut microbiome, unlike IT RC402. Oral RC402 induced antitumor immunity through Batf3+ cells, type I IFNs, and CD8+ T cells. Furthermore, the combination immunotherapy of oral RC402 with αPD-1(L1) and/or αCTLA-4 induced durable and protective antitumor immunity in colon cancer.
The gut microbiome is important in the regulation of antitumor immunity and impacts treatment response, resistance, and adverse events of ICB therapy52. Intestinal ASCs secrete IgA and regulate the balance between the host and gut microbiota49,53. ASCs express the α4β7 integrin and migrate to the intestinal lamina propria and gastrointestinal-associated lymphoid tissues through specialized venules that express MAdCAM-1, an α4β7 ligand54. According to a recent study by Tyler et al., MAdCAM-1 blockade depletes ASCs in the intestinal lamina propria, thus decreasing the alpha diversity of the gut microbiome and altering the microbiota’s composition. Moreover, MAdCAM-1 blockade raised plasma IgA while lowering fecal IgA levels49. Additionally, Fidelle et al. also reported that MAdCAM-1 modulates gut microbiome-mediated systemic antitumor immunity during ICB treatment55. Our study consistently demonstrated that systemic antitumor immunity is enhanced by oral RC402 treatment, and that MAdCAM-1 and ASCs appear to be involved in this process. Oral RC402 increased ASCs in the ileal lamina propria and altered the gut microbiota; MAdACM-1 was indispensable in this process. To validate these results in humans, additional analysis of RC402-induced alterations in the gut microbiome and in fecal or plasma IgA is warranted in oral RC402 clinical trials.
The mechanisms of how the gut microbiome affects distant TME are not fully elucidated. However, the potential mechanisms are postulated to be involved in this process. First, microbial-associated molecular patterns (MAMPs) secreted by commensal bacteria and the reovirus may have engaged various pattern recognition receptors (PRRs) expressed by host immune cells to stimulate the secretion of proinflammatory cytokines such as interferons in myeloid cells56,57. Second, specific epitopes expressed by RC402 or altered intestinal commensal bacteria may share homology with tumor-derived epitopes. For example, the SVY and TMP1 epitopes derived from bacteria exhibit homology to the tumor antigens SIY and PSMB458. Therefore, microbe-specific T cells may have recognized and attacked tumor cells expressing homologous antigens through this antigen mimicry. Thirdly, the gut microbiota-produced metabolites are presumed to modulate the host immune systems59. Therefore, the microbiome changes by oral reovirus therapy may have affected antitumor immunity through immunomodulatory metabolites.
Because reoviruses commonly infect humans, the prevalence of antireoviral antibodies has been reported to be as high as 24–50% in normal population studies60. However, it is not well understood how previous reovirus infection and the presence of antireoviral antibodies affect the outcomes of oncolytic reovirus treatment, such as RC402. In the phase I clinical trials of RC402, 33% of all patients had antireoviral antibodies prior to RC402 treatment35. Following RC402 treatment, antireoviral antibody titer increased in all patients, regardless of pre-treatment antireoviral antibody status, with a peak increase after 21 days of treatment, followed by a gradual decline thereafter. There were no significant differences in anti-reovirus antibody titers between responders and non-responders at baseline or during RC402 treatment in this phase I study. Therefore, the presence or absence of antireoviral antibodies does not appear to be related to the therapeutic efficacy of RC402; however, this needs to be confirmed in a larger number of patients.
Oral RC402 regulates tumor immunity through gut microbiome regulation. However, clinical indications of oral RC402 do not need to be limited to GI tract cancers. Oral RC402 induced systemic immune responses beyond the GI tract; it was effective as a monotherapy or in combination with αPD-L1 ICB in non-GI tract origin cancers such as melanoma in this study. Consistently, in a phase I study of RC402 showed that two patients with cutaneous squamous cell carcinoma experienced complete responses. Therefore, further preclinical and clinical validation need to be expanded to various cancer types of non-GI tract origin.
In this study, we focused on the effects of oral RC402 on distant tumors, but this does not mean that oral RC402 has no direct anti-tumor effects on GI tumors. Rather, the orthotopic colon cancer model demonstrated the possibility that RC402 could directly infect primary tumors in the GI tract and exert anti-tumor effects (Fig. 5K and Supplementary Fig. 3J). Therefore, future studies need to clarify the direct oncolytic effect of oral RC402 on primary GI cancers.
Our multifocal tumor model is a preliminary model for hypothesis generation and suggests that oral therapy may be as effective as IT therapy in certain conditions, but it has limitations. The treatment effect was the strongest in tumor 1, into which RC402 or PBS was IT injected directly, and the weakest in tumor 4, which was located on the same side as tumor 1. This was consistently observed when IT RC402 or IT PBS was injected. It is possible that the IT injection of the fluid itself increased the interstitial fluid pressure within the injected tumor. As tumors 1 and 4 are located on the same side and share a lymphatic drainage system (e.g., left lymphatic vessel), the increased interstitial pressure in tumor 1 may have affected the lymphatic drainage and tumor growth of tumor 4. It was recently reported that changes in lymphatic drainage can promote peritumoral edema and alter tumor immunity, thereby promoting cancer growth61. Therefore, we cannot exclude the possibility that the IT injection itself affected the tumors on the ipsilateral side, which could be a limitation of the multifocal tumor implantation model. Although this phenomenon has been observed in mice, further studies are needed to determine if it will be observed in humans, who have a much larger and more complex vascular-lymphatic system than mice.
In conclusion, our results indicate that oral administration of RC402 is an effective strategy to elicit robust antitumor immunity through extensive rewiring of the host immunity and the gut microbiome. Moreover, oral RC402 can enhance the efficacy of ICB, leading to tumor eradication and protective immune memory. Further preclinical and clinical investigations are warranted to optimize reovirus-based combination immunotherapies.
Methods
Mice and cell lines
All mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC approval No. #210124) of CHA University and were carried out in accordance with the approved protocols. Seven- to eight-week-old male BALB/c, BALB/c nude, or C57BL/6 mice were purchased from Orient Bio Inc. (Seongnam, Korea). Seven- to eight-week-old male B6.129S(C)-Batf3tm1Kmm/J (Batf3 KO) (#013755) mice were purchased from The Jackson Laboratory. Mice were housed in a specific pathogen-free facility of CHA University (Seongnam, Korea). All mice were fed standard autoclaved laboratory chow and water ad libitum under 12 h light/dark cycle, ambient temperature of 23–25 °C, and 50–70% humidity. CT26 and MC38 colon cancer cells and B16F10 melanoma cells were obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea, #80009 and #80008) and the National Cancer Center (Goyang, Korea). BHK-21 and RK13 cells were originally obtained from ATCC (#CCL-10 and #CCL-37). These cells were maintained at 37 °C under 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cell lines were authenticated by the company providing the cell lines, routine morphology was checked, and low-passage cells were used for all experiments.
Construction and production of virus
RC402 was provided by ViroCure Inc. (Seoul, Korea), and is the Dearing strain of reovirus serotype 3 derived from Korea Bank for Pathogenic Viruses31. This virus was propagated in BHK-21 cells and purified by cesium chloride ultracentrifugation according to the established protocol. Wild-type myxoma virus, the Lausanne strain derived from ATCC, was also provided by ViroCure Inc. and was produced in RK13 cells and purified by sucrose cushion ultracentrifugation62. The virus titer was measured using the median tissue culture infectious dose (TCID50) assay63. Briefly, RC402-infected BHK-21 or myxoma virus-infected RK13 cells were serially titrated, and the cytopathogenic effects were measured in each cell six days later and TCID50 calculated.
Tumor models and treatment regimens
Tumors were implanted via subcutaneous injection of 2 × 105 CT26 or 5 × 104 MC38 cells into the right flank of mice. Mice were orally and intratumorally administrated RC402 or the same volume of PBS daily when tumor volume exceeded 50 mm3. Orthotopic implantation involved injecting 1 × 106 CT26 cells into the cecum wall, or subcutaneously injecting 1 × 105 B16F10 cells into the right flank of mice. The tumors were measured with a digital caliper, and tumor volumes were calculated using the following modified ellipsoid formula: 1/2 × (length × width2). The mice were euthanized when the size of any single tumor exceeded 10% of body weight. AOM/DSS-induced colitis involved intraperitoneal injection of mice were with 12 mg/kg AOM (Sigma-Aldrich), followed by the administration of 2.5% DSS 7 days later. The 2.5% DSS administration was repeated four times during the entire treatment period (weeks 2, 5, 8, and 11). The Disease Activity Index (DAI) was calculated by assigning values ranging from 0 to 4 to weight loss, stool consistency, and hematochezia and then summing the scores of the three variables (Supplementary Table 1). The DAI score ranges from 0 to 12, with higher scores indicating more severe disease64. Immune activation experiments were performed using either oral administration of equivalent dose of inactivated RC402 (heated at 60 °C for 20 min) and myxoma virus or intraperitoneal injections of recombinant IFN-α (3000 U, R&D). The cell depletion study in mice involved intraperitoneal injections of anti-MAdCAM-1 (4 mg/kg, #BE0035, clone MECA-367, BioXCell), anti-IFNAR-1 (8 mg/kg, #BE0241, clone MAR1-5A3, BioXCell), anti-CD4 (8 mg/kg, #BE0003-1, clone GK1.5, BioXCell) or anti-CD8 (8 mg/kg, #BE0004-1, clone 53-6.72, BioXCell) antibody65. ICB experiments in mice were performed using intraperitoneal injections of anti-PD-1 (8 mg/kg, #BE0033-2, clone J43, BioXCell), anti-PD-L1 (2.5, 4, or 5 mg/kg, #BE0101, clone 10 F.9G2, BioXCell), or anti-CTLA-4 (4 mg/kg, #BE0164, clone 9D9, BioXCell) antibody at the given time points. Gut microbiota were depleted by subjecting mice to daily oral gavage with 200 μl of autoclaved water or autoclaved water supplemented with a broad spectrum antibiotic mixture consisting of ampicillin (1 mg/ml), neomycin (1 mg/ml), metronidazole (1 mg/ml), and vancomycin (0.5 mg/ml) for 22 days. The mice with complete tumor regression were rechallenged with 2 × 105 CT26 cells in the left flank. A multifocal tumor model was generated by subcutaneously implanting 2 × 105 CT26 cells into the right upper, left upper, right lower, and left lower quadrant of the dorsal skin of mice, respectively. In the IT virotherapy group, RC402 was administered by IT injection into tumors in the right upper quadrant, and PBS was given orally by a feeding needle. In the oral virotherapy group, RC402 was administered orally and PBS was injected IT into tumors in the right upper quadrant. Control mice received PBS both orally and IT. The total number of CT26 tumor cells injected and the total dose of RC402 administered per mouse were identical in the IT and oral treatment groups.
In vivo immunofluorescence imaging
Immunofluorescence imaging involved labeling RC402 with Alexa Fluor (AF) dye according to the manufacturer’s instructions. Briefly, AF647 succinimidyl ester (Molecular Probes, Invitrogen) was reconstituted in dimethyl sulfoxide (DMSO) and RC402 (2.4 × 1011 TCID50) was added to the AF647 solution to a final concentration of 100 μg/ml dye. RC402 and AF647 dye were incubated for 30 min at room temperature with gentle inversion every 10 min. Then, unbound AF647 dye was removed by an Amicon Ultra-2 centrifugal filter device (EMD Millipore). Mice were fasted for 12 h before treatment, and 100 μl of labeled RC402 was orally administered. The major organs (GI tract, heart, lung, kidney, liver, and spleen) were collected from mice at different time points, and the fluorescence images were recorded using a Fluorescence-labeled Organism Bioimaging Instrument (FOBI, NeoScience Co., Korea) by fixing the red channel.
Intestinal microbiota analysis
Fecal pellets from PBS and RC402-treated mice were collected and stored at −80 °C. Genomic DNA was extracted from fecal samples using a PowerSoil DNA isolation kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For the shotgun metagenomic sequencing, sequencing libraries were prepared according to the manufacturer’s instructions using a TruSeq Nano DNA High Throughput Library Prep Kit (Illumina). Paired-end (2 × 150 bp) sequencing was performed on individual samples using NovaSeq software (Illumina). The preprocessed data were analyzed using MetaPhlAn4 (v4.0.0) for approximately one million microorganisms composed of NCBI reference genomes and species-level genome bins (SGBs)66. Microbial diversity was calculated using MetaPhlAn4, and a metabolic pathway analysis was performed using HUMAnN3 (v3.5)67.
Quantitative PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) to evaluate expression of reovirus and tertiary lymphoid structure -related genes. The GoScript Reverse Transcription kit (Promega) was used to synthesize cDNA, and qPCR was performed using the FastStart Essential DNA Green Master (Roche). The primer sequences used for qPCR are listed in Supplementary Table 2. LightCycler 96 (Roche) was used for qPCR, and the results were analyzed using the LightCycler 96 SW 1.1 software (Roche). Relative fold differences in the expression levels were determined using the ΔCt method.
Immunoglobulin measurement
For protein extraction, fecal pellets were homogenized in PBS for 10 min and debris was removed by centrifugation at 13,000 g for 10 min twice. The supernatants were harvested, and IgA concentrations were measured using an ELISA kit (Abcam, CA, USA), following the manufacturer’s instructions. IgA concentrations were measured using Skanlt software v4.1 (Thermo Scientific) and normalized to the total fecal protein content as quantified by the Bradford Protein Assay (Bio-Rad, CA, USA).
Flow cytometry analysis of tumor-associated immune cells
The tumor tissues and spleens were dissociated into single cells for flow cytometry analysis following a previous protocol68. The cell clumps and red blood cells were removed by the addition of ACK lysis buffer (Gibco) for 3 min at room temperature prior to antibody staining. Next, the cells were incubated on ice for 30 min in Fixable Viability Dye eFluor 450 (1:1000, #65-0863-18, Invitrogen) to exclude the dead cells, followed by processing with mouse Fc receptor-binding antibody (1:100, #553141, 2.4G2, BD Bioscience) for 15 min at room temperature. Then, the cells were washed with FACS buffer (1% FBS in PBS) and incubated on ice for 30 min in FACS buffer containing surface antibodies against CD45 (1:100, #25-0451-82, 30-F11, Invitrogen), CD3 (1:100, #61-0031-82 or #47-0031-82, 17A2 or 145-2C11, Invitrogen), CD8a (1:100, #12-0081-82 or #MA5-16759, 53-6.7 or KT15, Invitrogen), CD4 (1:100, #11-0042-82, RM4-5, Invitrogen), CD25 (1:100, #69-0251-80, PC61.5, Invitrogen), CD62L (1:50, #56-0621-82, MEL-14, Invitrogen), or CD44 (1:50, #69-0441-82, IM7, Invitrogen). Cells were further permeabilized using a Foxp3 Staining Buffer kit (Invitrogen) and stained for Foxp3 (1:50, #12-5773-82, FJK-16s, Invitrogen) or granzyme B (1:50, #372222, NGZB, Invitrogen). Cells were stained with H-2Ld MuLV gp70 Tetramer-SPSYVYHQF or H-2Kb MuLV p15E Tetramer-KSPWFTTL (1:10, #021 or #052, MBL International, Woburn, MA, USA) (which are expressed in CT26 or MC38 tumor cells) to identify tumor-specific CD8 T lymphocytes. The gating strategy used for analyzing flow cytometry data is shown in Supplementary Fig. 4. Flow cytometry was performed using a CytoFLEX flow cytometer (Beckman Coulter), and the data was analyzed with CytExpert v2.4 and FlowJo software v10.8.1 (Tree Star Inc.).
Histological analysis
The tissue samples were fixed in 1% paraformaldehyde, dehydrated overnight in 20% sucrose solution, and frozen. The frozen blocks were sectioned into 50 μm-thick slices, which were permeabilized with 0.3% PBS-T (Triton X-100 in PBS), and blocked with 5% normal goat serum in 0.1% PBS-T for 30 min at room temperature. Next, the samples were incubated overnight with the following primary antibodies: anti-reovirus (1:100, rabbit, ViroCure), anti-CD31 (1:200, #MAB13982, hamster, clone 2H8, Millipore; 1:100, #ab28364, rabbit, Abcam), anti-CD45 (1:100, #14-0451-82, rat, clone 30-F11, Invitrogen), anti-B220 (1:100, #NB152224, rabbit, clone RA3-6B2, Novus Biologicals), anti-MAdCAM-1 (1:100, #550556, rat, clone MECA-367, BD Pharmingen), anti-IgA (1:100, #556969, rat, clone C10-3, BD Pharmingen), anti-CD8 (1:200, #553027, rat, clone 53-6.7, BD Pharmingen), anti-Caspase3 (1:200, #AF835, rabbit, R&D Systems), or anti-Granzyme B (1:200, #12-8898-82, rat, clone NGZB, Invitrogen). The samples were washed several times, and incubated for 2 h at room temperature with the following secondary antibodies: FITC-, Cy3-, or Cy5-conjugated anti-rabbit IgG (1:1000, #111-097-003, #111-165-144, #111-607-003, Jackson ImmunoResearch), FITC-, Cy3-, or Cy5-conjugated anti-rat IgG (1:1000, #112-097-003, #112-165-167, #112-607-003, Jackson ImmunoResearch), or FITC- or Cy3-conjugated anti-hamster IgG (1:1000, #127-545-160, #127-165-160, Jackson ImmunoResearch). Cell nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI, Invitrogen). Finally, samples were mounted with fluorescent mounting medium (DAKO), and images were acquired using a Zeiss LSM 880 microscope (Carl Zeiss).
Morphometric analysis
Density measurements of IgA+ ASCs, MAdCAM-1+ blood vessels, CD31+ blood vessels, T lymphocytes, apoptotic cells, and GzB+ cell area were performed using ZEN blue edition v8.0 and ImageJ software v1.53f51 (http://rsb.info.nih.gov/ij)69.
NanoString gene expression profiling
Total RNA was extracted from PP, mesenteric lymph node, and tumor tissue using TRIzol (Invitrogen) and purified using ethanol to perform NanoString gene expression profiling. RNA concentration and quality were confirmed using a Fragment Analyzer (Advanced Analytical Technologies, IA, USA). Immune profiling was performed with a digital multiplexed NanoString nCounter PanCancer Immune Profiling mouse panel (NanoString Technologies) using 100 ng of total RNA70. Data was collected using the nCounter Digital Analyzer and analyzed with nSolver software (NanoString Technologies). The Nanostring profiling data are shown in Supplementary Data 1.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software, La Jolla, California, USA) and PASW statistics 18 (SPSS). Values were presented as the mean ± SD unless otherwise indicated. The statistical differences were assessed using an unpaired 1- or 2-tailed Student’s t test, Fisher’s Exact test, Mann−Whitney U test, or ANOVA with Tukey post-hoc test. Waterfall plots were presented as the maximal percentage changes of each tumor at the end of the experiment compared with their baseline volume. Survival curves were generated using the Kaplan-Meier method, and statistical differences between curves were analyzed using the log-rank test. The level of statistical significance was set at P < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The NanoString data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE244046. The remaining data are available within the article, supplementary information, and source data file. Source data are provided with this paper.
References
Shalhout, S. Z., Miller, D. M., Emerick, K. S. & Kaufman, H. L. Therapy with oncolytic viruses: progress and challenges. Nat. Rev. Clin. Oncol. 20, 160–177 (2023).
Bell, J. C. Check and checkmate: battling cancer with multiplex immunotherapy. Mol. Ther. 28, 1236–1237 (2020).
Bourhill, T., Mori, Y., Rancourt, D. E., Shmulevitz, M. & Johnston, R. N. Going (Reo)viral: factors promoting successful reoviral oncolytic infection. Viruses 10, 421 (2018).
Kim, M. et al. Amplification of oncolytic vaccinia virus widespread tumor cell killing by sunitinib through multiple mechanisms. Cancer Res. 78, 922–937 (2018).
Oh, C. M., Chon, H. J. & Kim, C. Combination immunotherapy using oncolytic virus for the treatment of advanced solid tumors. Int. J. Mol. Sci. 21, 7743 (2020).
Parker, B. S., Rautela, J. & Hertzog, P. J. Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144 (2016).
Ilkow, C. S. et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 21, 530–536 (2015).
von Locquenghien, M., Rozalen, C. & Celia-Terrassa, T. Interferons in cancer immunoediting: sculpting metastasis and immunotherapy response. J. Clin. Investig. 131, e143296 (2021).
Lee, Y. S. et al. Oncolytic vaccinia virus reinvigorates peritoneal immunity and cooperates with immune checkpoint inhibitor to suppress peritoneal carcinomatosis in colon cancer. J. Immunother. Cancer 8, e000857 (2020).
Pikor, L. A., Bell, J. C. & Diallo, J. S. Oncolytic viruses: exploiting cancer’s deal with the devil. Trends Cancer 1, 266–277 (2015).
Prior, I. A., Lewis, P. D. & Mattos, C. A comprehensive survey of ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012).
Kim, M. et al. Attenuated reovirus displays oncolysis with reduced host toxicity. Br. J. Cancer 104, 290–299 (2011).
Kelly, K. et al. Reovirus-based therapy for cancer. Expert Opin. Biol. Ther. 9, 817–830 (2009).
Kim, M., Chung, Y. H. & Johnston, R. N. Reovirus and tumor oncolysis. J. Microbiol. 45, 187–192 (2007).
Abad, A. T. & Danthi, P. Recognition of reovirus RNAs by the Innate Immune System. Viruses 12, 667 (2020).
Stanifer, M. L., Kischnick, C., Rippert, A., Albrecht, D. & Boulant, S. Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci. Rep. 7, 10873 (2017).
Zhao, C. et al. Dysregulation of JAM-A plays an important role in human tumor progression. Int. J. Clin. Exp. Patho. 7, 7242–7248 (2014).
Alain, T. et al. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol. Ther. 15, 1512–1521 (2007).
Marcato, P., Shmulevitz, M., Pan, D., Stoltz, D. & Lee, P. W. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 15, 1522–1530 (2007).
DeAntoneo, C., Danthi, P. & Balachandran, S. Reovirus activated cell death pathways. Cells 11, 1757 (2022).
Clarke, P., Richardson-Burns, S. M., DeBiasi, R. L. & Tyler, K. L. Mechanisms of apoptosis during reovirus infection. Curr. Top. Microbiol. Immunol. 289, 1–24 (2005).
Goubau, D. et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature 514, 372–375 (2014).
Loo, Y. M. et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82, 335–345 (2008).
Clarke, P. et al. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 74, 8135–8139 (2000).
Kominsky, D. J., Bickel, R. J. & Tyler, K. L. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 9, 926–933 (2002).
Thirukkumaran, C. et al. PUMA and NF-kB are cell signaling predictors of reovirus oncolysis of breast cancer. PLoS ONE 12, e0168233 (2017).
Knowlton, J. J., Dermody, T. S. & Holm, G. H. Apoptosis induced by mammalian reovirus is beta interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-kappaB-dependent expression of Noxa. J. Virol. 86, 1650–1660 (2012).
Berger, A. K. et al. Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J. Virol. 91, e02404-16 (2017).
Thirukkumaran, C. M. et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy 9, 413–414 (2013).
Muller, L., Berkeley, R., Barr, T., Ilett, E. & Errington-Mais, F. Past, present and future of oncolytic reovirus. Cancers 12, 3219 (2020).
Kim, M. Naturally occurring reoviruses for human cancer therapy. BMB Rep. 48, 454–460 (2015).
Alain, T. et al. The oncolytic effect in vivo of reovirus on tumour cells that have survived reovirus cell killing in vitro. Br. J. Cancer 95, 1020–1027 (2006).
Kim, M. et al. Acquired resistance to reoviral oncolysis in Ras-transformed fibrosarcoma cells. Oncogene 26, 4124–4134 (2007).
Kim, M. et al. Composition for inhibiting angiogenesis, containing reovirus as active ingredient. Patent WO2014204275A1 https://patents.google.com/patent/WO2014204275A1/en (2014).
Voskoboynik, M. et al. A first-in-human phase 1 dose-escalation and expansion study of intratumoral oncolytic reovirus (RC402) as a monotherapy or in combination with pembrolizumab in advanced solid tumors. Cancer Res. 84, CT186 (2024).
Forsyth, P. et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol. Ther. 16, 627–632 (2008).
White, C. L. et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 15, 911–920 (2008).
Goldmacher, G. V. et al. Response Criteria for Intratumoral Immunotherapy in Solid Tumors: itRECIST. J. Clin. Oncol. 38, 2667–2676 (2020).
Aznar, M. A. et al. Intratumoral delivery of immunotherapy-act locally, think globally. J. Immunol. 198, 31–39 (2017).
Milling, L., Zhang, Y. & Irvine, D. J. Delivering safer immunotherapies for cancer. Adv. Drug Deliv. Rev. 114, 79–101 (2017).
Kim, K. J. et al. Antitumor effects of IL-12 and GM-CSF co-expressed in an engineered oncolytic HSV-1. Gene Ther. 28, 186–198 (2021).
Mehlen, P. & Puisieux, A. Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458 (2006).
Cho, E. et al. Characterization of oncolytic vaccinia virus harboring the human IFNB1 and CES2 transgenes. Cancer Res. Treat. 52, 309–319 (2020).
Adair, R. A. et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci. Transl. Med. 4, 138ra–177 (2012).
Chon, H. J. et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin. Cancer Res. 25, 1612–1623 (2019).
Guo, Y., Tian, X., Wang, X. & Xiao, Z. Adverse effects of immunoglobulin therapy. Front. Immunol. 9, 1299 (2018).
Wu, H. J. & Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 3, 4–14 (2012).
Fujisaka, S. et al. Antibiotic effects on gut microbiota and metabolism are host dependent. J. Clin. Investig. 126, 4430–4443 (2016).
Tyler, C. J. et al. Antibody secreting cells are critically dependent on integrin α4β7/MAdCAM-1 for intestinal recruitment and control of the microbiota during chronic colitis. Mucosal Immunol 15, 109–119 (2022).
Fuertes, M. B., Woo, S. R., Burnett, B., Fu, Y. X. & Gajewski, T. F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 34, 67–73 (2013).
Alain, T. et al. Reovirus decreases azoxymethane-induced aberrant crypt foci and colon cancer in a rodent model. Cancer Gene Ther. 14, 867–872 (2007).
Chelvanambi, M. & Wargo, J. A. MAdCAM-1: a newly identified microbial ‘gut check’ for T cells. Trends Immunol. 44, 568–570 (2023).
Brandtzaeg, P., Carlsen, H. S. & Halstensen, T. S. The B-cell system in inflammatory bowel disease. Adv. Exp. Med. Biol. 579, 149–167 (2006).
Mora, J. R. et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006).
Fidelle, M. et al. A microbiota-modulated checkpoint directs immunosuppressive intestinal T cells into cancers. Science 380, eabo2296 (2023).
Fessler, J., Matson, V. & Gajewski, T. F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 7, 108 (2019).
Li, D. & Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct. Target. Ther. 6, 291 (2021).
Fluckiger, A. et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 369, 936–942 (2020).
Villemin, C. et al. The heightened importance of the microbiome in cancer immunotherapy. Trends Immunol. 44, 44–59 (2023).
Tai, J. H. et al. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J. Infect. Dis. 191, 1221–1224 (2005).
Kataru, R. P. et al. Tumor lymphatic function regulates tumor inflammatory and immunosuppressive microenvironments. Cancer Immunol. Res. 7, 1345–1358 (2019).
Woo, J. K. et al. Dual-Armed oncolytic myxoma virus encoding IFN-γ and CD47 promotes Lymphocyte infiltration and tumor suppression of syngeneic murine melanoma. Cancers 15, 4703 (2023).
Igase, M. et al. The oncolytic effects of reovirus in canine solid tumor cell lines. J. Vet. Med. Sci. 77, 541–548 (2015).
Joo, H. Y., Lim, K. & Lim, K. T. Phytoglycoprotein (150 kDa) isolated from Solanum nigrum Linne has a preventive effect on dextran sodium sulfate-induced colitis in A/J mouse. J. Appl. Toxicol. 29, 207–213 (2009).
Lee, W. S. et al. Intratumoral immunotherapy using a TLR2/3 agonist, L-pampo, induces robust antitumor immune responses and enhances immune checkpoint blockade. J. Immunother. Cancer 10, e004799 (2022).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Go E. J., et al. Systemic delivery of a STING agonist-loaded positively charged liposome selectively targets tumor immune microenvironment and suppresses tumor angiogenesis. Small 19, e2300544 (2023).
Kim, J. H. et al. Deep learning model enables the discovery of a novel immunotherapeutic agent regulating the kynurenine pathway. Oncoimmunology 10, 2005280 (2021).
Yang, H. et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 129, 4350–4364 (2019).
Acknowledgements
This work was supported by the National Research Foundation of Korea [NRF] grants funded by the Korean government [MSIT] [NRF-2023R1A2C2006375 to C.K. and NRF-2023R1A2C2004339 to H.J.C.], and grants from the Canadian Cancer Society Research Institute (CCSRI) and Cancer Research Society (CRS) [CHA-#707351 and CRS-839202 to T.A.]. This work was also supported by Virocure, Inc.
Author information
Authors and Affiliations
Contributions
C.K. and H.J.C. conceived and supervised the study. W.S.L., S.J.L., H.J.L., T.A., C.K., and H.J.C. were involved in the conceptual design. W.S.L., S.J.L., H.J.L., H.Y., E-J.G., E.G., K-H.S., and X.X. performed the experiments. W.S.L., S.J.L., H.J.L., D.G.P., T.A., H.J.C., and C.K. interpreted the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
C.K. has a consulting or advisory role at Roche, MSD, BMS, ONO, Oncocross, Virocure, and Sillajen, and has received research grants from Panolos Bioscience, Boryung Pharmaceuticals, Oncocross, Sillajen, and Virocure. H.J.C. has a consulting or advisory role at Roche, Bayer, ONO, MSD, BMS, Celgene, Sanofi, Servier, AstraZeneca, Eisai, Sillajen, Menarini, GreenCross Cell, and Virocure and has received research grants from Panolos Bioscience, Roche and Boryung Pharmaceuticals. E.G., K-H.S. and D.G.P. are employees of Virocure. T.A. has a consulting and advisory role at Virocure, CuroV Science and Genvira Biosciences. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Toshiyoshi Fujiwara, Saman Maleki Vareki, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, W.S., Lee, S.J., Lee, H.J. et al. Oral reovirus reshapes the gut microbiome and enhances antitumor immunity in colon cancer. Nat Commun 15, 9092 (2024). https://doi.org/10.1038/s41467-024-53347-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53347-6
This article is cited by
-
Lactobacillus acidophilus potentiates oncolytic virotherapy through modulating gut microbiota homeostasis in hepatocellular carcinoma
Nature Communications (2025)
-
Targeting Metastasis: Exploring the Impact of Microbial Infections on Cancer Progression Through Innovative Biological Models
Current Microbiology (2025)
-
The evolving landscape of oncolytic virus immunotherapy: combinatorial strategies and novel engineering approaches
Medical Oncology (2025)
