
Abstract
The contamination of water by per- and polyfluorinated substances (PFAS) is a pressing global issue due to their harmful effects on health and the environment. This study explores a cationic covalent organic framework (COF), TG-PD COF, for the efficient detection and removal of perfluorooctanoic acid (PFOA) from water. Synthesized via a simple sonochemical method, TG-PD COF shows remarkable selectivity and sensitivity to PFOA, with a detection limit as low as 1.8 µg·L⁻¹. It achieves significant PFOA adsorption exceeding 2600 mg·g⁻¹ within seconds over several cycles in batch mode and complete removal at environmentally relevant concentrations in column adsorption. Results reveal unique adsorption behavior characterized by two phases, leveraging PFOA aggregation through hydrophobic interactions. Computer simulations elucidate the mechanisms underlying TG-PD COF’s sensing, adsorption, and charge transfer dynamics. Our findings position this COF design strategy as a promising solution for combating PFAS contamination in water bodies worldwide.
Similar content being viewed by others
Introduction
The emergence of the persistent, bio-accumulative, and toxic organic pollutants, perfluorinated and polyfluorinated alkyl substances (PFAS) and their derivatives, has led to widespread contamination of surface and groundwater worldwide, with detrimental consequences for human health and the environment1,2,3. Among these pollutants, perfluorooctanoic acid (PFOA) stands out, having been detected in the majority of human serum samples from exposed populations worldwide, with water being the primary exposure pathway4. Efficient detection and removal of PFOA from water resources is vital for minimizing human exposure. Material design plays a crucial role in developing systems that can simultaneously detect and adsorb PFOA.
Conventional sensing methods such as Liquid Chromatography-Mass Spectrometry (LC-MS) and High Precision Liquid Chromatography (HPLC), while accurate and reliable, are complex, expensive, and time-consuming5. Rapid and cost-effective PFOA sensing technologies are urgently needed to overcome these challenges6,7. Consequently, there’s been a significant increase in research and development focused on new materials for selective sensing of hazardous substances8,9,10. In the quest for efficient detection of water contaminants, fluorescence-based sensors have gained significant attention for their inherent advantages over their non-emitting counterparts6,11,12. These advantages include rapid response times13, high sensitivity at low contaminant concentrations14,15, and visual detection capabilities16. Designing an efficient fluorescent sensing material entails maximizing sensor-analyte interactions by optimizing the chemistry, density, and distribution of active groups within the material to mitigate diffusion limitations and improve analyte interactions17,18,19,20. These characteristics are essential for effective sensing and are equally important for developing an efficient adsorbent, highlighting the potential of simultaneously integrating sensing and adsorption properties in materials designed to mitigate water contamination issues.
Research on PFAS adsorption using common materials such as activated carbon and ion exchange resins has provided valuable insight into the main interaction mechanisms between PFAS and adsorbents. This knowledge is key for the development of materials that optimize these interactions while overcoming the challenges faced by conventional adsorbents21,22,23,24,25. These adsorbents rely primarily on hydrophobic interactions with the fluorinated alkyl chains of PFAS, which are particularly effective in activated carbon. Additionally, they rely on electrostatic interactions and ion exchange processes to target the polar functional groups of PFAS, especially in ion exchange resins26. Despite widespread use, these adsorbents have significant limitations, including slow adsorption kinetics, low PFAS uptake capacity, and harsh recycling conditions21,26,27. For this reason, the molecular design of adsorbents has been crucial in developing PFAS-specific materials that exploit both hydrophobic and electrostatic interactions21,28. Recent studies have focused on the development of hydrophobic aminated materials29 and charged fluorinated materials30,31, designed to maximize interactions with PFAS molecules. These studies have demonstrated promising results, significantly improving the adsorption capacity and efficiency compared to conventional materials. Such advancements highlight the potential of tailored adsorbents in effectively addressing PFAS contamination.
While designing adsorbents that harness both hydrophobic and electrostatic interactions is crucial, it is equally important that these adsorption sites are abundant and accessible to optimize the adsorption kinetics and capacity of emerging contaminants. Therefore, porous and open structures that are stable in aqueous environments are preferred for this purpose21,22,32,33,34. This need is particularly evident when developing adsorbents for long-chain PFAS like PFOA and perfluorooctane sulfonate (PFOS), which tend to form aggregates on the adsorbent surface via hydrophobic interactions of their fluorinated tails35,36,37,38,39. While these aggregates can block the adsorption sites within the pores, and hinder diffusion and accessibility36,37, they can alternatively boost PFAS adsorption capacity through cooperative adsorption35,38. Consequently, the challenge lies in engineering materials with tailored physicochemical properties, striking a balance between hydrophobic and electrostatic traits within an open, porous, and stable framework leveraging PFOA aggregation with an unimpeded access to adsorption sites inside the porous network.
Covalent organic frameworks (COFs) are a class of porous crystalline materials composed of light elements (for example, C, H, O, N, etc.) formed by covalent bonding of organic building blocks into extended porous and ordered 2D and 3D structures40,41. The structure of COFs can be tailored by the rational choice of their organic building blocks, which has contributed to their use in many applications such as gas storage42,43, water treatment44,45,46, sensing47,48, drug delivery49,50,51, and catalysis52,53. The inherent hydrophobic nature of COFs’ main backbone makes them suitable for the adsorption of PFAS via hydrophobic interactions. In addition, the introduction of cationic moieties into their building units allows for better sensing and adsorption through electrostatic host-guest interactions between the charged COF channels and the anionic head groups of PFOA and other anionic PFAS47,54. In a recent study54, cationic COFs were synthesized and used as adsorbents for the PFOA derivatives GenX (Qmax = 680 mg·g−1) and HFPO-TA (Qmax = 1076 mg·g−1) that reached adsorption equilibrium in 10–20 h. However, this is not suitable for real-life applications such as point-of-use gravity filtration systems that require fast kinetics54. Additionally, although there are few reports on the use of COFs in fluorescence sensing20,47,55,56,57,58,59, the synthesis and application of cationic COFs for the simultaneous fluorescence sensing and adsorption of PFOAs is rarely investigated.
In this study, we present a cationic COF, TG-PD COF, featuring guanidinium cores that introduce well-distributed positively charged adsorptive sites within its structure. These cationic sites are capable of creating electrostatic interactions with electron-rich species22,57 thereby enabling simultaneous fluorescence sensing and adsorption of PFOA from water. The uniformly distributed, abundant, and accessible cationic guanidinium moieties help to increase the TG-PD COF’s interactions with the anionic heads of PFOA and enhance selectivity. The applied design principle yielded a COF capable of detecting and removing PFOA molecules from water in seconds, even at environmentally relevant concentrations, with high uptake capacity leveraging cooperative adsorption through PFOA aggregation. In addition, we investigated the mechanisms underlying both detection, and rapid removal through computer simulations. Our results suggest that tailored cationic COFs represent a significant advancement in the detection and removal of persistent anionic pollutants from water.
Results
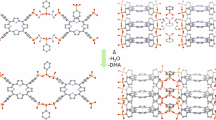
The synthesis of TG-PD COF (Fig. 1a) involved a simple sonochemical method, in which triamino guanidinium chloride TGH•Cl (42.3 mg, 0.30 mmol) and 2,9-diformyl 1, 10-phenanthroline PD (106.2 mg, 0.45 mmol) were combined in a mixture of 1,4-dioxane and water (1:1, v:v). The resulting mixture underwent ultrasonication for one hour at room temperature using a high power (40 %, 550 W) ultrasonic probe in continuous mode with a 3 mm microtip. The orange-colored product was subsequently purified by ethanol washing, and dried at 120 °C for 12 hours.

a Sonochemical synthesis of TG-PDCOF, b, c SEM and HRTEM images of TG-PD COF, d Experimental (black) and simulated PXRD (red) patterns of TGH+-PD COF, e Geometrically optimized crystal structure of TG-PDCOF.
Confirmation of the successful formation of the imine bond (-C=N) in TG-PD COF was achieved by Fourier transform infrared (FT-IR) spectroscopy and solid-state 13C cross-polarization magic-angle spinning (CP/MAS), and the results are shown in Figs. S1 and S2. The emergence of a new stretching vibration at 1625 cm−1, indicative of imine (–C=N) bonds, along with the disappearance of the band at 1701 cm−1, corresponding to −CHO groups in PD, provided evidence for the formation of TG-PD COF. Moreover, the 13C CP/MAS NMR spectrum of TG-PD COF showed a signal at ~147 ppm, providing additional confirmation of the formation of the –C=N bond (Fig. S2). Another signal observed at 152 ppm in the NMR spectrum was attributed to the C atom of the guanidinium moiety. The TG-PD COF demonstrated thermal stability up to approximately 200 °C (Fig. S3). The weight loss observed before 100 °C is mainly attributed to the desorption of water from the COF surface. Subsequent weight loss beyond 200 °C is attributed to the thermal decomposition of the COF network. The morphology of TG-PD was investigated using scanning electron microscopy (SEM) and high-resolution transmission-electron microscopy (HRTEM). SEM images revealed a hollow tube-like morphology with open-ended sides as shown in Fig. 1b and Fig. S4. HRTEM analysis further confirmed the hollow nature of TG-PD COF, as evidenced by the presence of an inner interior region covered by two outer rigid shells with a largely rough surface, as shown in Fig. 1c and Fig. S4.
PXRD analysis was conducted to verify the formation of the 2D organic network and confirm its periodic nature (Fig. 1d). For this purpose, a crystal structure model was simulated and geometrically optimized, based on the formation of extended layers through the covalent bonding between TG and DP building blocks (Fig. 1e, Fig. S5). The structure model consists of TG units acting as three connected nodes linked by DP moieties, forming honeycomb (hcb) type layers, with chlorine anions located in the pores. A model in the monoclinic P21 space group was optimized, with cell parameters a = 39.36 Å, b = 23.67 Å, c = 4.19 Å, β = 131.97°, where the covalent layers extend along the ab plane, and are stacked in an inclined fashion along the c direction. The calculated PXRD pattern is in excellent agreement with the experimental one. Thus, the experimental PXRD pattern show several diffraction lines, including a distinct peak centered at 2θ = 4.6°, which was assigned to the (110) diffraction plane of the COF network, followed by broader signals, spanning the 2θ range of 7 – 11°, that are mainly attributed to the (020), (220), and (310) planes. Additionally, a relatively sharp intense peak was observed at 2θ = 27°, which corresponds to the (11-1) reflexion. As expected for a two-dimensional, layered COF, where the interlayer interactions are not covalent, the peaks are broad, which is indicative of the limitations in the size crystalline domains. Nevertheless, this is comparable to most reported guanidinium-based 2D COFs47,56,60,61,62,63,64. Regarding the correspondence between the calculated and experimental pattern, the calculated position of the most intense reflections according to our model are in good agreement with the experimental pattern. The list of the most intense diffraction lines (I > 5%), and their corresponding d spacing values, hkl indexes, and 2θ position is shown in the following table (Table S1).
The BET surface area analysis of the TG-PD COF was conducted (Fig. S6), and the calculated surface area was around 13 m²·g−1. This is likely due to the presence of a large number of counter anions within the COF network, which may block the COF pores. Similar observations have also been noted in other reported guanidinium-based COFs47,61,62,63,64. Furthermore, the lower BET value can be attributed to several factors, such as disorderly distributed anions, and an imperfectly defined stacking sequence as a consequence of the use of phenanthroline units (Fig. S6).
Photophysical properties and PFOA detection
Despite its hydrophobicity, with a water contact angle of 112⁰ (Fig. S7), the TG-PD COF forms a relatively stable dispersion in water after just a few minutes of sonication, owing to its ionic nature, and its photophysical properties in water were investigated. In aqueous medium, TG-PD COF possesses an emission band with a maximum centered around 575 nm upon excitation at 365 nm. This emission is caused by intramolecular charge transfer (ICT) from the phenanthroline to the guanidinium moiety. After adding different concentrations of PFOA to the TG-PD COF dispersion, the fluorescence intensity showed a linear increase with increasing PFOA concentration (Fig. 2a). By fitting the linear data and using the 3σ/slope method, a limit of detection (LOD) for PFOA of 1.8 µg·L−1 was obtained, showing competitive PFOA sensitivity (Fig. 2b). While it is not the most sensitive of the materials listed in Table S265,66,67,68,69,70,71,72, it offers a detection capability in the environmentally relevant concentrations range, making it a viable option for the detection of PFOA in the environment.

a Normalized photoluminescence (PL) spectra (λex = 365 nm) of TG-PD COF dispersion in water with the stepwise addition of PFOA, b Linear plot of normalized PL intensity (λmax = 365 nm) as a function of PFOA concentration, c Bar graph illustrating the comparative fluorescence intensity of TG-PD COF in the presence of different analytes, where I0 is the initial intensity of TG-PD COF, and I is the final intensity in response to each analyte, d Solid state 13C CP/MAS NMR spectra of PFOA (black line), TG-PDCOF (green line) and TG-PD COF and PFOA (red line). Asterisks denote spinning side bands. e Overlay of the two-dimensional 1H−13C HETCOR solid-state NMR spectra of pure PFOA (red contour, red dotted circle) and PFOA bound to TG-PD COF (black contour, gray oval). Inset shows the simplified chemical structures of TG-PD COF and PFOA. Intramolecular 1H−13C correlation from pure PFOA is shown by green arrow and inter-molecular correlation between PFOA and TG-PD COF is shown by a red arrow. Upfield chemical shift change for the carbonyl carbon from PFOA upon binding to TG-PD COF is shown by a green arrow.
Interestingly, the TG-PD COF exhibits remarkable selectivity for the PFOA molecule. Control experiments were conducted to assess the sensing ability of TG-PD COF with other isostructural fluorinated and non-fluorinated strong acids (trifluoroacetic acid (TFA), propionic acid, and perfluorohexanoic acid). TG-PD COF showed a very weak response to these acids, probably due to their lower hydrophobic character (Fig. 2c, Fig. S8)68. We also tested NaCl and Na₂SO₄ for selectivity studies, considering that these salts might be present in contaminated water (Fig. 2c)66,69. However, they did not show any noticeable response with TG-PD COF in water. It is important to note that the PFOA molecules, with their long fluorinated tails, are highly hydrophobic in nature. The hydrophobic-hydrophilic interaction is the key factor in the selective sensing of PFOA by TG-PD COF. With its long alkyl fluorocarbon tail, PFOA exhibits a strong hydrophobic binding interaction with the TG-PD COF. In contrast, octanoic acid, which lacks fluorination, has lower hydrophobicity, which reduces its binding interaction with TG-PD COF68. To further understand the sensitivity of TG-PD, we conducted an additional experiment in which equal quantities of interfering analytes were added along with PFOA. As shown in Fig. S9, TG-PD exhibited a turn-on signal even in the presence of other analytes, indicating that the presence of interfering substances does not significantly affect its performance in PFOA sensing.
In water, when TG-PD COF and PFOA are in close proximity, large aggregates form as a result of the strong electrostatic interactions between the sensor and analyte molecules. In the aggregated state, the nonradiative intramolecular motions are restricted, which in turn leads to an enhancement in fluorescence. This phenomenon, known as aggregation-induced emission (AIE), is responsible for the observed “turn-on” fluorescence when TG-PD COF interacts with PFOA71,73,74. To confirm this hypothesis, zeta potential (ζ) measurements were performed to follow the change in surface charge of the TG-PD COF as the PFOA concentration increased (Fig. S16a). The zeta potential was calculated from DLS measurements. Initially, the COF suspension showed a positive zeta potential ( + 24 mV). Upon additions of PFOA, the surface charge significantly decreased to –19.95 mV. This notable change in the zeta potential values is strong evidence of the electrostatic interaction between the sensor and the analyte. It is known that in aggregates, the restriction of intramolecular rotation prevents nonradiative decay, causing fluorescence enhancement75,76,77.
To assess the practical applicability of our COF material, we also conducted solid-state fluorescence sensing experiments. In the solid state, we also observed an efficient fluorescence “turn-on” response to PFOA (Fig. S10). Solid-state PFOA sensing was conducted by depositing TG-PD COF powder onto a 2 cm portable filter paper. The paper-based sensor serves as a suitable substrate that allows for the uniform dispersion of the COF powder and the formation of a stable, uniform film. The emission intensity of TG-PD COF varied linearly with the concentration of PFOA, and a LOD of 12 μg L−1 was calculated in the solid state (Fig. S10b).
To further elucidate the sensing mechanism and determine the interaction sites, a thorough 13C CP/MAS NMR spectral analysis was performed (Fig. 2d) on TG-PD COF before and after exposure to PFOA. After binding of PFOA with the COF, the carbonyl carbon of PFOA is upfield shifted by ~ 3.4 ppm. The reason for this significant change in chemical shift could be attributed to the presence of strong hydrogen bonding or deprotonation of the carboxyl group as the electronic environment of the carbonyl carbon is changed. The loss of the proton and the formation of the carboxylate anion can decrease the electron-withdrawing effect, leading to increased shielding of the carbonyl carbon and, consequently, an upfield shift in the NMR signal. This was further supported by the observed ~ 3.5-fold fluorescence enhancement of the TG-PD COF upon interaction with PFOA, which clearly confirms the presence of strong interactions between the two species. This was also confirmed by two-dimensional 1H-13C HETCOR (Heteronuclear correlation) solid-state NMR experiments on pure PFOA and PFOA bound to TG-PD COF. Figure 2e shows the overlay of the two-dimensional 1H-13C HETCOR solid-state NMR spectra of pure PFOA (red contours) and PFOA bound to TG-PD COF (black contours). The spectrum of pure PFOA is mainly characterized by the presence of an intramolecular 1H−13C correlation peak (indicated by a red dotted circle) between the carboxyl carbon atom ( ~ 165.4 ppm) and the carboxyl proton ( ~ 9.6 ppm). On the other hand, the spectrum from PFOA bound to TG-PD COF is characterized by the presence of intramolecular 1H-13C correlation peaks appearing mainly from the aromatic protons from the TG-PD COF ( ~ 6.3 ppm) and the carbon atoms from the TG-PD COF (from 115 to 155 ppm) and intermolecular 1H-13C correlation peaks between the aromatic protons from the TG-PD COF ( ~ 6.3 ppm) and the carbon atoms from the bound PFOA (111 and 163 ppm). The presence of intermolecular 1H-13C correlation peaks between the aromatic protons from the TG-PD COF ( ~ 6.3 ppm) and the carbon atoms from the bound PFOA (111 and 163 ppm) indicate the presence of very strong interactions between PFOA and TG-PD COF and their close proximities in space. It is also worth noting that the spectrum from PFOA-bound TG-PD COF is characterized by the systematic absence of intramolecular 1H-13C correlation peak between the carboxyl carbon and the carboxyl proton (13C ~ 165.4/ 1H ~ 9.6 ppm) indicating the possible deprotonation of the carboxyl protons. Furthermore, 1H-13C HETCOR spectra also reveal significant upfield changes in the chemical shifts ( ~ 3.4 ppm) for the carbonyl carbon (highlighted by green arrow) of the PFOA between the free and bound state, indicating that the carbonyl group of PFOA is the major point of interaction between the PFOA and the TG-PD COF. To monitor the changes in 15N chemical shifts upon PFOA binding, DNP-enhanced 15N solid-state NMR experiments were performed on TG-PD COF before and after binding to PFOA (Fig. S11). The spectrum from pure TG-PD COF is characterized by the presence of peaks appearing mainly from the imine ( ~ 322.4 ppm) and from NH nitrogen atoms ( ~ 142.7 ppm). The spectrum from PFOA-bound TG-PD COF also reveals peaks appearing mainly from the imine ( ~ 315.5 ppm) and from NH nitrogen atoms ( ~ 144 ppm). However, 15N spectrum from PFOA-bound TG-PD COF reveals an upfield chemical shift of around 6.9 ppm for the imine nitrogen atoms and downfield chemical shift changes of about 1.3 ppm for the NH nitrogen atoms. The upfield shift of the imine nitrogen atoms can be attributed to the increase in electron density at these sites. When PFOA binds to the COF, the guanidine group can act as a hydrogen bond acceptor, interacting with the deprotonated carboxylate group of PFOA. This interaction can increase the electron density on the imine nitrogen atoms, leading to greater shielding and an upfield shift for imine nitrogen in the NMR spectrum. Conversely, the downfield shift of the NH nitrogen atoms indicates a decrease in electron density at these sites. The hydrogen bonding interaction between the guanidine group and the PFOA carboxylate can withdraw electron density from the NH nitrogen atoms, deshielding them and causing a downfield shift in the NMR signal.
Furthermore, 19F MAS solid-state NMR experiments were performed to better understand the interaction between PFOA and TG-PD COF. Figure S12 shows the 19F solid-state NMR spectra of pure PFOA (bottom, blue line) and TG-PD COF treated with PFOA (top, red line). The 19F spectrum obtained from the free PFOA is well resolved and reveals mainly chemical shifts from the terminal -CF3 groups ( ~ –83 ppm) and from the backbone -CF2 groups (between –120 to –130 ppm). Compared to the 19F spectrum of free PFOA, the spectrum of TG-PD COF treated with PFOA is relatively broad and shows chemical shifts from the terminal -CF3 groups ( ~ –82 ppm) and from the backbone -CF2 groups (between –110 to –130 ppm). The appearance of the well-resolved 19F peaks from the pure PFOA could be attributed to the presence of dynamics to sufficiently average out the anisotropic NMR interactions. On the other hand, the appearance of the broad 19F peaks from the TG-PD COF treated with PFOA could be ascribed to the lack of dynamics and aggregation of PFOA due to the preferential binding of the PFOA with the TG-PD COF.
Finally, to gain a deeper understanding of the sensing mechanism and the selectivity of TG-PD COF, we conducted computational analysis (Fig. S13) on the COF with PFOA in the gas phase (further details in SI). For band gap calculations, we selected octanoic acid due to its close structural similarity to PFOA. The band gap of TG-PD COF with octanoic acid is observed to be 1.92 eV, which is larger than both the pristine TG-PD COF and the TG-PD COF with PFOA. This larger band gap suggests a weaker interaction between TG-PD COF and OA compared to the interaction with PFOA.
PFOA Adsorption experiments
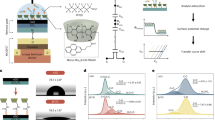
Adsorption studies were conducted to assess the efficiency of the cationic TG-PD COF in removing PFOA from water. Unless otherwise stated, all adsorption experiments were carried out at 24 °C using an adsorbent suspension (Fig. 3a). The concentrations of PFOA in these experiments were determined by HPLC-MS, and further details are outlined in the methods section.

a Schematic representation of the PFOA adsorption experiments with the COF suspension. Created in BioRender. (2023) BioRender.com/m60h920. b Adsorption kinetics at high (black) and low (blue) concentrations of PFOA and TG-PD COF and in the absence of COF (red). c Adsorption isotherm of PFOA onto TG-PD COF. d Schematic representation of the PFOA adsorption experiment with the TG-PD column. Created in Biorender (2024) BioRender.com/u27r837. e PFOA column adsorption at high (black) and low (blue) influent concentrations with respect to water filtrate volume using the TG-PD column. Control column adsorption experiments in the absence of TG-PD COF are also presented (red).
Kinetic studies were conducted at different PFOA and TG-PD COF concentrations to assess their influence on adsorption efficiency (Fig. 3b). Notably, at higher PFOA and adsorbent concentrations ([COF] = 400 mg·L−1, [PFOA] = 0.2 mg·L−1), PFOA was rapidly adsorbed by the TG-PD COF, with 96% of PFOA adsorbed within the seconds required for sampling and COF filtration from the solution. This remarkable performance of the TG-PD COF reflects rapid and efficient PFOA removal capabilities, outperforming previous studies with similar conditions (Table S3).
To evaluate the efficiency of the TG-PD COF in removing PFOA at environmentally relevant concentrations, a kinetic study was conducted at a lower concentration ([COF] = 100 mg·L−1, [PFOA] = 1 µg·L−1). Notably, even at such concentrations, nearly 50% of the PFOA was adsorbed within a minute, with an adsorption equilibrium corresponding to 83% PFOA removal achieved within one hour. The rapid adsorption kinetics achieved by the TG-PD COF highlights its potential for practical applications, where it is essential to swiftly adsorb PFAS contaminants during the brief contact period with the adsorption bed in adsorption columns.
The kinetic study data were fitted with pseudo-first-order and pseudo-second-order kinetic models (Fig. S14). As expected, the experimental data exhibited a better fit with the pseudo-second-order model, suggesting that chemisorption is the rate-limiting step in adsorption. The apparent rate constant was calculated to be 11470 g·mg−1·min−1 for the high concentration kinetics study, and 2535 g·mg−1·min−1 for the low-concentration study. Notably, the adsorption kinetics results reported herein at this low concentration exceed the exemplary reports on PFOA adsorption in the literature (Table S3). Nevertheless, direct comparisons with the existing literature are difficult, particularly with regard to the apparent rate constant, due to the prevalent use of highly concentrated PFOA solutions24,37,78,79,80,81.
To determine the maximum adsorption capacity of TG-PD COF, adsorption isotherm experiments were conducted across a wide range of initial PFOA concentrations from 0.2 mg·L−1 to 1000 mg·L−1, with the concentration of TG-PD COF remaining constant at 100 mg·L−1 in all experiments (Fig. 3c). The resulting adsorption isotherm showed two distinct regions separated by an equilibrium PFOA concentration of approximately 500 mg·L−1. Starting from the onset of the isotherm data up to around 500 mg·L−1, where the equilibrium adsorption capacity slightly exceeded 1000 mg·g−1, the adsorption capacity showed an asymptotic increase with increasing PFOA concentration. Interestingly, the trend of PFOA adsorption showed a significant increase at PFOA concentrations higher than 500 mg·L−1 instead of reaching a saturation point and plateau. This observation is consistent with the two-stage adsorption isotherms reported for the adsorption processes of ionic surfactants on oppositely charged adsorbents82,83. The initial phase involves “head-on” adsorption at lower PFOA concentrations, which is primarily driven by the electrostatic interactions between the anionic head of PFOA and the cationic guanidium moieties in the COF structure84,85. Due to the high surface charge, the PFOA anions must be oriented with their charged heads towards the oppositely charged COF surface, while their tails extend into the water mass84,85,86.
While the PFOA concentration in the bulk solution remains below the critical micellization concentration (CMC), a significant local concentration near the COF surface promotes the formation of PFOA molecular aggregates, attributed to hydrophobic interactions among the PFOA tails26,37. This localized aggregation phenomenon triggers a cooperative adsorption process, leading to a PFOA uptake exceeding 2600 mg·g−1, which is an exceptional capacity for PFOA uptake using COF-based adsorbents.
In order to assess the performance of the COF in removing PFOA from water under more realistic conditions, adsorption columns containing 3 mg of COF were prepared. Water containing two different concentrations of PFOA flowed through the columns at a rate of around 100 µL·min−1 (Fig. 3d, Fig. S15). Remarkably, complete removal of PFOA was achieved throughout the filtration of 20 mL of water with PFOA concentrations of 1 µg·L−1 and 200 µg·L−1 (Fig. 3e). The PFOA concentration in the filtrate was below the HPLC-MS detection limit, which was determined to be around 50 ppt. This further demonstrates the TG-PD COF’s effectiveness in the rapid and complete removal of PFOA in conditions similar to conventional gravity filters.
In an attempt to investigate the two-phase adsorption isotherm, other studies in the literature using various adsorbents have presented the effects of PFAS aggregation on increased adsorption over time and highlighted their influence on the adsorption isotherm21,39,87,88,89. However, the remarkable increase in the uptake capacity demonstrated in the isotherm in this study is not only more pronounced, but also results in a significantly higher adsorption capacity compared to these reports. The interplay of hydrophobic and electrostatic forces between the TG-PD COF and the PFOA molecules leads to a synergistic effect that uses the two most potent mechanisms known for the efficient extraction of PFOA from aqueous media. Moreover, the unique open architecture of the TG-PD COF prevents blockage of the internal adsorption sites by PFOA aggregates, a common limitation with other adsorbents36,37. Additionally, the periodic distribution of adsorption sites within the COF and its large pore size provide unobstructed access to these sites within the porous structure, optimizing the PFOA adsorption process.
To further understand the observed two-phase adsorption phenomenon shown in the isotherm (Fig. 3c), zeta-potential measurements were performed to follow the change in surface charge of the TG-PD COF as the initial PFOA concentration increased (Fig. S16a). Initially, the COF suspension exhibited a positive zeta potential ( + 24 mV), which is due to the presence of positively charged guanidinium moieties. After the introduction of a low PFOA concentration (1 µg·L–1), a significant decrease in the surface charge to about –0.8 mV was observed, indicating preferential adsorption on the outer surface of the TG-PD COF, leading to neutralization of the surface. As the PFOA concentrations continued to increase, the surface charge of the COF suspension remains almost neutralized, suggesting adsorption within the porous structure of the TG-PD COF following the saturation of the outer surface up to a PFOA concentration of around 200 mg·L−1. Notably, when the PFOA concentration was further increased up to 1000 mg·L−1, the surface charge decreased significantly to –19.95 mV. This phenomenon could be due to the aggregation of PFOA molecules on the COF surface via tail-to-tail hydrophobic interactions, induced by the increased concentration of PFOA molecules in the vicinity of the COF surface. This observation aligns well with the sudden increase in adsorption capacity observed in the isotherm, further suggesting that the formation of PFOA aggregates causes this abrupt increase in adsorption capacity.
Additionally, the pH of the solution (Fig. S16b) shows a progressive decrease with increasing initial PFOA concentration indicating the ionization of the PFOA molecules and the release of H3O+. Even at the highest PFOA concentration, the pH continued to decrease, indicating progressive ionization of the PFOA molecules, facilitating their adsorption onto the COF structure in their anionic state.
Scanning transmission electron microscopy (STEM), specifically utilizing high-angle annular dark field (HAADF) imaging, was used to study COF suspension at varying initial PFOA concentrations (0, 600, and 1000 mg·L−1). This technique provided detailed Z-contrast images, allowing accurate identification of the locations of the fluorine atoms within the COF structure (Fig. 4a–c). At a higher magnification, elemental mapping showed the dispersion of fluorine atoms throughout the COF material. At 600 mg·L−1, the fluorine atoms were evenly distributed across the COF, whereas at 1000 mg·L−1, while still uniformly distributed, fluorine also formed aggregates on the COF surface. The analysis showed a qualitative increase in fluorine content from 12.4% to 16.1% within the COF matrix as the PFOA concentration increased from 600 mg·L−1 to 1000 mg·L−1, as documented in Table S4 and Fig. S17. This is further supported by the stable chlorine concentrations observed by STEM, indicating that the adsorption of PFOA anions leads to an increased negative charge within the suspension. Aggregation, driven by hydrophobic interactions, marks a distinct phase in PFOA adsorption, as illustrated schematically in Fig. 4d.

a–c The elemental mapping of TG-PD COF at different PFOA concentrations d A graphical representation of PFOA adsorption onto the TG-PD COF at increasing PFOA concentrations in water e The effect of temperature increase on the equilibrium PFOA adsorption capacity f The removal efficiency of PFOA through 5 different regeneration cycle ([PFOA] = 0.2 mg·L−1, [COF] = 100 mg·L−1).
The effect of temperature on PFOA adsorption by the TG-PD COF was investigated at a PFOA concentration of 200 mg·L−1 and a COF concentration of 100 mg·L−1 (Fig. 4e). Increasing the adsorption temperature from 24 °C to 50 °C led to a significant decrease in COF adsorption capacity by almost 40 %. A further increase in the temperature to 80 °C led to a slight increase in adsorption, but still about 33 % lower than that at 24 °C. This decrease in adsorption capacity with increasing temperature indicates a predominant influence of enthalpic forces in the adsorption process. However, the subsequent increase in adsorption at higher temperatures implies that entropic contributions become more dominating, which is consistent with observations reported for surfactants85,90.
The adsorption isotherm data were fitted using both the Langmuir and Freundlich models, as shown in Fig. S18. Attempting to fit the entire data set to either the Langmuir or Freundlich model did not yield satisfactory linear fits, indicating that the adsorption process cannot be fully characterized as monolayer or multilayer adsorption over the entire range of PFOA concentrations. Upon dividing the adsorption isotherm dataset at a threshold of 600 mg·L−1, where aggregation is expected to occur, it was observed that the data points below 600 mg·L−1 fit the Langmuir model well. This suggests that monolayer adsorption predominates at lower PFOA concentration ranges. Conversely, data points above 600 mg·L−1 show a better fit with the Freundlich model, indicating a multilayer adsorption process within this PFOA concentration range83. Although the maximum adsorption capacity, Qmax, of the TG-PD COF is higher than 2600 mg·L−1, its calculation was not pursued due to safety concerns, which prevented experiments at higher PFOA concentrations. Further details on the safety considerations regarding experimental work involving PFOA can be found in the supporting information file and in literature91.
Regeneration experiments were carried out to evaluate the reusability of the TG-PD COF over several adsorption cycles. The COF was soaked in methanol overnight before being reused for PFOA adsorption (Fig. 4e). Notably, the COF effectively desorbed the PFOA in methanol and maintained its adsorption efficiency throughout five regeneration cycles, without any reduction in adsorption performance. Finally, we assessed the stability of the regenerated TG-PD COF. The unchanged PXRD pattern and SEM images after regeneration clearly confirm the stability of the TG-PD COF adsorbent (Fig. S19).
Computer simulations
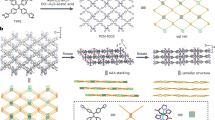
To gain deeper insights on the mechanism and the thermodynamic factors governing the adsorption of PFOA, Molecular Dynamics (MD) simulations were conducted. A 12-layer COF structure was constructed and solvated with water and ions to mimic experimental conditions, as depicted in Fig. 5a. Details regarding the parameters of the MD simulation setup can be found in the methods section. The convergence of the adsorption process was monitored by calculating the PFOA uptake, which stabilized after approximately 200 ns, as depicted in Fig. S20a. A time evolution of the adsorption process at various times is shown in (Fig. 5b).

a All-atom MD simulation setup of TG-PD COF with PFOA molecules. b Snapshot taken from simulation at different time points to show the process. c–e Time evolution of the density profiles along the long axis of the simulation box (z-axis) depicted in b for PFOA, water, and total charge, computed from simulation data averaged over a time interval of dt = 0.1 ns.
First, the accuracy of the molecular simulations was assessed by comparing the number of PFOA molecules adsorbed by the TG-PD COF with the experimental data. The simulations show that out of 290 PFOA molecules initially present in the simulation box, 280 were adsorbed by the COF upon reaching equilibrium. This translated to a removal capacity of 96% at a bulk PFOA concentration of 900 mg·L−1, in a COF concentration of 374 mg·L−1. This computational estimate closely matches the experimental findings at a corresponding PFOA concentration, where the equilibrium uptake was nearly 2300 mg·g−1 (Fig. 3b), compared to 2310 mg·g−1 obtained from the simulation.
Subsequently, simulations were employed to elucidate the mechanism of PFOA adsorption. Snapshots captured at various time points provide qualitative insights into the dynamics of the adsorption process (Fig. 5b). Changes occurring throughout the adsorption process were tracked by examining the density profiles of PFOA, water molecules, and the total charge along the long axis of the simulation box, as depicted in Fig. 5c–e. To guide the eye, the COF-solvent interface was highlighted with dashed lines in the density profiles.
Findings from the temporal evolution of the average density profiles reveal a significant shift in the distribution of components upon adsorption of PFOA by the TG-PD COF. The first notable change is observed when PFOA forms a layer on the surface of the COF between t = 1–10 ns, resulting in a peak in the PFOA density profile (compare Fig. 5b with Fig. 5c). This accumulation of PFOA at the interface leads to a drastic shift in water density (Fig. 5d). As the COF adsorbs PFOA molecules, a significant displacement of water molecules occurs from the COF to the bulk (Fig. 5d, Figure S20b). Upon reaching equilibrium, water in the bulk reaches a density of almost 0.98 g·cm−3, while the density of water inside the COF decreases to ~0.34 g·cm−3, indicating a substantial dehydration at the COF-PFOA interface. The binding of PFOA to the COF leads to the displacement of approximately 22 water molecules per adsorbed PFOA molecule. This displacement stems from PFOA molecules occupying positions previously held by water within the COF and stripping of water molecules from the solvation shell of PFOA (Fig. S20b). To interpret the zeta-potential measurements shown in Fig. S16a, we computed the average charge density along the longitudinal axis (z). Our findings reveal a positive surface charge for the COF at low PFOA loadings, which transitions to neutral and then to a negative surface charge distribution at high PFOA loadings (Fig. 5e), consistent with the observations in Fig. S16a.
To unravel the molecular mechanism underlying the exceptional PFOA uptake capacity, the specific interactions between PFOA, water, and the COF were examined using the radial distribution function (RDF). This analysis was conducted over the final 500 ns of the simulation, where equilibrium was achieved (Fig. 6a–g and Figs. S21–23). RDF peaks at shorter interatomic distances reveals specific binding sites between certain atom pairs, whereas the absence of peaks suggests a lack of interaction between pairs. Based on this, we observe a weak interaction between water and PFOA, with no specific binding observed between them (Fig. S21). In contrast, interactions between water and the COF were involved in weak hydrogen bonds between the aliphatic hydrogens of the COF and the oxygen atoms of water molecules (Fig. S22). The interactions between PFOA and the COF on the other hand were characterized by strong electrostatic forces at the binding sites, specifically between the positively charged nitrogen and aliphatic hydrogens of the COF and the negatively charged oxygen groups of PFOA, as well as the fluoride atoms (Fig. S23).To better understand the nature of the PFOA-COF interactions, we investigated the changes in the number of hydrogen bonds, \(\Delta {N}_{{hb}}\), and the changes in the non-bonded energy terms \(\Delta {E}_{{vdw}},{\rm{and}}\,\Delta {E}_{{elec}}\) using the first and last three nanoseconds of the simulation, representing the states of PFOA in its unbound and bound states respectively (Fig. 5b, and Figs. S24–26). We partitioned each term into the different species, namely water, PFOA, and COF. The results are summarized in Fig. 6h, i.

a Major surface atom types used for radial distribution function (RDF) analysis. b–g RDF of the COF-PFOA atom pairs. RDFs of water-PFOA and water-COF atom pairs are shown in Figs. S20 and S21. h Change in the number of hydrogen bonds between water-water, PFOA-water, COF-water, and the total system due to PFAO adsorption. i Non-bonded energy change during the adsorption process is partitioned into van der Walls and electrostatic terms. We consider total energy change and the energy change due to PFOA-COF interactions.
We observed that the process of PFOA adsorption causes a rise in the total number of hydrogen bonds, primarily governed by water-water hydrogen bonds. As PFOA molecules bind, the displaced water moving to bulk starts forming more hydrogen bonds with other water molecules. While the number of hydrogen bonds between water molecules increases, the count of COF-water and water-COF hydrogen bonds decreases (Fig. 6h and Figs. S24–26). Nonetheless, the acquired hydrogen bonds during water displacement outweigh the losses, providing an enthalpic and entropic contribution to the PFOA adsorption process. A second contributor to the PFOA-COF stability was found to be the change in the non-bonded interactions between PFOA and COF (Fig. 6i). The charge distribution of the PFOA and the cationic nature of the COF resulted in a dramatic reduction of the electrostatic energy upon PFOA adsorption into the pores in addition PFOA-COF form favorable interactions via dispersion forces. Overall our simulations propose that the enthalpic gain resulting from the interactions between PFOA and COF, supported by the increase in the number of hydrogen bonds of displaced water, serve as the driving force for PFOA adsorption by the COF.
Discussion
In conclusion, this study presents a novel approach to mitigate PFOA contamination by designing and synthesizing a multifunctional cationic covalent organic framework (COF), TG-PD COF, which seamlessly integrates PFOA sensing and removal capabilities. TG-PD COF exhibits high sensitivity for PFOA sensing, achieving a detection limit as low as 1.8 µg·L−1 through its fluorescence-based “Turn-On” sensing mechanism triggered by PFOA binding. Detailed investigations elucidate the charge transfer mechanism responsible for this performance. The adsorption performance is equally efficient, with rapid adsorption kinetics demonstrated even at environmentally relevant concentrations. The observed two-phase adsorption isotherm, attributed to the aggregation of PFOA molecules, culminates in a remarkable equilibrium uptake of over 2600 mg·g−1 at the highest PFOA concentration tested. Adsorption column tests have also revealed complete PFOA removal at environmentally relevant concentrations. The COF’s physicochemical properties, designed to harness both hydrophobic and electrostatic interactions, increase its efficiency in removing PFOA. In addition, the large pores and open structure prevent blockage of internal adsorption sites by PFOA aggregates and enable rapid adsorption kinetics. Molecular Dynamics simulations validate our experimental results and shed light on the intricate interactions between PFOA, water, and TG-PD COF at the atomic level. This study not only presents an innovative material but also provides profound insights into its interactions at the atomic-level, serving as a valuable guide for further research in this field.
Methods
Materials
All chemicals and starting materials were procured from Sigma-Aldrich and utilized without additional purification. The synthesis of 2,9-diformyl 1,10-phenanthroline (PD) and triamino guanidium hydrochloride salt (TGH•Cl) followed previously documented procedures in the literature92,93. Silica gel 60 F (Merck 9385, 0.040–0.063 mm) was employed for column chromatography. Amine analytes, including ammonium hydroxide (28.0–30.0%), methyl amine (2 M in THF), hydrazine hydrate (80%), pyridine (99%), 1-naphthylamine (99%), cadaverine (95%), diethylamine (99.5%), and triethylamine (99.5%) were obtained from Sigma Aldrich and utilized without further purification.
Characterization
Solution state nuclear magnetic resonance (NMR) spectroscopy was conducted at 25 °C using a Bruker Avance III spectrometer operating at frequencies of 500 MHz for 1H and 125.0 MHz for 13C nuclei. All chemical shifts are expressed in ppm relative to the signals corresponding to the residual non-deuterated solvents (CDCl3 = 7.26 ppm)94,95. Simultaneous decoupling of proton nuclei was employed for all 13C NMR spectra. Coupling constant values (J) are given in hertz (Hz). The proton spectrum multiplicity was denoted as follows: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), qt (quintet), sx (sextet), m (multiplet), and a broad signal is indicated by br (broad). Magic Angle Spinning (MAS) solid-state NMR experiments were carried out on a Bruker Avance-HD 600 MHz spectrometer operating at a static field of 14.1 T using a 4.0 mm MAS probe in the double channel mode. Samples were packed into 4.0 mm zirconia rotors and were spun at a MAS frequency of 14 kHz. Cross-Polarization Magic Angle Spinning (CP/MAS) NMR experiments were performed using a standard linearly ramped cross-polarization pulse sequence. 13C chemical shifts were externally referenced to the adamantane CH2 signal at 38.48 ppm on the TMS scale. NMR data were processed using TopSpin software.
Fourier transform infrared (FTIR) analyses were conducted using the Agilent 670-IR spectrometer. Thermogravimetric analysis (TGA) was carried out on the TA SDT Q600 instrument. Scanning electron microscopy (SEM) imaging was performed with the FEI Quanta 450FEG apparatus. Transmission electron microscopy (TEM) analysis of the COF material was conducted using a FEI-Titan 300 microscope, with samples prepared on carbon-coated copper grids and allowed to dry overnight after spotting a drop of dispersed TG-PD network. Surface area measurements were performed on a Micromeritics 3Flex gas sorption analyzer. Samples (20–50 mg) underwent degassing at 85 °C for 24 h followed by backfilling with N2. Adsorption isotherms were generated incrementally by exposure to ultrahigh-purity nitrogen up to 1 atm using ultrahigh-purity nitrogen in a liquid nitrogen bath, and surface area was determined employing BET adsorption models provided in the instrument software (Micromeritics ASAP 2020 V4.00). Powder X-ray diffraction (PXRD) analyses were carried out utilizing the PANalytical X’Pert PRO MP X-ray diffractometer equipped with a focusing elliptical mirror and a fast-high resolution detector (PIXCEL) using radiation with a wavelength of 0.15418 nm. UV–Visible spectroscopy (UV–Vis) was conducted on the Cary 5000 UV–Vis–NIR spectrophotometer. Emission spectra in water at room temperature were recorded using a Perkin Elmer LS55 Fluorescence Spectrometer. Dynamic light scattering measurements were executed with a Malvern Zeta sizer NanoSeries to determine the size and zeta-potential of the nanoparticles.
Quantum mechanical calculations
To compute point charges and the HUMO-LUMO levels, we utilized NWChem. We optimized the geometry of the structures using the Hartree Fock method and a 6-31 G* basis set. Charges were computed by fitting the electrostatic potential using the ESP module. To compute energy levels, we employed the B97-D functional by assigning a 6-311 G** basis set for all atoms.
PFOA adsorption experiments
Batch adsorption experiments of PFOA were performed using 20 mL glass scintillation vials at 24 °C. The experiments were conducted on a stirring hot plate at 500 revolutions per minute (rpm) stirring rate.
Prior to PFOA adsorption experiments, the vacuum-dried COF adsorbent was rehydrated by adding a precalculated mass of COFs to a scintillation vial with a precalculated volume of DI water to yield the required adsorbent concentration as a suspension. The COF suspensions were then sonicated for 30 min to disperse small aggregates. Then, the experiments were conducted by adding suitable volumes of the PFOA solution, DI water, and the COF adsorbent suspension to a 20 mL glass scintillation vial to generate the required concentrations of the PFOA solution and the COF suspension. Samples were collected in 1 mL volumes and filtered with a 0.2 μm syringe filter. Control experiments were conducted using samples that did not contain the COF adsorbent to account for PFOA losses due to other factors. These samples were collected for the control experiments in the same manner. All adsorption experiments were performed in triplicate.
Kinetic adsorption studies
Batch kinetic experiments were performed at both high and low concentrations of the PFOA and COF adsorbent. For experiments conducted at high concentration, the adsorbent dose was 400 mg L−1, while the PFOA concentration was 0.2 mg L−1. Prior to the measurements, the COF adsorbent was rehydrated by combining 8 mg of adsorbent with 16 mL of DI water in a 20 mL glass scintillation vial yielding a 500 mg·L−1 suspension. The suspension was sonicated for 30 minutes to disperse any small aggregates. Then, 4 mL of a 1 mg·L−1 PFOA solution was added to each suspension. 1 mL samples were collected in HPLC vials and filtered using a 0.2 µm syringe filter. Samples were collected at predetermined times (0, 0.1, 0.5, 1, 5, 10, 30 min and 22 h), where the time 0 sample was taken prior to the addition of the COF suspension and the 0.1 min sample was taken immediately following the addition of the COF suspension. Control experiments were performed in the exact same manner but without the addition of the COF suspension. The volume of DI water added to the control experiment was higher to compensate for the water not added from the COF suspension and to achieve the required final PFOA concentration.
The removal efficiency of PFOA at different experimental times was calculated by Eq. 1:
Where \({C}_{0}\) (mg·L−1) and \({C}_{t}\) (mg·L−1) are the initial and residual concentration of PFOA in the stock solution and filtrate, respectively. The adsorption capacity was determined by Eq. 2:
Where \({Q}_{t}\) (mg·g−1) is the PFOA uptake capacity by the COF adsorbent at any time t (min); \({C}_{0}\) (mg·L−1) is the PFOA average concentration in the control experiments; \({C}_{t}\) (mg·L−1) is the concentration of PFOA in the sample at time t (min); and \({C}_{A}\) (mg·L−1) is the concentration of adsorbent. The data from the adsorption kinetics were fitted against the pseudo-first-order and pseudo-second-order adsorption models in a linearized form represented by Eq. 3 and Eq. 4 respectively:
Where \(t\) is time, \({Q}_{e}\) (mg·g−1) is the PFOA uptake capacity by the COF adsorbent at equilibrium, \({K}_{1}\) (min−1) is the moduli of the pseudo-first order model and \({K}_{2}\) (g·mg−1·min−1) is the rate constant of adsorption.
Isotherm adsorption studies
Adsorption isotherm experiments were performed in 20 mL glass vials at 24 ̊C on a stirring hot plate (500 RPM) with magnetic stir bars. The COF adsorbent dose was fixed at 100 mg·L−1 for all the experiments and the initial concentration of the PFOA was varied with increments at 0.2, 1, 10, 20, 30, 50, 100, 200, 400, 600, 800, and 1000 mg·L−1. The experiments were stirred for 22 h to reach the equilibrium, then 1 mL samples were collected and filtered using 0.2 μm syringe filters to remove any remaining COF. Control experiments to account for PFOA losses were performed under the same conditions except for the addition of adsorbents, and samples were collected at 22 h. All adsorption isotherm experiments were performed in triplicate. Langmuir adsorption and Freundlich isotherm fits were generated by Non-linear Least Square Regression in Eq. 5 and Equation S:
Where \({C}_{e}\) (mg·L−1) is the equilibrium concentration of the PFOA in the solution following adsorption, \({Q}_{m}\) (mg·g−1) is the theoretical maximum adsorption capacity of the COF adsorbent, \({K}_{L}\) (L·mg−1) is a Langmuir-adsorption-affinity constant; \({K}_{f}\) is a Freundlich empirical constant which corresponds to the relative adsorption capacity of the adsorbents; and \(\frac{1}{n}\) is a Freundlich-adsorption-intensity constant. n is an indicator of the intensity of the adsorption.
Column adsorption studies
The COF column adsorption experiments were conducted using glass droppers as columns, allowing for controlled, small-scale experiments for safety, given the high concentrations of PFOA involved (Figure S15). A cotton layer was first inserted at the bottom of the column to support the COF and prevent it from exiting with the water. A 1000 mg·L−1 TG-PD COF suspension was used to form the COF bed. Specifically, 3 mL of the suspension was pipetted into the column to create a COF bed with exactly 3 mg of COF, and the suspension was allowed to settle. While the water exited through the cotton layer, the TG-PD COF particles settled on top of it. DI water was flushed through the column until clear water was collected from the filtrate end. Subsequently, 1 mL of the PFOA solution was pipetted into the dropper and discarded before sample collection. Then, 20 mL of each PFOA solution was continuously pipetted from the influent side, and the filtrate was collected in 1 mL volumes, placed in HPLC vials, and analyzed using HPLC-MS. This setup minimized contact and leak risks with the PFOA solution during the column tests.
Elemental mapping
High resolution transmission electron microscopy (HRTEM) images were collected using a Talos F200X STEM equipped with a CETA 16 M camera and a lattice-fringe resolution of 0.14 nm at an accelerating voltage of 200 kV. The samples were prepared on holey carbon film mounted on a copper grid. A drop of diluted particle solution was spotted on the grid and dried overnight at room temperature (298 K). The obtained images of periodic structures were analyzed using TIA software. Chemical mapping was carried out in STEM-EDAX mode while the energy-dispersive X-ray analysis (EDAX) was performed using a super-X EDS detector. The system has superior sensitivity with resolution of ≤136 eV/Mn-Kα for 10 kcps at zero-degree sample tilt. The detector provides quick data even for low intensity EDS signals. The data is the sum of 4 detectors and the collection time for the elemental maps in fast mapping mode can be reduced to minutes from hours. The data was analyzed using Velox analytical software. The samples for the HRTEM study were prepared on holey carbon film mounted on a copper grid.
Regeneration studies
Adsorption experiments
The regeneration studies began with batch adsorption experiments conducted as outlined in the isotherm adsorption experiments, using a PFOA concentration of 0.2 mg·L−1 and a COF concentration of 100 mg·L−1.
Desorption experiments
To desorb PFOA from the COF adsorbent, the COF powder was separated from the PFOA solution by centrifugation and subsequently soaked in methanol overnight. The PFOA concentration in methanol was analyzed by LC-MS to assess the desorption efficiency. The COF powder was then reused for another PFOA adsorption cycle following the previously described procedure.
Molecular modeling setup for the COF simulations
The COF unit cell was obtained from PXRD extended along each axis, forming a supercell of 2×2×12. The framework was then placed in a periodic box measuring 82.9×50.2×163.4 ų, with 63.4 Å extra space along the z-axis to create the bulk phase. 290 PFOA molecules were randomly added to the simulation box. Later, the box was solvated with water molecules, and Na+ ions were introduced to neutralize the system. The TIP3P model96 was employed to represent the water molecules, while the Dreiding force field97 was utilized for representing PFOA and COF. PFOA charges were derived from QM calculations implemented in NWchem98, while the Gasteiger method was adopted for the COF. For ion parameters, we used Ref 99. This force field combination provides an experimentally consistent depiction of the adsorption process, as demonstrated in our previous studies100,101.
Quantum mechanical calculations
To compute point charges and the HUMO-LUMO levels, we utilized NWChem. We optimized the geometry of the structures using the Hartree Fock method and a 6–31 G* basis set. Charges were computed by fitting the electrostatic potential using the ESP module.
Molecular dynamics simulations
The simulations were performed using GROMACS102. The simulation system was first optimized using the steepest descent for 500 steps103. Then the equations of motion were solved using the leapfrog integrator104 with a timestep of 1 fs in an NVT ensemble. The velocity rescaling was used to keep the temperature at 298 K. Long-range interactions were treated with a cut-off of 1 nm for both electrostatic and van der Waals interactions. The Particle Mesh Ewald Summation method105 was used to compute electrostatic interactions. Data recorded every 5 ps was used for the analysis. A detailed description of the simulation details can be found in our previous works100,101.
Data availability
The authors declare that the main data supporting the findings of this study are available within the article and its Supplementary Information files. The CIF File for TG-PD COF is available through Figshare (https://figshare.com/articles/dataset/TGPD_COF_cif/27222555?file=49774302). Extra data are available from the corresponding author upon request.
References
Ateia, M., Maroli, A., Tharayil, N. & Karanfil, T. The overlooked short- and ultrashort-chain poly- and perfluorinated substances: A review. Chemosphere 220, 866–882 (2019).
Zhang, X., Lohmann, R. & Sunderland, E. M. Poly- and perfluoroalkyl substances in seawater and plankton from the northwestern Atlantic margin. Environ. Sci. Technol. 53, 12348–12356 (2019).
Tokranov, A. K. et al. How do we measure poly- and perfluoroalkyl substances (pfass) at the surface of consumer products? Environ. Sci. Technol. Lett. 6, 38–43 (2019).
Lau, C. et al. Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicological Sci. 99, 366–394 (2007).
Sahu, S. P., Kole, S., Arges, C. G. & Gartia, M. R. Rapid and direct perfluorooctanoic acid sensing with selective ionomer coatings on screen-printed electrodes under environmentally relevant concentrations. ACS Omega 7, 5001–5007 (2022).
Basabe-Desmonts, L., Reinhoudt, D. N. & Crego-Calama, M. Design of fluorescent materials for chemical sensing. Chem. Soc. Rev. 36, 993–1017 (2007).
Ly, N. H., Kim, H. H. & Joo, S.-W. On-site detection for hazardous materials in chemical accidents. Bull. Korean Chem. Soc. 42, 4–16 (2021).
Albacete, P. et al. Chemical sensing of water contaminants by a colloid of a fluorescent imine-linked covalent organic framework. Chem. Commun. 55, 1382–1385 (2019).
Olorunyomi, J. F., Geh, S. T., Caruso, R. A. & Doherty, C. M. Metal–organic frameworks for chemical sensing devices. Mater. Horiz. 8, 2387–2419 (2021).
Kreno, L. E. et al. Metal–organic framework materials as chemical sensors. Chem. Rev. 112, 1105–1125 (2012).
Jia, R. et al. Amine-responsive cellulose-based ratiometric fluorescent materials for real-time and visual detection of shrimp and crab freshness. Nat. Commun. 10, 795 (2019).
Kumar, R. et al. Revisiting fluorescent calixarenes: from molecular sensors to smart materials. Chem. Rev. 119, 9657–9721 (2019).
EL-Mahdy, A. F. M., Lai, M.-Y. & Kuo, S. W. A highly fluorescent covalent organic framework as a hydrogen chloride sensor: roles of Schiff base bonding and π-stacking. J. Mater. Chem. C. 8, 9520–9528 (2020).
Zhu, M.-W. et al. The construction of fluorescent heteropore covalent organic frameworks and their applications in spectroscopic and visual detection of trinitrophenol with high selectivity and sensitivity. Chem. Commun. 54, 2308–2311 (2018).
Qian, H.-L., Dai, C., Yang, C.-X. & Yan, X.-P. High-crystallinity covalent organic framework with dual fluorescence emissions and its ratiometric sensing application. ACS Appl. Mater. Interfaces 9, 24999–25005 (2017).
Cui, W.-R. et al. Regenerable and stable sp2 carbon-conjugated covalent organic frameworks for selective detection and extraction of uranium. Nat. Commun. 11, 436 (2020).
Dalapati, S., Jin, E., Addicoat, M., Heine, T. & Jiang, D. Highly emissive covalent organic frameworks. J. Am. Chem. Soc. 138, 5797–5800 (2016).
Sun, R. et al. Flexible cyclosiloxane-linked fluorescent porous polymers for multifunctional chemical sensors. ACS Macro Lett. 9, 43–48 (2020).
Dong, J. et al. Fluorescent porous organic frameworks containing molecular rotors for size-selective recognition. Chem. Mater. 28, 7889–7897 (2016).
Das, G. et al. Chemical sensing in two dimensional porous covalent organic nanosheets. Chem. Sci. 6, 3931–3939 (2015).
Du, Z. et al. Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents—A review. J. Hazard. Mater. 274, 443–454 (2014).
Huang, N., Wang, P., Addicoat, M. A., Heine, T. & Jiang, D. Ionic covalent organic frameworks: design of a charged interface aligned on 1d channel walls and its unusual electrostatic functions. Angew. Chem. Int. Ed. 56, 4982–4986 (2017).
Kothawala, D. N., Köhler, S. J., Östlund, A., Wiberg, K. & Ahrens, L. Influence of dissolved organic matter concentration and composition on the removal efficiency of perfluoroalkyl substances (PFASs) during drinking water treatment. Water Res. 121, 320–328 (2017).
Chen, W., Zhang, X., Mamadiev, M. & Wang, Z. Sorption of perfluorooctane sulfonate and perfluorooctanoate on polyacrylonitrile fiber-derived activated carbon fibers: in comparison with activated carbon. RSC Adv. 7, 927–938 (2017).
Deng, S. et al. Sorption mechanisms of perfluorinated compounds on carbon nanotubes. Environ. Pollut. 168, 138–144 (2012).
Wang, Y., Darling, S. B. & Chen, J. Selectivity of per- and polyfluoroalkyl substance sensors and sorbents in water. ACS Appl. Mater. Interfaces 13, 60789–60814 (2021).
Xiao, F. Emerging poly- and perfluoroalkyl substances in the aquatic environment: A review of current literature. Water Res. 124, 482–495 (2017).
Liu, X. et al. Installation of synergistic binding sites onto porous organic polymers for efficient removal of perfluorooctanoic acid. Nat. Commun. 13, 2132 (2022).
Ateia, M., Alsbaiee, A., Karanfil, T. & Dichtel, W. Efficient PFAS removal by amine-functionalized sorbents: critical review of the current literature. Environ. Sci. Technol. Lett. 6, 688–695 (2019).
He, Y. et al. Fluorinated nonporous adaptive cages for the efficient removal of perfluorooctanoic acid from aqueous source phases. J. Am. Chem. Soc. 146, 6225–6230 (2024).
Román Santiago, A. et al. Imparting selective fluorophilic interactions in redox copolymers for the electrochemically mediated capture of short-chain perfluoroalkyl substances. J. Am. Chem. Soc. 145, 9508–9519 (2023).
Da, H.-J., Yang, C.-X. & Yan, X.-P. Cationic covalent organic nanosheets for rapid and selective capture of perrhenate: an analogue of radioactive pertechnetate from aqueous solution. Environ. Sci. Technol. 53, 5212–5220 (2019).
Wang, W. et al. Adsorption behavior and mechanism of emerging perfluoro-2-propoxypropanoic acid (GenX) on activated carbons and resins. Chem. Eng. J. 364, 132–138 (2019).
Jrad, A., Damacet, P., Yaghi, Z., Ahmad, M. & Hmadeh, M. Zr-Based metal–organic framework nanocrystals for water remediation. ACS Applied Nano Materials, https://doi.org/10.1021/acsanm.2c02128 (2022).
Chen, X., Xia, X., Wang, X., Qiao, J. & Chen, H. A comparative study on sorption of perfluorooctane sulfonate (PFOS) by chars, ash and carbon nanotubes. Chemosphere 83, 1313–1319 (2011).
Maimaiti, A. et al. Competitive adsorption of perfluoroalkyl substances on anion exchange resins in simulated AFFF-impacted groundwater. Chem. Eng. J. 348, 494–502 (2018).
Yu, Q., Zhang, R., Deng, S., Huang, J. & Yu, G. Sorption of perfluorooctane sulfonate and perfluorooctanoate on activated carbons and resin: Kinetic and isotherm study. Water Res. 43, 1150–1158 (2009).
Gagliano, E., Sgroi, M., Falciglia, P. P., Vagliasindi, F. G. A. & Roccaro, P. Removal of poly- and perfluoroalkyl substances (PFAS) from water by adsorption: Role of PFAS chain length, effect of organic matter and challenges in adsorbent regeneration. Water Res. 171, 115381 (2020).
McCleaf, P. et al. Removal efficiency of multiple poly- and perfluoroalkyl substances (PFASs) in drinking water using granular activated carbon (GAC) and anion exchange (AE) column tests. Water Res. 120, 77–87 (2017).
Côté, A. P. et al. Porous, crystalline, covalent organic frameworks. Science 310, 1166–1170 (2005).
Peng, Y. et al. Room temperature batch and continuous flow synthesis of water-stable covalent organic frameworks (COFs). Chem. Mater. 28, 5095–5101 (2016).
Furukawa, H. & Yaghi, O. M. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J. Am. Chem. Soc. 131, 8875–8883 (2009).
Huang, N., Chen, X., Krishna, R. & Jiang, D. Two‐dimensional covalent organic frameworks for carbon dioxide capture through channel‐wall functionalization. Angew. Chem. 127, 3029–3033 (2015).
Rasheed, T. Covalent organic frameworks as promising adsorbent paradigm for environmental pollutants from aqueous matrices: Perspective and challenges. Sci. Total Environ. 833, 155279 (2022).
Benyettou, F. et al. Tunable wettability of a dual-faced covalent organic framework membrane for enhanced water filtration. J. Am. Chem. Soc. 146, 23537–23554 (2024).
Abubakar, S. et al. Enhanced removal of ultratrace levels of gold from wastewater using sulfur-rich covalent organic frameworks. ACS Applied Materials & Interfaces, https://doi.org/10.1021/acsami.4c03685 (2024).
Das, G. et al. Fluorescence turn on amine detection in a cationic covalent organic framework. Nat. Commun. 13, 3904 (2022).
Dalapati, S. et al. An azine-linked covalent organic framework. J. Am. Chem. Soc. 135, 17310–17313 (2013).
Benyettou, F. et al. In vivo oral insulin delivery via covalent organic frameworks. Chem. Sci. 12, 6037–6047 (2021).
Fang, Q. et al. 3D porous crystalline polyimide covalent organic frameworks for drug delivery. J. Am. Chem. Soc. 137, 8352–8355 (2015).
Bai, L. et al. Nanoscale covalent organic frameworks as smart carriers for drug delivery. Chem. Commun. 52, 4128–4131 (2016).
Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208–1213 (2015).
Vyas, V. S. et al. A tunable azine covalent organic framework platform for visible light-induced hydrogen generation. Nat. Commun. 6, 1–9 (2015).
Wang, W. et al. Cationic covalent organic framework for efficient removal of PFOA substitutes from aqueous solution. Chem. Eng. J. 412, 127509 (2021).
Jiang, W., Cui, W.-R., Liang, R.-P. & Qiu, J.-D. Zwitterionic surface charge regulation in ionic covalent organic nanosheets: Synergistic adsorption of fluoroquinolone antibiotics. Chem. Eng. J. 417, 128034 (2021).
Mitra, S. et al. Self-exfoliated guanidinium-based ionic covalent organic nanosheets (iCONs). J. Am. Chem. Soc. 138, 2823–2828 (2016).
Singh, H. et al. Proton-triggered fluorescence switching in self-exfoliated ionic covalent organic nanosheets for applications in selective detection of anions. ACS Appl. Mater. Interfaces 12, 13248–13255 (2020).
Chen, H. et al. Cationic covalent organic framework nanosheets for fast li-ion conduction. J. Am. Chem. Soc. 140, 896–899 (2018).
Yahiaoui, O. et al. 3D anionic silicate covalent organic framework with srs topology. J. Am. Chem. Soc. 140, 5330–5333 (2018).
Zhang, Z. et al. Chemically stable guanidinium covalent organic framework for the efficient capture of low-concentration iodine at high temperatures. J. Am. Chem. Soc. 144, 6821–6829 (2022).
Zhang, P. et al. Covalent organic framework nanofluidic membrane as a platform for highly sensitive bionic thermosensation. Nat. Commun. 12, 1844 (2021).
Liang, Y. et al. Guanidinium-based ionic covalent organic frameworks for capture of uranyl tricarbonate. Adv. Compos. Hybrid. Mater. 5, 184–194 (2022).
Zhang, N., Li, X., Li, P. & Tang, S. Guanidinium cationic covalent organic nanosheets-based anion exchange composite membrane for fuel cells. Int. J. Hydrog. Energy 48, 25972–25983 (2023).
Tang, H., Kang, Y., Cao, S. & Chen, Z. Synthesis and performance of guanidinium-based cationic organic polymer for the efficient removal of TcO4-/ReO4. J. Hazard. Mater. 466, 133602 (2024).
Chen, B. et al. Screening and discrimination of perfluoroalkyl substances in aqueous solution using a luminescent metal–organic framework sensor array. ACS Appl. Mater. Interfaces 13, 47706–47716 (2021).
Zheng, Z. et al. Guanidinocalix[5]arene for sensitive fluorescence detection and magnetic removal of perfluorinated pollutants. Nat. Commun. 10, 5762 (2019).
Liang, J., Deng, X. & Tan, K. An eosin Y-based “turn-on” fluorescent sensor for detection of perfluorooctane sulfonate. Spectrochimica Acta Part A: Mol. Biomolecular Spectrosc. 150, 772–777 (2015).
Dalapati, R., Hunter, M., Sk, M., Yang, X. & Zang, L. Fluorescence turn-on detection of perfluorooctanoic acid (pfoa) by perylene diimide-based metal–organic framework. ACS Appl. Mater. Interfaces 16, 32344–32356 (2024).
He, Y. et al. Porous adaptive luminescent metallacage for the detection and removal of perfluoroalkyl carboxylic acids. Chem. 9, 93–101 (2023).
Yin, H.-Q. et al. A switchable sensor and scavenger: detection and removal of fluorinated chemical species by a luminescent metal–organic framework. Chem. Sci. 12, 14189–14197 (2021).
Cho, S. & Kim, Y. J-Aggregate-triggering bodipys: an ultrasensitive chromogenic and fluorogenic sensing platform for perfluorooctanesulfonate. Chem. – A Eur. J. 29, e202302897 (2023).
Concellón, A. & Swager, T. M. Detection of per- and polyfluoroalkyl substances (pfas) by interrupted energy transfer. Angew. Chem. Int. Ed. 62, e202309928 (2023).
Gogoi, B. & Sen Sarma, N. Curcumin–cysteine and curcumin–tryptophan conjugate as fluorescence turn on sensors for picric acid in aqueous media. ACS Appl. Mater. Interfaces 7, 11195–11202 (2015).
Concellón, A., Castro-Esteban, J. & Swager, T. M. Ultratrace PFAS detection using amplifying fluorescent polymers. J. Am. Chem. Soc. 145, 11420–11430 (2023).
Fang, C., Wu, J., Sobhani, Z., Amin, M. A. & Tang, Y. Aggregated-fluorescent detection of PFAS with a simple chip. Anal. Methods 11, 163–170 (2019).
Bera, N., Kiran Nandi, P., Hazra, R. & Sarkar, N. Aggregation induced emission of surface ligand controlled gold nanoclusters employing imidazolium surface active ionic liquid and pH sensitivity. J. Photochemistry Photobiol. A: Chem. 437, 114471 (2023).
Shulov, I. et al. Fluorinated counterion-enhanced emission of rhodamine aggregates: ultrabright nanoparticles for bioimaging and light-harvesting. Nanoscale 7, 18198–18210 (2015).
Hussain, F. A., Janisse, S. E., Heffern, M. C., Kinyua, M. & Velázquez, J. M. Adsorption of perfluorooctanoic acid from water by pH-modulated Brönsted acid and base sites in mesoporous hafnium oxide ceramics. iScience 25, 104138 (2022).
Zhang, D. et al. Sorption of perfluorooctanoic acid, perfluorooctane sulfonate and perfluoroheptanoic acid on granular activated carbon. Chemosphere 144, 2336–2342 (2016).
Xu, C., Chen, H. & Jiang, F. Adsorption of perflourooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) on polyaniline nanotubes. Colloids Surf. A: Physicochemical Eng. Asp. 479, 60–67 (2015).
Yao, Y., Volchek, K., Brown, C. E., Robinson, A. & Obal, T. Comparative study on adsorption of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) by different adsorbents in water. Water Sci. Technol. 70, 1983–1991 (2014).
Zhu, B.-Y. & Gu, T. Surfactant adsorption at solid-liquid interfaces. Adv. colloid interface Sci. 37, 1–32 (1991).
Kalam, S., Abu-Khamsin, S. A., Kamal, M. S. & Patil, S. Surfactant adsorption isotherms: a review. ACS Omega 6, 32342–32348 (2021).
Koopal, L. K., Lee, E. M. & Bohmer, M. R. Adsorption of cationic and anionic surfactants on charged metal oxide surfaces. J. Colloid Interface Sci. 170, 85–97 (1995).
Paria, S. & Khilar, K. C. A review on experimental studies of surfactant adsorption at the hydrophilic solid–water interface. Adv. Colloid Interface Sci. 110, 75–95 (2004).
Sahara, T. et al. Highly effective removal of perfluorooctanoic acid (PFOA) in water with DBD-plasma-enhanced rice husks. Sci. Rep. 13, 13210 (2023).
Karoyo, A. H. & Wilson, L. D. Investigation of the adsorption processes of fluorocarbon and hydrocarbon anions at the solid–solution interface of macromolecular imprinted polymer materials. J. Phys. Chem. C. 120, 6553–6568 (2016).
Karoyo, A. H. & Wilson, L. D. Tunable macromolecular-based materials for the adsorption of perfluorooctanoic and octanoic acid anions. J. Colloid Interface Sci. 402, 196–203 (2013).
Wang, Y. et al. Performance and mechanisms for removal of perfluorooctanoate (PFOA) from aqueous solution by activated carbon fiber. RSC Adv. 5, 86927–86933 (2015).
Pavan, P. C. et al. Adsorption of sodium dodecylsulfate on a hydrotalcite-like compound. Effect of temperature, pH and ionic strength. Colloids Surf. A: Physicochemical Eng. Asp. 154, 399–410 (1999).
Taylor, C. M., Breadmore, M. C. & Kilah, N. L. Good practices and practical considerations for research wiTH PERFLUOROALKYL Substances. Chem.–Methods n/a, e202300017 (2023).
Das, G. et al. Redox-triggered buoyancy and size modulation of a dynamic covalent gel. Chem. Mater. 31, 4148–4155 (2019).
Prakasam, T. et al. Dynamic stereoisomerization in inherently chiral bimetallic [2] catenanes. Chem. Commun. 51, 5840–5843 (2015).
Gottlieb, H. E., Kotlyar, V. & Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 62, 7512–7515 (1997).
Fulmer, G. R. et al. NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 29, 2176–2179 (2010).
Price, D. J. & Brooks, C. L. III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 121, 10096–10103 (2004).
Mayo, S. L., Olafson, B. D. & Goddard, W. A. DREIDING: a generic force field for molecular simulations. J. Phys. Chem. 94, 8897–8909 (1990).
Valiev, M. et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Computer Phys. Commun. 181, 1477–1489 (2010).
Joung, I. S. & Cheatham, T. E. III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 112, 9020–9041 (2008).
Garai, B. et al. Taming the topology of calix[4]arene-based 2d-covalent organic frameworks: interpenetrated vs noninterpenetrated frameworks and their selective removal of cationic dyes. J. Am. Chem. Soc. 143, 3407–3415 (2021).
Skorjanc, T. et al. Covalent organic framework based on azacalix[4]arene for the efficient capture of dialysis waste products. ACS Appl. Mater. Interfaces 14, 39293–39298 (2022).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
McSherry, T. M. A general steepest descent algorithm. IEEE Trans. Aerosp. Electron. Syst. AES-12, 12–22 (1976).
Van Gunsteren, W. F. & Berendsen, H. J. C. A leap-frog algorithm for stochastic dynamics. Mol. Simul. 1, 173–185 (1988).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Acknowledgements
This work was funded by New York University Abu Dhabi and the NYUAD Water Research Center funded by Tamkeen under the NYUAD Research Institute Award (project CG007), we thank them for their generous support. A.T. thanks Sandooq Al Watan (Grant No. SWARD-S22-014, Project ID: PRJ-SWARD-628), and ASPIRE (AARE20−116) for their generous support. The research work was carried out by using the Core Technology Platform resources at New York University Abu Dhabi. Computational research was carried out on the High-Performance Computing resources at New York University Abu Dhabi. The authors thank graphic designer Aisha Jrad for her contribution to the adsorption schemes.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.T., A.J., and G.D. Methodology: A.T., A.J., G.D., S.K., N.A., F.B., T.P., S.V., and F.G. Investigation: A.J., G.D., and N.A. Visualization: A.J., G.D., and M.O. Funding acquisition: A.T., A.J., F.B. Supervision: A.T. and S.K. Writing – original draft: A.J., G.D., and N.A. Writing – review & editing: A.T., S.K., M.O., A.J., G.D., N.A., S.V., F.G., F.B., and T.P.
Corresponding authors
Ethics declarations
Competing interests
Authors Ali Trabolsi, Asmaa Jrad, and Gobinda Das are named on the patent related to the technology and findings described in this manuscript (U.S. Patent Application No. 63/633,767). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rahul Banerjee and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jrad, A., Das, G., Alkhatib, N. et al. Cationic covalent organic framework for the fluorescent sensing and cooperative adsorption of perfluorooctanoic acid. Nat Commun 15, 10490 (2024). https://doi.org/10.1038/s41467-024-53945-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53945-4
This article is cited by
-
Nanoparticle-encapsulated organo-magnetogels: crosslinked network for broad-spectrum pollutant removal
npj Clean Water (2025)
-
Electro-activated dual-affinity membrane for efficiently removing per- and polyfluoroalkyl substances from drinking water
Nature Water (2025)
-
MoB QDs/N, F-CDs for ratiometric fluorescence sensing of perfluorooctanoic acid based on fluorine‑fluorine interaction
Microchimica Acta (2025)
