
Abstract
Flaviviruses strategically utilize the endoplasmic reticulum (ER) in their replication cycles. However, the role of ER autophagy (ER-phagy) in viral replication process remains poorly understood. Here, we reveal that prolonged Zika virus (ZIKV) infection results from the degradation of ER-phagy receptor FAM134B, facilitated by viral NS2A protein. Mechanistically, ER-localized NS2A undergoes K48-linked polyubiquitination at lysine (K) 56 by E3 ligase AMFR. Ubiquitinated NS2A binds to FAM134B and AMFR orchestrates the degradation of NS2A-FAM134B complexes. AMFR-catalyzed NS2A ubiquitination not only targets FAM134B degradation but also hinders the FAM134B-AMFR axis. Notably, a recombinant ZIKV mutant (ZIKV-NS2AK56R), lacking ubiquitination and ER-phagy inhibition, exhibits attenuation in ZIKV-induced microcephalic phenotypes in human brain organoids and replicates less efficiently, resulting in weakened pathogenesis in mouse models. In this work, our mechanistic insights propose that flaviviruses manipulate ER-phagy to modulate ER turnover, driving viral infection. Furthermore, AMFR-mediated flavivirus NS2A ubiquitination emerges as a potential determinant of viral pathogenecity.
Similar content being viewed by others


Introduction
The genus Flavivirus of the family Flaviviridae are vector-borne pathogens, comprising many viruses, including Zika virus (ZIKV), dengue virus (DENV), West Nile virus (WNV), Japanese encephalitis (JEV), yellow fever virus (YFV), and tick-borne encephalitis (TBEV)1. Flavivirus infections cause severe clinical manifestations in humans, such as encephalitis, hemorrhagic fever, and so on2,3. Especially, ZIKV infection can cause congenital neurological disorders and microcephaly in newborns4,5,6,7,8,9. The absence of clinically approved antivirals highlights the imperative for enhanced molecular comprehension of flavivirus replication and the precise virus–host factors that facilitate it, to pave the way for effective treatments of diseases caused by flaviviruses1,10,11.
Flaviviruses have a positive-sense, single-stranded RNA of ~11-kb nucleotides in length. The genomic RNA contains a 5′ untranslated region (UTR), a single open-reading frame (ORF), and a 3′ UTR. The ORF encodes a polyprotein that undergoes co-translational processing by both viral and host proteases, resulting in ten mature viral proteins, including three structural proteins (capsid [C], premembrane [prM], and envelope [E]) and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5)1,11. The structural proteins, along with genomic RNA, constitute the components of virions, while NS proteins primarily engage in viral RNA replication, virion assembly, and modulation of host processes12,13. Wherein, the flavivirus NS2A is a transmembrane protein resident in the endoplasmic reticulum (ER) and exhibits comparable functionalities in governing RNA replication, the assembly and secretion of viral particles, modulation of host antiviral responses, and the influence in viral pathogenicity14,15,16,17,18,19,20. The multifaceted functions observed underscore the significance of flavivirus NS2A as a pivotal target for antiviral strategies, emphasizing the need for a deeper understanding of the more prevalent interactions between flavivirus NS2A and host mechanisms.
The ER serves as the largest dynamic membrane-structured organelle for protein synthesis, folding, assembly, and transportation, as well as lipid metabolism and so on21. Disruption of ER homeostasis leads to the accumulation of misfolded and unfolded proteins in the ER lumen, resulting in ER stress, which in turn induces ER autophagy (ER-phagy)22. ER-phagy is a selective form of autophagy strictly mediated by ER-phagy receptors that selectively degrade the ER, which is an essential mechanism responsible for ER quality control23. ER-phagy receptors facilitate the connection of ER fragments to the autophagosome biogenesis machinery by binding to cellular LC3, promoting the recognition of scattered foci on the ER by autophagy24. Some mammalian ER membrane proteins, including FAM134B, SEC62, ATL3, RTN3, CCPG1, and TEX26425,26,27,28,29,30, have been reported as ER-phagy receptors.
Several RNA viruses, including flavivirus and coronavirus, exploit the ER as a source of membranes to establish their replication organelles and to facilitate their assembly and eventual virion maturation31,32,33,34,35, for example, ZIKV remodels ER membranes to accumulate the involvement of redox and methylation enzymes for efficient infection36. Consequently, the homeostasis and quantity of ER play a pivotal role in controlling viral infections. While numerous studies have consistently shown that viral infections trigger prolonged ER stress and activate the unfolded protein response to disrupt ER homeostasis, ultimately facilitating virus replication37,38,39,40,41, few studies have explored and linked the role of ER-phagy in the viral replication process. Furthermore, the mechanisms of how viruses subvert ER-phagy to promote the pathogenicity of viral diseases remain poorly understood.
Here, we show that ZIKV hijacks ER-located E3 ligase AMFR to ubiquitinate viral NS2A protein and AMFR orchestrates the degradation of NS2A-FAM134B complexes, thereby inhibiting the flux of ER-phagy. ZIKV-NS2A ubiquitination and its functional interplay with FAM134B are conserved across other flavivirus NS2A. Importantly, our mechanistic work shows ZIKV-NS2A-mediated ER-phagy inhibition promotes virus-induced microcephaly in human brain organoids and enhances viral pathogenicity in mouse models, which sheds light on the pathogenicity of viral infection by ER-phagy modulation.
Results
FAM134B undergoes degradation during several flavivirus infections
To determine how ZIKV hijacks host-protein signaling, a global quantitative proteomics experiment was performed in A549 cells. Cells were harvested in biological triplicate at four time points post-ZIKV infection (0, 24, 48, or 72 h) (Fig. 1a). Employing a tandem mass tag(TMT)-based proteomics approach, we partitioned and analyzed each sample for alterations in global protein abundance at 24, 48, and 72 h post-infection (hpi) in comparison to the baseline at 0 hpi, respectively. Successful infection was confirmed by the observation of a dramatic increase in viral protein abundance over the course of the infection period (Fig. 1b). Cellular component enrichment analysis unveiled a notable upregulation in organelle membrane components, particularly ER constituents, concomitant with the progression of viral infection (Fig. 1c and Supplementary Fig. 1a). Moreover, the mRNA levels of upregulated ER proteins were found to be minimally affected by ZIKV at 72 hpi (Supplementary Fig. 1b). In mammalian cells, the quantity and volume of the ER are tightly regulated through selective ER-phagy24. Thus, we hypothesized that ZIKV may exploit ER-phagy to elevate ER components, facilitating viral infection.

a Workflow for TMT-based quantitative proteomic analysis. b Median abundance of individual ZIKV proteins. c Cellular component analysis of significantly up-changing proteins (−log10 (P value)). d Western blotting analysis of A549 cells either mock-infected or infected with ZIKV at a MOI of 0.1. e Quantification of relative protein level of ER-phagy receptors. f Cells (A549, Huh7.0, and HeLa) stably expressing GFP-FAM134B were either mock-infected or infected with ZIKV at different MOIs (A549[0.1]; Huh7.0[1]; HeLa[1]) for 72 h, determined by flow cytometry. g–j Western blotting analysis of Huh7.0 (g) or A549 (i) were either mock-infected or infected with ZIKV at multiple time points with a MOI of 0.1 (g) or at multiple MOIs for 24 h (i) and the corresponding quantification of relative FAM134B level (h, j). k Cells (A549 and Huh7.0) stably expressing GFP-FAM134BR142A were either mock-infected or infected with ZIKV at different MOIs (A549[0.1]; Huh7.0[1]) for 72 h, determined by flow cytometry. l A549 stably expressing GFP-FAM134BR142A were either mock-infected or infected with ZIKV at multiple time points with an MOI of 0.1, determined by flow cytometry. m Viral titers of supernatants from Huh7.0 FAM134B WT cells, Huh7.0 FAM134B KO cells, Huh7.0 FAM134B KO cells stably expressing FAM134BWT or Huh7.0 FAM134B KO cells stably expressing FAM134BR142A infected with ZIKV at a MOI of 1 for 72 h, quantified by plaque assay. n Huh7.0 cells stably expressing GFP-FAM134BR142A were either mock-infected or infected with ZIKV, DENV, or JEV at a MOI of 1 for 72 h, determined by flow cytometry. o, p Viral titers of supernatants from Huh7.0 FAM134B WT, Huh7.0 FAM134B KO cells, or Huh7.0 FAM134B KO cells stably expressing FAM134BWT infected with DENV or JEV at a MOI of 1 for 72 h, quantified by plaque assay. Error bars of (e, f, h, j–p) present as the mean ± SEM (n = 3 independent experiments); Statistical analysis of (e, f, k, n) was performed using two-way ANOVA with Sidak’s multiple comparisons test and (h, j, l, m, o, p) was performed using one-way ANOVA with Tukey’s multiple comparisons test; Source data are provided as a Source Data file.
ER-phagy is reliant on receptor-mediated processes, and to date, six ER-phagy receptors have been identified, including FAM134B, SEC62, ATL3, RTN3, CCPG1, and TEX26425,26,27,28,29,30. Firstly, we assessed the expression of these six autophagic receptors in ZIKV-infected A549 cells via western blotting. Unexpectedly, only FAM134B exhibited significant degradation upon ZIKV infection (Fig. 1d, e), with no discernible impact on its mRNA levels (Supplementary Fig. 1c). In the proteomics analysis, the remaining five receptors exhibited no statistically significant changes throughout the course of the infection period, except that CCPG1 at 48 h was decreased (Supplementary Fig. 1d). Next, we utilized flow cytometry assay to further confirm ZIKV-induced FAM134B degradation and found A549, Huh7.0, and HeLa cells stably expressing GFP-FAM134B exhibited a diminished fluorescence signal of GFP upon ZIKV infection (Fig. 1f). Additionally, ZIKV selectively degraded endogenous FAM134B in A549 cells in a gradient manner at multiple time points and MOIs (Fig. 1g–j). Subsequently, we employed cycloheximide (CHX), a protein translation inhibitor, to carry out CHX chase experiments in order to evaluate the degradation kinetics of FAM134B. Our findings indicated that ZIKV infection resulted in diminished protein stability of FAM134B (Supplementary Fig. 2a). Notably, efficient degradation was also observed from other ZIKV strains, suggesting that virus-induced FAM134B degradation is a common regulation for ZIKV (Supplementary Fig. 2b). A prior study reported that ZIKV-NS2B3 protein cleaved the arginine (R) at position 142 of only exogenously expressed FAM134B in HEK293T cells42. Although the results lacked confirmations in live virus infection, we still checked whether ZIKV infection-induced FAM134B degradation, which we found, was a result of viral NS2B3-mediated cleavage. We successfully generated Huh7.0 and A549 cells stably expressing the FAM134B cleavage mutant GFP-FAM134BR142A, where the R was mutated to alanine (A) (Supplementary Fig. 2c, d). We found Huh7.0 and A549 cells expressing GFP-FAM134BR142A infected with ZIKV still resulted in a reduced fluorescence signal of GFP, as observed by flow cytometry (Fig. 1k), and a decline in FAM134BR142A protein levels, as indicated by western blotting (Supplementary Fig. 2e). ZIKV also gradually degraded FAM134BR142A in A549 cells at multiple time points and MOIs by flow cytometry assay (Fig. 1l and Supplementary Fig. 2f). Collectively, these findings demonstrated that ZIKV infection induces the degradation of the ER-phagy receptor FAM134B independently of viral NS2B3-mediated cleavage.
To determine the influence of FAM134B on ZIKV infection, we utilized CRISPR/Cas9 to knock out FAM134B in Huh7.0 cells and confirmed knockout (KO) efficiency by western blotting (Supplementary Fig. 3a). The depletion of FAM134B resulted in a significant elevation in ZIKV-Envelope (E) transcripts (Supplementary Fig. 3b) and ZIKV production (Supplementary Fig. 3c), which was consistent with the effects of FAM134B knockdown by siRNA conducted before42. This enhancing effect was reversed in Huh7.0 FAM134B KO cells stably expressing either FAM134BWT or FAM134BR142A (Fig. 1m). Remarkably, when the expression levels of FAM134BWT and FAM134BR142A were comparable, both exhibited no significant differences in inhibiting ZIKV-E protein levels (Supplementary Fig. 3d) and viral production (Fig. 1j). This suggests that the ZIKV-NS2B3-mediated cleavage of FAM134B, as elucidated solely at the protein level, may not exert a substantial influence on live virus infection. Within this context, a more precise and intricate mechanism governing the degradation of FAM134B during the ZIKV infection process remains to be elucidated.
Interestingly, when we tested the impact of other flaviviruses on FAM134B, we observed that infections with DENV or JEV similarly induced the degradation of FAM134B (Fig. 1n and Supplementary Fig. 2g, h). Additionally, the knockout of FAM134B heightened DENV and JEV infections, while the overexpression of FAM134B conversely restrained their infections (Fig. 1o, p). These findings paralleled the impact observed with ZIKV, suggesting that the degradation of FAM134B might represent a conserved signature across various flavivirus infections, and FAM134B served as a common restrictor for flaviviruses.
Flavivirus-NS2A targets FAM134B for degradation
To explore the molecular mechanisms behind ZIKV-induced degradation of FAM134B, we co-transfected equimolar amounts of FAM134B with each of the ten viral proteins of ZIKV in HEK293T cells. Intriguingly, ZIKV-NS2A protein emerged as the key player, resulting in reduced levels of FAM134B compared to other viral proteins (Fig. 2a, b). We further found exogenously expressed ZIKV-NS2A prompted dose-dependent degradation of both endogenous and exogenous FAM134B (Fig. 2c and Supplementary Fig. 4a), with no impact on other ER autophagic receptors ATL3, SEC62, and CCPG1 (Fig. 2c and Supplementary Fig. 4b, c). Furthermore, a gradual decline in GFP signal was observed with increasing doses of ZIKV-NS2A in Huh7.0-GFP-FAM134B cells through flow cytometry analysis (Fig. 2d). In order to determine that ZIKV-NS2A degrades FAM134B during infection, we utilized the recombinant virus ZIKV-HA-NS2A, bearing a hemagglutinin (HA)-tagged NS2A to mark NS2A20, to infect GFP-FAM134BR142A expressed Huh7.0 cells and found virus-infected cells resident in HA-NS2A showed a reduction of GFP fluorescence as evidenced by immunofluorescence assays (Fig. 2e, f). The above experiments demonstrated the specific degradation of FAM134B by ZIKV-NS2A protein.

a, b Western blotting analysis of HEK293T cells co-transfected for 48 h with indicated Myc-FAM134B and Flag-tagged ZIKV viral protein-expressing plasmids or empty vector. The red font highlighted the ZIKV-NS2A protein. c Western blotting analysis of A549 cells transfected for 48 h with increasing amounts of Flag-NS2A. d Huh7.0-GFP-FAM134B transfected for 48 h with increasing amounts of Flag-NS2A, determined by flow cytometry. e Huh7.0-GFP-FAM134BR142A cells were infected with recombinant virus ZIKV-HA-NS2A at a MOI of 1 for 72 h and assessed confocal microscopy analysis. The plus sign (+) represents virus-infected cells marked by HA-NS2A. Scale bars, 10 μm. f Quantification of mean intensity of GFP-FAM134BR142A, related to (e). g HEK293T cells were co-transfected with Flag-NS2A and HA-tagged ER-phagic receptor-expressing plasmids or empty vector. Cells were harvested for coIP using anti-HA beads. The asterisk (*) represents the indicated ER-phagy receptors. h Huh7.0 cells were either mock-infected or infected with ZIKV-HA-NS2A at a MOI of 1 for 72 h, and then cells were harvested for coIP using anti-HA beads and analyzed by western blotting. The asterisk (*) represents the co-immunoprecipitated endogenous FAM134B, and the band of HA-NS2Aα detected by western blotting represented a truncated form of NS2A20. i HEK293T cells were co-transfected with HA-FAM134B and Flag-tagged flavivirus-NS2A-expressing plasmids or empty vector. Cells were harvested for coIP using anti-flag beads. The asterisk (*) represents the indicated flavivirus-NS2A. j Schematic diagram of FAM134B mutants. k HEK293T cells were co-transfected with FAM134BWT and mutants with Flag-NS2A or empty vector. Cells were harvested for coIP using anti-flag beads. The asterisk (*) represents the indicated FAM134BWT and mutants. l, m Huh7.0-GFP-FAM134B△NTD were transfected for 48 h with increasing amounts of Flag-NS2A (l) or infected with ZIKV at a MOI of 1 (m) for 72 h, determined by flow cytometry. Error bars of (d, f, l, m) present as the mean ± SEM (d, l, m: n = 3; f: n = 32); Statistical analysis of (d, l, m) was performed using one-way ANOVA with Tukey’s multiple comparisons test and (f) was performed using two-tailed unpaired t-test analysis; Source data are provided as a Source Data file.
Since NS2A induces the reduction of FAM134B, we speculate that NS2A may interact with FAM134B. Next, co-immunoprecipitation (coIP) experiments were undertaken to elucidate the interactions between ZIKV-NS2A and six ER autophagic receptors. Our findings revealed that NS2A engaged with two receptors, exhibiting a robust interaction with FAM134B and a more modest interaction with ATL3 (Fig. 2g). The interaction of NS2A with FAM134B, but not with C, was verified through coIP using Flag-tagged viral proteins as bait (Supplementary Fig. 4d, e). Additionally, we employed purified Myc-FAM134B derived from HEK293T cells or purified GST-FAM134B from E. coli to successfully pull down purified Flag-NS2A and observed the direct interaction between NS2A and FAM134B in both instances (Supplementary Fig. 4f, g).
Furthermore, under viral infection conditions, the HA-tagged NS2A protein from the virus interacted with endogenous FAM134B, but not with another receptor, CCPG1, underscoring the specificity of the NS2A-FAM134B interaction during the infection process (Fig. 2h). In addition, we also detected FAM134B interaction with other flavivirus NS2A. CoIP studies showed that DENV, West Nile virus (WNV), and JEV-NS2A proteins all bound to FAM134B, and in the lysates, we found DENV, WNV, and JEV-NS2A all could degrade FAM134B (Fig. 2i), with similar efficiency as ZIKV-NS2A.
Next, we generated FAM134B truncations to identify the domain responsible for interaction with ZIKV-NS2A (Fig. 2j). Our results indicated that the N-terminal domain (NTD) of FAM134B is essential for interaction with NS2A in vivo and in vitro (Fig. 2k and Supplementary Fig. 4h, i). Disruption of FAM134B-NS2A interactions resulted in the failure of both exogenously expressed NS2A or ZIKV infection degrading FAM134B△NTD (Fig. 2l, m and Supplementary Fig. 4j).
Collectively, these data suggested the critical nature of the interaction between FAM134B and NS2A in ZIKV-induced FAM134B degradation and FAM134B-flavivirus NS2A interactions and subsequent FAM134B degradation was a conserved regulatory mechanism of ZIKV, DENV, WNV, and JEV.
AMFR mediates flavivirus-NS2A ubiquitinations which is required for NS2A-derived FAM134B degradation
Proteasomes and lysosomes are the primary loci for protein degradation in cells43,44. To unravel the mechanism behind ZIKV-NS2A-triggered FAM134B degradation, we treated HEK293T cells expressing NS2A and FAM134B with the proteasome inhibitor MG132 or the autophagosome-lysosome degradation inhibitor, chloroquine (CQ). When FAM134B was treated with MG132 alone, no significant impact was observed (Fig. 3a, compared lanes 1 and 2), but it showed an increase in the presence of CQ, indicative of the restoration of FAM134B due to inhibited ER-phagy (Fig. 3a, compared lanes 1 and 3), where overexpression of ER-phagy receptor alone can induce ER-phagy45. In contrast, NS2A proteins were markedly rescued by MG132 rather than CQ, irrespective of the presence of FAM134B (Fig. 3a, compared lanes 5 and 8). Additionally, NS2A proteins expressed in HEK293T cells were rapidly degraded upon treatment with CHX (Supplementary Fig. 5a), and this degradation was attenuated by MG132 (Supplementary Fig. 5b). These findings suggest that ZIKV-NS2A proteins undergo proteasome-dependent degradation, resulting in a short half-life in cells. Interestingly, when FAM134B and NS2A were co-expressed in HEK293T cells, ZIKV-NS2A failed to degrade FAM134B upon proteasome inhibition caused by MG132 treatment or in the CHX chase assay (Fig. 3a, compared lanes 7 and 8; Fig. 3b and Supplementary Fig. 5c). Additionally, FAM134B and autophagic marker LC3B exhibited colocalization under MG132 treatment. However, upon introduction of NS2A, neither FAM134B nor NS2A showed a negative correlation with LC3B and FAM134B-LC3B interaction was inhibited in the coIP assay, which suggested that NS2A-mediated FAM134B degradation was independent of the autophagic pathway (Supplementary Fig. 5d–f). In short, these observations suggested that ZIKV-induced FAM134B degradation relied on the NS2A-dependent proteasome pathway.

a, b, Western blotting analysis of HEK293T cells transfected for 30 h with the indicated plasmids and then treated with DMSO, MG132 (10 μM), and CQ (50 μM) for 8 h (a); Quantification of relative FAM134B level (b). c–f, h, j, HEK293T cells were transfected with the indicated plasmids. Cells were harvested for coIP using anti-Flag beads and analyzed by western blotting with a K48-linkage-specific polyubiquitin antibody to detect the ubiquitination of NS2A. g In vitro ubiquitination assays were measured by incubating purified Flag-NS2A with purified HA-AMFRWT or HA-AMFRC356G/H361A analyzed by western blotting with a K48-linkage specific polyubiquitin antibody to detect the ubiquitination of NS2A. i Huh7.0 cells were either mock-infected or infected with ZIKV-HA-NS2A at an MOI of 1 for 72 h and then cells were harvested for coIP using anti-HA beads and analyzed by western blotting. k A549 and Vero cells were mock-infected or infected with ZIKV-HA-NS2AWT (MOI = 0.1) or ZIKV-HA-NS2AK56R (MOI = 1) for 72 h and then harvested for coIP using anti-HA beads and analyzed by western blotting with a K48-linkage specific polyubiquitin antibody to detect the ubiquitination of NS2A and the input E was normalized for immunoprecipitation. l, m Western blotting analysis of HEK293T cells or HEK293T-AMFR knockdown cells transfected for 48 h with the indicated plasmids (l); Quantification of relative FAM134B level (m). n Huh7.0-GFP-FAM134BR142A cells were infected with ZIKV-NS2AWT (MOI = 1), ZIKV-NS2AK56R (MOI = 10), or mock-infected for 72 h, determined by flow cytometry. Error bars of (b, m, n) present as the mean ± SEM (n = 3 independent experiments); Statistical analysis of (b) was performed using two-way ANOVA with Sidak’s multiple comparisons test and (m, n) was performed using one-way ANOVA with Tukey’s multiple comparisons test; Source data are provided as a Source Data file.
Given that proteasomes preferentially target K48-linked ubiquitinated substrates for degradation46, we speculate that ZIKV-NS2A undergoes K48-linked polyubiquitination for ubiquitin-proteasome degradation, playing a pivotal role in mediating the degradation of FAM134B. Indeed, ubiquitination IP analysis revealed that NS2A undergoes strong endogenous K48-linked polyubiquitination, as opposed to K63-linked ubiquitination (Fig. 3c), and pulled down more HA-tagged K48-only ubiquitin (Supplementary Fig. 6a). To confirm NS2A ubiquitination modification, we screened some E3 ligases and found that ER-localized ligase AMFR, but not ligase-dead AMFRC356G/H361A, significantly enhanced K48-linked ubiquitination of NS2A and promoted NS2A degradation in HEK293T cells (Fig. 3d, e and Supplementary Fig. 6b). Notably, AMFR-mediated K48-linked polyubiquitination and degradation were also observed in DENV, WNV, and JEV-NS2A proteins (Fig. 3f). AMFRWT, but not ligase-dead AMFRC356G/H361A, promoted the K48-linked ubiquitination of NS2A in an in vitro ubiquitination assay (Fig. 3g). Silencing AMFR using short hairpin RNA (shRNA) reduced NS2A ubiquitination, and AMFRWT, but not AMFRC356G/H361A, rescued the downregulated effect (Fig. 3h). Furthermore, viral titers in the supernatants of ZIKV-infected Huh7.0 AMFR KO cells were lower than titers from wild-type Huh7.0 cells (Supplementary Fig. 6c, d). This decreasing effect was rescued by exogenous AMFRWT, but not AMFRC356G/H361A (Supplementary Fig. 6d) which suggested AMFR ligase activity played a great role in NS2A ubiquitination modification and ZIKV infection.
Next, the critical interaction between AMFR and NS2A was confirmed through coIP assays, demonstrating that ectopic AMFR could interact with ectopic NS2A (Supplementary Fig. 6e, f). This interaction was further supported by the confirmation of endogenous AMFR interactions with HA-tagged NS2A during ZIKV infection (Fig. 3i). Previous studies showed that FAM134B can interact with AMFR for ubiquitination to promote ER-phagy47. Thus, we guessed NS2A-AMFR interaction may interfere FAM134B-AMFR axis. Firstly, we used purified FAM134B, AMFR, and NS2A and detected the FAM134B-AMFR interaction by adding NS2A in vitro. We found FAM134B-AMFR interaction was weakened with the occurrence of NS2A (Supplementary Fig. 6g). Moreover, our study revealed that the deletion of AMFR 1-300aa led to the abolishment of NS2A-AMFR△1-300aa interaction, while FAM134B-AMFR△1-300aa interaction remained unaffected (Supplementary Fig. 6h, i). This prompted us to investigate whether NS2A could still inhibit the FAM134B-AMFR axis in the absence of NS2A-AMFR interaction. To address this question, we purified His-AMFR△1-300aa and GST-FAM134B from E. coli and examined their interaction by adding purified Flag-NS2A in vitro. Strikingly, we observed that the NS2A did not suppress the AMFR△1-300aa-FAM134B interaction when NS2A-AMFR interaction was abolished (Supplementary Fig. 6j). Furthermore, we performed immunofluorescence experiment to see the colocalization between these three proteins. Interestingly, we found that in the absence of NS2A, FAM134B and AMFR exhibited strong colocalization. However, upon the addition of NS2A, partial colocalization between AMFR and NS2A was observed, while the colocalization between AMFR and FAM134B was not as prominent (Supplementary Fig. 6k, l). Collectively, these findings suggest that ZIKV-NS2A usurps AMFR for ubiquitination, leading to the disturbance of the FAM134B-AMFR axis.
Furthermore, we aimed to identify the K48-linked ubiquitination site of NS2A mediated by AMFR. The ZIKV-NS2A protein comprises 226 amino acids, including ten lysine (K) residues (Supplementary Fig. 7a). Three ubiquitination sites of NS2A were identified through mass spectrometry analysis, comprising K56, K186, and K193 (Supplementary Fig. 7b–d). In order to confirm whether these three sites were all NS2A K48-linked ubiquitination sites, by individually mutating each of these ten K residues to R and examining the expression levels of these mutants in HEK293T cells, we identified K56 as a strong potential K48-linked ubiquitination site for NS2A, as the NS2AK56R mutant exhibited the highest expression level compared to the other two sites NS2AK186R and NS2AK193R (Supplementary Fig. 7e). Notably, subsequent ubiquitination analysis further confirmed that NS2AK56R had weakened K48-linked ubiquitination (Supplementary Fig. 7f), and AMFR failed to increase the ubiquitination level of NS2AK56R (Fig. 3j).
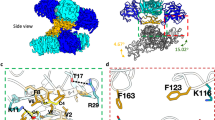
In order to clarify NS2AWT and NS2AK56R ubiquitination in the live virus infection, we generated a full-length infectious ZIKV clone encoding HA-NS2AK56R. We confirmed the full length of the genome sequence of the mutant virus (Supplementary Fig. 7g). Growth curves of ZIKV-HA-NS2AK56R were then assessed. ZIKV-HA-NS2AK56R displayed attenuation in Vero cells, with viral titer being ~10-fold lower than that of ZIKV-HA-NS2AWT at 5 dpi (Supplementary Fig. 7h). As expected, when the two viruses replicated at a comparable level in both A549 and Vero cells, NS2A ubiquitination was dramatically reduced in ZIKV-HA-NS2AK56R infection compared to ZIKV-HA-NS2AWT virus, which further convinced K56 was required for NS2A ubiquitination in live virus infections (Fig. 3k). These data support a model in which the E3 ligase AMFR interacts with ZIKV-NS2A protein and modifies the NS2A K56 site through K48-linked ubiquitination, leading to the degradation of NS2A via the ubiquitin-proteasome pathway. Remarkably, upon comparing the amino acid sequences of NS2A from other flaviviruses with that of ZIKV, a distinctive feature was observed: ZIKV-NS2A residue K56 is not conserved across other flaviviruses, where others are represented by the amino acid R (Supplementary Fig. 7i). We speculate that this unique site in ZIKV may be linked to ZIKV-induced congenital neurological disorders and microcephaly, distinguishing it from other flaviviruses. Based on the findings that flavivirus-NS2A interacted with FAM134B (Fig. 2i) and that AMFR mediated the ubiquitination of flavivirus-NS2A (Fig. 3f), we still posited that AMFR-mediated K48-linked ubiquitination of flavivirus-NS2A was responsible for FAM134B degradation, with the possibility that other lysine residues may be modified. Hypothesizing that the lysine residue nearest to AMFR in each respective NS2A may represent the ubiquitination site, we simulated the interactions between AMFR and DENV or JEV-NS2A utilizing AlphaFold3. Our analysis revealed that the K99 residue of DENV-NS2A is proximal to AMFR, with its side chain oriented towards AMFR (Supplementary Fig. 7j), whereas for JEV-NS2A, the K26 residue was similarly positioned (Supplementary Fig. 7k). These observations suggested that these lysine residues may be AMFR-modified ubiquitination sites for degrading FAM134B. Further investigations are warranted to validate these potential ubiquitination sites.
We then examined the impact of ZIKV-NS2A ubiquitination elements on FAM134B degradation. Firstly, we judged the role of AMFR in NS2A-regulated FAM134B degradation. A recent study reported that AMFR catalyzes the ubiquitination of 17 K residues in the RHD domain of FAM134B, thereby regulating the ER-phagy process47. To rule out the catalytic effect of AMFR on FAM134B, we constructed a mutant FAM134B with all 17 K residues mutated to R in the RHD domain, namely FAM134B17KR. AMFR fails to ubiquitinate FAM134B17KR and promote FAM134B-mediated ER-phagy47. However, FAM134B17KR still interacted with NS2A and was degraded by NS2A (Fig. 3l, m, compared lanes 1 and 2, and Supplementary Fig. 8a). When AMFR was downregulated, the expression of the NS2A-FAM134B17KR complex was reversed (Fig. 3l-m, compared lanes 2 and 3), and AMFRWT, but not AMFRC356G/H361A, could further degrade NS2A-FAM134B17KR (Fig. 3l, m, compared lanes 4 and 5). Furthermore, AMFR could also enhance other flavivirus-NS2A-mediated FAM134B degradation (Supplementary Fig. 8b). Additionally, exogenously expressed ubiquitination-compromised mutant ZIKV-NS2AK56R resulted in the failure of dose-dependent degradation of both endogenous and exogenous FAM134B (Supplementary Fig. 8c, d), although NS2AK56R still interacted with FAM134B (Supplementary Fig. 8e). To evaluate the impact of NS2A ubiquitination on FAM134B degradation during live virus infection, we generated a full-length infectious ZIKV clone encoding NS2AK56R without the HA tag (Supplementary Fig. 8f). Growth curves of ZIKV-NS2AK56R were similar with ZIKV-HA-NS2AK56R that ZIKV-NS2AK56R also displayed attenuation in Vero cells compared with ZIKV-NS2AWT (Supplementary Fig. 8g). Similarly, attenuated replication of ZIKV-NS2AK56R was observed in human neural progenitor cells (hNPCs) at 48 and 72 hpi (Supplementary Fig. 8h). To determine whether the decreased replication of ZIKV-NS2AK56R was attributable to a defect in virus replication or assembly rather than FAM134B-mediated ER-phagy, we infected A549 FAM134B KO cells with ZIKV-NS2AWT or ZIKV-NS2AK56R at a MOI of 0.1 and collected the supernatants at 24, 48, 72, and 96 hpi. We then assessed the growth curves of the two viruses and observed that ZIKV-NS2AK56R exhibited a similar growth trend to ZIKV-NS2AWT in Vero cells. Notably, there was no discernible difference between the two viruses at 48 hpi (Supplementary Fig. 8i), despite the wild-type virus displaying nearly a 10-fold increase over the mutant virus in A549 FAM134B wild-type cells at the same time point. These findings provided strong evidence that FAM134B-mediated ER-phagy played a significant role in the cytopathogenesis of ZIKV.
Then we sought to verify whether ZIKV-NS2AK56R reduced the ability to trigger FAM134B degradation. As expected, when the two viruses replicated at a comparable level, ZIKV-NS2AK56R greatly lost the ability to degrade FAM134B compared to ZIKV-NS2AWT virus by flow cytometry (Fig. 3n) and western blotting assays (Supplementary Fig. 8j).
Taken together, the above results revealed that ER-localized NS2A undergoes polyubiquitination at K56 catalyzed by the E3 ubiquitin ligase AMFR through K48-linked polyubiquitination. Ubiquitinated NS2A bound to FAM134B through interactions with the FAM134B N-terminal domain, and AMFR orchestrated the degradation of NS2A-FAM134B complexes. The mutant virus ZIKV-NS2AK56R failed to trigger FAM134B degradation, which emphasized the importance of ubiquitination in NS2A-drived regulation.
ZIKV subverts FAM134B to restrict ER-phagy process
Since ER-phagy is strictly reliant on receptor-mediated process, thus the content of receptor protein directly influences the efficiency of ER-phagy24. We reasoned NS2A-mediated FAM134B degradation may affect the flux of ER-phagy. To check our hypothesis, we used two validated reporter assays: mCherry-eGFP-RAMP4 tandem tagging assays and GFP-cleavage assays25,48. When ER-phagy is happened that ER fragments are degraded by autolysosomes, they will either appear as mCherry+eGFP--RAMP4 foci by confocal microscope because mCherry is more stable in an acidic environment (Fig. 4a) or they will produce free GFP visualized by western blotting analysis because of the partial digestion of GFP-SEC61B, an ER sheet resident protein (Fig. 4b). We chose U2OS cell which is a classic cell line in ER-phagy research to conduct the experiments. In mCherry-eGFP tandem tagging assays, U2OS cells expressing mCherry-eGFP-RAMP4 did not exhibit any ER-phagy activity and appeared as mCherry + eGFP + foci under confocal microscopy. However, upon activation of ER-phagy by FAM134B, the mCherry-eGFP-RAMP4 foci shifted to mCherry+eGFP- due to the increased stability of mCherry in an acidic environment. Subsequently, following infection of ER-phagy-activated cells with ZIKV-HA-NS2AWT or ZIKV-HA-NS2AK56R at similar levels of replication, we observed a significant reduction in mCherry + eGFP- foci in cells infected with the wild-type ZIKV compared to the ER-phagy inhibition-deficient virus (Fig. 4c, d). In GFP-cleavage assays, as expected, only ZIKV-NS2AWT infection or exogenously expressed NS2AWT decreased FAM134B-induced GFP fragment production (Fig. 4e, f).

a Diagram of mCherry-EGFP tandem tagging assays. b Diagram of GFP-cleavage assays. c Immunofluorescence analysis of U2OS cells expressing mCherry-eGFP- RAMP4 infected with ZIKV-HA-NS2AWT (MOI = 1), ZIKV-HA-NS2AK56R (MOI = 10), or mock-infected for 48 h. Scale bars, 10 μm. d ER-phagy flux was quantified by the number of mCherry+ eGFP– dots. Error bars present as the mean ± SEM (n = 21); Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test; Source data are provided as a Source Data file. e, g Western blotting analysis of U2OS cells transfected for 12 h with GFP-SEC61B and Myc-FAM134B (e) or Myc-FAM134B (g) and then infected with ZIKV-NS2AWT (MOI = 1), ZIKV-NS2AK56R (MOI = 10), or mock-infected for 48 h. f, i Western blotting analysis of U2OS cells transfected for 48 h with GFP-SEC61B and Myc-FAM134B and Flag-NS2AWT or Flag-NS2AK56R or empty vector (f) or Myc-FAM134B and Flag-NS2AWT or Flag-NS2AK56R or empty vector (h). h, j Quantification of relative CLIMP63 and REEP5 protein level, h corresponds to (g, j) corresponds to (i); Error bars of (h, j) present as the mean ± SEM (n = 3 independent experiments); Statistical analysis of (h, j) was performed using two-way ANOVA with Sidak’s multiple comparisons test; Source data are provided as a Source Data file.
FAM134B is responsible for the turnover of ER sheets degradation and detection of the content of ER sheet marker CLIMP63 can directly visualize the ER-phagy level28. Similarly, U2OS cells expressing FAM134B were infected by the two viruses, and we observed that only ZIKV-NS2AWT infection hindered the degradation of CLIMP63, as opposed to REEP5, which resides in ER tubules (Fig. 4g, h). The blockade of CLIMP63 degradation was also evident in cells expressing exogenous NS2AWT (Fig. 4i, j). The above results showed ZIKV-NS2A-induced FAM134B degradation results in the flux of ER-phagy inhibition.
ZIKV mutant (ZIKV-NS2AK56R) with ER-phagy inhibition-deficiency exhibits impaired microcephaly in human brain organoids
ZIKV prefers to infect and target hNPCs and causes fetal microcephaly upon maternal infection5,7. Given that FAM134B degradation-deficient ZIKV mutant exhibited significantly attenuated replication in hNPCs (Supplementary Fig. 8h), we speculate ER-phagy inhibition caused by ZIKV plays a great role in virus-induced fetal microcephaly. Human cerebral organoids have been extensively utilized as a model for studying ZIKV-associated microcephaly49. To assess the impact of wild-type ZIKV and the NS2AK56R mutant on early human brain development, we focused on day 10 brain organoids, exposing them to viral incubation for 24 h (Fig. 5a). After removing the inoculum, we examined organoids at 4 and 8 dpi. Both viruses demonstrated increasing expression of ZIKV-E RNA and the release of infectious particles at 4 and 8 dpi (Fig. 5b, c), indicating valid and productive infection. In comparison to mock-treated organoids, those infected with ZIKV-NS2AWT exhibited time-dependent growth attenuation (Fig. 5d, e). Conversely, ZIKV-NS2AK56R-infected organoids were significantly larger than those infected with ZIKV-NS2AWT at 8 dpi (Fig. 5d, e). Since hNPCs are particularly susceptible to ZIKV infection, and cerebral organoids contain ventricular zone (VZ)-like structures where hNPCs predominantly reside49, we firstly assessed the RNA levels of hNPC markers SOX1 and SOX2 at 4 and 8 dpi. SOX1 and SOX2 RNA levels in organoids infected by both viruses were significantly lower than those in mock-treated organoids (Fig. 5f, g), but the content of ZIKV-NS2AK56R-infected organoids was higher than that of ZIKV-NS2AWT-infected ones (Fig. 5f, g). Subsequently, we stained for SOX2+ hNPCs in the VZ-like region to directly observe virus-induced hNPCs depletion. We found that ZIKV-NS2AWT-infected organoids exhibited fewer and smaller VZ-like structures compared to mock-treated organoids (Fig. 5h, i). Although ZIKV-NS2AK56R-infected organoids were still smaller than mock-treated ones, they displayed larger and more complete morphological characteristics of VZ-like structures compared to ZIKV-NS2AWT-infected organoids (Fig. 5h, i). Furthermore, we observed FAM134B degradation and CLIMP63-degradation blockade in ZIKV-NS2AWT-infected organoids compared to ZIKV-NS2AK56R-infected organoids by western blotting analysis (Fig. 5j), which may suggest ZIKV-NS2AK56R failed to degrade FAM134B and inhibit ER-phagy in brain organoids.

a Diagram of viral exposure of brain organoids. b qPCR analysis of ZIKV-E RNA in organoids exposed to ZIKV-NS2AWT or ZIKV-NS2AK56R at 4 and 8 dpi. c Immunofluorescence analysis of Vero cells incubated with supernatants of ZIKV-infected organoids at 4 and 8 dpi. Scale bars, 75 μm. ZIKV-E (green), DAPI (blue). d, e Images and area measurements of organoids exposed to ZIKV-NS2AWT, ZIKV-NS2AK56R, or mock-treated at 4 and 8 dpi. Scale bars, 500 μm. f, g qRT-PCR analysis of SOX1 (f) and SOX2 (g) RNA in organoids exposed to ZIKV-NS2AWT, ZIKV-NS2AK56R or mock-treated at 4 and 8 dpi. h Immunofluorescence analysis of organoids exposed to ZIKV-NS2AWT, ZIKV-NS2AK56R or mock-treated. Scale bars, 75 μm. The area of two dashed lines represents ventricular zone (VZ)-like structures. ZIKV-E (green), SOX2 (red). i The measurement of VZ-like structures of (h). j Western blotting analysis of organoids exposed to ZIKV-NS2AWT (7 × 104 PFU/organoid), ZIKV-NS2AK56R (7 × 105 PFU/organoid) or mock-treated at 8 dpi. Error bars of (b, e, f, g, i) present as the mean ± SEM (b, f, g: n = 3; e: n = 13; i: n = 6); Statistical analysis of (b, e, f, g) was performed using two-way ANOVA with Sidak’s multiple comparisons test and (i) was performed using one-way ANOVA with Tukey’s multiple comparisons test; Source data are provided as a Source Data file.
In short, these results demonstrated the mutant virus ZIKV-NS2AK56R with ER-phagy inhibition-deficiency compromised to attenuate organoid growth, resulted in hNPCs depletion and caused a microcephaly-like phenotype in the early stages of human brain organoids compared with ZIKV-NS2AWT infection. Especially, ZIKV-NS2AK56R failed to restrain the flux of ER-phagy in brain organoids.
ZIKV-NS2AK56R infection causes weakened pathogenicity and lethality in mice
To assess the pathogenicity and lethality of the recombinant ZIKV carrying the NS2AK56R mutation in vivo, we established infection models in A129 (C57BL/6 Ifnar1−/−) mice with either ZIKV-NS2AWT or ZIKV-NS2AK56R. Monitoring the mice for body weight loss revealed that ZIKV-NS2AK56R-infected mice lost ~10% of their initial body weight at 8 dpi, while ZIKV-NS2AWT-infected mice exhibited more severe weight loss (Fig. 6a). Survival rates after intraperitoneal injection of the two viruses were also estimated. A129 mice infected with ZIKV-NS2AWT showed increased vulnerability to ZIKV-induced mortality, whereas those infected with ZIKV-NS2AK56R did not experience any deaths (Fig. 6b), indicating that ZIKV-NS2AK56R infection resulted in attenuated lethality. Correspondingly, A129 mice infected with ZIKV-NS2AK56R displayed a lower viral burden, as evidenced by reduced ZIKV-E RNA levels in various organs (lung, kidney, eye, brain, and liver) measured by qRT-PCR (Fig. 6c–g) and lower ZIKV-E protein levels in the brain assessed by immunofluorescence (Fig. 6h). Additionally, attenuated brain tissue damage was observed in A129 mice infected with ZIKV-NS2AK56R (Fig. 6i). We sought to determine whether the diminished replication of ZIKV-NS2AK56R in vivo resulted in deficiencies in FAM134B degradation and ER-phagy inhibition. Evaluation of mouse FAM134B (mFAM134B) protein levels in various organs upon ZIKV infections through western blotting revealed that mFAM134B was highly expressed in mice infected with ZIKV-NS2AK56R, while lower levels were observed in ZIKV-NS2AWT-infected mice, especially in the brain, liver, and lung organs (Fig. 6j). We also conducted immunohistochemistry analysis and found reduced FAM134B staining in the cerebral cortex of mice infected with ZIKV-NS2AWT, as compared to mice treated with PBS or those infected with ZIKV-NS2AK56R (Supplementary Fig. 9). In short, these findings suggested that the weakened replication of ZIKV-NS2AK56R in vivo led to deficiencies in mFAM134B degradation and compromise pathogenicity in mice.

a Daily weights of mice treated with PBS (n = 6), ZIKV-NS2AWT (n = 7), or ZIKV-NS2AK56R (n = 7) (i.p., 104 PFU/ mouse); Error bars present as the mean ± SEM; Source data are provided as a Source Data file. b Survival of mice from (a); Log-rank (Mantel-Cox) test. c–g qRT-PCR analysis of ZIKV-E RNA in different tissues of mice infected with ZIKV-NS2AWT or ZIKV-NS2AK56R (n = 6); Error bars of (c–g) present as the mean ± SEM; Statistical analysis of (c–g) was performed using two-tailed unpaired t-test analysis; Source data are provided as a Source Data file. h Immunofluorescence analysis of brains of mice infected with ZIKV-NS2AWT or ZIKV-NS2AK56R. Scale bars, 1000 μm. ZIKV-E (green), DAPI (blue). i Hematoxylin and eosin staining analysis of brains of mice infected with PBS, ZIKV-NS2AWT, or ZIKV-NS2AK56R. The black arrows represent the inflammation damage in the brain tissues. Scale bars, 100 μm. j Western blotting analysis of mFAM134B expression in mouse tissues infected with ZIKV-NS2AWT or ZIKV-NS2AK56R.
Discussion
Flaviviruses have utilized the host ubiquitin system to replicate efficiently in both a proteasome-dependent and -independent manner50,51, and ubiquitination of viral proteins has diverse roles in viral life cycle and pathogenesis52. For example, envelope protein ubiquitination of ZIKV provides the virus with an enhanced ability to enter new cells and is an important determinant of virus tropism and pathogenesis in the mammalian host53. DENV-NS3 ubiquitination facilitates the cleavage of STING to subvert the innate immune signaling for promoting viral replication54. IFN-induced ubiquitination of YFV-NS5 is required for binding human STAT2 to antagonize the interferon signaling55. Here, we unveil a novel aspect of ZIKV-NS2A protein dynamics, revealing its AMFR-mediated K48-linked polyubiquitination, leading to degradation via the ubiquitin-proteasome pathway and contributing to the observed short half-life of NS2A in ZIKV-infected cells (Fig. 3 and Supplementary Fig. 5a, b). Furthermore, we observed a dramatic increase in the abundance of nine ZIKV viral proteins except for NS2A over the infection period in quantitative proteomics experiments, and NS2A protein could not even be quantified in 72 hpi (Fig. 1b). This unexpected finding underscores the cunning adaptability of the virus, as ZIKV-NS2A, crucial to the viral life cycle (see Introduction), exhibits a specific instability serving distinct functions for viral infection. We speculate that ZIKV-NS2A may preferentially target the key host proteins participating in cellular pathways for degradation to promote virus infection. As expected, it has been reported that ZIKV-NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins18, inhibits interferon signaling by degradation of STAT1 and STAT256, and downregulates KPNA2, essential for embryonic stem cell maintenance and differentiation57. Among our investigation, ubiquitinated NS2A mediated by AMFR bound to FAM134B through interactions with its N-terminal domain and AMFR orchestrated the degradation of NS2A-FAM134B complexes, resulting in the flux of ER-phagy inhibition for promoting virus infection (Figs. 2, 3). This discovery enhances our comprehension of viral protein degradation, shedding light on promoting viral replications. Besides, polyubiquitination-mediated NS2A degradation and FAM134B-NS2A interactions and subsequent FAM134B degradation were conserved in ZIKV, DENV, WNV, and JEV (Figs. 2i, 3f and Supplementary Fig. 8b), which proposes flavivirus-NS2A ubiquitination emerges as a potential targeting agent as broad-spectrum antivirals. The viral protein NS2A specifically targeted FAM134B for degradation, leaving other ER-phagy receptors unaffected (Fig. 1d, e and Supplementary Fig. 1d). This observation suggested that the ER-phagy process was not entirely inhibited by ZIKV. Therefore, if flaviviruses aim to completely suppress the ER-phagy pathway, it is likely other alternative mechanisms may be employed to manipulate this process.
A previous study demonstrated that FAM134B engaged with AMFR to facilitate its ubiquitination, promoting ER-phagy47. In our investigation, the ubiquitination-deficient mutant FAM134B17KR was still subjected to degradation by NS2A (Fig. 3l, m). This finding indicated that the ubiquitination of FAM134B was not a prerequisite for its NS2A-mediated proteasomal degradation, and NS2A may function as a chaperone to facilitate the degradation of FAM134B. Furthermore, we observed a weakening of the FAM134B-AMFR interaction in the presence of NS2A (Supplementary Fig. 6g) through competitive coIP assays in vitro. This suggests that ZIKV not only utilizes AMFR to ubiquitinate NS2A for FAM134B degradation but also hinders the regulatory axis of FAM134B-AMFR-mediated ER-phagy. Of course, further experimental results, especially those related to virus infection and structural aspects, are necessary to fully elucidate the competitive relationships within the triple complexes.
Viruses employ various strategies to inhibit, evade, or manipulate the autophagy machinery, promoting their replication for survival and propagation58. In recent years, receptor-mediated organelle-specific selective autophagy has been one of the hot research areas in virus–host interactions. However, the role of ER-phagy in viral infection and pathogenicity remains incompletely explored, with limited studies expanding its scope. Ebola virus infection is limited by ER-phagy receptor FAM134B, but the restricted mechanism remains unknown59. SARS-CoV-2 ORF8 hijacks ATL3 and FAM134B into P62 condensates, resulting in ER-phagy inhibition and increased viral replication60. Additionally, depletion of BPIFB3 improves ER-phagy process and inhibits the replication of flavivirus61. Existing research on ER-phagy in viral infection is predominantly at the cellular level and lacks an understanding of its impact on the pathogenicity of viral infections under physiological conditions. In this study, we bridge this gap by combining in vivo analysis using mouse models and in vitro analysis utilizing human brain organoids to physiologically elucidate the basic mechanisms and viral pathogenicity by regulating ER-phagy under conditions of ZIKV-NS2AWT virus and the ER-phagy inhibition-deficient ZIKV-NS2AK56R mutant virus infections (Figs. 5, 6). ZIKV-NS2AK56R virus infection compromised to attenuate organoid growth (Fig. 5d, e) and cause hNPCs depletion (Fig. 5f–i) and failed to exhibit a microcephaly-like phenotype in human brain organoids. This highlights the potential involvement of the ER-phagy pathway in ZIKV-induced microcephaly. In the mouse model context, the reduced replicative potential of ZIKV-NS2AK56R was associated with decreased pathogenicity, evident through minimal weight loss and zero mortality rate (Fig. 6a, b). Notably, ZIKV-NS2AK56R-infected mice exhibited grossly attenuated brain tissue damage (Fig. 6i), correlating with lower ZIKV-E RNA (Fig. 6f) and ZIKV-E protein levels (Fig. 6h) in the brain. Importantly, the diminished replication of ZIKV-NS2AK56R in vivo led to deficiencies in FAM134B degradation in certain mouse tissues (Fig. 6j). Exploring these events sheds light on the role of the ER-phagy pathway in viral infections and contributes to a deeper understanding of the pathogenesis of viral diseases.
Several flavivirus NS3 proteases, including DENV and ZIKV, can cleave R at position 142 within the reticulon homology domain of exogenously expressed FAM134B in HEK293T cells which blocks the formation of viral NS3 and ER protein-enriched autophagosomes42. This intriguing observation was validated at the protein level, lacking confirmations in live virus infection-induced FAM134B cleavage and the flux of ER-phagy inhibition. In our system, when we generated FAM134B cleavage mutant FAM134BR142A, we found several flavivirus infections, including ZIKV, DENV, and JEV, still could degrade FAM134BR142A (Fig. 1k, l, n and Supplementary Fig. 2f–h), indicating FAM134B degradation-induced by flavivirus NS2A is independent of viral NS3-mediated cleavage. Importantly, expression of FAM134BWT and FAM134BR142A at comparable levels exhibited no significant differences in inhibiting ZIKV-E protein levels (Supplementary Fig. 3d) and viral production (Fig. 1m). This suggests that the cleavage of FAM134B, as elucidated solely at the protein level, may not exert a substantial influence on live ZIKV infection. Moreover, a critical amino acid, K56, in NS2A played a significant role in mediating FAM134B degradation. Infections with the recombinant ZIKV-NS2AK56R virus demonstrated attenuated replicative abilities in both Vero cells and hNPCs (Supplementary Fig. 8g, h), failing to induce FAM134B degradation (Fig. 3n and Supplementary Fig. 8j) and inhibit the flux of ER-phagy process (Fig. 4). Therefore, we made a profound study into the degradation of ER-phagy receptor by ZIKV infection, thereby inhibiting the occurrence of ER-phagy at the level of live virus infection, which provides substantive evidence for the authenticity of the viral manipulation of ER-phagy.
In summary, our study advances a model wherein viral-protein ubiquitination orchestrates the degradation of the crucial ER-phagy receptor protein through the ubiquitin-proteasome pathway, ultimately impeding the flux of ER-phagy. The mechanism we unveil is unique and novel, contributing to a deeper understanding of how viruses subvert ER-phagy to promote viral infection and the pathogenicity of viral diseases (Fig. 7).

Left: The ER-resident E3 ligase AMFR-mediated ubiquitination of ER-phagy receptor FAM134B promotes the dynamic flux of ER-phagy process to degrade ER sheets; Middle: During flavivirus infection, ZIKV hijacks AMFR to catalyze the polyubiquitination of ER-localized viral NS2A protein at K56 through K48-linked manner. Ubiquitinated NS2A binds to FAM134B, and AMFR orchestrates the degradation of NS2A-FAM134B complexes, ultimately impeding the flux of ER-phagy for ER accumulation and hindering the FAM134B-AMFR axis. Right: The ubiquitination and ER-phagy inhibition-deficient ZIKV-NS2AK56R infection exhibits weakened microcephaly and reduced replicative potential and decreased pathgenicity in mice.
Methods
Ethic statements
All the animal experiments were performed according to the Regulations of Hubei Province Laboratory Animal Management and approved by the Wuhan University Animal Experiment Ethics Committee under project licence SKLV-AE2022 007.
Cells
HEK293T (CRL-1573, ATCC), A549 (CCL-185, ATCC), Huh7.0 (obtained from Dr. Xianfeng Hui, Huazhong Agricultural University), Vero (obtained from Dr. Chao Shan, Wuhan Institute of Virology, Chinese Academy of Sciences), U2OS (HTB-96, ATCC), A549-GFP-FAM134B, A549-GFP-FAM134BR142A, HeLa-GFP-FAM134B, Huh7.0-GFP-FAM134B, Huh7.0-GFP-FAM134BR142A, Huh7.0-GFP-FAM134B△NTD, and Huh7.0 FAM134B KO cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, 11995065, Gibco) with 10% fetal bovine serum (FBS, 086150, WISENT). ReNcell CX cells, a cell line of hNPC (obtained from Dr. Shi Chen, Wuhan University), were cultured in DMEM/F12 (11330032, Gibco) containing N2 Supplement, B27 Plus Supplement, 20 ng/mL FGF-2 and 20 ng/mL EGF in 15 µg/mL laminin (354232, Corning)–coated dishes. All cells were cultured at 37 °C in a humidified incubator with 5% CO2.
Viruses
The ZIKV Cambodian strain FSS13025 and the mutant virus were produced from an infectious cDNA clone. The ZIKV Cambodian strain FSS13025 HA-tagged NS2A virus was produced from an infectious cDNA plasmid kindly provided by Dr. Xianwen Zhang from Shenzhen Bay Laboratory. ZIKV Puerto Rico strain PRVABC59 was generated from an infectious cDNA plasmid kindly provided by Dr. Zhijian Cao from Hubei University of Technology. ZIKV Asian strain SZ01 was kindly provided by Dr. Hanzhong Wang from the Wuhan Institute of Virology, Chinese Academy of Sciences. DENV strain TSV-01 and JEV strain SA-14-14-2 were kindly provided by Dr. Bo Zhang from the Wuhan Institute of Virology, Chinese Academy of Sciences.
Antibodies and reagents
Rabbit polyclonal anti-FAM134B (cat. no.21537-1-AP), rabbit polyclonal anti-ATL3 (cat. no.16921-1-AP), rabbit polyclonal anti-RTN3 (cat. no.12055-2-AP), rabbit polyclonal anti-TEX264 (cat. no.25858-1-AP), rabbit polyclonal anti-CCPG1 (cat. no. 13861-1-AP), rabbit polyclonal anti-AMFR (cat. no.16675-1-AP), rabbit polyclonal anti-CLIMP63 (cat. no.16686-1-AP), rabbit polyclonal anti-REEP5 (cat. no. 14643-1-AP), and mouse monoclonal anti-GAPDH (cat. no.60004-1-Ig) were purchased from Proteintech. Rabbit monoclonal anti-SOX2 (cat. no.23064), rabbit monoclonal anti-GFP (cat. no.2956), rabbit monoclonal anti-Ubiquitin (cat. no.43124), rabbit monoclonal anti-K48-linkage Specific Polyubiquitin (cat. no.8081), rabbit monoclonal anti-K63-linkage Specific Polyubiquitin (cat. no.5621), rabbit monoclonal anti-LC3 (cat. no.3868), rabbit monoclonal anti-His (cat. no.12698), rabbit monoclonal anti-GST (cat. no.2625) were purchased from Cell Signalling Technology. Rabbit polyclonal anti-ZIKV-E (cat.no.GTX133314), rabbit polyclonal anti-ZIKV-NS3 (cat.no. GTX133309) were purchased from GeneTex. Mouse monoclonal anti-Myc (cat.no.M192-3), mouse monoclonal anti-HA (cat.no.M180-3) were purchased from MBL. Mouse monoclonal anti-Flag (cat.no. A9469) was purchased from Sigma. Rabbit monoclonal anti-SEC62 (cat.no. A24001) was purchased from ABclonal. Goat anti-rabbit IgG HRP conjugated (cat. no.31460), goat anti-mouse IgG HRP conjugated (cat. no.31430), goat anti-rabbit IgG Rhodamine (cat. no.31670), and goat anti-mouse IgG Fluorescein (cat. no.31569) were purchased from Invitrogen. The inhibitors MG132 (HY-13259), CQ (HY-17589A), and CHX (HY-12320) were purchased from MedChemExpress (MCE). Earle’s balanced salt solution was purchased from GIBCO (24010043).
Plasmids
Myc-FAM134B was generated by inserting the corresponding cDNA into the pcDNA3.1-Myc vector. Myc-FAM134B△1-83aa, Myc-FAM134B△453-497aa, Myc-FAM134B△RHD, Myc-FAM134B△CTD, Myc-FAM134B△NTD, and Myc-FAM134B17KR were generated by performing overlap extension of PCR-mediated deletions or substitution on pcDNA3.1-Myc-FAM134B. GST-FAM134B was generated by subcloning into the vector pGEX-6P1. Flag-ZIKV-NS2A, DENV-NS2A, JEV-NS2A, WNV-NS2A, and other ZIKV viral proteins-encoded genes were generated by inserting the corresponding cDNA into the mammalian expression vector pCDH-CMV-IRES-MCS-SF-BLAST. Nine Flag-ZIKV-NS2A K to R mutants were generated by performing overlap extension of PCR-mediated substitution on pCDH-CMV-Flag-NS2A. HA-AMFR, HA-CCPG1, HA-FAM134B, HA-RTN3, HA-TEX264, HA-ATL3, HA-SEC62, and other encoding E3 ligase genes were cloned into the mammalian expression vector pCAGGS-MCS. HA-AMFRC356G/H361A and AMFR△1-300aa were generated by performing overlap extension of PCR-mediated deletions or substitution on pCAGGS-HA-AMFR. His–AMFR△1-300aa was generated by subcloning into the vector pET28a. GFP-SEC61B and pCDNA4-mCherry-eGFP-RAMP4 were kindly provided by Dr. Binbin Ding from Guangzhou Medical University. All constructs were confirmed by DNA sequencing.
Mice experiment
A129 mice (C57BL/6 Ifnar1−/−) were obtained from Dr. Yu Chen, Wuhan University62, and maintained under specific pathogen-free conditions. All animal experiments were conducted in an animal biosafety level 2 (ABSL-2) facility in accordance with the Regulations of Hubei Province Laboratory Animal Management and approved by the College of Life Sciences of Wuhan University. All mice were housed in a specifc-pathogen-free (SPF) at Wuhan University with a 12-h dark/12-h light cycle and fed with standard food and water.
About 4-week-old A129 mice (males and females) were infected with 104 PFU of ZIKV-NS2AWT or ZIKV-NS2AK56R (FSS13025 strain backbone) through the intraperitoneal route. PBS was given to the mock-infected mice through the same route. The body weight and survival data represent six mock-infected mice and eight mice infected with ZIKV-NS2AWT or ZIKV-NS2AK56R. The viremia data represents six mice infected with ZIKV-NS2AWT or ZIKV-NS2AK56R. Mouse tissues were collected at 8 days post-infection. The experiment was performed twice, and the results were combined.
H&E and Immunohistochemistry (IHC)
Brain samples were fixed in 4% (w/v) polyformaldehyde embedded in paraffin, and cut into 5-μm sections. For H&E staining, sections were deparaffinized, rehydrated, and stained with haematoxylin and eosin. For IHC staining, sections were deparaffinized, rehydrated, and then antigens were recovered in 10 mM citrate buffer (pH 6.0) for 10 min in a microwave oven. H2O2 (3%) was used to block endogenous peroxidase, followed by PBS wash. After sections were blocked in 3% BSA at room temperature for 30 min, they were incubated with primary antibodies for FAM134B overnight at 4 °C. The sections were then washed with PBS, incubated with HRP-labeled secondary antibodies at room temperature for 1 h, and washed with PBS. Sections were stained with 3,3’-diaminobenzidine and then counter-stained with hematoxylin. The quantification was performed using Image J software (1.53t/Java 1.8.0 345).
Immunoblotting and immunoprecipitation
Cell lysates were collected in lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 1% Triton X-100, 1 mM EDTA pH 8.0, 0.1% SDS) supplemented with protease inhibitor cocktail. Proteins in cell lysates were separated by SDS–PAGE and analyzed by immunoblotting.
For immunoprecipitation, cells were lysed by lysis buffer containing protease inhibitor cocktail for 30 min on ice and then centrifuged at 13,000×g for 30 min at 4 °C. The supernatants were immunoprecipitated by the indicated antibody for 12 h at 4 °C. The immunoprecipitants were washed by lysis buffer four times and then boiled in 1x SDS-loading buffer for immunoblotting.
RNA extraction, reverse transcription PCR, and real-time PCR
Total RNA was isolated by Trizol regent (15596026CN, Invitrogen) according to the manufacturer’s protocol. Reverse transcription PCR was carried out by using the ABScript II cDNA First-Strand Synthesis Kit (RK20400, ABclonal). The resulting cDNA was amplified by 2X Universal SYBR Green Fast qPCR Mix (RK21203, ABclonal). mRNA expression levels of the indicated genes were normalized to the expression levels of GAPDH as an endogenous reference control.
The following primers were used:
Human FAM134B forward: 5′-AGACTTTTCAGCTCTTTGTC-3′
Human FAM134B reverse: 5′-GATCATCAGAAGTGTCTGTC-3′
ZIKV-E forward: 5′-AGATGACTGCGTTGTGAAGC-3′
ZIKV-E reverse: 5′-GAGCAGAACGGGACTTCTTC-3′
Human SOX1 forward: 5′-AATAGCACAAACACGCGCAA-3′
Human SOX1 reverse: 5′-TTGTCCTTCTTGAGCAGCGT-3′
Human SOX2 forward: 5′-TTTGTCGGAGACGGAGAAGC-3′
Human SOX2 reverse: 5′-TAACTGTCCATGCGCTGGTT-3′
Human FNDC3A forward: 5′-GTGAAACAGAAAGCGGGAGC-3′
Human FNDC3A reverse: 5′-TCCAATTCTCCGAGTCACGC-3′
Human SLC3A2 forward: 5′-CGCATTGCGGCTTGGT-3′
Human SLC3A2 reverse: 5′-GAATGTGAGCCAGGCAACTG-3′
Human ALG5 forward: 5′-TCTAGAGAAGAGACAGAAACGAGA-3′
Human ALG5 reverse: 5′-ACTTTGTGGCTCCATCAGCA-3′
Human HS2ST1 forward: 5′-GTGTAGCAGAAGGTGGCTCA-3′
Human HS2ST1 reverse: 5′-CCTGAAAAACCGGGGCAATG-3′
Human CANX forward: 5′-GCCTCCGCCTCTCTCTTTAC-3′
Human CANX reverse: 5′-GGAGGAGCAGTGGTATCTGG-3′
Human GAPDH forward: 5′-GTCTCCTCTGACTTCAACAGCG-3′
Human GAPDH reverse: 5′-ACCACCCTGTTGCTGTAGCCAA-3′
Mouse GAPDH forward: 5′-ACGGCCGCATCTTCTTGTGCA-3′
Mouse GAPDH reverse: 5′-ACGGCCAAATCCGTTCACACC-3′
Knockout cell lines construction
Huh7.0 or A549 cells were transiently transfected with 5 μg PX459-FAM134B-KO plasmids and then the medium was replaced with fresh media containing 1 μg/ml puromycin (HY-B1743, MCE) after 48 h later. The surviving cells were sorted out as the monoclone 5 days later. Human FAM134B-targeted sgRNA: 5′-GAGCTCAGCAGCTCGTCGGCG-3′.
shRNA knockdown
HEK293T cells were co-transfected with psPAX2, pMD2.G, and PLKO.1-shAMFR plasmids to generate lentiviral particles. Forty-eight hours later, supernatants were collected and filtered with a 0.45-μm-pore filter to infect the indicated cells for 48 h and then selected with 1 μg/ml puromycin for 1 week. Human AMFR-targeted shRNA: 5′-CCTCGCTTAAACCAACACAAT-3′.
Generation of stable cell lines
HEK293T cells were transfected with psPAX2, pMD2.G, and pCDH-CMV-FAM134BWT or pCDH-CMV-FAM134BR142A or pCDH-CMV-GFP-FAM134BWT or pCDH-CMV-GFP-FAM134BR142A for 48 h to generate lentiviral particles. Supernatants were filtered with a 0.45-μm-pore filter and collected to infect the indicated cells (Huh7.0 or A549) for 48 h and then selected with 2 mg/ml Blasticidin S (cat. no. 3513-03-9, Yeasen) for 1 week.
Flow cytometry analysis
To determine the fluorescence intensity of GFP-FAM134BWT or GFP-FAM134BR142A in virus-mock or virus-infected cells, the cells were trypsinized and harvested by centrifugation at 4000 × g for 2 min at 4 °C. Cells were then resuspended in ice-cold PBS and analyzed on a CytoFLEX flow cytometer (Beckman) as soon as possible. Data analysis was performed using CytExpert software (Version 2.0.4.8).
Determination of the virus titer by plaque assay
Supernatants from infected cells were collected at the indicated time points and centrifuged for 10 min at 1000×g, 4 °C. The presence of infectious viral particles in the supernatant were serial-diluted by tenfold with DMEM. Then added 250 μL of each dilutes onto Vero cells monolayers in 24-well plates and incubated at 37 °C under 5% CO2. The supernatants were discarded, and 500 μL of fresh medium (containing 0.8% [g/mL] carboxymethyl cellulose, 2% [v/v] FBS, 1% [v/v] penicillin-streptomycin, and 0.5% [v/v] DMSO) was added to the cells. The cells were cultured for 5 days and discarded in the overlay. Then fixed cells with 4% paraformaldehyde, followed by staining with 0.8% crystal violet solution. Plaques were counted for the calculation of virus titers.
Generation of cerebral organoids
Human cerebral organoids were generated from H9 hESCs as previously described63. On day 0 hESCs were dissociated into single cells by accutase treatment. Cells were transferred to an ultra-low binding 96-well plate (3000 cells/well) in EB formation medium with low concentration bFGF (4 ng/ml) and ROCK inhibitor (50 mM). On day 6, the medium was replaced daily with Neural Induction Medium containing DMEM/F12, 1 × N2 supplement (17502001, Gibco), 1% non-essential amino acids (11140, Gibco), 2 mM GlutaMAX, and 1 ug/ml heparin. On day 11, the EBs were embedded into droplets of Matrigel and transferred into 6-cm dishes in neural expansion medium containing 50% DMEM/F12, 50% Neurobasal medium, 0.5× N2 supplement, 0.5 × B27 supplement without vitamin A, 2 mM GlutaMAX, 2.5 ng/ml human insulin, 0.5% non-essential amino acids, and 25 nM betamercaptoethanol (31350010, Gibco). On day 14, the medium was transferred to neural maturation medium containing 50% DMEM/F12, 50% neurobasal medium, 0.5× N2 supplement, 0.5×B27 supplement with vitamin A, 2 mM GlutaMAX, 2.5 ng/ml human insulin, 0.5% non-essential amino acids, and 25 nM betamercaptoethanol and organoids were cultured on an orbital shaker under 80 g rotating speed. The medium was changed every 2 days.
Infection of cerebral organoids
On day 10, organoids were incubated with ZIKV-NS2AWT or ZIKV-NS2AK56R diluted in 200 μL of neural induction medium (7 × 104 PFU/organoid) in ultra-low binding 96-well plates. Especially, for western blotting analysis, organoids were exposed to ZIKV-NS2AWT (7 × 104 PFU/organoid) or ZIKV-NS2AK56R (7×105 PFU/organoid) to analyze the degradation of FAM134B, CLIMP63, and REEP5. Then plates were incubated in the 5% CO2 incubator at 37 °C. After infection for one day, organoids were embedded in Matrigel, transferred to 6-cm dishes in an expansion medium, and cultured as described above.
Immunofluorescence assay
Cells were fixed with 4% (w/v) paraformaldehyde for 20 min at room temperature and were washed with PBS three times. Then, cells were permeated by 0.1% (v/v) Triton X-100 for 20 min at room temperature and were washed with PBS three times. Then cells were blocked by 3% (v/v) BSA for 30 min. After blocking, cells were incubated with indicated primary antibodies for 1 h and then washed with 1% (v/v) BSA three times. The second fluorescent antibodies were added for another 1 h. DAPI was used to stain the nucleus for 5 min.
Organoids were fixed with 4% (w/v) paraformaldehyde in 1×PBS at 4 °C overnight. Then organoids were incubated in a 30% (w/v) sucrose solution overnight at 4 °C, embedded in optical coherence tomography (O.C.T) compounds and frozen. Organoid blocks were sectioned at 10μm thickness using a freezing microtome (Leica). Organoid cryosections were fixed with 4% PFA for 15 min and washed with 1×PBS extensively.
Generation of recombinant ZIKV-NS2AWT, ZIKV-NS2AK56R, ZIKV-HA-NS2AWT, and ZIKV-HA-NS2AK56R
The pFLZIKV-FSS13025 ZIKV-NS2AWT or ZIKV-NS2AK56R mutant plasmids and inserted HA-tagged between NS1 and NS2A genes pFLZIKV-FSS13025 ZIKV-HA-NS2AWT or ZIKV-HA-NS2AK56R plasmids were amplified in E. coli Top10 and purified using MaxiPrep PLUS (12963, QIAGEN). For in vitro transcription, 10 μg plasmid was linearized with the restriction enzyme ClaI, and the mMESSAGE T7 kit (AM1344, Invitrogen) was used for transcription of RNA following the kit’s manual. The RNA was then precipitated with lithium chloride and quantitated by spectrophotometry. For transfection, 10 mg RNA was electroporated into Vero cells mixed with 0.8 ml of Ingenio® Electroporation Solution (MIR 50114, Mirus), in 4 mm cuvettes with the GenePulser apparatus (Gene Pulser Xcell, Bio-Rad) at settings of 0.45 kV and 25 μF, pulsing three times, with 3-s intervals. After a 10-min recovery at room temperature, the transfected cells were mixed with DMEM in T75 flasks and incubated in the 5% CO2 incubator at 37 °C. Recombinant viruses in cell culture media were harvested after transfection for 4 days.
Protein purification
For mammalian cells, HEK293T cells were transfected with indicated plasmids. After 48 h, the cells were collected and lysed by lysis buffer containing protease inhibitor cocktail for 30 min on ice and then centrifuged at 13,000×g for 30 min at 4 °C. The supernatants were immunoprecipitated by using anti-Flag beads or anti-HA beads for 12 h at 4 °C. The immunoprecipitants were washed by lysis buffer four times. Then, we utilized the elution protocols to elute Flag or HA-tagged proteins. Firstly, add 100 µL 0.1 M glycine, pH 2.0. Secondly, gently vortex to mix and incubate the sample at room temperature on a rotator for 5–10 min. Thirdly, magnetically separate the beads and save the supernatant containing the target antigen. Last, to neutralize the low pH, add 15 µL of 1 M Tris, pH 8.5 for each 100 µL of eluate.
For Escherichia coli, E.coli strain BL21, was transformed with the indicated plasmid encoding GST-FAM134B or His–AMFR△1-300aa. And then the monoclonal cell was first grown overnight in LB medium supplemented with 50 μg/mL ampicillin or kanamycin at 37 °C. About 5 mL overnight cultured bacteria were then inoculated into 100 mL LB medium for further culture. When the OD600 reached 0.6, protein expression was induced at 16–18 °C overnight by adding 0.5 mM IPTG. Cells were harvested after induction, and were sonicated in PBS buffer. The lysates were centrifuged at 8000×g for 15 min at 4 °C. After centrifugation, GST-FAM134B proteins or His–AMFR△1-300aa proteins were purified with glutathione resins (L00206, GenScript) or high-affinity Ni-NTA resins (L00250, GenScript) according to the manufacture’s protocols, respectively. The ExBlue was purchased from ZOMANBIO, Beijing, China (ZD305A).
In vitro ubiquitination assay
For in vitro ubiquitination assay, Flag-NS2A, HA-AMFRWT, and HA-AMFRC356G/H361A were purified from HEK293T cells using anti-Flag beads and anti-HA beads, respectively. Purified Flag-NS2A was incubated with HA-AMFRWT or HA-AMFRC356G/H361A in the presence of purified ubiquitin K48 only, E1 (UBA1), E2 (UBE2G2), Mg2+-ATP and DTT in a total reaction volume of 50 μL. The contents were mixed gently and incubated at 37 °C for 1 h. Quench assays by addition of 50 μL 2x SDS-loading buffer followed by heating to 95 °C for 5 min or 70 °C for 10 min. The K48-linked ubiquitination of NS2A was analyzed by immunoblotting with an anti-K48-linkage specific polyubiquitin antibody.
Tandem mass tags (TMT) mass spectrometry analyses and protein quantification
To determine the impact of ZIKV infection on cellular proteomics, a total of 12 protein samples were collected from mock-infected and ZIKV-infected A549 cells at 24, 48, and 72 h post-infection at an MOI of 0.1, three repeats. Proteins were digested into peptides by the SP3 (single-pot solid phase-enhanced sample preparation) method. In summary, peptides were eluted after digestion of the proteins with trypsin for 14 h at 37 °C. Twelve-plex TMT labeling was performed for mock and infected samples of the same time points following the manufacturer’s (Thermo Fisher Scientific, Waltham, CA, USA) instructions. An equal amount of twelve TMT labeled samples were mixed together, and 2D LC/MS/MS was performed using an Orbitrap Q Exactive HF-X instrument (Thermo Fisher Scientific, Bremen, Germany). For identification of the peptide/proteins, ZIKV (FSS13025 strain) and human (Uniprot 2016) databases were used as references. The intensities of TMT peptide level reporter tags were averaged over a ±0.1 Da window and corrected for isotopic overlap between channels using the batch-specific correction matrix provided. In this project experiment, a total of 647,400 secondary spectra were obtained through mass spectrometry analysis. After searching the protein theoretical data database for the secondary spectrum of mass spectrometry, the number of available effective spectra was 163,328, with a spectrum utilization rate of 25.2%. Through spectral analysis, a total of 103,636 peptide segments were identified, of which 100,493 were specific peptide segments. We identified a total of 6221 proteins, of which 5400 are quantifiable (quantitative proteins indicate that at least one comparison group has quantitative information).
Protein quantification calculation method: This project obtained the quantitative values of each sample in multiple repeated experiments of whole protein quantification. The first step is to calculate the differential expression level of proteins between two samples in the comparison group. First, calculate the average quantitative value of each sample in multiple replicates, and then calculate the ratio of the average values between the two samples. This ratio is used as the final differential expression level of the comparison group. The second step is to calculate the significant p value of the differential expression of the protein in two samples. Firstly, the relative quantitative values of each sample are taken as log2 (to make the data conform to a normal distribution), and then the p value is calculated using the two-sample two-tailed t-test method. When p value <0.05, a change in differential expression level exceeding 1.3 is considered a significant upregulation threshold, while a change below 1/1.3 is considered a significant downregulation threshold.
Mass spectrum
ZIKV-NS2A ubiquitination sites were detected by transient expression of NS2A in HEK293T cells. HEK293T cells were transfected with Flag-NS2A treating with 10 μM MG132. Thirty-six hours post transfection, cells were washed and lysed by lysis buffer, including 10 μM MG132. Following sonication and centrifugation, the supernatants were immunoprecipitated by Flag antibody for 12 h at 4 °C. The immunoprecipitants were washed by lysis buffer four times and then subjected to SDS–PAGE. Corresponding gel bands of ubiquitinated NS2A were excised and digested with trypsin. The digested peptides were analyzed by mass spectrometry.
Quantification and statistical analyses
All data were presented as means ± SEM and analyzed using GraphPad Prism software (Version 9.5.1). Two-tailed unpaired students’ t-tests were applied to data with two groups. ANOVA analyses were used for comparisons of data with greater than two groups. Sample sizes were indicated in the Figure legends. Data sets were considered significantly different if the P value was less than 0.05. Quantitation of western blotting was measured by Image J software (1.53t/Java 1.8.0 345).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data generated in this study have been deposited via the ProteomeXchange Consortium with the dataset identifier PXD050573. Source data are provided with this paper.
References
Pierson, T. C. & Diamond, M. S. The continued threat of emerging flaviviruses. Nat. Microbiol. 5, 796–812 (2020).
Bhatt, S. et al. The global distribution and burden of dengue. Nature 496, 504–507 (2013).
Maxmen, A. The hidden threat of West Nile virus. Nature 489, 349–350 (2012).
Platt, D. J. & Miner, J. J. Consequences of congenital Zika virus infection. Curr. Opin. Virol. 27, 1–7 (2017).
Mlakar, J. et al. Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958 (2016).
Rubin, E. J., Greene, M. F. & Baden, L. R. Zika virus and Microcephaly. N. Engl. J. Med. 374, 984–985 (2016).
Tang, H. et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18, 587–590 (2016).
Ho, C. Y. et al. Differential neuronal susceptibility and apoptosis in congenital Zika virus infection. Ann. Neurol. 82, 121–127 (2017).
Giraldo, M. I., Gonzalez-Orozco, M. & Rajsbaum, R. Pathogenesis of Zika virus infection. Annu. Rev. Pathol. 18, 181–203 (2023).
Schneider, W. M. & Hoffmann, H. H. Flavivirus-host interactions: an expanding network of proviral and antiviral factors. Curr. Opin. Virol. 52, 71–77 (2022).
Baharuddin, A. et al. Current approaches in antiviral drug discovery against the Flaviviridae family. Curr. Pharm. Des. 20, 3428–3444 (2014).
van den Elsen, K., Chew, B. L. A., Ho, J. S. & Luo, D. Flavivirus nonstructural proteins and replication complexes as antiviral drug targets. Curr. Opin. Virol. 59, 101305 (2023).
Mukhopadhyay, S., Kuhn, R. J. & Rossmann, M. G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3, 13–22 (2005).
Xie, X., Zou, J., Puttikhunt, C., Yuan, Z. & Shi, P. Y. Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J. Virol. 89, 1298–1313 (2015).
Leung, J. Y. et al. Role of nonstructural protein NS2A in flavivirus assembly. J. Virol. 82, 4731–4741 (2008).
Liu, W. J. et al. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 80, 2396–2404 (2006).
Wu, R. H. et al. Mutagenesis of dengue virus protein NS2A revealed a novel domain responsible for virus-induced cytopathic effect and interactions between NS2A and NS2B transmembrane segments. J. Virol. 91, e01836–16 (2017).
Yoon, K. J. et al. Zika-virus-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins. Cell Stem Cell 21, 349–358.e346 (2017).
Xie, X. et al. Dengue NS2A protein orchestrates virus assembly. Cell Host Microbe 26, 606–622.e608 (2019).
Zhang, X. et al. Zika virus NS2A-mediated virion assembly. mBio 10, e02375–19 (2019).
Schwarz, D. S. & Blower, M. D. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol. Life Sci. 73, 79–94 (2016).
Ferro-Novick, S., Reggiori, F. & Brodsky, J. L. ER-phagy, ER homeostasis, and ER quality control: implications for disease. Trends Biochem. Sci. 46, 630–639 (2021).
Mochida, K. & Nakatogawa, H. ER-phagy: selective autophagy of the endoplasmic reticulum. EMBO Rep. 23, e55192 (2022).
Chino, H. & Mizushima, N. ER-phagy: quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 30, 384–398 (2020).
Khaminets, A. et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358 (2015).
Fumagalli, F. et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 18, 1173–1184 (2016).
Chen, Q. et al. ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr. Biol. 29, 846–855.e846 (2019).
Grumati, P. et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6, e25555 (2017).
Smith, M. D. et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 44, 217–232.e211 (2018).
Chino, H., Hatta, T., Natsume, T. & Mizushima, N. Intrinsically disordered protein TEX264 mediates ER-phagy. Mol. Cell 74, 909–921.e906 (2019).
Romero-Brey, I. & Bartenschlager, R. Endoplasmic reticulum: the favorite intracellular niche for viral replication and assembly. Viruses 8, 160 (2016).
Roingeard, P. et al. The double-membrane vesicle (DMV): a virus-induced organelle dedicated to the replication of SARS-CoV-2 and other positive-sense single-stranded RNA viruses. Cell Mol. Life Sci. 79, 425 (2022).
Welsch, S. et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5, 365–375 (2009).
Arakawa, M. & Morita, E. Flavivirus replication organelle biogenesis in the endoplasmic reticulum: comparison with other single-stranded positive-sense RNA viruses. Int. J. Mol. Sci. 20, 2336 (2019).
Morita, E. & Suzuki, Y. Membrane-associated flavivirus replication complex-its organization and regulation. Viruses 13, 1060 (2021).
Denolly, S. et al. Zika virus remodelled ER membranes contain proviral factors involved in redox and methylation pathways. Nat. Commun. 14, 8045 (2023).
Li, S., Kong, L. & Yu, X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 41, 150–164 (2015).
Das, B. et al. Role of endoplasmic reticulum stress-related unfolded protein response and its implications in dengue virus infection for biomarker development. Life Sci. 329, 121982 (2023).
Prasad, V. & Greber, U. F. The endoplasmic reticulum unfolded protein response - homeostasis, cell death and evolution in virus infections. FEMS Microbiol. Rev. 45, fuab016 (2021).
Fung, T. S., Huang, M. & Liu, D. X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 194, 110–123 (2014).
Valadão, A. L., Aguiar, R. S. & de Arruda, L. B. Interplay between inflammation and cellular stress triggered by flaviviridae viruses. Front. Microbiol. 7, 1233 (2016).
Lennemann, N. J. & Coyne, C. B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 13, 322–332 (2017).
Ciechanover, A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6, 79–87 (2005).
Jackson, M. P. & Hewitt, E. W. Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem. 60, 173–180 (2016).
Jiang, X. et al. FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER-phagy. EMBO J. 39, e102608 (2020).
Pohl, C. & Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822 (2019).
Gonzalez, A. et al. Ubiquitination regulates ER-phagy and remodelling of endoplasmic reticulum. Nature 618, 394–401 (2023).
Khaminets, A., Behl, C. & Dikic, I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 26, 6–16 (2016).
Krenn, V. et al. Organoid modeling of Zika and herpes simplex virus 1 infections reveals virus-specific responses leading to microcephaly. Cell Stem Cell 28, 1362–1379.e1367 (2021).
Kumar, R., Mehta, D., Mishra, N., Nayak, D. & Sunil, S. Role of host-mediated post-translational modifications (PTMs) in RNA virus pathogenesis. Int. J. Mol. Sci. 22, 323 (2020).
Hay-McCullough, E. & Morrison, J. Contributions of ubiquitin and ubiquitination to flaviviral antagonism of type I IFN. Viruses 13, 763 (2021).
Gu, H. & Jan Fada, B. Specificity in ubiquitination triggered by virus infection. Int. J. Mol. Sci. 21, 4088 (2020).
Giraldo, M. I. et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 585, 414–419 (2020).
Liu, H. et al. Endoplasmic reticulum protein SCAP inhibits dengue virus NS2B3 protease by suppressing Its K27-linked polyubiquitylation. J. Virol. 91, e02234–16 (2017).
Laurent-Rolle, M. & Morrison, J. The role of NS5 protein in determination of host cell range for yellow fever virus. DNA Cell Biol. 38, 1414–1417 (2019).
Fanunza, E. et al. Zika virus NS2A inhibits interferon signaling by degradation of STAT1 and STAT2. Virulence 12, 1580–1596 (2021).
He, J. et al. Zika virus NS2A protein induces the degradation of KPNA2 (karyopherin subunit alpha 2) via chaperone-mediated autophagy. Autophagy 16, 2238–2251 (2020).
Chen, T. et al. The role of autophagy in viral infections. J. Biomed. Sci. 30, 5 (2023).
Chiramel, A. I., Dougherty, J. D., Nair, V., Robertson, S. J. & Best, S. M. FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of Ebola virus strains Makona and Mayinga. J. Infect. Dis. 214, S319–S325 (2016).
Tan, X. et al. Coronavirus subverts ER-phagy by hijacking FAM134B and ATL3 into p62 condensates to facilitate viral replication. Cell Rep. 42, 112286 (2023).
Evans, A. S., Lennemann, N. J. & Coyne, C. B. BPIFB3 regulates endoplasmic reticulum morphology to facilitate flavivirus replication. J. Virol. 94, e00029-20 (2020).
Pan, R. et al. N7-methylation of the coronavirus RNA cap is required for maximal virulence by preventing innate immune recognition. mBio 13, e0366221 (2022).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Acknowledgements
This research was supported by grants from the National Key R&D Program of China (2021YFC2300200, 2021YFC2300702), the National Natural Science Foundation of China (32200127, 82130064, and U22A20337), Key R&D Program Project of Hubei Province (2023BCB087), Key R&D of Biosafety in Hubei Jiangxia Laboratory (JXBS017), and Natural Science Foundation of Wuhan (2024040701010047).
Author information
Authors and Affiliations
Contributions
L.Z., H.W., C.H., Q.D., and J.Y. performed the experiments. L.Z. and M.C. designed the experiments and wrote the paper. W.G., C.S., W.Z., P.C., R.H., and Y.W. analyzed the results. Y.C., Y.Q., and M.C. supervised the study. All authors discussed the results and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, L., Wang, H., Han, C. et al. AMFR-mediated Flavivirus NS2A ubiquitination subverts ER-phagy to augment viral pathogenicity. Nat Commun 15, 9578 (2024). https://doi.org/10.1038/s41467-024-54010-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54010-w
This article is cited by
-
ER-phagy receptors: structural mechanisms in selective ER degradation and disease implications
Acta Pharmacologica Sinica (2026)
-
AMFR-mediated ER-phagy regulation and therapeutic targeting in osteosarcoma: a multifunctional nanoplatform strategy
Journal of Nanobiotechnology (2025)
-
Inhibition of STING-mediated antiviral innate immunity activation by CD97 via modulation of ER-phagy
Communications Biology (2025)
-
Zika virus disrupts steroidogenesis and impairs spermatogenesis by stalling the translation of CYP17A1 mRNA
Nature Communications (2025)
-
Neurovirulence of Zika virus–encoded proteins
Archives of Virology (2025)
