
Abstract
CO2 electroreduction is a potential pathway to achieve net-zero emissions in the chemical industry. Yet, CO2 loss, resulting from (bi)carbonate formation, renders the process energy-intensive. Acidic environments can address the issue but at the expense of compromised product Faradaic efficiencies (FEs), particularly for multi-carbon (C2+) products, as rapid diffusion and migration of protons (H+) favors competing H2 and CO production. Here, we present a strategy of tuning the 2-position substituent length on benzimidazole (BIM)-based copper (Cu) coordination polymer (CuCP) precatalyst – to enhance CO2 reduction to C2+ products in acidic environments. Lengthening the substituent from H to nonyl enhances H+ diffusion retardation and decreases Cu-Cu coordination numbers (CNs), favoring further reduction of CO. This leads to a nearly 24× enhancement of selectivity towards CO hydrogenation and C-C coupling at 60 mA cm−2. We report the highest C2+ product FE of more than 70% at 260 mA cm−2 on pentyl-CuCP and demonstrate a CO2-to-C2+ single-pass conversion (SPC) of ~54% at 180 mA cm−2 using pentyl-CuCP in zero-gap electrolyzers.
Similar content being viewed by others


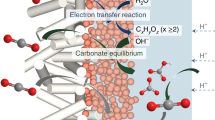
Introduction
Electrochemical CO2 reduction reaction (CO2RR) is an emerging technology that transforms CO2 into valuable chemicals and fuels1,2. Cu-based CO2RR catalysts are of particular interest for their considerable selectivity towards C2+ products such as ethylene (C2H4) and ethanol (EtOH)3,4. Alkaline and neutral environments were used in conventional CO2 electrolyzers, and high FEs for C2+ products of more than 90% were achieved in these systems5,6,7.
However, at the cathodes of alkaline and neutral CO2 electrolyzers, CO2 combines with hydroxide ions to form (bi)carbonates because of the strong local alkalinity caused by CO2RR8. This exergonic and spontaneous process, with an energy release of about −56 kJ mol−1 ref. 9, limits the SPC of CO2 to C2+ products (except for acetate, the limit of which is 36%) to less than 25%10. Part of the formed (bi)carbonate ions precipitate at the cathode as salts, and the other part migrates to the anode, releasing in the form of CO2 together with O2 generated by water oxidation11,12. The consequence of these two processes is increased energy consumption for regenerating CO2 loss, which can be higher than the energy stored in CO2RR products10,12.
To reclaim the energy efficiency and CO2 lost to (bi)carbonates, acidic CO2RR was proposed as a potential solution13,14,15. The idea is to use H+ flux from the bulk electrolyte or membrane to convert (bi)carbonates back to CO2. However, the H+ flux in an acidic CO2RR is typically in excess at the catalyst surface16, creating a microenvironment more favorable for H2 evolution and CO production17,18.
We posit that Cu CPs, when serving as (pre)catalysts, can potentially address the issue of oversupplied H+ flux and low C2+ product selectivity in acidic reaction environments. The organic building blocks of CuCPs can be designed to strongly repel H+ flux using hydrophobic carbon chain backbones19,20, suppressing the unwanted H2 evolution. At the same time, previous studies showcased that CuCP has a Cu-Cu distance that fits well with the geometry of the C-C coupling intermediate, which enhances C2+ product FEs21,22. Hence, the active sites in CuCPs can be customized for the desired products, which would also benefit C2+ product formation under acidic conditions.
Here, we explore the foregoing probability by investigating the impact of the 2-position substituent length of Cu-BIM CPs on their acidic CO2RR performance. By lengthening the substituent in the order of H <methyl <propyl <pentyl <heptyl <nonyl, we saw a volcano-like change in the FEs for C2+ products and a monotonic increase in the ratio between >2e− transfer products and CO. Specifically, pentyl-CuCP offers the highest C2+ FE of 73.4 ± 2.9% at 260 mA cm−2, and nonyl-CuCP increases the activity towards >2e− transfer products by 24 times compared to H-CuCP and Cu2O-derived Cu controls. Longer 2-position substituents endow the catalysts with (i) stronger impedance of H+ mass transfer and (ii) low-coordinated Cu sites to favor CO adsorption, hydrogenation, and coupling. These features are confirmed by our electrochemistry, in-situ scanning electrochemical microscopy (SECM), in-situ Raman spectroscopy, X-ray absorption spectroscopy (XAS) measurements, and density functional theory (DFT) calculations. Using pentyl-CuCP electrodes, we demonstrate a ~54% SPC for CO2RR to C2+ products at a current density of 180 mA cm−2 in acidic CO2RR using trilayer polymer electrolytes based MEAs (TPE-MEAs), breaking the theoretical limit in alkaline and neutral CO2 electrolyzers10. Compared to Cu2O-derived Cu, pentyl-CuCP enhances the total CO2 SPC by a factor of ~1.2, and doubles the CO2 SPC to C2+ products.
Results and discussion
CuCP synthesis and characterizations
The synthesis of CuCPs was conducted by reacting copper(I) oxide (Cu2O) with various benzimidazole (BIM) derivatives, specifically including 2-H-, 2-methyl-, 2-propyl-, 2-pentyl-, 2-heptyl- and 2-nonyl-BIM at 60 °C (Fig. 1a). These obtained materials were labeled H-CuCP, methyl-CuCP, propyl-CuCP, pentyl-CuCP, heptyl-CuCP, and nonyl-CuCP. The as-made CuCP materials exhibit two-dimensional structures according to scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, distinct from the Cu2O particle precursor (Supplementary Figs. 1–3). The uniform distribution of Cu was verified by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) analysis (Fig. 1b). X-ray diffraction (XRD) studies reveal a notable decrease or disappearance of Cu2O peaks in the as-made CuCPs (Supplementary Fig. 4). The synthesized CuCPs display intense characteristic peaks within 10°, for example, pentyl-CuCP at 6.3° (Fig. 1c). Note that peaks at 18° and 25° are attributed to the carbon paper substrate (GDE) (Supplementary Fig. 4b). The Fourier-transform infrared spectroscopy (FTIR) comparison between reactants and CuCPs also shows the disappearance of the Cu-O stretching mode peak at 607 cm−1 after reaction23 (Supplementary Fig. 5). This observation, alongside the presence of skeletal vibrations of benzene C-C bonds (1450–1600 cm−1) and stretching vibrations of C-H bonds in CuCPs (2800–3000 cm−1)24,25, confirms CP structures.

a Illustration of the synthesis process. The yellow spheres in BIM-based molecules stand for the substituent groups (R). b TEM, HADDF, and Cu EDS mapping images of pentyl-CuCP. c The XRD pattern and (d) Cu 2p XPS spectra of pentyl-CuCP. Source data are provided as a Source Data file.
Using X-ray photoelectron spectroscopy (XPS) analysis, we unveiled the presence of Cu(I) valence states in the obtained CuCPs (Fig. 1d and Supplementary Fig. 6). Before polymerization, BIM and all variants show two chemical environments for nitrogen atoms, as evidenced by two peaks in the N 1 s spectra; the spectra consolidate into a single peak in CuCPs, further indicating the formation of Cu-N bonds (Supplementary Fig. 7)25,26. The disappearance of the 530-eV peak in the O 1 s spectra of CuCPs corroborates the transformative chemical interactions within these compounds (Supplementary Fig. 8). Typically, the peak at 530 eV is attributed to lattice metal-O bonds, while the peaks at 532 and 533 eV are attributed to surface adsorbed oxygen species, including surface metal-OH bonds and other surface adsorbates (e.g., adsorbed water)27,28. The 532-eV peak may correspond to adsorbed water on the material’s surface. According to the Cu Auger LMM results (Supplementary Fig. 9), we confirmed the mixed valence of Cu(I) and Cu(II) in the formed CPs. The above characterizations demonstrated the successful preparation of CuCPs.
CO2RR performance comparison
We then evaluated the performance of CO2RR of the synthesized CuCPs. Gas diffusion electrodes (GDEs) were prepared by spray-coating CuCPs onto gas diffusion layers (GDLs). The resultant electrodes have increased hydrophobicity in the order of H-CuCP <methyl-CuCP <propyl-CuCP <pentyl-CuCP ≈ heptyl-CuCP ≈ nonyl-CuCP, verified by their contact angles with water drops (Supplementary Fig. 10). CO2RR was then measured in anion-exchange-membrane-(AEM)-equipped zero gap electrolyzers. Anolytes with a pH of ~1.7 were utilized to generate a weakly acidic membrane/catalyst interface at the cathode via the cation (i.e., H+) crossover through the AEM29,30,31. H+ crossover from the anode to the cathode can be confirmed by the observation of CO2 regeneration in AEM electrolyzers using KHCO3 (pH = 8.3) catholyte and H2SO4 (pH = 0.7) anolyte (Supplementary Fig. 11).
We found that with the increase in the substituent length from H to nonyl, the FEs for C2+ products and C2H4 exhibit a volcano-like distribution (Fig. 2a and Supplementary Fig. 12). At 260 mA cm−2, the FEs for C2+ products are 47.4 ± 1.8%, 53.1 ± 2.2%, 62.5 ± 2.2%, 73.4 ± 2.9%, 58.1 ± 1.4%, and 54.0 ± 1.6% for H-, methyl-, propyl-, pentyl-, heptyl- and nonyl-CuCP (Fig. 2a), respectively. For all CuCPs, C2H4 is the major C2+ product with an FE of 31.2 ± 0.6% for H-CuCP, 33.5 ± 0.8% for methyl-CuCP, 38.1 ± 0.7% for propyl-CuCP, 40.9 ± 1.2% for pentyl-CuCP, 25.1 ± 0.4% for heptyl-CuCP and 23.0 ± 0.7% for nonyl-CuCP (Supplementary Fig. 12). These trends were also seen at 60 mA cm−2. We observed enhanced CH4 FE caused by substituent length increase, especially at low current densities (Fig. 2b). Taking 60 mA cm−2 as an example, the CH4 FE is 0.5 ± 0.1%, 11.0 ± 0.9%, 14.2 ± 0.3%, 24.1 ± 1.2%, 32.7 ± 0.4% and 41.6 ± 0.5% in the order of H <methyl <propyl <pentyl <heptyl <nonyl. In contradistinction, CO production is suppressed by lengthening the 2-position substituent, embodied by the decrease of the FE from 61.0 ± 0.6% to 5.0 ± 0.5% at 60 mA cm−2 (Fig. 2c). Therefore, we confirmed that longer substituents facilitate CO hydrogenation and C-C coupling, which is validated by the relation between the substituent identity and the FE ratio of >2e- transfer products vs. CO (Fig. 2d). This ratio peaks at ~14 on nonyl-CuCP at 60 mA cm−2, 34 times as high as that on H-CuCP at the same current density. This gap remains 11 times at 260 mA cm−2, albeit with the CO and CH4 FEs decrease driven by increased overpotentials.

FEs of (a) C2+ products, (b) CH4, (c) CO, and (d) FE ratios of >2e- transfer products to CO under CuCPs at different current densities. Source data are provided as a Source Data file.
Compared to carbon-supported Cu2O-derived Cu with a similar Cu atomic fraction of ~35%, CuCPs are profoundly more selective for C2+ products (FE ratio: >50% vs. <40%) (Supplementary Tables 1 and 2). The C2+ FEs of Cu2O-derived Cu at 60 and 260 mA cm−2 are 11.9 ± 0.6% and 37.7 ± 2.2%, respectively. This means pentyl-CuCP is at least 1.9-fold more active for C2+ products than Cu2O-derived Cu. In addition, the overall activity for CO hydrogenation and C-C coupling Cu2O is greatly suppressed on Cu2O-derived Cu: the >2e− product vs. CO FE ratio ranges from 0.3 to 1.2 over the entire current range, similar to H-CuCP and much lower than pentyl-, heptyl- and nonyl-CuCPs.
The origin of the enhanced performance
We then sought to understand how increased substituent length promotes CO hydrogenation and C-C coupling selectivity and activity. As the increase in the anolyte pH was found to facilitate CO conversion to C2+ products (Supplementary Fig. 13 and Table 2), we first considered the probability that stronger suppression of H+ diffusion and migration by longer alkyl chains disfavors CO formation. This was assessed by measuring the linear sweep voltammetry of H2 evolution in 0.1 M H2SO4 solutions. On propyl-, pentyl-, heptyl- and nonyl-CuCP, the H+ reduction current density increases rapidly to a plateau of 60–80 mA cm−2 starting at ~−1.59 V vs. RHE (Supplementary Fig. 14). Given that the theoretical diffusion-limited current density for H+ reduction in 0.1 M H2SO4 is higher than 359 mA cm−2 (Supplementary Note 2), observing the H+ diffusion limit at lower current densities indicates that propyl-, pentyl-, heptyl- and nonyl-CuCP strongly reduce the H+ concentration near catalyst surfaces. The current density increases again at ~−1.93 V vs. RHE, corresponding to H2O reduction. For Cu2O, H-, and methyl-CuCP, the transfer-limited current density was not seen within the tested current range (Supplementary Fig. 13). This trend also corresponds with the static contact angles between water drops and the electrode surfaces: Cu2O < H- <methyl- <propyl- ≈ pentyl- ≈ heptyl- ≈ nonyl-CuCP (Supplementary Fig. 10). After the substituent is extended to propyl, the contact angle of ~140° is close to that of a superhydrophobic surface. The increase in the CO FE and reduction in C2+ product selectivity with shorter substituents suggest that high local H+ concentrations favor *CO desorption from the catalyst surface. This argument can be evidenced by CO2RR using pentyl-CuCP at a stronger acidity (pH = 0.7), where we saw higher CO and H2 FEs than tests under pH = 1.7 (Supplementary Fig. 14). The H+ diffusion retardation, when applied to acidic CO2RR in H-cells where the CO2 mass transfer is much less efficient than MEAs, reduces the H2 FE by ~10% at the same potentials and promotes CO hydrogenation and C-C coupling (Supplementary Fig. 15).
Seeking to gain insights into the impact of the substituent length on the active site structure, we explored the dynamic evolution of CuCPs under CO2RR conditions. H-CuCP, the sample with the shortest 2-position group, and pentyl-CuCP, the sample showing the highest C2+ FE, were taken as representatives. We utilized the SECM technique to study their morphology and activity evolution under CO2RR conditions (Supplementary Fig. 16 and Fig. 3a–d). The normalized feedback current (iT/iT,∞) distribution illustrates the surface reactivity heterogeneity, with a larger iT/iT,∞ representing higher local activity. Therefore, the feedback images of the electrode surfaces reveal the complete decomposition of H-CuCP during electrolysis, evidenced by the distinct patterns and normalized feedback current obtained before and after the CO2RR reaction (Fig. 3a, b). In stark contrast, the feedback current and electrochemical morphology of pentyl-CuCP remain unchanged before and after CO2RR (Fig. 3c, d). In line with the SECM results, the HADDF image and Cu EDS mapping reveal the structure decomposition and Cu agglomeration in H-CuCP after CO2RR (Supplementary Fig. 17a–d). In contrast, much less Cu exsolution and the uniform distribution of Cu in the cases of pentyl- and nonyl-CuCP were seen (Supplementary Fig. 17e–l). The in-situ Raman spectroscopies show the decomposition of H-CuCP after 1200 s and relatively more stable backbone structures for methyl-, propyl-, pentyl-, heptyl-, and nonyl-CuCPs (Fig. 3e, f, and Supplementary Figs. 18, 19) under both near-neutral and acidic conditions. The dissociation or decomposition of CuCPs does not contribute to CO2RR product FEs, which was verified by the fact that electrolysis under Ar only produces H2 with near-unity selectivity (Supplementary Fig. 20).

a, b SECM feedback images of H-CuCP catalyst before and after CO2RR. c, d SECM feedback images of pentyl-CuCP catalyst before and after CO2RR. e, f In-situ Raman spectra of H- and pentyl-CuCP. g The Cu K-edge XANES for H-, pentyl-CuCP, Cu foil, Cu2O, and CuO references. h The Cu-Cu CNs of H- and pentyl-CuCP at different current densities and times. i The free energy changes for the dissociation of in H-, pentyl- and nonyl-CuCPs. The electrolyte used in all electrochemical measurements is 0.1 M KHCO3. The potentials in (e) and (f) are non-iR corrected. Source data are provided as a Source Data file.
In-situ XAS was then employed to probe the real-time catalyst structure. At the open circuit potential (OCP), the X-ray absorption near edge structure (XANES) profiles suggest that the Average Cu valence state in H-CuCP and pentyl-CuCP is likely to be higher than that of Cu(I) but lower than that of Cu(II) (Fig. 3g). In combination with the results of the Cu LMM spectra analyses, the presence of a mixture of Cu(I) and Cu(II) in CuCPs was verified. The Cu white line peaks in the XANES spectra of H-CuCP shift rightward with increasing time and current density, indicating the reduction of the high-valence-state Cu to metallic Cu (Supplementary Fig. 21a). In contrast, the peak position for pentyl-CuCP does not show significant changes, implying its high chemical stability (Supplementary Fig. 21b). The extended X-ray absorption fine structure (EXAFS) in the R-space reveals the Cu-Cu signal centered at 2.5 Å for H-CuCP enhances with time and current density increase (Supplementary Fig. 22a–d). In contrast, the spectra for pentyl-CuCP have much weaker 2.5 Å peaks across the applied current density range (Supplementary Fig. 22e–h). The EXAFS fittings from OCP to 200 mA cm−2 show that the global Cu-Cu CN in the H-CuCP case increases from 1.32 to 12.00, similar to Cu foil, while that for pentyl-CuCP keeps below 2 during the measurement (Fig. 3h, Supplementary Fig. 23 and Table 3). Combining XRD analyses, in-situ Raman spectra, Cu EDS mappings, and XAS studies, we showed that longer substituents may provide highly undercoordinated CP-derived Cu due to the suppressed dissociation or decomposition of pentyl-CuCP. The undercoordinated Cu in pentyl-Cu would strengthen the adsorption of *CO, which is beneficial for producing products involving *CO hydrogenation and C-C coupling32,33. As all CuCPs possess comparable Cu atomic loadings ranging from 2.03 × 1016 to 2.81 × 1016 cm−2 (Supplementary Fig. 24), we excluded the contribution of active surface areas to the enhanced *CO hydrogenation and C-C coupling activities seen on CuCPs with longer 2-position substituents.
To have a more quantitative understanding of the impact of substituent lengths on the structural stability and CO2RR performance of these CuCPs, we carried out DFT calculations. Using H-, pentyl-, and nonyl-CuCPs as examples, combined with XRD refinement, we found that the Cu-Cu distances within the molecular chains are comparable for these CuCPs, slightly varying from 5.5 to 5.8 Å (Supplementary Figs. 25–27). However, the inter-chain Cu-Cu distances are 8.0 Å for H-CuCP, 14.0 Å for pentyl-CuCP, and 18.9 Å for nonyl-CuCPs (Supplementary Figs. 25–27). Under CO2RR conditions, the Cu-N bonds dissociate to form Cu metals, with required Gibbs free energy changes (ΔG) of 0.33 eV, 0.69 eV, and 0.89 eV (Fig. 3i), respectively. This indicates that increasing inner-chain Cu-Cu distance by lengthening the substituent impedes Cu exsolution from the CP backbones, and the Cu-Cu CNs of the formed Cu particles would decrease with longer substituents. The presence of undercoordinated active sites, together with the stronger H+ diffusion retardation offered by longer substituents, leads to the ascending C2+ product FEs from H to pentyl. However, the suppressed Cu exsolution also indicates more stable single-atom Cu sites in the presence of longer substituents. This increases the number of CH4-selective sites while decreasing that of C2+-selective sites34,35. Therefore, heptyl and nonyl are less favorable for C2+ products and more selective for CH4 than pentyl (Fig. 2a, b).
Achieving high SPC for CO2RR to C2+ products
To demonstrate carbon-efficient CO2RR to C2+ products reduction, we equipped a zero-gap electrolyzer with pentyl-CuCP and a trilayer polymer electrolyte (TPE) comprising a perforated anion-exchange layer (PAEL) and a bipolar membrane (BPM). In the TPE, the water dissociation (WD) catalyst offers H+ to convert (bi)carbonates produced by the cathode to CO2 at the interface between the PAEL and the cation-exchange layer (CEL) of the BPM (Fig. 4a). The regenerated CO2 then flows back to catalyst surfaces via the holes on the PAEL and is converted to products. Driven by the internal electric field and concentration difference, K+ cations can pass through the TPE from the anode to the cathode to provide the cation effect. This was verified by (bi)carbonate precipitation at 3 M KOH concentration (Supplementary Fig. 28).

a The Schematic of the membrane electrode assembly equipped with a TPE. b Comparison of CO2 SPC of pentyl-CuCP and Cu2O. c C2+ product partial current density obtained using pentyl-CuCP and Cu2O. d The FE of C2H4 using pentyl-CuCP in 7-h electrolysis at 180 mA cm−2. e FEC2H4 and full-cell voltage in CO2RR coupled with HzOR. The Cu content in the Cu2O control group was consistent with that in the pentyl-CuCP. The error bars in (b), (c), and (d) correspond to the standard deviations of three independent measurements. Source data are provided as a Source Data file.
With the optimized pentyl-CuCP electrode, we achieved an >80% total CO2RR single-pass conversion (SPC) and a ~ 54% SPC for C2+ products at a current density of 180 mA cm−2, surpassing the theoretical limits in alkaline and neutral CO2 electrolyzers36. The result also translates to a 2 × CO2 utilization improvement compared to carbon-supported Cu2O-derived Cu, of which the CO2 SPC to C2+ products is merely 25 ± 1% under the same condition (Fig. 4b). We achieved a ~ 150 mA cm−2 partial current density for total CO2RR and approximately 80 mA cm−2 for C2H4 (Fig. 4c). Detailed performance metrics are in Supplementary Tables 4–7. In 7-h CO2 electrolysis using the TPE membrane electrode assembly (TPE-MEA) at 180 mA cm−2, we saw a constant C2H4 FE of ~35% (Fig. 4d) and an average EtOH concentration of ~9% at the cathode (Supplementary Fig. 29). To reduce the full-cell voltage, we replaced the anodic oxygen evolution reaction (OER, standard potential 1.23 V vs. RHE) with the hydrazine oxidation reaction (HzOR, standard potential −0.33 V vs. RHE)14,37. We documented a full-cell voltage reduction of ~1 V across the applied current density range without compromising the C2H4 selectivity (Fig. 4e and Supplementary Table 8). Compared to the currently reported acidic CO2RR performances38,39,40,41, our CuCP catalyst in MEA achieves comparable C2+ product selectivity while further improving CO2 utilization (Supplementary Table 9).
In summary, this work develops a substituent tuning approach enhancing the performance of acidic CO2RR to C2+ products. By investigating the impact of the 2-position alkyl chain length of Cu-BIM CPs and their acidic CO2RR performance, we unveiled that longer substituents retard H+ diffusion and reduce the Cu-Cu CN, creating interface conditions beneficial for CO2RR to C2+ products. Compared to Cu2O-derived Cu and H-CuCP with the shortest 2-position substituent, pentyl-CuCP offers the highest C2+ FE of 73.4 ± 2.9% at 260 mA cm−2, and nonyl-CuCP increases the activity towards >2e− transfer products by 34 times. In an MEA reactor offering H+ flux by a TPE membrane, pentyl-CuCP breaks the conversion limit in alkaline and neutral electrolyzers and achieves a ~ 54% SPC for C2+ products and a total CO2 SPC of ~81%, together with a ~ 40% C2H4 FE and an average EtOH concentration of ~9%, at a current density of 180 mA cm−2. Our work highlights the promise of molecular design for achieving carbon-efficient and high-selectivity CO2 conversion to C2+ products.
Methods
Chemicals
Cuprous chloride (CuCl), potassium hydroxide (KOH), and potassium sulfate (K2SO4) were bought from Sigma Aldrich. Benzimidazole, 2-methylbenzimidazole, 2-propylbenzimidazole, titanium dioxide (TiO2), ferrocenemethanol (FcMeOH, 98%) and potassium chloride (KCl, 99.8%) were all purchased from Shanghai Macklin Biochemical Technology Co., Ltd. 2-pentylbenzimidazole, 2-heptylbenzimidazole and 2-nonylbenzimidazole were acquired from Sanjia Co., Ltd. Hydrochloric acid (HCl, 38%), sulfuric acid (H2SO4, 98%) and methanol were purchased from Qiangsheng Functional Chemical (Changshu) Co., Ltd. Potassium bicarbonate (KHCO3, ≥99.99%, metals basis) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. The glassy carbon films (10 × 10 × 2 mm3) were purchased from Wuhan Gaoss Union Technology Company, Ltd. Deionized (DI) water was used for all experiments. All commercial chemicals were utilized in their original state unless otherwise specified.
Synthesis of catalysts
Cu2O nanocrystals
In this experiment, copper(I) oxide was used both as a control group and as a precursor for the synthesis of copper coordination polymers (CuCPs). Firstly, 0.5 g of CuCl was dissolved in 2 M hydrochloric acid, and then 2 M KOH solution was added until no more yellow precipitate formed. Secondly, the supernatant was discarded, and the precipitate was washed three times with DI water and methanol. Finally, the precipitate was dried overnight in a vacuum oven at 50 °C.
Copper coordination polymers (CuCPs)
The synthesis procedure for all CuCP catalysts was identical. The preparation of pentyl-CuCP is described as an example.
Pentyl-CuCP catalysts
First, 0.072 g (0.0005 mol) of copper(I) oxide and 0.188 g (0.001 mol) of 2-pentylbenzimidazole were placed in a 350 ml glass bottle (the molar ratio of the substances was 1:1). Then, 200 ml of methanol was added, and the mixture was sonicated for one hour. Following sonication, the bottle was placed in an oven at 60 °C to react overnight. When the color of the precipitate in the bottle changed from yellow to gray-white, the reaction was considered complete. The supernatant was then decanted, and the precipitate was washed three times with methanol. Afterward, the precipitate was dried in an oven at 60 °C for 4 h.
Following the aforementioned method, H-CuCP, methyl-CuCP, propyl-CuCP, pentyl-CuCP, heptyl-CuCP, and nonyl-CuCP were synthesized.
Material characterizations
The scanning electron microscope (SEM) images were performed on a Zeiss G500 scanning electron microscope and a HITACHI SU8230 field-emission scanning electron microscope. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were captured using a field emission transmission electron microscope TALOS 200X operating at an accelerating voltage of 200 kV. The scanning transmission electron microscopy energy-dispersive X-ray spectroscopy (STEM-EDS) mapping images were acquired on TALOS 200X. The X-ray diffraction (XRD) patterns were collected on a PAN alytical X-ray diffractometer using Cu Kα radiation. The X-ray photoelectron spectroscopy (XPS) measurements were performed with a Thermo Scientific K-Alpha spectrometer equipped with an Al‒Kα X-ray source (1486.6 eV). The Fourier transform infrared spectroscopy (FTIR) measurements were conducted using an FTIR Spectrometer (V70 & Hyperion1000). Organic elemental analysis (EA) was carried out using an elementar unicube analyzer. Inductively coupled plasma optical emission spectrometry (ICP-OES) was performed with a Thermo Fisher iCAP PRO instrument. In-situ Raman spectroscopy measurements on all CuCPs were carried out by the Renishaw inVia Qontor Raman microscope in an open flow cell, using the 633 nm wavelength laser. In-situ X-ray absorption spectroscopy (XAS) measurements on prepared catalyst (Cu foil, Cu2O, CuO, H-CuCP, and pentyl-CuCP) were carried out in fluorescence mode at the Australian Synchrotron in Clayton, Victoria, Australia, using a multipole wiggler XAS beamline (12-ID).
Preparation of the electrodes
CuCP cathodes
In the AEM-MEA, 0.03 g of the CuCP catalyst, synthesized earlier, was combined with 0.01 g of acetylene black and 40 μL of an anion-exchange ionomer (Pention-D18, 5%, Xergy). This mixture was then subjected to ultrasonic agitation for an hour to create a homogeneous ink. Subsequently, this ink was evenly applied onto a 3 × 3 cm2 sheet of carbon paper (Sigracet 28BC) using a spray gun. After drying, the catalyst loading on the substrate was approximately 2 mg cm−2. In the TPE-MEA, the pentyl-CuCP electrode had a CP:carbon black ratio of 5:1, with a loading of 1 mg cm−2.
Cu2O cathodes
To match the Cu content (Supplementary Table 10), 12 mg of Cu2O was mixed with 0.01 g of acetylene black to ensure a catalyst active density consistent with that of CuCP. Additionally, 20 μL of Pention-D18 ionomer was blended into the mix. After ultrasonically agitating the combined ink for one hour, it was uniformly sprayed onto a 3 × 3 cm2 carbon paper (Sigracet 28BC) using a spray gun.
IrOx anodes
The preparation of the anode involved the deposition of iridium oxide (IrOx) onto a titanium substrate. The process was as follows: Initially, the titanium mesh (with a thickness of 0.002 inches, Fuel Cell Store) was cleaned with DI water and acetone. Subsequently, the cleaned titanium mesh was immersed in a 6 M HCl solution heated to 90 °C. A uniform solution was then prepared by dissolving 30 mg of IrCl3•xH2O in 10 mL of isopropanol containing 10% concentrated HCl. Following this, the etched titanium mesh was soaked in the IrCl3 solution for 10 min, removed, and then dried at 75 °C on a heating plate. Finally, the sample was calcined at 500 °C for 10 min. The final loading amount was approximately 2 mg cm−2.
NiFe anodes
A NiFe electrode was meticulously fabricated using an electrodeposition technique. This process involved immersing a 3 × 3 cm2 piece of pristine nickel foam (procured from Suzhou Suke Lean Instrument Co., Ltd.) in a solution containing 3 mM nickel nitrate hexahydrate and 3 mM iron nitrate nonahydrate, both from Sigma Aldrich. The nickel foam was adeptly connected as the working electrode. A platinum wire was employed as the counter electrode, and Ag/AgCl was utilized as the reference electrode. A voltage of −1 V vs. Ag/AgCl was applied for 5 min to synthesize the NiFeOx catalyst.
Treatments of ion exchange membranes
Pre-treatment of ion exchange membranes
The anion exchange membranes (Sustainion X37-50 Grade 60 and FAA-3−20 (Fuel Cell Store)) were both initially activated in 1 M KOH for 48 h. Additionally, the bipolar membrane (Fumasep FBM-PK, Fuel Cell Store) was activated in 1 M NaCl for 24 h.
Assembly of a TPE
A mixture of 10 mg TiO2 and 10 μL Nafion ionomer was prepared in 5 mL methanol and evenly sprayed onto cation exchange membranes (Nafion NR212, Chemours), sized 3 × 3 cm2 with a catalyst loading of 1 mg cm−2. The treated Nafion membrane was then adhered to one side of the pre-treated Sustainion membrane. The FAA-3−20 anion exchange membrane was rinsed with DI water to remove surface KOH, and small holes were punctured using a needle within the MEA rubber sealing ring. The membrane was then aligned and attached to the cation exchange side of the bipolar membrane, with air bubbles removed for a smooth surface. Finally, the assembled membrane was immersed in DI water to maintain hydration.
Electrochemical measurement
Linear sweep voltammetry (LSV)
In the LSV experiment, a three-electrode system was used, consisting of an Ag/AgCl reference electrode, a platinum wire counter electrode, and a gas diffusion electrode (GDE) with dimensions of 1 × 1 cm2, prepared according to the CuCP cathode method. The electrolyte used was 0.1 M sulfuric acid, which was purged with argon gas for 20 min prior to testing. The sweep rate during the experiment was set to 10 mV s−1.
Cyclic voltammetry (CV)
The same setup in LSV measurements was used, and the electrolyte was 0.1 M KHCO3.
AEM-MEA measurements
The MEA tests involved a mixed anolyte of 0.01 M sulfuric acid and 0.1 M potassium sulfate, using an IrO2 anode for the OER and an anion exchange membrane (Sustainion X37-50 Grade 60). The cathode gas flow was controlled with a digital mass flow controller (CS200A, from Seven Star Fluid (Beijing) Co., Ltd.), set at a CO2 flow rate of 30 standard cubic centimeters per minute (sccm). The anolyte was circulated at 60 ml/min, managed by a peristaltic pump. Electrochemical testing utilized a chronopotentiometry (V-t) method, operated by an electrochemical workstation from Donghua Analytical Instruments (Taizhou) Co., Ltd.
H-cell measurements
The testing catalyst electrodes were the same as described previously, with an Ag/AgCl reference electrode, a platinum wire counter electrode, and a graphite rod as the anode. The catalyst electrode area was 1 cm2. The electrolyte for both the cathode and anode consisted of 45 mL of 0.01 M H2SO4 and 0.5 M K2SO4. The CO2 flow rate was set to 40 sccm.
TPE-MEA measurements
The ion-exchange membrane employed was a homemade TPE membrane, with 1 M KOH solution as the anolyte, and a NiFe electrode as the anode for the OER. The cathodes were Cu2O and pentyl-CuCP. This phase included testing the single-pass conversion efficiency (SPC) of CO2, with the CO2 inlet flow rate being regulated using a gas flow meter. The electrochemical assessments continued using the constant current method with the electrochemical workstation. The CO2 flow rate was set to 20 sccm, and both the electrode plate and electrolyte were heated to 40 °C.
CO2RR coupled HzOR in TPE-MEA
The electrolyzer used was a TPE-MEA. The anolyte comprised 100 mL of 1 M KOH and 0.5 M hydrazine. The test electrode and conditions were consistent with previous experiments.
In all experiments, new materials were consistently used, and each test was replicated a minimum of three times for accuracy and to reduce potential errors. The gases produced at the cathode exit were subjected to analysis through a real-time gas chromatograph (GC2060, Volvo Information Technology (Shanghai) Co., Ltd.), which was fitted with both a Thermal Conductivity Detector (TCD) and a Flame Ionization Detector (FID). Additionally, liquid samples extracted from the electrolyte were analyzed using a high-performance liquid chromatograph (Vanquish Core, ThermoFisher) to identify the presence of liquid by-products.
The Faradaic efficiency (FE) of the gas product was calculated based on the following equation:
Here, i represents the total current, \({n}_{x}\) denotes the number of electrons transferred for producing 1 mole of product x, \({V}_{{gas}}\) is the flow rate of carbon dioxide (measured in sccm), \({c}_{x}\) represents the concentration of product x detected by gas chromatography (measured in ppm), F is the Faraday constant (96,485 C mol-1), and \({V}_{m}\) is the molar volume, which is 24.5 L mol−1 at room temperature (298.15 K).
The FE of the liquid product was calculated as follows:
where \({m}_{x}\) is the amount of liquid products in moles, and \(t\) is the duration of the measurement.
The partial current density for a given product was calculated as:
The CO2 single-pass conversion efficiency (SPC) was determined by calculating the ratio of the amount of CO2 that was transformed into the products to the amount of CO2 that entered into the MEA. The CO2 SPC was computed as follows:
The percentage concentration of EtOH (%EtOH) was determined by calculating the ratio of the mass of EtOH and the cathodic droplet:
Where cEtOH is the molar concentration of EtOH obtained by HPLC, Vcathode is the volume of liquid receiving the cathode tail gas, mcathode,i is the initial mass of the receiving liquid, and mcathode,t is the final mass of the receiving liquid.
All potentials vs. the Ag/AgCl electrode were converted to the potentials vs. RHE based on the following equation:
Scanning electrochemical microscopy (SECM)
Substrate preparation
To prepare the catalyst-coated electrode, 2 mg of the catalyst was dissolved in 1 mL of methanol. Then, this solution was sprayed onto the surface of a glassy carbon electrode, which was previously cleaned with methanol under ultrasonication and dried. After spraying, the electrode was heated at 50 °C to dry it.
SECM measurements
The SECM measurements were collected using a CHI 920D SECM from CH Instrument in a four-electrode setup. A platinum ultramicroelectrode (Pt-UME) (12.5 μm radius) with an RG of approximately 3 was applied as a working electrode, and spin-coated thin films of H-CuCP and Pentyl-CuCP on glassy carbon surfaces were used as target substrate electrodes to determine their stability changes before and after electrocatalytic carbon dioxide reduction reaction (CO2RR). The Ag/AgCl (3 M KCl) and Pt wire were used as the reference and the counter electrode, respectively. 0.1 M KHCO3 aqueous solution with 0.5 mM FcMeOH was applied as the test solution. The substrates were sealed inside a homemade Teflon cell with an exposed area of 0.20 cm2 (0.5 cm radius). During the test, the Pt-UME was placed at a 10-μm distance above the substrate. Pt-UME was held at 0.5 V vs. Ag/AgCl to oxidize FcMeOH. The substrate was held at open circuit potential (OCP) (~0.1 V vs. Ag/AgCl) to locally regenerate the reduced form. SECM feedback images of Pentyl-CuCP and H-CuCP substrate before and after CO2RR were obtained. The feedback current change reflects the variation in local electron transfer activity.
EXAFS data analysis
EXAFS data was analyzed using Demeter software (version 0.9.26.2) with Athena for energy calibration, spectral normalization and linear combination fitting (Cu foil as the standard). The EXAFS function was converted from energy space to k-space, where k represents the photoelectron wave vector. To enhance the EXAFS oscillations in the mid-k region, χ(k) was scaled by k². The Fourier transformation of the k2-weighted χ(k) from k to R space over a k range of 3–12 Å−1 differentiated Cu K-edge EXAFS oscillations across various coordination shells. EXAFS data in R space was then fitted using Artemis within Demeter, alongside the FEFF6 program. FEFF calculated phase and amplitude functions for Cu-O, Cu-N, and Cu-Cu interactions. This was followed by standard EXAFS fitting to derive the structure of the Cu samples.
In-situ Raman studies
In-situ Raman measurements under both near-neutral and acidic conditions were conducted in an open flow cell. The reference electrode was Ag/AgCl, the counter electrode was a Pt wire, and the working electrode area was 1 cm2. The near-neutral electrolyte was 0.1 M KHCO3 (pH ~8.3), and the acidic electrolyte was 0.01 M H2SO4 (pH ~1.7). The laser wavelength used was 633 nm.
Computational methods
All the density functional theory (DFT) calculations were implemented by the Vienna Ab initio Simulation Package (VASP). The electron exchange and correlation energies were handled using the Perdew-Burke-Ernzerhof (PBE) functionals. The projector-augmented wave (PAW) potentials were used to describe the interactions between the cores and valence electrons. The expansion of the Kohn-Sham valence states was carried out with a 400 eV plane-wave cut-off energy. For Brillouin zone integration, 5 × 5 × 15 Γ-centered Monkhorst–Pack grids were performed for H-CuCP, pentyl-CuCP, and nonyl-CuCP. The convergence criterion of structure optimization for energy and force were set as 10−4 eV and −0.05 eV Å−1, respectively. The overflow structure and energy barrier of Cu in the CuCP are calculated through the transition state searching method of climbing image-nudged elastic band (CI-NEB). The highest energy climbing point in the intermediate interpolation image is the structure corresponding to the transition state. The difference between the highest energy point and the initial energy of this stage is the reaction energy barrier. For the formation mechanism of the Cu2 on H-CuCP, pentyl-CuCP, and nonyl-CuCP, 5 intermediate images are set to discretize the reaction pathway during the CI-NEB calculation.
Data availability
All data generated in this study are provided in the Supplementary Information/Source data file. Source data are provided in this paper. Source data are provided with this paper.
References
Endrődi, B. et al. Continuous-flow electroreduction of carbon dioxide. Prog. Energy Combust. Sci. 62, 133–154 (2017).
Wang, Z. et al. Advanced catalyst design and reactor configuration upgrade in electrochemical carbon dioxide conversion. Adv. Mater. 35, 2303052 (2023).
Sun, B. et al. Challenges and strategies towards copper-based catalysts for enhanced electrochemical CO2 reduction to multi-carbon products. Fuel 332, 126114 (2023).
Woldu, A. R. et al. Electrochemical CO2 reduction (CO2RR) to multi-carbon products over copper-based catalysts. Coord. Chem. Rev. 454, 214340 (2022).
Zhuansun, M. et al. Promoting CO2 electroreduction to multi-carbon products by hydrophobicity-induced electro-kinetic retardation. Angew. Chem. Int. Ed. 62, e202309875 (2023).
Fang, M. et al. Hydrophobic, ultrastable Cuδ+ for robust CO2 electroreduction to C2 products at ampere-current levels. J. Am. Chem. Soc. 145, 11323–11332 (2023).
Qiu, X.-F. et al. A stable and conductive covalent organic framework with isolated active sites for highly selective electroreduction of carbon dioxide to acetate. Angew. Chem. Int. Ed. 61, e202206470 (2022).
Endrődi, B. et al. Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolysers. Nat. Energy 6, 439–448 (2021).
Dean, J. A. Lange’s Handbook of Chemistry. (McGraw-Hill Education, 1978)
Rabinowitz, J. A. et al. The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nat. Commun. 11, 5231 (2020).
Gabardo, C. M. et al. Continuous carbon dioxide electroreduction to concentrated multi-carbon products using a membrane electrode assembly. Joule 3, 2777–2791 (2019).
Ozden, A. et al. Cascade CO2 electroreduction enables efficient carbonate-free production of ethylene. Joule 5, 706–719 (2021).
Pan, B. et al. Close to 90% single-pass conversion efficiency for CO2 electroreduction in an acid-fed membrane electrode assembly. ACS Energy Lett. 7, 4224–4231 (2022).
Fang, W. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).
Gu, J. et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 5, 268–276 (2022).
Li, J. et al. Electrocatalytic carbon dioxide reduction in acid. Chem. Catal. 2, 29–38 (2022).
Ling, N. et al. Acidic media impedes tandem catalysis reaction pathways in electrochemical CO2 reduction. Angew. Chem. Int. Ed. 62, e202308782 (2023).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Liu, L. et al. The applications and prospects of hydrophobic metal–organic frameworks in catalysis. Dalton Trans. 50, 39–58 (2021).
Liu, S. et al. Proton-filtering covalent organic frameworks with superior nitrogen penetration flux promote ambient ammonia synthesis. Nat. Catal. 4, 322–331 (2021).
Liang, Y. et al. Stabilizing copper sites in coordination polymers toward efficient electrochemical C-C coupling. Nat. Commun. 14, 474 (2023).
Nam, D.-H. et al. Metal-organic frameworks mediate Cu coordination for selective CO2 electroreduction. J. Am. Chem. Soc. 140, 11378–11386 (2018).
Mikami, K. et al. Synthesis of Cu2O/CuO nanocrystals and their application to H2S sensing. Sensors 19, 211 (2019).
Mohan, S. et al. FTIR and Raman studies on benzimidazole. Spectrochim. Acta A Mol. Biomol. Spectrosc. 47, 1111–1115 (1991).
Pashchevskaya, N. V. et al. Effect of the condition of synthesis on the composition and structure of Copper(II) complexes with benzimidazole. Russ. J. Inorg. Chem. 55, 1425–1432 (2010).
Luo, M. et al. Coordination polymer electrocatalysts enable efficient CO-to-acetate conversion. Adv. Mater. 35, 2209567 (2023).
Eilert, A. et al. Subsurface oxygen in oxide-derived copper electrocatalysts for carbon dioxide reduction. J. Phys. Chem. Lett. 8, 285–290 (2017).
Wei, C. et al. Lattice oxygen-mediated electron tuning promotes electrochemical hydrogenation of acetonitrile on copper catalysts. Nat. Commun. 14, 3847 (2023).
El-Nagar, G. A. et al. Unintended cation crossover influences CO2 reduction selectivity in Cu-based zero-gap electrolysers. Nat. Commun. 14, 2062 (2023).
Sassenburg, M. et al. Zero-gap electrochemical CO2 reduction cells: challenges and operational strategies for prevention of salt precipitation. ACS Energy Lett. 8, 321–331 (2023).
Garg, S. et al. How membrane characteristics influence the performance of CO2 and CO electrolysis. Energy Environ. Sci. 15, 4440–4469 (2022).
Yao, K. et al. Mechanistic insights into OC–COH coupling in CO2 electroreduction on fragmented copper. J. Am. Chem. Soc. 144, 14005–14011 (2022).
Xu, Y. et al. Low coordination number copper catalysts for electrochemical CO2 methanation in a membrane electrode assembly. Nat. Commun. 12, 2932 (2021).
Zhao, Z. et al. Generalized surface coordination number as an activity descriptor for CO2 reduction on Cu surfaces. J. Phys. Chem. C. 120, 28125–28130 (2016).
Dai, Y. et al. Manipulating local coordination of copper single atom catalyst enables efficient CO2-to-CH4 conversion. Nat. Commun. 14, 3382 (2023).
Zhang, Y. et al. Coordination environment dependent selectivity of single-site-Cu enriched crystalline porous catalysts in CO2 reduction to CH4. Nat. Commun. 12, 6390 (2021).
Wang, T. et al. Progress in hydrogen production coupled with electrochemical oxidation of small molecules. Angew. Chem. Int. Ed. 61, e202213328 (2022).
Alkayyali, T. et al. Direct membrane deposition for CO2 electrolysis. ACS Energy Lett. 8, 4674–4683 (2023).
Perazio, A. et al. Acidic electroreduction of CO2 to multi-carbon products with CO2 recovery and recycling from carbonate. ACS Energy Lett. 8, 2979–2985 (2023).
Ma, Z. et al. CO2 electroreduction to multicarbon products in strongly acidic electrolyte via synergistically modulating the local microenvironment. Nat. Commun. 13, 7596 (2022).
Xie, K. et al. Bipolar membrane electrolyzers enable high single-pass CO2 electroreduction to multicarbon products. Nat. Commun. 13, 3609 (2022).
Acknowledgements
Y.W. acknowledges National Natural Science Foundation of China (22179088), the Natural Science Foundation of Jiangsu Province of China (BK20210699), the National Natural Science Fund for Excellent Young Scientists Fund Program (Overseas), the Program for Jiangsu Specially-Appointed Professors, the Program of Soochow Innovation and Entrepreneurship Leading Talents (ZXL2022450), the start-up supports of Soochow University, Suzhou Key Laboratory of Functional Nano & Soft Materials, the Collaborative Innovation Center of Suzhou Nano Science & Technology, and the 111 Project. J.H. acknowledges National Natural Science Foundation of China (22204115) and the Natural Science Foundation of Jiangsu Province of China (BK20220485). Y.S. acknowledges the “Young Talent Support Plan” of Xi’an Jiaotong University, and supercomputing facilities were provided by Hefei Advanced Computing Center and Computing Center in Xi’an. F.L. acknowledges the ARC Centre of Excellence for Green Electrochemical Transformation of Carbon Dioxide (CE230100017) funded by the Australian Government and the X-ray Absorption Spectroscopy (XAS) Beamlines at the Australian Synchrotron and part of ANSTO (M19559 and M20139). The authors thank Prof. Yanguang Li for providing the H-cell testing apparatus.
Author information
Authors and Affiliations
Contributions
Y.W., J.H., and Y.S. conceived the idea and supervised the project. H.D. designed and performed the experiments, and wrote the manuscript. T.L. performed the SECM tests. W.Z. performed the DFT calculations. J.W., Y.Z., W.T., and Z.C. provided help with the testing of the CO2RR. S.Z. performed the XAS tests. C.Y. assisted in the fitting of the EXAFS. Y.Y. provided XRD refinement suggestions. G.Z. and F.L. provided help with the manuscript writing. All authors discussed, commented on, and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xu Lu, Hongwen Huang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deng, H., Liu, T., Zhao, W. et al. Substituent tuning of Cu coordination polymers enables carbon-efficient CO2 electroreduction to multi-carbon products. Nat Commun 15, 9706 (2024). https://doi.org/10.1038/s41467-024-54107-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54107-2
This article is cited by
-
The promises and reality of metal–CO2 batteries
Nature Reviews Clean Technology (2025)
-
Selective C–C coupling via copper atom reconfiguration in CO2 electroreduction
Frontiers of Chemical Science and Engineering (2025)
