
Abstract
In the era of synthetic biology, design, construction, and utilization of synthetic chromosomes with unique features provide a strategy to study complex cellular processes such as aging. Herein, we successfully construct the 884 Kb synXIII of Saccharomyces cerevisiae to investigate replicative aging using these synthetic strains. We verify that up-regulation of a rRNA-related transcriptional factor, RRN9, positively influence replicative lifespan. Using SCRaMbLE system that enables inducible whole-genome rearrangement on synXIII, we obtain 20 SCRaMbLEd synXIII strains with extended lifespan. Transcriptome analysis reveal the expression of genes involve in global protein synthesis is up-regulated in longer-lived strains. We establish causal links between genotypic change and the long-lived phenotype via reconstruction of some key structural variations observed in post-SCRaMbLE strains and further demonstrate combinatorial effects of multiple aging regulators on lifespan extension. Our findings underscore the potential of synthetic yeasts in unveiling the function of aging-related genes.
Similar content being viewed by others

Introduction
With the continuous advancements in DNA editing and synthesis technologies, synthetic biology is revolutionizing our way to study, learn, and manipulate model organisms. The Synthetic Yeast Genome Project (Sc2.0) serves as a pioneering endeavor to construct the eukaryotic synthetic genome, aiming to develop a designed yeast genome with properties. Employing the iterative “design-build-test-learn” cycle, significant progress has been made in completing most chromosomes while gaining valuable insights into eukaryotic genome design, assembly methodologies, debugging strategies, and the intricate relationship between transcriptional regulation and 3D genome organization1,2,3,4,5,6,7,8,9,10,11,12. The introduction of a plethora of designers in the Sc2.0 genome has significantly enhanced the versatility of yeast, enabling synthetic chromosomes to be utilized for optimization of metabolic pathways13,14,15,16,17. However, their potential application in studying complex biological processes such as aging remains relatively unexplored. Meanwhile, as the integration of individual synthetic chromosomes into cells progresses towards completion, numerous opportunities for further investigation and discovery still lie ahead.
Aging, a universal trait observed across the evolutionary spectrum, is recognized as a critical risk factor for numerous human pathologies including Alzheimer’s disease, Parkinson’s disease, tumor progression, and cardiovascular dysfunction18,19. The intricate nature of potential interacting networks poses significant challenges in aging studies. Saccharomyces cerevisiae (budding yeast) has emerged as an influential chemical and genetic screening platform that is extensively employed as a genetic model to explore the mechanisms underlying aging due to its conservation of genes and pathways shared with humans20,21,22,23. However, despite the identification of well-known aging genes and pathways in yeast through genome-scale knockout or over-expression screening, it becomes even more important to analyze the co-effect of multiple genes on aging due to its complex nature involving various layers. Recently Zhou et al. successfully extended yeast lifespan by 82% through the co-regulation of HAP4 and SIR2 expression, surpassing the lifespan-extension rate achieved by any currently existing single-gene mutants24,25. Therefore, the generation of combinatorial mutant models for investigating yeast aging presents a substantial scientific value26,27, albeit accompanied by challenges to explore the very large combinatorial genetic space. As a distinctive feature of Sc2.0, the SCRaMbLE (Synthetic Chromosome Rearrangement and Modification by LoxPsym-mediated Evolution) system induces inversions, deletions, or duplications upon Cre recombinase induction occurs. The application of SCRaMbLE to synIXR (43 LoxPsym sites) resulted in the identification of 64 unique SCRaMbLEd strains containing 156 deletions, 89 inversions, 94 duplications, and 55 additional complex rearrangements28. This demonstrates that SCRaMbLE has the capability to generate diverse combinations through stochastic processes in synthetic chromosomes. Furthermore, this powerful tool also revealed functions for genomic and cell biology discovery using synthetic chromosomes. Given that many aging-related genes are located on synXIII and there are embedded 333 loxPsym sites, we hypothesize that the synthesis of Sc2.0 yeast synXIII provides a unique material for studying aging research purposes.
In this work, together with efforts of Sc2.0 consortium to build a eukaryotic organism with a synthetic genome, we described the design, construction, and characterization of an 883,749 bp synXIII. Despite the incorporation of thousands of designer features, synXIII strain exhibits near wild-type fitness and replicative lifespan compared to its wild-type counterpart under various growth conditions. Utilizing the unique resource of intermediate synthetic XIII strains, we found that an intentional design feature (synonymous codon replacement by PCRTag) in RRN9 gene resulted in an increase of its expression level and lifespan extension in the corresponding strain. We further deployed a HSP104 based reporter integrated into the synXIII strain and used the built-in SCRaMbLE system to generate a massive pool of mutant strains including those with extended lifespan. We identified 20 SCRaMbLEd long-lived yeast with various chromosomal rearrangements specific on synXIII. Global transcriptome analysis uncovered long-lived strains exhibiting up-regulation of genes involved in ribosomes, nucleolus, cytoplasmic translation and rRNA processing. Subsequently, whole-genome sequencing, coupled with the utilization of a microfluidic device and a time-lapse microscopy-based assay for replicative aging, unveiled six genes that could potentially play crucial roles in lifespan changes. In our investigation of the long-lived strain Ycz315, we identified multiple variants that potentially contribute to lifespan extension. Our study demonstrated that the Sc2.0 yeast and its built-in SCRaMbLE system could serve as a unique model system for studying aging
Results
Design and construction of synXIII
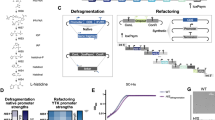
Based on the Sc2.0 project principles10, we designed and constructed the 883,749 bp synthetic chromosome XIII (synXIII) (Fig. 1a, Supplementary Table), containing 333 loxPsym sites insertion (loxPsym sequences are 34-bp nondirectional loxP sites capable of recombining in either orientation) and 68 deletions, a total of 100 TAG to TAA conversions, and introduction of 629 synthetic PCRtag pairs. According to the design principle, all tRNA genes on chromosome XIII were relocated onto the tRNA neochromosome29,30. However, one tRNA gene, tQ(CUG)M, is essential to maintain during the construction (Supplementary Fig. 1a). Thus, we relocated it to chrVI at this stage (Supplementary Fig. 1b) and it could be eliminated once the synXIII strain is consolidated into the final synthetic yeast strain carrying the tRNA neochromosome30. Overall, based on the design, synXIII is condensed by 4.4% in comparison with its native sequence.

a Overview of synXIII construction with the indicated designer features. The gray reprensents the native chromosome XIII, and the orangered represents the synthetic chromosome; The right table listed the difference between the chrXIII, synXIII and final constructed synXIII. b Native chromosome XIII replacement with synthetic chunks. On average 6~7 chunks were used to replace the native segments of chromosome XIII (gray). 9 and 6 step-by-step replacements were carried out in parallel from the left arm and right arm respectively with iterative selectable markers (LEU2 or URA3) through homologous recombination in yeast with homologous regions around 500 bp ~1 kb junctions (blue) produced by PCR using ligated chunks as template was designed to have 500 bp overlapping regions with two adjacent chunks to improve the replacement efficiency. I-SceI mediated synXIIIA-I and synXIIIJ-O integration. An I-SceI site (yellow) was introduced on both semi-synthetic chromosomes. After mating, I-SceI induction was performed to generate double strain breaks on both semi-synthetic chromosomes. 30 kb homologous region on both semi-synthetic chromosomes enabled the integration of complete synXIII.
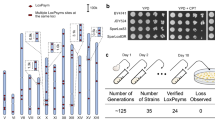
To speed up construction of synXIII, we adopt a hierarchical assembly strategy (Supplementary Fig. 2a)31. The minichunks were assembled to chunks in vitro using Gibson assembly strategy32. Adjacent minichunks share 40 base pairs of sequence identity, enabling the incorporation of 6 to 7 minichunks into DNA chunks ranging from 10 to 15 kilobases. This “one-pot” in vitro approach facilitates both minichunk digestion and chunk assembly, exhibiting comparable efficiency to purified minichunk assembly. The integrations were performed from both ends in vivo—along with two wild-type strains (BY4741 and BY4742) in parallel. The native chromosome was replaced by 6~7 chunks as a megachunk using the standard SwAP-In method33. Adjacent megachunks contained homologous fragments ranging from 500 to 800 bp, with the largest megachunk reaching up to 70 kb. Consequently, the entire synthetic chromosome XIII was efficiently divided into only 15 megachunks, thereby significantly reducing integration time. In our construction of an 884 kb chromosome, 9/6 rounds (A-I/J-O) transformation were carried out in BY4741/BY4742 respectively to generate left/right semi-synthetic strains of synXIII-I/J, and subsequently using meiotic recombination–mediated assembly (MAR) and I-SceI mediated chromosome integration strategies to combine two semi-synthetic chromosomes, resulting in the complete synthetic chromosome XIII (Fig. 1b). Karyotyping analysis of the strain revealed aberrations on chromosomes V and VIII, which were subsequently rectified through back-cross breeding with synXIII-I to obtain the final synXIII strain (strain ID: Ycz020) (Supplementary Fig. 2b). The synXIII was finally confirmed by whole genome sequencing (WGS) and revealed several “patchwork” variations produced by homologous recombination (Fig. 2a, Supplementary Fig. 2c and Supplementary Data 1), as reported in other finished synthetic chromosomes31,34.

a Scanning Electron Microscopy (SEM) images of BY4741 and synXIII strains. Scale bars: 5.00 μm. Source data are provided as a Source data file. b Growth curve analysis of BY4741 and synXIII. Each strain was analyzed with three biological replicates (n = 3), and each biological replicate consisted of three technical replicates. The error bands represent a total of nine replicates at each time point. Data are presented as mean values ±SD. Source data are provided as a Source data file c Doubling times of corresponding strains in YPD medium, each strain was conducted with three replicates (n = 3). Each replicate represents the mean doubling time of three technical replicates. Data are presented as mean values ±SD from three independent replicates. Source data are provided as a Source data file. d Cellular viability responses of synXIII upon exposure to normal and stress conditions. 10-fold serial dilutions of overnight cultures of synXIII and wild-type (BY4741 and BY4742) strains were used for plating. Conditions include: YPD at 30 °C; SC at 30 °C; YPD + Benomyl; YPD + H2O2 (1 mM, 2 h pretreatment); YPD + methyl methane sulfone (MMS, 0.01% v/v); YPD + Camptothecin (0.1 μg/mL); YPD + Rapamycin (0.2 μg/mL); YPD + Cycloheximide (10 μg/mL, 2 h pretreatment); YPD + Sorbitol (1 M); YPD, yeast extract peptone dextrose; YPEG, yeast extract peptone glycerol ethanol; SC, synthetic complete. e Survive curves of synXIII and BY4741. X-axis presents the number of daughter cells generated from the mother cell. Each strain was measured with three replicates (n = 3) and 40 cells were monitored for each replicate. Source data are provided as a Source data file. f Identified dysregulated genes of synXIII strain at the transcriptome level compared with BY4741. The P value was calculated by DESeq2 (v1.30.1). The differentially expressed genes were assessed at genome-wide significance P < 7.56 × 10−6 for 5% family-wise error rate based on 6607 genes with at least one mapped read and also corresponding to log2 (foldchange) ≥1 or ≤−1. Source data are provided as a Source data file. g Identified dysregulated genetic features of synXIII strain at the proteome level compared with BY474. The P value was calculated by IQuant (v2.0.1) in two-sided T-test. Source data are provided as a Source data file. f, g the up and down-regulated genes are labeled in red and green respectively.
Characterization of synXIII
To characterize synXIII, we carried out a series of phenotypic analyses to validate its fitness compared with the native counterparts (BY4741 and BY4742). The synXIII and native strain were visually indistinguishable under a scanning electron microscope (Fig. 2a). The synXIII strain exhibited a highly similar growth pattern under various growth conditions compared with the wild-type BY4741 and BY4742 strains (Fig. 2b–d, Supplementary Figs. 3 and 4). We also determined whether the sequence change made to chromosome XIII would affect the replicative lifespan by performing the lifespan measurement on both wild-type and synXIII strains. By employing a U-shape, high-pressure microfluidic chip previously developed in our lab (Supplementary Fig. 6a), we measured the replicative lifespan of synXIII in comparison with BY4741 and revealed a similar survival curve between these two strains (Fig. 2c). To analyze the cellular and molecular physiology of the synXIII strain, we explored the potential effect from the synthetic chromosome on the transcriptome and proteome. Transcriptome profiling revealed 10 differentially expressed genes (DEGs) out of 6606 ones, comprising three genes up-regulated and seven genes down-regulated (Fig. 2f). Proteomics analysis identified eight deferentially expressed proteins in synXIII compared to the native strain (Fig. 2g). Overall, our results demonstrate that synXIII has minimal effect on phenotypic fitness and cell physiology, it also displays a replicative lifespan that is comparable to that of the wild-type strain.
Identification of an aging related “bug” by using synthetic intermediate strains
During the progressive construction of synXIII, we observed that the intermediate strain synXIII-N shows an extended lifespan compared to its parental intermediate stain synXIII-O (Fig. 3a). By individually integrating each chunk of megachunk N into synXIII-O for lifespan measurement, we found that the synthetic N2 chunk integration strain showed an obvious increased lifespan relative to synXIII-O. Next, we further identified the modifications introduced in the RRN9 gene is responsible for the increased lifespan (Fig. 3b). The synthetic PCRTag is known to occasionally cause variations in expression levels and growth defect3. Thus, we introduced both synthetic and native versions of RRN9 gene (synRRN9 and wtRRN9) into the synXIII-O strain for RT-PCR analysis and found the mRNA level of synRRN9 was significantly up-regulated compared to that of wtRRN9 (Fig. 3c).

a Lifespan measurement of intermediate strains with three replicates (n = 3), synXIII-O and synXIII-N. The arrow presents the direction of construction. Source data are provided as a Source data file. b The lifespan measurement of all intermediate strains of synthetic megachunk N (highlight in orange) compared to synXIII-O. Synthetic megachunk N was segmented into N1~N6 chunks, which was integrated respectively using synXIII-O as the host strain. N2 chunk carried six synthetic genes (RSN2, PPA2, PRP24, TMA23, RRN9, URA10) which were integrated in the synXIII-O host strain respectively. Data are presented as mean values ±SD from three independent replicates (n = 3). Source data are provided as a Source data file. c The expression level of synthetic and native RRN9 gene measured by RT-PCR (normaried to native RRN9 in synXIII-O). The red dashed line represents the expression level of native RRN9 gene in BY4741. Each strain was conducted with three replicates (n = 3). Each replicate represents the expression level of three technical replicates. Data are presented as mean values ±SD from three independent experiments. The blue and gray bars present the synthetic and native PCRTags respectively. Source data are provided as a Source data file. d Dissection of the effect of synonymous recoding by PCRTags in RRN9 gene to its gene expression measured by RT-PCR, each strain was conducted with three replicates (n = 3). Each replicate represents the expression level of three technical replicates. Data are presented as mean values ±SD from three independent replicates. The synthetic and wildtype PCRTags are presented in blue and gray respectively. The right panel presents the corresponding sequence of synthetic and wildtype PCRtags. Source data are provided as a Source data file. e Replicative aging measurement of synXIII-O and BY4741 strains with wildtype and synthetic PCRTag2 with three independent replicates (n = 3). Data are presented as mean values ±SD from three independent replicates. Source data are provided as a Source data file. f Survival curves of the strains with increased doses of RRN9. The Ycz402 and Ycz403 strains express pRS416 (centromere-based empty vector) and pRS416-RRN9 in BY4741 respectively. Each lifespan measurement was performed with three replicates (n = 3) and 40 cells were monitored for each replicate. The statistical confidence is calculated by one-sided T-test, and error bars represent standard division. Source data are provided as a Source data file.
In the design of synXIII, RRN9 is synonymously recoded with two synthetic PCRtags. To determine which PCRTag was responsible for the increased mRNA abundance of synRRN9, we individually introduced the two PCRtags to synXIII-O strain. We found that the insertion of SYNtag2 was sufficient to cause increase mRNA level of RRN9 in synXIII-O (Fig. 3d). Meanwhile, the abundance of Rrn9 protein in the synXIII-O strain with SYNtag2 was found to be increased by 20% in comparison with that of synXIII-O strain with WTtag2 (Supplementary Fig. 5a). This could be partially explained by a mild increase in RNAPII occupancy via ChIP-qPCR9,35 in the strain carrying SYNtag2 in comparison with the strain carrying WTtag2 (Supplementary Fig. 5b).
In order to confirm the solo effect of SYNtag2 on replicative lifespan, we specifically introduced the SYNtag2 sequence into the genome of BY4741 and synXIII-O strains respectively. We found this single genomic change could significantly increase the replicative lifespan of both strains (Fig. 3e), suggesting that the SYNtag2 is responsible for the observed lifespan extension. To further validate the positive impact of enhanced RRN9 on extending lifespan, we augmented the copy number of RRN9 in the haploid BY4741 strain by introducing an additional copy of the RRN9 gene on a centromeric plasmid with medium copy number. Remarkably, this manipulation also resulted in a significant 37.5% increase in replicative lifespan (Fig. 3f, P value < 0.0001, P values were determined by two-sided, unpaired Student’s t test). Overall, we demonstrated that a single design feature would have great impact on yeast lifespan and Sc2.0 intermediate strains might serve as a valuable resource to identify key gene related to aging.
Leveraging SCRaMbLE system of synXIII to obtain long-lived strains
Given that 328 loxPsym sites were incorporated into synXIII, the induction of the SCRaMbLE system would generate a massive mutant library derived from Sc2.0 yeast and allow us to potentially screen for long-lived strains. To rapidly screen long-lived mutants from the pool, we adopted our previously reported method that use HSP104-eGFP as a reporter in single yeast cells36. Specifically, the long-lived strains exhibit reduced fluorescence and could be identified by fluorescence-activated cell sorting (FACS). To verify whether the HSP104-eGFP reporter is applicable across various yeast mutants, we measured both lifespan and HSP104-eGFP level in a series of well-studied mutants, including the well-known short-lived yeast (ted1Δ)37 and long-lived yeast (fob1Δ)38 as internal controls (Supplementary Fig. 6b, 6c). We observed a negative correlation between lifespan and the HSP104-eGFP level, suggesting that the HSP104-eGFP reporter system could be used to screen for long-lived strains. In addition, an auxotrophic markerURA3, was integrated between two loxPsym sites tagging YMR011W and YMR012W on synXIII (270,117~270,525 bp) to ensure a powerful positive selection for SCRaMbLEants from the library. To validate our approach, we developed a SCRaMbLE-mediated screening workflow to pinpoint strains with an augmented replicative lifespan through inducible genome rearrangements of synXIII (Fig. 4a). We initially obtained total 28 SCRaMbLEants and further identified 20 long-lived strains verified by microfluidic chip. Compared with the parental synXIII strain, these mutant strains exhibited different lifespan-extension rate ranging from 20.3% to 42.5% (Fig. 4b). Thus, we showed the inherent characteristics of synXIII provide a foundation to build a promising and easily accessible platform for efficiently screening long-lived strains.

a Schematic illustration of SCRaMbLE and screening process for long replicative aging strains. The HSP104 fused to eGFP (HSP104-eGFP) acts as a reporter for replicative lifespan in yeast. The URA3 reporter was randomly integrated into regions positioned between two loxPsym sites allowing for SCRaMbLEant selection by 5-FOA. SCRaMbLEants with lower fluorescence intensity were selected by FACS, followed by lifespan measurement using a microfluidic device. b Lifespan extension of 20 long-lived SCRaMbLEd strains compared to parental strain synXIII Ycz063, replicative lifespan measurement of each strain was conducted with three replicates (n = 3), each replicate was conducted with 40 cells. Each data presentation is derived from comparing the value of each sample with each control value respectively, generating nine data. Data are presented as mean values ±SD from three independent replicates. Source data are provided as a Source data file. c Transcriptome profiling of lifespan extended strains compared to synXIII strain Ycz063. The heatmap represents the statistical significance of Gene Ontology (GO) enrichment of differentially expressed genes. The number in the brackets indicates the Pearson correlation between the statistical significance and lifespan extension. red, upregulated; blue, down-regulated; green: relative lifespan compared to initial synXIII. Source data are provided as a Source data file.
To gain deeper insights into the mechanisms underlying the extended lifespans of these 20 synXIII strains, we performed a comprehensive transcriptome analysis. This analysis revealed a clear correlation between long-lived strains and transcriptional patterns. Specifically, a pronounced upregulation was observed in genes that encode cellular components of ribosomes (Gene Ontology, GO:0005840), rRNA processing (GO:0006364) and nucleolus (GO:0005730). In addition, expression level of genes involved in cytoplasmic translation were also increased (GO:0002181) (Fig. 4c). Interestingly, we further found significance level of these cellular processes showed a good correlation with lifespan extension (Fig. 4c). Overall, our data suggested that the increased capacity for protein synthesis in the longer-lived yeast strains, indicating a robust association between a heightened state of protein synthesis machinery and lifespan extension.
Deciphering aging regulators and combinational effects for lifespan extension based on multiple structural variations
We next conducted a whole-genome sequencing (WGS) analysis to decipher the genomic rearrangements that potentially contribute to the long-lived phenotype (Fig. 5a). A total of 118 recombination events including deletion, inversion and duplication were observed among these 20 long-lived strains at frequencies of 55.9% (66), 38.1% (45) and 5.9% (7), respectively (Supplementary Fig. 7a). Remarkably, all identified SCRaMbLE events encompassed 111 genes at various frequencies, representing ~22.5% of all genes on synXIII (Supplementary Data 1). Different types of structural variations including deletion, duplication, and inversion were observed. Among the inversion events, the coding domain sequence could be joined to a convergent CDS, noncognate UTR, or other noncoding sequence, which results in three sub-types called CDS-CDS, CDS-noncognate_UTR (CDS-NON_Native_UTR), and CDS-NC, respectively (Supplementary Data 2). Specifically, we observed 93, 35 and 119 variations with gene deleted, duplicated, or 3’-UTR changed (CDS-CDS, CDS-noncognate_UTR and CDS-NC) among 20 lifespan-extended strains (Supplementary Fig. 7b, c). A previous study using the yeast deletion collection discovered some gene deletions could lead to increased lifespan39. Interestingly, we also found many of these gene were removed in the post-SCRaMbLE strains. We verified that deleting six of these gene caused an extended lifespan with varying rates 19.1%~30.9% (Supplementary Fig. 8). Overall, these results showed that SCRaMbLE system allows the generations of abundant genomic rearrangements in synthetic yeast, which are potentially linked to phenotype of replicative lifespan.

a Schematic map of structural variations in 20 long-lived SCRaMbLEants. Y-axis shows the relative aging extension of each SCRaMbLEant in comparison to parental synXIII strain that of represented by color scale. The red rectangles indicate the loci of aging regulators on chromosome synXIII. Source data are provided as a Source data file. b Different variations were individually constructed in synXIII, and the lifespan was measured for each variant along with the calculation of the rate of lifespan change with three replicates (n = 3). In the figures, the upward arrow indicates an increase, the downward arrow indicates a decrease. The blue color indicates the sequence inverted, the orangered color indicates the sequence duplicated, and the vanished sequence is indicative of the deletion. Source data are provided as a Source data file.
To deepen our understanding of the causal links between genotypic and phenotypic change, we reconstructed some key structural variations observed in post-SCRaMbLE synXIII strains with extended lifespan. Among these, the strain Ycz315 stood out due to its significantly extended lifespan, and contained a diverse array of genomic changes. Specifically, compared with the parent strain, the genome of Ycz315 included 1 deletion, DEL1 (47,629 bp–50,039 bp), along with 3 inversions, INV1 (337,292 bp–346,702 bp), INV2 (176,041 bp–184,342 bp), and INV3 (353,392 bp–346,673 bp). Additionally, a large duplication, DUP1 (75,003 bp–88,593 bp), was also found in Ycz315. To verify the effect of each variant on lifespan, we individually reconstructed each structural variation in synXIII and measured the replicative lifespan of the resultant strains (strain ID: Ycz020) (Fig. 5c). We found INV1 had almost no effect on lifespan extension, and INV2 (5.9%), and DEL1 (8.6%) exhibited minor effects on lifespan extension. Reconstruction of some structural variations could produce notable effects. Specifically, DUP1 showed an increased lifespan of up to 11.1%, and INV3 showed an extension of up to 17.8%. Taken together, our finding not only demonstrates the potential of synthetic chromosomes to identify genomic targets in aging studies but also highlights the combinatorial effects of multiple genomic rearrangements on increased replicative lifespan of yeast cells, as exemplified by the Ycz315 strain.
Discussion
The construction of synXIII represents one pivotal milestone in achieving the first synthetic eukaryotic genome, accounting for approximately 8% of the yeast genome. synXIII itself serves as an invaluable platform for investigating fundamental scientific inquiries pertaining to genome evolution and cellular process control, among others. Additionally, the evenly distributed presence of 333 loxPsym offers a unique and innovative resource for studying aging through multiple unique reorganizations of the synXIII. Our efforts successfully identified 20 longer-lived strains and 6 targets that can contribute to yeast lifespan extension. These long-lived strains were assessed using a microfluidic chip device, which has shown convincing results compared to the traditional micromanipulator method40,41,42. We demonstrated the feasibility and efficiency of this approach for large-scale lifespan factor screening. Notably, the variations generated by SCRaMbLE provide another unique set of materials for studying replicative aging.
Specifically, we identified RRN9 as a lifespan regulator. We showed that synonymous codon substitution caused by a single PCRTag sequence led to substantial increases in expression level of RRN9 and consequent increase in replicative lifespan by 37.5%. RRN9 is necessary for upstream activation factor (UAF) complex integrity, which is a multifunctional transcription factor in Saccharomyces cerevisiae that plays dual roles in activating RNA polymerase I (Pol I) transcription and repression of RNA polymerase II (Pol II)43. We observed the UAF complex regulate SIR2 repression and regulate rDNA copy-number which affected on lifespan-extension. This is particularly intriguing, as enhancing the expression of a gene within the protein complex, UAF, also has an impact on lifespan extension the underlying mechanism is worthy of further study44,45,46.
In our study, 20 strains exhibited lifespan extension after SCRaMbLEd synthetic chromosome XIII. To investigate whether other genetic factors such as rDNA copy number might influence lifespan extension, we assessed rDNA copy numbers across all SCRaMbLEd strains. We found that 18 of these strains showing rDNA copy numbers similar to the original synXIII. However, strains Ycz372 and Ycz378 unexpectedly exhibited decreased rDNA copy numbers (Supplementary Fig. 9). Prior research by Manuel Hotz et al.47 has shown a significant positive correlation between rDNA copy number and replicative lifespan, suggesting that the reduced rDNA copy number in Ycz372 and Ycz378 likely contributes to their shortened lifespans. This finding implies that the phenotypic changes in these two strains could be attributed to rearrangements within the initial synXIII structure.
For replicative lifespan analysis, a longer replicative lifespan is usually associated with a shorter generation time, as is the case for fob1Δ36. A longer generation time indicates a greater division pressure, which is typically associated with a shorter lifespan. This suggests that the senescent phenotype, as manifested by lengthened generation time, is a dominant feature in yeast cells48. We studied the mean generation time of all 20 long-lived SCRaMbLEd strains (Supplementary Fig. 10a). The results showed that 17 out of strains showed shorter mean generation times compared to synXIII. The scatter plot of generation time and replicative lifespan also indicated a weak negative correlation. It suggested different mechanisms exist that can extend cell lifespans. Furthermore, we observed that the long-lived strains exhibited a longer mean chronological lifespan, suggesting an intricate interplay of factors influencing cellular aging (Supplementary Fig. 10b). Although aging gene identification has been successful, connecting these genes to the mechanisms driving aging has proven to be challenging. The long-lived SCRaMbLEd strains may offer a unique opportunity for studying aging mechanisms, perhaps even including chronological lifespan. In Fig. 4c, the longer-lived strains exhibited an upregulation in genes that encode cellular components of ribosomes, rRNA processing, nucleolus and cytoplasmic translation. Enhanced ribosomal function may contribute to lifespan extension by altering protein synthesis patterns, which can increase stress resistance and improve protein homeostasis49. Improved protein homeostasis enables cells to better maintain and repair themselves, thereby potentially extending lifespan. In our analysis, we identified 5 over-represented mutant genes—UBP8, AAC1. NAT4, BUL1 and ATG16—whose null mutants exhibited lifespan extension (Supplementary Fig. 8) and each of these null mutants also demonstrated enhanced ribosomal function (Supplementary Fig. 11). Additionally, we also noted that there are many other genes that are better ranked than the 6 selected genes. Within the structure of synthetic genes, where genes are located in the same loxPsym unit50. For example, the UBP8 gene is linked to the FSH2 gene, the SCRaMbLEd occurrence of the UBP8 gene consistently coincides with that of FSH2. Therefore, due to the constraints on testing capacity and the nature of gene linkage, we conducted additional screening based on the functionality of the identified genes. Particularly, complex combinations were present in longer-lived strains to promote the lifespan extension, validating the feasibility of utilizing SCRaMbLE technology on synthetic chromosomes in aging research, providing research models (synthetic yeast cell) and methods for aging research. In this study, our focus lies in the construction of synXIII and its potential application in aging research. Additionally, we have explored the utilization of synthetic Sc2.0 yeast through the integration of constructed synXIII and SCRaMbLE system to unravel regulatory genes and pathways involved in replicative aging. The elucidation of underlying mechanisms responsible for lifespan extension necessitates further comprehensive and rigorous investigation.
In brief, as a part of the Sc2.0 project, we have undertaken the design and synthesis of the complete 884 kb yeast chromosome XIII (synXIII) in this study. And in addition, like what has been done by other groups in this community, we further explored the potential application of synthetic Sc2.0 yeast by using constructed synXIII and SCRaMbLE system to uncover the regulator genes and networks in replicative aging. The SCRaMbLE system of Sc2.0 offered us the opportunity to shuffle multiple genes on chromosomal level and reveal potential long and short-lived mutants, as well as genetic or pathway interactions involving multiple target genes. Therefore, this system could serve as an effective means to identify factors associated with aging and provide a resource for delving into the intricate process of aging, thereby holding immense potential value for the advancement of anti-aging drugs, age-related diseases, and aging biomarkers.
Methods
Strains and growth media
The yeast strains used in this paper were derivatives of BY4741 or BY474250. Synthesis of the minichunks was outsourced to BGI TECH SOLUTIONS (BEIJING LIUHE) CO, LIMITED. The strains generated in this study are listed in Supplementary Data 3. Yeast culture and transformation were applied by using standard methods. The medium used in this study including two types: YPD and SC (synthetic complete medium) or synthetic medium without histidine (SC-His), synthetic medium without uracil (SC-Ura), synthetic medium without leucine (SC-Leu). Specifically, for replicative lifespan measurements, the cells were cultured on SC medium.
synXIII design and construction
The sequence of chromosome XIII was designed in silico and BioStudio following Sc2.0 design principles. The final version of designer chromosome XIII sequence (synXIII) was defined as yeast_chr13_3_40, with a total of 883,749 bp length and ~9% sequence modification, including removal of 21 tRNAs, insertion of 333 loxPsym sites, swap of TAG to TAA, and introduction of 529 pair synthetic PCRTags. More detailed information of synXIII can be accessed in Supplementary Table).
The sequence of synXIII was hierarchically segmented to 15 megachunks (~70 kb, named A-O), then to 84 chunks (~10 kb), and final 421 minichunks (~3 kb) for commercial synthesis. Gibson assembly strategy was used for assembly32. The minichunks were amplified and assembled in a single step process based on in vitro recombination. For assembly verification, single colonies were selected for overnight culture at 37 °C. The restriction enzyme digestion was performed to verify the assembly result. An 800−1000 bp of the homologous region between each chunk was designed for homologous recombination and 40-bp overlap between each minichunk designed for Gibson assembly. In each megachunk integration, 5–6 chunks (equivalent to 1 megachunk) were directly transformed with the same moles of each chunk as a pool into yeast to replace the corresponding wild-type sequence. For synthetic chunks transformation, we adapted lithium acetate transformation strategy and each chunk was needed 300~500 ng which was excised from plasmids and purified to screen the candidate colonies on the selective auxotroph medium plates. We integrated synthetic megachunks from both ends of chromosome XIII in parallel in strains BY4741 and BY4742.
Nine successive rounds of left synthetic semi-arm integration and six rounds of right in the synthetic right-semi arm were used to produce the semi-synthetic synXIII strains (synXIII-I and synXIII-J, strain ID: Ycz011, Ycz017) which fused into full-length synXIII by I-SceI mediated directed homologous recombination31,51, using the endonuclease I-SceI recognition site and the I-SceI enzyme to produce a break and enhance homologous recombination efficiency. All strains generated in this study are listed in (Supplementary Data 3).
Target colony selection
The colonies cultured on the selective plates were replicated onto the synthetic complete medium lacking uracil (SC–Ura) or leucine (SC–Leu). After overnight incubation at 30 °C, the clones can grow on one type of medium but not the other were identified and subjected to PCRtag analysis to verify the incorporation of the entire synthetic megachunk. To confirm the strains, two rounds of PCR were performed. At first, five or six pairs of PCRTags, distributed in each synthetic chunk, were chosen to screen the phenotypically desired clones. rTaq DNA polymerase (TaKaRa, DR001A) was used together with 300 ng of genomic DNA in a 10 mL reaction containing 1 mM of primer each. The PCR program was as following:1 cycle of 94 °C for 5 min, 35 cycles of 94 °C/30 s-55 °C/30 s-72 °C/30 s, 1 cycle of 72 °C for 5 min and 12 °C keep. The clones got through the first round of PCR test were subjected to next round of PCRTag analysis using all primers within the megachunk to identify the ones containing the entire synthetic DNA.
Scanning electron microscope
The samples were grown in OD = 0.1 for 4 hours to an A600 of 0.6, and then fixed in 2.5% glutaraldehyde (Sigma, catalog number: 340855, pH 7.4) for 2 h. After washing three times with 0.1 M phosphate buffer (pH 7.2) and fixation in 1% osmic acid (Sigma, catalog number: 104239) at 4 °C for 2 h, they were dehydrated through an ascending series of ethanol by ending in 100%, and then dried using Critical Point Dryer (Quorum, K850). Samples were coated with a very thin film of gold for 30 s using sputter coater (Cresstington, 108Auto). Samples were observed using an ultra-high resolution scanning electron microscope (Quasi-S, HITACHI Regulus 8100).
Immunoblot analysis
A single colony was picked and placed into 5 mL YPD liquid medium for overnight and next diluted into a total 5 mL fresh culture to A600 = 0.1. Until culturing at 30 °C for another 5 h, the cells were collected. Using 1 mL sterile water to resuspend the cells and the suspension was transferred to a new 1.5 mL EP tube, 10,000 g centrifugation for 1 min to collect the cells. 50 μL sterile water and 50 μL 0.2 M NaOH were added and maintained at room temperature for 5 min, followed by centrifugation for collected cells. Thereafter, 50 μL SDS sample buffer with 1% Triton X-20 was added, followed by heating to 95 °C for 5 min. The mixture was then centrifuged at 13,000 g for 10 min and the supernatant was aliquoted as the total protein extraction. 15 μL per lane for 10-lanes gel and mouse anti- Flag (F1804, Sigma; 1:1,000) was used to detect Flag-RRN9 and 2 μL per lane for 10-lanes gel to detect H3.
Growth measurement
A single colony of each strain was cultured in YPD liquid medium at 30 °C overnight. Approximately 300 colonies for each strain were then streaked onto corresponding agar plates and incubated at 30 °C for approximately 3 days. Subsequently, the colonies on the plates were automatically identified, photographed, and analyzed using software that measures colony diameter in millimeters, such as the Precision Colony Picker (model: PIXL711712, Singer Instruments). The relative colony size of each strain was determined by dividing the colony size (diameter) of individual colonies by the mean colony size of BY4741.
The growth curve measurements were conducted as previously reported5. Log-phase cells were diluted with fresh medium to an optical density (OD) of 0.1, and 100 µL of the diluted cells were dispensed into each well of Costar clear polystyrene 96-well plates. For each strain, three or four technical replicates were analyzed. The sealed 96-well plate with Breathe-Easy membrane (Sigma, MKBZ0331) was incubated in an Epoch2 microplate photometer (BioTek) at 30 °C for 24–48 h in the specified medium. The OD600 of each well was measured every 10 min. The doubling time (min) of each strain was calculated using GraphPad Prism 10 software.
ChIP-qPCR
The strains were cultured in YPD medium at 30 °C overnight. The culture as diluted with fresh YPD medium to A600 = 0.1 and further grown for 6 h with shaking until the A600 = 0.8–1.0. Then the Formaldehyde (1% final) was used to crosslink at 25 °C for 25 min. Next, Glycine (0.15 M final) was added to abort the crosslink. ~50 A600 of cells were collected. After breaking the cells by glass beads and the chromatin sonicated by 0.5 μL micrococcal nuclease (MNase), 1/10 of the sonicated chromatin was took as the input and 9/10 were incubated with 2 μL anti-flag antibody for above 8 h. The antibody-protein-DNA complex was put down by Protein A/G Magnetic beads. To reverse the protein-DNA crosslinks 2.5 μL 10 mg/mL Rnase A and 5 μL 20 mg/mL Proteinase K was added and incubated at 65 ˚C for another 8 h. The DNA was purified by the kit DNA Clean & Concentrator-5 (ZYMO, catalog number: D4004). RT- PCR was performed to calculate the protein occupancy. All the occupancy data were shown as the percentage of enrichment at target loci normalized by RPO2135.
For RT-PCR analysis, 50 μL of input samples were 10-diluted with TE as qPCR input samples (input versus chip dilution ratio was 100:1). RT-qPCR analyses were done with TB Green qPCR Kits (TAKARA, catalog number: AK8901). Each reaction with input or chip sample was performed under the following cycling condition: 95 °C 30 s and 39 cycles of 95 °C 5 s, 55 °C 10 s and 72 °C 30 s. Relative enrichments of ChIP versus input were quantified as input (N) = 2ΔCt (target gene)/2ΔCt (control gene). ChIP enrichment of each biological replicate (N = 3) was calculated from mean value of triplicated qPCR reactions. Primer sequences: RRN9 forward primer 5’-gcaataccttccggtatat-3’, reverse primer 5’- caaccgctgataaagagac; ACT1 forward primer 5’-atggattctgaggttgctgct-3’, reverse primer 5’- tggtgtcttggtctaccgac.
Mating type switch of yeast and sporulation
PJD147 (pGal-HO) was transferred into the synXIII strain using the LiOAc transformation method and colonies were selected on SC–Leu plates. The colony was picked into SC–Leu (2% glucose), medium cultured overnight at 30 °C, then diluted reached A600 = 0.1 in SC–Leu (2% galactose) fresh medium, followed by culturing for 4 hours, and then plating on YPD. The mating type was then confirmed by crossing MATa and MATα tester strains (Ycz050 and Ycz051) and examined by microscopy. A strain with MATa and another strain with MATα were recovered on YPD plates. The colonies from two strains were picked into YPD medium, cultured for 24 h and patched on YPD plates. Diploids verified by testing with the two mating type testers were cultured and cells were collected and washed with sterile water for four times, following sporulated in SPOR medium containing 1% potassium acetate (Sigma, 60035) and 0.125% yeast extract (Oxoid, LP0021B) for 3 days.
Replicative lifespan measurement using microfluidic assay
A microfluidic device based on published reports was used to monitor the number of mother cell divisions36,52, the replicative lifespan of each mother cell was determined by quantifying the number of daughter cells it produced. SC (synthetic complete) medium was used to grow all tested yeast strains. Cells incubated in 5 mL SC medium cultured overnight were diluted to an A600 of 0.1 in fresh medium, and incubated at 30 °C for 4~6 h until they reached an A600 of 0.6. The microfluidic device contains 8 micro-posts (side length in the range of 40−100 μm, depth of the chamber ranging between 3.8-4.2 μm) that clamp mother cells in place. In contrast, newly formed daughter cells are washed away by hydro-dynamically controlled flow of the surrounding liquid medium. Each micro-post tested one strain combined with time-lapse microscopy at the high temporal solution. Mother cells were continuously monitored for 60 h repeated microscopic imaging to measure produced daughter cells (Supplementary Fig. 6a)36. The number of daughter cells produced through cell division is used to assess the lifespan of a cell. For each strain, 40 mother cells were randomly selected, the number of daughter cells produced was detected, and the survival curve was drawn using Matlab.
SCRaMbLEant library preparation
An enhanced green fluorescent gene (eGFP) was inserted before the stop codon of HSP104 fused with LEU2 marker for selection. pSCW11-Cre/EBD-HIS3 was transferred into the strain and simultaneously a URA3 marker was integrated into the middle of two loxPsym sites between YMR011W and YMR012W on synXIII at position (270,117~270,525 bp). The colony was cultured in the SC–His medium overnight and then diluted to an A600 of 0.1 in the 5 mL fresh SC–His medium containing the inducer 1 µM estradiol at 200 rpm in a shaking flask, with induction for 8 h at 30 °C. Finally, 200 μL solution was collected and the cells were washed by sterile water, culturing on a SC + 5-FOA plates for 3 days. For each plate, 96 FOA-resistant single colonies were isolated from the plate for culturing with SC liquid medium in 96 deep-well plate. Then overnight culture for all plates were prepared for GFP fluorescence measurement. In this way, a pool of about 6000 SCRaMbLEant colonies was produced.
FACS testing
Each SCRaMbLEant colony was grown in 1 mL SC medium of a 2.2 mL deep well plate for 24 h. The following day, cells were diluted to an A600 of 0.001 in 1 mL fresh medium and grown up to an A600 of 0.8, and each strain was tested by flow cytometry (BD, FACSAria Cell Sorter; Beckman, CytoFLEX). Analysis and cell sorting with flow cytometry were conducted at a rate of 100,000 events per second. The 488 nm laser was used to excite the GFP protein and 528/29 filter was used to detect the fluorescence signal. An initial scatter-gating step was operated based on cell’s forward-scatter properties to collect data from single cells. Flow cytometry analysis was performed by analyzing 100,000 cells of each sample.
Omics analyses
Whole-genome sequencing was performed for the synXIII (yeast_chr13_9_2, strain ID: Yzc020) on the NextSeq 500 platform. For transcriptome and proteome, yeast cells Yzc030 and BY4741, with three biological replicates of each were cultured and collected until an A600 of ~0.8 was reached. The RNA sequencing libraries were constructed using NEB Next Ultra TMRNA Library Prep Kit (catalog number: E7770S, NEB). Then sequencing was performed on Illumina HiSeq PE150 Sequencing Systems by v2 kit (catalog number: 256782). For proteome, a free-labeled peptide method was used in this study. The peptides were loaded on an EASY-nLCTM 1200 UHPLC by the autosampler onto a 2 cm C18 trap column and an analytical C18 column packed in-house (0.075 × 150 mm column, 3 μm). The peptides were subjected to nano-electrospray ionization (nano ESI) followed by tandem mass spectrometry (MS/MS) in a benchtop Orbitrap Q Exactive mass spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled online to the HPLC. For nucleotide sequence analysis, the sequencing data quality control was performed using SOAPnuke; the clean data were aligned to reference sequence of synXIII yeast genome using Bowtie2 (version 2.2.5)53 with standard settings. And the variations were identified with both GATK (version 2.7)54 and SAMtools (version 0.1.19)55, using default parameters. The variants identified by either tool were merged with CombineVariants implemented in GATK. For transcriptomics, the reads were mapped to genomes by hisat2 (version 2.1.0)56 using default parameters, and the quantification and differential expression genes were analyzed by featureCounts (version 2.0.1)57 and DEseq2 (version v1.30.1)58. The statistical significance of differential expression genes was assessed if the P value fell below the threshold of the 5% Family Wise Error Rate (FWER) after Bonferroni correction (threshold = 7.56 × 10−6). For proteomics, MaxQuant (version v1.5.3.30) and Spectronau (version v12) were used for protein identification and quantification. False positive differentially expressed genes were inferred by the following rules and removed: dubious genes, transposable genes, 2-micron genes or genes with low coverage (<60%) for native and synthetic strains. Genes and metabolites with differential expression of log2 (fold-change) > 0 were considered up-regulated, while those with log2 (fold-change) <0 were considered down-regulated for enrichment analysis. Gene enrichment of KEGG pathways and Gene Ontology annotations were performed using the hyper-geometric and Chi-squared tests.
Quantification and statistical analysis
The data was analyzed using GraphPad Prism (version 9.4.0). One-tailed t tests were used to compare different groups in this paper. Error bars represent SD. Differences were considered as statistically significant at p value < 0.05. * represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001, **** represents P < 0.000.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study have been deposited into CNSA (CNGB Nucleotide Sequence Archive) under accession number CNP0003780. The reference genome of BY4741 is downloaded from the Saccharomyces Genome Database (http://sgd-archive.yeastgenome.org/sequence/strains/BY4741/BY4741_Toronto_2012/). The raw proteomics data is deposited at iProX under accession code PXD057330 [https://www.iprox.cn/page/project.html?id=IPX0010096000] and CNSA under accession number CNP0003780, available at https://ftp.cngb.org/pub/CNSA/data2/CNP0003780/Protein/. The results of sequencing data analysis in this study are provided in the Supplementary data and Source data file, which are provided with this paper. Source data are provided with this paper.
References
McCulloch, L. H. et al. Consequences of a telomerase-related fitness defect and chromosome substitution technology in yeast synIX strains. Cell Genom. 3, https://doi.org/10.1016/j.xgen.2023.100419 (2023).
Mercy, G. et al. 3D organization of synthetic and scrambled chromosomes. Science 355, https://doi.org/10.1126/science.aaf4597 (2017).
Wu, Y. et al. Bug mapping and fitness testing of chemically synthesized chromosome X. Science 355, https://doi.org/10.1126/science.aaf4706 (2017).
Lauer, S. et al. Context-dependent neocentromere activity in synthetic yeast chromosome VIII. Cell Genom. 3, https://doi.org/10.1016/j.xgen.2023.100437 (2023).
Shen, Y. et al. Dissecting aneuploidy phenotypes by constructing Sc2.0 chromosome VII and SCRaMbLEing synthetic disomic yeast. Cell Genom. 3, https://doi.org/10.1016/j.xgen.2023.100364 (2023).
Foo, J. L. et al. Establishing chromosomal design-build-test-learn through a synthetic chromosome and its combinatorial reconfiguration. Cell Genom. 3, https://doi.org/10.1016/j.xgen.2023.100435 (2023).
Williams, T. C. et al. Parallel laboratory evolution and rational debugging reveal genomic plasticity to S. cerevisiae synthetic chromosome XIV defects. Cell Genom. 3, https://doi.org/10.1016/j.xgen.2023.100379 (2023).
Xie, Z. X. et al. “Perfect” designer chromosome V and behavior of a ring derivative. Science 355, https://doi.org/10.1126/science.aaf4704 (2017).
Mitchell, L. A. et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science 355, https://doi.org/10.1126/science.aaf4831 (2017).
Dymond, J. S. et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature 477, 471–476 (2011).
Luo, J. et al. Synthetic chromosome fusion: effects on mitotic and meiotic genome structure and function. Cell Genom. 3, https://doi.org/10.1016/j.xgen.2023.100439 (2023).
Blount, B. A. et al. Synthetic yeast chromosome XI design provides a testbed for the study of extrachromosomal circular DNA dynamics. Cell Genom. 3, https://doi.org/10.1016/j.xgen.2023.100418 (2023).
Building better yeast. Nat. Commun. 9, https://doi.org/10.1038/s41467-018-04159-y (2018).
Coradini, A. L. V., Hull, C. B. & Ehrenreich, I. M. Building genomes to understand biology. Nat. Commun. 11, https://doi.org/10.1038/s41467-020−19753-2 (2020).
Shen, M. J. et al. Heterozygous diploid and interspecies SCRaMbLEing. Nat. Commun. 9, https://doi.org/10.1038/s41467-018-04157-0 (2018).
Hochrein, L., Mitchell, L. A., Schulz, K., Messerschmidt, K. & Mueller-Roeber, B. L-SCRaMbLE as a tool for light-controlled Cre-mediated recombination in yeast. Nat. Commun. 9, 1931 (2018).
Zhang, H. et al. Systematic dissection of key factors governing recombination outcomes by GCE-SCRaMbLE. Nat. Commun. 13, https://doi.org/10.1038/s41467-022-33606-0 (2022).
Niccoli, T. & Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 22, R741–R752 (2012).
He, C., Zhou, C. & Kennedy, B. K. The yeast replicative aging model. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2690–2696 (2018).
Moskalev, A. A. et al. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 12, 661–684 (2013).
Talens, R. P. et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell 11, 694–703 (2012).
Vendelbo, M. H. & Nair, K. S. Mitochondrial longevity pathways. Biochim. Biophys. Acta 1813, 634–644 (2011).
Lord, C. J. & Ashworth, A. The DNA damage response and cancer therapy. Nature 481, 287–294 (2012).
Zhou, Z. et al. Engineering longevity-design of a synthetic gene oscillator to slow cellular aging. Science 380, 376–381 (2023).
Olmez, T. T. et al. Sis2 regulates yeast replicative lifespan in a dose-dependent manner. Nat. Commun. 14, 7719 (2023).
Fontana, L., Partridge, L. & Longo, V. D. Extending healthy life span–from yeast to humans. Science 328, 321–326 (2010).
Lapierre, L. R. & Hansen, M. Lessons from C. elegans: signaling pathways for longevity. Trends Endocrinol. Metab. 23, 637–644 (2012).
Shen, Y. et al. SCRaMbLE generates designed combinatorial stochastic diversity in synthetic chromosomes. Genome Res. 26, 36–49 (2016).
Richardson, S. M. et al. Design of a synthetic yeast genome. Science 355, 1040–1044 (2017).
Schindler, D. et al. Design, construction, and functional characterization of a tRNA neochromosome in yeast. Cell 186, 5237–5253 e5222 (2023).
Shen, Y. et al. Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science 355, https://doi.org/10.1126/science.aaf4791 (2017).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Jovicevic, D., Blount, B. A. & Ellis, T. Total synthesis of a eukaryotic chromosome: redesigning and SCRaMbLE-ing yeast. Bioessays 36, 855–860 (2014).
Zhang, W. et al. Engineering the ribosomal DNA in a megabase synthetic chromosome. Science 355, https://doi.org/10.1126/science.aaf3981 (2017).
van Dijk, E. L. et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature 475, 114–117 (2011).
Xie, Z. et al. Molecular phenotyping of aging in single yeast cells using a novel microfluidic device. Aging Cell 11, 599–606 (2012).
Cui, H. J. et al. PMT1 deficiency extends the shortened replicative lifespan of TED1-deficient yeast in a Hac1p-dependent manner. FEMS Microbiol. Lett. 365, https://doi.org/10.1093/femsle/fny234 (2018).
Kaeberlein, M., Kirkland, K. T., Fields, S. & Kennedy, B. K. Genes determining yeast replicative life span in a long-lived genetic background. Mech. Ageing Dev. 126, 491–504 (2005).
McCormick, M. A. et al. A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 22, 895–906 (2015).
Lee, S. S., Avalos Vizcarra, I., Huberts, D. H., Lee, L. P. & Heinemann, M. Whole lifespan microscopic observation of budding yeast aging through a microfluidic dissection platform. Proc. Natl Acad Sci. USA 109, 4916–4920 (2012).
Zhang, Y. et al. Single cell analysis of yeast replicative aging using a new generation of microfluidic device. PLoS One 7, e48275 (2012).
Jo, M. C., Liu, W., Gu, L., Dang, W. & Qin, L. High-throughput analysis of yeast replicative aging using a microfluidic system. Proc. Natl. Acad. Sci. USA 112, 9364–9369 (2015).
Knutson, B. A., Smith, M. L., Belkevich, A. E. & Fakhouri, A. M. Molecular topology of RNA polymerase I upstream activation factor. Mol. Cell Biol. 40, https://doi.org/10.1128/MCB.00056-20 (2020).
Iida, T. & Kobayashi, T. RNA polymerase I activators count and adjust ribosomal RNA gene copy number. Mol. Cell 73, 645–654.e613 (2019).
Iida, T. & Kobayashi, T. How do cells count multi-copy genes?: “Musical Chair” model for preserving the number of rDNA copies. Curr. Genet. 65, 883–885 (2019).
Smith, J. S. The long and short of rDNA and yeast replicative aging. Proc. Natl. Acad. Sci. 119, https://doi.org/10.1073/pnas.2205124119 (2022).
Hotz, M. et al. rDNA array length is a major determinant of replicative lifespan in budding yeast. Proc. Natl. Acad. Sci. USA 119, e2119593119 (2022).
Jazwinski, S. M., Egilmez, N. K. & Chen, J. B. Replication control and cellular life span. Exp. Gerontol. 24, 423–436 (1989).
Clancy, D. J. et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292, 104–106 (2001).
Luo, Z. et al. Compacting a synthetic yeast chromosome arm. Genome Biol 22, 5 (2021).
Parenteau, J. et al. Deletion of many yeast introns reveals a minority of genes that require splicing for function. Mol. Biol. Cell 19, 1932–1941 (2008).
Casavant, B. P. et al. Suspended microfluidics. Proc. Natl. Acad. Sci. USA 110, 10111–10116 (2013).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFA0906003). Guangdong Special Support Program (2021JC06Y578). the Shenzhen Portion of the Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone (HTHZQSWS-KCCYB-2023060). the Shenzhen Medical Research Fund (No.B2402036). the Shenzhen Municipal Government of China (CJGJZD20200617102403009). the Guangdong Engineering Technology Research Center for clinical application of cancer genome (2023B191). The research fund from the Synthetic Biology Research Center of Shenzhen University.the Sanming Project of Shenzhen Health and Family Planning Commission (SZSM202011017SZ), the Shenzhen High-level Hospital Construction Fund, and the Shenzhen Institute of Synthetic Biology Scientific Research Program (ZTXM20214005). The National Natural Science Foundation of China (32170756, 31725002, 32322047), Shenzhen Science and Technology Program (KQTD20180413181837372), Shenzhen Outstanding Talents Training Fund and Bureau of International Cooperation, Chinese Academy of Sciences (172644KYSB20180022), and Jiangsu Provincial Department of Science and Technology (No.BM2023009). We thank the DNA assembly automation platform of China National GenBank for its support of synthetic chunk assembly. LAM and JSB were supported by NSF grants MCB-1026068, MCB-1443299, MCB-1616111, and MCB-1921641. YC is supported by UK Biotechnology and Biological Sciences Research Council (BBSRC) grants BB/M005690/1, BB/P02114X/1, and BB/W014483/1; a Volkswagen Foundation “Life? Initiative” Grant (Ref. 94 771); an Engineering and Physical Sciences Research Council (EPSRC) Fellowship EP/V05967X/1; and a European Research Council (ERC) Consolidator Award EP/Y024753/1.
Author information
Authors and Affiliations
Contributions
Conceptualization, W.H., Y.S.; funding and resources, W.H., Y.S, Z.C.; microfluidic platform used for aging screening: Z.X.; data production, C.Z., Y.H., Y.A., YL.W. data analyses, investigation, and visualization, Y.W., C.Z., J.Z.; writing—original draft, W.H., Y.S., C.Z, Y.W., X.F. and J.D.B.; writing—review & editing: all co-authors.
Corresponding authors
Ethics declarations
Competing interests
J.B. is a Founder and Director of CDI Labs, Inc., a Founder of and consultant to Neochromosome, Inc, a Founder, SAB member of and consultant to ReOpen Diagnostics, LLC and serves or served on the Scientific Advisory Board of the following: Sangamo, Inc., Logomix, Inc., Modern Meadow, Inc., Rome Therapeutics, Inc., Sample6, Inc., Tessera Therapeutics, Inc., and the Wyss Institute. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Julius Fredens, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, C., Wang, Y., Huang, Y. et al. The de novo design and synthesis of yeast chromosome XIII facilitates investigations on aging. Nat Commun 15, 10139 (2024). https://doi.org/10.1038/s41467-024-54130-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54130-3
