
Abstract
A bimetallic heterostructure has been shown effective to enhance the multi-carbon (C2+) product selectivity in CO2 electroreduction. Clarifying the interfacial structure under electrolysis and its decisive role in the pathway selection are crucial, yet challenging. Here, we conceive a well-defined Ag-Cu biphasic heterostructure to understand the interfacial structure-steered product selectivity: The Cu-rich interface prefers ethylene, while the dominant product switch to alcohols with an increasing Ag fraction, and finally to CO as Ag occupying the main surface. We unravel a *CO intermediate-regulated interfacial restructuring, and observe abundant of Cu atoms migrating onto the neighboring Ag surface under a locally high *CO concentration. The evolving structure alters the oxyphilic characteristic at the interface, which profoundly determines the hydrogenation energetics of CO2 and ultimately, the dominant C2+ product. This work explicitly links the evolving interfacial structure with distinct C2+ pathway, formulating design guidelines for bimetallic electrocatalysts with selectively enhanced C2+ yields.
Similar content being viewed by others

Introduction
Renewable electricity-powered carbon dioxide reduction (CO2R) into value-added chemicals, such as syngas, alcohols, and fuels, offers an appealing route to establish a sustainable, circular economy, and mitigate anthropogenic CO2 emission1,2,3,4. For cost-effective CO2 electrolysis at an industrial scale, selective electrocatalysts are indispensable. Particularly, multi-carbon (C2+) products-selective electrocatalysts are highly desirable because those C2+ chemicals, such as ethylene (C2H4), ethanol (C2H5OH), and n-propanol (n-C3H7OH), etc. are energy dense by volume and lever extensive existing infrastructure for the storage and distribution of carbon-based fuels5,6,7. With an optimal *CO binding energy (*CO represents the adsorbed CO intermediate), Cu has attracted particular attention due to their unique capability of reducing CO2 to valuable C2+ fuels with an adequate efficiency8,9, yet it accompanies poor selectivities toward C2+ due to the scaling relation of relevant carbon intermediates involved in the complex reduction pathway10,11,12. One strategy to break the scaling relation on monometallic Cu is to construct a bimetallic system, where Cu and the other metal component can function synergistically through electron density tuning13, lattice strain engineering14, asymmetric catalytic sites15 and tandem catalysis16,17, etc. Various secondary metals, e.g., Ag, Au, Sn, Zn, and Pd, etc., have been introduced onto Cu surface, thus far, to improve the C2+ production in a practical electrolyzer16,18,19,20,21, however fundamentally, it is relatively less explored how (if any) these bimetallic interfaces evolve under electrolysis and how the restructuring steers the selectivity toward a specific C2+ product.
A particular interesting scenario is the Ag-Cu heterostructure, which demonstrates substantially discrepant C2+ product distributions in different studies (see Supplementary Table 1). Compared to Cu, Ag was proved with a lower *CO affinity and thus a higher CO producing capability22. A tandem catalysis strategy has been proposed to improve C2+ fuels production on an Ag-Cu heterostructure, wherein CO2 can be reduced into *CO on Ag and then transferred to Cu for further reduction into C2+ products16,17. Although previous studies on AgCu alloy and Ag-Cu/CuOx heterostructures have established the important role of the bimetallic interface in promoting C2+ fuels23,24,25,26/C1 hydrocarbon production26,27 and suppressing hydrogen evolution28, precisely tracking the atomic-scale structural evolution during electrolysis remains lacking at the bimetallic interface, which is crucial for understanding the interfacial structure-steered CO2R pathway. Particularly, the impetus of structural evolution at such a biphasic interface has rarely been explored. Previous studies on monometallic Cu evidenced that, when exposing to CO gas, the Cu surface would decompose into lower-coordinated clusters, which would highly motivate the surface for water dissociation, a crucial step in the water-gas shift reaction29,30. On the other hand, there are additional studies showing that the negative potential field is a main driving force for the clustering phenomenon31. The limited understanding on the impetus of this restructuring behavior would heavily impede the rational design of catalyst and the fundamental understanding toward CO2-to-C2+ conversion.
Herein, a well-defined biphasic Ag-Cu heterostructure with controlled Ag molar fraction was assembled, and implemented as a model system to understand the interfacial dynamic restructuring and its critical role in the CO2R pathway selection. The Ag-Cu heterostructure was observed featuring a surface-sensitive C2+ product distribution: C2H4 was more pronounced on a Cu-rich interface, whereas the dominant product switched to alcohols (e.g., C2H5OH and n-C3H7OH) with an increasing Ag fraction, and finally to CO as Ag occupying the main surface. By probing the Ag-Cu interface down to an atomic scale, we unraveled that the *CO intermediate can strongly regulate the electrochemical restructuring behavior, e.g., Cu tends to migrate onto the neighboring Ag under a locally high *CO concentration, forming abundant of Ag-Cu interface. The evolving structure and enriched *CO intermediate strongly altered the oxyphilic characteristic at the interface, which profoundly determined the hydrogenation energetics of CO2, leading to disparate C2+ product distributions. This work establishes the empirical demonstration of dynamic structural reconstruction in a bimetallic model system, and sets up a paradigm to understanding the structure-function correlation in CO2R electrocatalysis.
Results and Discussions
Assembly of Ag-Cu biphasic heterostructure
A well-defined biphasic heterostructure interface without an intermetallic alloy serves as an exemplary system to track the dynamic restructuring during electrocatalysis. Here, Ag nanoparticles (Ag NPs) were assembled onto Cu nanowires (Cu NWs) using 4,4’-bipyridine (bipy) as a linker (see Experimental Procedures). The bipy molecules could successfully bring Cu and Ag together by forming strong coordination bonds between the nitrogen atoms of bipy ligands and the metal surface32,33,34,35. The aberration-corrected high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images in Fig. 1a, b showed the overview of an obtained Ag-Cu heterostructure, wherein Ag and Cu were interlinked by the bipy ligands, exhibiting a well-defined biphasic interface. The Cu NWs exhibited a diameter of ~50 nm, and the Ag NPs possessed an average size of ~13 nm (Supplementary fig. 1). This interface was further checked through Fast Fourier transform (FFT) of the selected area and energy dispersive X-ray spectroscopy (EDS) elemental maps, which confirmed the absence of interfacial alloy (Fig. 1c, d). We found the bipy functionalization of Cu rather than Ag was a prerequisite for assembling Ag-Cu heterostructure, otherwise Cu and Ag aggregates would be easily formed (Supplementary fig. 2,3). The successful exchange of oleylamine with bipy on Cu NWs can be confirmed via the infrared spectroscopy, and the peaks at 1400–1600 cm−1 (C = N, C = C stretching) and 3050 cm−1 ( = C−H stretching) were ascribed to the bipy ligand (Supplementary fig. 4).

a, b Low-(a) and high-(b) magnified HAADF-STEM images of Ag-Cu biphasic heterostructure. c FFT patterns taken from Ag and Cu regions in (b) along the zone axis of Ag < 110 > and Cu < 100 > , respectively. d Corresponding EDS elemental maps at Ag-Cu interface, showing the phase separation of Ag and Cu. e XRD patterns of as-synthesized Ag-Cu heterostructures with different constitution ratios. Two sets of characteristic peaks attributed to the fcc lattice of pure Ag (PDF-04-0783) and Cu (PDF-04-0836) were shown below. f, g XANES spectra (f) and EXAFS spectra (g) of variable Ag-Cu heterostructures with different constitution ratios at the Cu K-edge. Cu2O and Cu foil were listed as references.
To understand the structural influence on CO2R pathway, we prepared Ag-Cu heterostructure with six different constitution ratios, denoted as Agx-Cuy (x and y represent the mass fraction of Ag and Cu in the heterostructure, respectively). The Ag/Cu ratio can be well controlled by the input mass of Ag NPs and Cu NWs before assembly (Supplementary fig. 5), and was quantified using the inductively coupled plasma-optical emission spectroscopy (ICP-OES) (summarized in Supplementary Table 2). The crystallinity of Ag-Cu was checked by X-ray diffraction (XRD), showing two sets of characteristic peaks belonging to the face-centered cubic (fcc) lattice of pure Ag and Cu, respectively (Fig. 1e). Importantly, no obvious peak-shift of both Ag and Cu was observed, suggesting the absence of AgCu alloy, consistent with the STEM observation. In addition to the long-range order of crystal lattice, we further investigated the chemical states and local structures of Ag-Cu heterostructure through X-ray absorption spectroscopy (XAS), encompassing both X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). The Cu K-edge involved a transition from 1 s to 4p states, and the corresponding EXAFS spectra exhibited a discernible peak at ~2.3 Å ascribed to the Cu-Cu scattering pathway (Fig. 1f, g). Although a slight oxidation of Cu might happen on the surface of some Ag-Cu heterostructures due to air exposure, this naturally occurring oxide shell was rapidly reduced under the electrochemical operating conditions of CO2R (Supplementary fig. 6,7). Thus, this partially oxidation phenomenon would not impart strong influence to the conclusion of this paper. Collectively, these characterizations corroborate the successful fabrication of a well-defined phase-separated Ag-Cu bimetallic heterostructure.
Surface-sensitive CO2R product distribution
The electrochemical CO2R performance of Ag-Cu heterostructure was investigated within a CO2-saturated 0.1 M KHCO3 electrolyte (pH 6.8). For comparison, CO2R performances of Cu (after bipy-functionalization, denoted as Cu-bipy) and Ag NPs were also assessed under identical conditions, separately. Gaseous products were quantified via a gas chromatography online sampling system, while liquid products were meticulously collected and analyzed using 1H nuclear magnetic resonance (see Experimental Procedures). An extended one-hour constant potential experiment was employed to assess the performance across a broad range of potentials. Supplementary fig. 8,9 summarized the product distributions and current densities of different catalysts at each applied potential.
It was observed that the selectivities (Faradaic efficiencies, FEs) of C2+ products showed apparent discrepancies between Ag, Cu-bipy, and Ag-Cu heterostructure. Ag alone was not capable of producing any C2+ hydrocarbons or alcohols; instead, it exclusively generated CO and formic acid (HCOOH) (Fig. 2a). For instance, at −1.05 V versus reversible hydrogen electrode (vs. RHE), CO was a major product on Ag NPs with a highest FE of 73%, which can be ascribed to the weak binding energy between metal Ag and *CO intermediate. In contrast, the moderate binding energy between Cu and *CO facilitates further reduction of *CO and thereby the subsequent generation of C2+ fuels. As shown in Fig. 2a, Cu-bipy exhibited a wider product distribution, although bipy-functionalization slightly reduced the production of C2+ fuels, as compared to pure Cu36 (Supplementary fig. 10). Remarkably, Ag-Cu heterostructure demonstrated a distinctive CO2R behavior from monometallic Ag and Cu-bipy. In terms of C2+ fuels, Ag-Cu heterostructure was more advantageous than Cu-bipy, showing a ~ 250 mV more positive onset potential (Fig. 2a and Supplementary fig. 11). More importantly, the partial current density of C2+ products on Ag-Cu heterostructures were significantly enhanced in relative to primitive Cu-bipy (Fig. 2b), suggesting a synergistic catalytic behavior between Ag and Cu.

a The product distributions of CO2R at different potentials on Ag NPs (top), Cu-bipy (middle) and Ag0.5-Cu0.5 heterostructure (bottom). b The partial current density of C2+ fuels as a function of applied potentials on Cu-bipy, Ag NPs and Ag-Cu heterostructures with variable constitution ratios. c The FEs of C2H4, alcohols and CO as a function of Ag fraction. Error bars represent the standard deviation based on three separate measurements. d–f The corresponding contour maps for FEs of different products: C2H4 (d), alcohols (e) and CO (f) versus Ag fraction and applied potential, the potential is with 100% iR correction.
Notably, we observed the Ag-Cu heterostructure demonstrates a surface-sensitive product distribution. The FE of CO displayed an incremental trend with an increasing surface fraction of Ag, suggesting Ag plays a key role in enriching *CO. The generation of C2+ products got pronounced at approximately −0.85 V vs. RHE on Ag-Cu heterostructure, coinciding with the commencement of CO current lift on pure Ag (Fig. 2a and Supplementary fig. 12). Therefore, it is deducible that the increased local *CO population improved the production efficiency of C2+ fuels on neighboring Cu. Remarkably, the selectivity of C2+ strongly depended on the Ag/Cu constitution ratio. The production of C2H4 was more favorable at a Cu-rich (Ag-deficient) interface. As the fraction of Ag increasing, the dominant product switched from C2H4 to alcohols (e.g., C2H5OH and n-C3H7OH), and finally to CO as Ag occupying the main surface (Fig. 2c). Specifically, Ag0.25-Cu0.75 displayed an optimal C2H4 selectivity with a FE of ~32% at −1.15 V vs. RHE, while Ag0.5-Cu0.5 exhibited a superior selectivity for alcohols, attaining FEs of ~44% (Fig. 2d, e). However, an excess of Ag NPs would probably shield the active sites of Cu-bipy to perform carbon-carbon coupling, thus more *CO was released as CO molecules instead of getting further reduced when the fraction of Ag was higher than 70% (Fig. 2f). To further explore the vital role of local *CO production toward C2+ products enhancement, electrochemical CO reduction was performed under equivalent conditions. We found that such locally produced *CO could not be simply replaced by the CO-supplying reagent because no significant differences occurred in the C2+ selectivity and production rate between Cu-bipy and Ag0.5-Cu0.5 under multiple potentials in direct CO reduction (Supplementary fig. 13). These observations experimentally confirmed the promoted C2+ alcohols production on Ag-Cu biphasic heterostructure follows a tandem CO2R pathway. These observations experimentally demonstrated the importance of locally enriched *CO at the Ag-Cu interface. For the durability evaluation, we tested Ag0.5-Cu0.5 for time-dependent current density and product distribution as shown in Supplementary fig. 14. It was observed that such Ag-Cu heterostructure exhibited a slight decrease of current density with time, and the selectivity of alcohols decreased sharply in the first six hours, accompanied with increased production of C2H4 and hydrogen. Taken together, our electrochemical tests proved that the enhanced C2+ production resulted from tandem catalytic CO2R, wherein the accumulated *CO produced from Ag was crucial for the next-step conversion to other deep-reduced C2+ fuels.
*CO-regulated dynamic restructuring at the Ag-Cu biphasic interface
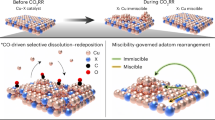
To understand the variable C2+ product distributions on the heterostructure with different constitution ratios, clarifying the interfacial structure is highly desirable. Aberration-corrected STEM was employed to reveal the biphasic interfaces of Ag0.25-Cu0.75, Ag0.33-Cu0.67, and Ag0.5-Cu0.5, respectively, after electrocatalysis at the potential of −1.05 V vs. RHE for one hour. To eliminate the influence from air exposure which might alter the surface states of Cu and Ag, all samples were prepared within an Ar-atmosphere and subsequently transferred to a transmission electron microscope using a vacuum transfer holder (see Experimental Procedures). Notably, all three heterostructures underwent significant restructuring during CO2R, as confirmed from the HAADF-STEM images and corresponding EDS elemental maps. In the case of Ag0.25-Cu0.75 (Fig. 3a and Supplementary fig. 15), Cu NWs exhibited a rough surface geometry, and Cu on the biphasic interface tended to form nanoparticles during electrolysis, separately from Ag NPs as shown from the EDS analysis. In contrast, in the case of Ag0.33-Cu0.67, Cu tended to migrate onto the surface of Ag and formed a thin overlayer (Fig. 3b). Intriguingly, Ag0.5-Cu0.5 manifested a distinctive and thicker porous core-shell nanostructure after electrocatalysis, with a shell thickness of ~2 nm (Fig. 3c). The EDS elemental maps further verified the location of Ag (core) and Cu (shell). Such a structural evolution occurred over the entire surface, and more results collected from other regions were summarized in Supplementary fig. 16. The migration of Cu onto Ag surface has also been observed in a flow cell geometry, where a large current density and higher *CO concentration was produced (Supplementary fig. 17). To unambiguously reveal the restructuring process, the HAADF-STEM and EDS characterization was performed on Ag0.5-Cu0.5 after a series of electrolysis time (Supplementary fig. 18). The enrichment of Cu on Ag surface could already be observed after 0.5 h electrolysis. While the electrolysis time extended to 3 hours, more Cu species migrated onto Ag, resulting in a thicker Cu layer (~4 nm). The growing Cu layer is responsible for the decreased alcohol selectivity with prolonged time (Supplementary fig. 14). As the reaction proceeds, the Cu layer became progressively thicker and fewer Ag-Cu interfaces were exposed, leading to a gradual drop yield of C2+ alcohols. Notably, for all samples, Ag and Cu tended to keep phase separation as the reaction proceeds. The EDS elemental mapping at the planar interface corroborated the lack of atomic interdiffusion between Ag and Cu atoms after electrocatalysis (Fig. 3a–c), excluding the formation of intermetallic alloy. This observation was consistent with the results from both operando and ex-situ XRD measurements, wherein both Ag0.25-Cu0.75 and Ag0.5-Cu0.5 exhibited no peak-shift after CO2R (Supplementary fig. 19,20), confirming the biphasic interface of Ag and Cu. The absence of alloy formation could potentially be attributed to the high immiscibility between Cu and Ag37,38,39. Different from the Au-Cu interface where intermetallic alloy is easily formed38,39,40, Ag-Cu interface exhibits a greater propensity to keep phase separate.

a–c HAADF-STEM images and corresponding EDS elemental maps of Ag0.25-Cu0.75 (a) Ag0.33-Cu0.67 (b) Ag0.5-Cu0.5 (c) after restructuring. Insets show schematics of the reconstructed Ag-Cu interface. d–f XPS spectra of Ag0.25-Cu0.75 (d) Ag0.33-Cu0.67 (e) and Ag0.5-Cu0.5 (f) before and after electrolysis. g A top-view of the concentration and flux distribution of *CO on Ag-Cu heterostructures with different Ag densities via the FEM simulation. h Proposed restructuring mechanisms of Ag-Cu heterostructures with different Ag densities.
What is noteworthy is that, the rich bimetallic interface from restructuring resulted in a discernable charge transfer between Ag and Cu. We compared the X-ray photoelectron spectra (XPS) of Ag0.25-Cu0.75, Ag0.33-Cu0.67, and Ag0.5-Cu0.5 before and after electrolysis (Fig. 3d–f). All three samples exhibited a Cu2+ signal at 935 eV and a Cu+/Cu signal at 932.5 eV. The peaks at 368.2 and 374.2 eV were ascribed to Ag 3d5/2 and 3d3/2, respectively. Notably, it was observed that the binding energies of Ag 3d5/2 and Cu 2p3/2 in Ag0.5-Cu0.5 were shifted by +0.2 eV and −0.17 eV, respectively, after the reaction, which can be attributed to the electron transfer from Cu to Ag41,42. In contrast, XPS analysis of Ag0.25-Cu0.75 and Ag0.33-Cu0.67 showed that the valence states of Cu and Ag remained intact. These results confirmed that, after surface restructuring, Ag0.5-Cu0.5 consequently exhibited more substantial biphasic interfaces between Ag and Cu, resulting in a stronger bimetallic interaction compared to other two counterparts.
We attempt to understand the more severe structural reconstruction of Ag-Cu heterostructure with a higher Ag surface fraction. Through the finite element method (FEM)-simulation, the *CO coverage was positively correlated with the density of Ag NPs on one Cu NW (Fig. 3g), e.g., the accumulated surface concentration of *CO increased from 0.06 to 0.2 mol m-3 as the surface fraction of Ag arising from 25% to 55%. We attributed the stronger restructuring tendency of Ag0.5-Cu0.5 to the higher local *CO coverage under operating conditions, and proposed a *CO-regulated restructuring behavior at the Ag-Cu bimetallic interface as schematically shown in Fig. 3h. A sequential Cu oxidation-reduction process is responsible for the dynamic surface restructuring. The Cu exposed to electrolyte is prone to be oxidized at an open circuit potential43,44, and some Cu atoms are leached to generate Cu ions, as verified by our ICP-OES analysis (Supplementary Table 3). Under a cathodic bias, the electrochemical reduction results in a redeposition of Cu ions from electrolyte, such redeposition, however, is observed prone to occur at different locations depending on the *CO-coverage. The general trend is that, with a higher local *CO concentration (on an Ag-rich surface), Cu ions tend to migrate onto the Ag domains, generating more abundant of Ag-Cu interface. To confirm our hypothesis, the electrochemical reconstruction of Ag0.5-Cu0.5 was investigated in gas mixtures with varying Ar and CO2 ratios (see Experimental Procedure and Supplementary fig. 21). In the absence of *CO, the surface Cu NWs after electrocatalysis even demonstrated a smoother surface than the reconstructed Ag0.25-Cu0.75 under CO2R wherein a low concentration of *CO was produced. The migration of Cu to Ag became more pronounced with an increasing CO2 composition (from Ar: CO2 = 9:1 to 1:1). We rationalized that the selective-deposition behavior of Cu ions stems from the distinct binding energies of *CO on Cu and Ag surfaces, e.g., comparing with Cu, Ag possesses a much lower binding energy toward *CO 22,45. Consequently, under CO2R operating condition, *CO is expected to desorb from Ag, and preferentially adsorbs on Cu, which would prevent the deposition of Cu ions on Cu NWs. We calculated and compared the free energy evolution upon the deposition of dissolved Cu ions on pure Cu and Cu-*CO. It was observed that the deposition on Cu was endothermic, and the degree of endothermicity increased when *CO adsorbed on Cu (Supplementary fig. 22). Therefore, higher *CO coverage would suppress the deposition of Cu ions on Cu NWs, instead, it promotes the migration of Cu to the neighboring clean Ag surface, leading to the formation of Ag-Cu interface. These results can well explain our experimental observations: With a higher local *CO concentration, Cu ions tend to migrate onto Ag NPs, generating more abundant of Ag-Cu interface. As far as we know, this is the first time to experimentally emphasize the critical role of *CO-coverage in the dynamic restructuring of bimetallic interface during CO2 electrolysis. Notably, some recent studies inspired that the adsorbed *CO can induce stronger reconstruction on monometallic Cu, consistent with our observations31,46,47.
Restructuring-steered C2+ product disparities
Next, we rationalize the C2+ product disparities on different reconstructed Ag-Cu heterostructures. Theoretically, we studied the reaction profiles along the hydrogenation of *OCHCH2 intermediate towards either C2H4 + O* or *OCH2CH3, a step suggested critical to control the differential production of C2H4 versus C2H5OH (see Fig. 4a, b, Supplementary fig. 23,24 and Supplementary Data 1)48,49,50. On the surface of pure Cu(111), the O-C bond in *OCHCH2 would more easily break, giving rise to the product C2H4 and adsorbed *O, which would be further hydrogenated to H2O (see the red path in Fig. 4a). Interestingly, at the Ag-Cu interface, the cleavage of O-C bond was prohibited because of an increased free energy evolution, ∆G ( ~ 0.27 eV), whereas a decreased ∆G (−0.08 eV) was observed on the competition hydrogenation pathway (see the blue path in Fig. 4b). Therefore, *OCHCH2 would prefer to be hydrogenated at the Ag-Cu interface and sequentially generate *OCHCH3, *OCH2CH3 and C2H5OH. These results are consistent with our electrochemical experimental observations: C2H4 is more favored on a Cu-rich surface without sever reconstruction, while the production of C2H5OH will be promoted at the reconstructed Ag-Cu interface. Beyond the structure of electrocatalyst, our DFT calculation showed that the elevated local *CO concentration at the Ag-Cu interface would further facilitate the hydrogenation instead of dissociation of *OCHCH2, and improve C2H5OH production. As shown in Fig. 4c, starting from *OCHCH2, the free energy for *OCHCH3 formation was reduced by 0.23 eV when the coverage of *CO increased from 0 to 2/9 monolayer (ML), whereas the free energy for the O-C bond cleavage was not observed much change (see more illustrations in Supplementary fig. 25,28).

a, b Reaction profile for C2H4 path and C2H5OH path on Cu(111) (a) and Ag-Cu interface (b). Insets show the schematic configurations of key intermediates, where the brown, sliver, gray, white and red circles represent for Cu, Ag, C, H and O atoms, respectively. c The free energy evolution of *OCHCH2 intermediate towards either C2H4 + O* (O-C bond cleavage) or *OCHCH3 (further hydrogenation) on Cu(111) with an increasing *CO coverage from 0 to 2/9 ML. d For *OCHCH2, the difference of ∆G between O-C bond cleavage and further hydrogenation, denoted as ∆∆G, as a function of the oxygen binding energy ∆G(O) on pure Cu and Cu-Ag interface with different *CO coverages. e The schematic illustration of the electrocatalytic pathway for C2H4 (left) and C2H5OH (right). f, g Operando ATR-SEIRAS spectra of Ag0.25-Cu0.75 (f) and Ag0.5-Cu0.5 (g) at OCP and variable cathodic potentials (V vs. RHE) in a CO2-saturated 0.1 M KHCO3.
For *OCHCH2, the difference of ∆G between O-C bond cleavage and further hydrogenation (*OCHCH3 formation), denoted as ∆∆G, can be implemented to represent the differential selection of C2H4 versus C2H5OH (see Experimental Procedures). C2H5OH becomes dominated when ∆∆G is greater than 0 eV, otherwise C2H4 is the major C2 product. Remarkably, ∆∆G was observed strongly correlated with the oxygen binding energy ∆G(O) (Fig. 4d). Interfacing Cu with Ag and increasing the *CO coverage would both decrease ∆G(O), leading to a positive ∆∆G and thus improving the production of C2H5OH. On the contrary, pure Cu with a low *CO coverage would result in a negative ∆∆G and subsequently C2H4 formation. These results from theoretical calculations further assist to understand the electrochemical performance of Ag-Cu bimetallic heterostructure with different Ag/Cu constitution ratios. Ag0.5-Cu0.5 electrochemically reconstructed to a large population of Ag-Cu interface, having a large *CO coverage and showing a weaker affinity to oxygen, which strongly prohibited the O-C bond cleavage of *OCHCH2. Therefore, *OCHCH2 kept being hydrogenated under this situation and finally generated C2H5OH. In contrast, Ag0.25-Cu0.75 with deficient Ag-Cu interface during electrocatalysis showed a lower *CO coverage and a stronger oxygen affinity, which facilitated the O-C bond cleavage of *OCHCH2 and thus the formation of C2H4. This mechanism was schematically shown in Fig. 4e.
Operando attenuated total reflectance surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) was performed on Ag0.25-Cu0.75 and Ag0.5-Cu0.5 across varying CO2R potentials to confirm the different intermediate adsorption kinetics, as shown in Fig. 4f–g. The signals at ~1650 ~ 1420 and ~1480 cm−1 were ascribed to surface-accumulated H2O, CO32- and HCO3-, respectively51,52,53,54. The *COOH intermediate responsible for CO production could be identified at ~1265 cm−1 55. Some other reactive oxygen intermediates were also observed. For instance, the peaks emerging at ~1160 cm−1 was attributed to the *C-O-H stretching of the *OCCOH intermediate52,54,56. The signal at ~1060 cm−1 corresponded to the *OCCHO intermediate adsorbed on catalysts54. Intriguingly, we detected a unique signal corresponding to *OCH2CH3 ( ~ 1330 cm−1)57,58 on the surface of Ag0.5-Cu0.5 rather than Ag0.25-Cu0.75, which is a key intermediate along the production way of C2H5OH, consistent with our electrochemical performance and theoretical calculation that Ag0.5-Cu0.5 is more selective for C2H5OH. Collectively, our observations demonstrate that the evolution of the intrinsic intermediate adsorption kinetics from electrochemical restructuring is an important cause for the C2+ selectivity disparities at the bimetallic Ag-Cu interface.
In summary, we demonstrated an exemplary Ag-Cu biphasic heterostructure for tandem electrochemical CO2R, and correlated the variable C2+ preferences with the distinguished bimetallic restructuring behaviors and their interplay with the inherent intermediate adsorption energetics. The evolving structure and enriched *CO intermediate strongly altered the oxyphilic characteristic at the interface, which profoundly determined the hydrogenation energetics of CO2, leading to the disparate C2+ product distributions. This work establishes the empirical demonstration of dynamic structural reconstruction in the bimetallic model system, and sets up a paradigm to understanding the tandem CO2 electroreduction selectivity from an atomic level.
METHODs
Materials and chemicals
Oleylamine (OAm, > 70%), copper(I) chloride (CuCl, 98%), 4, 4’-bipyridine (C10H8N2, 99%) were purchased from Sigma Aldrich. Silver(I) nitrate (AgNO3, 99.995%) was purchased from Admas-beta. Isopropanol and hexanes were purchased from Sinopharm Chemical Reagent Co., Ltd. Acetone and chloroform were purchased from Shanghai Dahe Chemicals Co., Ltd. Nafion solution was purchased from Sigma-Aldrich Co., Ltd. Nafion N-117 membrane was provided by DuPont Co., Ltd. Carbon paper (P75T) was purchased from Avcarb Co., Ltd. Carbon support (Ketjen black, EC-300J) was purchased from LION. Potassium bicarbonate (KHCO3) was purchased from Macklin Co., Ltd. All aqueous solutions were prepared by Milli-Q water (18.2 MΩ cm). High-purity CO2 (99.99%), N2 (99.999%) and Ar (99.999%) were provided by Shanghai Tomoe gases Co., Ltd.
Synthesis of Cu NWs
0.2 g of CuCl was dissolved in 6 mL of OAm at 25 °C under a N2 flow and vigorous magnetic stirring for 15 min. The solution was heated at 100 °C for 30 min to degas, and subsequently at 200 °C for 90 min. After cooling down to room temperature, 15 mL of hexane was added to collect Cu NWs via centrifugation (4000 rpm, 5 min). The product was purified by adding 15 mL of hexane and centrifugation for 3 times. The obtained Cu NWs were re-dispersed in hexane.
Synthesis of Ag NPs
0.17 g of AgNO3 was dissolved in 20 mL of OAm at 60 °C under a N2 flow. The solution was then heated to 180 °C for 1 h and cooled down naturally to the room temperature. The obtained sample was washed with 40 mL of acetone via centrifugation (6000 rpm,10 min) and redispersed in 10 mL of hexane, wherein the acetone was added dropwise until the colloid became turbid. The Ag NPs were then precipitated by centrifugation (3000 rpm, 5 min) and redispersed in hexane; the process was carried out twice.
Assembly of Ag-Cu bimetallic heterostructure
Ag NPs were assembled onto Cu NWs using 4, 4’-bipyridine (bipy) as a linker, as inspiring from previous reports35. 20 mg of as-synthesized Cu NWs were added in 20 mL chloroform solution of 100 mM bipy. The mixture solution was then stirred for 12 h at room temperature to exchange the surfactant of Cu NWs. The obtained bipyridine functionalized Cu NWs (Cu-bipy) were washed with chloroform twice via centrifugation (1500 rpm, 3 min). 10 mg of Ag NPs was dispersed in 10 mL hexane, and mixed with 10 mL of chloroform dispersed Cu -bipy. The mixture was stirred for 6 h at room temperature and then washed with 20 mL of chloroform for 3 times, and re-dispersed in chloroform finally.
TEM samples preparation and characterization
We conducted the aberration-corrected scanning transmission electron microscopy (AC-STEM) characterization using a ThermoFisher Themis Z microscope, which featured two aberration correctors and operated at 300 kV. The HAADF-STEM images were collected with a convergence semi-angle of 11 mrad, along with inner and outer collection angles set at 59 mrad and 200 mrad, respectively. We performed the EDS analysis utilizing four in-column Super-X detectors. Prior to analysis, all samples were meticulously prepared by dispersing the catalyst in ethanol and subsequently deposited onto molybdenum grids backed with carbon films within a glove box. To avoid any alteration in the valence states of Cu and Ag due to air exposure, a vacuum transfer holder (Model 2550, Fischione Instruments, USA) was employed.
Other physical characterizations
Powder XRD measurement was carried out on a Bruker D2 PHASER diffractometer with Cu Kα radiation (λ = 1.5406 Å). IR spectroscopy was performed on Nicolet iS10. TEM was carried out on HT7800. ICP-OES was performed on a Thermo Scientific iCAP 7400. XPS measurements were acquired by a PHI 5000 C spectrometer with a Mg Kα X-ray source (1253.6 eV), with the pressure inside the chamber maintained below 4 × 10−9 Torr and spectra were collected at a pass energy of 17.9 eV. The highest peak in C 1 s spectra was shifted to 284.8 eV for charge correction.
Operando XRD characterization
The operando XRD measurement was performed at the BL14B (18 eV) beamline in Shanghai Synchrotron Radiation Facility (SSRF). The wavelength of X-rays was calibrated to 0.6887 Å by using the LaB6 standard from NIST (660b), and the normal XRD patterns were performed on a Bruker D8 Advance with Cu Kα radiation (1.54 Å). The XRD measurements were performed using a surface detector, a home-customized electrochemical cell, and a computer-controlled Gamry Interface 1010 potentiostat. The electrochemical cell was equipped with an optical window in the center of the cell, which allows the penetration of X-ray. Pt and Ag/AgCl electrode (CHI) were used as the counter and reference electrode, respectively. A 0.1 M KHCO3 solution was used as the electrolyte (pH =6.8), which was purged with CO2 at a flow rate of 5 sccm for 15 min before and during each measurement while stirring.
Operando XAS characterization
The XAS data was recorded at the BL11B beamline of Shanghai Synchrotron Radiation Facility (SSRF) in the fluorescence mode, with a Gamry Interface 1010 potentiostat. Platinum served as the counter electrode, and an Ag/AgCl (CHI) electrode was used as the reference. We used a 0.1 M KHCO3 electrolyte (pH=6.8), which was continuously purged with CO2 at 5 sccm before and during each measurement. Monochromatization of the X-ray beam was achieved with a Si (111) double-crystal monochromator, and higher harmonic suppression was managed using Rh-coated mirrors. Photon energy calibration referenced the first inflection of the Cu K-edge at 8980 eV. The data processing of XANES and EXAFS spectra utilized the ATHENA and ARTHEMIS programs from the IFEFFIT software suite.
Working electrode preparation
10 mg of catalyst material and 100 μL of 5 wt% Nafion solution were mixed and dispersed in 2.5 mL of isopropanol with 1 h sonication to obtain a well-mixed ink. Afterward, 67 μL of ink was drop-casted onto a carbon paper with a 0.5 cm2 geometric area and was dried overnight at the room temperature.
Electrochemical CO2R measurement and product analysis
The catalytic performance of electrocatalysts was initially assessed in a two-compartment H-cell, separated by a Nafion 117 proton exchange membrane. The Nafion membrane underwent an immersion treatment in a 5% hydrogen peroxide solution at 80 °C for an hour, followed by rinsing in deionized water for 30 minutes, then treatment in 5% sulfuric acid (by mass) at 80 °C for another hour, and a final deionized water rinse for 30 minutes. Each compartment held 6 mL of 0.1 M KHCO3 solution, and the compartment with the working electrode was sealed to ensure the accurate gas product analysis. No special storage conditions were needed for the electrolyte beyond room temperature. CO2 was introduced at 5 sccm for 20 minutes using a mass flow controller, both before and during measurements, with constant stirring; flow rate verification was conducted using a bubble flowmeter at the cathode chamber’s outlet. Ag/AgCl (saturated KCl, Gaoss Union, Inc.) and Pt mesh served as the reference and counter electrodes, respectively. Potentials were converted to the RHE scale using the equation:
where pH represents the electrolyte pH, ~6.8 ± 0.05 in a CO₂-saturated solution. Potentials include a full iR compensation, with the uncompensated resistance (RΩ) determined by extrapolating high-frequency impedance data, averaging around 10 ± 0.5 Ω in 0.1 M KHCO3. The calibration of Ag/AgCl reference was conducted in a H₂-saturated 0.1 M KHCO3 with polished Pt electrodes as both working and counter electrodes.
Electrochemical experiments were carried out using a CHI 760E potentiostat. Pre-electrolysis linear sweep voltammetry was performed at 10 mV/s before applying constant potential electrolysis (CA) for one hour. Gas products were analyzed every 20 minutes via gas chromatography (Agilent 7890B, HP-PLOT and MS-5A columns) with TCD for H₂ and FID for CO, methane, and ethylene. Liquid-phase products were subsequently analyzed by 400 MHz NMR (AVANCE III HD), using dimethyl sulfoxide and phenol as internal standards, with solvent pre-saturation applied to minimize water interference.
Faradaic efficiency (FE) and production rates were calculated accordingly:
Calculation of the FE of gas products:
Calculation of the FE of liquid products:
FE: faraday efficiency, %
N: number of transferred electrons for certain product
F: faradaic constant 96485, C·mol−1
n: amount of substance of target product, mol
V: volume ratio of gas product, determined by on-line GC
v: flow rate of CO2, mL·min−1
I: steady-state cell current, mA
t: electrolysis time, s
S: integral area of liquid product after 1 h of electrolysis, determined by NMR
Hnum: the number of H of the target product at a certain chemical shift
VDMSO: volume of DMSO in a nuclear magnetic tube, mL (CDMSO = 10 mM)
Vi: volume of the electrolyte in a nuclear magnetic tube, mL
Vtot: volume of the electrolyte in the working cell, mL
Electrochemical measurement in a gas mixtures having variable CO2 and Ar fractions
The test methodology was identical to the above CO2R measurement except changing the feeding gas supply. The gas flow of CO2 and Ar was controlled by the mass flow controller and well mixed before entering the electrochemical cell. We applied a potential of −1.05 V vs. RHE (this potential is optimal for alcohols production) to the catalyst Ag0.5-Cu0.5 for one hour.
Finite-element simulation
The finite-element simulations were performed using the COMSOL Multiphysics. The chemical mass transport model for Ag-Cu heterostructures was developed using three modules. First, “Chemistry” module was used to defined the CO2R step. The generation of CO intermediate from CO2 on the surfaces of Cu and Ag was calculated and defined as feedstock for ethanol generation from Cu substrates. Next, the “Dilute Species Transport” module was used to solve for the mass transport of the three species. We determined the equilibrium constants and rate constants of the chemical reactions by scanning these parameters over a large range (2 orders of magnitude) and fitting these electrochemical CO2R data. We swept six parameters: Keq CO2, Keq CO, and Keq ethanol equilibrium constants for the adsorption and desorption of three species at the surface, as well as kf1 and kf2 for CO2 to CO on Ag and Cu surface and kf3 for CO to ethanol on Cu surface. Finally, the “Surface Reaction” module defines the surface on which the catalytic reaction takes place.
Operando ATR-SEIRAS experiments
Operando ATR-SEIRAS experiments were performed in a custom spectroelectrochemical cell using a Nicolet 6700 spectrometer, equipped with a liquid nitrogen-cooled mercury cadmium telluride detector and an ATR accessory. A thin Au film was chemically deposited onto the surface of a silicon prism to prepare the substrate. Catalyst inks were then applied to the Au film and air-dried to form the working electrode. The reference electrode was Ag/AgCl, and the counter electrode was a graphite rod. The electrolyte used was 0.1 M KHCO3 with continuous CO2 bubbling. Electrochemical tests were conducted using a Gamry Interface 1010 potentiostat. Spectra were presented in absorbance units, collected at a resolution of 4 cm⁻¹ over 64 scans per spectrum.
DFT calculation
Spin-polarized density functional theory (DFT) calculations were conducted on Vienna Ab-initio Simulation Package (VASP)59,60 for total energies of all considered structures. The projector augmented-wave (PAW) method61,62 and the revised Perdew, Burke, and Ernzerhof (rPBE) generalized gradient approximant (GGA) functional63,64 were employed. The kinetic energy for plane waves smaller than 400 eV and 600 eV were included to describe the surface and the bulk optimization, respectively. The dispersion interaction was described by the DFT-D365. All the calculations reach the convergence criteria when the Hellmann-Feynman forces on each relaxed ion are less than 0.05 eV/Å and the energy difference between two steps are smaller than 10−5 eV.
The Cu and Ag unit cells were optimized with Monkhorst-Pack k-points mesh of (15 × 15 × 15). The 4 layers (3 × 3) Cu(111) surface was cleaved from the optimized cell, where the bottom two layers were frozen to simulate the behaviors of bulk phase. The (4 × 1 × 3) Ag and (4 × 1 × 3) Cu (111) were combined for cell optimization with k-points mesh (2 × 14 × 6). The 4 layers (8 × 4) Ag-Cu (111) interface surface was cleaved, where the bottom two layers were frozen to simulate the behaviors of bulk phase.
The free adsorption energy of each intermediate was calculated using VASPKIT66 by
where E is DFT total energy, and Ezpe is the zero-point energy with referenced to H2(g), CO2(g) and H2O(l), along with the temperature T and the entropy S. In order to evaluate the electrochemical activities of electrodes, the computational hydrogen electrode model was applied at 0 V. In this model, the reference electrode is
which occurs at 0 V with respect to reversible hydrogen electrode (RHE).
The difference of ∆G between O-C bond cleavage, ∆∆G, is defined as
Data availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Source data are provided with this paper.
References
Varela, A. S. et al. Electrochemical reduction of CO2 on metal-nitrogen-doped carbon catalysts. ACS Catal. 9, 7270–7284 (2019).
Bushuyev, O. S. et al. What should we make with CO2 and how can we make it? Joule 2, 825–832 (2018).
Hepburn, C. et al. The technological and economic prospects for CO2 utilization and removal. Nature 575, 87–97 (2019).
Ross, M. B. et al. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2, 648–658 (2019).
Arán-Ais, R. M., Gao, D. & Roldan Cuenya, B. Structure- and electrolyte-sensitivity in CO2 electroreduction. Acc. Chem. Res. 51, 2906–2917 (2018).
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
Zhu, C. et al. Product-specific active site motifs of Cu for electrochemical CO2 reduction. Chem. 7, 406–420 (2021).
Kuhl, K. P., Cave, E. R., Abram, D. N. & Jaramillo, T. F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 5, 7050–7059 (2012).
Garza, A. J., Bell, A. T. & Head-Gordon, M. Mechanism of CO2 reduction at copper surfaces: pathways to C2 products. ACS Catal. 8, 1490–1499 (2018).
Ma, M., Djanashvili, K. & Smith, W. A. Controllable hydrocarbon formation from the electrochemical reduction of CO2 over Cu nanowire arrays. Angew. Chem. Int. Ed. 55, 6680–6684 (2016).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Zhou, Y. et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 10, 974–980 (2018).
Lee, C. W. et al. Defining a materials database for the design of copper binary alloy catalysts for electrochemical CO2 conversion. Adv. Mater. 30, 1704717 (2018).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Chen, C. et al. Cu-Ag tandem catalysts for high-rate CO2 electrolysis toward multicarbons. Joule 4, 1688–1699 (2020).
Lum, Y. & Ager, J. W. Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu. Energy Environ. Sci. 11, 2935–2944 (2018).
Gu, J. et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 5, 268–276 (2022).
Ren, D., Ang, B. S.-H. & Yeo, B. S. Tuning the selectivity of carbon dioxide electroreduction toward ethanol on oxide-derived CuxZn catalysts. ACS Catal. 6, 8239–8247 (2016).
Zhu, C., Zhao, S., Shi, G. & Zhang, L. Structure-function correlation and dynamic restructuring of Cu for highly efficient electrochemical CO2 conversion. ChemSusChem 15, e202200068 (2022).
Huang, J., Mensi, M., Oveisi, E., Mantella, V. & Buonsanti, R. Structural sensitivities in bimetallic catalysts for electrochemical CO2 reduction revealed by Ag-Cu nanodimers. J. Am. Chem. Soc. 141, 2490–2499 (2019).
Vasileff, A., Xu, C., Jiao, Y., Zheng, Y. & Qiao, S. Z. Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction. Chem. 4, 1809–1831 (2018).
Kim, J.-Y. et al. Selective hydrocarbon or oxygenate production in CO2 electroreduction over metallurgical alloy catalysts. Nat. Synth. 3, 452–465 (2024).
Du, C. et al. Cascade electrocatalysis via AgCu single-atom alloy and Ag nanoparticles in CO2 electroreduction toward multicarbon products. Nat. Commun. 14, 6142 (2023).
Chang, C. J. et al. Quantitatively unraveling the redox shuttle of spontaneous oxidation/electroreduction of CuOx on silver nanowires using in situ X-ray absorption spectroscopy. ACS Cent. Sci. 5, 1998–2009 (2019).
Wei, D. et al. Decrypting the controlled product selectivity over Ag-Cu bimetallic surface alloys for electrochemical CO2 reduction. Angew. Chem. Int. Ed. 62, e202217369 (2023).
Chang, C. J. et al. Dynamic reoxidation/reduction-driven atomic interdiffusion for highly selective CO2 reduction toward methane. J. Am. Chem. Soc. 142, 12119–12132 (2020).
Clark, E. L., Hahn, C., Jaramillo, T. F. & Bell, A. T. Electrochemical CO2 reduction over compressively strained CuAg surface alloys with enhanced multi-carbon oxygenate selectivity. J. Am. Chem. Soc. 139, 15848–15857 (2017).
Eren, B. et al. Activation of Cu(111) surface by decomposition into nanoclusters driven by CO adsorption. Science 351, 475–478 (2016).
Eren, B. et al. One-dimensional nanoclustering of the Cu(100) surface under CO gas in the mbar pressure range. Surf. Sci. 651, 210–214 (2016).
Huang, J. et al. Potential-induced nanoclustering of metallic catalysts during electrochemical CO2 reduction. Nat. Commun. 9, 3117 (2018).
Whiteford, J. A., Lu, C. V. & Stang, P. J. Molecular architecture via coordination: self-assembly, characterization, and host−guest chemistry of mixed, neutral-charged, pt−pt and pt−pd macrocyclic tetranuclear complexes. X-ray crystal structure of cyclobis[[cis- Pt(dppp)(4-ethynylpyridine)2][cis-Pd2+(PEt3)22-OSO2CF3. J. Am. Chem. Soc. 119, 2524–2533 (1997).
Sun, D. et al. Encapsulated diverse water aggregates in two Ag(I)/4,4′-bipyridine/dicarboxylate hosts: 1D water tape and chain. Cryst. Growth Des. 10, 4642–4649 (2010).
Sun, J.-K. & Zhang, J. Functional metal–bipyridinium frameworks: self-assembly and applications. Dalton Trans. 44, 19041–19055 (2015).
Fu, J. et al. Bipyridine-assisted assembly of Au nanoparticles on Cu nanowires to enhance the electrochemical reduction of CO2. Angew. Chem. Int. Ed. 58, 14100–14103 (2019).
Huang, Y., Handoko, A. D., Hirunsit, P. & Yeo, B. S. Electrochemical reduction of CO2 using copper single-crystal surfaces: effects of CO* coverage on the selective formation of ethylene. ACS Catal. 7, 1749–1756 (2017).
Valodkar, M., Modi, S., Pal, A. & Thakore, S. Synthesis and anti-bacterial activity of Cu, Ag and Cu-Ag alloy nanoparticles: A green approach. Mater. Res. Bull. 46, 384–389 (2011).
Yang, C. et al. Overcoming immiscibility toward bimetallic catalyst library. Sci. Adv. 6, eaaz6844 (2020).
Rapallo, A. et al. Global optimization of bimetallic cluster structures. I. Size-mismatched Ag-Cu, Ag-Ni, and Au-Cu systems. J. Chem. Phys. 122, 194308 (2005).
Zhu, C. et al. Dynamic restructuring of epitaxial Au-Cu biphasic interface for tandem CO2-to-C2+ alcohols conversion. Chem. 8, 3288–3301 (2022).
Lv, X. et al. Electron-deficient Cu sites on Cu3Ag1 catalyst promoting CO2 electroreduction to alcohols. Adv. Energy Mater. 10, 2001987 (2020).
Zhou, B. et al. Highly efficient binary copper-iron catalyst for photoelectrochemical carbon dioxide reduction toward methane. Proc. Natl Acad. Sci. Usa. 117, 1330–1338 (2020).
Ren, D. et al. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper(i) oxide catalysts. ACS Catal. 5, 2814–2821 (2015).
Lee, S. H. et al. Oxidation state and surface reconstruction of Cu under CO2 reduction conditions from in situ X-ray characterization. J. Am. Chem. Soc. 143, 588–592 (2021).
Shi, C., Hansen, H. A., Lausche, A. C. & Nørskov, J. K. Trends in electrochemical CO2 reduction activity for open and close-packed metal surfaces. Phys. Chem. Chem. Phys. 16, 4720–4727 (2014).
Amirbeigiarab, R. et al. Atomic-scale surface restructuring of copper electrodes under CO2 electroreduction conditions. Nat. Catal. 6, 837–846 (2023).
Zhang, W., Yang, Y., Tang, Y. & Gao, Q. In-situ reconstruction of catalysts in cathodic electrocatalysis: New insights into active-site structures and working mechanisms. J. Energy Chem. 70, 414–436 (2022).
Zhi, X., Vasileff, A., Zheng, Y., Jiao, Y. & Qiao, S. Z. Role of oxygen-bound reaction intermediates in selective electrochemical CO2 reduction. Energy Environ. Sci. 14, 3912–3930 (2021).
Zhuang, T. T. et al. Steering post-C-C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat. Catal. 1, 421–428 (2018).
Santatiwongchai, J., Faungnawakij, K. & Hirunsit, P. Comprehensive mechanism of CO2 electroreduction toward ethylene and ethanol: the solvent effect from explicit water-Cu(100) interface models. ACS Catal. 11, 9688–9701 (2021).
Firet, N. J. & Smith, W. A. Probing the reaction mechanism of CO2 electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 7, 606–612 (2017).
Pérez-Gallent, E., Figueiredo, M. C., Calle-Vallejo, F. & Koper, M. T. M. Spectroscopic observation of a hydrogenated CO dimer intermediate during CO reduction on Cu(100) electrodes. Angew. Chem. Int. Ed. 56, 3621–3624 (2017).
Kim, Y. et al. Time-resolved observation of C-C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction. Energy Environ. Sci. 13, 4301–4311 (2020).
Chernyshova, I. V., Somasundaran, P. & Ponnurangam, S. On the origin of the elusive first intermediate of CO2 electroreduction. Proc. Natl Acad. Sci. Usa. 115, 9261–9270 (2018).
Zhu, S., Li, T., Cai, W. B. & Shao, M. CO2 electrochemical reduction as probed through infrared spectroscopy. ACS Energy Lett. 4, 682–689 (2019).
Zhou, X. et al. Stabilizing Cu2+ ions by solid solutions to promote CO2 electroreduction to methane. J. Am. Chem. Soc. 144, 2079–2084 (2022).
Wang, P. et al. Boosting electrocatalytic CO2-to-ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022).
Katayama, Y. et al. An in-situ surface-enhanced infrared absorption spectroscopy study of electrochemical CO2 reduction: selectivity dependence on surface C-bound and O-bound reaction intermediates. J. Phys. Chem. C. 123, 5951–5963 (2019).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Wang, V., Xu, N., Liu, J. C., Tang, G. & Geng, W. T. VASPKIT: A user-friendly interface facilitating high-Throughput computing and analysis using VASP code. Comput. Phys. Comm. 267, 108033 (2021).
Acknowledgements
This work is funded by the National Key R&D Program of China (2022YFA1505200), Natural Science Foundation of China (Grants 22072030, 22272029, and 22376062), Science and Technology Commission of Shanghai Municipality (Grant 22520711100, 22ZR1415700, and 23ZR1406900), the Fundamental Research Funds for the Central Universities (20720220008), Shanghai Rising-star Program (20QA1402400), Academic Research Fund Tier 1 (No. RG5/22), Academic Research Fund Tier 2 (MOE-T2EP10220-0005), Academic Research Fund Tier 2 (MOE-T2EP20221-0004) and the computing resources from National Supercomputing Center Singapore (NSCC). Additional support was provided by the Frontiers Science Center for Materiobiology and Dynamic Chemistry and the Feringa Nobel Prize Scientist Joint Research Center at East China University of Science and Technology.
Author information
Authors and Affiliations
Contributions
L.Z. designed and conceived the experiment. X.G., G.S., Q.X., Y.Y., Y.S., M.C., C.Z., T.L., Y.W., and G.D. fabricated the electrodes and performed the electrochemical characterization and/or data analysis. Y.J. and S.D. carried out the TEM characterizations. J.L. and S.L. contributed to the DFT calculations. C.Y. helped with the FEM modeling and data fitting. X.G., J.L., and L.Z. wrote the manuscript. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jiangzhou Xie, Bijandra Kumar and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, X., Jiang, Y., Liu, J. et al. Intermediate-regulated dynamic restructuring at Ag-Cu biphasic interface enables selective CO2 electroreduction to C2+ fuels. Nat Commun 15, 10331 (2024). https://doi.org/10.1038/s41467-024-54630-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54630-2
